

DRM2 induces substrate deformation to underlie its methylation preference for CHH DNA in plants.
Abstract
DNA methylation is a major epigenetic mechanism critical for gene expression and genome stability. In plants, domains rearranged methyltransferase 2 (DRM2) preferentially mediates CHH (H = C, T, or A) methylation, a substrate specificity distinct from that of mammalian DNA methyltransferases. However, the underlying mechanism is unknown. Here, we report structure-function characterization of DRM2-mediated methylation. An arginine finger from the catalytic loop intercalates into the nontarget strand of DNA through the minor groove, inducing large DNA deformation that affects the substrate preference of DRM2. The target recognition domain stabilizes the enlarged major groove via shape complementarity rather than base-specific interactions, permitting substrate diversity. The engineered DRM2 C397R mutation introduces base-specific contacts with the +2-flanking guanine, thereby shifting the substrate specificity of DRM2 toward CHG DNA. Together, this study uncovers DNA deformation as a mechanism in regulating the specificity of DRM2 toward diverse CHH substrates and illustrates methylome complexity in plants.
INTRODUCTION
DNA methylation at cytosines is an evolutionarily conserved epigenetic mechanism that is required for gene expression and genome stability (1–3). Dysregulation of DNA methylation leads to developmental defects and various diseases in animals, most notably cancer and pleiotropic developmental defects in plants (4–6), highlighting an essential role for DNA methylation in both kingdoms (3, 7). Nevertheless, the mechanisms of DNA methylation have diverged between plants and animals (3). In animals, de novo DNA methyltransferases DNMT3A and DNMT3B primarily mediate methylation of CpG dinucleotides (8, 9), with appreciable levels of CH (H = A, T, or C) methylation identified in oocytes, embryonic stem cells, and neural cells (10). Subsequently, CG methylation is maintained by DNA methyltransferase 1 (DNMT1) in a replication-dependent manner (11). In contrast, DNA methylation in plants is prevalent in all sequence contexts: CG, CHG, and CHH (3). Domains rearranged methyltransferase 2 (DRM2) mediates the establishment of DNA methylation in all three sequence contexts, whereas plant DNA methyltransferase 1 (MET1) and chromomethylase 3 (CMT3) maintain CG and CHG methylation, respectively (12–14). chromomethylase 2 (CMT2) and DRM2 are jointly responsible for maintaining CHH methylation in long heterochromatic transposable elements (TEs) and short euchromatic TEs, respectively (15). However, the molecular mechanism underlying the divergent methylation patterns between plants and animals remains unclear.
DRM2-mediated methylation is achieved through an RNA-directed DNA methylation pathway, which involves the biogenesis and enrichment of 24-nucleotide small interfering RNAs, mostly at short TEs and edges of long TEs (16). Targeting DRM2 to specific genomic loci depends on many factors, including small RNAs, long noncoding RNAs, histone modifications, and the action of DRM2-interacting proteins (17–20). Emerging evidence has implicated local chromatin environment and the sequence context of DNA substrates in regulating DRM2-mediated methylation. For instance, genome-wide methylation analysis has revealed strong context-dependent methylation in Arabidopsis, with a >900-fold difference between the highest and lowest levels of CHH methylation in the 7-mer sequence context (21). In the example of nucleotide repeats (CCCTAAA)n, the third cytosine has a greater proportion of methylation than the cytosines in the first and second positions (21), showing a CHH subcontext specificity. Genomic meta-analysis has also shown that certain trinucleotide contexts, such as CAA and CTA, have a greater methylation frequency than others (e.g., CCC and CCT) (22), supporting a role for sequence context in shaping genomic methylation.
Structures of the DNA methyltransferase-substrate complexes from bacteria and mammals have been reported (23–28), providing insights into their sequence-specific DNA methylation. The methyltransferase domains of these evolutionarily diverse enzymes are commonly composed of a catalytic core and a target recognition domain. While the catalytic core is highly conserved throughout evolution and harbors the active site, the target recognition domain is divergent in both sequence and structure and serves as an essential element for sequence-specific substrate recognition (29, 30). Distinct from DNA methyltransferases that recognize substrates with a specified DNA sequence, DRM2 and its paralog DRM1 are active on DNA substrates with diverse sequence contexts (31, 32), with a preference for CHH and CHG over CG substrates (32, 33). Consistently, the structure of tobacco DRM2 (NtDRM) in apo form reveals a structurally conserved catalytic core but a unique target recognition domain (19). How DRM2 interplays with substrate sequences for CHH methylation remains unknown.
To elucidate the molecular basis of DRM2-mediated CHH methylation, we performed comprehensive structural characterizations of DRM2-substrate complexes and functional validation analysis in vivo. Residue R595 from the catalytic core intercalates into the nontarget strand, resulting in large DNA deformation. The target recognition domain stabilizes the deformed DNA major groove via shape complementarity rather than the canonical, base-specific interaction mechanism observed for DNMT3A and other DNA methyltransferases. Biochemical and genome-wide methylation analyses reveal that this DNA deformation mechanism limits DRM2 from methylating CG DNA, whereas it permits its high methylation efficiency on targets with AT-rich flanking sequences populated in TEs. Substitution of residue C397 with arginine introduces the base-specific contacts between the target recognition domain and the +2-flanking guanine of the CHG motif, thereby shifting the substrate preference of DRM2 toward the CHG DNA sequence context and consequently reshaping the genome-wide DNA methylation patterns. Collectively, this study identified a previously unknown substrate recognition paradigm for DNA methylation, underpinned by DNA deformation, with strong implications in sequence-specific DNA methylation establishment and maintenance in plants.
