

Abstract
Protease, serine, 3 (PRSS3), a member of the trypsin family of serine proteases, has been shown to be aberrantly expressed in several cancer types and to play important roles in tumor progression and metastasis. However, the expression and function of PRSS3 gene in hepatocellular carcinoma (HCC) remain unclear. Here we found that PRSS3 expression was decreased in human HCC cell lines and HCC surgical specimens. This was associated with intragenic methylation of PRSS3 gene. Treatment with DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine and/or histone deacetylase inhibitor trichostatin A restored PRSS3 expression in HCC cell lines. Ectopic overexpression of PRSS3 gene in HCC cell lines significantly suppressed cell proliferation and colony formation and arrested cell cycle at G1/S phase, accompanied with downregulation of cyclin D1 (CCND1)/CDK4 and cyclin E1 (CCNE1)/CDK2 complexes. Moreover, PRSS3 overexpression in HCC cells inhibited HCC cell migration and invasion with downregulation of matrix metallopeptidase 2 (MMP2). Further study showed that PRSS3 overexpression diminished the phosphorylation of mitogen-activated protein kinase/extracellular-signal-regulated kinase signaling protein, mitogen-activated protein kinase kinase 1 (MEK1)/mitogen-activated protein kinase kinase 2 (MEK2) and extracellular-signal related kinase 1 (ERK1)/extracellular-signal related kinase 2 (ERK2), in HCC cells. In contrast, knockdown of PRSS3 by small interfering RNA resulted in opposite effects on an HCC cell line SNU-387 which constitutively expresses PRSS3. These results demonstrate that downregulation of PRSS3 by intragenic hypermethylation provides growth and metastasis advantage to HCC cells. The clinical relevance of PRSS3 to human HCC was shown by the intragenic methylation of PRSS3 in HCC specimens and its association with poor tumor differentiation in patients with HCC. Thus, PRSS3 is a potential prognostic biomarker and an epigenetic target for intervention of human HCC.
Keywords: Protease, serine, 3; Hepatocellular carcinoma; Intragenic methylation; Growth; Metastasis; Tumor suppressor
Introduction
Hepatocellular carcinoma (HCC) is one of the deadliest malignant tumors worldwide with increasing incidence in many developing countries [1]. Although considerable effort has been made to improve the effect of surgical and chemotherapeutic intervention, the prognosis of HCC patients generally remains poor with a 5-year survival rate below 12%, mainly due to cancer recurrence and metastasis [1]. Therefore, it is highly desirable to delineate the molecular mechanisms of HCC progression and develop effective strategies for early detection.
Recently, a growing body of genes and signaling pathways has been implicated in hepatocellular carcinogenesis and metastasis [2, 3].The accumulation of both genetic variations and epigenetic disruptions, resulting in amplification or mutation of oncogenes and loss of tumor suppressor genes, plays an important role in HCC development and progression [2–5]. In contrast to genetic mutations, epigenetic alterations are progressive and reversible processes during tumorigenesis, thus having significant implications for early detection and targeted treatment of HCC.
Trypsin is a member of the serine protease family, which plays critical roles in multiple pathophysiological processes including carcinogenesis [6–10]. Trypsinogens are produced by the pancreas as inactive precursors to trypsins and activated in the gastrointestinal tract [8]. In human, protease serine, 1 gene (PRSS1) encodes cationic trypsinogen 1 (PRSS1); PRSS2 encodes anionic trypsinogen 2 (PRSS2); and PRSS3 encodes a minor constituent isoenzyme trypsinogen 3 (PRSS3) [9, 10]. In contrast to PRSS1 and PRSS2, as major digestive isoenzymes in pancreas, PRSS3 is an inhibitor-resistant trypsin isoform capable of digesting common trypsin inhibitors [8, 9, 11]. PRSS3 is represented to all isoforms of trypsinogen 3 protein, encoded by different transcript variants of PRSS3 gene. For instance, PRSS3 was originally identified as brain trypsinogen 4 (TRY4) [12] and pancreatic trypsinogen 3 or mesotrypsinogen (MTG) [11, 13], encoded by trypsinogen transcript variant 1 (PRSS3-V1, referred to as “TRY4”) and 2 (PRSS3-V2), respectively. MTG is functionally characterized as a secreted protein found in human pancreatic tissue and fluid [8, 9]. The signal peptide of MTG is replaced in TRY4 by a leader sequence containing 72 amino acids [11, 12]. Therefore, TRY4 is an intracellular protein and is considered as an “extrapancreatic” trypsin-3 isoform with a tissue-specific distribution [9, 14, 15], suggesting a different biofunction of TRY4 compared to MTG.
Due to a high sequence similarity between different trypsinogens, the expression of PRSS3 transcripts in different tissues and body fluids has not yet been illustrated. The expression of PRSS3 is thought to be mainly restricted to pancreas when it was first discovered [12, 13]. Recent studies revealed that PRSS3 gene was widely expressed in tissues including brain, liver, pancreas and keratinocytes [15], indicating that PRSS3 may play an important role in physiological processes in addition to its digestive activity. However, the expression of PRSS3 and its role in tumor progression have been inconclusive [6, 8, 16], with some studies showing upregulation of PRSS3 associated with cancer metastasis and recurrence [16–23], while others suggesting PRSS3 as a tumor suppressor due to epigenetic silencing of PRSS3 gene through DNA hypermethylation [24–26]. Nevertheless, the expression, regulation, and function of PRSS3 in hepatocellular carcinoma (HCC) remain unknown.
In this study, we report that epigenetic silencing of PRSS3 gene by intragenic hypermethylation facilitates the growth, migration, and invasion of human hepatocellular carcinoma (HCC), suggesting that PRSS3 exerts a tumor suppressor gene function against HCC growth and metastasis.
Materials and methods
Cell lines and reagents
Human HCC cell lines (HepG2, PLC/PRF/5, Bel-7402, SMMC-7721, HBXF-344, SNU-387, and SNU-449), human pancreatic cancer cell lines (PANC 504, SW1990, MIAPaCa-2, PANC-1), and human embryo liver cell line L02 were grown as described [27, 28] in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS; Hyclone, Logan, UT). The cells were split to low density (30% confluence) for overnight culture and were then treated with 2 μM of 5-AZA (Sigma-Aldrich) for 96 h with the medium exchanged every 24 h or with 4 μM of trichostatin A (TSA) (Sigma-Aldrich) for 24 h. For combined treatment, the cells were initially exposed to 5-AZA for 72 h followed by 5-AZA and TSA for 24 h.
The primary antibodies were used against the following proteins for Western blot: PRSS3 from R&D Systems (Cat. no.: MAB3710); p-MEK1/2 from Cell Signaling Technology (Cat. no.: 9121); cyclin D1 from Proteintech Group, Inc. (Cat. no.: 60186–1-Ig); and cyclin-dependent kinase 2 (CDK2), CDK4, cyclin E1, matrix metallopeptidase 2 (MMP2), MEK1/2, ERK1/2, p-ERK1/2, and β-actin from Bioworld Technology, Inc. (Cat. nos.: BS1050, MB0027, BS1085, BS1236, BS3599, BS1112, BS5016, and BS6007M, respectively).
All oligonucleotide sequences are listed in Supplementary Table 1.
Establishment of stable cell lines
Human PRSS3 complementary DNA (cDNA) (sequence identification number NM_007343.3) was amplified by PCR and cloned into the plenti6-GFP vector (Invitrogen). PRSS3-expressing lentiviral or empty vectors were packaged using the ViraPower™ lentiviral expression system (Invitrogen, San Diego, CA, USA). The resulting lentivirus was used to infect PLC/PRF/5 or HepG2 cells and was subjected to blasticidin selection (2 μg/ml, Invitrogen) for 2 weeks to generate stable cell lines expressing PRSS3.
RNA interference knockdown
Small interfering RNA (siRNA) oligonucleotides specific for PRSS3 (siPRSS3–1 and siPRSS3–2) and RNAi Negative Control Duplex (siNC) [20] were synthesized by Gene Pharma Co. (Shanghai, China) (Supplementary Table 1). The siRNAs were transfected into HCC cells with Lipofectamine® RNAiMAX according to the manufacturer’s instructions (Invitrogen, USA). After the knockdown efficiency was assessed by Western blot, the transfected cells were used in future experiments.
