

Abstract
In the past, in vitro studies of invasion and tumor progression were performed primarily using cancer cells cultured on a flat, two‐dimensional (2D) surface in a monolayer. In recent years, however, many studies have demonstrated differences in cell signaling and cell migration between 2D and 3D cell cultures. Traditional 2D monolayer cancer cell invasion models do not fully recapitulate 3D cell‐to‐cell and cell−to−extracellular matrix interactions that in vivo models can provide. Moreover, although in vivo animal models are irreplaceable for studying tumor biology and metastasis, they are costly, time‐consuming, and impractical for answering preliminary questions. Thus, emergent and evolving 3D spheroid cell culture models have changed the way we study tumors and their interactions with their surrounding extracellular matrix. In the case of breast cancer, metastasis of breast cancer tumors results in high mortality rates, and thus development of robust cell culture models that are reproducible and practical for studying breast cancer progression is important for ultimately developing preventatives for cancer metastasis. This article provides a set of protocols for generating uniform spheroids with a thin sheet of basement membrane for studying the initial invasion of mammary epithelial cells into a surrounding collagen‐rich extracellular matrix. Details are provided for generating 3D spheroids with a basement membrane, polymerizing collagen I, embedding the spheroids in the 3D collagen gel, and immunostaining the spheroids for invasion studies. Published 2020. U.S. Government.
Basic Protocol 1: Growth of uniformly sized tumor spheroids with an encapsulating basement membrane
Basic Protocol 2: Polymerization and embedding of tumor spheroids in a 3D type I collagen gel
Alternate Protocol: Embedding of tumor spheroids in collagen gels using a sandwich method
Basic Protocol 3: Fixing and immunostaining of tumor spheroids embedded in 3D collagen gels
Keywords: spheroid, basement membrane, invasion, 3D‐model
INTRODUCTION
In the normal mammary gland, epithelial cells that line the lumen are adjacent to contractile myoepithelial cells that surround them peripherally. The epithelial and adjacent contractile cells are encased in a supportive basement membrane. At the initial stage of a mammary tumor, ductal carcinoma in situ (DCIS) premalignant epithelial cells proliferate into the ductal lumen but remain confined within the basement membrane (Kaushik, Pickup, & Weaver, 2016; Lee et al., 2019). In contrast, during mammary tumor progression into invasive ductal carcinoma (IDC), there is a loss of the myoepithelial cell layer and a marked breaching of malignant cells through the basement membrane (Kaushik et al., 2016).
Cell migration is a critical step in early morphogenesis and in cancer cell invasion, which occurs through dynamic processes of synchronized cell adhesion and contractility, resulting in degradation and remodeling of the extracellular matrix (ECM) (Wolf et al., 2007). Invasive cancer cells are able to migrate away from the original tumor mass, traversing the confining basement membrane and moving through the three‐dimensional (3D) ECM by means of single‐cell migration or collective cell migration (Deakin & Turner, 2011; Friedl et al., 1995). Basement membrane penetration by cancer cells is an important initial step in invasion and hematogenous dissemination, leading to tumor metastasis and decreased patient survival. Since tumor metastasis is responsible for the high mortality rates in breast cancer and other malignancies, studying the initial steps of invasion through the basement membrane is important for therapeutic discoveries and prevention of metastasis.
To understand the initial stages of cancer cell invasion and migration, many researchers have started to use 3D hydrogels to mimic the ECM. For years, 2D cell cultures in monolayers were the standard for studying cancer cell biology and testing drug treatments and immunotherapies in vitro (Tevis, Colson, & Grinstaff, 2017). For decades, 2D cell cultures provided valuable insights for deciphering the mechanisms of cellular migration and identifying the key players involved in unicellular and multicellular cell adhesion and cell motility (Cukierman, Pankov, Stevens, & Yamada, 2001; Ridley et al., 2003). However, 2D cultures fail to fully recapitulate the dimensionality of the breast cancer microenvironment and those of other malignancies. Moreover, studies have demonstrated differences in cellular morphologies and cellular signaling between 2D and 3D culture systems (Yamada & Sixt, 2019). To overcome this obstacle, in vitro 3D tumor models have emerged in recent years as a superior method for studying the mechanisms of cancer initiation and progression. Many laboratories have used a mixture of Matrigel and collagen I gel to model basement membrane−ECM−cell interactions (Carey, Martin, & Reinhart‐King, 2017; Lee et al., 2019). Here we describe a protocol for generating uniform mammary tumor spheroids with an intact basement membrane embedded in a 3D collagen gel for invasion studies. Since DCIS is shown to form overwhelmingly in the areas of the breast that are more radiologically dense, with increased ECM stiffness and higher collagen I expression (DeFilippis et al., 2012; Ursin, Hovanessian‐Larsen, Parisky, Pike, & Wu, 2005), we use type I collagen gels, extracted from rat tail, to model the collagen‐rich ECM.