RESULTS
Crystal structure of the DRM2-CHH DNA complex reveals substrate deformation
To understand how DRM2 mediates CHH methylation, we determined the crystal structure of DRM2 in complex with CHH DNA, formed by the methyltransferase domain of DRM2 from Arabidopsis thaliana (Fig. 1A and fig. S1A) and an 18-mer, AT-rich DNA duplex harboring a central CTT motif, in which the cytosine was replaced by a 5-fluorocytosine (Fig. 1B). Introduction of the 5-fluorocytosine into the DNA substrate permits the formation of a stable, covalent complex between DRM2 and DNA, as described previously (27, 34). The crystal structure of the DRM2-CTT complex bound to the S-adenosyl-homocysteine (SAH) was solved at 2.1 Å resolution (Fig. 1, C and D, and table S1).
Fig. 1. Structure of DRM2 in complex with an 18-mer CTT DNA.
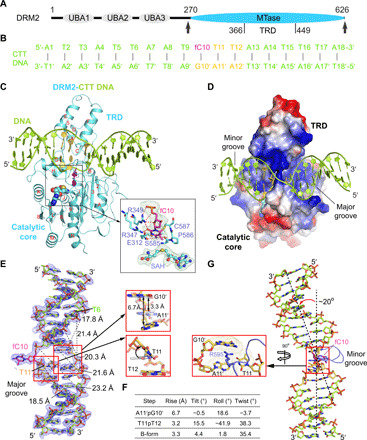
(A) Domain architecture of DRM2, with the methyltransferase (MTase) domain that harbors a target recognition domain (TRD) marked with arrowheads. UBA, ubiquitin-associated domain. (B) The sequence of CTT DNA used for the structural study. fC, 5-fluorocytosine. (C and D) Ribbon (C) and electrostatic surface (D) representations of DRM2 bound to DNA and SAH. DRM2 and bound DNA are colored in aquamarine and limon, respectively. The CTT motif is colored in yellow or purple (fC10). The SAH molecule is shown in sphere representation. The active site is shown in expanded view, with the Fobs-Fcalc omit map (cyan) of fC10 and SAH contoured at 2.0 σ level and hydrogen-bonding interactions depicted as dashed lines. The α helices and β strands are counted in alphabetic and numeric orders, respectively, in (C). The color scheme in (C) is applied to subsequent figures, unless otherwise indicated. (E) The Fobs-Fcalc omit map (blue) of the CTT DNA, contoured at 2.0 σ level. The major groove widths of the deformed DNA upon binding of DRM2 are indicated by dashed lines. Structural alignments of the A11′pG10′ and T11pT12 steps with B-form DNA (gray) are shown in expanded views. (F) Geometric parameters for the DNA base steps boxed in (E). (G) Kinked conformation of DRM2-bound DNA, with the R595 intercalation shown in expanded view.
We were able to trace the entire methyltransferase domain of DRM2 and the DNA molecule. DRM2 is composed of a catalytic core adopting a Rossmann fold and a target recognition domain (Fig. 1C and fig. S1B), as previously observed for DNA-free NtDRM (19). The DNA duplex is embedded in the cleft formed by the target recognition domain and catalytic core of DRM2, resulting in ~1747 Å2 of buried surface area (Fig. 1D). The target 5-fluorocytosine, fC10, breaks its Watson-Crick base pairing with Gua10′ and inserts into the catalytic pocket of DRM2, where it is trapped through covalent linkage with the catalytic cysteine C587 and hydrogen-bonding interactions with other catalytic residues (Fig. 1C). Comparison of DNA-bound DRM2 with DNA-free NtDRM reveals a notable structural difference in the C587-containing catalytic loop (residues 584 to 598), which is disordered in DNA-free NtDRM but well defined in DNA-bound DRM2 (fig. S1C), indicating a DNA binding–induced folding. In comparison with B-form DNA, the DRM2-bound DNA undergoes a large unwinding (Fig. 1, E to G, and fig. S1D), showing increased interstrand distances at the segment spanning from Thymine 6 (Thy6) to Thy11 (Fig. 1E). Most notably, the side chain of R595 on the catalytic loop intercalates into the base step between unpaired Gua10′ and the +1-flanking Ade11′ of the nontarget strand (Fig. 1G), which increases the helical rise of the Ade11′-Gua10′ step by 3.4 Å [6.7 Å for Ade11′-Gua10′ versus 3.3 Å for B-form DNA in Fig. 1 (E and F)] and kinks the DNA by ~20° (Fig. 1, E to G). The R595-mediated DNA intercalation also introduces a large roll (−41.9°) and tilt (15.5°) to the +1 fC10-flanking nucleotide, Thy11 (Fig. 1, E and F), which increases the propeller twist of the Thy11·Ade11′ pair by ~23° (8.8° for Thy11·Ade11′ versus –14.5° for B-form DNA), leading to a reduced base stacking of the Thy11-Thy12 step (fig. S1D).
Interaction between DRM2 and CHH DNA
The interaction between DRM2 and CTT DNA involves both the major and minor grooves, spanning 13 base pairs (bp; Fig. 2, A and B). The minor groove is contacted by a subset of residues from the catalytic loop and the loop harboring the rearranged motif IV (E312-N313-V314; rearranged loop: residues 312 to 320; Fig. 2B) (35). The major groove is embraced by a loop–helix (αE)–helix (αF) (LHH) motif and helix αH from the target recognition domain, both of which span the widened DNA strands (Fig. 2B).
Fig. 2. Intermolecular interactions between DRM2 and DNA.
