

Abstract
The germinal center (GC) reaction is a key feature of adaptive humoral immunity. GCs represent the site where mature B cells refine their B-cell receptor (BCR) and are selected based on the newly acquired affinity for the antigen. In the GC, B cells undergo multiple cycles of proliferation, BCR remodeling by immunoglobulin somatic hypermutation (SHM), and affinity-based selection before emerging as effector memory B cells or antibody-secreting plasma cells. At least 2 histologically and functionally distinct compartments are identified in the GC: the dark zone (DZ) and the light zone (LZ). The proliferative burst and immunoglobulin remodeling by SHM occur prevalently in the DZ compartment. In the LZ, GC B cells undergo an affinity-based selection process that requires the interaction with the antigen and accessory cells. GC B cells are also targeted by class switch recombination, an additional mechanism of immunoglobulin remodeling that ensures the expression of diverse isotype classes. These processes are regulated by a complex network of transcription factors, epigenetic modifiers, and signaling pathways that act in concert with mechanisms of intra-GC B-cell trafficking. The same mechanisms underlying the unique ability of GC B cells to generate high affinity antibodies and ensure immunological memory are hijacked during lymphomagenesis and become powerful weapons for malignant transformation. This review will summarize the main processes and transcriptional networks that drive GC B-cell development and are relevant for human B-cell lymphomagenesis.
The germinal center reaction
Germinal centers (GCs) are histological structures that develop upon engagement of the B-cell receptor (BCR) on naive B cells by a cognate antigen. Activated naive B cells migrate into the T cell zones of the secondary lymphoid organs where they are stimulated to proliferate and to acquire features of GC B cells. The GC reaction aims to generate B cells with high affinity BCRs and to foster the production of effector memory B cells and antibody-secreting plasma cells.1,2 The process of BCR affinity maturation relies on the introduction of somatic mutations in the variable region of the immunoglobulin genes by the somatic hypermutation (SHM) machinery followed by affinity-based selection.
The GC comprises at least 2 compartments that are histologically and functionally distinct: the dark zone (DZ) and the light zone (LZ).3 The DZ is the site of active proliferation and where SHM occurs. The LZ is the site of affinity-based selection, which drives the DZ re-entry, commitment to post-GC differentiation, or elimination by apoptosis2,3 (Figure 1). GC B cells undergo class switch recombination (CSR) in order to acquire the ability to express different antibody isotype classes with diverse effector functions.4 Although, CSR was considered a mechanism associated with the LZ compartment, recent data suggest that this process may start early in naive B cells primed to become GC B cells.5 In addition, CSR is not an obligatory consequence of transit through the GC: post-GC cells may exit as not switched lymphocytes, especially if they reside in the GC for a short time.6,7
Figure 1.
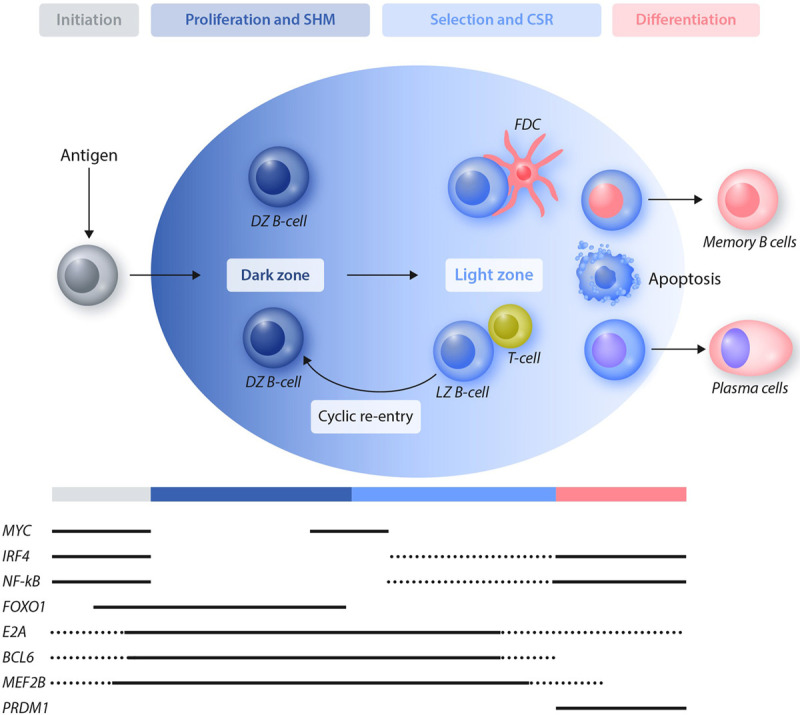
The GC reaction and the expression pattern of key transcription factors. Antigen-activated B cells differentiate into DZ GC B cells that are characterized by a high rate of proliferation and are subject to SHM. B cells then move into the LZ compartment and undergo activation, affinity-based selection and CSR. GC B cells will undergo multiple cycles of re-entry from the LZ to the DZ before terminally differentiating into memory B cells or plasma cells. Cells that have not acquired a favorable B-cell receptor will be negatively selected and eliminated by apoptosis. Multiple transcription factors are involved in regulating the GC initiation, expansion, and exit toward terminal differentiation. The pattern of expression of selected transcription factors that are relevant for the GC reaction and are also hijacked during lymphomagenesis is displayed. Dotted lines refer to expression in a subset of the cells. BCL6 = BCL6 transcription repressor; CSR = class switch recombination; DZ = dark zone; E2A = transcription factor E2-alpha; FDC = follicular dendritic cell; FOXO1 = forkhead box O1; GC = germinal center; IRF4 = interferon regulatory factor 4; LZ = light zone; MEF2B = myocyte enhancer factor 2B; MYC = MYC proto-oncogene, bHLH transcription factor; NF-kB = nuclear factor kappa B; PRDM1 = PR/SET domain 1; SHM = somatic hypermutation.
The simplified view of a bi-compartmental GC structure has been expanded by recent data showing that a more granular assessment of GC B cells by single-cell transcriptomic analyses identifies a much broader spectrum of subpopulations, including an additional “intermediate zone” between the DZ and the LZ.8–10 These data suggest that GC B cells are likely to move along a continuum of states rather than between 2 defined and uniform populations.