In this article, we describe three main protocols for mammary tumor invasion studies (Fig. 1): (1) generation of uniform mammary tumor cell spheroids with an encapsulating basement membrane (see Basic Protocol 1), (2) polymerization of collagen type I from rat tail and embedding of the tumor spheroid in the 3D collagen gel (see Basic Protocol 2), and (3) fixing and immunostaining of the spheroid (Basic Protocol 3). Although described for mammary cells, these protocols could be readily extended to other cancer cell types.
Figure 1.
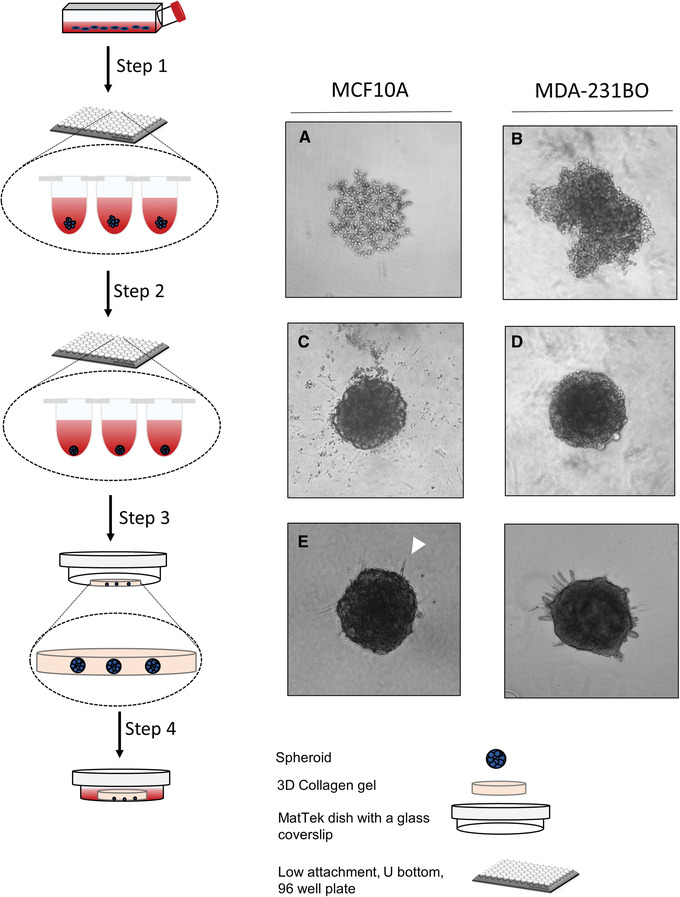
Schematic representation of procedures for generating uniform spheroids with intact basement membranes and embedding them in 3D collagen gels for invasion studies. (1) Seed 2000 cells per well in a low‐attachment 96 well U‐bottom plate. After centrifugation, aggregates form in the wells (A and B). (2) Adding 5% Matrigel solution results in a conversion of the aggregates into well‐formed spheroids in the wells (C and D). (3) After neutralizing the collagen gel, embed the spheroids in the collagen gels in MatTek dishes. Allow the gel to polymerize at 37°C for 30 min. (4) Add fresh medium to the dishes and allow cells to invade into the collagen‐rich ECM after 48 hr (E and F).
Basic Protocol 1. GROWTH OF UNIFORMLY SIZED TUMOR SPHEROIDS WITH AN ENCAPSULATING BASEMENT MEMBRANE
During DCIS, a premalignant stage in breast cancer, cells remain within the basement membrane and the myoepithelial cell layer, but invasive cells start to grow into the duct. This stage is also accompanied by deposition, linearization, and thickening of collagen I fibers, resulting in increased ECM stiffness (Kaushik et al., 2016). Progression to IDC from DCIS involves the breach of tumor cells through the basement membrane and further stiffness of the surrounding ECM. To model DCIS and the first stage in mammary cancer progression, we demonstrate a method for growing uniform spheroids with an intact basement membrane surrounding the spheroid. For the purpose of this protocol, we use two cell lines (MCF10A and MDA‐231BO) to show the method and the possible variability of the basement membrane surrounding the spheroids. MDA‐231BO cells (Yoneda, Williams, Hiraga, Niewolna, & Nishimura, 2001) were a gift from Dr. Kandice Tanner.
Materials
-
Cell lines and culture media:
Matrigel, growth factor–reduced, phenol red–free (Corning, cat. no. 356231)
200‐μl multichannel pipettor (e.g., Gilson) with reservoirs
Costar ultra‐low‐attachment U‐bottom 96‐well plates (Sigma‐Aldrich, cat. no. CLS7007)
Humidified incubator at 37°C with 10% CO2
Benchtop centrifuge for plates (e.g., Eppendorf 5810R) at room temperature
Day 1
-
1
Using a multichannel pipettor, seed 2000 cells per well in sterile ultra‐low‐attachment U‐bottom 96‐well plates. Culture for 8‐16 hr in a humidified 37°C, 10% CO2 incubator.
Calculate the number of cells needed based on a final volume of 200 μl per well. Include enough for at least ten extra wells when using a multichannel pipettor.
Day 2
-
2
Centrifuge plates for 5 min at 18 × g, 25°C to initiate aggregation of cells within each well.
