

With a combined prevalence of approximately 20%, mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2 are among the most recurrent genetic abnormalities in newly diagnosed acute myeloid leukemia (AML).1 The high frequency of IDH mutations and the fact that IDH mutations induce a gain in enzymatic activity have led to the development of targeted inhibitors of mutated IDH proteins. These efforts have shown to be successful with the development of several, and clinical approval of 2, small molecules in a matter of years.2,3 In addition to dedicated IDH inhibitors, other modes of targeted AML therapy may be effective in eradicating IDH-mutated AML. Ongoing research is directed toward pinpointing optimal combination strategies of IDH inhibitors with other modalities and toward dissecting factors that determine primary or secondary resistance. In this perspective article, an update on targeting IDH-mutated AML is provided.
IDH1 and IDH2 mutations in AML
IDH1 and IDH2 encode enzymes with functions in cellular metabolism. They both catalyze the oxidative decarboxylation of isocitrate to alpha-ketoglutarate (α-KG), but have distinctive physiological roles and a different cellular localization (IDH1 is cytoplasmic while IDH2 is mitochondrial).4 AML-associated IDH mutations affect specific arginine residues (IDH1 R132 and IDH2 R140 or R172), are typically heterozygous, and are somatically acquired. IDH1 and IDH2 mutations are mutually exclusive in most cases. IDH mutations are thought to be early events in leukemogenesis that tend to be stable, presenting again at the time of possible relapse.1 They arise predominantly in the context of a normal karyotype. Frequently co-mutated genes include NPM1, DNMT3A, SRSF2, and FLT3.1,5
Mutant IDH proteins acquire neomorphic enzyme activity through which they reduce α-KG to R-2-hydroxyglutarate (R-2-HG, further abbreviated as 2-HG), which acts as a competitive inhibitor of α-KG-dependent enzymes, including tet methylcytosine dioxygenase 2 (TET2) and the jumonji domain containing family of histone lysine demethylases.6–8 Consequently, IDH-mutated AML is characterized by a genome-wide increase in DNA hypermethylation and a block of myeloid differentiation (Figure 1).9 While epigenetic dysregulation induced by 2-HG has been relatively well-studied, the oncogenic consequences of mutated IDH proteins may be more diverse and several additional mechanisms have been proposed, many of which are also linked to 2-HG.4,10 Biological insights do not only come from AML, but also from solid tumors, as IDH mutations can be found in a variety of other cancer types including glioma, chondrosarcoma, and cholangiocarcinoma.10,11
Figure 1.
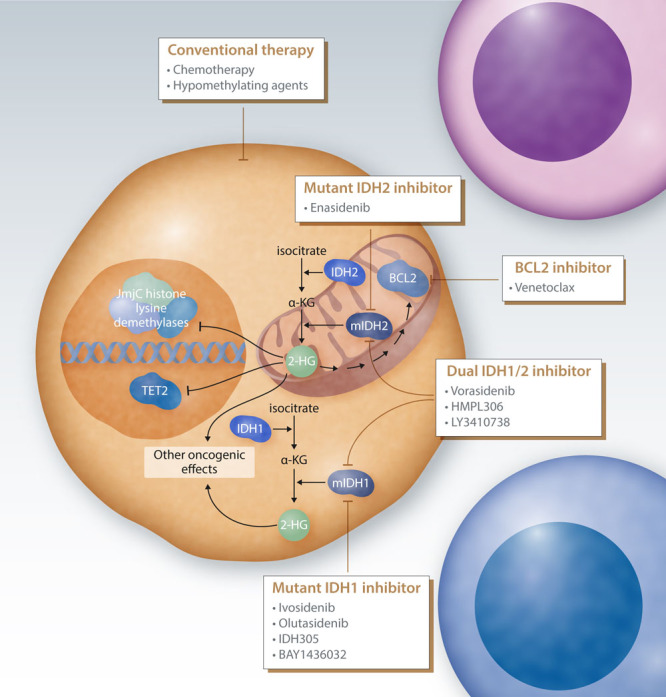
Strategies to target IDH mutated AML. Mutant IDH1 (mIDH1) and mutant IDH2 (mIDH2) proteins catalyze the conversion of α-KG to R-2-HG (2-HG), which mediates much of their oncogenic potential. Important effects of 2-HG include the inhibition of α-KG-dependent enzymes including epigenetic regulators such as tet methylcytosine dioxygenase 2 (TET2) and jumonji domain containing histone lysine demethylases. In addition, mIDH1/mIDH2 may have other oncogenic effects that may also be driven by 2-HG. Mutant IDH proteins can be specifically targeted by small-molecule compounds. IDH1 inhibitors that are or have been in clinical development include ivosidenib, olutasidenib, IDH305, and BAY1436032. Enasidenib is a specific inhibitor of mutant IDH2, while several compounds have been developed that target both mutant IDH1 and IDH2 (vorasidenib, HMPL306, and LY3410738). 