Immunoglobulin remodeling
The BCR is formed by the immunoglobulins associated with co-factors (CD79A and CD79B) that participate in signal transduction. Antibodies are composed of 4 polypeptides: 2 identical heavy chains (immunoglobulin heavy locus [IGH]) and 2 identical light chains (immunoglobulin lambda locus [IGL] or immunoglobulin kappa locus [IGK]). Each polypeptide consists of an amino terminal variable segment (V) and a constant carboxyterminal region (C). Each B cell acquires the ability to express a specific BCR on the surface during the early stages of maturation occurring in the bone marrow. The production of a functional and unique BCR occurs through V(D)J recombination, a process that leads to rearrangements in the IGH locus of the genes encoding for the variable (V), diversity (D), and joining (J) regions, followed by the rearrangements in one of the light chain loci (IGL or IGK) of the genes encoding for the V and J regions (Figure 2A). As a result of productive V(D)J recombination in the IGH and IGL or IGK loci, immature B cells express BCRs with the immunoglobulin heavy constant mu (IGHM) isotype and are ready to undergo counter selection for autoreactivity.11 Positively selected cells become mature naive B cells that co-express IGHM and immunoglobulin heavy constant delta isotypes and leave the bone marrow to circulate in the peripheral blood and in the secondary lymphoid organs.
Figure 2.
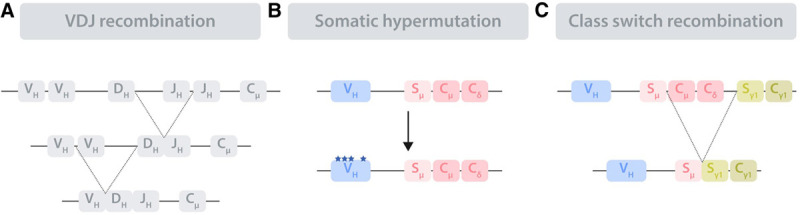
Schematic representation of the immunoglobulin remodeling processes. The immunoglobulin loci undergo diverse remodeling processes in order to encode a functional, high-affinity BCR. (A), Immunoglobulin remodeling starts in bone marrow precursor B cells by V(D)J recombination, a process that assembles 3 (V, D, and J) or 2 (V and J) genes in the immunoglobulin loci encoding for the immunoglobulin heavy locus (IGH) and light chain (not shown), respectively. (B), Somatic hypermutation takes place in the GC DZ compartment and introduces point mutations, deletions, and duplications in the rearranged V regions of the immunoglobulin loci. (C), Class switch recombination targets the constant (C) region of the IGH, leading to the replacement of the original Cμ and Cδ with one of several other C regions (in the scheme Cγ1), each resulting in an antibody with different effector functions. The exchange occurs at the switch (S) regions. BCR = B-cell receptor; D = diversity; DZ = dark zone; GC = germinal center; J = joining; V = variable.
The encounter with cognate antigens triggers naive B cells to proceed into the differentiation process and become GC B cells. In order to expand BCR diversity and therefore increase the probability to generate high affinity antibodies, GC B cells undergo SHM: a process that introduces point mutations, small deletions, insertions, and duplications in the rearranged variable genes of the IGH and IGL or IGK loci12 (Figure 2B). It affects a defined region of 1–2 Kb downstream the V gene promoters and it depends on the enzyme activation-induced cytidine deaminase (AICDA) that converts cytidine into uracil triggering an error-prone DNA repair mechanism.12 Several nonimmunoglobulin loci are also targeted by SHM, although with a much lower mutational rate.13
Most GC B cells undergo an additional step of IGH remodeling by CSR. During CSR, the C region of the IGH chain can be replaced by a mechanism of DNA recombination depending on AICDA and DNA double-strand breaks in specialized switch regions upstream of the C genes4 (Figure 2C).
Role of SHM and CSR in GC lymphomagenesis
GC B cells are the cell-of-origin for the majority of mature B-cell lymphomas. The most common GC-derived B-cell lymphomas include Burkitt lymphoma (BL), follicular lymphoma (FL), and diffuse large B-cell lymphoma (DLBCL).14 The latter comprises at least 2 distinct subtypes: the GC B-cell like (GCB) and the activated B-cell like (ABC) DLBCL.15 The footprints of their GC transit are detectable in the immunoglobulin loci of these malignant cells that carry evidence of SHM.
The physiologic mechanisms of recombination and SHM targeting the immunoglobulin loci are also involved in the generation of genetic alterations identified in B-cell lymphomas. A hallmark of many B-cell lymphomas are chromosomal translocations involving an immunoglobulin locus and a proto-oncogene. These translocations have features consistent with the involvement of V(D)J recombination, CSR, and SHM, supporting the notion that the DNA breaks associated with each of these processes carry a risk of generating translocations.16 The t(14;18) translocation involving BCL2 apoptosis regulator (BCL2) and IGH displays structural features related to V(D)J recombination, thus occurring in bone marrow precursors, although representing an hallmark of FL that is a GC-derived B-cell malignancy. Therefore, V(D)J recombination is involved in the generation of translocations associated with lymphomas that originate at later time in the differentiation process. This observation suggests that seeding oncogenic events can happen early in the hematopoietic development, but require further differentiation to take place before additional occurrences lead to malignant transformation.
The process of SHM introduces nucleotide exchange, but also a significant fraction of insertion and deletions that are associated with DNA breaks.12 Along this line, SHM has been involved in the generation of a subset of translocations targeting MYC proto-oncogene, bHLH transcription factor (MYC) and BCL6 transcription repressor (BCL6) and displaying breakpoints in the V and J regions of the immunoglobulin loci.16 In addition, DLBCL display features of an aberrant SHM mechanism affecting genes that are not physiologic targets of SHM in GC B cells.17
The footprint of CSR is evident in a number of translocations involving the IGH switch regions and various oncogenes, including: MYC, resulting in the t(8;14) translocation, a hallmark of sporadic BL; BCL6, leading to the t(3;14) translocation often detected in DLBCL; MAF bZIP transcription factor in the t(14;16) and interferon regulatory factor 4 (IRF4) in the t(6;14) associated with multiple myeloma. These translocations lead to heterologous promoters and/or enhancers being juxtaposed to an oncogene, leading to dysregulated or ectopic expression of the oncogene.16
In conclusion, the physiologic mechanisms that empower the generation of antibody diversity come with the risk of promoting genetic alterations.
BCR signaling in normal and malignant GC B cells
A key step in the early differentiation of B cells is the acquisition of a functional and nonautoreactive BCR that, during the GC reaction, is further refined to improve affinity (Figure 2). BCR expression is essential for B-cell survival18,19 and GC B cells are no exception.11
BCR signaling can occur independently of antigen engagement. This antigen-independent or “tonic” BCR signaling depends on phosphatidylinositol 3-kinase (PI3K)-mediated activation of the AKT serine/threonine kinase (AKT), which affects several downstream survival pathways.20 Tonic BCR signaling does not engage nuclear factor kappa B (NF-κB) activation, which requires antigen-dependent or “active” BCR signaling. DZ B cells do not display evidence of AKT or NF-κB activation,21,22 while LZ B cells that express high affinity BCR experience active BCR signaling and NF-κB activation as essential mechanisms toward cell fate determination.