-
3
Incubate at 37°C for 2 days to allow cells to grow and form more compact aggregates.
Day 3
-
4
Thaw Matrigel basement membrane matrix by leaving it overnight at 4°C in the back of a refrigerator or in a cold room.
Matrigel can begin to polymerize and form small clumps if not thawed slowly overnight. The process described above ensures that the Matrigel will thaw gradually and yield a homogeneous solution when added to the wells. Leaving pipette tips in the refrigerator overnight for handling the Matrigel also helps ensure that the Matrigel will not polymerize prematurely during pipetting.
Day 4
-
5
Remove 100 μl medium from each well using a multichannel pipettor, being careful to touch the pipet tips to the inside wall of each well before slowly aspirating.
-
6
Prepare a 10% (v/v) Matrigel solution by diluting the stock in ice‐cold supplemented DMEM/F12 medium (concentration 7.1 mg/ml).
-
7
Pipet 100 μl of 10% Matrigel into each well, making the final Matrigel concentration 5% compared to the original stock.
-
8
Centrifuge plates for 5 min at 18 × g 25°C.
It is important to centrifuge plates at 25°C. Centrifuging at 4°C during this step will result in patchy assembly of basement membrane around the spheroids.
-
9
Return to incubator and allow spheroids to grow for 2 days after addition of Matrigel.
You should see formation of very round spheroids the next day, but 2 days are needed to ensure adequate assembly of basement membrane components around the spheroids. If you let spheroids grow for only 1 day, the basement membrane will look patchy when immunostained.
Basic Protocol 2. POLYMERIZATION AND EMBEDDING OF TUMOR SPHEROIDS IN A 3D TYPE I COLLAGEN GEL
During cancer progression, invasive cancer cells invade through the basement membrane and migrate through the ECM microenvironment, which generally consists primarily of collagen type I protein. DCIS has been shown to form in more mammographically dense tissue with increased collagen density (Ursin et al., 2005). Collagen I is known to be a cellular signaling and proliferative molecule, as shown by the Keely lab (Provenzano et al., 2008), but linear collagen type I in the ECM can also act as a highway on which invasive cancer cells can crawl and migrate (Fang, Yuan, Peng, & Li, 2014; Provenzano et al., 2006). Here, we describe a method for embedding 3D tumor spheroids with an intact basement membrane in a collagen type I gel for invasion studies.
Before beginning, the detailed protocols of Dr. Andrew Doyle are used to isolate rat tail collagen I (see Current Protocols article: Doyle, 2016), label collagen gels (Doyle, 2018), and acid wash and activate MatTek dishes by silanization (see Current Protocols article: Doyle, 2009) to ensure that the collagen gels remain adherent to the bottom of the dish and that cells do not preferentially adhere directly to the dish. It is critical that these protocols be followed exactly.
Materials
Dulbecco's PBS with calcium and magnesium (DPBS; HyClone, cat. no. SH30264.02)
6.0 mg/ml rat tail collagen stock solution in 20 mM acetic acid (see Current Protocols article: Doyle, 2016)
10× DMEM (see recipe)
10× reconstitution buffer (RB; see recipe)
1 N NaOH (see recipe)
96‐well U‐bottom plate with spheroids (see Basic Protocol 1)
Hanks’ balanced salt solution (HBSS; Current Protocols, 2001)
Culture medium (see Basic Protocol 1)
1.5‐ml microcentrifuge tube
Ceramic tile (large enough for a MatTek dish)
Positive‐displacement pipettors and pipet tips
pH strips (e.g., MColorpHast, pH range 6.5‐10, Millipore‐Sigma, cat. no. 1.09543.0001)
Inverted microscope
MatTek dishes (35 mm, no. 1.5 coverslip, 20‐mm opening; cat. no. P35G‐1.5‐20‐C), acid‐washed and silanized (see Current Protocols article: Doyle, 2009)
Humidified incubator at 37°C with 10% CO2
Prepare neutralized collagen
-
1
Place a 1.5‐ml microcentrifuge tube and a ceramic tile in a rectangular ice dish to chill. Also place DPBS, collagen stock solution, and aliquots of 10× DMEM, 10× RB, 1 N NaOH, and 1 N HCl on ice.
The tile will provide a smooth surface on which the MatTek dishes will be placed on ice and kept cold. The tube will be used for neutralizing the acidic collagen solution.
-
2Calculate the volume of 6.0 mg/ml stock collagen (V collagen) needed to give 200 μl per dish of 4 mg/ml neutralized collagen. For two dishes:
- V collagen × 6.0 mg/ml = 400 μl × 4.0 mg/ml
- V collagen = 266.7 μl collagen
Transfer this amount of stock collagen to the prechilled 1.5‐ml tube using a positive‐displacement pipettor and tip.
IMPORTANT NOTE: Be sure to keep the tube of collagen on ice at all times.
-
3
Add 1/10th volume of 10× DMEM and 10× RB (40 μl each), and mix well by pipetting up and down.
-
4
Centrifuge by pulsing for 10 s to remove any bubbles.