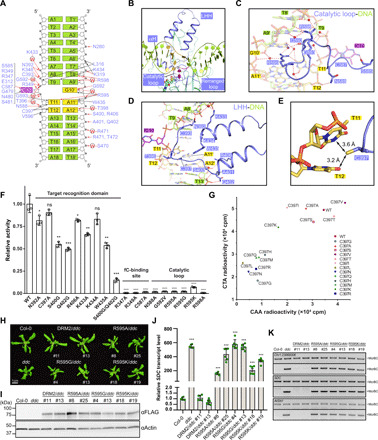
(A) Schematic view of the intermolecular interactions between DRM2 and CTT DNA. The hydrogen-bonding and van der Waals contacts are represented by red and black arrows, respectively. Water-mediated hydrogen bonds are labeled with the letter “W”. (B) Close-up view of the DNA-binding regions of DRM2, colored in slate. (C and D) Close-up views of the DNA interactions of the catalytic loop (C) and LHH (D). The hydrogen-bonding interactions are shown as dashed lines. (E) Close-up view of the DNA interactions of DRM2 C397. The van der Waals radii of the side chain of C397 and the DNA bases are indicated by dots. (F) In vitro methylation assay of wild-type (WT) and mutant DRM2 on the (TAC)12 DNA duplex. n = 3 replicates; Data are means ± SD. Statistical analysis used two-tailed Student’s t test for the difference from WT: ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (G) In vitro methylation assay of DRM2, WT, or C397 mutants, on two CHH DNA duplexes. CTA, (TAC)12; CAA, (AAC)12. (H) Phenotypes of 3-week-old plants. Col-0 (Columbia-0) and ddc (drm1 drm2 cmt3) serve as controls. (I) Western blot of FLAG-tagged DRM2 proteins from the same lines listed in (H). (J) Reverse transcription quantitative polymerase chain reaction (RT-qPCR) of SDC relative transcript level. Data are means ± SD. Statistical analysis used two-tailed Student’s t test for the difference from Col-0. ***P < 0.001. (K) McrBC digestion of three DRM2 target sites. “−McrBC” represents no enzyme and serves as a control.
Toward the minor groove, the catalytic loop extends into the DNA cavity vacated by base flipping, with R595 engaging in DNA intercalation with Gua10′ and Ade11′ on the nontarget strand (Fig. 2C), as described above. Stacking of R595 with the unpaired Gua10′ is further supported by a network of water-mediated hydrogen bonds bridging the guanidium group of R595 with the backbone and side chain of Gua10′ (Fig. 2C). In addition, the Nη2 atom of R595 and the O4 atom of Thy11 are within a distance (3.4 Å) that permits hydrogen bond formation (Fig. 2C). The unpaired Gua10′ is further stabilized by a direct hydrogen bond between its N2 atom and the backbone carbonyl of G592 (Fig. 2C). On the rearranged loop, residue E312 forms a hydrogen bond with fC10, while residue K319 interacts with the backbone phosphates of the nontarget strand (T7′ and A6′) through hydrogen bonding and electrostatic contacts (fig. S1E). The rearranged loop also inserts residue L316 into the center of the minor groove to interact with the backbone of both DNA strands through van der Waals contacts (fig. S1E). Additional contacts between the catalytic core and DNA involve the N-terminal loop and β7, which interact with the DNA backbone via hydrogen-bonding interactions (fig. S1F).
Toward the major groove, the LHH motif extends along the target strand and then diverts by ~90° at Thy11 to approach the nontarget strand (Fig. 2D), resulting in an L-shaped conformation that complements well with the shape of the deformed DNA (Figs. 1D and 2D). Consequently, the LHH motif engages both DNA strands for polar and nonpolar interactions, involving residues from the two α helices (S400, A401, Q402, R406, K433, K434, and W435) and the preceding loop (N392, C393, and T396) (Fig. 2D). Among these, the sulfhydryl group of DRM2 C397 is in a position for van der Waals contacts with the base rings of Thy11 and Thy12 (Fig. 2, D and E). Next to the LHH motif, helix αH spans both DNA strands, with its N-terminal end (S470-T472) hydrogen bonded to the backbone of the nontarget strand and its C-terminal end (G479-S481) engaging in van der Waals contacts with fC10 (fig. S1G).
We mutated key DNA-contacting residues for enzymatic assays on CHH DNA. Mutation of the catalytic loop residues or the fC10-binding site largely abolishes the activity of DRM2 (Fig. 2F). Mutation of single target recognition domain residues leads to a modest reduction of the enzymatic activity of DRM2, whereas the introduction of multisite mutations (e.g., S400G/Q402G) severely impairs its activity (Fig. 2F). These data suggest that the catalytic loop plays an essential role in enzymatic catalysis, while the residues within target recognition domain collectively stabilize substrate deformation.
Adaption of C397 to DRM2-mediated DNA methylation
The close proximity between DRM2 C397 and the +1- to +2-flanking bases (Fig. 2E) coincides with the fact that DRM2 proteins from diverse plant species universally contain a small residue (e.g., C, A, or V) on the corresponding site (fig. S2A). Through mutation of C397 into differently sized amino acids, we observe that the activity of DRM2 on CHH substrates (CTA and CAA in Fig. 2G) largely falls into a trend that the size of amino acid replacement inversely correlates with the activity, with the naturally occurring C397A and C397V substitutions corresponding to the high-activity group (Fig. 2G). Note that a large reduction of the C397 side chain, as shown by the C397G mutant, also severely impairs the DRM2 activity. These observations support the notion that the C397-corresponding site of DRM2 has been evolutionarily adapted for efficient DNA methylation in plants.