Most GC-derived lymphomas retain expression of the BCR, and their need for BCR-driven survival signals is supported by the fact that translocations into the immunoglobulin loci are virtually always found on the nonproductively rearranged alleles.23 Specific subtypes of lymphomas appear to rely on different BCR mode of action. BL are dependent on tonic BCR signaling that is promoted by multiple genetic alterations, including disruption of the transcription factor E2-alpha (E2A)-inhibitor of DNA binding 3, HLH protein (ID3) module.24,25 Consistently, the constitutive activation of the PI3K signaling pathway, together with ectopic MYC expression, is required for development of BL in mice.26 Conversely, the ABC subtype of DLBCL depends on a chronic form of active BCR signaling that is associated with self-reactive BCRs and is sustained by numerous oncogenic aberrations mostly targeting proximal members of the pathway.27–29
GC transcriptional networks hijacked by lymphomagenesis
The transcriptional networks wiring the GC reaction rely on multiple transcription factors and epigenetic modifiers. While transcription factors are key players in establishing defined transcriptional programs, epigenetic chromatin remodeling ensures coordinated and rapid switching between discrete transcriptional programs. Recent genomic analyses have shown that a large fraction of these molecules are targeted by genetic alterations in lymphoma. This section focuses on selected proteins that are involved in driving the GC reaction and are recurrently targeted by genetic lesions in GC-derived lymphomas.
MYC
MYC is a pleiotropic protein that functions as a transcription factor regulating an extremely large network of targets and a broad range of cellular programs,30 but also acts as a modulator of DNA replication through mechanisms that are independent of its transcriptional activity.31
The very limited expression of MYC in the GC has remained a puzzling observation that has been addressed only in the recent years.32,33 MYC is required at the initial stages of the GC formation, but it is then downregulated by mechanisms that include repression by BCL6.34,35 In GC B cells, expression of MYC is restricted to a subset of LZ B cells that are primed to re-enter the DZ34,35 (Figure 1).
The ectopic expression of MYC is a common feature of GC-derived lymphoma. In particular, translocations juxtaposing MYC to the immunoglobulin loci represent the hallmark of BL. The contribution of MYC to malignant transformation in GC B cells is most likely related to its constitutive and ectopic expression that disrupts the tightly regulated bimodal pattern of expression observed in normal GC B cells.
FOXO1
Forkhead box O1 (FOXO1) is a transcription factor that acts at different stages of B-cell differentiation.36 In the GC, FOXO1 is expressed in the DZ compartment consistent with the low activity of PI3K-AKT signaling that promotes cytoplasmic translocation and degradation of FOXO1.22 FOXO1 transcriptional network acts to sustain the DZ program while repressing LZ features such as activation and differentiation.22 Consistently, GC-specific deletion of Foxo1 in mice leads to the development of GC containing only LZ cells.22,37
In GC-derived lymphomas, FOXO1 nuclear expression is detected in the vast majority of BL and a subset of DLBCL, regardless of PI3K-AKT activity in these tumors. In a fraction of cases, the fact that FOXO1 is retained in the nucleus in the presence of PI3K signaling has been linked to mutations that prevent phosphorylation and negative regulation of FOXO1.38,39
BCL6
BCL6 acts as a master regulator of the GC reaction and it is indeed required for GC formation and maintenance.40,41 BCL6 is a transcriptional repressor that controls a wide set of targets aiming to restrain the DNA damage response, modulate apoptosis, and hamper the premature activation and differentiation of GC B cells.42 BCL6 repression activity is dependent on its ability to bind specific DNA motifs and to recruit class I and class II histone deacetylase complexes either directly or through interaction with multiple co-repressors.43–45 BCL6 expression in mature B cells is restricted to the GC stage (Figure 1). The signals inducing the expression of BCL6 at the initiation of the GC remain elusive, although several transcription factors including interferon regulatory factor 8,46 IRF4,47 and myocyte enhancer factor 2B (MEF2B)48 have been shown to contribute to BCL6 expression. Expression of BCL6 in GC B cells is finely tuned by an auto-regulatory mechanism through which BCL6 binds to its own promoter to negatively regulate its transcription.49
In the LZ, BCL6 expression and activity are negatively modulated by multiple signaling pathways. Indeed, post-GC differentiation requires BCL6 silencing, thus multiple pathways coordinately contribute to negatively modulate BCL6 at different levels.42 IRF4-mediated transcriptional repression of BCL6 occurs upon NF-κB activation following BCR, CD40, or Toll-like receptor signaling.50 In addition, CD40 signaling triggers cytoplasmic translocation of the BCL6 co-repressor nuclear receptor corepressor 2, leading to a rapid inactivation of BCL6 transcriptional activity on a subset of targets.51 Interaction with co-repressors is also impaired by BCL6 acetylation, which provides an additional relatively fast and reversible mechanism to inactivate BCL6 prior to its transcriptional and post-transcriptional down-regulation.52 BCR signaling, induced by high-affinity interactions with the antigen, leads to mitogen-activated protein kinase-mediated BCL6 phosphorylation and degradation.53 BCL6 protein degradation is mediated by F-box protein 11 (FBXO11), a specific adaptor protein that recruits BCL6 into the S-phase kinase associated protein 1-Cullin 1 ubiquitin ligase complex and targets it for ubiquitylation and proteasome-mediated degradation.54
BCL6 dysregulated expression and activity is a common feature of GC-derived lymphomas and contributes to lymphomagenesis by enforcing a proliferative phenotype, suppressing DNA damage responses and blocking differentiation. The oncogenic potential of BCL6 dysregulated constitutive expression has been confirmed in mouse models.55
An expanding number of mechanisms converge to dysregulate BCL6 expression and activity in lymphoma. BCL6 was first identified for its involvement in translocations that replace its regulatory region with heterologous promoters including the IGH locus.56–58 Mutations targeting the auto-regulatory circuit49 or the IRF4 binding sites50 in BCL6 promoter interfere with basal or CD40-induced negative modulation. In addition, several indirect mechanisms affect BCL6 during lymphomagenesis, including gain-of-function mutations in MEF2B, a positive modulator of BCL6; inactivation of FBXO11, involved in BCL6 protein degradation; and loss-of-function alterations in the acetyl transferases CREB binding protein (CREBBP) and E1A binding protein p300 (EP300), which are involved in acetylation-mediated inactivation of BCL6.14
MEF2B
MEF2B is a member of the MEF2 family of transcription factors, which includes 3 additional members (MEF2A, MEF2C, and MEF2D) originally identified as regulators of myocyte differentiation.59 Similarly to BCL6, MEF2B expression in mature B cells is restricted to the GC stage. However, MEF2B induction, upon T cell-dependent antigen activation, occurs slightly earlier than BCL648 (Figure 1). This observation is consistent with BCL6 transactivation following MEF2B binding in regulatory regions of the BCL6 locus.48,60,61 In mice, GC-specific loss of Mef2b leads to a partial GC impairment, most likely due to compensatory mechanisms through other Mef2 family members.62 Mef2c, which is expressed at all stages of mature B-cell differentiation, modulates B-cell proliferation by acting downstream of BCR signaling and its deficiency impacts GC development.63 Although Mef2b and Mef2c appear to have different functions in GC B cells, they could partially compensate for each other, as shown by the fact that only their co-deletion fully abrogates GC formation in vivo.62,63
MEF2B binds to a wide range of regulatory regions in the genome of GC B cells and contributes to the modulation of a large network of genes involved in cell cycle, DNA replication and repair, apoptosis, GC confinement, differentiation, and chromatin remodeling.62 This transcriptional network is consistent with a role in promoting the GC, and in particular the DZ, program. Similarly to BCL6, MEF2B expression is negatively regulated in the LZ compartment in order to allow differentiation (Figure 1). However, the mechanisms leading to MEF2B silencing at the end of the GC reaction remain to be identified.