It is okay to have bubbles when the collagen is mixed, as long as they are completely removed by centrifugation.
-
5
Add an initial 6 μl of 1 N NaOH, mix well, and test the pH by putting a small drop on a pH strip and waiting 1‐2 min for the pH to change. Adjust the pH until it is between 7.0 and 7.4 and the solution has a peach tint.
The solution will turn bright red when NaOH is first added, but should turn orange/peach as the solution is mixed. If the color goes back to yellow, add 1 N NaOH in 1‐μl increments until it is peach color. If the pH exceeds 7.4, lower it using 1 N HCl. Keep track of the volumes of NaOH and HCl added, as the volume of DPBS will need to be adjusted accordingly.
The required volume of NaOH will depend on the particular collagen preparation as well as the final desired concentration and volume of collagen.
-
6Calculate the volume of DPBS (V DPBS) needed to reach the desired collagen concentration:
- V DPBS = V final – V collagen – V DMEM – V RB – V NaOH
- V DPBS = 400 – 266.7 – 40 – 40 – V NaOH
Add this volume of ice‐cold DPBS to the neutralized collagen.
Embed spheroids in collagen gel
-
7
Remove a 96‐well U‐bottom plate with spheroids from the incubator and examine all spheroids under an inverted microscope to ensure that they have formed tight, compact spheres rather than loose aggregates. Mark wells in which the cells are not spherical using a lab marker to avoid transferring them to the collagen gel.
-
8
Gently aspirate 100 μl medium from one well, making sure to gently touch the pipette tip to the wall of the well without disturbing the spheroid. Add 100 μl HBSS to the well to wash the spheroid.
-
9
Repeat two more times to ensure that the spheroid is adequately washed.
-
10
Cut off the tip of a 200‐μl pipette tip using a sharp, sterile razor blade.
The blade can be sprayed with 70% ethanol to sterilize it.
-
11
Place an acid‐washed and silanized MatTek dish on the cold tile for ∼1 min to cool the bottom of the dish.
Cooling the dish will keep the gel from starting to polymerize right away. The dish should not be left on the cold tile for more than 3‐4 min, however, because it will start to accumulate condensation.
-
12
Gently aspirate the washed spheroid into the cut pipette tip, bring it to the very tip by expelling the medium between the spheroid and the tip, and then expel the spheroid into the neutralized collagen.
This process minimizes the volume of medium transferred to the neutralized collagen.
Multiple spheroids can be added to one MatTek dish. To keep the concentration of collagen consistent, an equal number of spheroids should be added to each dish.
-
13
Using the same cut tip, transfer 200 μl neutralized collagen with spheroid(s) to the center of an activated MatTek dish. Spread the gel with the pipet tip so that it covers the entire bottom of the dish and check under an inverted microscope to ensure that there are no air bubbles at the edges of the glass circle.
-
14
Place the dish in a 37°C incubator for 30 min to allow the collagen gel to polymerize.
-
15
When the gel is opaque (indicating that it has polymerized), slowly add 2 ml warm medium to the dish by touching the pipette tip to the wall of the dish and slowly releasing the medium.
Do not add less than 1 ml medium, as the medium can evaporate quickly in the incubator. Be careful not to disrupt the gel, because liquid can travel under the gel through small gaps, causing the collagen gel to pop off the coverslip.
-
16
Change the medium every 2 days by removing 2 ml and adding 2 ml fresh warm medium. Be very careful when aspirating medium from the dishes to avoid dislodging the collagen gel.
EMBEDDING OF TUMOR SPHEROIDS IN COLLAGEN GELS USING A SANDWICH METHOD
For an easier approach to embedding the spheroids and controlling the distance at which the spheroids are situated above the glass bottom of the dish, one can sandwich the spheroid between two layers of collagen gel. Here, the bottom layer is fully polymerized, the spheroid is placed on top of this layer, and then the top layer is added over the spheroid and polymerized. It is important to note that some cell types can anomalously migrate along the horizontal plane between the two layers of collagen, rather than migrating outward in all dimensions through the gel. Other cell types, such as fibroblasts, will migrate outward easily into the collagen gels.
See Basic Protocol 2 for all materials.
-
1
Prepare neutralized collagen (see Basic Protocol 2, steps 1‐6).
-
2
Place a MatTek dish on the cold tile for ∼1 min to chill.
-
3
Place 60 μl neutralized collagen in the center of the dish and spread it with the pipet tip so that it completely covers the glass surface.
-
4
Place the dish in the 37°C incubator for 30 min for the collagen to polymerize.
-
5
Check spheroids and wash three times with HBSS (see Basic Protocol 2, steps 7‐9).
-
6
Using a cut 200‐μl pipette tip, gently aspirate a spheroid and place it in the center of the MatTek dish, on top of the polymerized collagen (see Basic Protocol 2, steps 10 and 12).
-
7
Pipette 140 μl neutralized collagen onto the spheroid and tilt the dish to spread the collagen around the surface of the dish.
-
8
Return the dish to the incubator for 30 min for the collagen to polymerize.
-
9
Add warm medium to the dish and culture cells, changing the medium every 2 days (see Basic Protocol 2, steps 15‐16).
