

Abstract
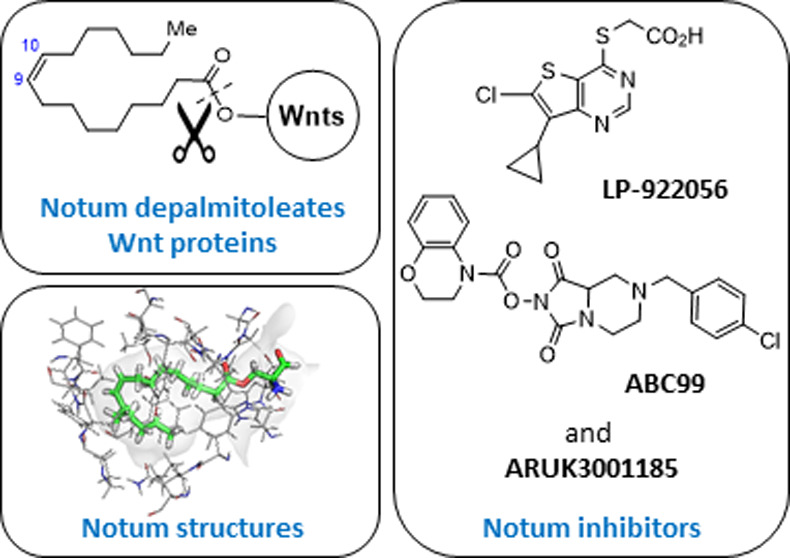
Regulation of the Wnt signaling pathway is critically important for a number of cellular processes in both development and adult mammalian biology. This Perspective will provide a summary of current and emerging therapeutic opportunities in modulating Wnt signaling, especially through inhibition of Notum carboxylesterase activity. Notum was recently shown to act as a negative regulator of Wnt signaling through the removal of an essential palmitoleate group. Inhibition of Notum activity may represent a new approach to treat disease where aberrant Notum activity has been identified as the underlying cause. Reliable screening technologies are available to identify inhibitors of Notum, and structural studies are accelerating the discovery of new inhibitors. A selection of these hits have been optimized to give fit-for-purpose small molecule inhibitors of Notum. Three noteworthy examples are LP-922056 (26), ABC99 (27), and ARUK3001185 (28), which are complementary chemical tools for exploring the role of Notum in Wnt signaling.
Introduction to Wnt Signaling
During development and adult homeostasis, cells use secreted proteins to communicate and coordinate their activities. Most of the known signaling proteins fall into a relatively small number of families. One such family encompasses the Wnt proteins (pronounced “wint”), which were discovered independently by developmental biologists and cancer biologists. Indeed, the name Wnt is a fusion of wingless, a gene required for Drosophila wing specification1 and embryo patterning,2 with int-1, a viral integration site associated with mammary tumors.3 Thus, from the start, Wnt genes embody the molecular links between normal development and tumorigenesis. Since their original identification, Wnt proteins have been found across metazoans ranging from worms to humans.4 Subsequent work to identify all the key signal transduction components further highlighted the highly conserved nature of the Wnt signaling pathway. Canonical signaling is initiated by binding of a Wnt to a member of the Frizzled (FZD) family of receptor, leading to recruitment of the co-receptor LRP5/65 and inactivation of a protein complex that normally degrades β-catenin.6 Thus, in the presence of Wnt, β-catenin accumulates and, along with a number of cofactors,7 triggers the transcriptional activation of target genes. Wnt also activates noncanonical signaling, for example, the planar cell polarity pathway, which is also initiated by binding to a FZD receptor but does not involve LRP5/6 and β-catenin.8 In a minor pathway, Wnts can also exert their influence independently of FZD receptors, via other receptors such as ROR or RYK.9 Here we focus on FZD-dependent Wnt signaling, which accounts for most of Wnt proteins’ activities (Figure 1).
Figure 1.

Overview of the canonical Wnt signaling pathway.
A characteristic feature of Wnts (including all 19 human Wnts) is that they are post-translationally appended with a palmitoleate moiety.10,11 Without this lipid, the affinity of Wnts for FZD is greatly reduced, an observation that can be readily understood from structural studies, which showed that the Wnt lipid establishes close contact with a hydrophobic groove present in the extracellular domain of FZD.12 As expected therefore, Wnt mutant proteins lacking the conserved serine that is normally palmitoleoylated display low activity in vivo.13,14 Likewise, genetic abrogation of Porcupine, a membrane-bound O-acyl transferase (MBOAT) solely devoted to Wnt palmitoleoylation, phenocopies inactivation of all Wnts.11,15,16 The same effect is achieved by chemical inhibitors of Porcupine,17 which are currently undergoing clinical trials for the treatment of cancers caused by Wnt overexpression. Thus, classical and chemical genetic approaches have highlighted the importance of the Wnt lipid.
Loss of function studies have shown that Wnt proteins control a wide range of physiological functions such as vertebrate axis specification, growth control, bone regeneration, synaptogenesis, stem cell maintenance, and many more.18−20 Conversely, excess or ectopic Wnt signaling has been associated with various diseases, including cancer, vascular disease, Alzheimer’s disease, and developmental defects. Most prominently, overactivation of Wnt signaling is thought to account for the majority of colorectal cancers and to be a key contributor to many other cancers.21 It is clear therefore that Wnt signaling needs to be finely balanced in time and space such that it is high enough to control essential functions such as stem cell maintenance, while avoiding the deleterious effects of excess signaling. This is achieved by an array of feedback mechanisms that modulate the spread of Wnt ligands, their activity, receptor availability, or the function of intracellular signal transduction components.22 Here, we consider one such mechanism, that mediated by the secreted protein Notum.
This Perspective will provide a summary of current and emerging therapeutic opportunities in modulating Wnt signaling, especially through inhibition of Notum carboxylesterase activity. Notum is emerging as a druggable target to modulate Wnt signaling. Our emphasis will be on structural biology, modern hit finding approaches, and medicinal chemistry strategies to deliver fit-for-purpose chemical tools. Three noteworthy examples of inhibitors of Notum activity are LP-922056 (26), ABC99 (27), and ARUK3001185 (28), which are complementary chemical tools for exploring the role of Notum in Wnt signaling.
Wnt Pathway and Determination of Notum Function
Notum was identified independently by two groups studying the regulation of Wingless signaling in Drosophila. In the group of Gerlitz and Basler, an enhancer trap screen for genes that are activated by Wingless signaling led to the identification of a mutation that caused an expansion of presumptive wing tissue.23 The corresponding gene was hence named wingful. At the same time, Giraldez et al. found that overexpression of what turned out to be the same gene caused the opposite phenotype, the loss of wing tissue,24 as well as enlargement of the notum, an anatomical structure at the back of the fly. The latter phenotype led the authors to name the gene notum, a name that somehow superseded wingful. Both studies showed that Notum is a target of Wingless signaling and that its protein product is a potent inhibitor of Wingless signaling. In Drosophila, Notum is probably a universal feedback inhibitor since its expression is activated at all known locations of Wingless signaling.25 In mice, Notum is also expressed at many,26 though not all, sites of Wnt signaling, including in β-catenin-driven tumors.27 Therefore, in vertebrates as in flies, Notum is a feedback inhibitor that contributes to dampening Wnt signaling.
How does Notum inhibit Wnt signaling? The presence of a signal peptide indicated at the outset that Notum acts in the extracellular space. Protein sequence similarity between Notum and plant pectin acetylesterases suggested that Notum could modify the glycosaminoglycans of glypicans, glycosylphosphatidylinositol (GPI)-anchored proteoglycans to which Wnts and other growth factors are known to bind.24 However, subsequent biochemical experiments cast doubt on this model and suggested instead that Notum could be a phospholipase that cleaves the GPI anchor of glypicans thus releasing them from the cell surface, along with any bound Wnt.28 If the molecular target of Notum was really a glypican, one would expect more pleiotropic effects since a number of extracellular proteins besides Wnts bind to glypicans (e.g., Hedgehog, FGF, BMP). Yet, genetic analysis showed that Notum is a Wnt-specific inhibitor.29 The phospholipase model could be unambiguously excluded by structural analysis and enzymatic assays, which showed that Notum is a carboxylesterase (Figure 2).29
Figure 2.