In FL and DLBCL, MEF2B is targeted by genetic alterations, which comprise mostly of recurrent missense mutations targeting the N-terminus, and scattered missense, nonsense, and frame-shift mutations affecting the C-terminus of the protein. Most N-terminus mutants abrogate the interaction with co-repressors leading to an enhanced and de-regulated MEF2B activity, which has been shown to promote malignant transformation in vivo.48,62 Mutations in the C-terminus appear to function through different mechanisms converging to provide escape from inactivation mediated by post-translational modifications.48 Overall, lymphoma-associated genetic alterations of MEF2B appear to interfere with diverse layers of negative modulation leading to a de-regulated activity of this protein.
E2A (TCF3)
The TCF3 gene encodes for the E2A transcription factor that is highly expressed in the DZ compartment64 (Figure 1) and activates expression of cyclin D3 (CCND3), a key player in the proliferative expansion of DZ GC B cells.65–67 In addition, E2A regulates the expression of BCR components (immunoglobulin heavy- and light-chains) and of protein tyrosine phosphatase non-receptor type 6, a negative regulator of BCR signaling.25 E2A activity is negatively modulated by heterodimerization with ID3, which impairs E2A binding to DNA.68 The E2A-ID3 axis appears of relevance in GC B cells, as shown by the requirement of Tcf3 for GC formation in mice.69
In BL, TCF3 and ID3 genes are frequently targeted by genetic alterations, which impair interaction of E2A with ID3 or lead to ID3 inactivation, respectively.24,25 Impaired negative modulation of E2A by ID3 contributes to sustain the survival of lymphoma cells by promoting tonic (antigen-independent) BCR signaling and transactivation of CCND3.25 In addition, CCND3 is often stabilized at protein level due to mutations that impair its ubiquitin-mediated degradation, suggesting that expression of CCND3 is relevant for BL cell proliferation.24,25
NF-κB
NF-κB transcriptional activity is critical for the generation of normal mature B cells and for antigen-dependent B-cell activation.70 The NF-κB transcription factor transduces the expression of a broad network of genes sustaining many cellular processes, including cell survival and proliferation.70 The nuclear translocation of NF-κB is induced at the early stages of the GC initiation, but it is efficiently blocked in the DZ compartment and re-induced later in a subset of LZ GC B cells (Figure 1).3,21,33 During affinity-based selection, B cells that display high-affinity BCR are sustained by the engagement of additional receptors including CD40, TNF receptor superfamily member 13C, and Toll-like receptors. These interactions trigger numerous signaling pathways and converge on the activation of NF-κB. Different NF-κB subunits are associated with distinct functions in GC B cells: REL proto-oncogene, NF-κB subunit is required for GC initiation and maintenance, while activation of the RELA proto-oncogene, NF-κB subunit becomes indispensable at later stages for the generation of plasma cells.71 Consistently, GC-specific deletion of RelA in mice impairs plasma cell differentiation,71 while constitutive activation of NF-κB leads to plasma-cell accumulation.72,73 A key target of NF-κB in LZ GC B cells is represented by IRF4, the induction of which represents an early event toward plasma cell differentiation. IRF4 downregulates BCL6,50 thus favoring termination of the GC program and expression of the plasma cell master regulator B lymphocyte-induced maturation protein 1 (BLIMP1).74
Constitutive NF-κB activation is a feature of the ABC subtype of DLBCL that is required for lymphoma cell survival and sustained by a variety of genetic lesions.75 These lesions act on multiple levels: by promoting chronic, active BCR signaling (through activating mutations in CD79A and CD79B), which feeds into NF-κB activation27,28; by mutating the MYD88 adaptor molecule, which is part of a signaling hub for NF-κB activation76–78; and by removing negative modulators of the NF-κB signaling pathway, such as TNF alpha induced protein 3.79,80
IRF4
IRF4 is a member of the interferon-regulatory factor family of transcription factors that can function both as repressor or activator due to its alternative interactions with diverse partners. In GC B cells, IRF4 displays a bimodal pattern of expression, being induced at the initiation stage when it contributes to BCL6 induction,47,81 repressed in the DZ, and re-expressed in a subset of LZ B cells that are committed to plasma cell differentiation82 (Figure 1). IRF4 is required for plasma cell differentiation as shown by the fact that its deletion in mice leads to lack of plasma cells.83,84 In the LZ, IRF4 downregulates BCL650: the molecular mechanisms by which IRF4 acts to negatively regulate BCL6 in the LZ, while it induces its expression in early GC B cells, are not known and may involve the recruitment of distinct co-transcriptional complexes. IRF4 promotes plasma-cell differentiation by activating PR/SET domain 1 (PRDM1) directly,84,85 as well as by releasing the BCL6-mediated repression on the PRDM1 promoter.86
IRF4 was originally identified as a proto-oncogene involved in chromosomal translocations in multiple myeloma,87 but it is targeted by genetic alterations also in DLBCL, and the ABC subtype depends on IRF4 expression.88
BLIMP1 (PRDM1)
The transcriptional repressor BLIMP1, encoded by the PRDM1 gene, is a master regulator of plasma cell differentiation, a process that requires termination of the GC program.74,89 BLIMP1 contributes to extinguish the GC B-cell identity,90,91 while it implements a plasma cell-specific transcriptional program,92 which includes repression of cell proliferation and the acquisition of an antibody secretory phenotype dependent upon the expression of X-box binding protein 1.93,94 In the GC, PRDM1 is induced in LZ cells that have committed to plasma cell differentiation (Figure 1).
