

Abstract
Background:
The recent development of metagenomic sequencing makes it possible to massively sequence microbial genomes including viral genomes without the need for laboratory culture. Existing reference-based and gene homology-based methods are not efficient in identifying unknown viruses or short viral sequences from metagenomic data.
Methods:
Here we developed a reference-free and alignment-free machine learning method, DeepVirFinder, for identifying viral sequences in metagenomic data using deep learning.
Results:
Trained based on sequences from viral RefSeq discovered before May 2015, and evaluated on those discovered after that date, DeepVirFinder outperformed the state-of-the-art method VirFinder at all contig lengths, achieving AUROC 0.93, 0.95, 0.97, and 0.98 for 300, 500, 1000, and 3000 bp sequences respectively. Enlarging the training data with additional millions of purified viral sequences from metavirome samples further improved the accuracy for identifying virus groups that are under-represented. Applying DeepVirFinder to real human gut metagenomic samples, we identified 51,138 viral sequences belonging to 175 bins in patients with colorectal carcinoma (CRC). Ten bins were found associated with the cancer status, suggesting viruses may play important roles in CRC.
Conclusions:
Powered by deep learning and high throughput sequencing metagenomic data, DeepVirFinder significantly improved the accuracy of viral identification and will assist the study of viruses in the era of metagenomics.
Keywords: metagenome, deep learning, virus identification, machine learning
Author summary:
We developed a reference-free and alignment-free machine learning method, DeepVirFinder, for identifying viral sequences in metagenomics using deep learning. Sequences from viral and prokaryotic genomes are used for training the model. The neural network is composed by a convolutional layer, a max pooling layer, two dense layers to generate the prediction score between 0 and 1. DeepVirFinder outperformed the state-of-the-art method VirFinder at all contig lengths, achieving AUROC 0.93, 0.95, and 0.97 for 300, 500, and 1000 bp sequences respectively, and it will greatly assist the study of viruses in the era of metagenomics.
INTRODUCTION
Viruses infecting microbes have great impact on both human health and ecosystems, but studies of the effect of those types of viruses on their host communities as well as on public health are in their earliest stages. It has only recently become possible to identify viruses in large metagenomic datasets using next-generation sequencing (NGS) technologies coupled with computational advances. Unlike traditional methods of isolating viruses through laboratory cultures, metagenomic sequencing technology effectively sequences all types of genetic materials in a microbial community regardless of their cultivability, making it possible to exhibit the true viral diversity of a sample. Studies using metagenomic sequencing to study viruses in the human gut have revealed important associations between viruses and human diseases, such as inflammatory bowel disease (IBD) [1], severe acute malnutrition (SAM) [2], and type II diabetes [3].
Identifying viral sequences from metagenomic samples is the first crucial step for all downstream analyses for viruses. A few methods have been developed to tackle the virus identification problem in metagenomic samples [4–6]. Besides those methods, tools for metaviromic or metagenomic composition analysis that characterizes the taxonomy of sequences in a sample [7–13] can also be used for identifying viral sequences, although they are not designed for this purpose. Before the metagenomic era, several tools for identifying proviruses from within prokaryotic genomes were developed [14–17]. While identifying proviruses in prokaryotic genomes is a different problem than identifying viral sequences in metagenomics, provirus finding tools laid the groundwork for virus identification approaches in metagenomic era. In general, all the above methods characterize viruses using features from the following three major aspects:(1) sequence alignment-based metrics for comparing query sequences with virus refernece genomes, (2) gene homology-based metrics for comparing genes in query sequences and viral gene database, and (3) alignmnet-free k-mer based metrics that use genomic signatures for virus prediction.
For sequence alignment-based methods, the tools, Metavir [7] and ViromeScan [8], classify viral metagenomics reads based on sequence alignment and homology searches against known viral reference genomes. Kraken[10], Centrifuge [11], and MetaPhlAn [12], which were designed mainly for bacteria composition analysis though, can rapidly map reads to known viral reference genomes. However, the current virus genome database are markedly biased towards certain types whose hosts are cultivable in the lab. The current virus references are far from being a complete representation of the whole viral diversity. It is estimated that only about 15% of viruses in human gut have similarity to known viruses in the database [18].
Gene homology-based methods, VIROME [9], DIAMOND [13], VirSorter [4], JGI Earth Virome Pipeline[19], MARVEL [6], and most provirus finding tools, Prophinder [15], Phage-Finder [14], PhiSpy [16], and PHASTER [17], determine viral sequences mainly by comparing genes in the query sequence against viral gene databases, and see if there is enough evidence showing the query sequences carries viral proteins. Since some mutations in nucleotide sequences are not appearing in the amino acide sequences, comparing sequences at the amino acid level instead of at the nucleotide level improves prediction stability given the high mutation rate in viral genomes. In addition, VIROME, VirSorter, and JGI Earth Virome Pipeline enlarged the viral gene database by adding viral genes from metavirome datasets to detect unknown viruses. Metavirome is viruslike particle (VLP)-derived metagenomes where cellular organisms like bacteria are filtered out physcially before sequencing and mostly viruses are sequenced. The gene-based methods normally require query sequences to contain complete genes, so they are not able to predict sequences from non-coding regions. VirSorter in particular requires query sequences to have at least three genes to make predictions. Since metagenomics reads and the majority of metagenomically assembled sequences (also called contigs) are only hundred base pairs long, the method however fails to recall the majority of metagenomic viral sequences.
Recently we developed a program, VirFinder, to identify viral sequences using a machine learning method based on k-mer frequency features [5]. VirFinder automatically learned k-mer patterns that are enriched or depleted in viruses, and built a powerful classifier to predict viral sequences. Since VirFinder characterizes sequences using k-mer frequencies, it can predict contigs from both coding and non-coding regions. Moreover, VirFinder does not depend on gene finding and homology-based searches so that it can predict short viral contigs that contain few or even only partial genes. VirFinder recalls 78-, 2.4-, 1.8-, and 1.2-times more viral contigs of 1, 3, 5, and 10 kb, respectively, than the gene-based method VirSorter, at the same false positive rates. The success of VirFinder demonstrates that the k-mer based machine learning method is more powerful than traditional gene-based methods for short contigs (≤10k).
Deep learning is one type of advanced machine learning algorithms that uses deep artificial neural networks to learn features from the input and predict the output. Deep learning techniques have been successfully used to solve various problems in computational biology, such as predicting protein binding specificity using deep learning [20–23], predicting the effect of non-coding variants [24,25], predicting chromatin accessibility [26], calling genetic variants from millions of short reads in NGS samples [27], predicting methylation quantitative trait loci [28], identifying evolutionary conserved sequences [29], and identifying enhancers and promoters[30] and their interactions [31]. Yue et al. [32] gave a detailed review of deep learning applications in genomics. Significant improvements have been achieved for the above problems by deep learning methods over traditional methods, especially when training was driven by sufficient data. Given the importance of virus recognition problem and the urgency of utilizing the existing massive amount of metagenomic data, it is crucial to develop a powerful deep learning method for accurately identifying viruses in metagenomics.
In this study we developed a novel method, DeepVirFinder, to identify viral sequences from metagenomic data using deep learning techniques. DeepVirFinder designed convolutional neural networks (ConvNets) to automatically learn viral genomic signatures, and simultaneously built a predictive model based on those genomic signatures to predict if a sequence is from a viral genome. Trained with a large number of sequences, DeepVirFinder outperformed the previous state-of-the-art method VirFinder at all contig lengths. DeepVirFinder achieved AUROCs of 0.95, 0.97, and 0.98 for 500, 1000 and 3000 bp viral sequences respectively. For short sequences, decent prediction accuracy was obtained (AUROC 0.93 for 300 bp), suggesting DeepVirFinder can be directly applied to raw sequencing reads. To further elevate the prediction accuracy for sequences from under-represented viral groups, we enlarged the training data tremendously by adding millions of unknown viral sequences in metavirome datasets. As a case study, we applied DeepVirFinder to identify viruses in gut microbial communities for patients with CRC. Ten virus bins associated with the cancer status were discovered, suggesting potential roles that viruses play in human disease.
