

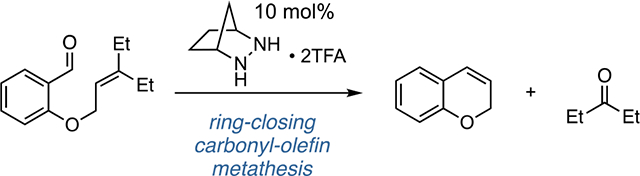
Abstract
The catalytic ring-closing carbonyl-olefin metathesis (RCCOM) of O-allyl salicylaldehydes to form 2H-chromenes is described. The method utilizes a [2.2.1]-bicyclic hydrazine catalyst and operates via a [3+2]/retro-[3+2] metathesis manifold. The nature of the allyl substitution pattern was found to be crucial, with sterically demanding groups such as adamantylidene or diethylidene offering optimal outcomes. A survey of substrate scope is shown along with a discussion of mechanism supported by DFT calculations. Steric pressure arising from syn-pentane minimization of the diethylidene moiety is proposed to facilitate cycloreversion.
Keywords: Carbonyl-olefin metathesis, hydrazines, chromenes, cycloaddition, cycloreversion
Graphical Abstract
The 2H-chromene architecture and its derivatives are common motifs in biologically relevant molecules,1,2 including for example the antiviral compound BW683C (1),3 the pesticide hildgardtene (2),4 and the antidiabetic compound 3 (Figure 1A).5 Accordingly, many synthetic strategies have been devised to access 2H-chromenes 7,6 and the development of novel strategies remains a valuable undertaking. One conceptually attractive approach to 2H-chromene synthesis is via ring-closing double-bond metathesis,7,8 particularly because such strategies can capitalize on the widespread availability of salicylaldehydes 49 and the trivial nature of their conversion to allyl ethers 5 (Figure 1B).10 Most notably, Grubbs has reported the use of olefin metathesis to convert dienes to the corresponding 2H-chromenes.7 However, a drawback to this approach is that it requires an additional step to convert the aldehyde moiety of 5 into an alkene, which thus incurs the costs of additional time, waste, and potential substrate incompatibility. A more desirable strategy would be to convert aldehydes 5 directly into chromenes 7 via catalytic ring-closing carbonyl-olefin metathesis (RCCOM). Unfortunately, carbonyl-olefin metathesis (COM) technologies remain relatively underdeveloped, and only in recent years have the first catalytic strategies been reported.11,12–13 Indeed, the first catalytic RCCOM reactions were described by Schindler in 2016 using Lewis acid catalysis,14 while others have subsequently shown that a variety of Lewis15 and Brønsted acids16 can catalyze such reactions with certain substrates. While these works have greatly expanded the scope of carbonyl-olefin metathesis, acid catalysis is not compatible with all substrates and COM modalities. In fact, during the course of this work, we have found that the attempted use of acids to achieve the RCCOM of O-allyl salicylaldehydes 5 was unproductive and resulted primarily in deallylation reactions (vide infra). Thus, alternative strategies are required if this and other COM reactions are to be fully realized.
Figure 1.
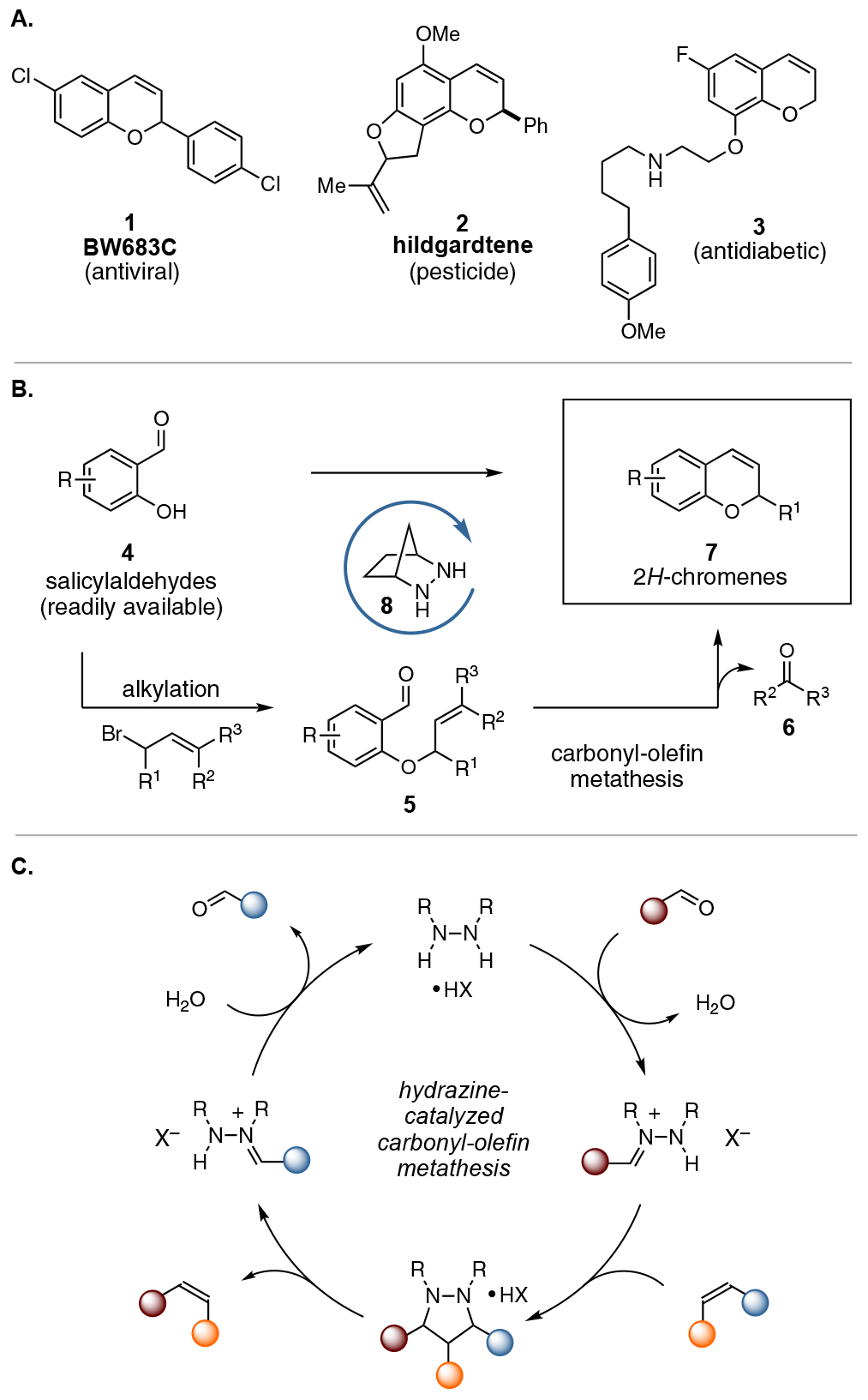
(A) Bioactive 2H-chromenes. (B) Conversion of salicylaldehydes to chromenes. (B) Hydrazine-catalyzed carbonyl-olefin metathesis.