Basic Protocol 3. FIXING AND IMMUNOSTAINING TUMOR SPHEROIDS EMBEDDED IN 3D COLLAGEN GELS
One of the advantages of 3D spheroids embedded in a hydrogel is that the spheroid can be easily immunostained to study the individual or collective migration of its cells into its surrounding ECM. Staining and imaging of 3D spheroids make it possible to study multiple variables that could play a role in tumor invasion and metastasis.
Materials
16% paraformaldehyde in aqueous solution (Electron Microscopy Sciences, VWR cat. no. 100503‐914)
Dulbecco's PBS with calcium and magnesium (DPBS) chilled to 4°C (HyClone, cat. no. SH30264.02)
Spheroids embedded in collagen gel in MatTek dishes (Basic Protocol 2 or Alternate Protocol)
Triton X‐100 (Sigma, cat. no. T9284)
Donkey serum (Sigma‐Aldrich, cat. no. D9663)
Goat anti−collagen IV primary antibody (Millipore, cat. no. AB769)
Donkey anti‐goat 647 immunofluorescent secondary antibody (Jackson ImmunoResearch, cat. no. 705‐605‐003)
Invitrogen DAPI (ThermoFisher Scientific, cat. no. D1306)
Invitrogen Alexa Fluor 488 Phalloidin (ThermoFisher Scientific, cat. no. A12370)
Rocker
Confocal microscope
Day 1
-
1
Before you start, prepare a 4% paraformaldehyde fixation solution by diluting the 16% paraformaldehyde solution in 1× DPBS, and warm the mixture up to 37°C. Make sure to handle paraformaldehyde in a fume hood.
Prepare only the amount that you will use for each day's experiments and properly discard the rest in a chemical liquid waste container.
-
2
Remove the medium from each MatTek dish slowly by tilting the dish to one side, and, using a 1‐ml pipette tip, carefully aspirate and remove the medium completely from the dish.
Do not keep the dishes out too long at room temperature. After removal of the growth medium, immediately move to the next step. Cells can retract their protrusions if they are left out of 37°C for too long.
-
3
Immediately add the fixative solution from step 1 by slowly pipetting 2 ml down the wall of the MatTek dishes. Use a P1000 single‐channel pipettor and 1000‐µl pipet tip. Leave for 1 hr at 37°C.
-
4
Aspirate the fixative solution and discard properly.
-
5
Wash with DPBS three times for 15 min each and leave the dishes on the rocker each time.
-
6
Freshly prepare a 0.5% Triton X‐100 permeabilization solution in DPBS, add this to the MatTek dish, and leave on the dishes overnight in a 4°C cold room on the rocker.
-
7
Wash three times with DPBS. Each time gently rock the dishes using the rocker.
-
8
Prepare a blocking solution consisting of 5% donkey serum in DPBS, and add 500 μl per well of the blocking solution in each of the MatTek dishes.
-
9
Prepare a primary antibody solution based on the manufacturer's recommended concentration. Here we prepare a 1:200 solution of collagen IV antibody in DPBS (optional: add 5% donkey serum to the antibody solution). Leave on a rocker for 4 hr at room temperature or at 4°C in cold room, overnight.
-
10
Aspirate and wash away unbound primary antibody with DPBS three times, 15 min each time on the rocker, at room temperature.
-
11
Prepare a secondary antibody solution by diluting donkey anti‐goat 647 immunofluorescent secondary antibody at 1:200 dilution in DPBS. Also add DAPI (1:200 dilution) and phalloidin Alexa Fluor 488 (1:200 dilution) to this secondary antibody immunostaining solution. Leave the secondary antibody in the MatTek dishes for 4 hr at room temperature or overnight at 4°C in cold room.
-
12
Wash three times with DPBS. Each time gently rock the dishes using the rocker. After the last wash, put them in 1‐2 ml of DPBS.
You can store the dishes in 4°C for 3‐4 days, but make sure to place them in a petri dish with the lid on, and parafilm the outside to keep the gels from drying out.
-
13
Image the spheroids using a confocal microscope.
REAGENTS AND SOLUTIONS
DMEM, 10×
Dissolve 1 packet of powdered DMEM with phenol red (Sigma‐Aldrich, cat. no. D2429) in 50 ml distilled water and stir at ∼50°C until the powder goes into solution. While the solution is still warm, filter sterilize using a 0.2‐μm filter (e.g., Steriflip, Millipore). Prepare aliquots of 1.0 ml and freeze using powdered dry ice. Store indefinitely at –20°C or up to 1 month at 4°C.
NaOH, 1 N
Dissolve 0.5 g NaOH pellets in 12.5 ml distilled water. Mix well, filter sterilize, and divide into 500‐μl aliquots. Store indefinitely at −20°C.
Reconstitution buffer (RB), 10×
Measure out 2.2 g sodium bicarbonate and 4.8 g HEPES or 20 ml of 1 M HEPES stock solution for a 0.2 M final buffer concentration. Bring to 100 ml using distilled water and dissolve using a stir bar. Filter sterilize and prepare aliquots of 0.5 ml. Store indefinitely at −20°C or up to 1 month at 4°C.