2-HG indirectly induces an increased dependency on the anti-apoptotic protein BCL2, which can be therapeutically leveraged by targeting with the BCL2 inhibitor venetoclax. In addition to those novel targeted approaches, IDH1/2-mutated AML may be treated by conventional therapy, including intensive chemotherapy and hypomethylating agents. 2-HG = 2-hydroxyglutarate; AML = acute myeloid leukemia; IDH = isocitrate dehydrogenase.
IDH-mutated AML and response to conventional therapy
While randomized studies with IDH-specific therapy in the frontline setting are still ongoing, recommended first-line therapy for fit patients with either IDH1 or IDH2 mutations is intensive induction therapy, typically cytarabine and anthracycline-based, followed by postremission therapy. The prognostic relevance of IDH mutations in this context is controversial, which is due to inconsistent findings in various patient series. An emerging picture from meta-analyses and larger patient series is that there may be a context-dependent prognostic relevance of IDH mutations.5,12 The risk may vary between IDH1 and IDH2 and between the various mutation hot spots, and is most likely dependent on the cytogenetic and co-mutational situation. It has been suggested that IDH2 R172 may define a specific subtype with its own co-mutational spectrum, higher levels of 2-HG, and a possibly favorable outcome.1,5 A single-center retrospective study reported on IDH-mutated AML to exhibit specific cardiac toxicity through 2-HG–dependent effects on cardiomyocytes when exposed to intensive chemotherapy, highlighting a possibly increased risk of treatment-related morbidity and mortality.13 Overall, given the inconsistencies between studies, larger patient series will be needed to conclusively address the prognostic impact of IDH1/2 mutations.
It has been hypothesized that the profound DNA hypermethylation of IDH-mutated AML could render these leukemias more sensitive to hypomethylating agents (HMAs). Retrospective studies cannot uniformly confirm this.14 On the one hand, this may be explained by the heterogeneous mode of action of HMAs, and on the other hand, the fact that IDH mutations also have methylation-independent effects.
Targeting IDH mutations by small molecule IDH inhibitors
IDH inhibitors selectively inhibit mutant IDH proteins and block the aberrant production of 2-HG. Consequently, exposure of AML cells to IDH inhibitors induces myeloid differentiation ex vivo, in vivo, and in human patients with IDH-mutated AML (Figure 1).15–17
Several small molecule oral IDH inhibitors have either been developed or are in clinical development. These include the IDH1 inhibitors ivosidenib, olutasidenib, IDH305, and BAY1436032; the IDH2 inhibitor enasidenib; and the dual IDH1/2 inhibitors vorasidenib (AG-881), HMPL-306, and LY3410738 (Table 1). Most advanced results have been obtained with ivosidenib and enasidenib, which will be discussed in more detail below. Results of a phase 1 trial for BAY1436032 were published and showed only modest activity of this compound in AML, which led to the decision to stop further clinical development.21 Although interim results of trials involving other IDH inhibitors have in some cases been presented at scientific meetings, final results for those studies have not yet been published at this time and are not discussed here.