In 2012 we described a novel approach to carbonyl-olefin metathesis that employed hydrazine catalysts and proceeded via a thermally-allowed [3+2] cycloaddition / cycloreversion sequence (Figure 1C).11 Using this strategy, we developed a catalytic ring-opening carbonyl-olefin metathesis (ROCOM) of cyclopropenes. That chemistry was greatly facilitated by the release of ring strain during both the cycloaddition and cycloreversion events,17 and it was thus unclear whether the strategy could be extended to realize the RCCOM of O-allylsalicylaldehydes 5 to form 2H-chromenes 7.
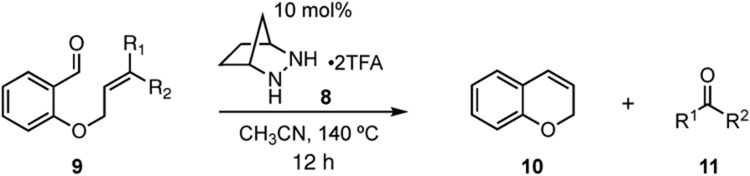
To investigate this possibility, we began by examining the reaction of a series of O-allyl salicylaldehydes 9, in which the substitution of the alkylidene fragment was varied (Table 1). For this screen, we used 10 mol% [2.2.1]-bicyclic hydrazine 8•(TFA)2 as the catalyst,18 with acetonitrile solvent heated to 140 °C in a sealed vial for 12 h. The main products observed in these reactions included the desired 2H-chromene 10 and salicylaldehyde (not shown) resulting from deallylation. The ketone products 11 were not isolated. In varying the allyl fragment, we found that an allyl group itself (9a) led to little conversion and no desired product (entry 1). Cinnamyl (9b) and 3,3-diphenylallyl (9c) groups were similarly unproductive (entries 2 and 3). On the other hand, the use of a prenyl group (9d) led to appreciable consumption of starting material, albeit with only 13% yield of the desired chromene 10 (entry 4). A cyclohexylidene ethyl ether (9e) resulted in a notable improvement in consumption of starting material and in the generation of the desired product (entry 5), although the yield of 10 was limited by competing intramolecular ene reaction (see supporting information for details). Changing the ring size offered no improvements in this regard (9f, 9g, entries 6 and 7). More productively, the use of an adamantylidene ethyl ether (9h) resulted in reasonable yield (30%) in relation to the amount of starting material conversion (40%, entry 8). The most effective group proved to be 3,3-diethylallyl (9i, entry 9), which resulted in the highest yield of 10, with minimal amounts of starting material lost to deallylation. Extension to isopropyl groups was not beneficial however (9j, entry 10). The efficiency of the diethylallyl substrate is striking, particularly in comparison to the dimethyl (entry 4) and cyclohexylidene (entry 5) substrates. These results suggest that the ethyl groups play an important role in facilitating this catalytic process, and we propose a hypothesis for their impact in the mechanistic discussion section.
Table 1.
Allyl group screen.a
 | ||||
|---|---|---|---|---|
| entry | substrate | 2 consumption (%) | 3 yield (%) | salicylaldehyde yield (%) |
| 1 |  |
13 | 0 | 3 |
| 2 | 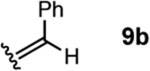 |
29 | 0 | 5 |
| 3 | 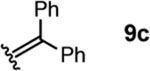 |
32 | 0 | 15 |
| 4 | 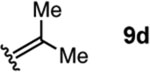 |
51 | 13 | 5 |
| 5 | 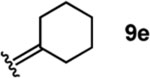 |
86 | 31 | 5 |
| 6 | 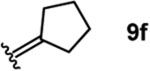 |
90 | 10 | 10 |
| 7 | 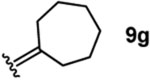 |
80 | 30 | 7 |
| 8 | 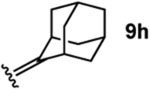 |
40 | 30 | 2 |
| 9 | 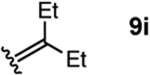 |
65 | 48 | 2 |
| 10 | 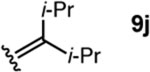 |
27 | 3 | 3 |
Conditions: substrate 9 (0.2 mmol) and catalyst 8•(TFA)2 (10 mol %) in 1.0 mL of solvent in a 5 mL sealed tube were heated to 140 °C for 12 h. Conversions and yields were determined by 1H NMR using CH2Br2 as an internal standard.
Next, we conducted an optimization study of the RCCOM of salicylaldehyde ether 9i in order to maximize the yield of chromene (10) and to minimize unwanted side products (Table 2). First, a screen of solvents revealed that the reaction occurred in a variety of media (entries 1–4), but EtOH proved to be optimal in terms of conversion and formation of the desired product 10. At a lower temperature of 120 °C, a lower conversion and product yield were obtained in the same reaction time (entry 5). Because it has been shown that certain substrates can undergo carbonyl-olefin metathesis reactions under Brønsted acid catalysis,16 we examined this reaction in the presence of TFA alone (entry 6). While we observed 100% conversion of starting material, no desired chromene 10 was obtained, demonstrating that the hydrazine plays a crucial role in the optimized process. We also examined whether Lewis acid conditions14,15 would promote this reaction (entry 7). Again, no desired product was formed, and only deallylated salicylaldehyde was obtained.