Role of R595 in DRM2-mediated DNA methylation
To examine the role of R595 intercalation in DRM2-mediated DNA methylation, we compared the enzymatic activities of wild-type and R595-mutated DRM2 in vitro. The R595G and R595A mutations largely abolish the activities of DRM2 in all sequence contexts (Fig. 2F and fig. S2, B to D). Unexpectedly, even the R595K mutation, which minimally perturbs the side chain of R595, leads to a severe reduction of the methylation efficiency on CHH, CHG, and CG DNA (Fig. 2F and fig. S2, B to D), confirming the critical role of R595 side chain in the enzymatic activity of DRM2. We further introduced wild-type or R595-mutated DRM2 into the drm1 drm2 cmt3 (ddc) triple knockout mutant, which shows a global reduction in CHH and CHG methylation and a curled leaf phenotype due to the reactivation of suppressor of DRM1 DRM2 CMT3 (SDC) (36). SDC has seven tandem repeats in its promoter and is silent in the wild-type plants but becomes demethylated and transcriptionally reactivated when both DRM2 and CMT3 pathways are inactivated (36). Compared to the wild-type DRM2 transgene that rescues the curled leaf phenotype of ddc, R595G, R595A, and R595K all fail to rescue (Fig. 2H), despite having similar overall protein levels (Fig. 2I). Consistently, R595A, R595G, and R595K show similarly elevated SDC transcript levels to ddc (Fig. 2J). We next examined the DNA methylation levels of SDC and two other DRM2 targets by digestion with McrBC, which cuts DNA in a methylation-dependent manner. We found similar amplification levels in R595A, R595G, R595K, and ddc at all these loci (Fig. 2K), indicating that these regions lack DNA methylation. These data highlight the importance of R595 for DRM2 activity, in line with its strict sequence conservation among plant species (fig. S2A).
Substrate deformation regulates the enzymatic preference of DRM2
To elucidate how the interaction between DRM2 and substrates interplays with DNA sequence, we further solved the structures of DRM2 complexed with CTG, CCG, CAT, and CCT DNAs (Fig. 3A and table S1). The CHG DNAs (CTG and CCG) were derived from the CTT DNA by introducing three or four additional C·G base pairs, while the new CHH DNAs (CAT and CCT) were derived from the CTT DNA by introducing six or seven additional C·G pairs (Fig. 3B). These DRM2-DNA complexes were crystallized in two different fashions, with DRM2-CCT and DRM2-CAT belonging to space group C2 and DRM2-CTT, DRM2-CTG, and DRM2-CCG belonging to the space group C2221. Nevertheless, the structures of DRM2-CCT, DRM2-CAT, DRM2-CCG, and DRM2-CTG complexes reveal highly conserved protein-DNA interactions, including R595-mediated DNA intercalation (figs. S3 to S5). Among these DRM2-CHH/CHG complexes, the +1-flanking base pairs undergo substantial deviations from coplanarity, resulting in similar DNA deformation around the CHH/CHG motif (Fig. 3, A to D, and fig. S5). For comparison, we also generated a structural model of DRM2 with CG-containing DNA (fig. S3, F and G), on the basis of the structure of the DRM2-CAT complex. The structural model of the DRM2-CG complex indicates a similar R595-DNA intercalation mechanism (fig. S4F). These observations, therefore, suggest that DRM2-induced substrate deformation occurs for all sequence contexts, involving disruption of base-stacking interactions between the target C·G pair and the +1-flanking site on both strands and an impairment of stacking between the +1- and +2-flanking sites on the target strand.
Fig. 3. Shape analysis of DRM2-bound DNAs.
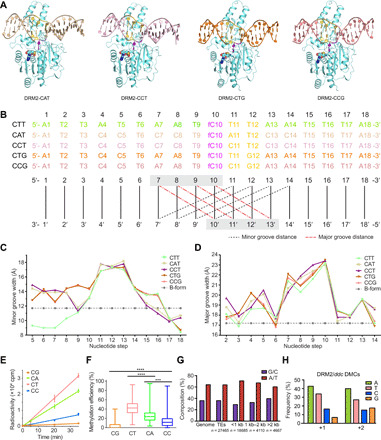
(A) Crystal structures of DRM2 in complex with CAT, CCT, CTG, and CCG DNAs, respectively. (B) DNA sequences of the target strand for methylation in each structure. Minor groove widths (dashed lines in black) and major groove widths (dashed lines in red) are determined by measuring the cross-strand distances of the two phosphate groups of the nucleotides at indicated positions, as illustrated in the schematic below the DNA sequences. (C and D) DNA sequence–dependent minor groove widths (C) and major groove widths (D) of the DRM2-bound DNAs in the DRM2-CTT, DRM2-CAT, DRM2-CCT, DRM2-CTG, and DRM2-CCG complexes. Dashed lines show the canonical groove widths for B-form DNA. (E) In vitro methylation kinetics of DRM2 on the 30-mer DNA containing a single CG, CA, CC, or CT site. Data are means ± SD (n = 3 replicates). (F) Box plot comparing the methylation efficiencies of DRM2 on the CG/CH sites of a 637-bp DNA fragment based on bisulfite sequencing analysis, with 25 to 75% in the box, entire range for the whiskers, and median indicated. In total, 56 clones, including 28 for the upper strand and 28 for the lower strand, were analyzed. Two-tailed Student’s t tests were performed to compare distributions between different groups. ***P < 0.001 and ****P < 0.0001. (G) Histogram showing the percent A/T versus G/C composition of the Arabidopsis genome in comparison to all TEs and TEs of different sizes. (H) Quantification of the nucleotide frequency of the first base pair downstream (+1) and the second base pair downstream (+2) of all hyper differentially methylated cytosines (DMCs) called against ddc.
The base-stacking interactions are knowingly dependent on DNA sequence (37, 38). We therefore asked whether the sequence composition of the flanking sequence affects the activity of DRM2. We first interrogated the enzymatic activity of DRM2 on a 30-bp AT-rich DNA duplex harboring a central CG, CC, CT, or CA motif in vitro. DRM2 is highly active on CT- and CA-containing DNAs but least active on CG-containing DNA (Fig. 3E). This observation, consistent with a previous report that DRM2 prefers methylation of CHH and CHG sites over CG sites (32), correlates well with the order of the base-stacking interactions among the four sequences (fig. S6A) (37). Next, we measured the activity of DRM2 on a 637-bp DNA containing multiple CG, CC, CT, and CA sites via bisulfite sequencing (Fig. 3F and fig. S6, B to D). Analysis of the methylated substrates, with the overall methylation efficiency ranging from 25% to 29%, again indicates that DRM2 is highly efficient on AT-rich regions (fig. S6B) but least efficient on the CG sites (Fig. 3F and fig. S6C). Consistently, analysis of the Arabidopsis genome reveals that TEs, including small TEs that were previously identified as the DRM2 targets (19), are AT rich in sequence composition (Fig. 3G). Furthermore, inspection of the hyper differentially methylated cytosines for DRM2-complemented ddc reveals that G is the least frequent nucleotide at the +1- and +2-flanking positions (Fig. 3H). Together, these observations support a role for base-stacking interactions in the substrate preference of DRM2. In addition, the packing between R595 side chain and the +1 base on the nontarget strand (fig. S4, A to F), as well as the differential DNA deformability of AT and GC sequences (39, 40), may also shape the flanking sequence preference of DRM2.