(A) Chemical reaction of the carboxylesterase activity of Notum acting on Wnt to remove the palmitoleate group. (B) Two-dimensional schematic of binding interactions. The Gly127–Trp128 amide participates in formation of the oxyanion hole in addition to the canonical Ser232–Ala233 and Gly126–Gly127 amides. Catalytic triad (Ser232, His389, Asp340) in bold.
The crystal structure of Notum also revealed a hydrophobic pocket that could accommodate cis-palmitoleate, an observation that naturally led to the hypothesis that Notum could hydrolyze the O-linkage of palmitoleate to Wnts. This was indeed verified in vitro by MALDI analysis of palmitoleoylated peptides treated with recombinant Notum. Interestingly, structural analysis did uncover several heparin binding sites at the surface of Notum. Such sites not only account for Notum’s ability to bind glypicans, they also provide a molecular framework for the documented genetic interactions between Notum and glypicans. It is now accepted that Notum is a glypican-dependent Wnt deacylase.29
Small Molecule Approaches to Modulate Wnt Signaling
The strong links between aberrant Wnt signaling and multiple human diseases has promoted significant interest in the Wnt pathway machinery as targets for drug therapies. To date, most efforts have been directed at inhibiting Wnt signaling, in particular signaling through the canonical pathway and ultimately β-catenin regulated gene transcription.30 The most appropriate target(s) for intervention in the Wnt signaling cascade has been the topic of much debate as there needs to be clear therapeutic benefit (efficacy) with a suitable safety index.31,32
A number of recent reviews have summarized the state of the art with regard to Wnt inhibition with a focus on cancer therapeutics.33−36 As such, discussion in this section will be limited to Wnt pathway inhibitors either that are in clinical trials or for which there is structural information on the ligand–protein interaction. Small molecule approaches toward activating or enhancing Wnt signaling are also described. The application of biological agents that modulate the Wnt pathway that have progressed to clinical trials will be briefly discussed for comparison (Tables 1 and 2; Figure 3).
Table 1. Selected Small Molecule Tools Targeting Wnt Pathway Components.
| compound identifier | molecular target | effect on canonical Wnt signaling | structural information in public domain | furthest progress to date | refs |
|---|---|---|---|---|---|
| ICG-001, 5 | CBP | inhibit | no | preclinical | (45) |
| IWP-2, 9 | Porcupine | inhibit | no | preclinical | (51) |
| endo-IWR-1, 11 | TNKS1/2 | inhibit | yes | preclinical | (51) |
| XAV939, 12 | TNKS1/2 | inhibit | yes | preclinical | (57,59) |
| G007-LK | TNKS1/2 | inhibit | yes | preclinical | (60,63) |
| NVP-TNKS656 | TNKS1/2 | inhibit | no | preclinical | (61) |
| MSC2504877 | TNKS1/2 | inhibit | no | preclinical | (62) |
| BML-268, 13 | DVL3 | inhibit | yes | early discovery | (65) |
| NPL-4011, 14 | DVL3 | inhibit | yes | early discovery | (66) |
| NSC654259, 15 | FZD8 | inhibit | yes | early discovery | (67) |
| carbamazepine, 16 | FZD8 | inhibit | yes | preclinicala | (68) |
| SRI37892, 17 | FZD7 | inhibit | yes | early discovery | (70) |
| gallocyanine, 21 | LRP6 | enhance | yes | early discovery | (77) |
| WAY-362692, 22 | sFRP-1 | enhance | no | preclinical | (82−84) |
| WAY-262611, 23 | DKK1 | enhance | no | preclinical | (85) |
| LP-922056, 26 | Notum | enhance | yes | preclinical | (95) |
| ABC99, 27 | Notum | enhance | no | preclinical | (118) |
| ARUK3001185, 28 | Notum | enhance | no | preclinical | (126) |
Carbamazepine is an approved anticonvulsant drug used clinically in the treatment of epilepsy and neuropathic pain.
Table 2. Summary of Agents in Clinical Trials.
| compound identifier | molecular target | clinical phase | NCT reference | indications |
|---|---|---|---|---|
| SM04690, 1 | CLK2/DYRK1A | phase 2 | NCT03727022 | knee osteoarthritis |
| phase 2 | NCT03706521 | knee osteoarthritis | ||
| SM04755, 2 | CLK2/DYRK1A | phase 2 | NCT03229291 | tendinopathy |
| phase 1 | NCT02191761 | colorectal, gastric, hepatic and pancreatic cancer | ||
| phase 1 | NCT03502434 | tendinopathy | ||
| SM08502, 3 | pan-CLKs/DYRK1A | phase 1 | NCT03355066 | advanced solid tumors |
| SM04554, 4 | not reported | phase 2/3 | NCT03742518 | androgenetic alopecia |
| phase 2 | NCT02503137 | androgenetic alopecia | ||
| phase 2 | NCT02275351 | androgenetic alopecia | ||
| PRI-724, 6 | CBP | phase 2 | NCT03620474 | hepatitis b and c, liver cirrhosis |
| NCT04047160 | primary biliary cholangitis, liver cirrhosis | |||
| C-82, 7 | CBP | phase 2 | NCT02349009 | systemic scleroderma |
| NCT02432027 | psoriasis | |||
| CWP-291, 8 | Sam68 | phase 1/2 | NCT03055286 | acute myeloid leukemia |
| phase 1 | NCT02426723 | multiple myeloma | ||
| phase 1 | NCT01398462 | acute myeloid leukemia, myelofibrosis | ||
| LGK974, 10 | Porcupine | phase 1 | NCT02278133 | metastatic colorectal cancer |
| phase 1 | NCT01351103 | multiple solid malignancies | ||
| phase 2 | NCT02649530 | squamous cell carcinoma (withdrawn) | ||
| OMP-54F28, 18 | Wnts | phase 1 | NCT02069145 | liver cancer |
| phase 1 | NCT02092363 | ovarian cancer | ||
| phase 1 | NCT02050178 | pancreatic cancer | ||
| phase 1 | NCT01608867 | solid tumors | ||
| OMP-18R5, 19 | FZD 1,2,5,7,8 | phase 1 | NCT01345201 | solid tumors |
| phase 1 | NCT02005315 | pancreatic cancer | ||
| phase 1 | NCT01957007 | solid tumors | ||
| phase 1 | NCT01973309 | metastatic breast cancer | ||
| BI-905677, 20 | LRP5/LRP6 | phase 1 | NCT03604445 | solid tumors |
| DKN-01, 24 | DKK1 | phase 2 | NCT03645980 | liver cancer |
| phase 2 | NCT04166721 | metastatic esophageal and gastric cancer | ||
| phase 2 | NCT03837353 | prostate cancer | ||
| phase 2 | NCT03395080 | gynecological cancers | ||
| Foxy-5, 25 | Wnt5a | phase 1 | NCT02655952 | metastatic breast, colon and prostate cancer |
| phase 2 | NCT03883802 | colon cancer |
Figure 3.
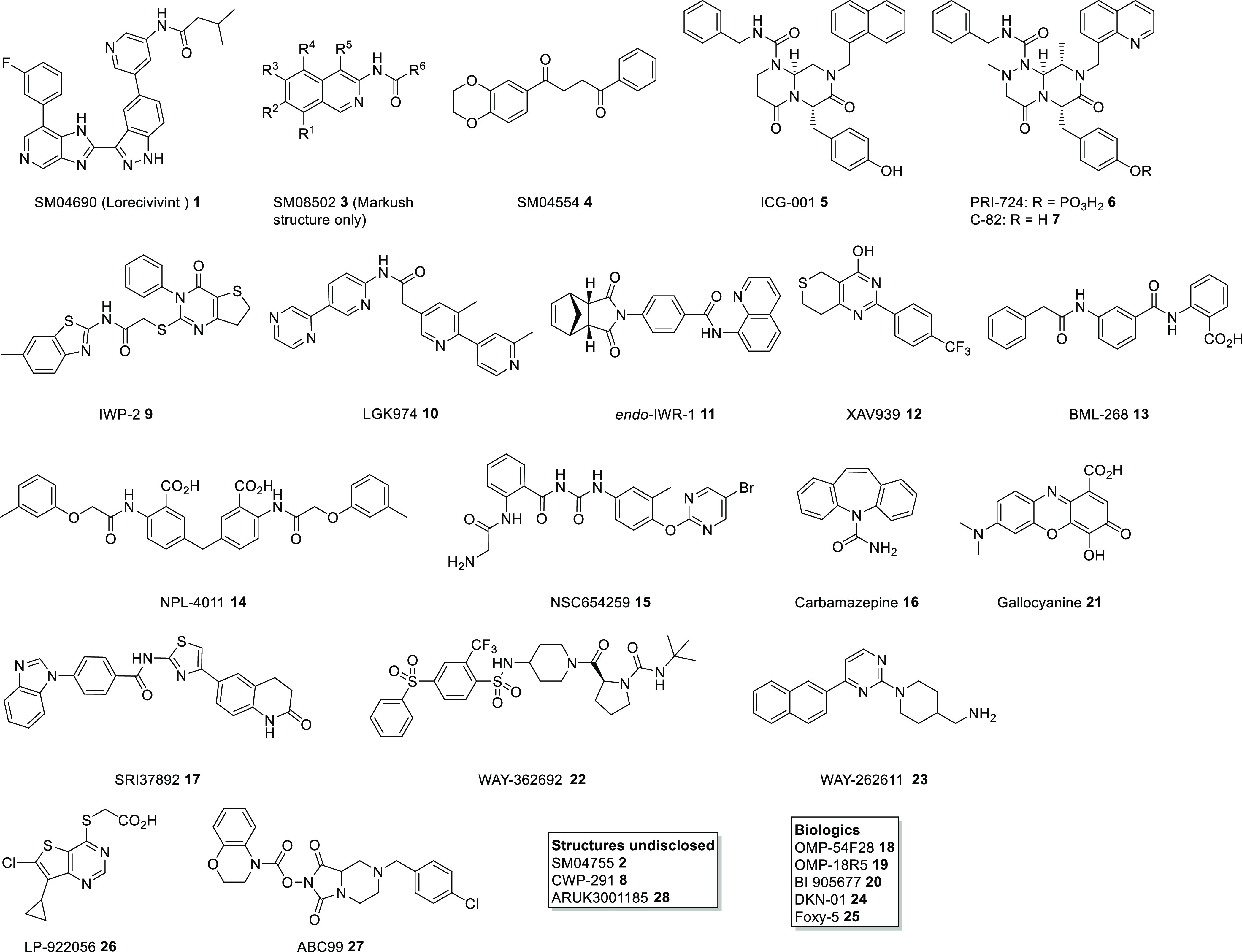
Chemical structures of selected small molecules modulating the Wnt pathway.
Wnt Pathway Inhibitors and Activators
A popular approach for investigating small molecule mediated modulation of Wnt activity has been through cell-based functional assays, such as TOP-flash, in which cells transiently or stably express luciferase proteins under the TCF/LEF promoter.37 A large number of compounds have been reported to inhibit Wnt signaling using this approach, in particular natural products,38 many without apparent follow-up or target identification. Compounds of interest without disclosed molecular targets will be discussed here. A number of compounds that modify Wnt signaling have been disclosed by Samumed LLC and are currently in clinical trials. SM04690 (1, lorecivivint) was recently reported as a highly potent (EC50 20 nM), selective Wnt pathway inhibitor in a TCF/LEF reporter system in SW480 colon cancer cells.39 It was shown to be effective in a number of in vitro cellular models of osteoarthritis (OA) and efficacious in a rodent model of OA and is currently being tested in two phase 2 clinical trials for knee OA. Early reports on efficacy and safety from these trials appears promising.40 Samumed recently reported a novel mechanism of action for Wnt pathway inhibition in human mesenchymal stem cells for SM04690 via dual inhibition of intranuclear kinases, CLK2 (IC50 8 nM) and DYRK1A (IC50 30 nM).41 SM04755 (2) and SM08502 (3) (undisclosed structures) have also been reported as Wnt inhibitors with activity against CLK kinases and are currently in clinical trials for various disease indications.42 Conversely, SM04554 (4) is reported as a small molecule activator of Wnt signaling that has been shown to increase total and nuclear β-catenin and increase hair growth and follicle number in a depilated murine model.43 It is currently in phase 2/3 clinical trials as a topical agent for androgenetic alopecia.
Targeting Wnt Signaling in the Nucleus
Targeting the machinery of the β-catenin/TCF response elements involved in regulating transcription provides a direct method for modulating Wnt-dependent gene expression. In general, drugging transcription factors is considered challenging,44 and although there have been reports of molecules directly binding to β-catenin,36 currently the most advanced strategies have targeted transcriptional coactivators. The first generation Wnt signaling inhibitor ICG-001 (5) was discovered through a screen with a modest 5000 compound library using a TOP-flash assay in the colon cancer SW480 cell line (IC50 3 μM). Follow-up target identification studies showed that it antagonized β-catenin/TCF dependent transcription by binding the N-terminus of the transcriptional coactivator protein CBP, blocking its interaction with β-catenin while not binding the related transcriptional coactivator p300.45 ICG-001 was found to selectively induce apoptosis in transformed colon cells but not normal colon cells and showed efficacy in xenograft mouse models of colon cancer. Structurally related second generation phosphate prodrug PRI-724 (6), developed by PRISM Pharma Co., was entered into phase 1/2 clinical trials for multiple anticancer indications. Preclinical studies in mice suggested that the β-catenin/CBP interaction was important for the onset of liver fibrosis, and this was suppressed by treatment with PRI-724.46 Clinical trials with PRI-724 are ongoing for the treatment of liver cirrhosis. C-82 (7), the active form of PRI-724, entered phase 1/2 clinical trials in 2015 as a topical agent for the treatment of the skin conditions psoriasis and systemic sclerosis.47 CWP-291 (8, formerly CWP232291) is a peptidomimetic small molecule drug precursor to the active metabolite CWP232204 (structures undisclosed) developed by JW Pharmaceuticals that reduces β-catenin levels in a TOP-flash reporter assay in HEK293 cells (IC50 273 nM) and is currently in phase 2 clinical trials for acute myeloid leukemia.48 The active form CWP-291 binds to Sam68 (an RNA-binding protein that regulates alternative splicing of the TCF-1 transcription factor in a complex with CBP) and induces β-catenin degradation.49
Targeting Wnt Signaling in the Cytoplasm
The post-translational acylation of Wnt ligands is considered to be a key step prior to its secretion and activation.11 The endoplasmic reticulum MBOAT enzyme Porcupine specifically acylates Wnt proteins at a conserved serine residue (Ser209 in hWnt3a). Inhibition of Porcupine has been shown to impair the correct processing and secretion of Wnts resulting in decreased Wnt-dependent signaling and downstream gene expression. A number of highly potent Porcupine inhibitors have been reported, and the therapeutic potential of targeting Porcupine, particularly in oncology, has been recently reviewed elsewhere.17,50 Briefly, the first inhibitors of Porcupine were discovered by Chen and co-workers who utilized a chemical genetics approach to identify two distinct class of inhibitors of Wnt signaling, one class that inhibited Wnt production (IWPs) and another group that inhibited the Wnt response (IWRs) downstream of LRP6 and Dishevelled.51 Using biochemical markers of Wnt/β-catenin pathway activation, it was confirmed that the IWPs blocked key events across the canonical Wnt signaling cascade and follow-up target identification and validation experiments (primarily using IWP-2, 9), demonstrated that this activity was due to specific, on-target inhibition of Porcupine. Despite IWPs’ clear value as tool molecules for probing Wnt signaling and Porcupine biology in an in vitro setting, their use in vivo has been more limited. LGK974 (10, WNT974), one of the first clinical candidate Porcupine inhibitors, developed by Novartis, was discovered through a ligand-based medicinal chemistry program and was shown to display exquisite potency (IC50 0.4 nM) in a Wnt reporter assay in T3 cells. LGK974 also showed dose dependent efficacy as measured by tumor growth delay and regression in a Wnt-driven murine tumor model.52 Despite the near universal importance of Wnt acylation for Wnt signaling, LGK974 was found to be well-tolerated at the efficacious dose in both preclinical animal models and phase 1 clinical trials for solid tumors.53,54 Other Porcupine inhibitors to enter clinical trials include ETC-1922159, RXC004, and CGX1321. Recent identification of key binding residues for Porcupine mediated Wnt acylation55 and the publication of the first crystal structure of a homologous MBOAT enzyme56 should aid in the future identification of next generation Porcupine modulators; however management of on-target toxicity remains an ongoing challenge for Porcupine inhibitors delivered systemically.
In the case of the IWRs, Chen et al. noted that IWR-1-endo (11) inhibited Wnt signaling in a reporter assay (IC50 180 nM) and showed that this occurred by increased degradation of β-catenin through increased protein levels of the key β-catenin destruction complex protein Axin.51 Following the identification of 11, Huang et al. used a chemical genetic screen to identify Xav939 (12) that also antagonized Wnt signaling through the stabilization of Axin.57 It was shown that this stabilization occurred through inhibition of the poly-ADP-ribosylating (PARP) enzymes tankyrase 1 and 2 (TNKS 1 and 2), and that both IWR-1-endo and Xav939 were potent dual TNKS 1 and 2 inhibitors. PARylation of Axin leads to its degradation resulting in more free β-catenin in the cytosol; inhibiting PARylation reduces Axin’s turnover, which in turn increases sequestration of cytosolic β-catenin into the destruction complex. While bound in the destruction complex, β-catenin is tagged for degradation through sequential phosphorylation by CK1α and GSK3β.58 Since the first crystal structures of the human TNKS2 in complex with Xav939 was reported in 2009,59 there has been a steady stream of more potent and selective tankyrase inhibitors generated through structure-based approaches, for example, G007-LK,60 NVP-TNKS656,61 MSC2504877,62 and others.35 However, despite showing efficacy in preclinical Wnt-driven cancer models, on-target toxicity has hampered progression of tankyrase inhibitors into the clinic.63 Dishevelled (Dvl; 3 human orthologues) also forms part of the Wnt cytosolic machinery, and upon extracellular binding of a Wnt ligand to a FZD receptor, Dvl is recruited to the cell membrane, where it binds to the FZD intracellular C-terminal domain through a PDZ domain and to Axin through a DIX domain. Dvl binding to Axin effectively sequesters Axin out of the destruction complex resulting in increased levels of free cytosolic β-catenin and enhanced transmission of the Wnt signal.64 Thus, targeting Dvl provides another opportunity for influencing Wnt signaling with a small molecule. To date, most efforts have focused on Wnt inhibition by blocking the Dvl–FZD interaction through targeting the PDZ domain of Dvl. Following on from the weak hit BML-268 (13) against mouse Dvl-PDZ domain described by Grandy,65 Hori and co-workers used a combination of virtual screening and NMR titration experiments against human Dvl1 to identify five new binders of the hDvl1PDZ. The most potent binder, NPL-4011 (14) (KD 34.5 ± 6.6 μM), a pseudo-dimeric structural analogue of 13, was reported to bind much more strongly than 13 (KD 954 ± 403 μM), which was rationalized, in part, to be due to structural differences between the mouse and human DvlPDZ domains.66 It remains to be seen whether more potent, drug-like DVLPDZ inhibitors can be developed, but given the ubiquity of PDZ domains in proteins throughout the proteome, acceptable selectivity profiles will need to be carefully considered.
Targeting Membrane Components of Wnt Signaling
There are currently no small molecules inhibitors in clinical trials that target the cell membrane bound extracellular machinery of the Wnt signaling cascade. Considering that there are 19 human Wnts and 10 FZD receptors, targeting these highly complex extracellular receptor–ligand interactions can be somewhat daunting. Functional redundancy between FZD receptors means that strategically targeting individual receptors is challenging with potential pitfalls in terms of efficacy and off-target effects. At the same time, it also offers opportunities to selectively modulate certain portions of the Wnt pathway or modulate it in a more restricted fashion, ideally in disease relevant, localized cell populations only. Such an approach may then offer advantages in terms of safety and therapeutic window compared to targeting a globally essential pathway component such as Porcupine.
Lee and co-workers used a combination of structure-based drug design (SBDD), molecular modeling, and virtual screening to construct a focused-library to screen for inhibitors against the Wnt-binding site of the extracellular FZD8 cysteine-rich domain (CRD).67 This approach led to a small collection of compounds, exemplified by NSC654259 (15), that displayed single digit micromolar activity in a Wnt 3a-dependent reporter assay in 3T3 cells. Biolayer interferometery was used to confirm binding to the FZD8CRD domain (KD 2.9 ± 2.4 μM). The authors acknowledged that the key binding residues (Leu97, Met149, Asp150) are conserved across most of the FZD receptors, suggesting that further work is required to determine their selectivity profiles. Zhao et al. recently reported that the antiepileptic drug carbamazepine (16) binds into a novel allosteric binding pocket of the CRD of FZD8.68 Initially discovered by a small molecule screen using surface plasmon resonance (SPR), the binding site of carbamazepine was definitively confirmed by X-ray crystallography. The structure revealed a well-defined mostly hydrophobic pocket but for a water bridged hydrogen bond between Tyr52 and the carboxamide of the ligand. This hydrogen bond might be important for selectivity as Tyr52 is not conserved across the FZD family. Follow-up studies using SPR showed 16 to have a KD of 16.8 μM against FZD8CRD with no apparent binding to FRZ5CRD and FRZ7CRD, the most closely related FZD receptor CRDs. Using a modified TOP-flash reporter assay, it was shown that 16 could inhibit Wnt signaling in an engineered FZD8 HEK293 cell line, suggesting that this novel allosteric binding site could be druggable. Zhang and co-workers used the recently reported crystal structure of the transmembrane domain (TMD) of the hedgehog signaling pathway protein Smoothened, bound to a small molecule ligand,69 to develop a homology model for the TMD of FZD7.70 Through this approach they executed a structure-based virtual screen and identified a series of N-aryl benzimidazoles antagonists. Further design and screening led to SRI37892 (17), which displayed sub-micromolar activity against Wnt/β-catenin signaling in LRP-6 expressing HEK293 cells (IC50 = 0.78 μM) and low micromolar antiproliferation activity against the triple negative breast cancer cell lines HS578T and BT549. Most approaches for targeting FZD receptors with small molecules are in the early stages of development; however the feasibility of targeting this family of receptors has recently been enhanced by the publication of a crystal structure of the human FZD8CRD–Wnt3 complex.71
In comparison, biological approaches to targeting the membrane components of the Wnt pathway are more advanced. OMP-54F28 (18, ipafricept), developed by OncoMed, is a fusion protein between a truncated FZD8 receptor that contains the Wnt-binding CRD domain and the immunoglobulin IgG1 FC region.72 OMP-54F28 thus acts as a FZD8 decoy receptor that binds to and sequesters Wnt proteins preventing binding to endogenous FZD receptors; the net result is to functionally antagonize Wnt signaling. OMP-54F28 has completed a number of phase 1 clinical trials for various malignancies.73 OncoMed also has a humanized monoclonal antibody OMP-18R5 (19, vantictumab) in clinical trials for oncology indications. Originally designed against FZD7, it was found to bind to 4 additional FZD receptors (FZD1, 2, 5, and 8) and is thought to block Wnt signaling by direct inhibition of Wnt binding.74,75 Recently, Zinzalla et al. reported BI905677 (20) as a first-in-class potent inhibitor of Wnt signaling. BI905677 is a biparatopic antibody comprising two modules that bind unique, nonoverlapping epitopes of LRP5/6 and presumably blocks Wnt signaling by preventing formation of the FZD–Wnt–LRP5/6 trimeric complex.76 The extracellular secreted protein Dickkopf-1 (DKK1) competes with Wnt proteins for binding to LRP5/6 and functions as a negative regulator of Wnt signaling.77 Gallocyanine (21, NCI8642) was shown to block the LRP6–DKK1 interaction by SPR through binding to LRP6 (KD 0.47 μM). Gallocyanine functionally restored Wnt signaling in a concentration dependent manner in a β-catenin intracellular accumulation and nuclear translocation assay, carried-out in L-cells treated with Wnt3a and DKK1 conditioned media (IC50 12.6 μM).78 Mpousis et al. followed-up this work and have reported a series of small molecules, based on the gallocyanine core, that inhibit the LRP6–DKK1 interaction and enhance Wnt signaling with micromolar activities.79 It was also reported that these gallocyanine derivatives could reduce Tau phosphorylation at serine 396 in a DKK1 induced Tau phosphorylation assay in SH-SY5Y cells.80
Targeting Secreted Modulators of Wnt Signaling
Secreted frizzled-related proteins (sFRPs) form a family of 5 glycoproteins that contain a CRD domain homologous to the Wnt-binding sites of FZD receptors and are generally considered to be antagonists of Wnt signaling. This antagonism has been proposed to be conferred by their CRD domain and to occur through two main mechanisms: (1) direct competition with FZD receptors for Wnt binding sites and (2) directly forming extracellular inactivating complexes with FZD receptors.81 Researchers at Wyeth, interested in activating Wnt signaling as a potential therapeutic mechanism for treating bone loss disorders, designed a high-throughput screen (HTS) based around an optimized TCF-luciferase reporter assay looking for activation of canonical Wnt signaling; specifically, looking for Wnt activation through sFRP-1 inhibition. Of the 685 initially confirmed hits, 65 were found to inhibit Wnt signaling in an sFRP-1 specific manner.82 A ligand-based drug design approach was then used to optimize hit WAY-316606 (not shown) cumulating in the potent lead compound WAY-362692 (22) with activity in both a fluorescent polarization binding assay (IC50 20 nM) and a Wnt-activation reporter assay (EC50 30 nM). Additionally, 22 was demonstrated to increase total bone area by 75% in an ex vivo model of bone formation.83,84 As discussed in the previous section, DKK1 is a secreted extracellular negative regulator of Wnt signaling. Again, researchers at Wyeth looking for Wnt agonists, used a HTS and ligand-based optimization approach to discover WAY-262611 (23), a small molecule inhibitor of DKK1 as measured by a TCF-luciferase reporter assay in an osteosarcoma cell line treated with Wnt3a and DKK1 conditioned media (EC50 0.63 μM). WAY-262611 was optimized to be highly selective over GSK-3β, have favorable pharmacokinetic (PK) properties, and display efficacy in an in vivo rat model of bone formation when dosed orally.85
The first-in-class humanized monoclonal antibody DKN-01 (24), developed by Leap therapeutics, binds to and inhibits DKK1, which in turn has been shown to restore Wnt signaling. Elevated levels of circulating DKK1 has been associated with a number of neoplastic diseases, and DKN-01 is currently undergoing multiple clinical trials for various malignancies.86 WNT Research has developed Foxy-5 (25), a synthetic 6 amino acid peptide fragment of Wnt5a that mimics the effects of Wnt5a. Wnt5a is a ligand of the noncanonical Wnt signaling pathway that can also effect canonical signaling in a context and cell-type dependent manner. In certain cancers, low Wnt5a expression has been linked to increased metastasis and a poorer disease prognosis.87 Foxy-5 has completed phase 1 clinical trials for metastatic breast, colon, and prostate cancer and is currently recruiting for a phase 2 study for colon cancer.
Notum, a secreted carboxylesterase, has recently been identified as a negative modulator of Wnt signaling.29 Notum acts through the removal of an essential palmitoleoyl moiety from Wnt proteins, thereby rendering them inactive. Signaling by Wnt proteins is finely balanced and tightly regulated by a sophisticated network of modulators and feedback processes including secreted inhibitory proteins, and it is of note that Notum performs the reverse biochemical process to the intracellular enzyme Porcupine.
As an enzyme with a defined high-resolution crystal structure and druggable pocket, Notum presents a tractable target for modulating Wnt signaling. There are now a number of reports of inhibitors of Notum carboxylesterase activity providing chemical tools for use in drug target validation experiments, and a few of these inhibitors 26–28 have been now evaluated in PK and disease models.
Insights from Structural Biology
Several key members of the Wnt signaling pathway have had their structures determined, allowing an enhanced understanding of their function as well as providing ways to rationally design modulators. The key signal transduction event, Wnt binding to FZD, has been captured in this manner. While the structure of the FZD CRD that serves as the Wnt binding site has been known for some time,88 the first structure showing Wnt binding was reported in 2012 (PDB 4F0A)12 and subsequent work has shown the structures also for a mammalian complex (PDB 6AHY).71 An interesting aspect of the Wnt–FZD complex is the direct involvement of Wnt lipidation in the binding by interaction with a hydrophobic groove present in the extracellular domain of FZD. It has long been known that post-translational addition of palmitoleate is a required feature for fully functional Wnt,10 but the molecular basis for the enhanced activity on the receptor is clearly illustrated by the structural information. Additional structural studies have shown that while FZD receptors display certain hallmark properties of G-protein coupled receptors (GPCRs) such as seven transmembrane regions, they are at the same time distinct from other GPCRs with large differences in the ligand binding region, possibly indicating that new approaches are required for the modulation of FZD receptors.89 Recently, a structure of FZD-8 bound to carbamazepine (16, PDB 6TFB) revealed an allosteric site that offer opportunities for future targeting of this receptor.68
Also, part of the mechanism for downstream signal transduction has been elucidated through the determination of various Dishevelled (Dvl) domain structures. From the first NMR structure offering a putative mechanism of signal transduction,90 more structures have followed including the PDZ domain with bound inhibitory peptides,91 the latter offering a potential approach for the development of small molecule Wnt signaling inhibitors. Additional components related to the signal transduction have also had their structures determined, including β-catenin/TCF92 and LRP6 in complex with DKK1.93
The Wnt proteins themselves are about 40 kDa in size and across the family contain a conserved serine that serves as an anchor point for a palmitoleic ester post-translational modification. Regulating the lipidation of Wnt is primarily done by the enzymes Porcupine and Notum, acylating and deacylating Wnt, respectively. No structural information is available for Porcupine, although related enzymes have been reported.56 However, for Notum several structures have recently been reported, offering a molecular level insight into Wnt regulation.
The mechanism of Notum mediated hydrolysis is well understood.29 The basis for the ability of Notum to selectively cleave palmitoleate from Wnt is a well-defined hydrophobic pocket (∼380 Å3) in proximity to a typical hydrolase Ser-His-Asp catalytic triad (Ser232, His389, and Asp340 in hNotum) (Figure 2). The size of the pocket is optimal to accommodate a cis-unsaturated lipid at C9–C10 with a total length up to 16 carbon atoms. The structure of O-palmitoleoyl serine bound to Notum (PDB 4UZQ) shows the fatty acid chain occupying the pocket, positioning the ester bond in close proximity to the catalytic triad and in an ideal position for a tetrahedral transition state to be stabilized by the oxyanion hole formed by backbone amides (Gly127–Trp128, Ser232–Ala233, and Gly126–Gly127). Another feature of the Notum structure is the presence of sulfate binding sites that have been implicated in glypican binding. This binding is suggested to retain Notum at the cell surface. Several structures of drug-like molecules bound to Notum are also reported in the literature, giving valuable insight on inhibitor binding; these are discussed in more detail in later sections of this review. The available structures of Notum in the Protein Data Bank (PDB) are summarized in Table 3 and Figure 4.
Table 3. X-ray Crystal Structures of Notum.
Figure 4.