Human tissue specimens
A total of 66 cases of primary HCC tumor tissues were collected immediately after surgical resection at the Chinese PLA General Hospital, among which, 25 pairs of tumors and tumor-adjacent tissue samples were obtained as paired paraffin blocks. Normal hepatic tissue specimens (20 cases) were obtained from the edge of resected hemangiomas of the liver. Tumor staging was determined according to the American Joint Committee on Cancer (AJCC) Cancer Staging Manual, 2010 (seventh ed.) (Table 1). All specimens were collected under the guidelines approved by Institutional Review Board of the Chinese PLA General Hospital and with informed consent from patients. The samples were also confirmed by histopathological examination of hematoxylin and eosin (H&E)-stained sections [28].
Table 1.
Relationship between methylation status of PRSS3 gene and clinicopathological features in human HCC
| Clinicopathologic factor | n (%) | PRSS3 methylation status |
p value* | |
|---|---|---|---|---|
| Methylation n (%) | Unmethylation n (%) | |||
| Age (years) | ||||
| < 50 | 22 (33.3) | 19 (35.8) | 3 (23.1) | 0.815 |
| ≥ 50 | 44 (66.7) | 34 (64.2) | 10 (79.2) | |
| Sex | ||||
| Male | 52 (78.8) | 42 (79.2) | 10 (76.9) | 0.845 |
| Female | 14 (21.2) | 11 (20.8) | 3 (23.1) | |
| HBsAg | ||||
| Negative | 17 (25.8) | 13 (24.5) | 4 (30.8) | 0.971 |
| Positive | 49 (74.2) | 40 (75.5) | 9 (69.2) | |
| Liver cirrhosis | ||||
| Negative | 25 (37.3) | 21 (38.9) | 4 (30.8) | 0.823 |
| Positive | 42 (62.7) | 33 (61.1) | 9 (69.2) | |
| Size (diameter) | ||||
| < 5 cm | 20 (30.3) | 15 (28.3) | 5 (38.5) | 0.475 |
| ≥ 5 cm | 46 (69.7) | 38 (71.7) | 8 (61.5) | |
| Differentiation | ||||
| Well | 6 (9.1) | 3 (5.7) | 3 (23.1) | 0.05 |
| Moderate and poor | 60 (90.9) | 50 (94.3) | 10 (76.9) | |
| TNM | ||||
| I/II | 34 (51.5) | 26 (49.1) | 8 (61.5) | 0.42 |
| III/IV | 32 (48.5) | 27 (50.9) | 5 (38.5) | |
| Metastasis | ||||
| Negative | 43 (65.2) | 33 (62.3) | 10 (76.9) | 0.993 |
| Positive | 23 (34.8) | 20 (37.7) | 3 (23.1) | |
RNA isolation and RT-PCR
Total RNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen Hilden, Germany), and first-strand cDNA was synthesized using the Roche’s Transcriptor First Strand cDNA Synthesis Kit (Roche Life Tech, USA). Semi-quantitative RT-PCR was run 25–31 cycles to analyze the expression of PRSS3 variant RNA using the oligonucleotide sequences listed in Supplementary Table 1. PCR products wereseparated and stained with ethidium bromide.
MSP and BS
Total DNA was prepared by the proteinase K method. MSP and BS were performed as previously described [27, 28]. The in vitro methylation DNA (IVD) serving as a positive control was the genomic DNA from HepG2 cells treated in vitro with Sss I methyltransferase (New England Biolabs, MA, USA), and the negative control was the DNA from normal human peripheral lymphocytes as described. MSP products were analyzed using a 2% agarose gel electrophoresis. Bisulfite-treated sample DNA was amplified using BS primers, and the PCR products were gel purified and ligated into pEASY-T1 vector (TransGen Biotech, Beijing, China). Colonies were randomly selected for plasmid isolation using Wizard miniprep kits (Promega, Shanghai, China) and were subjected to sequence with the M13 reverse primer via automated sequencing (BGI Sequencing, Beijing, China) as described [27].
Western blot
Total protein was prepared from HCC cells and transferred onto PVDF membranes using a Bio-Rad Mini PROTEAN 3 system. The membranes were blocked with TBS containing 5% milk and 0.1% Tween-20 at room temperature for 1 h. The membranes were then incubated with desired primary antibodies followed by horseradish peroxidase conjugated secondary antibodies. Immunoreactive bands were visualized by using enhanced chemiluminescence Kit (Sigma, USA) according to the manufacturer’s instructions.
IHC
IHC was performed on 5-mm-thick serial sections of formaldehyde fixed, paraffin embedded tissue blocks derived from liver cancer with paired adjacent tissue as previously described [28, 29]. The sections were incubated overnight at 4 °C with 1:50 dilution of the antibody against PRSS3 (1 mg/ml) (AFAF3586, R&D Systems). The staining intensity and extent of the stained area were graded according to the German semi-quantitative scoring system: staining intensity of the nucleus, cytoplasm, and/or membrane (no staining = 0; weak staining = 1; moderate staining = 2; strong staining = 3), as well as the percentage of stained cells (0% = 0, 1–24% = 1, 25–49% = 2, 50–74% = 3, 75–100% = 4). The final immunoreactive score (0 to 12) was determined by multiplying the intensity score with the percentage of stained cells.
Cell viability
HCC cells were seeded into 96-well plates at 2 × 103 cells/ well. Cell viability was measured every day by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay Kit (KeyGEN Biotech, Jiangsu, China). The absorbance at 490 nm wavelength was detected using a microplate reader (Thermo Multiskan MK3, Thermo Fisher Scientific Inc., USA).
Colony formation
HCC cells were seeded in soft agar in 6-well tissue culture plates (0.3–1 × 103 cells/well) in triplicate. Colonies with more than 50 cells were counted after 2 weeks. The cells were fixed with 75% ethanol for 30 min and stained with 0.2% crystal violet (Beyotime Ltd., Jiangsu Province, China) for 20 min.
Flow cytometry
Trypsinized HCC cells (1 × 106) were fixed, incubated with 100 μg/ml RNase A, and stained with a solution containing 40 μg/ml propidium iodide (KeyGEN Biotech, Jiangsu, China) at 4 °C for 30 min. The cells were then sorted by FACS Caliber (BD Biosciences, San Jose, CA) and analyzed by ModFit software. For apoptosis analysis, the Annexin V-FITC Apoptosis Detection Kit (KeyGen Biotech) was used according to the manufacturer’s protocol.
Cell monolayer scratching, migration, and invasion
The cell monolayer scratching assay was performed with serum-starved cells, and images were captured using an inverted microscope at 10 and 24 h after cell monolayer scratching. Transwell apparatus was used with 8 μm pore inserts (Corning Inc., Corning, NY, USA). For migration assay, the upper chambers were seeded with 200 μl of serum-free medium containing 1 × 104 of serum-starved cells in PLC/PRF/5, 2.5 × 104 of HepG2, or 4 × 104 of SNU-387 cells, respectively. The lower chambers were filled with 500 μl of 10% FBS-RPMI 1640.After 7 h for PLC/PRF/5, 9 h for HepG2, or 24 h for SNU-387 cells, the cells that migrated to the lower chamber were fixed and stained with 0.2% crystal violet (Beyotime). For invasion assay, the upper chamber of the apparatus was coated with Matrigel (BD Biosciences, San Jose, CA) and seeded with 0.5 × 105 of PLC/PRF/5, or HepG2, or 2 × 105 of SNU-387 cells for 20, 18, or 48 h.
Statistical analysis
The data are expressed as the means ± standard deviation (SD) of at least three independent experiments. The correlation between DNA methylation and clinicopathologic features of human liver cancer was analyzed by χ2 test for independence dichotomous variables and Student’s t test for continuous variables. All analyses were carried out using SPSS 19.0 software (SPSS Inc., Chicago, IL). The results were considered to be statistically significant at p < 0.05 (*).
Results
Decreased expression of PRSS3 in human HCC cell lines and primary tumors
Human PRSS3 gene, located on chromosome 9p12, is formed by arrangement of interchromosomal duplications from the PRSS1 and PRSS2 gene locus on chromosome 7q35 and the frequently altered LOC120224 gene region on chromosome 11q24, which gives rise to four different known alternative splice variants and gene products through alternative splicing and/or alternative promoter [9, 30], referred to as trypsinogen transcript variants 1, 2, 3, and 4 (PRSS3-V1 or TRY4, PRSS3-V2, PRSS3-V3, and PRSS3-V4), encoding trypsin-3 isoform 1 (TRY4), 2 (MTG), 3, and 4, respectively (Supplementary Table 2) [9, 15]. These PRSS3 transcript variants contain identical sequences in their central regions and 3′ regions but are various in 5′ regions, resulting in variants transcribed from different transcription start sites as well as different ATG start code (Supplementary Fig. 1A and B).