Table 2.
Optimization studies.a
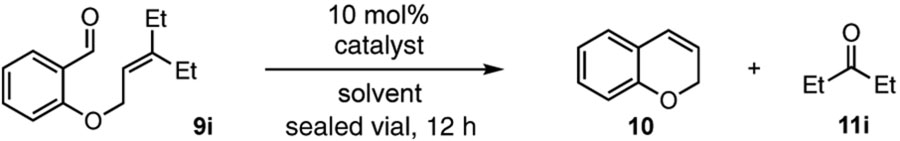 | |||||
|---|---|---|---|---|---|
| entry | catalyst | solvent | temp. (°C) | conv. (%) | 3 yield (%) |
| 1 | 8• (TFA)2 | CH3CN | 140 | 65 | 48 |
| 2 | 8• (TFA)2 | DCE | 140 | 49 | 20 |
| 3 | 8• (TFA)2 | MeOH | 140 | 100 | 40 |
| 4 | 8• (TFA)2 | EtOH | 140 | 100 | 78 (73)b |
| 5 | 8• (TFA)2 | EtOH | 120 | 85 | 47 |
| 6c | TFA | EtOH | 140 | 100 | 0 |
| 7 | FeCl3 | DCE | rt | 100 | 0 |
Conditions: substrate 9i (0.2 mmol) and 10 mol % catalyst in 1.0 mL of solvent in a 5 mL sealed tube were heated to the temperature indicated for 12 h. Conversions and yields were determined by 1H NMR using CH2Br2 as an internal standard.
Isolated yield;
20 mol % TFA was used.
Before discussing our scope studies, we note that substrate synthesis is straightforward and scalable (Scheme 1). Thus, 3-pentanone (11i) could be converted to ester 13 in 85% yield by Horner-Wadsworth-Emmons reaction. Reduction of 13 with DIBAL followed by treatment with PBr3 then furnished the allyl bromide 14 in 82% yield. Notably, this three-step procedure requires no chromatography and provides access to 14 on preparative scale (24.7 g). Finally, reaction of salicylaldehyde (15) with 14 in the presence of K2CO3 yielded the metathesis substrate 9i in 92% yield.
Scheme 1.
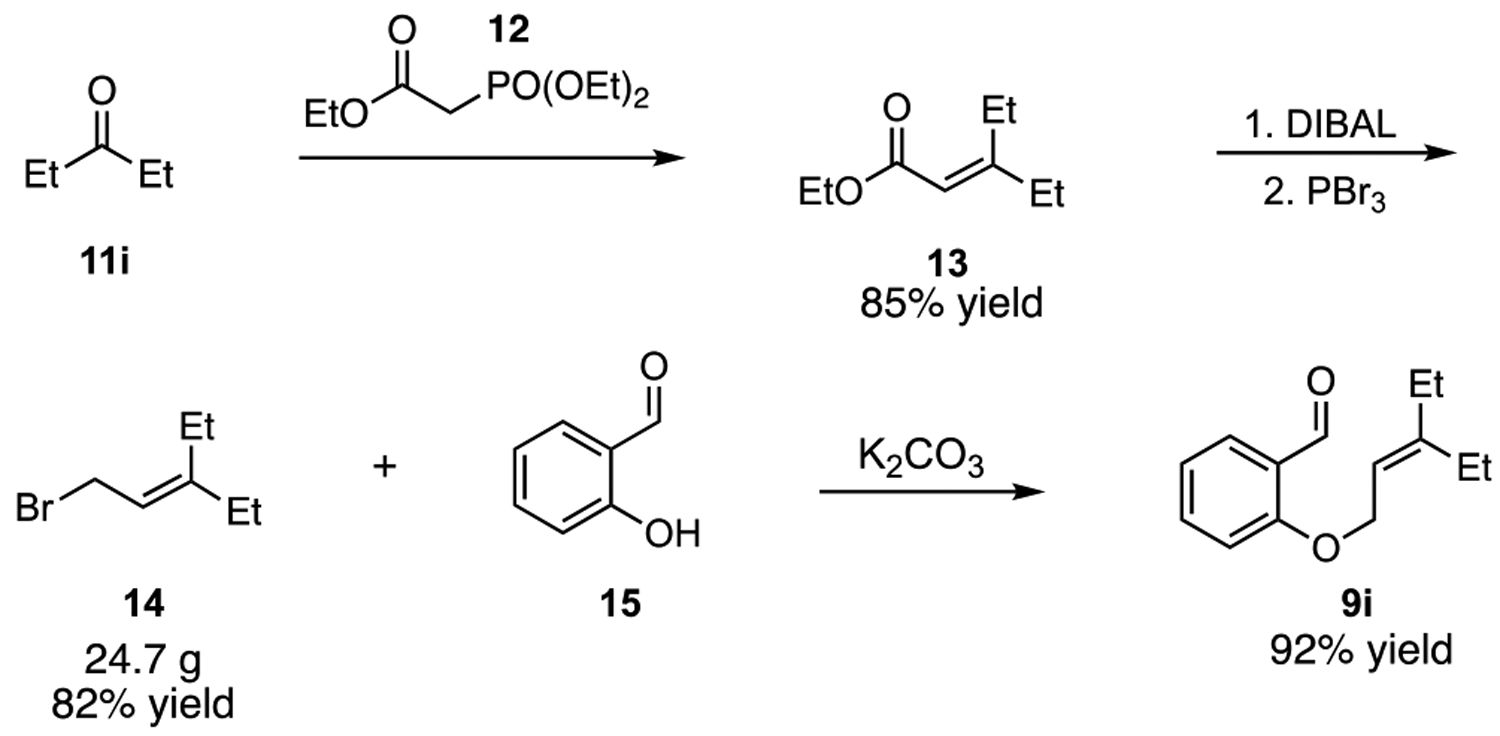
Synthesis of metathesis substrate 9i.