Active site of Notum structures with bound ligand published to date. Residues are shown within 4 Å of the bound ligand(s) with the pocket surface highlighted (gray). Bold number in parentheses refers to chemical structure.
Overall, the available structural data allow us to get a fairly complete picture of the molecular mechanism of Wnt signaling and offer several exciting possibilities to leverage SBDD approaches to discover new modulators of the pathway.
Notum and Disease
Understanding of the role of Notum in mammalian biology and disease is predicated upon the premise that loss of Notum, either through genetic manipulation or through pharmacological inhibition, leads to an increase in Wnt signaling. While certainly this is true, it remains a possibility that Notum may have other substrate(s), and therefore its manipulation may theoretically lead to changes in other extracellular signaling systems. Notum’s function is also informed by its tissue and cellular level expression: our knowledge of Notum expression and distribution remains incomplete, but at a tissue level it appears to be expressed in bladder, brain, liver, kidney, lung, skeletal muscle, and skin.94 As described below, more detailed studies are now defining cellular level expression.
The first clues regarding Notum function in mammals, and thereby any association with disease, came from genetically modified mice where Notum had been deleted.95 The Notum knockout mice were stated to have increased bone formation on endocortical (marrow-facing) bone surface, presumably due to increased local Wnt signaling. Interestingly, the knockout mice were also reported to have a defect in dentin morphogenesis, indicating a role for Notum in tooth development.96 The adult Notum knockout mice had slightly reduced body weight and lean body mass and body fat compared to wild-type mice, and histological analysis of 40 soft tissues revealed no phenotypic changes.95 The role of Notum in modulating bone was further validated using small molecule inhibitors of Notum, for example, LP-922056 (26), where increases in femur cortical bone thickness were reported following chronic dosing of mice.95 Subsequent mechanistic investigation using more sophisticated conditional and cell lineage specific Notum knockouts lead to the conclusion that osteoblast-derived Notum is a local regulator of cortical bone mass through effects on periosteal bone formation.97 These authors also reported that variants in the Notum gene are associated with bone density variation in adult humans. A subsequent study98 also described the expression of Notum in bone osteoblasts and the use of both small molecule Notum inhibitors and a neutralizing anti-Notum antibody to chronically deplete Notum activity in adult mice, leading to increased cortical bone thickness and strength. Together these reports support the concept of therapies targeting osteoblast-derived Notum to strengthen cortical bone and prevent nonvertebral fractures.
A liver specific Notum knockout mouse has also been described.99 Given that Wnt signaling plays a critical role in liver development, a profound phenotype may have been predicted. However, these mice did not appear to have any changes in liver development or zonation. The only phenotype reported was the development of obesity and glucose intolerance, curiously restricted to male mice. Note that this finding is in contrast to the global Notum knockout mouse.95 The relevance of this observation to human biology and disease is unclear.
Wnt signaling has long been recognized to play a central role in the maintenance of adult stem cell populations in tissues as diverse as the intestine, skin, and brain,100 particularly in the context of the so-called “stem cell niche”.101 Given the role of many adult stem cell populations in the ongoing regeneration and replacement of specific differentiated cell types, this leads to the notion that modulation of Wnt signaling in niche–stem cell interactions could provide an approach to therapeutically targeting tissue renewal and age-related regeneration. To date, two examples have described an important role for Notum in modulating Wnt signaling in the adult stem cell niche:
-
(1)
In the intestine, ongoing regeneration of the epithelium is critical. This is known to be a Wnt dependent system that declines with age. Wnt signaling is essential to maintain the so-called intestinal stem cell niche, thereby enabling the ongoing generation of new epithelium. Pentinmikko et al. have found a critical role for Notum in regulating this system.102 It was found that Paneth cells, located in the intestinal stem cell niche, express Notum. Upon aging, the expression of Notum increases, leading to decreased Wnt signaling and subsequent reduction in stem cell maintenance and regeneration. Excitingly, the authors found that application of a small molecule Notum inhibitor, ABC99 (27), normalized Wnt signaling and restored epithelial regeneration. These observations open up the possibility of the therapeutic use of Notum inhibitors, for instance, to mitigate against adverse effects of chemotherapeutic agents on intestine structure and function, particularly in older patients.
-
(2)
In the adult mammalian brain, two regions have been described where there is ongoing generation of new neurons from stem cell populations (“neurogenesis”): the subventricular zone and the subgranular zone. The existence of these in the human brain has, at times, proven controversial.103 Similarly, the biological role (and thereby the opportunity for therapeutic manipulation), particularly in humans, has proven difficult to investigate.104 As in the intestine, it is recognized that Wnt signaling is important in regulation of stem cell populations in the adult brain.105 Mizrak and colleagues have now discovered a role for Notum in modulating Wnt signaling, and thereby neurogenesis, in the adult mouse subventricular zone.106 They found that Notum, secreted by specific neural stem cell (NSC) intermediates, inhibits the proliferation of nearby NSC progeny (presumably by inhibiting Wnt signaling), thereby possibly providing a favorable environment for their cellular progeny. The downstream implications of inhibiting Notum on the migration, integration, and function of progeny neurons, and thereby any regenerative potential to treat acute and chronic neurological and neurodegenerative disease, remains to be elucidated. Additionally, whether Notum modulates neurogenesis in the other stem cell population in the brain (the subgranular zone of the hippocampus) is currently unknown.
Dysfunction of Wnt signaling is well recognized as a component of many cancer types,107 and indeed its modulation is considered as a valid approach for the development of targeted therapies.108 The role of Notum as a potential contributor to aberrant Wnt signaling in various forms of cancer is at an early stage of investigation. Nevertheless, there are some reports emerging that implicate a role. In 2008, Torisu et al. reported that Notum was overexpressed in primary hepatocellular cancer samples and that this was associated with increased nuclear β-catenin, consistent with increased intracellular Wnt signaling.109 The role of Notum in cancer biology or whether the upregulation of Notum represented simply a negative feedback mechanism of increased Wnt tone was not investigated. A similar upregulation of Notum was reported in tissue from an animal model of colorectal cancer and human biopsy material.110 Indeed, this upregulation of Notum in certain cancers has led to the suggestion that Notum levels in the plasma may be a useful biomarker of disease.111 An important point to consider is that the role of Notum in the development of a cancer may be analogous to its role in the adult stem cell niche described above; that is, rather than a cell autonomous role for Notum, it is possible that there is a more complex interplay between tumorigenic cells and their surrounding niche, with Notum playing a key role in aberrant Wnt signaling.
Small Molecule Inhibitors of Notum Activity
A number of complementary strategies have been applied to discover inhibitors of Notum activity. Successful approaches that have identified drug-like small molecules include high-throughput screening (HTS) campaigns, activity-based protein profiling (ABPP), natural products, and screening of fragment libraries. Several of these inhibitors have been soaked into crystals of Notum, and the structures have been solved to determine the inhibitor binding modes (Table 3 and Figure 4). Hence, under suitable conditions, Notum is very amenable to SBDD to help guide progress toward the discovery of potent inhibitors.
There are several screening assay formats that have been used to identify and characterize inhibitors of Notum. Inhibition of Notum carboxylesterase activity has been routinely measured in a cell-free biochemical assay with synthetic fluorescent substrates (Figure 5). Test compounds are incubated with Notum and trisodium 8-octanoyloxypyrene-1,3,6-trisulfonate (OPTS, 29a) as the substrate, and fluorescence is recorded; an inhibitor of Notum (IC50) would suppress fluorescence by binding to Notum and preventing hydrolysis of OPTS. This assay can also be performed with PPTS (29b), which incorporates the palmitoleate group, but this requires a custom synthesis of the substrate and shorter incubation times.
Figure 5.
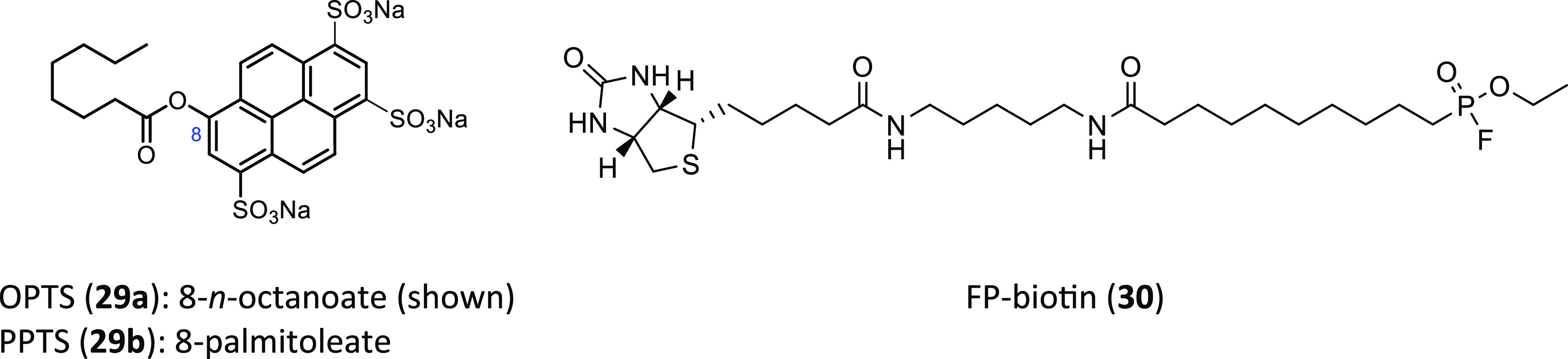
Reagents used in the identification of Notum inhibitors.
Inhibitors can be screened in cell-based TCF/LEF reporter gene assays to assess their ability to restore Wnt/β-catenin signaling when activated by an exogenous recombinant Wnt in the presence of Notum. An inhibitor of Notum should show activation of Wnt signaling (EC50) in this model system. Performing this assay in the absence of Notum should show a maximal Wnt response at all concentrations of test compounds to confirm the outcome was due to direct inhibition of Notum activity and not related to assay interference or cell toxicity.
Inhibitors of Notum have also been tested in a Notum occupancy assay using FP-biotin (30),112 a covalent serine hydrolase activity-based probe, whereby labeling of Ser232 of Notum with FP-biotin can be blocked by an inhibitor occupying the active site of Notum. Biophysical screening methods, such as differential scanning fluorimetry (DSF) (thermal shift assay, ΔTm) and SPR, can provide additional information on affinity and binding kinetics.
Notum Inhibitors Identified by High-Throughput Screening
The first report of small molecule inhibitors of Notum activity in 2012 came from Lexicon Pharmaceuticals, Inc. (US) as patent applications based upon fused heterocyclic scaffolds.113,114 These initial patent disclosures gave some understanding of structure–activity relationships (SAR), which would be expanded upon in detail in later publications.
Using a HTS campaign with a TCF/LEF Cell sensor reporter assay, Han et al. identified a range of fused heterocyclic compounds as potent inhibitors of Notum (Figure 6).115 The initial lead carboxylic acid 31 was selected for further development with modifications of the carboxyl group, the tricyclic core and substitution of the aromatic rings. Interestingly, while secondary and tertiary carboxamides were tolerated, primary amides showed significant improvements in potency over the parent acid. However, profiling of these amides in liver microsomes showed them to have poor metabolic stability, and so amides were not investigated further. SARs then focused upon retaining the carboxylic acid and exploring the tricyclic motif.
Figure 6.