RESULTS
DeepVirFinder: viral sequences prediction using convolutional neural networks
We developed a powerful deep learning model for predicting viral sequences using convolutinoal neural networks. Our model takes DNA sequences as input, without using any pre-defined features such as k-mers as before [5], and simultaneously learns features that are useful for virus prediction. The model consists of a convolutional layer, a max pooling layer, a fully connected layer, and several dropout layers, and outputs a prediction score between 0 to 1 for a binary classification between virus and prokaryote (Fig. 1). See Section of “Predicting viral sequences using convolutional neural networks” for the details of the model. Considering DNA sequences are double stranded, and the prediction should be identical for either the forward strand or the backward strand, we apply the same network to both the original sequence and its reverse complement, and define the final prediction score as the average of the predictions from both sequences.
Figure 1. The deep learning framework of DeepVirFinder.
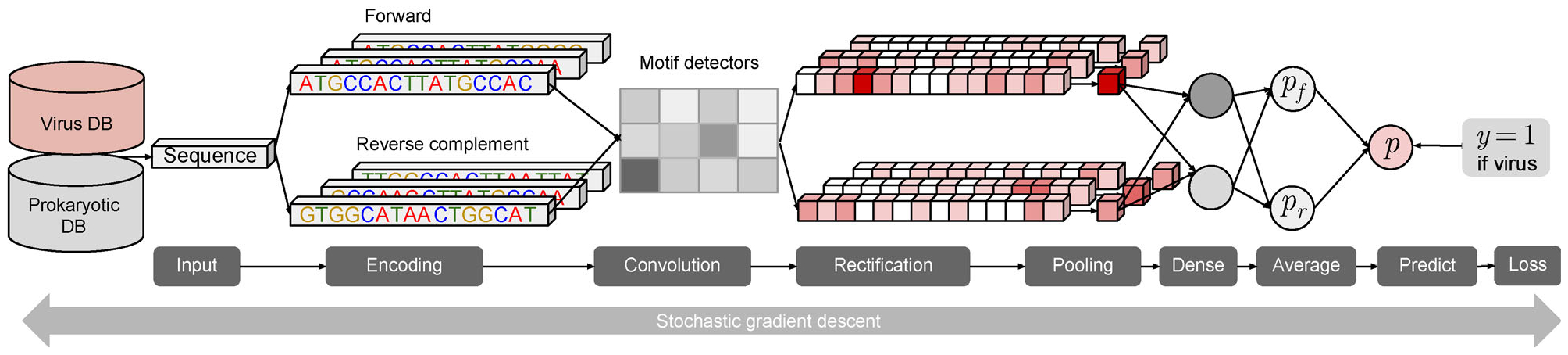
Sequences from viral genomes and prokaryotic genomes are used for training the model. The neural network is composed by a convolutional layer, a max pooling layer, a dense layer with ReLU activation function, and a final dense layer with sigmoid function to generate the prediction score between 0 and 1. The higher score indicates the more likely a sequence is from viral genomes. For each sequence, both forward and its reverse complementary are fed into the same neural networks, and the final prediction score is the average of the two corresponding prediction scores.
The model was trained and evaluated using a curated large dataset containing hundred thousands viral sequences and prokaryotic sequences. We downloaded 2,314 reference genomes (also known as RefSeq) of viruses infecting prokaryotes (bacteria and archaea) from National Center for Biotechnology Information (NCBI). The dataset was partitioned into three parts based on the dates when the genomes were discovered. We used the genomes discovered before January 2014 for training, those between January 2014 and May 2015 for validation, and those after May 2015 for test. The partitioning of the dataset not only avoids the overlaps between the training, validation, and test datasets, but also helps to evaluate the methods ability for predicting future new viruses based on the previously discovered viruses. The viral genomes are fragmented into nonoverlapping short sequences to mimic the real metagenomic contigs. The virus dataset is paired with the same number of prokaryotic sequences, fragmented from 38,234 RefSeq and partitioned by the exact same dates. See Section of “Viruses and prokaryotic genomes used for training, validation and testing” for details. To further enlarge the dataset for training, we collected a large number of metavirome samples which contain mostly viral sequences including many uncultivated viruses. After carefully filtering out of the possible contamination of prokaryotic DNA in the samples, we added up to 1.3 million sequences to training. See Section of “Collection of metavirome datasets” for details.
The model was trained using training dataset, and optimal hyperparameters were selected based on the performance of the model on validation dataset. The area under the receiver operating characteristic curve (AUROC) was used as the metric for performance evaluation. AUROC provides a quantitative measure for predictive performance, with higher values indicating higher prediction power. Once the optimal hyperparameters were detemined, the final model was trained using all sequences in both training and validation dataset, and evaluated on the final test dataset which was never touched during training. Our model was compared with the other method using the same trainig, validation, and test datasets, to ensure a fair comparison.
Determining the optimal model for DeepVirFinder
We implemented a series of training and validation experiments to search for the optimal paramter setting for the model of DeepVirFinder. In convolutional neural networks, two critical hyperparameters, the length of motifs (or filters) and the number of motifs, determine the complexity of the model. To find the best parameter settings, we trained the model with different combinations of the two parameters using the training data, and evaluated the model performance using AUROC on the validation dataset. We studied the motif length ranging from 4 to 18 and the number of motifs from 100, 500, 1000, and 1500. We observed that as motif length increased from 4 to 8, the validation AUROC increased rapidly. The highest AUROC achieved when the motif length was around 10, and the value kept in the same level as the motif length further increased (Supplementary Fig. S1A, red curves). For example, for the model trained with 500 bp sequences, when fixing the model having 1000 motifs, the validation AUROC increased from 0.7747 to0.9464 as motif length increased from 4 to 8, and achieved the highest value of 0.9496 when motif length is10. This trend was similar for all other sequence sizes and numbers of motifs. Thus, we set the motif length as 10 in the final model. Note that the optimal k-mer length is 8 in VirFinder, a similar value as the motif length chosen here.
We next studied the effect of the number of motifs on the model performance by fixing the motif length as 10 and increasing the number of motifs from 100 to 1,500. The validation AUROC gradually increased with the number of motifs (Supplementary Fig. S1B). For example, for the 500 bp model, the validation AUROC was 0.8990, 0.9402, 0.9497, and 0.9500 for models using 100, 500, 1000, and 1,500 motifs, respectively. The number of neurons in the dense layer had a similar effect on the model performance. Considering the model simplicity and the computational intensity, we chose to use 1,000 motifs in the convolutional layer and 1,000 neurons in the dense layer in the final model.
The model was trained based on stochastic gradient descent and back-propagation. Training for more epochs produced a higher training accuracy but it could also cause overfitting. We observed the validation AUROC increased quickly in the first 20 epochs and was stabilized after 30 epochs (Supplementary Fig. S1C). Thus we train the final model for 30 epochs.
Comparing models across different sequence lengths, we observed that longer sequences had higher prediction accuracies. For example, the models with 1000 motifs of 10 bp and trained using 30 epochs had the validation AUROCs of 0.8635, 0.9210, 0.9496, 0.9668, and 0.9784 for 150, 300, 500, 1000, and 3000 bp sequences, respectively. Longer sequences contain more information and thus are easier to make predictions. The low AUROC for the model of 150 bp sequences was due to the inherent difficulty of predicting very short sequences.
Once the parameters were determined, we trained the model using all sequences before May 2015 (training plus validation datasets). We evaluated model performance on sequences after May 2015, independent from all the training, validaton and parameter tuning process, to obtain an unbiased evaluation of model performance.