We first examined the messenger RNA (mRNA) expression of PRSS3 gene in human HCC cell lines by RT-PCR using a set of primers annealing to the sequence common to all PRSS3 splicing variants (Supplementary Fig. 1B). We found that PRSS3 mRNA was completely absent in five cell lines (HepG2, PLC/PRF/5, HBXF344, BEL-7402, SMMC-7721) but was weakly expressed in the cell line SNU-449 and relatively highly expressed in the cell line SNU-387 compared with an immortalized liver embryonic cell line L02 (Fig. 1a). Further RT-PCR analysis using specific primers for each transcript of PRSS3 gene showed that TRY4 was preferentially expressed in SNU-449 HCC and L02 cells in comparison to the spliced variants of PRSS3 gene expressed in pancreatic cancer cell lines [15] (Supplementary Fig. 1B and C), suggesting that PRSS3 mRNA expressed in normal liver cells and some HCC cells was mostly contributed by the transcription of TRY4. Western blotting confirmed that PRSS3 protein was undetectable in HepG2 and PLC/PRF/5 cells but was highly expressed in SNU-387 cell line (Fig. 3g). Immunohistochemistry (IHC) results revealed that the expression of PRSS3 was significantly decreased in HCC tumor tissues compared with adjacent non-tumor tissues from 25 patients (p<0.05;Fig. 1b, c). These results suggest that a decreased expression of PRSS3 may be associated with HCC development.
Fig. 1.
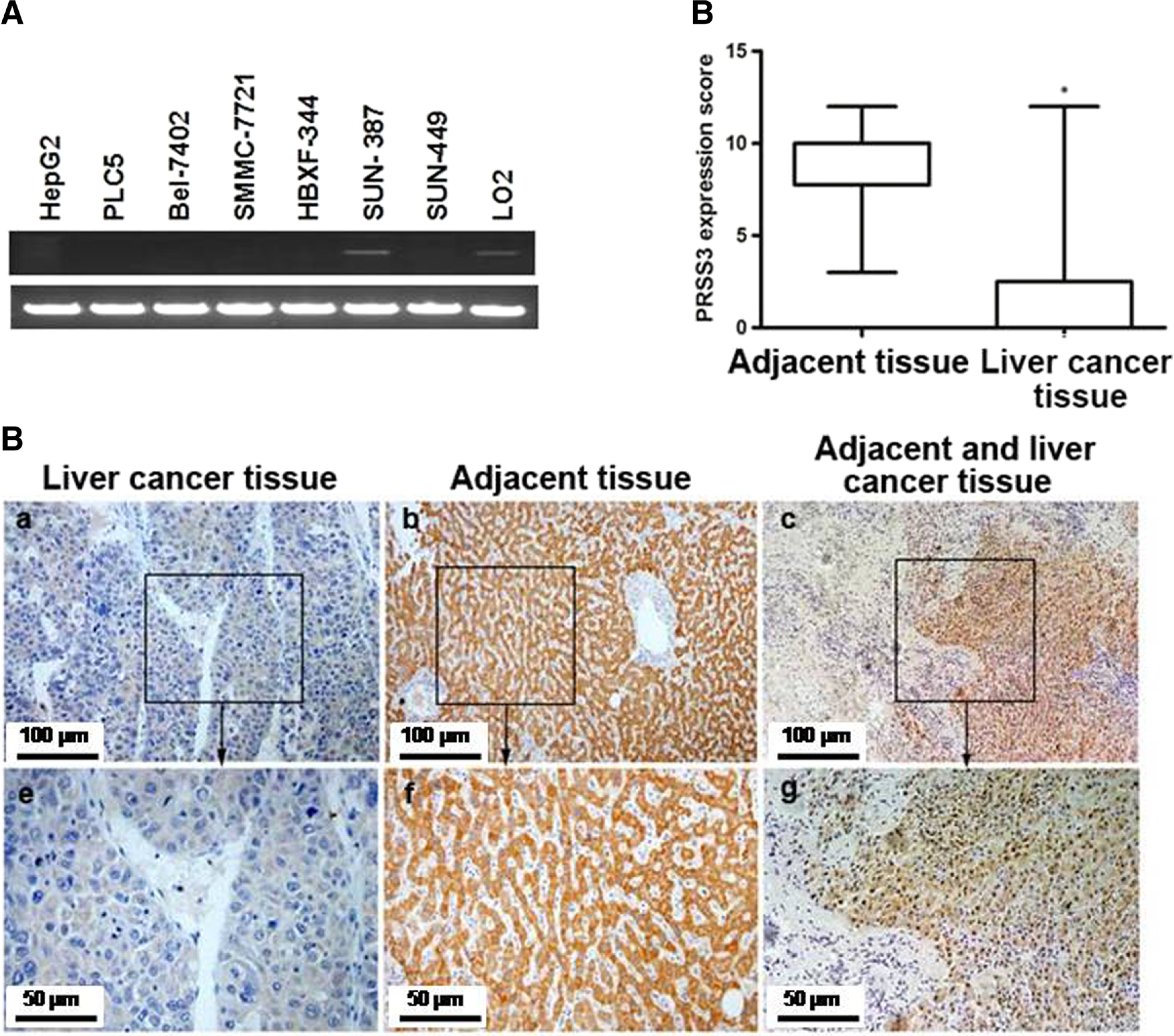
PRSS3 expression in human HCC cell lines and tissues. a PRSS3 mRNA expression was analyzed by semi-quantitative RT-PCR using a pair of primers targeting the sequences common to all PRSS3 splicing variants (Supplementary Fig. 1A). The PCR amplicons were shown by agarose gel electrophoresis (the top lane: PRSS3; the bottom lane: GAPDH). b IHC staining for PRSS3 protein expression in tumor and adjacent tissue samples from 25 patients with primary liver cancer. IHC analysis of PRSS3 expression was scored and shown as box plots according to the German semi-quantitative scoring system (see the “Materials and methods” section). The boxes represent the upper and lower quartiles of the data. Vertical bars represent the range of data. *: p < 0.05. c Representative macroscopic images of IHC staining of PRSS3 expression in tumor tissue (a, e), adjacent tissue (b, f), and the junction between tumor and adjacent tissue (c, g). a–c The images with × 200 magnification. e–g The images with × 400 magnification from boxed regions in the a–c, respectively
Fig. 3.