Tricyclic heterocyclic acids as first generation Notum inhibitors from Lexicon Pharmaceuticals, Inc. IC50 and EC50 values presented in Figures 6–20 refer to the Notum OPTS and TCF/LEF assays, respectively, unless stated otherwise.
The S1 atom of the thiophene ring was shown to be crucial for activity, as was the sp2 C3 atom, and neither could be replaced. The importance of the distal phenyl ring was also demonstrated, with analogues bearing heteroaromatics having greatly reduced activities. The 5-position of the dihydronaptho ring was shown to be tolerant of changes in heteroatom, with potency activity in the order of S > C > O. A thorough SAR investigation of substitutions on the distal phenyl ring of the dihydronaptho analogue 31 showed that a range of substitution patterns and substituents were tolerated and superior to R6–R9 = H. The most active compound in this set was 32, which gave potent inhibition of both human and mouse Notum. The corresponding chromene analogues, for example, 33, were generally less potent, although similar SAR trends were observed.
The effect of substitution on the phenyl ring of the thiochromene core also followed the same general trend with multiple halogen substituents generally improving potency, which led to the development of the 6-F,8-F analogue 34. Compound 34 was evaluated in mouse PK studies and, when dosed orally (10 mg/kg), showed good exposure (Cmax 16 μM; AUC 56 μM·h) and moderate clearance (Cl 19 mL/min/kg). Compound 34 was then advanced into a mouse model of bone growth where F1 male hybrid (129xC57) mice were treated for 28 days with compound administration in their diet (5 and 20 mg/kg). Mice treated with 34 showed dose-dependent increases in midshaft femur cortical thickness of 4% and 8% over control.
Lexicon then published a second lead series from their HTS with [2,3-d]pyrimidine acid 35 as a hit that showed activity in both the OPTS and TCF/LEF assays, which offered a good starting point for optimization (Figure 7).95 A range of alternative 5,6-heterocycles were synthesized and evaluated, with significant loss in potency. The only tolerated change was transposition of the thiophene ring to give the isomeric thieno[3,2-d]pyrimidine 36, and so 35 and 36 were selected for further development.
Figure 7.

Lexicon [2,3-d]pyrimidine acid 35 HTS hit and isomer [3,2-d]pyrimidine acid 36.
A systematic investigation in the [2,3-d]pyrimidine acid series 35 at the 5- and 6-positions showed that Notum inhibition could be improved to EC50 ca. 100 nM by suitable choice of substituents with R5 = Cl and R6 = Me (37) or nPr (38) being the most potent from this set (Figures 8 and 9). The acid of the original lead 35 was converted to a number of carbonyl derivatives (esters, amides, ketones, and N-acylsulfonamides) but these modifications offered no advantage, and the carboxamides again suffered from poor metabolic stability. In contrast, the parent carboxylic acids had good ADME properties.
Figure 8.

General SAR trends of the [2,3-d]pyrimidine acid template 35.
Figure 9.

Lexicon optimized leads LP-914822 (37), LP-922056 (26), and LP-935001 (39).
The thieno[3,2-d]pyrimdine scaffold 36 was explored in a similar manner to 35, which identified 26 as one of the most potent inhibitors prepared to date (Figure 9). Compound 26 was evaluated in mouse PK studies (10 mg/kg po) and showed high exposure (Cmax 129 μM; AUC 1533 μM·h) and low clearance (Cl 0.49 mL/min/kg) that was superior to thiochromene 34. Compound 26 was also assessed in the same mouse model of bone growth, and mice treated daily with 26 (3, 10, 30 mg/kg, po) for 25 days showed increases in midshaft femur cortical thickness at all doses tested.
LP-914822 (37), LP-922056 (26), and LP-935001 (39) (Figure 9) emerged as three advanced leads for the program and were used to demonstrate in rodent pharmacology studies, along with complementary approaches, that inhibition of Notum activity is a potential novel anabolic therapy for strengthening cortical bone and preventing nonvertebral fractures.98
Additional mouse PK data for 26 was generated to evaluate brain penetration.116 Following a single oral dose (10 mg/kg, po), the plasma parameters from these experiments (Cmax, AUC, and t1/2) were consistent with published data.95 Brain penetration of 26 is very low with brain/plasma concentration ratios ∼0.01 at all time points measured up to 24 h and also 0.01 based on AUC(0→inf). Hence, 26 is unsuitable for use in models of disease where brain penetration is an essential requirement.
Scaffold-Hopping Approaches
The thienopyrimdine scaffolds 37 and 26 were revisited by Atkinson et al. at University College London (UCL) in an attempt to introduce brain permeability.117 A small library of diverse carboxamides was prepared from acids 37 and 26 to explore if they could be modified to deliver a central nervous system (CNS) penetrant tool by capping off the acid as an amide. This strategy was guided by SBDD with the determination of Notum structures with 37 (PDB 6T2H) and 26 (PDB 6T2K) (Figure 10). Both 37 and 26 place the thienopyrimidine group in the palmitoleate pocket but with the thiophene ring at a slightly different position to accommodate the substituents; the remainder of these templates adopt a similar position. From a design perspective, these structures show significant space at the mouth of the pocket to accommodate a suitable group as an amide derivative of 37 or 26.
Figure 10.

Scaffold-hopping, supported by X-ray structure determination, identified furano[2,3-d]pyrimidine amide 41 with reasonable brain permeability.
Although significant Notum inhibition activity could be achieved (IC50 < 10 nM), none of these amides demonstrated the required combination of metabolic stability along with cell permeability without evidence of P-glycoprotein (P-gp) efflux to varying degrees. The most advanced compound from this set, 40, was assessed in mouse PK (10 mg/kg, po) and showed low plasma exposure, which was attributed to high clearance and highlighted the need to further improve metabolic stability.
Scaffold-hopping from thienopyrimdine 40 identified furano[2,3-d]pyrimidine amide 41 as a potent inhibitor of Notum with improved stability in mouse liver microsomes (MLM). Crucially, it was found that by combining the furano[2,3-d]pyrimidine heterocycle with the preferred N-methylimidazolidin-4-one amide gave 41 with the best in vitro ADME profile of the compounds tested. The lower lipophilicity of 41 (clogP 2.1, logD7.4 1.6) compared to 40 (ΔclogP = −0.5) offered an explanation for the higher microsomal stability (MLM, Cli 6.9 v 25 μL/min/mg protein, respectively). In addition, 41 was stable in mouse plasma and did not inhibit CYP450 enzymes. Compound 41 had a modest efflux ratio (ER = 2.4) in the MDCK–MDR1 permeability assay suggesting some recognition by P-gp mediated efflux transport.
PK experiments with 41 were performed in mouse with a single oral dose (10 mg/kg, po) to determine plasma exposure and brain penetration. The plasma half-life was modest (t1/2 0.6 h), which was somewhat unexpected based on the in vitro MLM and plasma stability data. Brain penetration was reasonable with a brain/plasma concentration ratio of 0.29. The incomplete brain penetration was probably due to P-gp mediated efflux as shown in the asymmetry in the MDCK-MDR1 permeability assay.
Covalent Inhibitors Identified by Activity-Based Protein Profiling (ABPP)
In 2018 Cravatt et al. described the development of a series of potent and selective irreversible Notum inhibitors discovered using gel-based activity-based protein profiling (ABPP).118 ABPP is a chemical proteomics approach that uses chemical probes to investigate the functional state of enzymes directly in native systems. ABPP probes have been developed that react selectively with most members of specific enzyme classes such as serine hydrolases (SH).119,120
Having established that Notum activity could be assayed with SH-directed fluorophosphonate (FP) ABPP probes, Cravatt et al. screened a diverse set of inhibitors containing triazole urea and N-hydroxyhydantion (NHH) carbamate reactive groups. A series of NHH carbamates, exemplified by ABC28 (42) (Figure 11), were taken forward for optimization, due to their comparatively modest serine hydrolase promiscuity. Benzomorpholine replacement of the 4-phenylpiperidine “staying group” and SAR investigation of the phenyl ring resulting in a direct chlorine replacement of the bromine moiety provided significant improvements in selectivity and potency to give ABC99 (27). During the course of developing ABC99, it was discovered that oxidation of the benzylic position provided amide ABC101 (43) as a useful inactive control. In addition to gel-based ABPP, the selectivity of ABC99 was characterized using quantitative mass spectrometry (MS)-based ABPP in conditioned media and SW620 in situ treated cells. These data showed that ABC99 was highly selective for Notum over the 64 other serine hydrolases quantified.
Figure 11.

Irreversible inhibitor of Notum activity, 27, along with structure-matched inactive control 43 and clickable probe 44. aNotum IC50 determined by competitive gel-based ABPP.
The authors were interested to use these probe compounds in order to evaluate the activity of Notum in native biological systems and, for that purpose, designed ABC99yne (44) as a clickable probe, which incorporated an alkyne into the staying group. ABC99yne exhibited a sub-micromolar IC50 value and highly selective labeling of Notum in the conditioned media of SW620 cells, when samples were treated with a rhodamine-azide reporter tag under copper mediated click conditions.
ABC99 has been used to investigate the role of Notum produced by Paneth cells in regulating the intestinal stem cell niche of the small intestine.102 As aged Paneth cells exhibit significant Notum overexpression, it was hypothesized that increased Notum secretion was inhibiting ISC Wnt signaling and ultimately reducing intestinal regeneration. Application of ABC99 normalized Wnt signaling and restored epithelial regeneration.
Screening of the XChem Diversity Fragment Library
In order to identify new small molecule inhibitors of Notum, Zhao and Jones performed a crystallographic fragment screen using the XChem platform at Diamond Light Source (Oxford, UK).121 Crystals of C-terminal His-tagged Notum (Ser81-Thr451 Cys330Ser) were soaked with the DSi-Poised library (XChem, 768 fragments).122 Notum has a well-defined, large, hydrophobic active-site pocket adjacent to the catalytic triad that accommodates the palmitoleate group of Wnt, and the 60 fragments observed to bind in this pocket were all resupplied as solid samples by compound purchase or resynthesis. Inhibition of Notum carboxylesterase activity of these hits was then measured in a biochemical assay. To date, three of these hits have been disclosed along with descriptions of their optimization to more potent inhibitors.123−125
2-Phenoxyacetamides
Using this XChem screening platform, Atkinson et al. developed a series of 2-phenoxyacetamides as inhibitors of Notum activity.123 The fragment hit 2-(2-methylphenoxy)-N-(pyridine-3-yl)acetamide (45; IC50 33 μM) was selected for further investigation for several reasons including excellent hit-like properties, synthetic tractability to explore SAR, and the Notum–45 structure to guide compound design (Figures 12 and 13). The SAR studies systematically explored each structural feature of the inhibitor and most changes were found to be detrimental to activity. There was limited scope to modify the substituents on the phenoxy ring with small lipophilic groups in the 2-position preferred (2-Cl; 2-CF3) but offering minimal improvement over 45 (2-Me). These results were consistent with the structural information showing 45 (PDB 6R8P) sitting deep in the palmitoleate pocket with minimal space to accommodate a group larger than H or F at the 3- or 4-positions. In contrast, modification of the N-acetamide group gave the most potent inhibitors in this series. Large increases in potency over 45 could be achieved by replacement of the original 3-pyridine with benzo-fused heterocycles such as indole 46 and quinoxaline 47. Further investigation of these 6,5- and 6,6-ring systems achieved gains in potency with indazole 48 and isoquinoline 50 as the most potent inhibitors in this series.
Figure 12.