Table 1.
Clinical Development of Targeted IDH Inhibitors in Adult Patients With AML
| Drug | Target | ClinicalTrials.gov Identifier | Phase | Description | Ref. |
|---|---|---|---|---|---|
| Ivosidenib (AG-120) | IDH1 | NCT02074839 | 1 | Monotherapy | 3 |
| NCT02632708 | 1 | With intensive chemotherapy | 18 | ||
| NCT03839771 | 3 | With intensive chemotherapy vs placebo | — | ||
| NCT03471260 | 1b/2 | With venetoclax, with or without azacitidine | — | ||
| NCT03173248 | 3 | With azacitidine vs placebo | — | ||
| NCT03564821 | 1 | Monotherapy maintenance | — | ||
| NCT02677922 | 1b/2 | With azacitidine | 19 | ||
| NCT04493164 | 2 | With CPX-351 | — | ||
| NCT04250051 | 1 | With combination chemotherapy/FLAG | — | ||
| NCT04044209 | 2 | With nivolumab | — | ||
| NCT04774393 | 1b/2 | With decitabine/cedazuridine and venetoclax | — | ||
| NCT04655391 | 1 | With glasdegib | — | ||
| Enasidenib (AG-221) | IDH2 | NCT01915498 | 1/2 | Monotherapy | 2,20 |
| NCT02632708 | 1 | With intensive chemotherapy | 18 | ||
| NCT03839771 | 3 | With intensive chemotherapy vs placebo | — | ||
| NCT02577406 | 3 | Monotherapy vs conventional care | — | ||
| NCT04092179 | 1b/2 | With venetoxlax | — | ||
| NCT03683433 | 2 | With azacitidine | — | ||
| NCT03515512 | 1 | Monotherapy maintenance after allo-SCT | — | ||
| NCT03728335 | 1 | Monotherapy maintenance after allo-SCT | — | ||
| NCT02677922 | 1b/2 | With azacitidine | — | ||
| NCT03825796 | 2 | With CPX-351 | — | ||
| NCT04774393 | 1b/2 | With decitabine/cedazuridine and venetoclax | — | ||
| NCT04655391 | 1 | With glasdegib | — | ||
| Olutasidenib (FT-2102) | IDH1 | NCT02719574 | 1/2 | Monotherapy, or with azacitidine or low dose cytarabine | — |
| NCT04013880 | 1b/2 | With decitabine/cedazuridine | — | ||
| IDH305 | IDH1 | NCT02381886 | 1 | Monotherapy | — |
| NCT02826642 | 1 | With standard of care | — | ||
| BAY1436032 | IDH1 | NCT03127735 | 1 | Monotherapy | 21 |
| Vorasidenib (AG-881) | IDH1/2 | NCT02492737 | 1 | Monotherapy | — |
| HMPL-306 | IDH1/2 | NCT04764474 | 1 | Monotherapy | — |
| LY3410738 | IDH1/2 | NCT04603001 | 1 | Monotherapy | — |
For studies of which results have been published, references are included in the last column. For some other studies, (interim) results have been presented but not published yet; because of space limitations, no references have been included for those.
AML = acute myeloid leukemia; IDH = isocitrate dehydrogenase.