Using the conditions defined in entry 4 of Table 2, we explored the substrate scope of this process (Table 3). In addition to the unsubstituted case 10 (entry 1), we found alkyl (18, entry 2), halogen (19-21, entries 3–5), and methoxy substitution (22, entry 6) at the 7-position of the chromene system to be well-tolerated. Substitution at the 6-position also proved possible, as alkyl (23 and 24, entries 7 and 8), halogen (25 and 26, entries 9 and 10), and trifluoromethoxy (27, entry 11) substituted products were accessed in good to excellent yields. Electron-withdrawing groups in the form of a nitro or carbomethoxy substituent could also be accommodated (28 and 29, entries 12 and 13), albeit in lower yields. Meanwhile, substitution of the 8-position with either a methyl or an allyl group proved possible (30 and 31, entries 14 and 15), demonstrating that encumbrance of the ether oxygen is not detrimental to reactivity and that the presence of a distal olefin is compatible with the reaction. Substitution next to both the ether and carbonyl functionalities was also tolerated (entry 16). Two RCCOM reactions on the same substrate was possible using increased catalyst loading (33, entry 17), and a fused aromatic ring system in the form of 3H-benzo[f]chromene (34) could also be generated with this method (entry 18).
Table 3.
Substrate scope studies for hydrazine-catalyzed RCCOM of salicylaldehyde allylic ethers.a
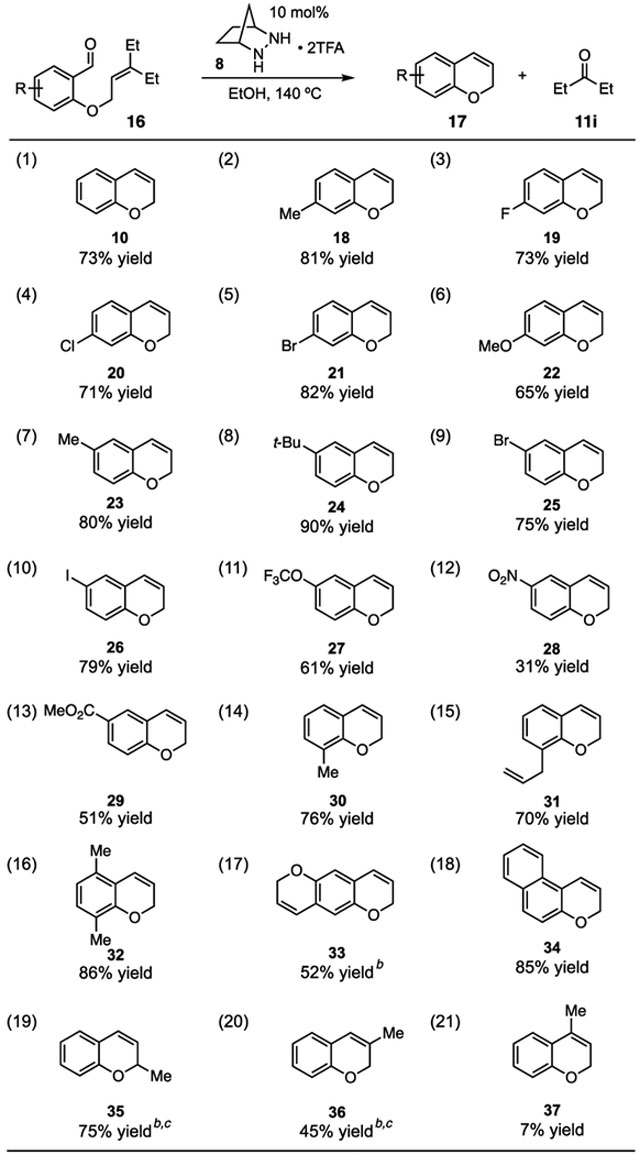
|
Conditions: substrate (0.1 mmol) and catalyst 8•(TFA)2 (10 mol %) in 0.5 mL of ethanol were heated to 140 °C in a 5 mL sealed tube for 12 h. Yields were determined on purified products.
20 mol% catalyst used.
Isopropanol was used as solvent.
Although the standard conditions were less accommodating to substitution on the pyran ring, we found that slight modifications enabled productive reactions to occur. Thus, while 2-methyl chromene 35 was generated in only 18% yield with the standard procedure, switching to i-PrOH as solvent and the use of 20 mol% catalyst increased the yield to 75% (entry 19). Similarly, a low yield of less than 20% of 36 was improved to 45% yield with these same modifications (entry 20). On the other hand, the reaction of a ketone instead of an aldehyde-containing substrate remained problematic (37, entry 21). We suspect that condensation of the hydrazine and/or cycloaddition is quite challenging with this substrate, and thus the design of catalysts with less steric demand may be necessary for this class of substrates.
Although the elevated reaction temperature for this method might raise questions about functional group compatibility, we note that the conditions are relatively mild. To illustrate this point, we conducted a functional group compatibility screen19 as shown in Table 4, using the ROCOM reaction of 9i as our standard. We found that electron-rich alkyne and alkene functionality were well-tolerated (38 and 39, entries 1 and 2), as was a ketone (40, entry 3). Notably, a basic pyridine did not undergo decomposition and did not inhibit the reaction to any appreciable extent (41, entry 4). Potentially sensitive heterocycles including indole, benzofuran, and benzothiophene were innocuous in the reaction (42-44, entries 5–7). Similarly, an unprotected alcohol, carboxylic acid, and amide were unscathed in the reaction and offered no inhibition to the metathesis (45-47, entries 8–10). Two functionalities were found to not be unscathed: an unprotected aniline (48, entry 11) and an alkyl bromide (49, entry 12). Although the metathesis product 10 was still produced, the yield was diminished somewhat in both cases, and the additives were recovered in low yield. These last two examples notwithstanding, the functional group compatibility of this method appears to be reasonably broad.
Table 4.