Optimization of 2-phenoxyacetamide fragment hit 45 through modification of the N-acetamide identified 46–50.
Figure 13.
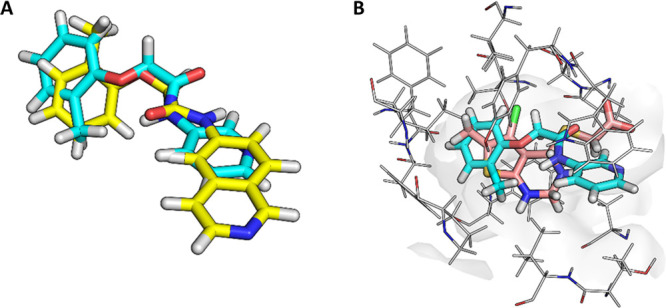
(A) Overlay of 45 (teal) and 50 (yellow). Isoquinoline 50 shows a flipped binding mode when compared to 45 and 49. (B) Overlay of 37 (pink) and 45 (teal) Notum structures showing the different orientations of these templates.
Structural studies with 49 (PDB 6R8Q) and 50 (PDB 6R8R) showed some unexpected features in their binding modes with Notum despite their similar potencies. Both 49 and 50 form π–π stacking with Trp128 through their heterocyclic rings, but there was a marked change in the orientation of their phenoxyacetamide backbones. Benzotriazole 49 binds in a similar mode to fragment 45, whereas 50 shows a rotation of 180° around the amide bond, positioning the methyl of the tolyl group on the opposite side of the pocket and the carbonyl oxygen pointing away from the oxyanion hole.
Progress in this series was limited to improving potency of fragment hit 45 into 48 (1000-fold increase in activity) because it was not possible to combine Notum inhibition activity with metabolic stability in liver microsomes. All examples from this 2-phenoxyacetamide series screened in HLM and MLM were rapidly metabolized in an NADPH-independent manner with short half-lives (t1/2 < 12 min) limiting their use to in vitro models.
Melatonin and Related Structures
The XChem platform output was explored by Zhao et al. to identify inhibitors of Notum, and fragment hit N-[2-(5-fluoro-1H-indol-3-yl)ethyl]acetamide (51; IC50 37.2 μM; PDB 6TR7) was highlighted due to its structural similarity to the brain hormone melatonin (52) (Figure 14).124 Additional structural studies were performed with 52 (PDB 6TR5) and N-acetylserotonin (53; PDB 6TR6) by soaking into Notum crystals, and high-resolution structures of their complexes were obtained. In each of these structures, two molecules bind with Notum: one at the enzyme’s catalytic pocket and the other at the edge of the pocket opposite the substrate entrance.
Figure 14.

Notum inhibitors related to the hormone melatonin.
The structural information reported may guide the design new of potent inhibitors of Notum although it will be essential to develop selectivity over the melatonin receptors (MT1–3).
Development of 5-Phenyl-1,3,4-oxadiazol-2(3H)-ones and 1-Phenyl-1,2,3-triazoles
A standout hit from this fragment set was (1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methanol (54; IC50 11.5 μM), which was selected as a starting point for a hit-to-lead program (Figure 15).125 The X-ray structure of 54 (PDB 6ZUV) showed occupation of the palmitoleate pocket by the 4-chlorophenyl ring, forming a π–π stacking interaction with residue Phe268. The triazole headgroup shows possible π–π stacking interactions with Trp128 and hydrogen bonding between N2 of the triazole and peptidic backbone of Trp128. Residue Trp128 is also involved in a hydrogen bond to the oxygen of the methyl alcohol group. Optimization of 54 by modification of the heterocyclic headgroup identified two complementary leads: oxadiazole 55 and triazole 56.
Figure 15.

Fragment hit 54 yielded two complementary leads in oxadiazole 55 and triazole 56.
Further investigation of the oxadiazole series 55 by exploring substitution on the aryl ring, which binds deep in the palmitoleate pocket, identified 57 as a preferred example from the 37 analogues synthesized (Figure 16). Compound 57 has physicochemical properties consistent with drug-like chemical space and contains a weakly acidic proton with a measured pKa = 6.7. Compound 57 demonstrated good metabolic stability and cell permeability with no evidence for P-gp mediated efflux. Compound 57 restored Wnt/β-catenin signaling in a cell-based TCF/LEF reporter gene assay and prevented labeling by FP-biotin, confirming competitive binding to Notum. PK studies with 57 were performed in vivo in mouse with single oral administration of 57 showing good plasma exposure and partial brain penetration (brain/plasma ratio, 0.16).
Figure 16.
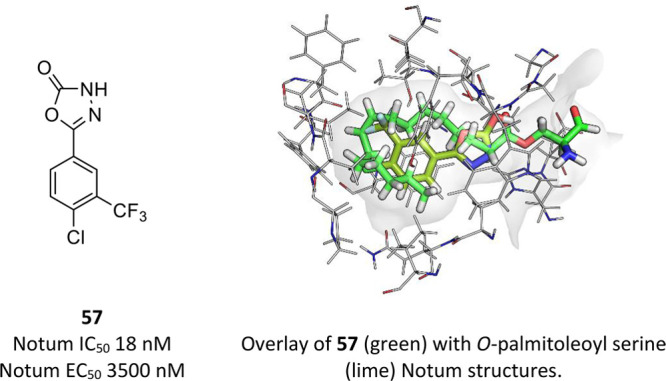
Optimized 1,3,4-oxadiazol-2(3H)-one 57 forms H-bonds with the oxyanion hole and fills the palmitoleate pocket.
An X-ray crystallographic structure determined the binding mode of 57 (PDB 6ZVL) in Notum. Inhibitor 57 makes an effective interaction with the oxyanion hole with hydrogen bonds to three separate amino acids (Gly127, Trp128, Ala223) while still filling the hydrophobic palmitoleate pocket (Figure 16).
Significant progress was made in developing X-ray fragment hit 54 into lead 57 (>600-fold increase in activity), which showed good plasma exposure in mouse PK experiments. Furthermore, contemporaneous studies with alternative lead triazole 56 identified ARUK3001185 (28, only Markush structure disclosed), which was shown to be a potent, selective, and brain penetrant inhibitor of Notum activity suitable for use in both cellular and in vivo models of CNS disease (Figure 17).125−127
Figure 17.

Markush structure and representative preferred compounds (4 out of 15 shown) from patent application WO 2020043866.
Custom-Designed Fragment Libraries
In parallel to the application of the XChem platform to find fragment hits, Mahy et al. designed and screened a second library to identify alternative chemical scaffolds as inhibitors of Notum.128 Whereas the original DSi-Poised fragment library was a diversity subset (768 compounds), this second library was a custom-designed fragment library of carboxylic acids (250 compounds) where each member incorporated the following: a carboxylic acid to interact with the catalytic triad; a lipophilic group to be presented into the palmitoleate pocket; and a linker of various lengths to connect the acid to the lipophilic group.
Inhibition of Notum activity of this library was measured in a biochemical assay with OPTS as the substrate, and 20 compounds were identified as fragment hits (IC50 < 25 μM). All 20 hits were soaked into crystals of Notum, and X-ray structure determination showed that 14 fragments bound in the palmitoleate pocket. From this set, two preferred hit series were selected for further optimization: pyrrole-3-carboxylic acids (e.g., 58; PDB 6YV4) and pyrrolidine-3-carboxylic acids (e.g., 59; PDB 6YV2) (Figure 18).
Figure 18.

Representative hits 58 and 59 from a custom-designed acid fragment library.
Optimization of pyrrole 58 by SAR studies guided by SBDD identified 1-phenylpyrrole 60 (clogP 5.5) as the most potent compound from this series, albeit with relatively high lipophilicity (Figure 19). Focus then switched to the pyrrolidine series 59 as this template offered the advantage of significantly lower lipophilicity when compared to the matched pyrrole but with weaker activity. Optimization of 1-phenylpyrrolidine 59 gave acid (S)-61, amide 62, and oxadiazolone 63. Once again, the 4-chloro-3-(trifluoromethyl)phenyl group was preferred (cf. 57) and a Notum–61 (PDB 6YSK) structure showed that the inhibitor fully occupied the palmitoleate pocket while making effective interactions with the oxyanion hole.
Figure 19.

Notum inhibitors pyrrole 60 and pyrrolidines 61–63 derived from fragment hits 58 and 59, respectively.
This set of inhibitors, 60–63, were assessed in in vitro ADME assays to compare their aqueous solubility, microsomal stability, and cell permeability. These inhibitors were selected based on their Notum activity but also their chemical structural diversity and complementary physicochemical properties (clogP, pKa). On balance, pyrrolidine-3-acid 61 demonstrated a superior profile. The design and screening of a second fragment library further confirms that inhibition of Notum activity can be achieved by small, drug-like molecules possessing favorable in vitro ADME profiles.
Natural Products
Notum activity can be inhibited by caffeine (64) and, to a lesser degree, by theophylline (65) (Figure 20).129 The caffeine–Notum interaction was thoroughly characterized by both biochemical and biophysical methods. High-resolution structures of 64 (PDB 6TV4) and 65 (PDB 6TUZ) show that both compounds bind at the center of the palmitoleate pocket but with quite different binding modes. This structural information may guide the design of more potent Notum inhibitors.
Figure 20.