To interrogate the role of the R595-mediated DNA deformation on DRM2 activity in vivo, we performed whole-genome bisulfite sequencing on R595K/ddc and wild-type DRM2/ddc transgenic plants. We found greatly reduced CHG and CHH methylation levels in R595K/ddc compared to DRM2/ddc (fig. S7A). Inspection of the methylation distribution over TEs, which are the preferential target sequences of DRM2 in vivo (15, 16), also reveals a severe loss of CHH methylation in R595K/ddc as that in ddc (fig. S7B). Furthermore, we determined the number of differentially methylated cytosines induced by R595K against ddc and identified 1180 CG, 18 CHG, and 107 CHH hyper differentially methylated cytosines mediated by R595K, which are much lower than those of DRM2 (6792 CG, 16,087 CHG, and 76,234 CHH; tables S2 and S3). Consequently, analysis of the flanking sequence of hyper differentially methylated cytosines mediated by DRM2 indicates that the R595K mutation leads to an A/T-to-G shift at the +1-flanking position (fig. S7, C and D). The mechanistic factors underlying this +1-flanking nucleotide shift of differentially methylated cytosines remain to be determined. Nevertheless, these results suggest an important role of R595 in DRM2-mediated CHH methylation, reinforcing the notion that R595-induced DNA deformation underlies the differential substrate preference of DRM2 for DNAs with various +1- and +2-flanking nucleotides.
DNA sequence flanking the CHH/CHG motif fine-tunes the DRM2-DNA interaction
Structural analysis of the DRM2-CHH/CHG complexes also reveals that the DNA conformation beyond the CHH/CHG motif is sequence dependent (Fig. 3, B to D). The conformation of CTT DNA differs from those of CCG and CTG only at the 5′ flanking region but differs from those of CCT and CAT at both flanking regions, in line with the fact that the sequence of the CTT differs from those of CCG/CTG DNAs only at 5′ flanking region and those of CCT/CAT DNAs at both 5′ and 3′ flanking regions (Fig. 3, B to D). Note that the changes in DNA conformations also lead to altered protein-DNA interactions outside the CHH/CHG motifs, involving the differential minor-groove contacts at the 5′ flanking region by residues in the N-terminal loop (N280) and rearranged loop (L316 and K319) (fig. S8A) and major-groove contacts at the 3′ flanking region by the LHH (S400, A401, Q402, and R406) and αH (S470-T472) (fig. S8B). These observations point to a role for DNA shape in fine-tuning the DRM2-DNA contact by the CHH/CHG-flanking sequence, supporting DNA shape as an important factor in regulating protein-DNA interactions (41).
Distinct substrate recognition mechanism between DRM2 and DNMT3A
Protein interaction–induced DNA deformation is recurrently observed for protein-DNA complexes (42), including bacterial and mammalian DNA methyltransferases (23–28). To identify how DRM2 diverges from other DNA methyltransferases in substrate recognition, we first compared the structure of DRM2-CTT with that of the human DNMT3A/DNMT3L-CGT DNA complex that we reported previously (28). The DRM2-CTT complex aligns well with the DNMT3A-CGT complex, with a root mean square deviation (RMSD) of 1.5 Å over 328 aligned Cα atoms (Fig. 4A). The two complexes show a similar catalytic loop conformation (Fig. 4, A to D) and partially aligned target recognition domain (Fig. 4, A and E to G). Nevertheless, they exhibit distinct modes of substrate recognition: DRM2 disrupts the nontarget strand through R595-mediated intercalation, while DNMT3A presents a smaller V716 to stack against the CpG guanine of the target strand, resulting in much less DNA deformation (Fig. 4, B to D, and table S4). Furthermore, the target recognition domains of DRM2 and DNMT3A interact with the major groove differently. In DNMT3A, target recognition domain residue R836 forms hydrogen bonds with the CpG guanine in the CGT-containing DNA (Fig. 4, E and G) (28) or the +1- to +3-flanking nucleotides in the CGA-containing DNA (43). In contrast, the structurally aligned DRM2 C397 does not form any base-specific hydrogen-bonding interaction with DNA substrates (Fig. 4, E and F). These differences in protein-DNA interaction lend an explanation to the fact that DNMT3A is highly specific for CG sites (28, 44), whereas DRM2 discriminates against CG sites (32). To further compare the effects of base-specific contacts on DRM2 and DNMT3A, we measured the activities of these two enzymes on modified CG DNA, in which the CpG guanine on the target strand is replaced with an abasic site. DNMT3A is highly active on the CG DNA but largely inactive on the DNA with an abasic site (fig. S8C), confirming the critical role of the base-specific interaction of the CpG guanine in DNMT3A activity (28). In contrast, the activity of DRM2 on the abasic site–containing DNA is only reduced by ~20% when compared to the unmodified form (fig. S8C), suggesting a less substantial role of the +1-flanking base-protein interaction on DRM2 activity.
Fig. 4. Structural comparison of the DRM2-DNA and DNMT3A-DNA complexes.