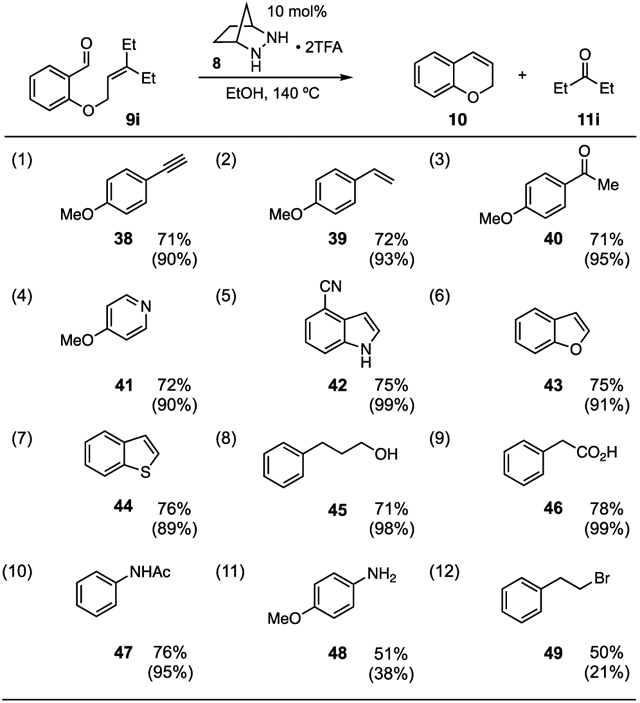
|
Yields of 10 were determined by 1H NMR.
Values in parentheses refer to amount of recovered additive as determined by 1H NMR.
A description of the catalytic cycle for this reaction is shown in Figure 2. Hydrazine catalyst 8•(TFA)2 condenses with aldehyde 9i to generate hydrazonium intermediate 50. Intramolecular 1,3-dipolar cycloaddition between the hydrazonium moiety and the olefin then furnishes the pentacyclic intermediate 51a. After proton migration between nitrogen atoms to generate 51b, cycloreversion reveals the chromene adduct 10 and hydrazonium 52. Finally, hydrolysis of 52 unveils 3-pentanone (11i) and returns the hydrazine 8 to the catalytic cycle.
Figure 2.
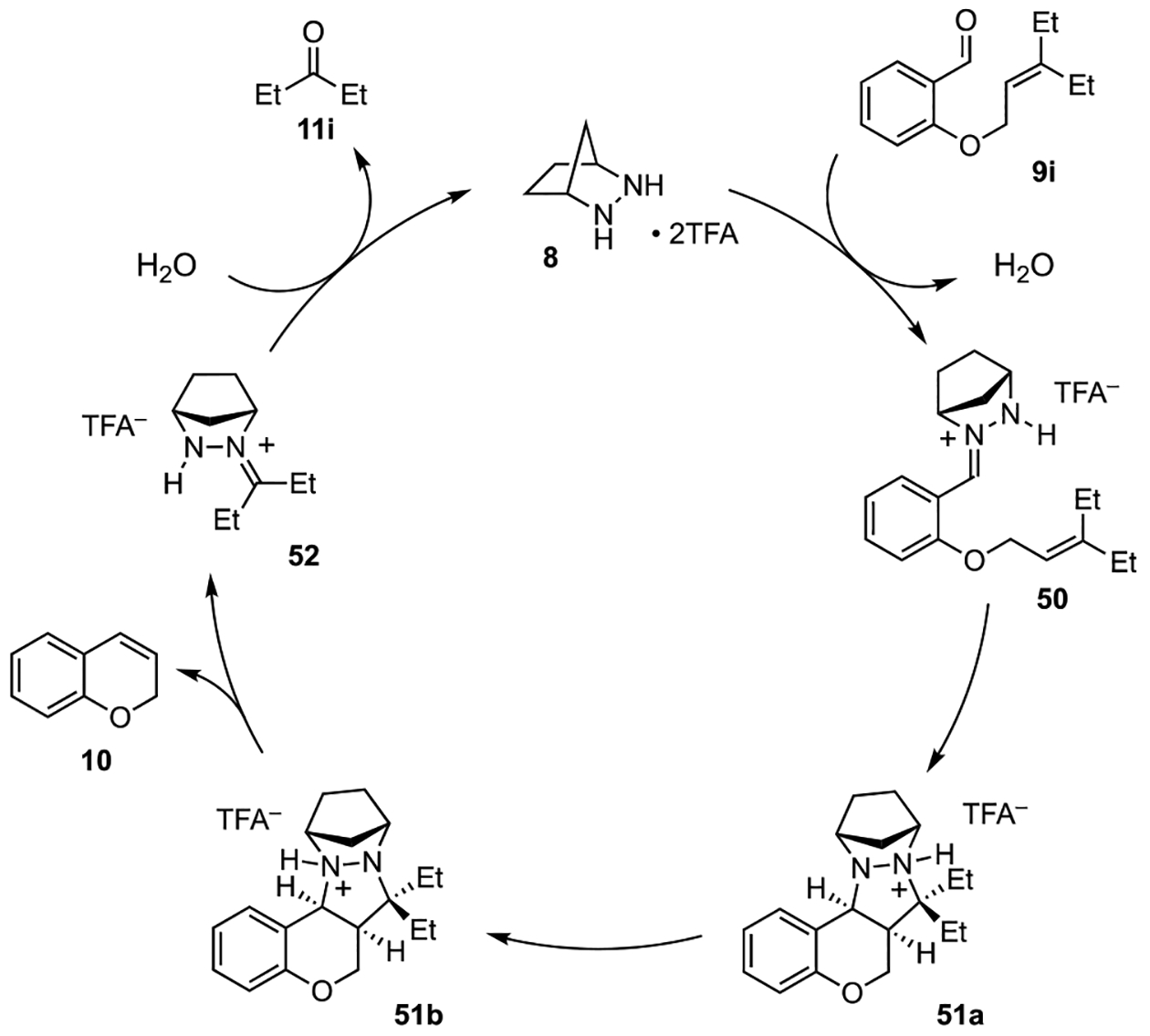
Mechanistic rationale for hydrazine 8-catalyzed RCCOM of O-allyl salicylaldehyde 9i.