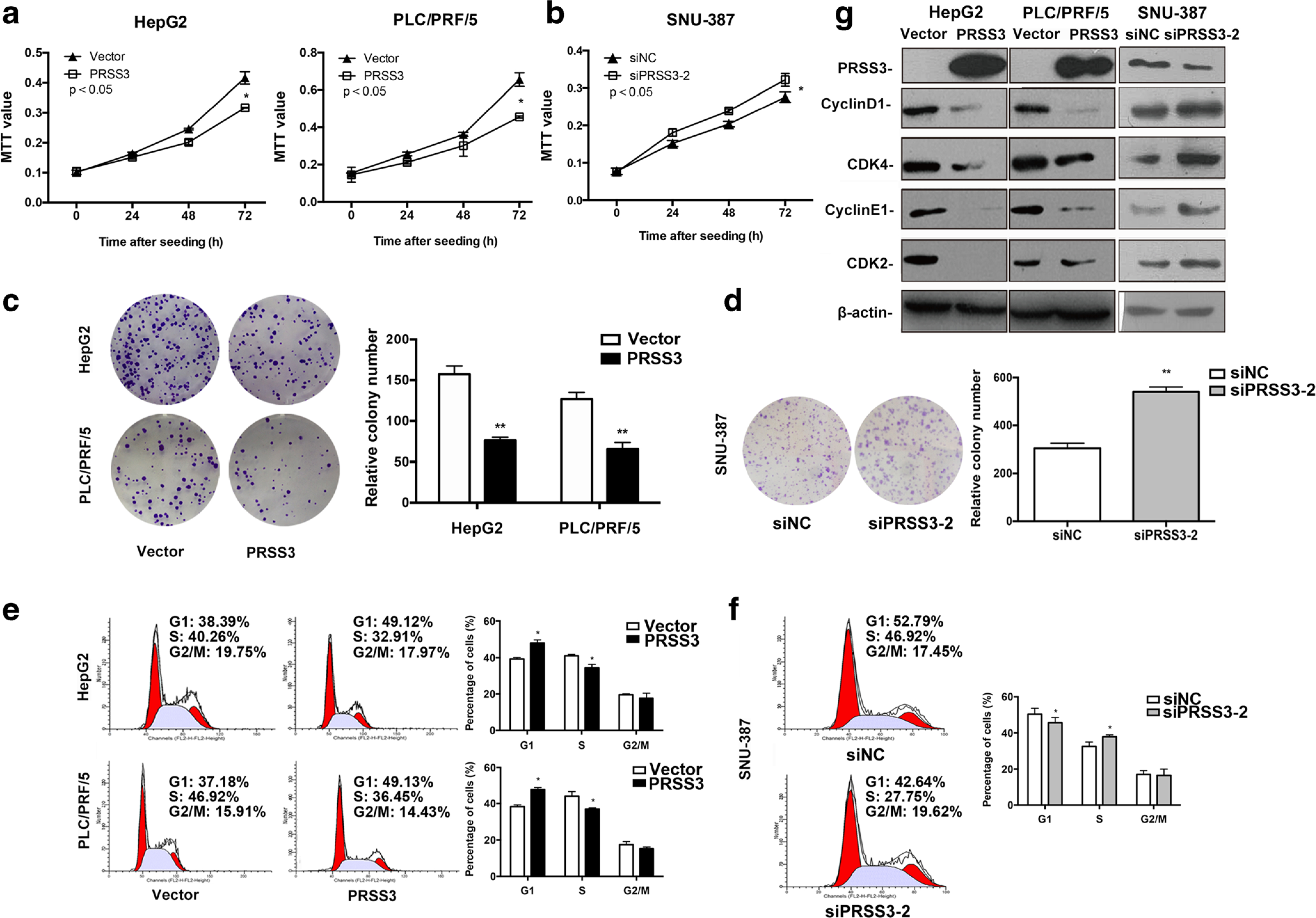
Effect of PRSS3 on the growth properties of human HCC cells. HCC HepG2 and PLC/PRF/5 cell lines with overexpression of PRSS3, and SNU-387 cell line with knockdown of PRSS3 expression were established to evaluate the potential roles of PRSS3 in HCC development. a, b MTT assays showed the viability of HCC HepG2 and PLC/PRF/5 cells with PRSS3 overexpression (PRSS3) or vector control (Vector) (a), or SNU-387 cells transfected with either siRNA against PRSS3 (siPRSS3–2) [20] or RNAi Negative Control Duplex (siNC) (b). c, d. Colony formation in soft agar for 2 weeks by HCC HepG2 and PLC/PRF/5 cells with PRSS3 overexpression, or control (C), or SNU-387 cells transfected with siPRSS3–2 or siNC (d). Left panel: representative image; Right panel: quantitative analysis. e, f FACS analysis of cell cycle distribution of HepG2 and PLC/PRF/5 cells with or without ectopic expression of PRSS3 (E), or SNU-387 cells transfected with siPRSS3–2 or siNC (f). Left panel: representative FACS histograms with the percentage of cells in each cell cycle phase (Left red peak: G0/G1; right red peak: G2/M; hatched peak: S; coefficient of variation of G1 peak: % CV); right panel: quantitative graphs. g Western blotting analysis of the levels of PRSS3, cyclin D1, CDK4, cyclin E1, and CDK2 in the transfected HCC cell lines. The experiments were repeated at least three times, and the results were presented as the mean ± SD, *p < 0.05, versus control
Epigenetic silencing of PRSS3 gene in HCC cells due to intragenic methylation
To elucidate whether the downregulation of PRSS3 expression in HCC was due to epigenetic silencing, we analyzed the methylation status of PRSS3 gene in HCC cell lines. The primers for MSP and BS were designed to cover the intragenic region of PRSS3 gene for TPY4 transcript, where there is a typical CpG island rather than in its promoter region (Fig. 2c). As shown in Fig. 2a, in the intragenic region, PRSS3 DNA was methylated in HepG2 and PLC/PRF/5 cell lines but was partially methylated in SNU-449 cell line and L02 cell line. No methylation was found at the intragenic region of PRSS3 gene in SNU-387 cell line with high expression of PRSS3 at both mRNA and protein levels (Figs. 1a and 3g). BS analysis further showed that PRSS3 intragenic DNAwas almost fully methylated at the examined CpG sites in HepG2 and PLC/PRF/5 cell lines, whereas these CpG sites were partial methylation in L02 cell line and barely methylated in SNU-387 cell line (Fig. 2b). The data suggests that the downregulation of PRSS3 expression is associated with the intragenic methylation of PRSS3 gene.
Fig. 2.
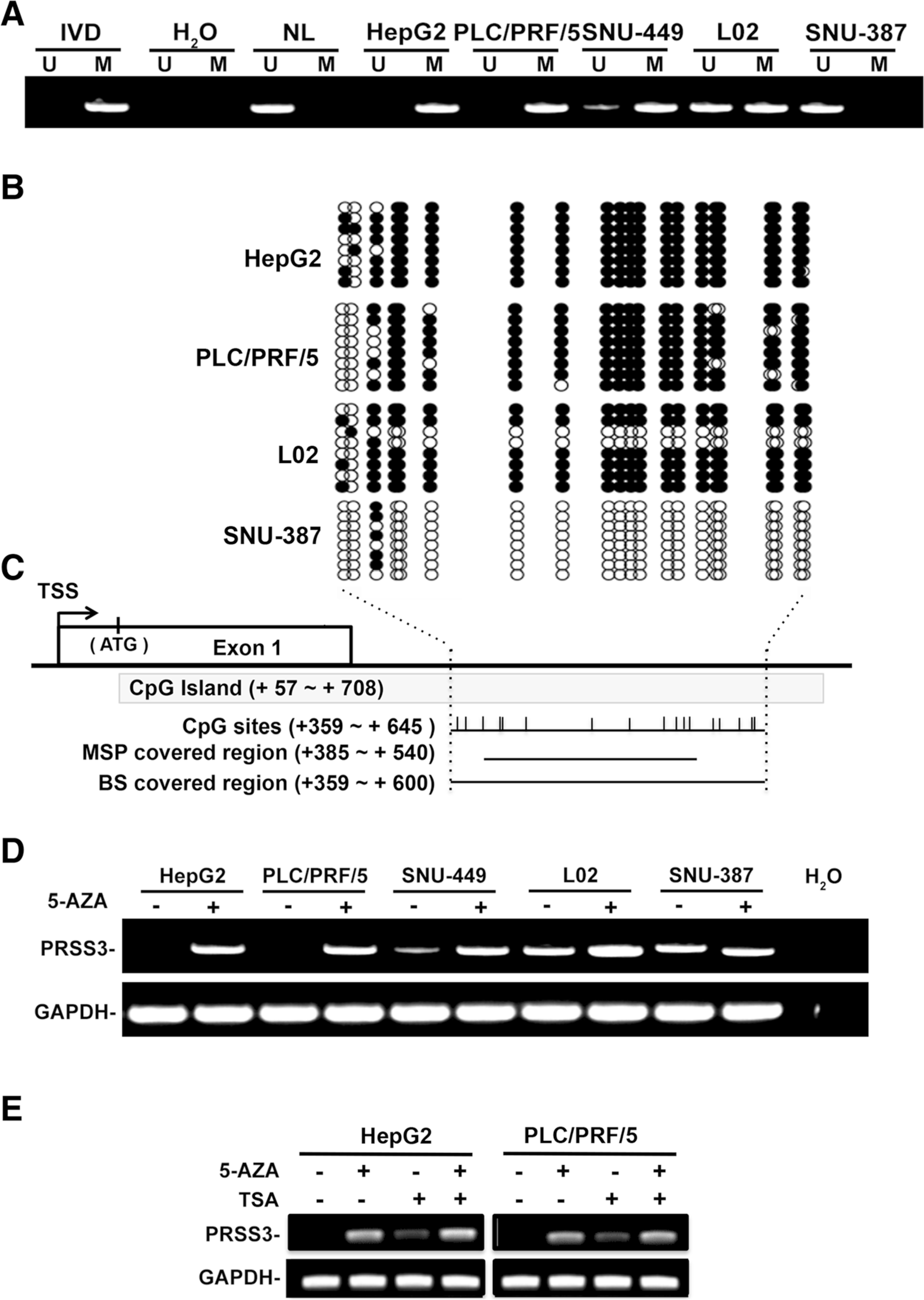
Intragenic DNA methylation status of PRSS3 gene in HCC cells. a The intragenic DNA methylation status of PRSS3 in HCC cell lines was analyzed by MSP. The MSP primers covered region shown as c in below. U: unmethylated; M: methylated; IVD: in vitro methylated DNA served as MSP positive control; NL: normal blood lymphocyte DNA as negative control; MSP: methylation-specific PCR; BS: bisulfite sequencing. b BS analysis of PRSS3 intragenic methylation. The sequencing results were aligned as rows in the grid. Each row represents a sequencing result of the PRSS3 intragenic CpG methylation status from an individual allele of one colony. Circles represent CpG sites, with black as methylated ones and white as unmethylated ones. c The schematic diagram of the intragenic region closed to the first exon of PRSS3 gene encoding for TYR4.The CpG island (+ 57 ~ + 708 from the transcription start site) is shown in gray box, in which MSP and BS covered regions are shown as black lines. Vertical bars represent CpG sites in the intragenic region examined by BS and MSP assays as indicated. TSS: transcription start site. d PRSS3 expression in human HCC cells after treatment with 5-AZA. e PRSS3 expression in human HCC cells after treatment with AZA and/or TSA treatment. 5-AZA: 5-aza-2′-deoxycytidine; TSA: trichostatin A; GAPDH: the internal control of RT-PCR
To investigate whether silencing PRSS3 might be attributable to its epigenetic alteration, HCC cell lines were treated with demethylation agent 5-AZA. Upon 5-AZA treatment, the expression of PRSS3 mRNA was restored in HepG2 and PLC/PRF/5 cell lines, and was weakly upregulated in SNU-449 and L02 cell lines, but was not affected in SNU-387 cells (Fig. 2d), suggesting that the downregulation of PRSS3 gene in HCC might be resulted from its DNA hypermethylation. In addition, the expression of PRSS3 in HepG2 and PLC/PRF/5 cell lines was also restored by TSA, a histone deacetylase inhibitor, and further was enhanced by the combination of TSA with 5-AZA (Fig. 2e). These findings suggest that the epigenetic silencing of PRSS3 gene in HCC is attributable to DNA methylation and to histone acetylation.