DeepVirFinder outperforms VirFinder at all sequence lengths
We compared the newly developed model DeepVirFinder with the previous state-of-the-art method VirFinder [5]. To make a fair comparison, both methods were trained using the sequences before May 2015 and assessed on data after May 2015. DeepVirFinder outperformed VirFinder at all sequence lengths, where the ROC curves for DeepVirFinder were always above those for VirFinder (Fig. 2A and Supplementary Table S1). The improvement in AUROC was more remarkable for short sequences of length < 1000 bp. For example, DeepVirFinder had AUROC of 0.8766, 0.9272, and 0.9494 for 150, 300, and 500 bp sequences, respectively, while the corresponding scores for VirFinder were 0.8101, 0.8771, and 0.9163, reflecting 8.2%, 5.7%, and 3.6% increase, respectively. For 1000 bp sequences, DeepVirFinder improved the AUROC from 0.9471 to 0.9735 (2.8% increase), and for sequence of size 3000 bp, the increase from 0.977 to 0.9847 was minimal but still significant (p-value for one-sided t-test, 5.896e–16). DeepVirFinder can predict sequences as short as 300 bp with a decent accuracy (AUROC 0.9272). With this increased prediction power, DeepVirFinder can be used to predict viral sequences directly at the read level in metagenomic samples without assembly.
Figure 2. Comparison of DeepVirFinder with VirFinder and the effect of contig length and mutation rates on the performance of DeepVirFinder.
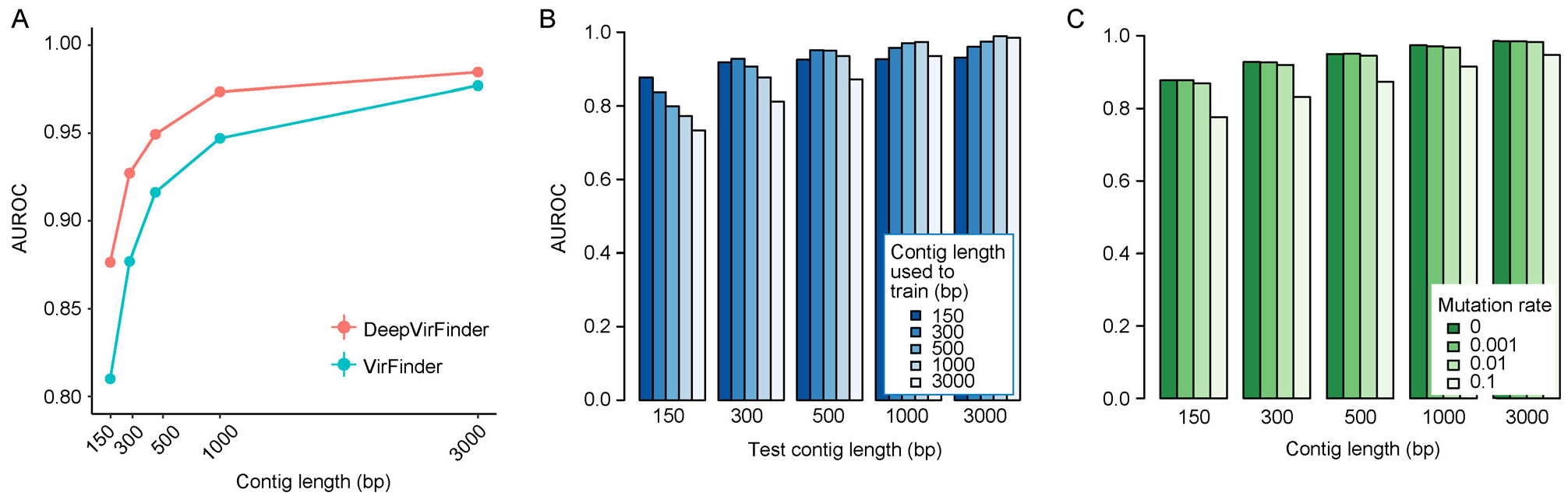
(A) AUROCs for VirFinder and DeepVirFinder when trained on sequences before May 2015, and tested on sequences after May 2015. See Supplementary Fig. S1 for the exact numbers and the standard errors. (B) AUROCs for different combinaions of sequence lengths used for training and testing. (C) AUROCs for prediction when adding mutations at different rates.
Predicting sequences of various lengths
We trained the deep learning models using fixed length sequences of 150 bp, 300 bp, 500 bp, 1000 bp and 3000 bp independently, while real metagenomic samples may contain sequences of varible lengths, especially for samples whose reads are assembled into contigs. Given a query sequence, a natural question is which model we should use for prediction. We evaluated the performance of different trained models for predicting sequences of variable lengths. In particular, we predicted 150 bp sequences after May 2015 using models trained by 150 bp, 300 bp, 500 bp, 1000 bp, and 3000 bp sequences before May 2015, respectively. The highest AUROC was achieved when using the model trained by 150 bp sequences (Fig. 2B). Similarly, using the model trained by 300 bp sequences had the best performance for predicting 300 bp sequences, and the same conclusion holds for 500 and 1000 bp sequences. For sequences of length >1000, there was no obvious difference between models. Therefore, we decided to use the model trained by 150 bp sequences for predicting any sequences < 300 bp. Similarily, we used the model trained by 300 bp sequences for predicting sequences of the length 300–500 bp, the model trained by 500 bp sequences for predicting 500–1000 bp sequences, and the 1000 bp model to predict sequences >1000 bp.
Model robustness to mutations
Considering viruses have a higher mutation rate than bacteria, we tested the model’s robustness to genetic mutations. This also served as a test of the sensitivity of the models to sequencing errors. For the sequences in the test set, we randomly introduced mutations by replacing the original letter at each position by another different letter with equal probability at the rate of 0.001, 0.01, and 0.1. We compared the AUROC for predicting the mutated sequences with that for the original sequences with no mutations. We observed that the AUROC scores dropped less than 0.06% at 0.001 mutation rate, 0.66% at 0.01 mutation rate, and 7.96% at 0.1 mutation rate (Fig. 2C). Thus, our models are not sensitive to either the typical viral mutation rate of ≤0.001 as suggested in the previous study [33] or the sequencing error rate of 0.001 by Illumina Platform [34].
Enlarging the training dataset by adding millions of metavirome contigs
Though a large number of training sequences were obtained from viral RefSeq, the existing RefSeq database represents mostly cultivated viruses. To represent a more diverse viral population and to couple the deep learning algorithm with even larger dataset, viral sequences from metavirome datasets were added to the training. We collected a large set of metavirome samples from several large-scaled metagenomic sequencing projects, and we carefully selected the samples that had high quality to reduce the possibility of contamination of prokaryotic DNA. Reads from those samples were assembled and the resulting millions of viral contigs were used to generate more viral sequences for training. See Section of “Collection of metavirome datasets” for details. The model trained using the enlarged dataset was evaluated using the test sequences from RefSeq after May 2015, and compared with the original model trained based only on RefSeq.
We investigated the AUROC for predicting viruses from different host phyla. The new model trained using the enlarged dataset had significantly higher AUROC scores for the viral groups that are under-represented in the RefSeq database, compared with the original model with all p-values for one-sided t-test < 2.2e–16 (Fig. 3A). For example, only 2.08% viruses in RefSeq infect Bacteroidetes, and because of the low representation, the original model trained using only RefSeq had a relatively low AUROC of 0.8287 for predicting viruses infecting Bacteroidetes, even though Bacteroidetes is of the two dominant phyla in human gut. After adding the metavirome contigs, the AUROC was improved to0.9591. Similarly, the new model improved the AUROC from 0.8272 to 0.8952 for viruses infecting Crenarchaeota, and from 0.9714 to 0.9847 for viruses infecting Cyanobacteria, which only represent 2.23% and 4.39% of the viruses in RefSeq database, respectively. The above results were for 500 bp contigs, and the conclusion holds for other sequence lengths (data not shown). The results also confirmed the distribution of viruses in RefSeq database is different from that in the real environment, possibly due to the fact that most viruses in RefSeq were obtained by the procedure of cultivation and isolation in the lab so that the viruses in RefSeq database are greatly biased towards the limited cultivable viruses against those uncultivable majority. Thus, adding viral sequences from metavirome dataset effectively corrected the sampling bias, and improved the prediction accuracy for the viruses under-represented in the RefSeq.