IDH inhibitors as monotherapy
The IDH1 inhibitor ivosidenib and the IDH2 inhibitor enasidenib were both evaluated in patients with advanced hematological disorders, predominantly either refractory or relapsed (R/R) AML. Two comparable single arm phase 1 studies in which the respective oral IDH inhibitor was administered continuously as monotherapy showed similar efficacy results.2,3,20 Overall response rates in the primary efficacy populations were 41.6% for ivosidenib and 38.8% for enasidenib, with 34.4% and 28.9% of patients, respectively, achieving a complete remission (CR) or complete remission with incomplete hematologic recovery (CRi) or incomplete platelet recovery (CRp).3,20 Median durations of response were 6.5 and 5.6 months, respectively, and median overall survival was 8.8 months in both studies. Median time to the first response was 1.9 months for both inhibitors.2,3,20 Correlative studies demonstrated a strong reduction of 2-HG levels in almost all patients, confirming the on-target effect of therapy. The fact that only a subset of those patients achieved a clinical response indicates that other factors besides 2-HG reduction determine response or resistance (see below). It was recently suggested that enasidenib may also directly stimulate erythroid differentiation independently of IDH2.22 In both trials, complete molecular clearance of IDH1 or IDH2 mutation was predictive for achieving a CR.
Based on these two single arm studies, ivosidenib and enasidenib received US Food and Drug Administration (FDA) approval for R/R IDH-mutated AML. In addition, ivosidenib received FDA approval for newly diagnosed IDH1-mutated AML in patients unfit for intensive therapy.23 In Europe, no approval was obtained so far. While comparison of the results of these trials to overall historical data on outcome of R/R AML seem favorable, retrospective data indicate that conventional salvage therapy has the potential to achieve remissions in some patients with R/R IDH1- or IDH2-mutated AML.24,25 To directly compare enasidenib to conventional care regimens in patients with R/R AML, a randomized phase 3 trial was performed (NCT02577406). Although final results of this trial are pending, it was announced that this study failed to meet its primary endpoint: overall survival.26 To better understand the implications of these findings, it will be important to assess the full analyses once available.
IDH inhibitors in combination therapy
Several clinical trials have been initiated to investigate the possibility to combine IDH inhibitors with orthogonal treatment modalities, including conventional chemotherapy, HMAs, or other small molecules targeting other oncogenes (Table 1). Importantly, some of these trials are assessing the possible value of IDH inhibitors in newly diagnosed disease.
A phase 1 trial combining either ivosidenib or enasidenib with intensive chemotherapy-based regimens in newly diagnosed AML demonstrated the feasibility to combine IDH inhibitors with a backbone of “7 + 3” (cytarabine/anthracyclines), with limited additional toxicity (see below).18 End of induction CR/CRi/CRp rates were 72% and 63% for the ivosidenib group and enasidenib group, respectively, with molecular measurable residual disease (MRD) negativity of 39% and 23% in responsive patients. The trial included maintenance therapy after induction and consolidation therapy. Patients could alternatively proceed to allogeneic stem cell transplantation at any point, but in that case, went off-protocol. With a relatively short follow-up, median overall survival was not reached in the ivosidenib group and was 25.6 months in the enasidenib group. An international, randomized, placebo-controlled phase 3 trial coordinated by the Hemato Oncology Foundation for Adults in the Netherlands (HOVON) and the German-Austrian AML Study Group (AMLSG) is currently evaluating the efficacy of ivosidenib or enasidenib when added to a backbone of intensive chemotherapy, followed by consolidation therapy and maintenance therapy with IDH inhibitor (or placebo) for up to 2 years (NCT03839771). The primary endpoint of this trial is event-free survival.
Combination of IDH inhibitors with HMAs may be synergistic. A small phase 1/2 study of ivosidenib in combination with azacitidine in newly diagnosed AML showed a response rate of 78.3% that was durable.19 Patients who achieved a CR had a high clearance rate of IDH1 mutation (71.4%). In the same trial, IDH2-mutated AML patients were enrolled and treated with the combination of enasidenib and azacitidine. Results of those patients have not been published yet. A randomized placebo-controlled trial evaluating the addition of ivosidenib to azacitidine in newly diagnosed IDH1-mutated AML is ongoing.
Combination of IDH inhibitors with small molecules targeting either cooperating mutations or other pathways could be rational and could circumvent resistance mechanisms. Several of such trials are ongoing (Table 1).