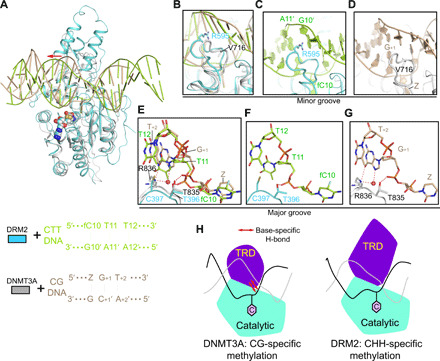
(A) Structural overlay of DRM2-CTT complex (cyan) and DNMT3A-CGT DNA complex (gray; Protein Data Bank: 5YX2). For clarity, only one DNMT3A molecule and associated DNA are shown. Z, cytosine analog zebularine. The differential major groove distortion between DRM2- and DNMT3A-bound DNAs is indicated by a red arrow. (B) Distinct catalytic loop–DNA contact between DRM2-CTT and DNMT3A-CGT complexes. (C) Close-up view of the R595-mediated intercalation in DRM2-CTT complex. (D) Close-up view of the V716-CG DNA contact in DNMT3A-CGT complex. (E) Structural comparison of the TRD loop–DNA contact between DRM2-CTT and DNMT3A-CGT complexes. Hydrogen bonds are shown as dashed lines. (F) Close-up view of the DNA contacts by T396 and C397 in the DRM2-CTT complex. (G) Close-up view of the DNA contacts by T835 and R836 in the DNMT3A-CGT complex. (H) Model for the distinct substrate recognition mechanisms between DRM2 and DNMT3A. The TRD of DNMT3A engages base-specific hydrogen-bonding interactions with CG site (left), whereas the TRD of DRM2 interacts with deformed major groove via shape complementarity (right), thereby accommodating substrate diversity. The target cytosine is shown as a pink hexagon.
Structural analysis of the enzyme-substrate complexes of other reported bacterial and mammalian DNA methyltransferases reveals that these enzymes all present the target recognition domain to engage in base-specific hydrogen-bonding interactions with their target DNA sequences (Fig. 4G and fig. S9). These base-specific interactions presumably provide a mechanism of energetic compensation for the DNA deformation–associated base pair disruption and/or rearrangements upon catalysis, thereby underpinning the sequence-specific DNA methyltransferase-substrate recognition (Fig. 4H). Together, these observations suggest that DRM2 has evolved with a unique substrate recognition mechanism for its preference for diverse CHH substrates (Fig. 4H).
The C397R mutation alters the substrate preference of DRM2
The observation that DNMT3A R836 differs from its structurally aligned DRM2 C397 in forming base-specific substrate contacts implies that this difference may partially account for the lack of a base-specific hydrogen-bonding interaction by the DRM2 target recognition domain. We therefore asked whether the replacement of C397 with an arginine would affect the substrate recognition and preference of DRM2. To address this, we first compared the activities of wild-type and mutant DRM2 via in vitro enzymatic assay. The C397R mutation leads to reduced enzymatic efficiency toward CHH DNA but a substantially increased activity toward CHG DNA (Fig. 5A). In contrast, mutation of DRM2 C397 into histidine (C397H) or alanine (C397A) does not change the relative methylation efficiency of DRM2 on CHH and CHG substantially: The C397H mutation decreases the methylation efficiency of DRM2 on CG, CHH, and CHG DNAs to a similar extent, whereas the C397A mutation does not affect the DRM2 activity appreciably (fig. S10A versus Fig. 5A). These data reinforce the notion that DRM2-mediated CHH methylation prefers small amino acids at the position of C397 (Fig. 2G and fig. S2A) and suggest that the C397R mutation alters the substrate recognition of DRM2.
Fig. 5. The C397R mutation boosts DRM2-mediated CHG methylation.
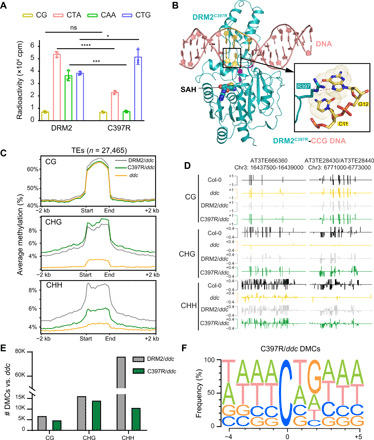
(A) In vitro methylation assay of DRM2 or C397R on DNA with different sequence contexts. CG, (GAC)12; CTA, (TAC)12; CAA, (AAC)12; CTG, (TGC)12. Data are means ± SD (n = 3 replicates). Statistical analysis used two-tailed Student’s t test for the difference from WT: *P < 0.05, ***P < 0.001, and ****P < 0.0001. (B) Ribbon representation of DRM2C397R bound to CCG DNA and SAH. Hydrogen-bonding interactions formed between the side chain of R397 and G12 are depicted as dashed lines in expanded view. The bases of C11 and G12 in the expanded view are colored yellow. (C) Metaplots showing average methylation level of DRM2, C397R, and ddc over TEs for CG, CHG, and CHH contexts. (D) Representative genomic regions of two TEs on chromosome 3 (AT3TE666360 and AT3TE28430/AT3TE28440) showing the methylation levels of Col-0, ddc, DRM2/ddc, and C397R/ddc. (E) Bar chart showing the total number of DMCs in each context of DRM2/ddc and C397R/ddc called against ddc. (F) Motif of the 4 nucleotides upstream and 5 nucleotides downstream of hyper DMCs in C397R called against ddc (n = 29,347).