PRSS3 suppressed human HCC cell growth associated with G1/S cell-cycle arrest
To investigate the functional role of PRSS3 in HCC cells, we stably overexpressed PRSS3 in HepG2 (PRSS3-HepG2) and PLC/PRF/5 cell lines (PRSS3-PLC/PRF/5) or knocked down PRSS3 expression in SNU-387 by siRNA (siPRSS3-SNU-387) cell line. The efficiency of overexpression or knockdown of PRSS3 in these HCC cell lines was validated by Western blotting analysis of PRSS3 protein (Fig. 3g). As shown in Fig. 3a, the viability of HCC cells was significantly reduced in PRSS3-overexpressing cell lines as compared with vector transfected control cells. Knockdown of PRSS3 expression in SNU-387 cell line increased cell growth (Fig. 3b). The results were confirmed by colony formation assays, in which the PRSS3-overexpressing HCC cells formed significantly fewer colonies than the control cells (Vector) (Fig. 3c), whereas siPRSS3-SNU-387 cells formed more colonies than the cells transfected with control RNAi (siNC-SNU387) (Fig. 3d). Thus, PRSS3 suppresses the proliferation of human HCC cells.
We further monitored the influence of PRSS3 on the cell cycle of HCC cells by flow cytometry. The results showed that the percentage of cells in the G1 phase was increased concomitantly with the decrease of cells in S phase after the overexpression of PRSS3 in HepG2 and PLC/PRF/5 cells (Fig. 3e). This was in contrast to observation in siPRSS3-SNU-387 cells (Fig. 3f). Western blotting showed that the expression levels of cyclin D1 (CCND1), CDK4, cyclin E1 (CCNE1), and CDK2, key proteins regulating G1 phase cell cycle, were reduced in PRSS3-overexpressing HCC cells but were elevated in siPRSS3-SNU-387 cell line (Fig. 3g). These results indicate that PRSS3 suppresses the growth of HCC cells by causing G1/S phase cell-cycle arrest via downregulation of CCND1/CDK4 and CCNE1/CDK2 complexes.
PRSS3 inhibited the invasion and migration capabilities of human HCC cells
We next examined the function of PRSS3 in the metastatic ability of HCC cells. Cell monolayer scratching assay showed that PRSS3 overexpression in HepG2 and PLC/PRF/5 cells caused a marked inhibition of cell migration ability at both 10 and 24 h assay period (Fig. 4a). Analysis of HCC cell migration using transwell assay showed that the number of migrated cells was significantly decreased in PRSS3 overexpressing HepG2 and PLC/PRF/5 cells (Fig. 4b). Consistent with this, the invasion ability of HCC cells in Matrigel was significantly reduced by overexpression of PRSS3 in HepG2 and PLC/PRF/5 cells as shown in Fig. 4c. In contrast, the motility of SNU-387 cells transfected with siPRSS3 was markedly increased (Fig. 4d). Moreover, Western blotting demonstrated that the expression of matrix metallopeptidase 2 (MMP2), an important regulator of cancer invasion and metastasis, was markedly decreased in PRSS3 transfected HCC cells and increased in PRSS3 knockdown SNU-387 cells (Fig. 4e). These results suggest that PRSS3 inhibits the in vitro invasion and metastasis potential of HCC cells associated with downregulation of MMP2.
Fig. 4.
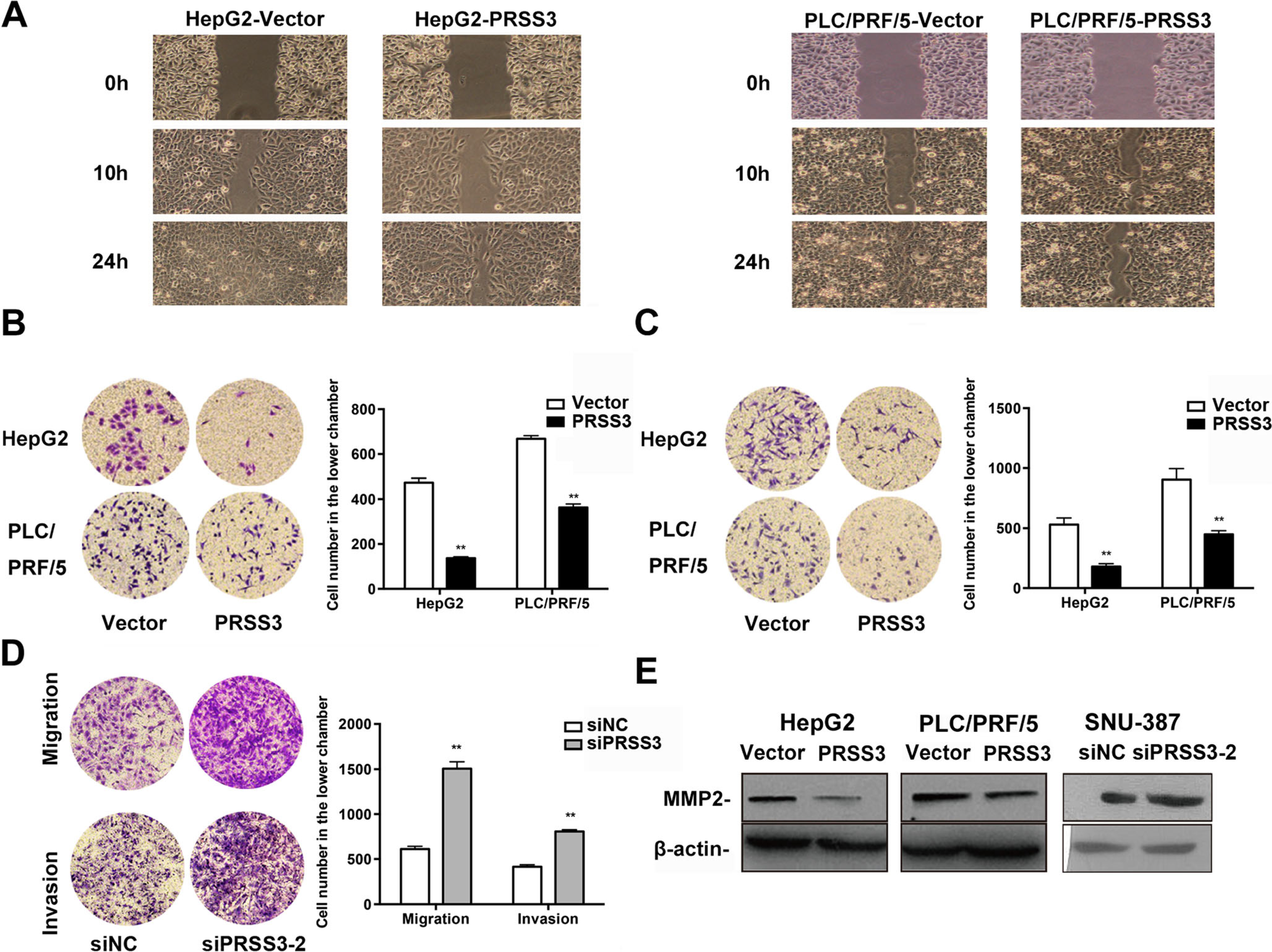
PRSS3 effect on the migration and invasion capabilities of HCC cells. a Cell monolayer scratching assay was performed to monitor the cell migration ability of HepG2 and PLC/PRF/5 cells with or without ectopic PRSS3 overexpression. b, c Cell movement ability of HepG2 and PLC/PRF/5 cells with or without PRSS3 overexpression was assessed using transwell migration assay (b) and invasion assay (c), respectively. Left panel: representative image; right panel: quantitative analysis. d Transwell migration and invasion assays of SUN387 cells transfected with siPRSS3–2 or siNC. Left panel: representative image; right panel: quantitative analysis. e Western blotting of the levels of MMP2 in the transfected HCC cell lines. Data from at least three times independent experiments is presented as the mean ± SD, *p < 0.05, versus control
PRSS3 was involved in MAPK/ERK signaling in HCC cells
In many types of cancer cells, including HCC, cell cycle progression from G0/G1 to S phase was associated with activation of CCND1/CDK4 and CCNE1/CDK2 complexes via mitogen-activated protein kinase/extracellular-signal-regulated kinase (MAPK/ERK) pathway [2, 3, 31–34]. The biofunctions of MMPs and proteases have been shown to be involved in MAPK/ERK pathway [7, 16]. To explore the molecular mechanisms of the functions of PRSS3 in HCC, we tested the effect of PRSS3 on the expression and phosphorylation of MEK1/2 and ERK1/2, important regulators in MAPK/ERK pathway. As shown in Fig. 5, overexpression of PRSS3 in HCC cells reduced the phosphorylation of MEK1/2 and ERK1/2 (p-MEK1/2 and p-ERK1/2), without affecting the expression of total proteins. Knocking down PRSS3 in SNU-387 cell line increased p-MEK1/2 and p-ERK1/2 without changing total MEK1/2 and ERK1/2. These results indicate that PRSS3 gene exerts suppressive functions in HCC cell proliferation, migration, and invasion possibly mediated by inhibition of MAPK/ERK signal pathway.