Figure 3. Comparison of AUROCs between the model trained using only viral RefSeq, and the model trained using the enlarged dataset including millons of sequences from metavirome.
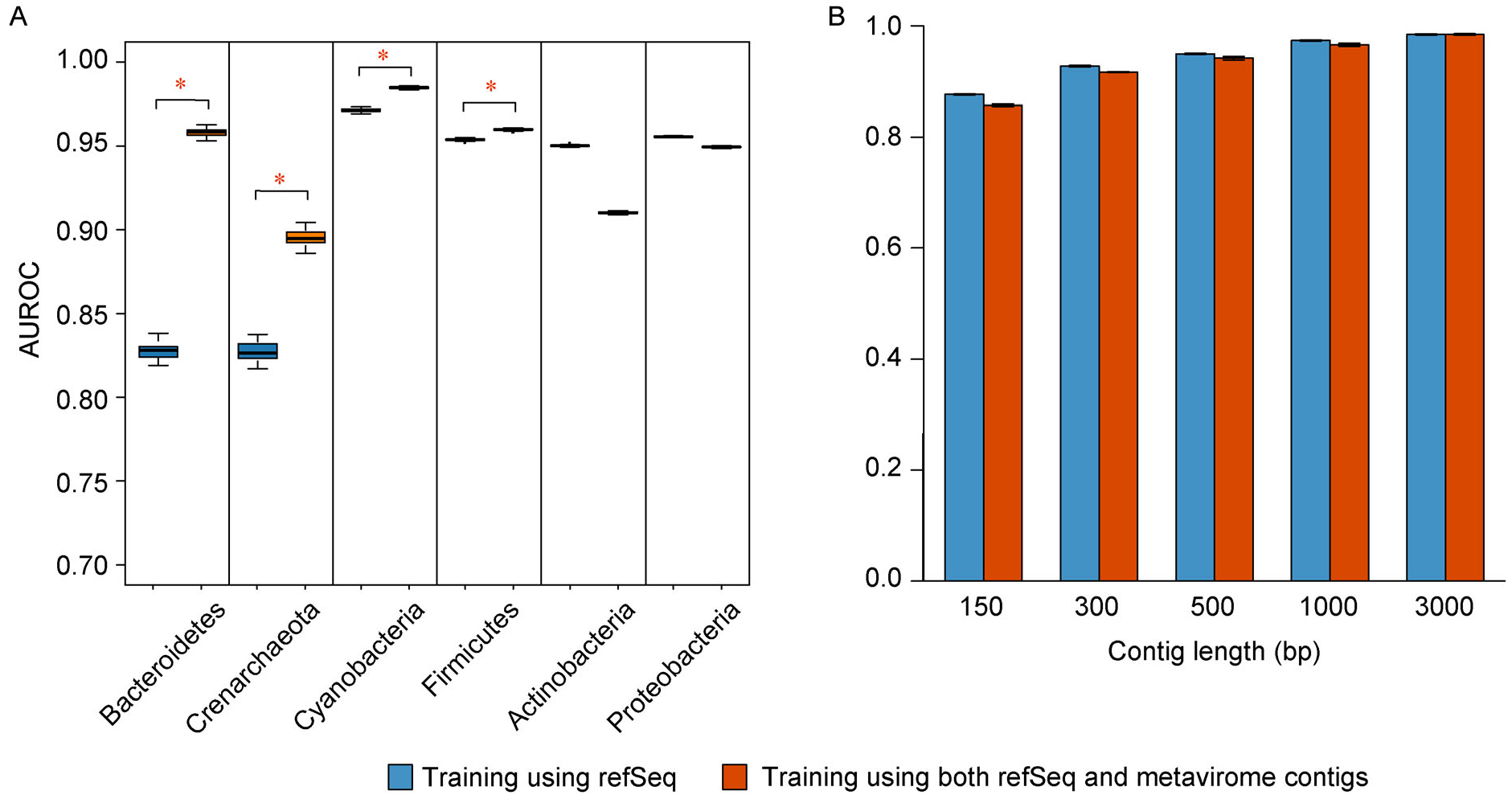
(A) The AUROCs for predicting 500 bp viral sequences from different host phyla. The under-represented viruses groups, viruses infecting Crenarcheota, Bacteroidetes (B) the overall AUROCs between the two models at different sequence lengths.
We also noticed that the viruses infecting Proteobacteria and Actinobacteria, the two most abundant virus types taking up to 63% in RefSeq, had decreased AUROCs when predicting using the model trained with the enlarged dataset. Due to the decrease in AUROC for the two major viral groups, the overall AUROC was also slightly decreased for the new model (Fig. 3B). Note that we tested the model based on sequences from RefSeq after May 2015. This may disfavor the evaluation of the model trained using the enlarged dataset, because the model was trained using RefSeq plus metavirome dataset while the distribution of the test data was shifted towards only RefSeq. Though metavirome samples represent viruses closer to the real virus distribution, considering that the test data needs to be clean and those metavirome-derived viral sequences may contain unavoidable contamination, we did not use metavirome-derived viral sequences to test the model. We expect the model trained with the enlarged dataset will have better performance if the testing data was from the true viral distribution. Overall, we suggest to use the model trained with the enlarged data to predict viruses from under-represented groups, and use the original model if the viruses are mainly from the common groups in RefSeq.
Evaluation of DeepVirFinder on simulated metagenomic contigs of variable lengths
To test the performance of DeepVirFinder on predicting viral contigs in metagenomics data, we simulated several metagenomics samples based on the abundance profile of a real human gut metagenomic sample, and evaluated performance of DeepVirFinder on identifying viral contigs in the simulated metagenomic samples. Considering the viral fraction differs based on the experimental exampling strategy, we simulted three metagenomic samples with the viral fractions of 10%, 50% and 90%, while keeping the relative abundance within virus and host groups the same. The simulated contig was of variable length between hundreds of base pairs to thousands of base pairs, with the majority ranging between 300–000 bp. We used the model trained with RefSeq to predict contigs of different lengths.
In general, AUROC scores increased as the contig length increases, having the same trend as in Fig. 2. For example, the AUROC scores for contigs of length < 300 bp, 300–500 bp, 500–1000 bp, and >1000 bp were about 0.8317, 0.8767, 0.8966, and 0.9451 on average. When predicting contigs of length across multiple intervals, AUROCs were 0.8829 for all contigs, 0.8952 for contigs >300 bp, and 0.9129 for contigs >500 bp (Fig. 4B). Thus, in the real data application, we are able to predict contigs >300 bp in order to achieve the overall AUROC around 0.90.
Figure 4. Evaluation of the performance of DeepVirFinder on viral contigs of variable lengths in simulated metagenomic samples with various viral fractions.