To determine the mechanism by which the C397R mutation affects the substrate preference of DRM2, we solved the crystal structure of C397R DRM2 in complex with a CCG DNA (DRM2C397R-CCG) at 2.25 Å resolution (Fig. 5B and table S1). The structure of DRM2C397R-CCG aligns well with the DRM2-CCG complex, with an RMSD of 0.15 Å over 329 aligned Cα atoms (fig. S10B). Nevertheless, we observe distinct protein interactions involving the +2 guanine (Gua12) between the two complexes. Unlike the DRM2-CCG complex where Gua12 only engages in van der Waals contact with C397 (fig. S4H), the DRM2C397R-CCG complex involves base-specific hydrogen-bonding interactions between Gua12 and R397: The N7 and O6 atoms of Gua12 are both in close proximity (3.2 to 3.4 Å) with the side chain of R397, permitting the formation of a hydrogen bond between the N7 atom of Gua12 and the guanidinium group of R397, as well as a C-H-O hydrogen bond between the O6 atom of Gua12 and the Cγ atom of R397 (Fig. 5B). These observations suggest a role for base-specific hydrogen bond in the C397R DRM2-substrate recognition, providing an explanation to the shift of substrate preference by the C397R mutation.
To verify the change of substrate preference, we compared DRM2- and C397R-mediated DNA methylation in vivo. The C397R mutation does not affect the overall DRM2 protein level, SDC expression level, or leaf phenotype in vivo appreciably (fig. S10, C to F). Next, we examined the DNA methylation levels at the SDC locus by performing bisulfite Sanger sequencing and found much higher CHG methylation in the two independent C397R transgenic lines compared to the two DRM2 lines (fig. S10, G and H), consistent with the increased enzymatic activity of C397R toward CHG DNA (Fig. 5A). We further performed the whole-genome bisulfite sequencing and found a relatively increased abundance of CHG methylation accompanied by reduced CHH methylation level in the C397R transgenic line (fig. S10I). In contrast, the C397H mutation leads to a decrease in the CHG methylation level (fig. S10, I and J). In addition, the C397A mutation, which naturally occurs at the equivalent position in NtDRM, leads to a DRM2-like methylation preference (figs. S2A and S10, I and J). We further plotted DNA methylation over TEs and found more CHG methylation in C397R than DRM2, C397A, or C397H, accompanied by decreased CHH methylation (Fig. 5, C and D, and fig. S10J). We next called hyper differentially methylated cytosines for C397R and DRM2 against ddc and found that the number of differentially methylated cytosines in each non-CG context differs greatly between them, with 14,002 hyper CHG and 10,623 hyper CHH in C397R but 16,087 hyper CHG and 76,234 hyper CHH in DRM2 (Fig. 5E and table S3). Further examination of the flanking sequences around all C397R methylated cytosines reveals a notable difference in the +2 position, with 52% being G in C397R (Fig. 5F) but only 18% G in DRM2 (Fig. 3H), suggesting that introducing the base-specific interactions by the C397R mutation reshapes the substrate specificity of DRM2.
DISCUSSION
DNA methylation is a widespread epigenetic mechanism that is essential for cell survival and differentiation. While mammalian DNA methylation predominantly occurs in the CG context, DNA methylation in plants is prevalent in CG, CHG, and CHH contexts (8, 9). How DNA methylation machineries have evolved to account for the evolutionary dynamics of methylomes across life kingdoms remains a fundamental and longstanding question. In plants, DRM2 is not only responsible for de novo methylation in all sequence contexts but also critical for maintaining CHH methylation. Through comprehensive structural, biochemical, and functional analyses, this study uncovers a previously unknown substrate recognition mechanism for DRM2.
Distinct from NtDRM and mammalian de novo methyltransferases DNMT3A and DNMT3B that function in a dimeric form (19, 23, 28), Arabidopsis DRM2 methylates DNA in a monomeric form. Sequence comparison between NtDRM and DRM2 reveals that those residues mediating dimerization of NtDRM are largely preserved in DRM2 (fig. S11), with the exception of a few divergent sites, which may give rise to their distinct oligomeric states.
As a plant de novo DNA methyltransferase, DRM2 is active on cytosines in all sequence contexts, as demonstrated by the silencing of an FWA transgene, which is controlled by CG methylation (35, 45). However, as a maintenance enzyme, DRM2 has been shown to have much higher activity on CHH compared to CG substrates by in vitro biochemical assays (32). Consistent with this observation, genetic analysis revealed that DRM2 prefers methylation of CHH over CG sites as thousands of loci lose CHH, but not CG, methylation in the absence of DRM2 (31). In this regard, this study reveals an arginine-DNA intercalation triggering substantial DNA deformation during DRM2-mediated methylation, most notably at the +1-flanking position that contributes to the substrate discrimination of DRM2 against CG sites. This substrate recognition mechanism reinforces the RNA-directed DRM2 methylation of CHH sites, preferably those with AT-rich flanking sequences, with implication for sequence-specific DNA methylation in plants.
This study also provides a molecular explanation for the broad methylation activity of DRM2 toward CHH substrates. In contrast to bacterial and mammalian DNA methyltransferases that present the target recognition domain for sequence-specific substrate recognition (29, 30), DRM2 does not engage target recognition domain for base-specific interactions with the substrates. Instead, it is poised to stabilize the deformed DNA major groove caused by the R595 intercalation. This lack of base-specific DNA interaction presumably permits DRM2 to accommodate substrates with a variety of sequence contexts. Consistent with this notion, our engineered DRM2 C397R mutation introduces base-specific interactions between the DRM2 target recognition domain and CHG DNA, thereby shifting the substrate preference of DRM2 toward CHG sites.
Our genomic analysis reveals DNA methylome complexity in plants. Besides the conventional CG, CHG, and CHH components, local sequence context is shown to be an additional player in shaping genomic DNA methylation patterns. The genomic location and local chromatin environment are both implicated in sequence-specific DNA methylation establishment and maintenance in plants (22). For DRM2, both the N-terminal ubiquitin-associated (UBA) domains and the C-terminal methyltransferase domain are required for its RNA-directed DNA methylation (46). This study shows that DRM2 preferentially methylates substrates with AT-rich flanking sequences, thereby adding another layer of methylome complexity via the interplay of DNA methyltransferases with substrate sequence. How these multilayered mechanisms cooperate in regulating DRM2-mediated DNA methylation awaits further investigation.