Caffeine (64) and theophylline (65) are inhibitors of Notum activity. aDetermined by surface plasmon resonance (SPR).
Future Perspectives and Conclusion
Regulation of the Wnt signaling pathway is critically important for a number of cellular processes in both development and adult mammalian biology. Conversely, dysregulation of Wnt signaling can disrupt normal development and has been associated with a number of human diseases. The Wnt pathway has attracted significant interest for the development of new drugs, although it is proving to be challenging to identify new chemical entities or biologics that have progressed to advanced clinical trials. Modulation of the Wnt pathway requires careful selection of the molecular target to give therapeutic benefit in disease without safety issues linked to the target and, possibly, pathway. In addition, some of these targets would not be defined as “druggable” with a clearly defined site or pocket for binding a small molecule. Despite this, a number of potential drugs in the Wnt pathway have now progressed beyond human PK and safety studies, and evaluation of their potential benefit in treating disease (cancer and others) is ongoing in a number of phase 2 and 3 clinical trials.
Recently, Notum was shown to act as a negative regulator of Wnt signaling through the removal of an essential palmitoleate group; this group is required for binding of the Wnt proteins to the FZD receptors. Hence, inhibition of Notum carboxylesterase activity may represent a new approach to treat disease where dysregulation of Wnt signaling is an underlying cause and Notum has been identified as the source.
Notum is emerging as a druggable target to modulate Wnt signaling. A number of reliable screening technologies are available to identify inhibitors of Notum and these have been successfully combined with complementary screening strategies (HTS, ABPP, fragment libraries) to identify hits. Structural biology is also making an important contribution as several of these hits have been soaked into crystals of Notum and the structures have been solved to determine their binding modes. Hence, Notum can be very amenable to SBDD to help guide progress toward the discovery of potent inhibitors. A selection of these hits have been optimized to give fit-for-purpose small molecule inhibitors of Notum.
Three noteworthy examples are LP-922056 (26), ABC99 (27), and ARUK3001185 (28) (Table 4), which are valuable chemical tools for exploring the role of Notum in Wnt signaling at a cellular level and for use in animal disease models, that is, target validation experiments. These chemical tools have complementary profiles, and we suggest that they should be properly aligned for application in animal models. All three have been used successfully to demonstrate modulation of function in cellular and animal models at reasonable doses. Acid 26 has excellent plasma exposure upon oral dosing but very high plasma protein binding in mouse that will reduce free drug levels. In addition, 26 has negligible brain penetration in wild-type mouse. N-Hydroxyhydantion carbamate 27 is an irreversible inhibitor of Notum and has been dosed chronically by intraperitoneal injection although no drug levels were reported. ARUK3001185 (28) is a potent, selective, and brain penetrant inhibitor of Notum activity suitable for use in both cellular and in vivo models of CNS disease. However, available reports on the properties of 28 are currently limited to the patent literature and presentations at conferences.
Table 4. Summary of Properties of Notum Inhibitors LP-922056 (26), ABC99 (27), and ARUK3001185 (28).
| LP-922056 (26) | ABC99 (27) | ARUK3001185 (28) | |
|---|---|---|---|
| Physicochemical Properties | |||
| mol wt | 300 | 456 | 281 |
| clogP | 3.1 | 4.3 | 3.9 |
| logD7.4 | nda | nd | 1.3 |
| Notum Inhibition | |||
| OPTS, IC50 (nM) | 1.1 | 170b,c | 6.5 |
| gel-based ABPP, IC50 (nM) | nd | 13 | nd |
| TCF-LEF, EC50 (nM) | 23 | 89 | 110 |
| Selectivity | |||
| serine hydrolases (number screened) | nd | yes (64) | yes (49) |
| drug targets (number screened) | nd | nd | yes (47) |
| kinases (number screened) | nd | nd | yes (485) |
| Mouse Pharmacokinetics (1 mg/kg iv and 10 mg/kg po) | |||
| half-life (t1/2, h) | 8.3 | nd | 2.4 |
| oral bioavailability (Fo, %) | 65 | nd | 68 |
| exposure (Cmax) (po) | 129 μM | nd | 2300 ng/mLd |
| exposure (AUC∞) (po) | 1533 μM*h | nd | 10 800 (ng·h)/mLd |
| mouse plasma protein binding (mPPB) (fu, %) | 0.1 | nd | 4.2 |
| brain/plasma ratio (Kp) (po) | <0.01 | nde | 1.08 |
| Mouse In Vivo Studies | |||
| route of administration and dosing regime | 3, 10, 30 mg/kg, po, 25 days | 10 mg/kg, ip, 7 days | 2 × 30 mg/kg bid, po, 30 days |
| Rat In Vivo Studies | |||
| route of administration and dosing regime | 30 mg/kg, po, 126 days | nd | nd |
| refs | (95, 98, 116) | (102, 106, 118) | (125−127) |
nd, not determined or not disclosed.
Notum IC50 data presented for comparison in a common assay format.
As a covalent inhibitor, the IC50 value will be time dependent.
For ease of comparison of Cmax and AUC∞ data, 2300 ng/mL is equivalent to 8.2 μM and 10 800 (ng·h)/mL to 38 μM*h, respectively.
ABC99 is reported to be brain penetrant in mouse; see, ref (106).
For a molecular target to be druggable, it will need to show an appropriate safety window (therapeutic index) in the patient population. As small molecule inhibitors of Notum activity are still relatively early in the drug discovery pipeline (preclinical), a preliminary drug safety assessment of modulating Notum has to be based on available mouse genetic knockout data and in vivo rodent toleration information.95−99 These studies do not raise any significant safety issues at this time. Ultimately, the safety of inhibiting Notum will need to be tested in toxicology studies where any on-target effects have been disconnected from any compound related toxicity.
Looking ahead, based on current precedent, it seems feasible to generate additional inhibitors of Notum activity. These will be of most value if they have significantly improved profiles (activity, selectivity, PK) or from alternative chemotypes with a different risk–benefit profile. Fragment screening seems to have been a successful strategy to discover new hits, although this growing body of screening data, structural information and chemical structures will facilitate virtual screening (VS) and artificial intelligence (AI) design approaches as well.
As these first generation inhibitors of Notum show utility in disease models and are then progressed to safety studies, it will possible to determine if Notum proves to be an attractive molecular target in modulating Wnt signaling that will ultimately deliver new drug candidates for use in clinical studies.
Acknowledgments
We thank our colleagues Stefano Benvegnu, Magda Bictash, Jamie Bilsland, Dustin Flanagan, Sarah Frew, Sarah Jolly, E. Yvonne Jones, Svend Kjaer, William Mahy, Amy Monaghan, Ernest Palomer, Morgan Roberts, Reinis R. Ruza, Patricia Salinas, Owen Sansom, Laura Schuhmacher, James Sipthorp, Ed Tate, Luca Vecchia, Matthias Zebisch, and Yuguang Zhao of our Notum Consortium for their support and advice. The Notum X-ray structures presented in this Perspective were determined by E. Yvonne Jones, Yuguang Zhao, and colleagues (STRUBI, University of Oxford, U.K.).
Glossary
Abbreviations Used
- ABPP
activity-based protein profiling
- ADME
absorption, distribution, metabolism, and elimination
- BBB
blood–brain barrier
- CNS
central nervous system
- CRD
cysteine-rich domain
- DKK1
Dickkopf
- Dsh
Dishevelled (Drosophila)
- Dvl
Dishevelled (mammalian)
- ER
efflux ratio
- FP
fluorophosphonate
- FZD
Frizzled
- HLM
human liver microsomes
- HTS
high-throughput screen
- LRP5
low-density lipoprotein receptor-related protein 5
- MLM
mouse liver microsomes
- OPTS
trisodium 8-octanoyloxypyrene-1,3,6-trisulfonate
- PDB
Protein Data Bank
- P-gp
P-glycoprotein
- PD
pharmacodynamics
- PK
pharmacokinetic
- SAR
structure–activity relationship
- SBDD
structure-based drug design
- SPR
surface plasmon resonance
- TMD
transmembrane domain
Biographies
Elliott D. Bayle completed his Ph.D. in organic chemistry at the University of Cambridge and postdoctoral research at University College London School of Pharmacy before working for a year as a medicinal chemist at Charles River laboratories. Since 2016, he has been a medicinal chemist at the Alzheimer’s Research UK UCL Drug Discovery Institute where he is currently a Senior Research Associate.
Fredrik Svensson is a Senior Research Associate at the Alzheimer’s Research UK UCL Drug Discovery Institute. He was awarded his Ph.D. in 2015 from Uppsala University, Sweden, after which he obtained a postdoctoral fellowship from the Swedish Pharmaceutical Society for research in cheminformatics at the University of Cambridge with Dr. Andreas Bender. He has published on several structure-based drug discovery projects and his research focuses on the use of computational methods in drug discovery.
Benjamin N. Atkinson attained his Ph.D. from University of Bath under Professor Jonathan M. J. Williams on metal catalyzed acyl transfer reactions. Following this, he was a Research Fellow for 2 years in the Translational Research Office’s Drug Discovery Group at UCL, which included 4-month secondments at GSK and ESC Newhouse. His current research at Alzheimer’s Research UK UCL Drug Discovery Institute is around the workup and assessment of HTS hits from phenotypic and target based projects as well as fragment based X-ray crystallography screens.
David Steadman completed his Ph.D. in organic chemistry at University College London in 2012. As a Postdoctoral researcher in both academia and biotech, he has contributed to medicinal chemistry programs in the fields of bacterial antivirulence therapies, immuno-oncology, and neurodegeneration. He is currently a Research Associate at the Alzheimer’s Research UK UCL Drug Discovery Institute working on novel inhibitors of the Notum enzyme as modulators of Wnt signaling.
Nicky J. Willis is a Research Associate at the Alzheimer’s Research UK UCL Drug Discovery Institute. Nicky started his career in medicinal chemistry at the Novartis Institute for Biomedical Research, investigating novel anti-inflammatory agents. He then moved to the University of Oxford, supporting the development of Summit Therapeutics Duchenne Muscular Dystrophy clinical candidate ezutromid and fast follower compounds. Nicky also has a background in early stage process chemistry from appointments at Piramal Healthcare and GlaxoSmithKline.
Hannah L. Woodward has been a Research Associate within the Alzheimer’s Research UK UCL Drug Discovery Institute since 2016. She carried out her Ph.D. studies in synthetic organic chemistry at the University of Bristol. After receiving her Ph.D. in 2009, she spent 2.5 years working at Novartis Institute for BioMedical Research before taking up a Postdoctoral Training Fellow position at The Institute of Cancer Research in 2011. Hannah has been involved in the discovery of quality preclinical drug candidates and chemical probes across several therapeutic disease areas.
Paul Whiting is Chief Scientific Officer for the Alzheimer’s Research UK UCL Drug Discovery Institute and Group Leader at the UK Dementia Research Institute. Paul is a neuroscientist with a career in drug discovery in both the pharma and academic sectors. He has lead drug and cell therapy programs from basic research through to clinical trials and has coauthored around 200 scientific publications.
Jean-Paul Vincent is a Senior Group Leader at The Francis Crick Institute where he investigates how intracellular trafficking and other processes modulate the activity of Wingless, an important signaling molecule. He was elected to EMBO in 2006. He is a fellow of the Academy of Medical Sciences (FMedSci) and the Royal Society (FRS).
Paul V. Fish is Head of Chemistry for the Alzheimer’s Research UK UCL Drug Discovery Institute and Senior Group Leader at The Francis Crick Institute (University Attachment). Paul is a medicinal chemist with a career in drug discovery, and through his research he has helped to codiscover and deliver a number of drug candidates across several therapeutic disease areas, some of which have gone on to advanced clinical studies. He acts as a scientific advisor to a number of charitable organizations.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by Alzheimer’s Research UK (ARUK) and The Francis Crick Institute. The ARUK UCL Drug Discovery Institute is core funded by Alzheimer’s Research UK (520909). The Francis Crick Institute receives its core funding from Cancer Research UK (FC001002), the UK Medical Research Council (FC001002), and the Wellcome Trust (FC001002).
The authors declare the following competing financial interest(s): E.D.B., B.N.A., N.J.W., H.L.W., and P.V.F. are co-inventors of patent application WO 2020043866, which describes inhibitors of Notum. The authors have no other relevant affiliations or financial involvement apart from those disclosed.
References
- Sharma R. P.; Chopra V. L. Effect of the Wingless (wg1) on wing and haltere development in Drosophila melanogaster. Dev. Biol. 1976, 48, 461–465. 10.1016/0012-1606(76)90108-1. [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C.; Wieschaus E. Mutations affecting segment number and polarity in Drosophilia. Nature 1980, 287, 795–801. 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Nusse R.; Varmus H. E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- Loh K. M.; van Amerongen R.; Nusse R. Generating cellular diversity and spatial form: Wnt signaling and the evolution of multicellular animals. Dev. Cell 2016, 38, 643–655. 10.1016/j.devcel.2016.08.011. [DOI] [PubMed] [Google Scholar]
- MacDonald B. T.; Tamai K.; He X. Wnt//-catenin signaling: components, mechanisms, and disease. Dev. Cell 2009, 17, 9–26. 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. E.; Huang H.; Zhao M.; Zhang A.; Semonov M. V.; MacDonald B. T.; Zhang X.; Abreu J. G.; Peng L.; He X.; Zhang X. Wnt stabilization of. -catenin reveals principles for morphogen receptor-scaffold assemblies. Science 2013, 340, 867–870. 10.1126/science.1232389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Städeli R.; Basler K. Dissecting nuclear Wingless signalling: recruitment of the transcriptional co-activator Pygopus by a chain of adaptor proteins. Mech. Dev. 2005, 122, 1171–1182. 10.1016/j.mod.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Humphries A. C.; Mlodzik M. From instruction to ouput: Wnt/PCP signaling in development and cancer. Curr. Opin. Cell Biol. 2018, 51, 110–116. 10.1016/j.ceb.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J.; Nusse R.; van Amerongen R. The role of Ryk and Ror receptor tyrosine kinases in Wnt signal transduction. Cold Spring Harbor Perspect. Biol. 2014, 6, a009175 10.1101/cshperspect.a009175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K.; Brown J. D.; Danenberg E.; Duncan A. W.; Weissman I. L.; Reya T.; Yates J. R.; Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- Takada R.; Satomi Y.; Kurata T.; Ueno N.; Norioka S.; Kondoh H.; Takao T.; Takada S. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev. Cell 2006, 11, 791–801. 10.1016/j.devcel.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Janda C. Y.; Waghray D.; Levin A. M.; Thomas C.; Garcia K. C. Structural basis of Wnt recognition by Frizzled. Science 2012, 337, 59–64. 10.1126/science.1222879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena-López L. A.; Franch-Marro X.; Vincent J.-P. Wingless promotes proliferative growth in a gradient-independent manner. Sci. Signal. 2009, 2, ra60 10.1126/scisignal.2000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Wu Y.; Belenkaya T. Y.; Huang Q.; Ray L.; Qu J.; Lin X. Roles of N-glycosylation and lipidation in Wg secretion and signaling. Dev. Biol. 2012, 364, 32–41. 10.1016/j.ydbio.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki T.; Wilder E.; Klingensmith J.; Zachary K.; Perrimon N. The segment polarity gene Porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 1996, 10, 3116–3128. 10.1101/gad.10.24.3116. [DOI] [PubMed] [Google Scholar]