Fig. 5.

PRSS3 impairs MAPK/ERK signaling in HCC cells. Western blotting analysis of the levels of p-ERK1/2, ERK1/2, p-MEK1/2, and MEK1/2 proteins in PLC/PRF/5 and HepG2 cells with or without ectopic expression of PRSS3 or in SNU-387 cells transfected with siPRSS3–2 or control siNC
PRSS3 gene methylation in primary HCC was associated with tumor differentiation
To explore the clinical relevance of PRSS3 methylation in human primary HCC, we examined the intragenic methylation of PRSS3 gene in 66 cases of primary HCC tumor tissues (Table 1) and 20 cases of normal hepatic tissue specimens. Among 66 HCC tissues, 25 cases were available with paired adjacent non-tumor tissues and were determined by IHC for PRSS3 expression (Fig. 1b, c). As shown in Fig. 6a, b, we found that the intragenic region of PRSS3 gene was unmethylated in all 20 cases of normal hepatic tissues but was densely or partially methylated in 80.30% (53 of 66) of primary HCC tissues (Fig. 6c, p < 0.05). In the 25 paired tissue samples, 16 of 18 HCC samples with PRSS3 intragenic methylation were associated with low or reduced expression of PRSS3 protein (Fig. 6d, p < 0.05), suggesting that in primary HCC, decreased PRSS3 expression correlated with PRSS3 intragenic methylation. Analysis of the correlation between PRSS3 methylation and clinicopathologic features in 66 patients with HCC revealed that PRSS3 intragenic methylation was significantly associated with tumor differentiation (p < 0.05), but not with gender, age, liver cirrhosis, tumor size, and TNM stage (Table 1). These data indicate that epigenetic silencing of PRSS3 gene by its intragenic hypermethylation may play an important role in the development of HCC.
Fig. 6.

Methylation of PRSS3 in association with downregulation of PRSS3 in primary liver cancer. a, b MSP analysis of PRSS3 intragenic methylation in HCC tissues. Agarose gel electrophoresis showed partial results from MSP analysis of PRSS3 intragenic methylation in human normal liver tissues (a) and primary liver cancer specimens (b). c MSP analysis of PRSS3 methylation in the specimens from human HCC patients (n = 66) or in human normal liver tissues (n = 20). The clinicopathologic data of the patients are shown in Table 1. d Correlation analysis between PRSS3 methylation and its expression in the tumor and paired adjacent tissue samples from 25 patients with primary HCC. *p < 0.05
Discussion
Although PRSS3 has been implicated in cancer progression [6, 7, 10, 16, 35], the full spectrum of the expression and molecular functions of PRSS3 gene has not been established. In the present study, we showed for the first time that PRSS3 gene was epigenetically silenced in HCC cell lines and clinical specimens due to its intragenic methylation. Clinicopathologically, the intragenic methylation of PRSS3 gene in primary HCC tissues was significantly associated with the loss or reduced expression of PRSS3, and corresponded with tumor differentiation. Moreover, gain- and loss-function studies support a tumor suppressor gene function of PRSS3 in HCC cells potentially through downregulation of MAPK/ERK signal pathway.
PRSS3 transcripts consist of a mixed population of mRNAs and appear to be upregulated in some epithelial cancers, such as cancers of pancreas [20], colon [16], prostate [18], breast [36], ovary [21, 37], and neuroblastoma [23], in which PRSS3 plays an essential role in facilitating tumor cell growth, invasion and metastasis. However, several recent studies showed that PRSS3 gene was epigenetically silenced and may function as a tumor suppressor [24, 26, 38, 39]. Moreover, there were controversial reports on the expression of trypsinogens even in the same type of cancer, such as non-small cell lung cancer (NSCLC) [25, 36]. In these reports, PRSS3 gene aberrantly expressed was in fact of different splice variants (Supplementary Table 2 and Supplementary Fig. 1A). In this study, we showed that TRY4, but not other PRSS3 variants, was preferentially silent in HCC cells. Despite the fact that the high similarity to the structures of different trypsinogen isoenzymes made it difficult to distinguish their effects from the distributions in different tissues, the conflicting results reported suggest that the protumor or antitumor properties of PRSS3 depend on the cellular source and cancer microenvironment, as well as the differential expression of PRSS3 variants at both mRNA and protein levels [6].
Among multiple steps attributable to aberrant gene expression and hepatocarcinogenesis is DNA methylation associated epigenetic silencing of tumor suppressor genes leading to malignant development [4, 5]. In addition to DNA hypermethylation in the promoter region associated with gene repression and cancer development, recent studies have shown that intragenic or gene body methylation plays a role in transcriptional regulation and efficiency and is tightly associated with splicing variants by altering chromatin structure and elongation efficiency potentially in a cell-/tissue-type specific manner [40–44]. Most recently, we reported that silencing of H+, K+-ATPase Beta gene (ATP4B) due to intragenic epigenetic alteration tissue specifically in human gastric cancer cells represents a promising target for future efforts to develop diagnostic biomarkers as well as chemotherapeutic intervention of gastric cancer [27]. Although the biological significance of the differentially expressed PRSS3 variants across tissues remains unclear, the epigenetic silencing of PRSS3 gene by intragenic methylation observed in HCC suggests its potential tumor suppressor role. In addition, the unexpected results of PRSS3 expression in SUN-387 cells distinguished from other HCC cell lines, as well as immortalized liver embryonic cell line L02 in present study, may imply a cell-type specific epigenetic alteration effect on cancer cells to acquire stemness properties during cancer progression such as invasion and metastasis.
In evaluating the putative role of PRSS3 in HCC cell lines, we found that PRSS3 exerts tumor-suppressive functions in human HCC by inducing G1/S cell cycle arrest and associated with suppressing the complexes of CCND1/CDK4 and CCNE1/CDK2. The CCND1 is a key cell cycle regulator by forming a complex with CDK4 to control cell cycle progression through the G1 phase and to promote the cell cycle transition into S phase by activation of CCNE1/CDK2 complex [31–33]. The disruption of CCND1/CDK4 or CCNE1/CDK2 in mouse hepatocytes led to impaired S-phase progression [33]. Thus, the tumor suppressor role of PRSS3 in hepatocellular carcinogenesis may be attributed to its capacity to reduce G1/S cell cycle transition.