To better understand the reaction pathway and to help rationalize the optimal performance of the diethyl allyl moiety, we conducted DFT calculations for each step of the presumed reaction pathway for substrate 9i (Figure 3). Condensation of catalyst 8•(TFA)2 with 9i to form hydrazonium 50 is mildly exergonic. The energy barrier for cycloaddition (cf. 53) is modest at 19.8 kcal/mol, and indeed we have found this reaction proceeds, albeit slowly, at room temperature. A favorable proton migration event between the two nitrogens following the cycloaddition leads to 51b, which is overall 20.2 kcal/mol more stable than the cycloaddition precursor 50. As anticipated, the cycloreversion event (cf. 54) is the rate-determining step, with an energy barrier for the diethyl substrate 54i of 38.8 kcal/mol. Interestingly, while the transition state energies for the allyl (54a) and prenyl (54d) substrates were found to be higher (42.2 and 40.4 kcal/mol respectively), the cyclohexylidene (54e) and adamantylidene (54h) substrates had significantly lower barriers (33.1 and 35.0 kcal/mol) than the diethyl substrate. The reason why the cyclohexylidene substrate is less effective is due to the competition of an intramolecular ene reaction (see supporting information for details). Both the adamantylidene and diethyl substrates offer high yields under the optimal conditions, which we believe is a combination of accessible cycloreversion activation energies and steric protection from deallylation of the starting material. Interestingly, the cycloreversion to generate 2H-chromene (10) is calculated to be slightly endergonic (+2.9 kcal/mol), as is the subsequent hydrolysis of hydrazonium 52 to form 3-pentanone (11i) and catalyst 8 (+1.9 kcal/mol). Overall, each catalyst turnover only becomes irreversible once the next cycloaddition has taken place, and thus the resting state of the catalyst is the cycloadduct 51b.
Figure 3.
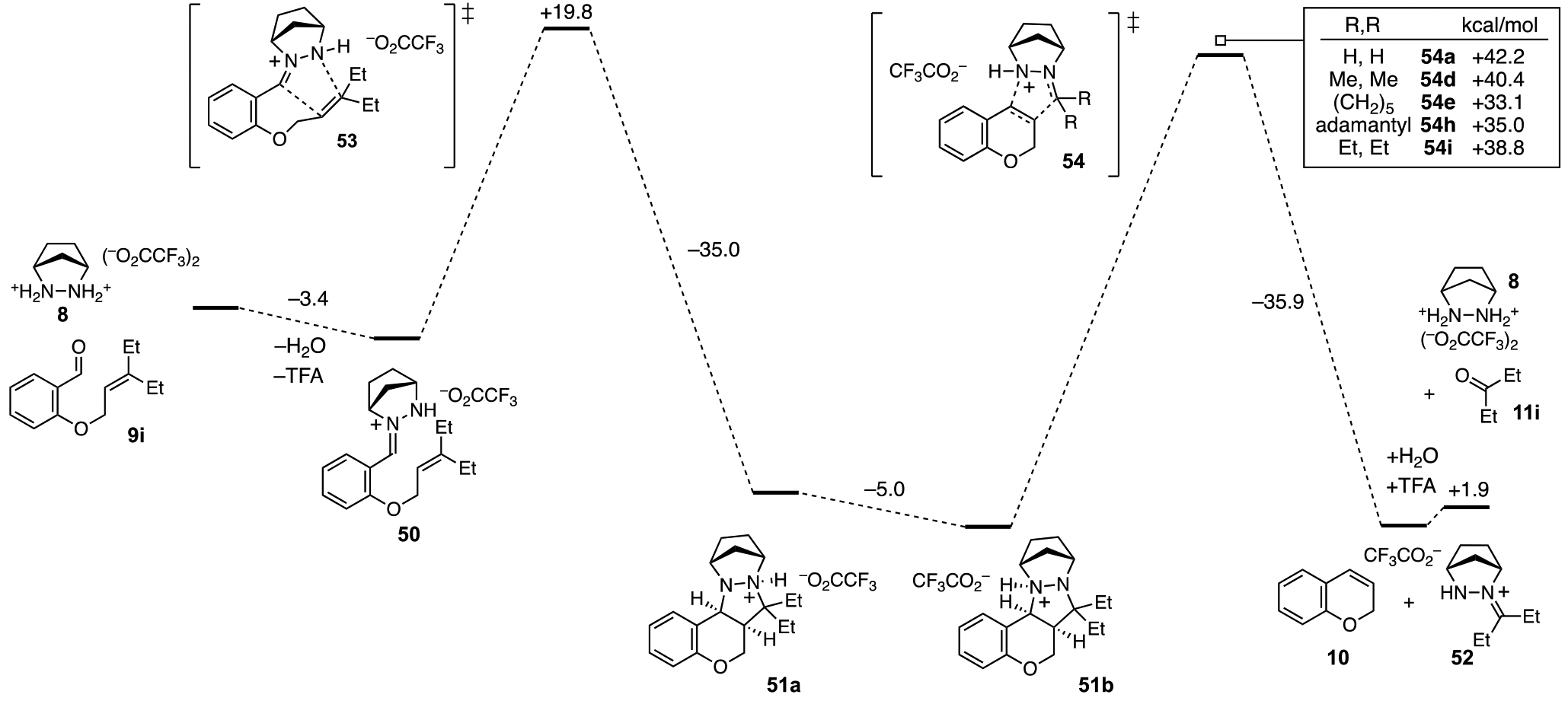
Calculated energies for hydrazine 8-catalyzed RCCOM of salicylaldehyde ether 9i. Energies were calculated at the M06-2X/6-311+G(d,p)/PCM(ethanol) level of theory. All energies are given in kcal/mol.
In further support of this analysis, we were able to prepare the cycloadduct 51b in 90% yield by combining an equimolar mixture of salicylaldehyde ether 9i and hydrazine 8•(TFA)2 in EtOH at 80 °C (Scheme 2). The molecular structure of this cycloadduct obtained by X-ray crystallography confirms the expected stereochemistry.20 When this material was heated to 140 °C in EtOH for 2 h, 2H-chromene (10) was produced in 75% isolated yield, thus confirming that the cycloadduct 51b is a viable intermediate for this catalytic procedure. We note that the gem-diethyl group of cycloadduct 51 adopts a classic syn-pentane minimized conformation (blue arrow). We speculate that in doing so, this structural feature exerts a steric pressure (red arrows) that facilitates cycloreversion. Such steric pressure could rationalize the even greater impact of the adamantylidene group as well.