- Nile A. H.; Hannoush R. N. Fatty acylation of Wnt proteins. Nat. Chem. Biol. 2016, 12, 60–69. 10.1038/nchembio.2005. [DOI] [PubMed] [Google Scholar]
- Ho S. Y.; Keller T. H. The use of Porcupine inhibitors to target Wnt-driven cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5472–5476. 10.1016/j.bmcl.2015.10.032. [DOI] [PubMed] [Google Scholar]
- Clevers H.; Nusse R. Wnt//-catenin signaling and disease. Cell 2012, 149, 1192–1205. 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Moon R. T.; Kohn A. D.; De Ferrari G. V.; Kaykas A. WNT and W-catenin signalling: diseases and therapies. Nat. Rev. Genet. 2004, 5, 691–701. 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Marzo A.; Galli S.; Lopes D.; McLeod F.; Podpolny M.; Segovia-Roldan M.; Ciani L.; Purro S.; Cacucci F.; Gibb A.; Salinas P. C. Reversal of synapse degeneration by restoring Wnt signaling in the adult hippocampus. Curr. Biol. 2016, 26, 2551–2561. 10.1016/j.cub.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T.; Rindtorff N.; Boutros M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat C.-M.; Niehrs C. Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081 10.1101/cshperspect.a015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlitz O.; Basler K. Wingful, an extracellular feedback inhibitor of Wingless. Genes Dev. 2002, 16, 1055–1059. 10.1101/gad.991802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez A. J.; Copley R. R.; Cohen S. M. HSPG modification by the secreted enzyme Notum shapes the Wingless morphogen gradient. Dev. Cell 2002, 2, 667–676. 10.1016/S1534-5807(02)00180-6. [DOI] [PubMed] [Google Scholar]
- Chang M. V.; Chang J. L.; Gangopadhyay A.; Shearer A.; Cadigan K. M. Activation of Wingless targets requires bipartite recognition of DNA by TCF. Curr. Biol. 2008, 18, 1877–1881. 10.1016/j.cub.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos I.; Asare A.; Levorse J.; Ouspenskaia T.; de la Cruz-Racelis J.; Schuhmacher L.-N.; Fuchs E. Progenitors oppositely polarize WNT activators and inhibitors to orchestrate tissue development. eLife 2020, 9, e54304 10.7554/eLife.54304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. H.; Kim D.; Kim J.; Lee H.; Ghim J.; Kang B. J.; Song P.; Suh P.-G.; Ryu S. H.; Lee T. G. NOTUM is involved in the progression of colorectal cancer. Cancer Genomics Proteomics 2018, 15, 485–497. 10.21873/cgp.20107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J.; Perez L.; Giraldez A. J.; Cohen S. M. Opposing activities of Dally-like glypican at high and low levels of Wingless morphogen activity. Dev. Cell 2004, 7, 503–512. 10.1016/j.devcel.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Kakugawa S.; Langton P. F.; Zebisch M.; Howell S.; Chang T.-H.; Liu Y.; Feizi T.; Bineva G.; O’Reilly N.; Snijders A. P.; Jones E. J.; Vincent J.-P. Notum deacylates Wnt proteins to supress signalling activity. Nature 2015, 519, 187–192. 10.1038/nature14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger S.; Willert K. Mechanisms of Wnt signaling and control. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1422 10.1002/wsbm.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn M. Can we safely target the WNT pathway?. Nat. Rev. Drug Discovery 2014, 13, 513–532. 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C. Wnt is back in drugmakers’ sights, but is it druggable?. Nat. Biotechnol. 2018, 36, 1028–1029. 10.1038/nbt1118-1028. [DOI] [PubMed] [Google Scholar]
- Jung Y.-S.; Park J.-I. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond. -catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. 10.1038/s12276-020-0380-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B.; Green B. A.; Farr J. M.; Lopes F. C. M.; Van Raay T. J. Wnt drug discovery: weaving through the screens, patents and clinical trials. Cancers 2016, 8, 82. 10.3390/cancers8090082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S.; Liu J.; Wu Y.; Huang T. L.; Wang G. Small-molecule inhibitors of Wnt signaling pathway: towards novel anticancer therapeutics. Future Med. Chem. 2015, 7, 2485–2505. 10.4155/fmc.15.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C.; Zhou X.; Zhang W.; Qu Y.; Ke X. Is. -catenin a druggable target for cancer therapy?. Trends Biochem. Sci. 2018, 43, 623–634. 10.1016/j.tibs.2018.06.003. [DOI] [PubMed] [Google Scholar]
- Behrens J.; Von Kries J. P.; Kühl M.; Bruhn L.; Wedlich D.; Grosschedl R.; Birchmeier W. Functional interaction of. -catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Sferrazza G.; Corti M.; Brusotti G.; Pierimarchi P.; Temporini C.; Serafino A.; Calleri E. Nature-derived compounds modulating Wnt//-catenin pathway: a preventive and therapeutic opportunity in neoplastic diseases. Acta Pharm. Sin. B 2020, 10, 1814–1834. 10.1016/j.apsb.2019.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh V.; Hu H.; Barroga C.; Bossard C.; Kc S.; Dellamary L.; Stewart J.; Chiu K.; Ibanez M.; Pedraza M.; Seo T.; Do L.; Cho S.; Cahiwat J.; Tam B.; Tambiah J. R. S.; Hood J.; Lane N. E.; Yazici Y. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthritis and Cartilage 2018, 26, 18–27. 10.1016/j.joca.2017.08.015. [DOI] [PubMed] [Google Scholar]
- Yazici Y.; McAlindon T. E.; Gibofsky A.; Lane N. E.; Latterman C.; Skrepnik N.; Swearingen C. J.; DiFrancesco A.; Tambiah J. R.; Hochberg M. C. Efficacy and safety from a phase 2B trial of SM04690, a novel, intra-articular, WNT pathway inhibitor for the treatment of osteoarthritis of the knee. Osteoarthritis and Cartilage 2019, 27, S503. 10.1016/j.joca.2019.02.566. [DOI] [PubMed] [Google Scholar]
- Deshmukh V.; O’Green A. L.; Bossard C.; Seo T.; Lamangan L.; Ibanez M.; Ghias A.; Lai C.; Do L.; Cho S.; Cahiwat J.; Chiu K.; Pedraza M.; Anderson S.; Harris R.; Dellamary L.; Kc S.; Barroga C.; Melchior B.; Tam B.; Kennedy S.; Tambiah J.; Hood J.; Yazici Y. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthritis and Cartilage 2019, 27, 1347–1360. 10.1016/j.joca.2019.05.006. [DOI] [PubMed] [Google Scholar]
- Tam B. Y.; Chiu K.; Chung H.; Bossard C.; Nguyen J. D.; Creger E.; Eastman B. W.; Mak C. C.; Ibanez M.; Ghias A.; Cahiwat J.; Do L.; Cho S.; Nguyen J.; Deshmukh V.; Stewart J.; Chen C.-W.; Barroga C.; Dellamary L.; Kc S. K.; Phalen T. J.; Hood J.; Cha S.; Yazici Y. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020, 473, 186–197. 10.1016/j.canlet.2019.09.009. [DOI] [PubMed] [Google Scholar]
- Deshmukh V.; Pedraza M.; Barroga C.; Seykora J.; Yazici Y. Small molecule Wnt pathway inhibitor (SM04755) as a potential topical psoriasis treatment. J. Invest. Dermatol. 2017, 137, S122. 10.1016/j.jid.2017.02.735. [DOI] [Google Scholar]
- Yan C.; Higgins P. J. Drugging the undruggable: transcription therapy for cancer. Biochim. Biophys. Acta, Rev. Cancer 2013, 1835, 76–85. 10.1016/j.bbcan.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami K. H.; Nguyen C.; Ma H.; Kim D. H.; Jeong K. W.; Eguchi M.; Moon R. T.; Teo J.-L.; Oh S. W.; Kim H. Y.; Moon S. H.; Ha J. R.; Kahn M. A small molecule inhibitor of. -catenin/cyclic AMP response element-binding protein transcription. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12682–12687. 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa Y.; Oboki K.; Imamura J.; Kojika E.; Hayashi Y.; Hishima T.; Saibara T.; Shibasaki F.; Kohara M.; Kimura K. Inhibition of cyclic adenosine monophosphate (cAMP)-response element-binding protein (CREB)-binding protein (CBP)//-catenin reduces liver fibrosis in mice. EBioMedicine 2015, 2, 1751–1758. 10.1016/j.ebiom.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafyatis R.; Mantero J. C.; Gordon J.; Kishore N.; Carns M.; Dittrich H.; Spiera R.; Simms R. W.; Varga J. Inhibition of. -catenin signaling in the skin rescues cutaneous adipogenesis in systemic sclerosis: a randomized, double-blind, placebo-controlled trial of C-82. J. Invest. Dermatol. 2017, 137, 2473–2483. 10.1016/j.jid.2017.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha J. Y.; Jung J.-E.; Lee K.-H.; Briaud I.; Tenzin F.; Jung H. K.; Pyon Y.; Lee D.; Chung J. U.; Lee J. H.; Oh S.-W.; Jung K. Y.; Pai J. K.; Emami K. Anti-tumor activity of novel small molecule Wnt signaling inhibitor, CWP232291, in multiple myeloma. Blood 2010, 116, 3038. 10.1182/blood.V116.21.3038.3038. [DOI] [Google Scholar]
- Briaud I.; Tenzin F.; Chun J. K.; Oh S.-W.; Chung J. U.; Jung K. Y.; Jeong M. W.; Cha J. Y.; Pyon Y.; Lee D.; Jung J. E.; Emami K. H. Therapeutic potential of small molecule inhibitor of Wnt signaling in acute myelogenous leukemia. Blood 2008, 112, 4001. 10.1182/blood.V112.11.4001.4001. [DOI] [Google Scholar]
- Torres V. I.; Godoy J. A.; Inestrosa N. C. Modulating Wnt signaling at the root: Porcupine and Wnt acylation. Pharmacol. Ther. 2019, 198, 34–45. 10.1016/j.pharmthera.2019.02.009. [DOI] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C.-W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth M. G.; Amatruda J. F.; Chen C.; Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Pan S.; Hsieh M. H.; Ng N.; Sun F.; Wang T.; Kasibhatla S.; Schuller A. G.; Li A. G.; Cheng D.; Li J.; Tompkins C.; Pferdekamper A.; Steffy A.; Cheng J.; Kowal C.; Phung V.; Guo G.; Wang Y.; Graham M. P.; Flynn S.; Brenner J. C.; Li C.; Villarroel M. C.; Schultz P. G.; Wu X.; McNamara P.; Sellers W. R.; Petruzzelli L.; Boral A. L.; Seidel H. M.; McLaughlin M. E.; Che J.; Carey T. E.; Vanasse G.; Harris J. L. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20224–20229. 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F.; Connolly R.; LoRusso P.; de Jonge M.; Vaishampayan U.; Rodon J.; Argilés G.; Myers A.; Hsu Schmitz S.-F.; Ji Y.; McLaughlin M.; Palmer M. R.; Morawiak J. Phase 1 study of WNT974, a first-in-class Porcupine inhibitor, in advanced solid tumors. Mol. Cancer Ther. 2015, 14, C45. 10.1158/1535-7163.TARG-15-C45. [DOI] [Google Scholar]
- Rodon J.; Argilés G.; Connolly R. M.; Vaishampayan U.; de Jonge M.; Garralda E.; Giannakis M.; Smith D. C.; Dobson J. R.; McLaughlin M.; Seroutou A.; Ji Y.; Dolan S.; Morawiak J.; Moody S.; Janku F. Biomarker analyses from phase 1 study of WNT974, a first-in-class Porcupine inhibitor, in patients (pts) with advanced solid tumors. Cancer Res. 2018, 78, CT175. 10.1158/1538-7445.AM2018-CT175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios-Esteves J.; Haugen B.; Resh M. D. Identification of key residues and regions important for Porcupine-mediated Wnt acylation. J. Biol. Chem. 2014, 289, 17009–17019. 10.1074/jbc.M114.561209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D.; Wang Z.; Merrikh C. N.; Lang K. S.; Lu P.; Li X.; Merrikh H.; Rao Z.; Xu W. Crystal structure of a membrane-bound O-acyltransferase. Nature 2018, 562, 286–290. 10.1038/s41586-018-0568-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-M. A.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. Tankyrase inhibition stabilizes Axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Mariotti L.; Pollock K.; Guettler S. Regulation of Wnt/β-catenin signalling by Tankyrase-dependent poly(ADP-ribosyl)ation and scaffolding. Br. J. Pharmacol. 2017, 174, 4611–4636. 10.1111/bph.14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlberg T.; Markova N.; Johansson I.; Hammarström M.; Schütz P.; Weigelt J.; Schüler H. Structural basis for the interaction between Tankyrase-2 and a potent Wnt-signaling inhibitor. J. Med. Chem. 2010, 53, 5352–5355. 10.1021/jm100249w. [DOI] [PubMed] [Google Scholar]
- Voronkov A.; Holsworth D. D.; Waaler J.; Wilson S. R.; Ekblad B.; Perdreau-Dahl H.; Dinh H.; Drewes G.; Hopf C.; Morth J. P.; Krauss S. Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J. Med. Chem. 2013, 56, 3012–23. 10.1021/jm4000566. [DOI] [PubMed] [Google Scholar]
- Shultz M. D.; Cheung A. K.; Kirby C. A.; Firestone B.; Fan J.; Chen C. H.; Chen Z.; Chin D. N.; Dipietro L.; Fazal A.; Feng Y.; Fortin P. D.; Gould T.; Lagu B.; Lei H.; Lenoir F.; Majumdar D.; Ochala E.; Palermo M. G.; Pham L.; Pu M.; Smith T.; Stams T.; Tomlinson R. C.; Touré B. B.; Visser M.; Wang R. M.; Waters N. J.; Shao W. Identification of NVP-TNKS656: the use of structure-efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J. Med. Chem. 2013, 56, 6495–6511. 10.1021/jm400807n. [DOI] [PubMed] [Google Scholar]
- Menon M.; Elliott R.; Bowers L.; Balan N.; Rafiq R.; Costa-Cabral S.; Munkonge F.; Trinidade I.; Porter R.; Campbell A. D.; Johnson E. R.; Esdar C.; Buchstaller H.-P.; Leuthner B.; Rohdich F.; Schneider R.; Sansom O.; Wienke D.; Ashworth A.; Lord C. J. A novel tankyrase inhibitor, MSC2504877, enhances the effects of clinical CDK4/6 inhibitors. Sci. Rep. 2019, 9, 201.and references therein 10.1038/s41598-018-36447-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau T.; Chan E.; Callow M.; Waaler J.; Boggs J.; Blake R. A.; Magnuson S.; Sambrone A.; Schutten M.; Firestein R.; Machon O.; Korinek V.; Choo E.; Diaz D.; Merchant M.; Polakis P.; Holsworth D. D.; Krauss S.; Costa M. A novel Tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. 10.1158/0008-5472.CAN-12-4562. [DOI] [PubMed] [Google Scholar]
- Mlodzik M.The Dishevelled protein family: still rather a mystery after over 20 years of molecular studies. In Current Topics in Developmental Biology; Wassarman P. M., Ed.; Academic Press, 2016; Vol. 117, pp 75–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandy D.; Shan J.; Zhang X.; Rao S.; Akunuru S.; Li H.; Zhang Y.; Alpatov I.; Zhang X. A.; Lang R. A.; Shi D.-L.; Zheng J. J. Discovery and characterization of a small molecule inhibitor of the PDZ domain of Dishevelled. J. Biol. Chem. 2009, 284, 16256–16263. 10.1074/jbc.M109.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K.; Ajioka K.; Goda N.; Shindo A.; Takagishi M.; Tenno T.; Hiroaki H. Discovery of potent Disheveled/Dvl inhibitors using virtual screening optimized with NMR-based docking performance index. Front. Pharmacol. 2018, 9, 983. 10.3389/fphar.2018.00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-J.; Bao J.; Miller A.; Zhang C.; Wu J.; Baday Y. C.; Guibao C.; Li L.; Wu D.; Zheng J. J. Structure-based discovery of novel small molecule Wnt signaling inhibitors by targeting the cysteine-rich domain of Frizzled. J. Biol. Chem. 2015, 290, 30596–30606. 10.1074/jbc.M115.673202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Hillier J.; Lu W.; Jones E. Y. Antiepileptic drug carbamazepine binds to a novel pocket on the Wnt receptor Frizzled-8. J. Med. Chem. 2020, 63, 3252–3260. 10.1021/acs.jmedchem.9b02020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Wu H.; Katritch V.; Han G. W.; Huang X.-P.; Liu W.; Siu F. Y.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human Smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Lu W.; Ananthan S.; Suto M. J.; Li Y. Discovery of novel Frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459–91470. 10.18632/oncotarget.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H.; Matoba K.; Mihara E.; Arimori T.; Takagi J. Crystal structure of a mammalian Wnt-Frizzled complex. Nat. Struct. Mol. Biol. 2019, 26, 372–379. 10.1038/s41594-019-0216-z. [DOI] [PubMed] [Google Scholar]
- Yeung P.; Beviglia L.; Cancilla B.; Dee-Hoskins C.; Evans J. W.; Fischer M. M.; Yen W.-C.; Gurney A.; Lewicki J.; Hoey T.; Kapoun A. M. Wnt pathway antagonist OMP-54F28 (FZD8-Fc) inhibits tumor growth and reduces tumor-initiating cell frequency in patient-derived hepatocellular carcinoma and ovarian cancer xenograft models. Cancer Res. 2014, 74, 1907. 10.1158/1538-7445.AM2014-1907. [DOI] [Google Scholar]
- Jimeno A.; Gordon M.; Chugh R.; Messersmith W.; Mendelson D.; Dupont J.; Stagg R.; Kapoun A. M.; Xu L.; Uttamsingh S.; Brachmann R. K.; Smith D. C. A first-in-human phase 1 study of the anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for Wnt ligands, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 7490–7497. 10.1158/1078-0432.CCR-17-2157. [DOI] [PubMed] [Google Scholar]