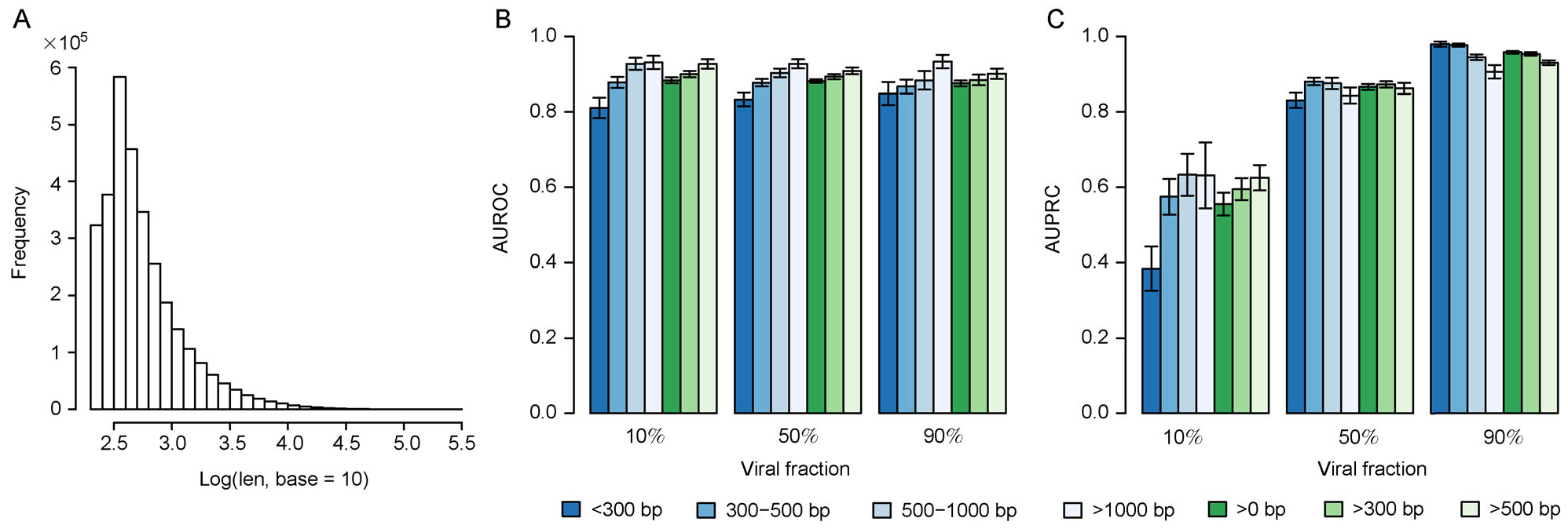
(A) The distribution of contig length used for simulating metagenomic samples, and the (B) AUROC and (C) AUPRC for predicting viral sequences with various viral fractions (10%, 50% and 90%) for contigs of different lengths.
Different viral fraction does not markedly affect AUROC, since the true positive rate and the false positive rate are defined based on the relative proportions within the viral group and the host group independently. As a complementary method to AUROC, we considered the metric of the area under the precision-recall curves (AUPRC) which is more sensitive when assessing the effect of viral fraction on the prediction accuracy. For example, the AUPRC for contigs of length >500 bp is0.9296 for the sample with 90% viral fraction, and are0.8638 and 0.6437 for samples with 50% and 10% viral fractions, respectively (Fig. 4C). We also observed that AUPRC had large variations for the sample with 10% viral fraction, compared to that for the sample with 50% and 90% viral fractions. This may be caused by the fact of a small number of viruses in the 10% viral fraction sample.
Case study: identifying viruses in the gut microbiome associated with CRC
CRC is among the top second most frequently diagnosed cancer in women and the third in men as of 2012 [35,36]. Several studies have shown the effect of human gut bacteria on CRC [37–40], but the association between gut viruses and CRC has not been investigated. As viruses play important roles in controlling host population and altering host metabolism, it is of significance to investigate the gut viruses in patients with CRC. To showcase the use of DeepVirFinder in real applications, we used DeepVirFinder to identify viruses in the gut microbiome, and assessed the association between virus and the disease status.
We collected 114 metagenomic samples in a previous study [40]. After spliting the dataset into training and test, the metagenomic samples in the training set were cross-assembled, resulting in 1,335,046 contigs of length greater than 500 bp. DeepVirFinder identified 51,138 viral contigs and the false discovery rate was controlled at the rate 0.01. Those contigs were then grouped into 175 contig bins based on their k-mer similarity and abundance correlation using the software COCACOLA [41]. See Section of “Viral analysis of human gut metagenomics from patients with colorectal cancer” for details of the analysis. Using the average reads per kilobase per million mapped reads (RPKM) as the feature for each bin and the disease status as the response variable, we built a logistic Lasso regression classifier to predict cancer status based on the viral abundance. The AUROC score was 0.7557, and 10 viral bins selected by the Lasso classifier were associated with the CRC status (Supplementary Table S2). Six of the bins had positive coefficients (high abundance in CRC) and four bins had negative coefficients (low abundance in CRC).
Comparing the sequences in the bins to RefSeq database, six bins had similar sequences to known viruses. Most of the viruses infect bacteria (called phages), while we also noticed one endogenous virus infecting human cells. Eukaryotic viruses can potentially share similar motifs with prokaryotic viruses, since we previously observed a decent predictive accuracy of VirFinder for predicting eukaryotic viruses (data not shown). In addition, Bin7, with a negative coefficient––0.0193 in the regression model, contains sequences similar to crAssphage. This is consistent with the fact that crAssphage is a highly abundant virus in healthy guts[42]. The remaining four bins with no similarity to RefSeq viruses are possibly new viruses. We also searched the proteins in contigs against Pfam database [43]. On average 65.68% of contigs contained proteins, demonstrating DeepVirFinder’s ability of identifying virus sequences from non-coding regions over the other gene homology-based method. Seven of the 10 bins contained phage associated proteins, such as tail, capsid, integrase, connector, holin, and portal, indicating those bins were trully from viruses. In addition, all bins except Bin188 had proteins with domains of unknown function (DUF). This also indicates those bins were most likely to be viruses because viral proteins are less characterized than bacteria in Pfam database [4], and this was in fact a criterion used for viral prediction in Roux et al. [4].
DISCUSSION AND CONCLUSIONS
Prokaryotic viruses are the most abundant biological entities on earth. They play crucial roles in regulating microbial communities, but our knowledge of prokaryotic viruses has been limited by available experiment techniques and the computational methods for a long time. Identification of viral sequences is not trivial due to viruses’ high mutation rates and incomplete reference database, and the existing methods could not achieve high recall rates for short viral sequences.
We developed a novel method, DeepVirFinder, to identify viral sequences in metagenomic data. To the best of our knowledge, it is the first deep learning based program for identifying viral sequences from metagenomic data. Powered by the deep learning techniques, and trained with a large number of viral sequences, DeepVirFinder outperformed VirFinder at all contig lengths. Enlarging the training data with millions of additional viral sequences from environmental samples futher improved the prediction accuracy for under-represented viral groups. The improvements are remarkable for healthcare researchers, since the false predictions may cause serious consequence. We demonstrated the usefulness of the method by a case study where we applied DeepVirFinder to the real human gut metagenomic samples and discovered a group of key viruses associated with CRC, which can motivate futher investigation of viruses’ roles in human disease.
As more RefSeq and environmental metavirome samples for training are generated, we expect DeepVirFinder will keep increasing its power to identify viruses in metagenomic data. Though the usage of metavirome dataset for training requires careful quality control for bacterial sequences contaminations, fully utilizing metavirome datasets for training will help to further improve the prediction accuracy especially for under-represented and unknown viruses.
With the ability of accurately predicting viral sequences as short as read length, our tool can potentially be used to improve assembly pipelines for viral genomes. Individual reads could be classified as virus and then do assembly on those reads to help simplify the complexity in assembly and reduce computing resources, and hopefully improve assembly accuracy. Alternatively, viral contigs can be predicted from millions of assembled contigs, and the reads mapping to those viral contigs can be pulled out and re-assemble. Evaluation and comparison of the two approaches for assembling viral genomes is of our interest in the future.
Deep learning models is designed for end-to-end solutions with no need for feature engineering. It is believed that features can be automatically learned through large number of training examples. Because of the our limited understanding of viral genomes, deep learning model can fit better and achieve higher accuracy than other pre-defined feature-based machine learning models, such as k-mer frequencies logistic regression [5], and gene-based random forest model [6,44]. On the other hand, it will be interesting to compare the motifs learned from the deep learning model with the real biological motifs, though it is not trivial to collect a database of viral motifs.