The inability to terminally differentiate is a common process in GC lymphomagenesis. In ABC-DLBCL it appears to depend on 2 mechanisms that are largely mutually exclusive and converge on the negative regulation of BLIMP1. PRDM1 is targeted either by deletions that are often bi-allelic95–97 or it is constitutively repressed by BCL6 dysregulation.86 In addition, a subset of ABC DLBCL display gain-of-function alterations of SPIB,29,98 a transcription factor that directly represses PRDM1 transcription.88,99,100 Mouse models, carrying Prdm1 genetic inactivation in GC B cells develop ABC-DLBCL that display constitutive NF-κB activation, demonstrating the cooperation of these pathways in malignant transformation.95,101
Enhancer of zeste homolog 2
The enhancer of zeste homolog 2 (EZH2) methyl transferase is a component of the polycomb repressive complex-2 that negative regulates gene transcription by inducing trimethylation of the lysine 27 of histone H3.102–104 In mature B cells, EZH2 expression is restricted to the GC stage105,106 and it is required for GC development.107,108 EZH2 promotes GC B-cell proliferation, through epigenetic silencing of cyclin dependent kinase inhibitor 1A, and release of E2F transcription factor 1, and it prevents terminal differentiation by affecting IRF4 and PRDM1 expression.109,110 Overall, EZH2 acts to maintain key features associated with GC B cells and in particular, with the DZ compartment.
The EZH2 gene is targeted by gain-of-function mutations in GCB DLBCL.111 Mice engineered to express the Ezh2 mutant develop GC hyperplasia that, in the presence of dysregulated Bcl2 expression, evolves into lymphomas, consistent with the co-occurrence of these genetic lesions in GCB DLBCL.107,108
CREBBP and EP300
CREBBP and EP300 are acetyl transferases that are ubiquitously expressed and catalyze lysine acetylation of both histone and non-histone proteins.112 In the GC, CREBBP and EP300 display partially overlapping functions, as shown by the fact that only their co-deletion abrogates GC formation in mice.113 Although CREBBP and EP300 appear to compensate for each other during GC formation, they also affect distinct transcriptional programs. CREBBP appears to be more involved in the modulation of pathways related to the LZ, while EP300 affects preferentially DZ-associated genes.113 In particular, CREBBP appears to contrast the BCL6 program by promoting acetylation, and thus transcriptional activation, at the promoters and enhancers of virtually all BCL6 targets.114,115 CREBBP-mediated BCL6 acetylation and inactivation provides an additional layer of negative regulation on the GC program.52
CREBBP and, less frequently, EP300, are genetically inactivated in FL and DLBCL. The genetic alterations usually target only 1 allele of either CREBBP or EP300 in a mutually exclusive fashion.116,117 Although GC-specific loss of Crebbp in mice is insufficient to induce lymphomagenesis, it leads to lymphoma development when combined with Bcl2 dysregulated expression, consistent with the co-occurrence of these genetic lesions in the human malignancies.114,115
KMTs2D
Lysine methyltransferase 2D (KMT2D) is a histone methyl transferase that catalyzes mono- and di-methylation of lysine at position 4 of histone H3.118 These modifications characterize competent enhancers and transcriptionally active chromatin. In GC B cells, KMT2D is involved in modulating the expression of genes related to apoptosis, proliferation, CD40 signaling, and cell migration.119,120
Inactivation of KMT2D is a highly recurrent genetic lesion in FL (about 80% of cases) and DLBCL (about 30% of cases).116,121 Interestingly, KMT2D inactivation appears to be a very early event in FL pathogenesis, suggesting that prior changes in the epigenome, perhaps in combination with the dysregulated expression of BCL2, may facilitate malignant transformation in the GC.122,123 Consistently, conditional loss of Kmt2d by expression of cre recombinase driven by CD19, but not by the GC-specific Cγ1 promoter, leads to expansion of the GC compartment in mice.120
Lymphoma cell-of-origin relating to GC B cells
The attempts to associate lymphoma cells to certain stages of normal B-cell development were initially based on the analysis of morphological and immune phenotypic features and were later complemented by BCR characterization. Based on these observations, most types of B-cell lymphoma have been shown to originate from either GC or post-GC B cells, and they consistently carry somatically hypermutated BCRs. Further advances in the characterization of the transcriptome, genome, and epigenome of B-cell lymphomas have led to a more detailed characterization of lymphoma cell-of-origin, expanding the number of lymphoma subtypes that are identified.
BL, FL, and DLBCL are the most common B-cell lymphomas, the normal counterpart of which is found in the GC. BL originate from the malignant transformation of DZ GC B cells, of which they retain the fast proliferative rate, the highly mutated BCR, and a defined transcriptional signature64 (Figure 3).
Figure 3.
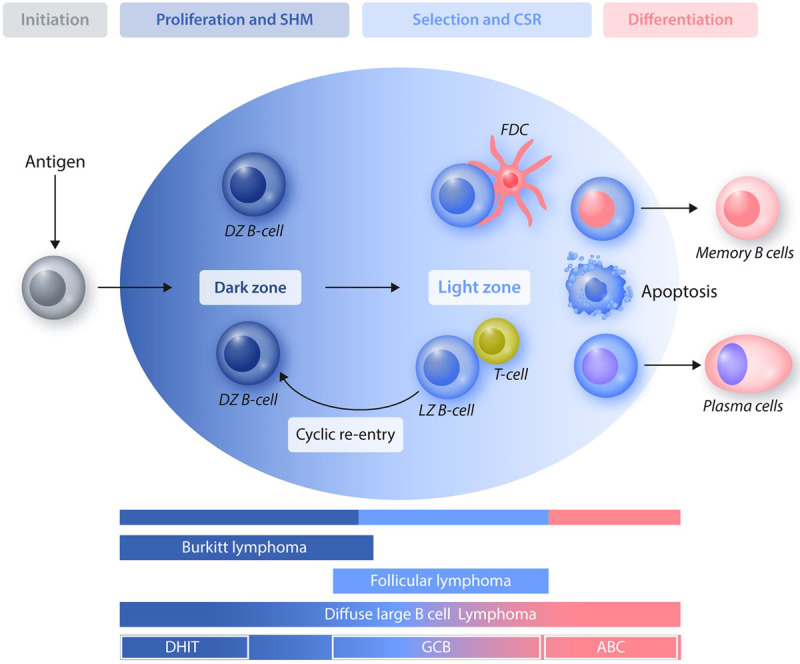
Cell-of-origin of GC-derived lymphomas. Distinct types of GC-derived lymphomas originate from cells that are blocked at different stages of maturation. Burkitt lymphomas resemble DZ B cells, follicular lymphomas and GCB-DLBCL originate from LZ B cells, and ABC-DLBCL display characteristics of late GC B cells that are committed to post-GC differentiation. A subset of DLBCL carrying a DHIT signature related to aberrant expression and/or activity of MYC proto-oncogene, bHLH transcription factor and BCL2 apoptosis regulator (and/or BCL6 transcription repressor) appear to originate from DZ GC B cells. ABC = activated B-cell like; CSR = class switch recombination; DHIT = double-hit; DLBCL = diffuse large B-cell lymphoma; DZ = dark zone; GC = germinal center; GCB = GC B-cell like; LZ = light zone; SHM = somatic hypermutation.