Scheme 2.
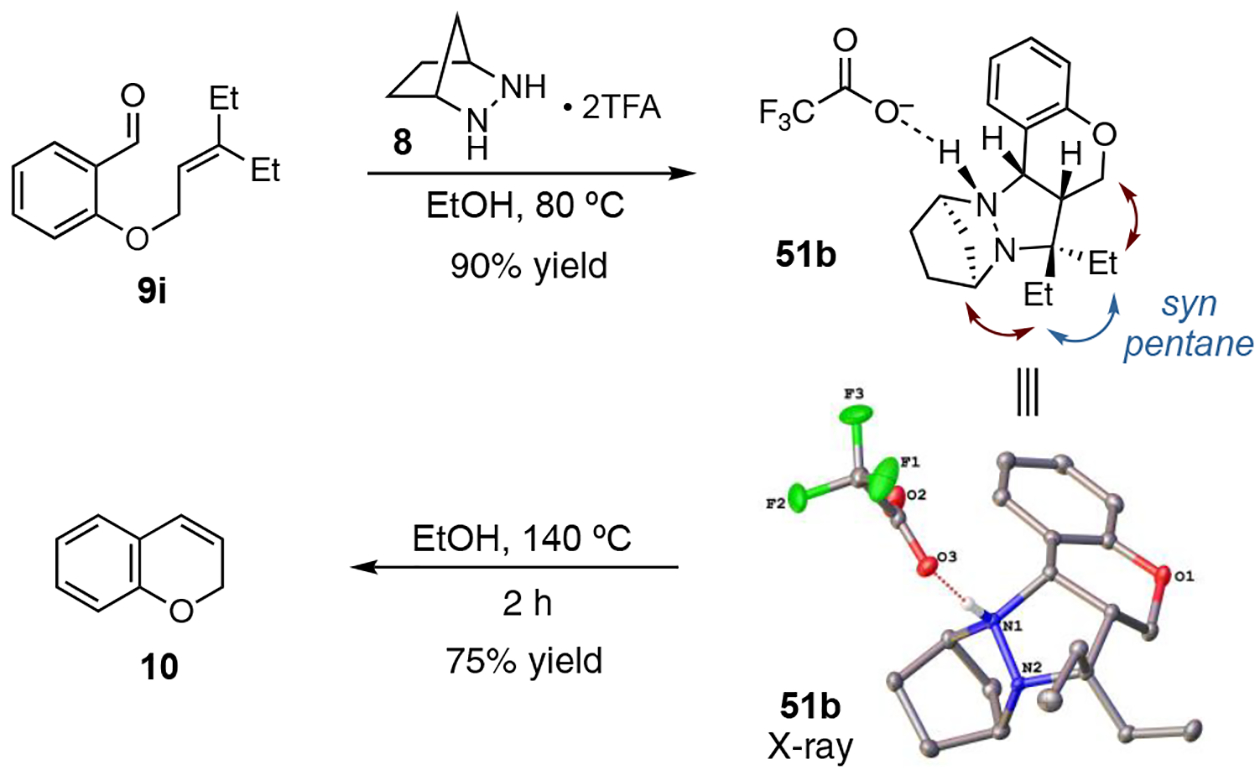
Synthesis, X-ray structure, and cycloreversion of cycloadduct 51b. Atoms are shown with an ellipsoid probability of 50%, and all hydrogen atoms except for the one bound to N1 have been omitted for clarity.
The current method offers an efficient new approach to synthesize 2H-chromenes from readily available salicylaldehyde ethers. More broadly, this work advances the use of hydrazine catalysis for carbonyl-olefin metathesis to the context of ring-closing reactions. The optimal behavior of sterically demanding groups for these reactions provides important guidance for the pursuit of other RCCOM methods.
Supplementary Material
Acknowledgement:
Financial support for this work was provided by NIH R35 GM127135.
Footnotes
Supporting Information Available: Experimental procedures and product characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pratap R; Ram VJ Natural and Synthetic Chromenes, Fused Chromenes, and Versatility of Dihydrobenzo[h]chromenes in Organic Synthesis. Chem. Rev 2014, 114, 10476–10526. [DOI] [PubMed] [Google Scholar]
- 2. For example:; (a) Rochfort S; Metzger R; Hobbs L; Capon R New Chromenols From a Southern Australian Tunicate, Aplidium solidum. Aust. J. Chem 1996, 49, 1217–1219. [Google Scholar]; (b) Covington AD Modern Tanning Chemistry. Chem. Soc. Rev 1997, 26, 111–126. [Google Scholar]; (c) Jankun J; Selman SH; Swiercz R; Skrzypczak-Jankun E Why Drinking Green Tea Could Prevent Cancer. Nature 1997, 387, 561–561. [DOI] [PubMed] [Google Scholar]; (d) Rensburg H. v.; Heerden P. S. v.; Bezuidenhoudt BCB; Ferreira D Enantioselective Synthesis of the Four Catechin Diastereomer Derivatives. Tetrahedron Lett. 1997, 38, 3089–3092. [Google Scholar]
- 3.Bauer DJ; Selway JWT; Batchelor JF; Tisdale M; Caldwell IC; Young DAB 4’,6-Dichloroflavan (BW683C), a new anti-rhinovirus compound. Nature 1981, 292, 369–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Monache FD; Labbiento L; Marta M; Lwande W 4β-Substituted Flavans from Tephrosia Hildebrandtii. Phytochemistry 1986, 25, 1711–1713. [Google Scholar]; (b) Machocho AK; Lwande W; Jondiko JI; Moreka LVC; Hassanali A Three New Flavanoids from the Root of Tephrosia Emoroides and Their Antifeedant Activity Against the Larvae of the Spotted Stalk Borer Chilo Partellus Swinhoe . Int. J. Pharmacogn 1995, 33, 222–227. [Google Scholar]
- 5.Yasunaga T; Kimura T; Naito R; Kontani T; Wanibuchi F; Yamashita H; Nomura T; Tsukamoto S.-i.; Yamaguchi T; Mase T Synthesis and Pharmacological Characterization of Novel 6-Fluorochroman Derivatives as Potential 5-HT1A Receptor Antagonists. J. Med. Chem 1998, 41, 2765–2778. [DOI] [PubMed] [Google Scholar]
- 6. For a review on 2H-chromene synthesis, see:; Majumdar N; Paul ND; Mandal S; Bruin B. d.; Wulff WD Catalytic Synthesis of 2H-Chromenes. ACS Catal. 2015, 5, 2329–2366. [Google Scholar]
- 7.Chang S; Grubbs RH A Highly Efficient and Practical Synthesis of Chromene Derivatives Using Ring-Closing Olefin Metathesis. J. Org. Chem 1998, 63, 864–866. [DOI] [PubMed] [Google Scholar]
- 8.Bera K; Sarkar S; Biswas S; Maiti S; Jana U Iron-Catalyzed Synthesis of Functionalized 2H-Chromenes via Intramolecular Alkyne-Carbonyl Metathesis. J. Org. Chem 2011, 76, 3539–3544. [DOI] [PubMed] [Google Scholar]
- 9. Salicylaldehydes can be prepared by the ortho formylation of phenols.; Olah GA; Ohannesian L; Arvanaghi M Formylating Agents. Chem. Rev 1987, 87, 671–686. [Google Scholar]
- 10.Rohlmann R; Daniliuc C-G; Mancheño OG Highly Enantioselective Synthesis of Chiral 7-Ring O- and N-Heterocycles by a One-Pot Nitro-Michael–Cyclization Tandem Reaction. Chem. Commun 2013, 49, 11665–11667. [DOI] [PubMed] [Google Scholar]