The high prediction accuracies of VirFinder and DeepVirFinder suggest the two groups, viruses and prokaryotic hosts, possess distinguishing k-mer or more general motif patterns. On the other hand, previous studies showed that virus and their infecting hosts share kmer similarity and utilized this phenomenon to predict hosts of viruses based on genomic sequences [18,45,46]. This phenomenon is likely due to the evolutionary pressure on viruses to adopt similar codons used by their hosts since they are dependent on host machinery for replication [47–50]. The two seemly contradictive observations are not mutually exclusive, because viruses can share higher similarity with each other than that with their hosts so that k-mer or more general motif features can be used for both viral sequence identification and virus-host predictions effectively.
DeepVirFinder is designed for identifying prokaryotic viruses in metagenomics which is a mixture of genomes from prokaryotic cells and prokaryotic viruses. We note that some metagenomic samples may contain contamination of sequences from host eukaryotic genomes such as human genome [51]. Users who have the concerns of eukaryotic contaimination should first filter out eukaryotic host sequences by mapping the reads to host reference genomes before applying Deep-VirFinder, as DeepVirFinder may potentially misidentify those eukaryotic sequences as viral, since eukaryotic sequences were not included in our training dataset. For more complicated cases where host reference genomes are not available, computational methods for detecting sequence contamination is of our interest in the future. In fact this problem is closely related to a general problem of out-of-distribution detection for AI safety [52]. Various methods based on discriminative models or generative models have been developed for this purpose [53–60] and worth our future investigation.
Overall, DeepVirFinder markedly improves the accuracy for identifying viruses in metagenomic data, and it will assist the study of viruses in various environments.
MATERIALS AND METHODS
Viruses and prokaryotic genomes used for training, validation and testing
We collected 2,314 RefSeq of viruses infecting prokaryotes (bacteria and archaea) from NCBI (https://www.ncbi.nlm.nih.gov/genome/browse). The dataset was partitioned into three parts based on the dates when the genomes were discovered. We used the genomes discovered before January 2014 for training, those between January 2014 and May 2015 for validation, and those after May 2015 for testing. The partitioning of the dataset not only avoids the overlaps between the training, validation, and test datasets, but also helps to evaluate the methods ability for predicting future new viruses based on the previously discovered viruses. We previously used the data before May 2015 in Ren et al. [5]. For this study, we updated the dataset to include new viruses after May 2015 and it was natural to use them as the test data.
Since sequences in real metagenomics data are of various lengths ranging from hundreds to thousands of base pairs, we fragmented the genomes into nonoverlapping sequences of different sizes, L = 150, 300, 500, 1000, and 3000 bp. We then built models for sequences of each size, respectively. In particular, the models for 150 and 300 bp were designed for the next generation sequencing technology, which commonly generates sequences of those fixed lengths. Table 1 shows the numbers of sequences in different sizes that were used for training, validation, and test. The dataset is paired with the same number of prokaryotic sequences, fragmented from 38,234 RefSeq and partitioned by the exact same dates.
Table 1.
The number of viral sequences of various sizes from viral genomes discovered before January 2014, between January 2014 and May 2015, and after May 2015
| Length | Training (Before 1/2014) | Validation (1/2014–5/2015) | Test (After 5/2015) | Total |
|---|---|---|---|---|
| 150 bp | 505,259 | 164,918 | 355,204 | 705,697 |
| 300 bp | 252,630 | 82,458 | 177,416 | 512,504 |
| 500 bp | 154,640 | 50,350 | 106,298 | 311,288 |
| 1000 bp | 77,014 | 25,087 | 52,956 | 155,057 |
| 3000 bp | 25,263 | 8,246 | 17,385 | 50,894 |
The three parts of the dataset partitioned by dates were used for training, validation, and testing, respectively.
Predicting viral sequences using convolutional neural networks
We used deep learning techniques and developed a powerful framework for predicting viral sequences. Given a query sequence, the framework outputs a score between 0 and 1, with a larger score indicating a higher possibility of being a viral sequence. Previously k-mer frequencies were used as features to a machine learning model that distinguishes viral sequences from prokaryotes [55]. The success of the method confirmed that viruses and their prokaryotic hosts have different preferences in k-mer usage. Those k-mers can be easily generalized as motifs, which are commonly represented using position weight matrices (PWM) of size 4 by k where each column specifies the probabilities of having A/C/G/T at a position. We expected that using motifs as the model features could increase the model flexibility and would improve prediction accuracy. Thus, we designed a deep learning methods using ConvNets, where the filters in ConvNets are able to capture sequence patterns. The filters are represented in the form of weight matrices of size 4 by k, where k is the filter length. This representation is similar to PWM. The generalization from k-mers to ConvNets provides a more general model and potentially gives better prediction accuracy.
We call our method DeepVirFinder. The model consists of a convolutional layer, a max pooling layer, a fully connected layer, and several dropout layers (Fig. 1). A DNA sequence of length L, X = X1… Xl… XL, Xl∈{A,C,G,T}, is first encoded using the one-hot encoding method, resulting in a 4 × L matrix . Ambiguous nucleotide “N” is encoded as a vector of . The encoded DNA sequences are fed into a convolutional layer with the rectifier activation, where the convolutional layer contains M motifs of length K. The m-th motif can be represented using a matrix of size 4 × K, Um, with tunable coefficients Um, i, k, i=1, …, 4, k= 1, …, K. Each motif scans the sequence Z(1) from the beginning to the end, obtaining a series of the motif intensities. Motif intensity is computed as the cross-correlation between subsequences of length K in Z(1) and each motif. The resulting motif intensities for all M motifs can be represneted using a vector Z(2) of size M × (L – K + 1), where . A rectified linear unit (ReLU) is applied for each motif, resulting a matrix Z(3) of the same size as Z(2), where . A max pooling layer reduces the dimension by keeping only the highest intensity for each motif, resulting in an M × 1 matrix Z(4), where .
The output is then fed into a dense layer containing N fully connected neurons with weight vector Wn and bias bn for the n-th neuron. The resulting output matrix is of dimension N × 1, Z(5), where . Another ReLu is applied for each neuron, resulting . The output is finally summarized using a dense layer with sigmoid function to generate a prediction score ranging from 0 to 1, i.e., Z(6)= σ(z), where , is the sigmoid function.
In summary, the input sequence X in fed into the neural networks, and the resulting output score Y is
Since the DNA sequence is double stranded, and the contigs in real data can come from both strands, the prediction score should be identical for either the forward strand or the backward strand. Thus, we apply the same network to the reverse complement of the original sequence, and the final prediction score is the average of the predictions from the original and the reverse complement sequences. That is, . Similar techniques were used in Quang et al. and Wang et al. [22,23].
The objective function is to minimize the binary cross-entropy loss between the predicted score Yfinal and the true labels (0 for prokaryotic sequences, and 1 for virus sequences). The training dataset is iteratively fed into the model in batches of size 150. One iteration of finishing feding batches of the whole training dataset is called one epoch. The parameters in the neural networks were updated through back-propagation using Adam optimization algorithm for stochastic gradient descent with learning rate 0.001 [61]. Dropout regularization of rate0.1 are applied after the max pooling layer, and after the fully connected layer, to reduce overfitting in neural networks by randomly dropping out a few dimensions.
This convolutional neural network has three critical hyper parameters, the length of motifs (or filters), the number of motifs, and the number of epochs for training. The first two determine the complexity of the model, and the third one controls the balance between overfitting and underfitting. To find the best parameter settings, we trained the model with different combinations of the three parameters using the data before January 2014, and evaluated the model performance using AUROC on the validation dataset. We studied the motif length ranging from 4 to 18, the number of motifs from 100, 500, 1000 to 1500, and the number of epochs up to 60.