The GC origin of FL is indicated by several features including: its appearance as a follicular-like structure, at least in the early stages; the expression of specific GC markers, such as CD10 and BCL6; the presence of mutated BCR with evidence of ongoing SHM activity; and the presence of a transcriptional signature related to LZ GB B cells64 (Figure 3). Although FL clearly arise from the malignant transformation of GCB cells, it is recognized that the transformation process starts at earlier stages of differentiation and requires multiple events before the final step of clonal expansion occurring in the GC. The t(14;18) translocation, representing the hallmark of FL, is a byproduct of V(D)J recombination occurring in bone marrow B-cell precursors that then move through the differentiation process and enter the GC reaction. In the GC, they display a survival advantage due to the ectopic BCL2 expression and are selected as memory B cells that can undergo multiple GC transits and be exposed to the risk of accumulating additional genetic alterations ultimately driving malignant transformation.124
DLBCL are the most heterogeneous group of GC-derived lymphomas that are characterized by diverse phenotypic, molecular, and clinical features. Based on transcriptional signatures, DLBCL have been classified in at least 2 subgroups: the GCB- and the ABC-DLBCL that appear to originate from either cells at the LZ stage or from GC B cells committed to post-GC differentiation, respectively (Figure 3).8,15,64 The GCB and ABC subtypes are also associated with distinct genetic lesions,125–127 suggesting that they rely on different mechanisms of malignant transformation. Indeed, common pathways altered in GCB DLBCL aim to enhance the GC program, to restrain apoptosis, and to interfere with GC confinement, while ABC DLBCL rely on promoting activation of NF-κB and impairing differentiation. These DLBCL subtypes also display different responses to therapy, with a significantly more favorable prognosis in the GCB patients.15 Based on these numerous evidences, GCB- and ABC-DLBCL are now identified as distinct molecular subtypes in the World Health Organization classification of lymphoid neoplasms. Nonetheless, more heterogeneity appears to be embedded under the DLBCL umbrella. A subset of DLBCL are characterized by a double-hit (DHIT) signature that is driven by MYC and BCL2 (and/or BCL6) dysregulation. Although often classified as GCB, the DHIT DLBCL appear to have a DZ cell-of-origin and display poor outcome128 (Figure 3). In addition, recent data dissecting DLBCL cell-of-origin based on single-cell transcriptomic analysis of GC B cells suggest that DLBCL normal counterparts can be found at every stage during GC development.8 A full integration of the recent efforts aiming to classify DLBCL based on their transcriptomic and genomic125–127 features will be instrumental to fully characterize this heterogeneous group of malignancies.
The malignant transformation of GC B cells
Several characteristics of GC B cells are easily found as features of tumor cells (Figure 4). GC B cells implement risky behaviors to acquire high affinity BCR, and the GC can become a supportive environment for malignant transformation.
Figure 4.
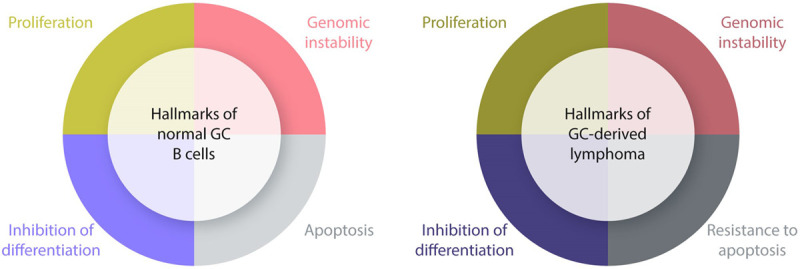
Hallmarks of normal and malignant GC B cells. This scheme summarizes the key features of normal (left) and malignant (right) GC B cells. The proliferative phenotype, the propensity to tolerate DNA breaks, and mechanisms aiming to inhibit terminal differentiation represent features that are often found in GC-derived lymphomas. The sensitivity of GC to apoptosis is overcome in lymphomas by diverse mechanisms including dysregulated expression of anti-apoptotic molecules (ie, BCL2 apoptosis regulator) and enforced activation of signaling pathways that promote survival. GC = germinal center.
DZ GC B cells display an extremely high rate of proliferation, a feature shared by aggressive lymphomas, in particular BL, which displays an extremely rapid duplication rate.
GC B cells are targeted by SHM and CSR that are necessary to refine the affinity and functionality of the BCR but, at the same time, these physiologic DNA breaks provide a potentially deleterious environment for the generation of genetic lesions. In addition, for the immunoglobulin remodeling to occur, GC B cells implement a program aiming to impair the DNA damage response, providing the ideal setting for the accumulation of genetic lesions.
In the context of their risky behavior, GC B cells are efficiently programmed to die by apoptosis unless rescued by specific signals in the LZ. In order for lymphomagenesis to occur, cells must acquire mechanisms to overcome the pro-apoptotic GC program either by ectopic expression of anti-apoptotic molecules (ie, BCL2) or by enforcing the delivery of pro-survival signals that mimic those provided in the LZ.
Finally, GC B cells put on hold the process of terminal differentiation until the affinity-based selection is accomplished. Hacking the program that physiologically restrains GC B cells from differentiating is a dominant mechanism of malignant transformation.
Overall, GC B cells provide a suitable base for malignant transformation to occur, and lymphomagenesis has succeeded in figuring out ways to ensure that every detail is taken care of.
Disclosures
The author has no conflicts of interest to disclose.