- Gurney A.; Axelrod F.; Bond C. J.; Cain J.; Chartier C.; Donigan L.; Fischer M.; Chaudhari A.; Ji M.; Kapoun A. M.; Lam A.; Lazetic S.; Ma S.; Mitra S.; Park I.-K.; Pickell K.; Sato A.; Satyal S.; Stroud M.; Tran H.; Yen W.-C.; Lewicki J.; Hoey T. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11717–11722. 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S.; Beilschmidt M.; To M.; Lin K.; Lui F.; Jmeian Y.; Ng M.; Fernandez M.; Fu Y.; Mascall K.; Duque A.; Wang X.; Pan G.; Angers S.; Moffat J.; Sidhu S. S.; Magram J.; Sinclair A. M.; Fransson J.; Julien J.-P. Structure-guided design fine-tunes pharmakinetics, tolerability, and antitumor profile of multispecific Frizzled antibodies. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 6812–6817. 10.1073/pnas.1817246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinzalla V.; Drobits-Handl B.; Savchenko A.; Rinnenthal J.; Bauer M. J.; Sanderson M.; Blake S. M.; Schweifer N.; Vries R. G. J.; Clevers H.; Kraut N. BI 905677: a first-in-class LRP5/6 antagonist targeting Wnt-driven proliferation and immune escape. Cancer Res. 2019, 79, DDT01-01. 10.1158/1538-7445.AM2019-DDT01-01. [DOI] [Google Scholar]
- Bafico A.; Liu G.; Yaniv A.; Gazit A.; Aaronson S. A. Novel mechanism of Wnt signalling inhibition mediated by Dikkopf-1 interaction with LRP6/Arrow. Nat. Cell Biol. 2001, 3, 683–686. 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- Iozzi S.; Remelli R.; Lelli B.; Diamanti D.; Pileri S.; Bracci L.; Roncarati R.; Caricasole A.; Bernocco S. Functional characterization of a small-molecule inhibitor of the DKK1-LRP6 interaction. ISRN Mol. Biol. 2012, 2012, 823875. 10.5402/2012/823875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpousis S.; Thysiadis S.; Avramidis N.; Katsamakas S.; Efthimiopoulos S.; Sarli V. Synthesis and evaluation of gallocyanine dyes as potential agents for the treatment of Alzheimer’s disease and related neurodegenerative tauopathies. Eur. J. Med. Chem. 2016, 108, 28–38. 10.1016/j.ejmech.2015.11.024. [DOI] [PubMed] [Google Scholar]
- Thysiadis S.; Katsamakas S.; Mpousis S.; Avramidis N.; Efthimiopoulos S.; Sarli V. Design and synthesis of gallocyanine inhibitors of DKK1/LRP6 interactions for the treatment of Alzeimer’s disease. Bioorg. Chem. 2018, 80, 230–244. 10.1016/j.bioorg.2018.06.018. [DOI] [PubMed] [Google Scholar]
- Bafico A.; Gazit A.; Pramila T.; Finch P. W.; Yaniv A.; Aaronson S. A. Interaction of Frizzled Related Protein (FRP) with Wnt ligands and the Frizzled receptor suggests alternative mechanisms for the FRP inhibition of Wnt signaling. J. Biol. Chem. 1999, 274, 16180–16187. 10.1074/jbc.274.23.16180. [DOI] [PubMed] [Google Scholar]
- Bodine P. V. N.; Stauffer B.; Ponce-de-Leon H.; Bhat R. A.; Mangine A.; Seestaller-Wehr L. M.; Moran R. A.; Billiard J.; Fukayama S.; Komm B. S.; Pitts K.; Krishnamurthy G.; Gopalsamy A.; Shi M.; Kern J. C.; Commons T. J.; Woodworth R. P.; Wilson M. A.; Welmaker G. S.; Trybulski E. J.; Moore W. J. A small molecule inhibitor of the Wnt antagonist secreted Frizzled-Related Protein-1 stimulates bone formation. Bone 2009, 44, 1063–1068. 10.1016/j.bone.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Moore W. J.; Kern J. C.; Bhat R.; Commons T. J.; Fukayama S.; Goljer I.; Krishnamurthy G.; Magolda R. L.; Nogle L.; Pitts K.; Stauffer B.; Trybulski E. J.; Welmaker G. S.; Wilson M.; Bodine P. V. N. Modulation of Wnt signaling through inhibition of secreted Frizzled-Related Protein 1 (sFRP-1) with N-substituted piperidinyl diphenylsulfonyl sulfonamides. J. Med. Chem. 2009, 52, 105–116. 10.1021/jm801144h. [DOI] [PubMed] [Google Scholar]
- Moore W. J.; Kern J. C.; Bhat R.; Bodine P. V. N.; Fukyama S.; Krishnamurthy G.; Magolda R. L.; Pitts K.; Stauffer B.; Trybulski E. J. Modulation of Wnt signaling through inhibition of secreted Frizzled-Related Protein 1 (sFRP-1) with N-substituted piperidinyl diphenylsulfonyl sulfonamides: part II. Bioorg. Med. Chem. 2010, 18, 190–201. 10.1016/j.bmc.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Pelletier J. C.; Lundquist J. T.; Gilbert A. M.; Alon N.; Bex F. J.; Bhat B. M.; Bursavich M. G.; Coleburn V. E.; Felix L. A.; Green D. M.; Green P.; Hauze D. B.; Kharode Y. P.; Lam H.-S.; Lockhead S. R.; Magolda R. L.; Matteo J. J.; Mehlmann J. F.; Milligan C.; Murrills R. J.; Pirrello J.; Selim S.; Sharp M. C.; Unwalla R. J.; Vera M. D.; Wrobel J. E.; Yaworsky P.; Bodine P. V. N. (1-(4-(Naphthalen-2-yl)pyrimidin-2-yl)piperidin-4-yl)methanamine: a wingless. -catenin agonist that increases bone formation rate. J. Med. Chem. 2009, 52, 6962–6965. 10.1021/jm9014197. [DOI] [PubMed] [Google Scholar]
- Kagey M. H.; He X. Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology. Br. J. Pharmacol. 2017, 174, 4637–4650. 10.1111/bph.13894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asem M. S.; Buechler S.; Wates R.; Miller D. L.; Stack M. S. Wnt5a signaling in cancer. Cancers 2016, 8, 79. 10.3390/cancers8090079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann C. E.; Hsieh J.-C.; Rattner A.; Sharma D.; Nathans J.; Leahy D. J. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature 2001, 412, 86–90. 10.1038/35083601. [DOI] [PubMed] [Google Scholar]
- Yang S.; Wu Y.; Xu T.-H.; de Waal P. W.; He Y.; Pu M.; Chen Y.; DeBruine Z. J.; Zhang B.; Zaidi S. A.; Popov P.; Guo Y.; Han G. W.; Lu Y.; Suino-Powell K.; Dong S.; Harikumar K. G.; Miller L. J.; Katritch V.; Xu H. E.; Shui W.; Stevens R. C.; Melcher K.; Zhao S.; Xu F. Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature 2018, 560, 666–670. 10.1038/s41586-018-0447-x. [DOI] [PubMed] [Google Scholar]
- Wong H. C.; Mao J.; Nguyen J. T.; Srinivas S.; Zhang W.; Liu B.; Li L.; Wu D.; Zheng J. Structural basis of the recognition of the Dishevelled DEP domain in the Wnt signaling pathway. Nat. Struct. Biol. 2000, 7, 1178–1184. 10.1038/82047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Appleton B. A.; Wiesmann C.; Lau T.; Costa M.; Hannoush R. N.; Sidhu S. S. Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat. Chem. Biol. 2009, 5, 217–219. 10.1038/nchembio.152. [DOI] [PubMed] [Google Scholar]
- Graham T. A.; Weaver C.; Mao F.; Kimelman D.; Xu W. Crystal structure of a. -catenin/TCF complex. Cell 2000, 103, 885–896. 10.1016/S0092-8674(00)00192-6. [DOI] [PubMed] [Google Scholar]
- Cheng Z.; Biechele T.; Wei Z.; Morrone S.; Moon R. T.; Wang L.; Xu W. Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 2011, 18, 1204–1210. 10.1038/nsmb.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Portal . https://gtexportal.org/home/gene/NOTUM (accessed Nov 16, 2020).
- Tarver J. E. Jr.; Pabba P. K.; Barbosa J.; Han Q.; Gardyan M. W.; Brommage R.; Thompson A. Y.; Schmidt J. M.; Wilson A. G. E.; He W.; Lombardo V. K.; Carson K. G. Stimulation of cortical bone formation with thienopyrimidine based inhibitors of Notum Pectinacetylesterase. Bioorg. Med. Chem. Lett. 2016, 26, 1525–1528. 10.1016/j.bmcl.2016.02.021. [DOI] [PubMed] [Google Scholar]
- Vogel P.; Read R. W.; Hansen G. M.; Powell D. R.; Kantaputra P. N.; Zambrowicz B.; Brommage R. Dentin dysplasia in Notum knockout mice. Vet. Pathol. 2016, 53, 853–862. 10.1177/0300985815626778. [DOI] [PubMed] [Google Scholar]
- Movérare-Skrtic S.; Nilsson K. H.; Henning P.; Funck-Brentano T.; Nethander M.; Rivadeneira F.; Coletto Nunes G.; Koskela A.; Tuukkanen J.; Tuckermann J.; Perret C.; Souza P. P. C.; Lerner U. H.; Ohlsson C. Osteoblast-derived NOTUM reduces cortical bone mass in mice and the NOTUM locus is associated with bone mineral density in humans. FASEB J. 2019, 33, 11163–11179. 10.1096/fj.201900707R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brommage R.; Liu J.; Vogel P.; Mseeh F.; Thompson A. Y.; Potter D. G.; Shadoan M. K.; Hansen G. M.; Jeter-Jones S.; Cui J.; Bright D.; Bardenhagen J. P.; Doree D. D.; Movérare-Skrtic S.; Nilsson K. H.; Henning P.; Lerner U. H.; Ohlsson C.; Sands A. T.; Tarver J. E.; Powell D. R.; Zambrowicz B.; Liu Q. NOTUM inhibition increases endocortical bone formation and bone strength. Bone Res. 2019, 7, 2. 10.1038/s41413-018-0038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal F.; Charawi S.; Grimber G.; Houbron C.; Drouet V.; Colnot S.; Terris B.; Cavard C.; Perret C. Generation of mice with hepatocyte-specific conditional deletion of Notum. PLoS One 2016, 11, e0150997 10.1371/journal.pone.0150997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S. H.; Barker N. Wnt signaling in adult epithelial stem cells and cancer. Prog. Mol. Biol. Transl. Sci. 2018, 153, 21–79. 10.1016/bs.pmbts.2017.11.017. [DOI] [PubMed] [Google Scholar]
- Clevers H.; Loh K. M.; Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. 10.1126/science.1248012. [DOI] [PubMed] [Google Scholar]
- Pentinmikko N.; Iqbal S.; Mana M.; Andersson S.; Cognetta A. B.; Suciu R. M.; Roper J.; Luopajärvi K.; Markelin E.; Gopalakrishnan S.; Smolander O. P.; Naranjo S.; Saarinen T.; Juuti A.; Pietiläinen K.; Auvinen P.; Ristimäki A.; Gupta N.; Tammela T.; Jacks T.; Sabatini D. M.; Cravatt B. F.; Yilmaz O. H.; Katajisto P. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 2019, 571, 398–402. 10.1038/s41586-019-1383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckervordersandforth B.; Rolando C. Untangling human neurogenesis to understand and counteract brain disorders. Curr. Opin. Pharmacol. 2020, 50, 67–73. 10.1016/j.coph.2019.12.002. [DOI] [PubMed] [Google Scholar]
- Kuhn H. G.; Toda T.; Gage F. H. Adult hippocampal neurogenesis: a coming-of-age story. J. Neurosci. 2018, 38, 10401–10410. 10.1523/JNEUROSCI.2144-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota Y.; Sawada M.; Huang S. H.; Ogino T.; Ohata S.; Kubo A.; Sawamoto K. Roles of Wnt signaling in the neurogenic niche of the adult mouse ventricular-subventricular zone. Neurochem. Res. 2016, 41, 222–230. 10.1007/s11064-015-1766-z. [DOI] [PubMed] [Google Scholar]
- Mizrak D.; Bayin N. S.; Yuan J.; Liu Z.; Suciu R. M.; Niphakis M. J.; Ngo N.; Lum K. M.; Cravatt B. F.; Joyner A. L.; Sims P. A. Single-cell profiling and SCOPE-Seq reveal lineage dynamics of adult ventricular-subventricular zone neurogenesis and NOTUM as a key regulator. Cell Rep. 2020, 31, 107805. 10.1016/j.celrep.2020.107805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z.; Yu J.; Virshup D. M.; Madan B. Wnts and the hallmarks of cancer. Cancer Metastasis Rev. 2020, 39, 625–645. 10.1007/s10555-020-09887-6. [DOI] [PubMed] [Google Scholar]
- Carreira-Barbosa F.; Nunes S. C. Wnt signaling: paths for cancer progression. Adv. Exp. Med. Biol. 2020, 1219, 189–202. 10.1007/978-3-030-34025-4_10. [DOI] [PubMed] [Google Scholar]
- Torisu Y.; Watanabe A.; Nonaka A.; Midorikawa Y.; Makuuchi M.; Shimamura T.; Sugimura H.; Niida A.; Akiyama T.; Iwanari H.; Kodama T.; Zeniya M.; Aburatani H. Human homolog of NOTUM, overexpressed in hepatocellular carcinoma, is regulated transcriptionally by c-catenin/TCF. Cancer Sci. 2008, 99, 1139–1146. 10.1111/j.1349-7006.2008.00814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis M.; Arigoni M.; Loiacono L.; Riccardo F.; Calogero R. A.; Feodorova Y.; Tashkova D.; Belovejdov V.; Sarafian V.; Cavallo F.; Signori E. Novel insights into Notum and glypicans regulation in colorectal cancer. Oncotarget 2015, 6, 41237–41257. 10.18632/oncotarget.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan B.; Ke Z.; Lei Z. D.; Oliver F. A.; Oshima M.; Lee M. A.; Rozen S.; Virshup D. M. NOTUM is a potential pharmacodynamic biomarker of Wnt pathway inhibition. Oncotarget 2016, 7, 12386–12392. 10.18632/oncotarget.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Patricelli M. P.; Cravatt B. F. Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 14694–14699. 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa J.; Carson K. G.; Gardyan M. W.; He W.; Lombardo V.; Pabba P.; Tarver J. Jr.. Inhibitors of Notum Pectinacetylesterase and Methods of Their Use. US Patent Application 0065200, Mar 15, 2012.
- Barbosa J.; Carson K. G.; Gardyan M. W.; Healy J. P.; Han Q.; Mabon R.; Pabba P.; Tarver J. Jr.; Terranova K. M.; Tunoori A.; Xu X.. 4H-Thieno[3,2-c]chromene-Based Inhibitors of Notum Pectinacetylesterase and Methods of Their Use. US Patent Application 0302562, Nov 29, 2012.
- Han Q.; Pabba P. K.; Barbosa J.; Mabon R.; Healy J. P.; Gardyan M. W.; Terranova K. M.; Brommage R.; Thompson A. Y.; Schmidt J. M.; Wilson A. G. E.; Xu X.; Tarver J. E. Jr.; Carson K. G. 4H-Thieno[3,2-c]chromene based inhibitors of Notum pectinacetylesterase. Bioorg. Med. Chem. Lett. 2016, 26, 1184–1187. 10.1016/j.bmcl.2016.01.038. [DOI] [PubMed] [Google Scholar]
- Willis N. J.; Bayle E. D.; Papageorgiou G.; Steadman D.; Atkinson B. N.; Mahy W.; Fish P. V. An improved, scalable synthesis of Notum inhibitor LP-922056 using 1-chloro-1,2-benziodoxol-3-one as a superior electrophilic chlorinating agent. Beilstein J. Org. Chem. 2019, 15, 2790–2797. 10.3762/bjoc.15.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson B. N.; Steadman D.; Mahy W.; Zhao Y.; Sipthorp J.; Bayle E. D.; Svensson F.; Papageorgiou G.; Jeganathan F.; Frew S.; Monaghan A.; Bictash M.; Jones E. Y.; Fish P. V. Scaffold-hopping identifies furano[2,3-d]pyrimidine amides as potent Notum inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126751. 10.1016/j.bmcl.2019.126751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suciu R. M.; Cognetta A. B.; Potter Z. E.; Cravatt B. F. Selective irreversible inhibitors of the Wnt-deacylating enzyme NOTUM developed by activity-based protein profiling. ACS Med. Chem. Lett. 2018, 9, 563–568. 10.1021/acsmedchemlett.8b00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin D. A.; Cravatt B. F. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discovery 2012, 11, 52–68. 10.1038/nrd3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Tian Y.; Wang M.; Wang M.; Sun G.-B.; Sun X.-B. Advanced activity-based protein profiling application strategies for drug development. Front. Pharmacol. 2018, 9, 353. 10.3389/fphar.2018.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond Light Source. https://www.diamond.ac.uk/Home (accessed Nov 16, 2020).
- DSi-Poised Library . https://www.diamond.ac.uk/Instruments/Mx/Fragment-Screening/Fragment-Libraries0/DSi-Poised-Library.html (accessed Nov 16, 2020).
- Atkinson B. N.; Steadman D.; Zhao Y.; Sipthorp J.; Vecchia L.; Ruza R. R.; Jeganathan F.; Lines G.; Frew S.; Monaghan A.; Kjaer S.; Bictash M.; Jones E. J.; Fish P. V. Discovery of 2-phenoxyacetamides as inhibitors of the Wnt-depalmitoleating enzyme NOTUM from an X-ray fragment screen. MedChemComm 2019, 10, 1361–1369. 10.1039/C9MD00096H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Hillier J.; Jones M.; Lu W.; Jones E. Y. Structural characterization of melatonin as an inhibitor of the Wnt deacylase Notum. J. Pineal Res. 2020, 68, e12630 10.1111/jpi.12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahy W.; Willis N. J.; Zhao Y.; Woodward H. L.; Svensson F.; Sipthorp J.; Vecchia L.; Ruza R. R.; Hillier J.; Kjaer S.; Frew S.; Monaghan A.; Bictash M.; Salinas P. C.; Whiting P.; Vincent J.-P.; Jones E. Y.; Fish P. V. 5-Phenyl-1,3,4-oxadiazol-2(3H)-ones are potent inhibitors of Notum carboxylesterase activity identified by the optimization of a crystallographic fragment screening hit. J. Med. Chem. 2020, 63, 12942–12956. 10.1021/acs.jmedchem.0c01391. [DOI] [PubMed] [Google Scholar]
- Fish P. V.; Mahy W.; Willis N. J.; Woodward H. L.; Atkinson B. N.; Bayle E. D.; Sipthorp J.; Jones E. Y.; Zhao Y.; Vecchia L.; Ruza R. R.. Inhibitors of Secreted Carboxylesterase Notum and Their Use in Increasing Wnt Protein Signalling and Treatment of Central Nervous and Other Disorders. WO Patent Application 2020043866, Mar 5, 2020.
- Fish P. V.Chemical Biology Enabling Drug Discovery. Small Molecule Chemical Tools of the Wnt-Depalmitoleating Enzyme NOTUM; ChemBiOx 2019: Oxford, U.K., Jul 30–31, 2019. https://chembiox.web.ox.ac.uk/home (accessed Nov 16, 2020).
- Mahy W.; Patel M.; Steadman D.; Woodward H. L.; Atkinson B. N.; Svensson F.; Willis N. J.; Flint A.; Papatheodorou D.; Zhao Y.; Vecchia L.; Ruza R. R.; Hillier J.; Frew S.; Monaghan A.; Costa A.; Bictash M.; Walter M.; Jones E. Y.; Fish P. V. Screening of a custom-designed acid fragment library identifies 1-phenylpyrroles and 1-phenylpyrrolidines as inhibitors of Notum carboxylesterase activity. J. Med. Chem. 2020, 63, 9464–9483. 10.1021/acs.jmedchem.0c00660. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Hillier J.; Lu W.; Jones E. Y. Caffeine inhibits Notum activity by binding at the catalytic pocket. Commun. Biol. 2020, 3, 555. 10.1038/s42003-020-01286-5. [DOI] [PMC free article] [PubMed] [Google Scholar]