Toxicity of IDH inhibitors
The (added) hematological toxicity of IDH inhibitors is relatively limited, both as monotherapy and in combination with intensive chemotherapy.2,3,18,20 The most prominent and specific common toxicity of selective IDH inhibitors is a differentiation syndrome (IDH-DS) associated with signs and symptoms such as fluid retention, fever, dyspnea, and/or leukocytosis. IDH-DS was reported in 11% of patients in the ivosidenib and enasidenib monotherapy trials, of which 4% and 7%, respectively, were grade 3 or higher.2,3,20 Post-hoc analyses by the FDA using a standardized classification scheme reported a higher incidence.27 IDH-DS is rare when combined with intensive chemotherapy.18 IDH-DS can result in mortality, but can be properly managed if recognized, which should include immediate treatment with corticosteroids, and exclusion and treatment of possible alternative explanations (eg, infections).28
Other forms of specific toxicity of IDH inhibitors include a Gilbert syndrome type of indirect hyperbilirubinemia due to inhibition of UGT1A1 (enasidenib) and prolongation of QT time (mostly ivosidenib). The latter can be challenging in combination with other essential (prophylactic) medication with a possible effect on the QT time, for example, imidazole derivatives. With proper monitoring of electrocardiograms and electrolytes, concurrent administration of ivosidenib and such co-medication appears feasible.3,19
Targeting IDH-mutated AML by other agents
BCL2 inhibition (venetoclax)
IDH1/2-mutated AML has an increased dependency on the antiapoptotic protein BCL2. This is due to a 2-HG mediated decrease in cytochrome C oxidase activity, which results in a lowered apoptosis threshold (Figure 1).29 Consistently, IDH1/2-mutated leukemias were among the most responsive subtypes of AML in clinical trials evaluating the BCL2 inhibitor venetoclax. In the pivotal VIALE-A trial (venetoclax or placebo added to azacitidine), patients with IDH mutations had a particularly high likelihood of benefitting from the combination of azacitidine with venetoclax, with estimated hazard ratios for death of 0.28 and 0.34 compared to 0.64 for all patients in the trial.30 Thus, the approval by FDA and European Medicines Agency of venetoclax-based regimens has brought an interesting new treatment option to IDH-mutated AML patients who are unfit for intensive therapy.
To further establish the role of BCL2 inhibition in treatment of IDH1/2-mutated AML, several combination trials of IDH inhibitors with venetoclax have been initiated (Table 1). As the sensitivity of IDH1/2-mutated AML to venetoclax is predicted to be particularly dependent on mutant IDH1/2-induced production of 2-HG, it will be interesting to learn what the net effect of co-administration of venetoclax with IDH inhibitors will be.
Possible other agents
In addition to dedicated IDH inhibitors and venetoclax, preclinical studies have identified other potential vulnerabilities of IDH1/2-mutated AML. IDH1/2-mutated AMLs may be sensitive to poly ADP ribose polymerase inhibitors, BRD4 inhibitors, glutaminase inhibitors, or acute promyelocytic leukemia-like therapy. Furthermore, it has been proposed that IDH1/2 mutations are among the more immunogenic abnormalities in AML.31–35 Further clinical evaluation will need to clarify whether these preclinical data translate to human disease.