References
- 1.De Silva NS, Klein U. Dynamics of the germinal center B-cell reaction. Nat Rev Immunol. 2015; 15:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012; 30:429–457. [DOI] [PubMed] [Google Scholar]
- 3.Victora GD, Schwickert TA, Fooksman DR, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010; 143:592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008; 26:261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roco JA, Mesin L, Binder SC, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity. 2019; 51:337–350.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budeus B, Schweigle de Reynoso S, Przekopowitz M, et al. Complexity of the human memory B-cell compartment is determined by the versatility of clonal diversification in germinal centers. Proc Natl Acad Sci U S A. 2015; 112:E5281–E5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seifert M, Küppers R. Molecular footprints of a germinal center derivation of human IgM+(IgD+)CD27+ B cells and the dynamics of memory B cell generation. J Exp Med. 2009; 206:2659–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes AB, Corinaldesi C, Shen Q, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020; 217:e20200483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kennedy DE, Okoreeh MK, Maienschein-Cline M, et al. Novel specialized cell state and spatial compartments within the germinal center. Nat Immunol. 2020; 21:660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milpied P, Cervera-Marzal I, Mollichella ML, et al. Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol. 2018; 19:1013–1024. [DOI] [PubMed] [Google Scholar]
- 11.Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996; 381:751–758. [DOI] [PubMed] [Google Scholar]
- 12.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007; 76:1–22. [DOI] [PubMed] [Google Scholar]
- 13.Pasqualucci L, Migliazza A, Fracchiolla N, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci U S A. 1998; 95:11816–11821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. 2015; 15:172–184. [DOI] [PubMed] [Google Scholar]
- 15.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403:503–511. [DOI] [PubMed] [Google Scholar]
- 16.Küppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001; 20:5580–5594. [DOI] [PubMed] [Google Scholar]
- 17.Pasqualucci L, Neumeister P, Goossens T, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001; 412:341–346. [DOI] [PubMed] [Google Scholar]
- 18.Kraus M, Alimzhanov MB, Rajewsky N, et al. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004; 117:787–800. [DOI] [PubMed] [Google Scholar]
- 19.Lam KP, Kühn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997; 90:1073–1083. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasan L, Sasaki Y, Calado DP, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009; 139:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basso K, Klein U, Niu H, et al. Tracking CD40 signaling during germinal center development. Blood. 2004; 104:4088–4096. [DOI] [PubMed] [Google Scholar]
- 22.Dominguez-Sola D, Kung J, Holmes AB, et al. The FOXO1 transcription factor instructs the germinal center dark zone program. Immunity. 2015; 43:1064–1074. [DOI] [PubMed] [Google Scholar]
- 23.Küppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005; 5:251–262. [DOI] [PubMed] [Google Scholar]
- 24.Richter J, Schlesner M, Hoffmann S, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012; 44:1316–1320. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012; 490:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sander S, Calado DP, Srinivasan L, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012; 22:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010; 463:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenz G, Davis RE, Ngo VN, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008; 319:1676–1679. [DOI] [PubMed] [Google Scholar]
- 29.Lenz G, Wright GW, Emre NC, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008; 105:13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008; 22:2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dominguez-Sola D, Ying CY, Grandori C, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007; 448:445–451. [DOI] [PubMed] [Google Scholar]
- 32.Klein U, Tu Y, Stolovitzky GA, et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A. 2003; 100:2639–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaffer AL, Rosenwald A, Hurt EM, et al. Signatures of the immune response. Immunity. 2001; 15:375–385. [DOI] [PubMed] [Google Scholar]
- 34.Calado DP, Sasaki Y, Godinho SA, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol. 2012; 13:1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominguez-Sola D, Victora GD, Ying CY, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol. 2012; 13:1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013; 14:83–97. [DOI] [PubMed] [Google Scholar]
- 37.Sander S, Chu VT, Yasuda T, et al. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity. 2015; 43:1075–1086. [DOI] [PubMed] [Google Scholar]
- 38.Kabrani E, Chu VT, Tasouri E, et al. Nuclear FOXO1 promotes lymphomagenesis in germinal center B cells. Blood. 2018; 132:2670–2683. [DOI] [PubMed] [Google Scholar]
- 39.Trinh DL, Scott DW, Morin RD, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013; 121:3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dent AL, Shaffer AL, Yu X, et al. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997; 276:589–592. [DOI] [PubMed] [Google Scholar]
- 41.Ye BH, Cattoretti G, Shen Q, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997; 16:161–170. [DOI] [PubMed] [Google Scholar]
- 42.Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev. 2012; 247:172–183. [DOI] [PubMed] [Google Scholar]
- 43.Hatzi K, Jiang Y, Huang C, et al. A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep. 2013; 4:578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang C, Gonzalez DG, Cote CM, et al. The BCL6 RD2 domain governs commitment of activated B cells to form germinal centers. Cell Rep. 2014; 8:1497–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C, Hatzi K, Melnick A. Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms. Nat Immunol. 2013; 14:380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee CH, Melchers M, Wang H, et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med. 2006; 203:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ochiai K, Maienschein-Cline M, Simonetti G, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. 2013; 38:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ying CY, Dominguez-Sola D, Fabi M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013; 14:1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasqualucci L, Migliazza A, Basso K, et al. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003; 101:2914–2923. [DOI] [PubMed] [Google Scholar]
- 50.Saito M, Gao J, Basso K, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007; 12:280–292. [DOI] [PubMed] [Google Scholar]
- 51.Polo JM, Ci W, Licht JD, et al. Reversible disruption of BCL6 repression complexes by CD40 signaling in normal and malignant B cells. Blood. 2008; 112:644–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bereshchenko OR, Gu W, Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002; 32:606–613. [DOI] [PubMed] [Google Scholar]
- 53.Niu H, Ye BH, Dalla-Favera R. Antigen receptor signaling induces MAP kinase-mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes Dev. 1998; 12:1953–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duan S, Cermak L, Pagan JK, et al. FBXO11 targets BCL6 for degradation and is inactivated in diffuse large B-cell lymphomas. Nature. 2012; 481:90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cattoretti G, Pasqualucci L, Ballon G, et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell. 2005; 7:445–455. [DOI] [PubMed] [Google Scholar]
- 56.Chen W, Iida S, Louie DC, et al. Heterologous promoters fused to BCL6 by chromosomal translocations affecting band 3q27 cause its deregulated expression during B-cell differentiation. Blood. 1998; 91:603–607. [PubMed] [Google Scholar]
- 57.Ye BH, Chaganti S, Chang CC, et al. Chromosomal translocations cause deregulated BCL6 expression by promoter substitution in B cell lymphoma. EMBO J. 1995; 14:6209–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ye BH, Lista F, Lo Coco F, et al. Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science. 1993; 262:747–750. [DOI] [PubMed] [Google Scholar]
- 59.Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007; 134:4131–4140. [DOI] [PubMed] [Google Scholar]