Supplemented DMEM (for MDA‐231BO cells)
Dulbecco's Modified Eagle Medium (DMEM; Gibco, cat. no. 11965‐092) containing:
10% fetal bovine serum (Life Technologies)
100 U/ml penicillin (Life Technologies)
100 μg/ml streptomycin (Life Technologies)
1% l‐glutamine (Life Technologies)
Store up to expiration date on medium bottle at 4°C
Supplemented DMEM/F12 (for MCF10A cells)
DMEM/F12 medium (Life Technologies)
5% horse serum (Life Technologies)
20 ng/ml hEGF (Life Technologies)
100 U/ml penicillin (Life Technologies)
100 μg/ml streptomycin (Life Technologies)
0.5 μg/ml hydrocortisone (Sigma‐Aldrich)
10 μg/ml insulin (Sigma‐Aldrich)
Store up to expiration date on medium bottle at 4°C
COMMENTARY
Background Information
Three‐dimensional spheroid assays represent a recent approach to modeling tumors that has been shown to be superior to single‐cell monolayer studies of tumor cells. Although a 3D in vitro collagen hydrogel is not an exact replicate of original tissues such as mammary tumor tissue, it provides valuable insight into the morphological and signaling behavior of tumor cells during single‐cell and collective cell migration in a 3D microenvironment. Migration studies in 3D are also more accurate because tumor cells tend to migrate outward in three dimensions in a more physiological ECM environment, instead of single‐layer 2D migration on a stiff surface. Mouse models are valuable for modeling human disease, but they can be expensive, time consuming, and require more effort than 3D spheroids, which can be formed as uniform spheroids that can be compared directly.
Other current 3D spheroid models do not possess an intact, well‐formed basement membrane surrounding the spheroid. Most 3D models model the 3D microenvironment by mixing Matrigel and collagen I (Berens, Holy, Riegel, & Wellstein, 2015) or by using a mixture of agarose and Matrigel to from spheroids (Li et al., 2011)—or they form a thicker and more diffuse layer around the spheroids that is not organized into a discrete, thin sheet of basement membrane (Guzman, Sanchez Alemany, Nguyen, Zhang, & Kaufman, 2017). With the protocols in this article, collagen IV and laminin are observed by immunostaining to be co‐localized, with fibronectin decorating collagen I everywhere in the ECM hydrogel, surrounding the spheroid. We have used this technique to study single‐cell and multicellular migration from a spheroid into 3D collagen.
Critical Parameters
Basic Protocol 1
After the addition of Matrigel, it is important to centrifuge the plate at room temperature. If you centrifuge at 4°C, the basement membrane may result in a “patchy” collagen IV surrounding the spheroid. Please note that the roundness of the spheroid also depends on the cell type itself. Some cells do not form perfect spheres even if you follow these exact steps.
Basic Protocol 2
Do not skip the washing step with HBSS, since this will ensure that the spheroids do not have excess Matrigel around them that will subsequently create an empty zone between the collagen I ECM and the spheroid. The cells in the spheroid will not migrate into such an empty area where the collagenous ECM is not immediately adjacent to the basement membrane and cells of the spheroid. Also make sure to perform the acid wash and activate MatTek dishes using silanization to ensure that the collagen gel will remain adherent to the bottom of the MatTek dish, and that cells will not drop to the bottom of the dish and will remain in the gel.
Basic Protocol 3
Be very careful when aspirating and releasing different solutions to the 3D gels, to ensure that you do not disturb the gel. Use a P1000 micropipetter and slowly aspirate and release medium by touching the tip to the wall of the MatTek dish, away from the 3D gel, to ensure that the gel does not pop off the glass coverslip.
Understanding Results
In Basic Protocol 1, you should observe compact spheroids. The cells first form an aggregate because they are in a low‐attachment plate, which results in the cells attaching to one another. If they have cell‐cell adhesive capability, they will successfully aggregate in a lump of cells that may seem spherical, but which does not have a defined edge and is not compacted. These cells prefer to attach to one another rather than to the low‐attachment plate, and they form a spherical aggregate because that is the lowest energy state; nevertheless, they do not compact well. After the addition of Matrigel, the cells will form a more compact sphere, because they are confined within a tight basement membrane with laminin and collagen IV acting as a barrier to keep them enclosed. Leaving the spheroids for 2 days will allow the sphere to become compacted and the cells to start multiplying. If you do not see this after following the troubleshooting steps in Table 1, your cells may simply not be able to form spheroids due to a lack of sufficient cell‐to‐cell adhesion.
Table 1.