Metastasis and recurrence are the main contributors to the poor outcome of HCC patients. In the present study, we showed significant association of epigenetic silencing of PRSS3 gene with poor differentiation of HCC, indicating an important role of PRSS3 gene in control of tumor cell invasion and metastasis. Further investigation showed that the in vitro invasion and migration of HCC were attenuated by overexpressing PRSS3 but enhanced by the downregulation of PRSS3, in which the expression of PRSS3 was accompanied with an opposite alteration of MMP2. MMPs and serine proteases are important proteins associated with cancer invasion and metastasis due to their ability to degrade extracellular matrix to promote the dissemination of cancer cells into adjacent normal tissues [7, 16, 34, 45]. Increased expression of MMP2 mediated by MAPK/ERK signaling pathway was detected in the tissues samples of patients with HCC, as a prognostic factor for poor outcomes of HCC patients [34, 45, 46]. It is also well known that tumor-associated trypsins, such as PRSS1 and 2, contribute to tumor proliferation, invasion, and metastasis by breaking down extracellular matrix and cellular adhesion molecules, inducing chemokines, growth factors, growth factor receptors, and other signaling receptors. The tumor-suppressive function of PRSS3 shown in our study may be attributable to the different expression of PRSS3 variants regulated by epigenetic modifications in tissues, among which, the expression of TRY4 and its functions might be relevant to the liver other than the pancreas due to the lack of a signal peptide for extracellular secretion [6, 9, 10]. Our results thus provide additional evidence to support the dual roles of proteases in carcinogenesis [6].
MAPK/ERK signaling plays important roles in the development of HCC [2, 3, 31, 34]. For example, phosphorylation of ERK enhances cancer cell proliferation and invasion via regulation of CDK4/CCND1, which, in turn, promote the cell cycle [2, 32]. PRSS3 has been shown to exert its proteolytic enzyme activity after secretion in many physiological and pathological processes through activation of protease-activated receptors (PARs) [14, 47–49]. Despite conflicting studies showing that mesotrypsin cannot activate epithelial PARs [50], supportive evidence suggests that mesotrypsin-mediated PAR activation is implicated in triggering the phosphorylation of MAPK/ERK pathway, resulting in tumor cell proliferation, migration, invasion, and metastasis [17,20, 47, 51]. Recently, Lau et al. showed that PAR-2 presenting on intestinal epithelium apically or basolaterally directed distinct signaling events through promoting or delaying ERK1/2 activation [52], suggesting the dual role of PAR2 in inflammation and tumor metastasis attributable to its localization. In this study, phosphorylated MEK1/2 and ERK1/2 were reduced following PRSS3 overexpression suggesting the effect of PRSS3 on MAPK/ERK signaling pathway. Thus, PRSS3 plays a tumor-suppressor role in HCC via inhibiting MAPK/ERK pathway. This leads to downregulation of CDK4/CCND1 and G1/S phase arrest in HCC cells.
Our study showed that epigenetic silencing of PRSS3 gene by intragenic hypermethylation facilitates the growth, migration, and invasion of HCC, suggesting an epigenetic dependent effect on tissue/cell-type specific expression of PRSS3 variants in HCC. Identification of PRSS3 variants regulated by specific interplay between epigenetic marks could add value towards diagnostics and treatment of HCC. Also, the tumor-suppressor function of PRSS3 gene linked to MARK/ERK pathway provides a molecular basis for the ability of PRSS3 to suppress progression and metastasis in human HCC. Therefore, further research is needed to delineate the precise molecular mechanisms for the effects of PRSS3 on HCC in relation to epigenetic regulation of PRSS3 variants.
Supplementary Material
Key messages.
PRSS3 is downregulated by intragenic hypermethylation in HCC.
Epigenetic silencing of PRSS3 facilitates growth, migration, and invasion of HCC.
PRSS3 intragenic methylation has implication in diagnosis of HCC.
Acknowledgements
We thank Dr. Keqiang Chen for help with the data interpretation. This work was supported by the following grants: National Key Scientific Instrument Special Program of China (Grant No.2011YQ03013405); National Science Foundation of China (NSFC No.8167318, U1604281, 81230047, 81490753, 81121004, 81402345); Beijing Science Foundation of China (BJSFC No. 7171008); and Translational foundation of Chinese PLA General Hospital (2016ZHJJMS-GMZ). JH and JMW were also funded in part by Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and were supported in part by the Intramural Research Program of the NCI, NIH. JH was also supported by the Intramural Research funding of Beijing Jiaotong University (S12RC00030).
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Compliance with ethical standards
Ethics approval This study was conducted with the approval of the Institutional Review Board of the Chinese PLA General Hospital and with informed consent from patients.
Electronic supplementary material The online version of this article (doi:10.1007/s00109-017-1578-5) contains supplementary material, which is available to authorized users.
References
- 1.El-Serag HB (2011) Hepatocellular carcinoma. N Engl J Med 365: 1118–1127 [DOI] [PubMed] [Google Scholar]
- 2.Feo F, Frau M, Pascale RM (2008) Interaction of major genes predisposing to hepatocellular carcinoma with genes encoding signal transduction pathways influences tumor phenotype and prognosis. World J Gastroenterol 14:6601–6615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frenette C, Gish RG (2011) Hepatocellular carcinoma: molecular and genomic guideline for the clinician. Clin Liver Dis 15:307–321, vii-x [DOI] [PubMed] [Google Scholar]
- 4.Mann DA (2014) Epigenetics in liver disease. Hepatology 60: 1418–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pogribny IP, Rusyn I (2014) Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett 342:223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Matrisian LM (2007) Emerging roles of proteases in tumour suppression. Nat Rev Cancer 7:800–808 [DOI] [PubMed] [Google Scholar]
- 7.Nyberg P, Ylipalosaari M, Sorsa T, Salo T (2006) Trypsins and their role in carcinoma growth. Exp Cell Res 312:1219–1228 [DOI] [PubMed] [Google Scholar]
- 8.Itkonen O (2010) Human trypsinogens in the pancreas and in cancer. Scand J Clin Lab Invest 70:136–143 [DOI] [PubMed] [Google Scholar]
- 9.Sahin-Toth M (2005) Human mesotrypsin defies natural trypsin inhibitors: from passive resistance to active destruction. Protein Pept Lett 12:457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salameh MA, Radisky ES (2013) Biochemical and structural insights into mesotrypsin: an unusual human trypsin. Int J Biochem Mol Biol 4:129–139 [PMC free article] [PubMed] [Google Scholar]
- 11.Nyaruhucha CN, Kito M, Fukuoka SI (1997) Identification and expression of the cDNA-encoding human mesotrypsin(ogen), an isoform of trypsin with inhibitor resistance. J Biol Chem 272: 10573–10578 [DOI] [PubMed] [Google Scholar]
- 12.Wiegand U, Corbach S, Minn A, Kang J, Muller-Hill B (1993) Cloning of the cDNA encoding human brain trypsinogen and characterization of its product. Gene 136:167–175 [DOI] [PubMed] [Google Scholar]
- 13.Tani T, Kawashima I, Mita K, Takiguchi Y (1990) Nucleotide sequence of the human pancreatic trypsinogen III cDNA. Nucleic Acids Res 18:1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cottrell GS, Amadesi S, Grady EF, Bunnett NW (2004) Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem 279:13532–13539 [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi J, Yamamoto M, Koyama J, Sato J, Hibino T (2010) Keratinocytes synthesize enteropeptidase and multiple forms of trypsinogen during terminal differentiation. J Invest Dermatol 130:944–952 [DOI] [PubMed] [Google Scholar]
- 16.Soreide K, Janssen EA, Korner H, Baak JP (2006) Trypsin in colorectal cancer: molecular biological mechanisms of proliferation, invasion, and metastasis. J Pathol 209:147–156 [DOI] [PubMed] [Google Scholar]