- 60.Chu CS, Hellmuth JC, Singh R, et al. Unique immune cell coactivators specify locus control region function and cell stage. Mol Cell. 2020; 80:845–861.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryan RJ, Drier Y, Whitton H, et al. Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov. 2015; 5:1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brescia P, Schneider C, Holmes AB, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell. 2018; 34:453–465.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilker PR, Kohyama M, Sandau MM, et al. Transcription factor Mef2c is required for B cell proliferation and survival after antigen receptor stimulation. Nat Immunol. 2008; 9:603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Victora GD, Dominguez-Sola D, Holmes AB, et al. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012; 120:2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cato MH, Chintalapati SK, Yau IW, et al. Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol Cell Biol. 2011; 31:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pae J, Ersching J, Castro TBR, et al. Cyclin D3 drives inertial cell cycling in dark zone germinal center B cells. J Exp Med. 2021; 218:e20201699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peled JU, Yu JJ, Venkatesh J, et al. Requirement for cyclin D3 in germinal center formation and function. Cell Res. 2010; 20:631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kee BL. E and ID proteins branch out. Nat Rev Immunol. 2009; 9:175–184. [DOI] [PubMed] [Google Scholar]
- 69.Kwon K, Hutter C, Sun Q, et al. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity. 2008; 28:751–762. [DOI] [PubMed] [Google Scholar]
- 70.Kaileh M, Sen R. NF-κB function in B lymphocytes. Immunol Rev. 2012; 246:254–271. [DOI] [PubMed] [Google Scholar]
- 71.Heise N, De Silva NS, Silva K, et al. Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J Exp Med. 2014; 211:2103–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chu Y, Vahl JC, Kumar D, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011; 117:2227–2236. [DOI] [PubMed] [Google Scholar]
- 73.Tavares RM, Turer EE, Liu CL, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010; 33:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, et al. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity. 2003; 19:607–620. [DOI] [PubMed] [Google Scholar]
- 75.Davis RE, Brown KD, Siebenlist U, et al. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001; 194:1861–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011; 470:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knittel G, Liedgens P, Korovkina D, et al. B-cell-specific conditional expression of Myd88p.L252P leads to the development of diffuse large B-cell lymphoma in mice. Blood. 2016; 127:2732–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Phelan JD, Young RM, Webster DE, et al. A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature. 2018; 560:387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Compagno M, Lim WK, Grunn A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009; 459:717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009; 459:712–716. [DOI] [PubMed] [Google Scholar]
- 81.Willis SN, Good-Jacobson KL, Curtis J, et al. Transcription factor IRF4 regulates germinal center cell formation through a B cell-intrinsic mechanism. J Immunol. 2014; 192:3200–3206. [DOI] [PubMed] [Google Scholar]
- 82.Falini B, Fizzotti M, Pucciarini A, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000; 95:2084–2092. [PubMed] [Google Scholar]
- 83.Klein U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006; 7:773–782. [DOI] [PubMed] [Google Scholar]
- 84.Sciammas R, Shaffer AL, Schatz JH, et al. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006; 25:225–236. [DOI] [PubMed] [Google Scholar]
- 85.Kwon H, Thierry-Mieg D, Thierry-Mieg J, et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity. 2009; 31:941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tunyaplin C, Shaffer AL, Angelin-Duclos CD, et al. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004; 173:1158–1165. [DOI] [PubMed] [Google Scholar]
- 87.Iida S, Rao PH, Butler M, et al. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet. 1997; 17:226–230. [DOI] [PubMed] [Google Scholar]
- 88.Yang Y, Shaffer AL, 3rd, Emre NC, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012; 21:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kallies A, Hasbold J, Fairfax K, et al. Initiation of plasma-cell differentiation is independent of the transcription factor Blimp-1. Immunity. 2007; 26:555–566. [DOI] [PubMed] [Google Scholar]
- 90.Lin KI, Angelin-Duclos C, Kuo TC, et al. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. 2002; 22:4771–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shaffer AL, Lin KI, Kuo TC, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002; 17:51–62. [DOI] [PubMed] [Google Scholar]
- 92.Nutt SL, Taubenheim N, Hasbold J, et al. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011; 23:341–349. [DOI] [PubMed] [Google Scholar]
- 93.Hu CC, Dougan SK, McGehee AM, et al. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009; 28:1624–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001; 412:300–307. [DOI] [PubMed] [Google Scholar]
- 95.Mandelbaum J, Bhagat G, Tang H, et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell. 2010; 18:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pasqualucci L, Compagno M, Houldsworth J, et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med. 2006; 203:311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tam W, Gomez M, Chadburn A, et al. Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood. 2006; 107:4090–4100. [DOI] [PubMed] [Google Scholar]
- 98.Lenz G, Nagel I, Siebert R, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. J Exp Med. 2007; 204:633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Care MA, Cocco M, Laye JP, et al. SPIB and BATF provide alternate determinants of IRF4 occupancy in diffuse large B-cell lymphoma linked to disease heterogeneity. Nucleic Acids Res. 2014; 42:7591–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmidlin H, Diehl SA, Nagasawa M, et al. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood. 2008; 112:1804–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Calado DP, Zhang B, Srinivasan L, et al. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010; 18:580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298:1039–1043. [DOI] [PubMed] [Google Scholar]
- 103.Czermin B, Melfi R, McCabe D, et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002; 111:185–196. [DOI] [PubMed] [Google Scholar]
- 104.Müller J, Hart CM, Francis NJ, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002; 111:197–208. [DOI] [PubMed] [Google Scholar]
- 105.Su IH, Basavaraj A, Krutchinsky AN, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003; 4:124–131. [DOI] [PubMed] [Google Scholar]
- 106.van Galen JC, Dukers DF, Giroth C, et al. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur J Immunol. 2004; 34:1870–1881. [DOI] [PubMed] [Google Scholar]
- 107.Béguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013; 23:677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Caganova M, Carrisi C, Varano G, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013; 123:5009–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Velichutina I, Shaknovich R, Geng H, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010; 116:5247–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Béguelin W, Rivas MA, Calvo Fernández MT, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun. 2017; 8:877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010; 42:181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000; 14:1553–1577. [PubMed] [Google Scholar]
- 113.Meyer SN, Scuoppo C, Vlasevska S, et al. Unique and shared epigenetic programs of the CREBBP and EP300 acetyltransferases in germinal center B cells reveal targetable dependencies in lymphoma. Immunity. 2019; 51:535–547.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jiang Y, Ortega-Molina A, Geng H, et al. CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discov. 2017; 7:38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang J, Vlasevska S, Wells VA, et al. The CREBBP acetyltransferase is a haploinsufficient tumor suppressor in B-cell lymphoma. Cancer Discov. 2017; 7:322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011; 476:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011; 471:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012; 81:65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ortega-Molina A, Boss IW, Canela A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015; 21:1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang J, Dominguez-Sola D, Hussein S, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015; 21:1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011; 43:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Okosun J, Bödör C, Wang J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014; 46:176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pasqualucci L, Khiabanian H, Fangazio M, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014; 6:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pasqualucci L. Molecular pathogenesis of germinal center-derived B cell lymphomas. Immunol Rev. 2019; 288:240–261. [DOI] [PubMed] [Google Scholar]
- 125.Chapuy B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018; 24:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schmitz R, Wright GW, Huang DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018; 378:1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wright GW, Huang DW, Phelan JD, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020; 37:551–568.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ennishi D, Jiang A, Boyle M, et al. Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol. 2019; 37:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]