Troubleshooting Guide for Generating Uniform Spheroids with an Intact, Thin, Single‐Layer Basement Membrane, Embedding the Spheroids in Collagen Type I Gels, and Immunostaining
| Procedure | Problem | Possible cause | Solution |
|---|---|---|---|
| Uniform spheroid formation (Basic Protocol 1) | Spheroids did not form (cells are still in aggregates and there is no defined edge) | Did not centrifuge the plates after seeding and/or after adding Matrigel | Centrifuge plates the day after seeding and after adding Matrigel. Note that this is also dependent on the cell type, and some cells may form spheroids that are irregular in shape |
| Polymerizing collagen and embedding spheroid with basement membrane in collagen gels (Basic Protocol 2) | Matrigel is spread around the spheroid in an unorganized fashion | Did not wash the spheroid sufficiently with HBSS to remove excess Matrigel | Wash spheroids with HBSS and make sure you are removing more and more Matrigel/medium from the well with each wash |
| Collagen gel pops off the coverslip | There were bubbles present at the edge of the gel at the junction of the coverslip and the plastic MatTek dish | Always check under a microscope after putting the neutralized collagen solution into the MatTek dish to ensure you have spread the gel uniformly over the coverslip with no holes or bubbles present | |
| Cells die in the gel | The gel has a low pH and it was not adjusted to neutral pH | Make sure to wait 1‐2 min for the pH strip to show the correct pH and for the gel mixture to turn a peach hue | |
| Embedding the spheroids in collagen gels using the “sandwich” method (Alternate Protocol) | The cells migrate in between the two layers of gel | This can vary with different cells and their normal migration patterns in 3D. Some cells may find it easier to migrate through the two collagen layers instead of through the collagen fibers. | Follow Basic Protocol 1 to pre‐mix the collagen gel and the spheroid in a tube before adding it into the MatTek dish |
| Fixing and immunostaining the 3D spheroid model (Basic Protocol 3) | There is no staining in the middle of the spheroid | The spheroid was not properly permeabilized | If permeabilized for only 2 hr, extend the permeabilization step for another 2‐4 hr |
In Basic Protocol 2, the spheroids will be embedded in the collagen gel, and after 1 day they will start migrating out of the sphere and into the surrounding ECM, depending on the density of the collagen gel.
In Basic Protocol 3, you can immunostain spheroids, and upon imaging with confocal microscopy, you may see that the spheroids can be bumpy and irregular, as shown in Figure 2.
Figure 2.

Spheroids with encapsulating basement membrane in 3D collagen gels. (A) MCF10A spheroids confined by a basement membrane stained with collagen IV antibody and imaged using a confocal microscope. (B) MDA‐231BO spheroid stained for collagen IV is shown as a confocal slice and a max‐projection of the collagen IV surrounding the 3D spheroid. (C) High‐resolution images of MDA‐231BO spheroids with an intact basement membrane before and after initial break through the basement membrane. (D) Six days after spheroids were incubated in 3D collagen gels, cells migrated into the 3D ECM. Note that the absence of a basement membrane or collagen IV staining is because the cells have broken out and migrated away from the initial spheroid, which is depicted by dashed lines.
Time Considerations
Seeding cells for the spheroid assay takes 30 min. However, it requires approximately 5 days to generate spheroids with an encapsulating basement membrane (Basic Protocol 1). The time required for polymerizing collagen and embedding spheroids in collagen gels depends on the number of spheroids you are embedding. For about five spheroids in one MatTek dish, it will take approximately 30 min to complete, and then another 30 min to polymerize the gel in the incubator (Basic Protocol 2).
Fixing the spheroids and washing them takes about 2 hr. Permeabilizing the spheroids can be done at room temperature for 2 hr, or it can be done overnight to ensure that permeabilization occurs sufficiently deep in the spheroid. Blocking nonspecific sites in the spheroids takes 2 hr, and immunostaining the spheroids using primary and secondary antibodies requires 8 hr, or 2 days if left overnight, since diffusion of the antibodies into the interior of a chemically fixed spheroid is slow (Basic Protocol 3).
Acknowledgments
This research was fully supported by the Intramural Research Program and the NIDCR imaging core (ZIC DE000750‐01) of the NIH, NIDCR.