- 11.Griffith AK; Vanos CM; Lambert TH Organocatalytic Carbonyl-Olefin Metathesis. J. Am. Chem. Soc 2012, 134, 18581–18584. [DOI] [PubMed] [Google Scholar]
- 12. For reviews of carbonyl-olefin metathesis, see:; (a) Lambert TH Development of a Hydrazine-Catalyzed Carbonyl-Olefin Metathesis Reaction. Synlett, 2019, DOI: 10.1055/s-0039-1689924. [DOI] [Google Scholar]; (b) Ludwig JR; Schindler CS Lewis Acid Catalyzed Carbonyl–Olefin Metathesis. Synlett 2017, 28, 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ravindar L; Lekkala R; Rakesh KP; Asiri AM; Marwani HM; Qin H-L Carbonyl–Olefin Metathesis: A Key Review. Org. Chem. Front 2018, 5, 1381–1391. [Google Scholar]; (d) Groso EJ; Schindler CS Recent Advances in the Application of Ring-Closing Metathesis for the Synthesis of Unsaturated Nitrogen Heterocycles. Synthesis 2019, 51, 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becker MR; Watson RB; Schindler CS Beyond Olefins: New Metathesis Directions for Synthesis. Chem. Soc. Rev 2018, 47, 7867–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludwig JR; Zimmerman PM; Gianino JB; Schindler CS Iron(III)-Catalysed Carbonyl–Olefin Metathesis. Nature 2016, 533, 374–379. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ma L; Li W; Xi H; Bai X; Ma E; Yan X; Li Z FeCl3-Catalyzed Ring-Closing Carbonyl–Olefin Metathesis. Angew. Chem. Int. Ed 2016, 55, 10410–10413. [DOI] [PubMed] [Google Scholar]; (b) McAtee CC; Riehl PS; Schindler CS Polycyclic Aromatic Hydrocarbons via Iron(III)-Catalyzed Carbonyl–Olefin Metathesis. J. Am. Chem. Soc 2017, 139, 2960–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Groso EJ; Golonka AN; Harding RA; Alexander BW; Sodano TM; Schindler CS 3-Aryl-2,5-Dihydropyrroles via Catalytic Carbonyl-Olefin Metathesis. ACS Catal. 2018, 8, 2006–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ni S; Franzén J Carbocation Catalysed Ring Closing Aldehyde–Olefin Metathesis. Chem. Commun 2018, 54, 12982–12985. [DOI] [PubMed] [Google Scholar]; (e) Tran UPN; Oss G; Breugst M; Detmar E; Pace DP; Liyanto K; Nguyen TV Carbonyl–Olefin Metathesis Catalyzed by Molecular Iodine. ACS Catal. 2018, 912–919. [Google Scholar]; (f) Tran UPN; Oss G; Pace DP; Ho J; Nguyen TV Tropylium-Promoted Carbonyl–Olefin Metathesis Reactions. Chem. Sci 2018, 9, 5145–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Albright H; Riehl PS; McAtee CC; Reid JP; Ludwig JR; Karp LA; Zimmerman PM; Sigman MS; Schindler CS Catalytic Carbonyl-Olefin Metathesis of Aliphatic Ketones: Iron(III) Homo-Dimers as Lewis Acidic Superelectrophiles. J. Am. Chem. Soc 2019, 141, 1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catti L; Tiefenbacher K Brønsted Acid-Catalyzed Carbonyl-Olefin Metathesis inside a Self-Assembled Supramolecular Host. Angew. Chem. Int. Ed 2018, 57, 14589–14592. [DOI] [PubMed] [Google Scholar]
- 17.Hong X; Liang Y; Griffith AK; Lambert TH; Houk KN Distortion-Accelerated Cycloadditions and Strain-Release-Promoted Cycloreversions in the Organocatalytic Carbonyl-Olefin Metathesis. Chem. Sci 2014, 5, 471–475. [Google Scholar]
- 18.Lambert TH; Griffith AK 2,3-Diazabicyclo[2.2.1]heptane. Encyclopedia of Reagents for Organic Synthesis 2014, doi: 10.1002/047084289X.rn01779. [DOI] [Google Scholar]
- 19.Collins KD; Glorius F Intermolecular Reaction Screening as a Tool for Reaction Evaluation. Acc. Chem. Res 2015, 48, 619–627. [DOI] [PubMed] [Google Scholar]
- 20.Crystallographic data for compound 51b are available free of charge from the Cambridge Crystallographic Data Centre under CCDC 1919817.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.