Primary and secondary resistance to IDH inhibitors
Factors that are associated with primary resistance to monotherapy with ivosidenib or enasidenib in R/R AML include the presence of receptor tyrosine kinase-pathway mutations or other signaling mutations as well as a high overall mutational burden.15,36 A somewhat counterintuitive observation has been that the variant allele frequency of the IDH mutation itself is not clearly predictive for response to IDH inhibitors, and that patients with mutations in subclonal populations can achieve a complete hematological remission.3,15,36
Several correlative studies have investigated mechanisms of secondary resistance to treatment with IDH inhibitors. The largest study to date was performed in the cohort of patients with R/R IDH1-mutated AML enrolled in the ivosidenib monotherapy trial.3,36 Longitudinal next-generation sequencing data were available for 74 patients at relapse or disease progression. In 27% of those patients, newly acquired mutations in receptor tyrosine kinase receptor genes were identified, while 23% of patients harbored new mutations in IDH2 and/or second-site mutations in IDH1 that restored 2-HG levels. These findings corroborate earlier observations that there are largely 2 main mechanisms at play in secondary resistance to monotherapy with IDH inhibitors, one being 2-HG dependent and the other being 2-HG-independent. In a series of 16 patients with IDH2-mutated AML who relapsed on enasidenib therapy, sustained suppression of plasma 2-HG was observed in 14/16 patients, implying an ongoing on-target effect of enasidenib.37 Indeed, in these patients the appearance of subclones with additional mutations in other recurring AML genes was documented. The 2 patients who displayed newly elevated 2-HG levels had acquired IDH1 mutations to circumvent inhibition of IDH2. This so-called isoform switching, from IDH2 to IDH1 or vice versa, leading to restoration of elevated 2-HG levels, had also been reported in an earlier study.38 Similar to IDH1, second-site mutations in IDH2 have been reported, which prevented binding of enasidenib to the IDH2 dimer and caused relapse during enasidenib treatment.39 Single cell sequencing experiments have shown that relapse patterns may be complex and polyclonal.36
Conclusion and future perspectives
Treatment options for patients with IDH1/2-mutated AML have increased significantly in the past few years with the clinical development of IDH inhibitors and the clinical confirmation of the increased sensitivity of these AMLs to venetoclax-based regimens.
Important questions that lay ahead are how the enlarged armamentarium available for patients with IDH1/2-mutated AML can be optimally employed. The place for IDH1/2 inhibitors in newly diagnosed AML will need to be established, as will the most viable combination strategies. Several clinical trials are currently addressing these questions.
Further work will be dedicated to a better understanding of the clonal dynamics of IDH1/2-mutated AML under therapeutic pressure and factors that determine primary and secondary resistance. It is likely that optimal combination therapy will be instrumental in maximizing the chance of achieving MRD negativity and minimizing the risk of relapse. The stable and frequently ancestral nature of IDH1/2 mutations suggests that complete molecular clearance will be key. Prospective evaluation of (molecular) MRD will therefore be a critical aspect in ongoing and future clinical trials.
Disclosures
BJW received speaker’s fees and/or travel fees from Celgene, Roche, and BMS and is principal investigator of the HOVON-150 trial for which HOVON, the sponsor of the trial, received research funding from Agios and BMS/Celgene.
References
- 1.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016; 374:2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017; 130:722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018; 378:2386–2398. [DOI] [PubMed] [Google Scholar]
- 4.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010; 102:932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meggendorfer M, Cappelli LV, Walter W, et al. IDH1R132, IDH2R140 and IDH2R172 in AML: different genetic landscapes correlate with outcome and may influence targeted treatment strategies. Leukemia. 2018; 32:1249–1253. [DOI] [PubMed] [Google Scholar]
- 6.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010; 17:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011; 19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012; 483:474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010; 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Cadoux-Hudson T, Schofield CJ. Isocitrate dehydrogenase variants in cancer - cellular consequences and therapeutic opportunities. Curr Opin Chem Biol. 2020; 57:122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark O, Yen K, Mellinghoff IK. Molecular pathways: isocitrate dehydrogenase mutations in cancer. Clin Cancer Res. 2016; 22:1837–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Q, Li Y, Lv N, et al. Correlation between isocitrate dehydrogenase gene aberrations and prognosis of patients with acute myeloid leukemia: a systematic review and meta-analysis. Clin Cancer Res. 2017; 23:4511–4522. [DOI] [PubMed] [Google Scholar]