Nazari, S. S. (2020). Generation of 3D tumor spheroids with encapsulating basement membranes for invasion studies. Current Protocols in Cell Biology, 87, e105. doi: 10.1002/cpcb.105
Literature Cited
- Berens, E. B. , Holy, J. M. , Riegel, A. T. , & Wellstein, A. (2015). A cancer cell spheroid assay to assess invasion in a 3D setting. Journal of Visualized Experiments, 2015(105), e53409. doi: 10.3791/53409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey, S. P. , Martin, K. E. , & Reinhart‐King, C. A. (2017). Three‐dimensional collagen matrix induces a mechanosensitive invasive epithelial phenotype. Scientific Reports, 7, 42088. doi: 10.1038/srep42088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman, E. , Pankov, R. , Stevens, D. R. , & Yamada, K. M. (2001). Taking cell‐matrix adhesions to the third dimension. Science, 294(5547), 1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- Current Protocols . (2001). Laboratory stock solutions and equipment. Current Protocols in Cell Biology, 00, A.2A.1–A.2A.10. doi: 10.1002/0471143030.cba02as00. [DOI] [Google Scholar]
- Deakin, N. O. , & Turner, C. E. (2011). Distinct roles for paxillin and Hic‐5 in regulating breast cancer cell morphology, invasion, and metastasis. Molecular Biology of the Cell, 22(3), 327–341. doi: 10.1091/mbc.E10-09-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis, R. A. , Chang, H. , Dumont, N. , Rabban, J. T. , Chen, Y. Y. , Fontenay, G. V. , … Tlsty, T. D. (2012). CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discovery, 2(9), 826–839. doi: 10.1158/2159-8290.CD-12-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, A. D. (2009). Generation of micropatterned substrates using micro photopatterning. Current Protocols in Cell Biology, 45, 10.15.1–10.15.35. doi: 10.1002/0471143030.cb1015s45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, A. D. (2016). Generation of 3D collagen gels with controlled diverse architectures. Current Protocols in Cell Biology, 72, 10.20.11–10.20.16. doi: 10.1002/cpcb.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, A. D. (2018). Fluorescent labeling of rat‐tail collagen for 3D fluorescence imaging. Bio Protocol, 8(13), e2919. doi: 10.21769/BioProtoc.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, M. , Yuan, J. , Peng, C. , & Li, Y. (2014). Collagen as a double‐edged sword in tumor progression. Tumour Biology, 35(4), 2871–2882. doi: 10.1007/s13277-013-1511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl, P. , Noble, P. B. , Walton, P. A. , Laird, D. W. , Chauvin, P. J. , Tabah, R. J. , … Zanker, K. S. (1995). Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in‐vitro. Cancer Research, 55(20), 4557–4560. [PubMed] [Google Scholar]
- Guzman, A. , Sanchez Alemany, V. , Nguyen, Y. , Zhang, C. R. , & Kaufman, L. J. (2017). A novel 3D in vitro metastasis model elucidates differential invasive strategies during and after breaching basement membrane. Biomaterials, 115, 19–29. doi: 10.1016/j.biomaterials.2016.11.014. [DOI] [PubMed] [Google Scholar]
- Kaushik, S. , Pickup, M. W. , & Weaver, V. M. (2016). From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer and Metastasis Reviews, 35(4), 655–667. doi: 10.1007/s10555-016-9650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. Y. , Chang, J. K. , Dominguez, A. A. , Lee, H. P. , Nam, S. , Chang, J. , … Chaudhuri, O. (2019). YAP‐independent mechanotransduction drives breast cancer progression. Nature Communications, 10(1), 1848. doi: 10.1038/s41467-019-09755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Chen, C. , Kapadia, A. , Zhou, Q. , Harper, M. K. , Schaack, J. , & LaBarbera, D. V. (2011). 3D models of epithelial‐mesenchymal transition in breast cancer metastasis: High‐throughput screening assay development, validation, and pilot screen. Journal of Biomolecular Screening, 16(2), 141–154. doi: 10.1177/1087057110392995. [DOI] [PubMed] [Google Scholar]
- Provenzano, P. P. , Eliceiri, K. W. , Campbell, J. M. , Inman, D. R. , White, J. G. , & Keely, P. J. (2006). Collagen reorganization at the tumor‐stromal interface facilitates local invasion. BMC Medicine, 4(1), 38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano, P. P. , Inman, D. R. , Eliceiri, K. W. , Knittel, J. G. , Yan, L. , Rueden, C. T. , … Keely, P. J. (2008). Collagen density promotes mammary tumor initiation and progression. BMC Medicine, 6, 11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley, A. J. , Schwartz, M. A. , Burridge, K. , Firtel, R. A. , Ginsberg, M. H. , Borisy, G. , … Horwitz, A. R. (2003). Cell migration: Integrating signals from front to back. Science, 302(5651), 1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Tevis, K. M. , Colson, Y. L. , & Grinstaff, M. W. (2017). Embedded spheroids as models of the cancer microenvironment. Advanced Biosystems, 1(10), 1700083. doi: 10.1002/adbi.201700083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursin, G. , Hovanessian‐Larsen, L. , Parisky, Y. R. , Pike, M. C. , & Wu, A. H. (2005). Greatly increased occurrence of breast cancers in areas of mammographically dense tissue. Breast Cancer Research, 7(5), R605–R608. doi: 10.1186/bcr1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, K. , Wu, Y. I. , Liu, Y. , Geiger, J. , Tam, E. , Overall, C. , … Friedl, P. (2007). Multi‐step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nature Cell Biology, 9(8), 893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- Yamada, K. M. , & Sixt, M. (2019). Mechanisms of 3D cell migration. Nature Reviews Molecular Cell Biology, 20(12), 738–752. doi: 10.1038/s41580-019-0172-9. [DOI] [PubMed] [Google Scholar]
- Yoneda, T. , Williams, P. J. , Hiraga, T. , Niewolna, M. , & Nishimura, R. (2001). A bone‐seeking clone exhibits different biological properties from the MDA‐MB‐231 parental human breast cancer cells and a brain‐seeking clone in vivo and in vitro. Journal of Bone and Mineral Research, 16(8), 1486–1495. doi: 10.1359/jbmr.2001.16.8.1486. [DOI] [PubMed] [Google Scholar]