Collection of metavirome datasets
To achieve high prediction accuracy, a deep learning algorithm needs a large amount of training data. Though a large number of training sequences were obtained from RefSeq, there is a potential to enlarge the training dataset by including viral sequences from metavirome sequencing data. Metavirome sequencing targets at sequencing mainly viruses by removing prokaryotic cells in samples using the physical 0.22 μm filters. Metavirome sequencing does not rely on culturing viruses in the lab, so it is able to capture both cultivated and uncultivated viruses, representing the true viral diversity. A few studies have used this technique to extract viruses and sequenced viral genomes in human gut and ocean samples [1,2,62,63]. Normal et al. sequenced virome in the human gut sample from IBD patients using Illumina sequencing technology[1]. Reyes et al. studied viruses in fecal samples from Malawian twins with Severe Acute Malnutrition (SAM) using Roche 454 sequencing technology [2]. Minot et al. and Kim et al. investigated virome in healthy human gut using Roche 454 [11,62]. For marine virome, the Tara Ocean Virome project collected the largest number of virome samples from both surface- and deep-ocean sites over the world [63].
We collected the metavirome samples from those studies and aimed to add more viral diversity, especially adding viruses not- or under-represented in RefSeq, to the training data. We were careful in quality control of the samples because it is likely that the sample can be contaminated by prokaryotic DNA, since the physical filters may not exclude small sized prokaryotic cells. The details of preparation of metavirome data and quality control can be found in Supplementary Materials and Supplementary Table S3. Up to 1.3 million of sequences were generated from the metavirome data, and they were combined with sequences derived from viral RefSeq before May 2015 for training. The same number of prokaryotic sequences were paired with the viral sequences in the enlarged dataset for training. The new model was evaluated and compared with the original model trained based on RefSeq only, using the test sequences from RefSeq after May 2015.
Simulation of metagenomic datasets
To assess the performance of DeepVirFinder trained previously using RefSeq on predicting viruses in metagenomic samples, we generated sythetic metagenomic samples based on organism abundance profiles derived from a real human gut metagenomic sample (accession ID SRR061166, Platform: Illumina). Given a total budget of base pairs in a sample, the number of base pairs in contigs sampled from each genome was computed proportionally to the abundance profile. For each reference genome, contigs were sampled randomly and independently from the genome, where the contig length follows the same distribution as that in a real human metagenomics dataset for CRC patients (Fig. 4A), until the number of base pairs reaches the total budget. The details can be found in Supplementary Materials.
We constructed metagenomic samples with different viral fractions and evaluated DeepVirFinder on each of them. In metagenomic sequencing experiments, there are two major types of genome sampling strategies. One is referred to as cellular metagenomes in which all the genetic materials, including bacteria, archaea, and viruses, are sampled and sequenced. Another type of data is metavirome where cellular organisms like bacteria are filtered out first before sequencing and mostly viruses are sequenced. To mimic the different viral fractions in real metagenomic data, the abundance profile was rescaled to make samples of three different viral fractions 10%, 50%, and 90%, while keeping the relative abundance within viruses and that within hosts the same.
Viral analysis of human gut metagenomics from patients with colorectal cancer
Human gut metagenomics samples from patients with CRC and the control group were downloaded from European Nucleotide Archive (ENA) database (see the website:www.ebi.ac.uk/ena) with accession number ERP005534. Samples from 53 cancer patients and 61 normal individuals were randomly split into 2/3 for training and 1/3 for testing. The patient ID and the disease status can be found in the Supplementary Materials. The metagenomics samples from training were combined and cross-assembled. To guarantee high accuracies in the downstream analysis including virus contig identification and contig binning, we filtered contigs smaller than 500 bp. DeepVirFinder was then applied to predict viral contigs in the remaining dataset. To control the false discovery rate, the predicted p-value for each contig was converted to a q-value. The q-value is an estimation of the proportion of false prediction if the prediction is made at the level of the corresponding p-value. Contigs were sorted by q-values from the smallest to the largest, and the contigs having q-values < 0.01 were predicted as viruses. The viral contigs predicted by DeepVirFinder were then grouped into contig bins, and the abundance of contig bins was derived based on the read mapping results. To study the association between the viruses and the cancer status, we built a logistic regression classifier with Lasso penalty to predict the CRC status based on the bin abundance on training data, and evaluated the perfor mance on test data. The details can be found in the Supplementary Materials.
Data and code availability
The software DeepVirFinder is available at the website (github.com/jessieren/DeepVirFinder). The NCBI accession numbers of the viral and prokaryotic RefSeq, the species abundance profile for simulated metagenomic samples, and the sample IDs used for identifying viruses in CRC patients can be found at the website (github.com/jessieren/DeepVirFinder/tree/master/supplementary_tables).
Supplementary Material
ACKNOWLEDGEMENTS
The research was supported by the U.S. National Institutes of Health R01GM120624, National Science Foundation DMS-1518001, National Natural Science Foundation of China (11701546), and the Simons Collaboration on Computational Biogeochemical Modeling of Marine Ecosystems (CBIOMES; grant ID 549943). We thank Drs. Michael S. Waterman, Gesine Reinert, Ying Wang, Rui Jiang, Yang Lu, Lizzie Dorfman, Mr. Weili Wang, and Mr. Luigi Manna for helpful discussions and suggestions. We thank USC Center for High Performance Computing (HPC) for helping us use their cluster computers.
Footnotes
SUPPLEMENTARY MATERIALS
The supplementary materials can be found online with this article at https://doi.org/10.1007/s40484-019-0187-4.
The authors Jie Ren, Kai Song, Chao Deng, Nathan A. Ahlgren, Jed A. Fuhrman, Yi Li, Xiaohui Xie, Ryan Poplin and Fengzhu Sun declare that they have no conflicts of interest.
All procedures performed in studies were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
NOTE TO RELATED WORK OF FANG et al. [64]
A preliminary version of this manuscript was put in arXiv (arxiv.org/abs/1806.07810) on June 20, 2018. During the process of the submission to regular journals, Fang et al. [64] used deep learning to classify metagenomic fragments to chromosomal, viral and plasmid sequences. Similar prediction accuracy for viruses using nucleotide base encoding as presented in this paper was obtained. The two studies should be considered independent.