- 17.Han S, Lee CW, Trevino JG, Hughes SJ, Sarosi GA Jr (2013) Autocrine extra-pancreatic trypsin 3 secretion promotes cell proliferation and survival in esophageal adenocarcinoma. PLoS One 8: e76667. doi: 10.1371/journal.pone.0076667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hockla A, Miller E, Salameh MA, Copland JA, Radisky DC, Radisky ES (2012) PRSS3/mesotrypsin is a therapeutic target for metastatic prostate cancer. Mol Cancer Res 10:1555–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hockla A, Radisky DC, Radisky ES (2010) Mesotrypsin promotes malignant growth of breast cancer cells through shedding of CD109. Breast Cancer Res Treat 124:27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang G, Cao F, Ren G, Gao D, Bhakta V, Zhang Y, Cao H, Dong Z, Zang W, Zhang S et al. (2010) PRSS3 promotes tumour growth and metastasis of human pancreatic cancer. Gut 59:1535–1544 [DOI] [PubMed] [Google Scholar]
- 21.Ma R, Ye X, Cheng H, Ma Y, Cui H, Chang X (2015) PRSS3 expression is associated with tumor progression and poor prognosis in epithelial ovarian cancer. Gynecol Oncol 137: 546–552 [DOI] [PubMed] [Google Scholar]
- 22.Ghilardi C, Silini A, Figini S, Anastasia A, Lupi M, Fruscio R, Giavazzi R, Bani MR (2015) Trypsinogen 4 boosts tumor endothelial cells migration through proteolysis of tissue factor pathway inhibitor-2. Oncotarget 6:28389–28400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei JS, Greer BT, Westermann F, Steinberg SM, Son CG, Chen QR, Whiteford CC, Bilke S, Krasnoselsky AL, Cenacchi N et al. (2004) Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res 64:6883–6891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marsit CJ, Karagas MR, Schned A, Kelsey KT (2006) Carcinogen exposure and epigenetic silencing in bladder cancer. Ann N YAcad Sci 1076:810–821 [DOI] [PubMed] [Google Scholar]
- 25.Marsit CJ, Okpukpara C, Danaee H, Kelsey KT (2005) Epigenetic silencing of the PRSS3 putative tumor suppressor gene in non-small cell lung cancer. Mol Carcinog 44:146–150 [DOI] [PubMed] [Google Scholar]
- 26.Yamashita K, Mimori K, Inoue H, Mori M, Sidransky D (2003) A tumor-suppressive role for trypsin in human cancer progression. Cancer Res 63:6575–6578 [PubMed] [Google Scholar]
- 27.Lin S, Lin B, Wang X, Pan Y, Xu Q, He JS, Gong W, Xing R, He Y, Guo L et al. (2017) Silencing of ATP4B of ATPase H+/K+ transporting beta subunit by intragenic epigenetic alteration in human gastric cancer cells. Oncol Res 25:317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu H, Wu K, Yan W, Hu L, Yuan J, Dong Y, Li Y, Jing K, Yang Y, Guo M (2013) Epigenetic silencing of DACH1 induces loss of transforming growth factor-beta1 antiproliferative response in human hepatocellular carcinoma. Hepatology 58:2012–2022 [DOI] [PubMed] [Google Scholar]
- 29.Pan Y, Lin S, Xing R, Zhu M, Lin B, Cui J, Li W, Gao J, Shen L, Zhao Y et al. (2016) Epigenetic upregulation of metallothionein 2A by diallyl trisulfide enhances chemosensitivity of human gastric cancer cells to docetaxel through attenuating NF-kappaB activation. Antioxid Redox Signal 24:839–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowen L, Williams E, Glusman G, Linardopoulou E, Friedman C, Ahearn ME, Seto J, Boysen C, Qin S, Wang K et al. (2005) Interchromosomal segmental duplications explain the unusual structure of PRSS3, the gene for an inhibitor-resistant trypsinogen. Mol Biol Evol 22:1712–1720 [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C (2006) Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 66:11851–11858 [DOI] [PubMed] [Google Scholar]
- 32.Massague J (2004) G1 cell-cycle control and cancer. Nature 432: 298–306 [DOI] [PubMed] [Google Scholar]
- 33.Jena N, Deng M, Sicinska E, Sicinski P, Daley GQ (2002) Critical role for cyclin D2 in BCR/ABL-induced proliferation of hematopoietic cells. Cancer Res 62:535–541 [PubMed] [Google Scholar]
- 34.Yoshida T, Hisamoto T, Akiba J, Koga H, Nakamura K, Tokunaga Y, Hanada S, Kumemura H, Maeyama M, Harada M et al. (2006) Spreds, inhibitors of the Ras/ERK signal transduction, are dysregulated in human hepatocellular carcinoma and linked to the malignant phenotype of tumors. Oncogene 25:6056–6066 [DOI] [PubMed] [Google Scholar]
- 35.Radisky ES (2013) PRSS3/mesotrypsin in prostate cancer progression: implications for translational medicine. Asian J Androl 15: 439–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diederichs S, Bulk E, Steffen B, Ji P, Tickenbrock L, Lang K, Zanker KS, Metzger R, Schneider PM, Gerke V et al. (2004) S100 family members and trypsinogens are predictors of distant metastasis and survival in early-stage non-small cell lung cancer. Cancer Res 64:5564–5569 [DOI] [PubMed] [Google Scholar]
- 37.Hirahara F, Miyagi Y, Miyagi E, Yasumitsu H, Koshikawa N, Nagashima Y, Kitamura H, Minaguchi H, Umeda M, Miyazaki K (1995) Trypsinogen expression in human ovarian carcinomas. Int J Cancer 63:176–181 [DOI] [PubMed] [Google Scholar]
- 38.Marsit CJ, Karagas MR, Danaee H, Liu M, Andrew A, Schned A, Nelson HH, Kelsey KT (2006) Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis 27:112–116 [DOI] [PubMed] [Google Scholar]
- 39.Martinez I, Wang J, Hobson KF, Ferris RL, Khan SA (2007) Identification of differentially expressed genes in HPV-positive and HPV-negative oropharyngeal squamous cell carcinomas. Eur J Cancer 43:415–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y et al. (2010) Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466:253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Jadhav RR, Liu J, Wilson D, Chen Y, Thompson IM, Troyer DA, Hernandez J, Shi H, Leach RJ et al. (2016) Roles of distal and genic methylation in the development of prostate tumorigenesis revealed by genome-wide DNA methylation analysis. Sci Rep 6:22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK (2012) On the presence and role of human gene-body DNA methylation. Oncotarget 3:462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM (2011) DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One 6:e14524. doi: 10.1371/journal.pone.0014524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lorincz MC, Dickerson DR, Schmitt M, Groudine M (2004) Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol 11:1068–1075 [DOI] [PubMed] [Google Scholar]
- 45.Daniele A, Divella R, Quaranta M, Mattioli V, Casamassima P, Paradiso A, Garrisi VM, Gadaleta CD, Gadaleta-Caldarola G, Savino E et al. (2014) Clinical and prognostic role of circulating MMP-2 and its inhibitor TIMP-2 in HCC patients prior to and after trans-hepatic arterial chemo-embolization. Clin Biochem 47:184–190 [DOI] [PubMed] [Google Scholar]
- 46.Golubnitschaja O, Yeghiazaryan K, Stricker H, Trog D, Schild HH, Berliner L (2016) Patients with hepatic breast cancer metastases demonstrate highly specific profiles of matrix metalloproteinases MMP-2 and MMP-9 after SIRT treatment as compared to other primary and secondary liver tumours. BMC Cancer 16:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darmoul D, Gratio V, Devaud H, Laburthe M (2004) Protease-activated receptor 2 in colon cancer: trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J Biol Chem 279:20927–20934 [DOI] [PubMed] [Google Scholar]
- 48.Su S, Li Y, Luo Y, Sheng Y, Su Y, Padia RN, Pan ZK, Dong Z, Huang S (2009) Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene 28:3047–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knecht W, Cottrell GS, Amadesi S, Mohlin J, Skaregarde A, Gedda K, Peterson A, Chapman K, Hollenberg MD, Vergnolle N et al. (2007) Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J Biol Chem 282:26089–26100 [DOI] [PubMed] [Google Scholar]
- 50.Grishina Z, Ostrowska E, Halangk W, Sahin-Toth M, Reiser G (2005) Activity of recombinant trypsin isoforms on human proteinase-activated receptors (PAR): mesotrypsin cannot activate epithelial PAR-1, −2, but weakly activates brain PAR-1. Br J Pharmacol 146:990–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV (2015) Protease-activated receptors (PARs)-biology and role in cancer invasion and metastasis. Cancer Metastasis Rev 34:775–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lau C, Lytle C, Straus DS, DeFea KA (2011) Apical and basolateral pools of proteinase-activated receptor-2 direct distinct signaling events in the intestinal epithelium. Am J Physiol Cell Physiol 300:C113–C123 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.