- 13.Kattih B, Shirvani A, Klement P, et al. IDH1/2 mutations in acute myeloid leukemia patients and risk of coronary artery disease and cardiac dysfunction-a retrospective propensity score analysis. Leukemia. 2020 September 18. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willekens C, Rahme R, Duchmann M, et al. Effects of azacitidine in 93 patients with IDH1/2 mutated acute myeloid leukemia/myelodysplastic syndromes: a French retrospective multicenter study. Leuk Lymphoma. 2021; 62:438–445. [DOI] [PubMed] [Google Scholar]
- 15.Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017; 130:732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017; 7:478–493. [DOI] [PubMed] [Google Scholar]
- 17.Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013; 340:622–626. [DOI] [PubMed] [Google Scholar]
- 18.Stein EM, DiNardo CD, Fathi AT, et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: a phase 1 study. Blood. 2021; 137:1792–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DiNardo CD, Stein AS, Stein EM, et al. Mutant isocitrate dehydrogenase 1 inhibitor ivosidenib in combination with azacitidine for newly diagnosed acute myeloid leukemia. J Clin Oncol. 2021; 39:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019; 133:676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heuser M, Palmisiano N, Mantzaris I, et al. Safety and efficacy of BAY1436032 in IDH1-mutant AML: phase I study results. Leukemia. 2020; 34:2903–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutta R, Zhang TY, Köhnke T, et al. Enasidenib drives human erythroid differentiation independently of isocitrate dehydrogenase 2. J Clin Invest. 2020; 130:1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roboz GJ, DiNardo CD, Stein EM, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood. 2020; 135:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Largeaud L, Bérard E, Bertoli S, et al. Outcome of AML patients with IDH2 mutations in real world before the era of IDH2 inhibitors. Leuk Res. 2019; 81:82–87. [DOI] [PubMed] [Google Scholar]
- 25.Largeaud L, Bertoli S, Bérard E, et al. Outcome of relapsed/refractory AML patients with IDH1R132 mutations in real life before the era of IDH1 inhibitors. Leuk Lymphoma. 2020; 61:473–476. [DOI] [PubMed] [Google Scholar]
- 26.Bristol Myers Squibb Provides Update on Phase 3 IDHENTIFY Trial in Patients with Relapsed or Refractory Acute Myeloid Leukemia [press release]. 2020. Princeton, NJ, USA: Bristol Myers Squibb. Available at: https://news.bms.com/news/details/2020/Bristol-Myers-Squibb-Provides-Update-on-Phase-3-IDHENTIFY-Trial-in-Patients-with-Relapsed-or-Refractory-Acute-Myeloid-Leukemia/default.aspx. Accessed April 17, 2021. [Google Scholar]
- 27.Norsworthy KJ, Mulkey F, Scott EC, et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration Systematic Analysis. Clin Cancer Res. 2020; 26:4280–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fathi AT, DiNardo CD, Kline I, et al. ; AG221-C-001 Study Investigators. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase ½ study. JAMA Oncol. 2018; 4:1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015; 21:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020; 383:617–629. [DOI] [PubMed] [Google Scholar]
- 31.Molenaar RJ, Radivoyevitch T, Nagata Y, et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin Cancer Res. 2018; 24:1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mugoni V, Panella R, Cheloni G, et al. Vulnerabilities in mIDH2 AML confer sensitivity to APL-like targeted combination therapy. Cell Res. 2019; 29:446–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roerden M, Nelde A, Walz JS. Neoantigens in hematological malignancies-ultimate targets for immunotherapy? Front Immunol. 2019; 10:3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boutzen H, Saland E, Larrue C, et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J Exp Med. 2016; 213:483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Liu Y, Lu C, et al. Cancer-associated IDH2 mutants drive an acute myeloid leukemia that is susceptible to Brd4 inhibition. Genes Dev. 2013; 27:1974–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choe S, Wang H, DiNardo CD, et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020; 4:1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quek L, David MD, Kennedy A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med. 2018; 24:1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018; 8:1540–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018; 559:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]