REFERENCES
- 1.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al. (2015) Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell, 160, 447–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reyes A, Blanton LV, Cao S, Zhao G, Manary M, Trehan I, Smith MI, Wang D, Virgin HW, Rohwer F, et al. (2015) Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA, 112, 11941–11946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma Y, You X, Mai G, Tokuyasu T and Liu C (2018) A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome, 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roux S, Enault F, Hurwitz BL and Sullivan MB (2015) VirSorter: mining viral signal from microbial genomic data. PeerJ, 3, e985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren J, Ahlgren NA, Lu YY, Fuhrman JA and Sun F (2017) VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome, 5, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amgarten D, Braga LPP, da Silva AM and Setubal JC (2018) Marvel, a tool for prediction of bacteriophage sequences in metagenomic bins. Front. Genet, 9, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roux S, Faubladier M, Mahul A, Paulhe N, Bernard A, Debroas D and Enault F (2011) Metavir: a web server dedicated to virome analysis. Bioinformatics, 27, 3074–3075 [DOI] [PubMed] [Google Scholar]
- 8.Rampelli S, Soverini M, Turroni S, Quercia S, Biagi E, Brigidi P and Candela M (2016) ViromeScan: a new tool for metagenomic viral community profiling. BMC Genomics, 17, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wommack KE, Bhavsar J, Polson SW, Chen J, Dumas M, Srinivasiah S, Furman M, Jamindar S and Nasko DJ (2012) VIROME: a standard operating procedure for analysis of viral metagenome sequences. Stand. Genomic Sci, 6, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood DE and Salzberg SL (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol, 15, R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D, Song L, Breitwieser FP and Salzberg SL (2016) Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res, 26, 1721–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C and Segata N (2015) MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods, 12, 902–903 [DOI] [PubMed] [Google Scholar]
- 13.Buchfink B, Xie C and Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat. Methods, 12, 59–60 [DOI] [PubMed] [Google Scholar]
- 14.Fouts DE (2006) Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res, 34, 5839–5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima-Mendez G, Van Helden J, Toussaint A and Leplae R (2008) Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics, 24, 863–865 [DOI] [PubMed] [Google Scholar]
- 16.Akhter S, Aziz RK and Edwards RA (2012) PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res, 40, e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y and Wishart DS (2016) PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res, 44, W16–W 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux S, Hallam SJ, Woyke T and Sullivan MB (2015) Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife, 4, e08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paez-Espino D, Pavlopoulos GA, Ivanova NN and Kyrpides NC (2017) Nontargeted virus sequence discovery pipeline and virus clustering for metagenomic data. Nat. Protoc, 12, 1673–1682 [DOI] [PubMed] [Google Scholar]
- 20.Alipanahi B, Delong A, Weirauch MT and Frey BJ (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol, 33, 831–838 [DOI] [PubMed] [Google Scholar]
- 21.Zeng H, Edwards MD, Liu G and Gifford DK (2016) Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics, 32, i121–i127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quang D and Xie X (2019) Factornet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods,166, 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang M, Tai C, E, W. and Wei L (2018) DeFine: deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Res, 46, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J and Troyanskaya OG (2015) Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods, 12, 931–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quang D and Xie X (2016) DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res, 44, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley DR, Snoek J and Rinn JL (2016) Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res, 26, 990–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poplin R, Chang P-C, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, et al. (2018) A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol, 36, 983–987 [DOI] [PubMed] [Google Scholar]
- 28.Zeng H and Gifford DK (2017) Predicting the impact of non-coding variants on DNA methylation. Nucleic Acids Res, 45, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Quang D and Xie X (2017) Understanding sequence conservation with deep learning. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, pp. 400–406. ACM [Google Scholar]
- 30.Li Y, Shi W and Wasserman WW (2018) Genome-wide prediction of cis-regulatory regions using supervised deep learning methods. BMC Bioinformatics. 19, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh S, Yang Y, Poczos B and Ma J (2019) Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quant. Biol 7, 122–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yue T and Wang H (2018) Deep learning for genomics: A concise overview. arXiv:1802.00810 [Google Scholar]
- 33.Lauring AS, Frydman J and Andino R (2013) The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol, 11, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glenn TC (2011) Field guide to next-generation DNA sequencers. Mol. Ecol. Resour, 11, 759–769 [DOI] [PubMed] [Google Scholar]
- 35.World Health Organization. (2014) World Cancer Report 2014. Stewart B, Wild CP, eds., IAIC [Google Scholar]
- 36.Hawk ET and Levin B (2016) Colorectal cancer prevention. J. Clinic. Oncolo 23, 378–391 [DOI] [PubMed] [Google Scholar]
- 37.Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, et al. (2015) Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun, 6, 6528. [DOI] [PubMed] [Google Scholar]
- 38.Vogtmann E, Hua X, Zeller G, Sunagawa S, Voigt AY, Hercog R, Goedert JJ, Shi J, Bork P and Sinha R (2016) Colorectal cancer and the human gut microbiome: reproducibility with whole-genome shotgun sequencing. PLoS One, 11, e0155362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, Ng SC, Tsoi H, Dong Y, Zhang N, et al. (2015) Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun, 6, 8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, Amiot A, Böhm J, Brunetti F, Habermann N, et al. (2014) Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol, 10, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu YY, Chen T, Fuhrman JA and Sun F (2017) COCACOLA: binning metagenomic contigs using sequence COmposition, read CoverAge, CO-alignment and paired-end read LinkAge. Bioinformatics, 33, 791–798 [DOI] [PubMed] [Google Scholar]
- 42.Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GG, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, et al. (2014) A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun, 5, 4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. (2018) The pfam protein families database in 2019. Nucleic Acids Res. D427–D432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng T, Li J, Ni Y, Kang K, Misiakou M-A, Imamovic L, Chow BKC, Rode AA, Bytzer P, Sommer M, et al. (2019) Mining, analyzing, and integrating viral signals from metagenomic data. Microbiome, 7, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edwards RA, McNair K, Faust K, Raes J and Dutilh BE (2016) Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev, 40, 258–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahlgren NA, Ren J, Lu YY, Fuhrman JA and Sun F (2017) Alignment-free d2 oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res, 45, 39–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gouy M and Gautier C (1982) Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res, 10, 7055–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharp PM, Rogers MS and McConnell DJ (1985) Selection pressures on codon usage in the complete genome of bacteriophage T7. J. Mol. Evol, 21, 150–160 [DOI] [PubMed] [Google Scholar]
- 49.Pride DT, Wassenaar TM, Ghose C and Blaser MJ (2006) Evidence of host-virus co-evolution in tetranucleotide usage patterns of bacteriophages and eukaryotic viruses. BMC Genomics, 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carbone A (2008) Codon bias is a major factor explaining phage evolution in translationally biased hosts. J. Mol. Evol, 66, 210–223 [DOI] [PubMed] [Google Scholar]
- 51.Ponsero AJ and Hurwitz BL (2019) The promises and pitfalls of machine learning for detecting viruses in aquatic metagenomes. Front. Microbiol, 10, 806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Amodei D, Olah C, Steinhardt J, Christiano P, Schulman J and Man’e D (2016) Concrete problems in AI safety. arXiv:1606.06565 [Google Scholar]
- 53.Hendrycks D and Gimpel KA (2017) A baseline for detecting misclassified and out-of-distribution examples in neural networks. In: Proceedings of International Conference on Learning Representations 2017. Toulon [Google Scholar]
- 54.Lakshminarayanan B, Pritzel A and Blundell C (2017) Simple and scalable predictive uncertainty estimation using deep ensembles. In: Proceedings of Advances in Neural Information Processing Systems, pp. 6402–6413 [Google Scholar]
- 55.Liang S, Li Y and Srikant R (2017) Enhancing the reliability of out-of-distribution image detection in neural networks. arXiv: 1706.02690 [Google Scholar]
- 56.Hendrycks D, Mazeika M and Dietterich TG (2018) Deep anomaly detection with outlier exposure. arXiv:1812.04606 [Google Scholar]
- 57.Shafaei A, Schmidt M and Little JJ (2018) Does your model know the digit 6 is not a cat? a less biased evaluation of outlier detectors. arXiv:1809.04729 [Google Scholar]
- 58.Ren J, Liu PJ, Fertig E, Snoek J, Poplin R, DePristo MA, Dillon JV and Lakshminarayanan B (2019) Likelihood ratios for out-of-distribution detection. arXiv:1906.02845 [Google Scholar]
- 59.Ovadia Y, Fertig E, Ren J, Nado Z, Sculley D, Nowozin S, Dillon JV, Lakshminarayanan B and Snoek J (2019) Can you trust your model’s uncertainty? Evaluating predictive uncertainty under dataset shift. arXiv:1906.02530 [Google Scholar]
- 60.Nalisnick E, Matsukawa A, Teh YW and Lakshminarayanan B (2019) Detecting out-of-distribution inputs to deep generative models using a test for typicality. arXiv:1906.02994 [Google Scholar]
- 61.Kingma DP and Ba J (2015) Adam: A method for stochastic optimization. In: Proceedings of the 3rd International Conference for Learning Representations. San Diego [Google Scholar]
- 62.Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD and Bushman FD (2011) The human gut virome: inter-individual variation and dynamic response to diet. Genome Res, 21, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roux S, Brum JR, Dutilh BE, Sunagawa S, Duhaime MB, Loy A, Poulos BT, Solonenko N, Lara E, Poulain J, et al. (2016) Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature, 537, 689–693 [DOI] [PubMed] [Google Scholar]
- 64.Fang Z, Tan J, Wu S, Li M, Xu C, Xie Z and Zhu H (2019) PPR-Meta: a tool for identifying phages and plasmids from metagenomic fragments using deep learning. Gigascience, 8, giz066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The software DeepVirFinder is available at the website (github.com/jessieren/DeepVirFinder). The NCBI accession numbers of the viral and prokaryotic RefSeq, the species abundance profile for simulated metagenomic samples, and the sample IDs used for identifying viruses in CRC patients can be found at the website (github.com/jessieren/DeepVirFinder/tree/master/supplementary_tables).