The observation on the DRM2 R595-triggered substrate deformation is reminiscent of the DNA deformations induced by a large group of DNA binding proteins, most well known for histones and transcription factors (47, 48). These DNA deformations often occur at AT-rich regions associated with reduced helical stability (38, 39, 48). Coincidently, the Arabidopsis genome, especially small TEs and other regions that are the preferential targets of DRM2, is AT-rich (Fig. 3G) (19, 31). This observation raises an interesting possibility that DRM2-mediated DNA deformation may be an adaptive mechanism for such a high AT chromatin environment. Note that, although DNMT3A/DNMT3B induces much less DNA deformation around the CpG sites in their respective DNA comethylation complexes, the DNA segment arching over the homodimeric interface of DNMT3A or DNMT3B shows an evident curvature (23, 28). It remains to be investigated whether the nucleotide composition of this segment of DNA affects the DNMT3A/DNMT3B-mediated DNA comethylation. Comparative studies between different plant species and animal systems will be important to reveal how DNA methylation machineries have evolved divergent mechanisms to account for the evolutionary dynamics of methylomes for genome regulation.
MATERIALS AND METHODS
Protein expression and purification
A synthetic DNA fragment encoding the methyltransferase domain of A. thaliana DRM2 (residues 270 to 626) was cloned into pRSFDuet-1 vector (Novagen), preceded by an N-terminal His6-SUMO tag. The expression plasmid was transformed into Escherichia coli BL21 DE3 (RIL) cells, and the cells were grown at 37°C. After the cell density reached an optical density at 600 nm of 0.8, the temperature was lowered to 16°C. Subsequently, the cells were induced by 100 μM isopropyl-β-D-thiogalactopyranoside and continued to grow overnight. The cells were collected and resuspended in lysis buffer [50 mM tris-HCl (pH 8.0), 1 M NaCl, 25 mM imidazole, and 1 mM phenylmethylsulfonyl fluoride] and lysed using an Avestin Emulsiflex C3 homogenizer. After centrifugation, the supernatant was applied to a Ni2+–nitrilotriacetic acid affinity column and the His6-SUMO-DRM2 fusion protein was eluted with elution buffer [20 mM tris-HCl (pH 8.0), 300 mM NaCl, and 300 mM imidazole]. The His6-SUMO tag was then removed by ubiquitin-like protease 1–mediated cleavage. The tag-free protein was further purified through ion-exchange chromatography on a Heparin HP column (GE Healthcare) and size exclusion chromatography on a 16/600 Superdex 200 pg column (GE Healthcare). The final protein sample was concentrated and stored in −80°C freezer for future use.
To generate covalent DRM2-DNA complexes, DRM2-methyltransferase, wild type, or C397R reacted with a synthesized 18-mer DNA duplex (Keck Biotechnology Resource Laboratory, Yale University) containing a central CTT, CCT, CAT, CCG, or CTG motif, in which the target cytosine is replaced by 5-fluorodeoxycytosine (CTT DNA, 5′-ATTATTAATXTTAATTTA-3′; CCT DNA, 5′-ATTCCTCCTXCTCCTTTA-3′; CAT DNA, 5′-ATTCCTCCTXATCCTTTA-3′; CCG DNA, 5′-ATTCCTAATXCGAATTTA-3′; and CTG DNA, 5′-ATTCCTAATXTGAATTTA-3′; X = 5-fluorodeoxycytosine), in a buffer containing 25 mM tris-HCl (pH 8.0), 25% glycerol, 50 mM dithiothreitol (DTT), and 30 μM S-adenosyl-l-methionine (SAM) at room temperature. The reaction products were sequentially purified through a HiTrap Q XL column (GE Healthcare) and a 16/600 Superdex 200 pg column. The final protein samples were concentrated to ~0.5 mM in a buffer containing 20 mM tris-HCl (pH 8.0), 250 mM NaCl, 5 mM DTT, and 5% glycerol.
Crystallization conditions and structure determination
For crystallization, the DRM2-DNA complexes were each mixed with 1 mM SAH. Crystals for all the DRM2-DNA complexes were generated using sitting-drop vapor-diffusion method at 4°C. Each drop was prepared by mixing 0.5 μl of DRM2-DNA complex sample with 0.5 μl of precipitant solution [for DRM2-CTT, DRM2-CCG, and DRM2C397R-CCG complexes: 2% v/v Tacsimate (pH 6.0), 0.1 M bis-tris (pH 6.5), and 20% w/v polyethylene glycol 3350; for DRM2-CAT complex: 0.1 M sodium acetate trihydrate (pH 7.0) and 12% w/v polyethylene glycol 3350; for DRM2-CCT complex: 0.1 M sodium formate (pH 7.0) and 12% w/v polyethylene glycol 3350; and for DRM2-CTG: 0.2 M potassium iodide and 20% w/v polyethylene glycol 3350 (pH 7.0)]. The crystal quality was further improved using the microseeding method. To harvest crystals, the crystals were soaked in cryoprotectants made of mother liquor supplemented with 30% glycerol before being flash-frozen in liquid nitrogen.
X-ray diffraction datasets for the DRM2-CTT, DRM2C397R-CCG, DRM2-CCG, and DRM2-CCT complexes were collected on beamline 5.0.1 or 5.0.2 at the Advanced Light Source, Lawrence Berkeley National Laboratory. X-ray diffraction datasets for the DRM2-CAT and DRM2-CTG complexes were collected on the 24-ID-E and 24-ID-C NE-CAT beamlines, respectively, at the Advanced Photon Source, Argonne National Laboratory. The diffraction data were indexed, integrated, and scaled using the HKL-3000 program (49). The structures of the complexes were solved by molecular replacement with the PHASER program (50) using the structure of the methyltransferase domain of NtDRM (Protein Data Bank: 4ONJ) as search model. The structural models of the DRM2-DNA and DRM2C397R-DNA complexes were then subjected to modification using COOT (51) and refinement using the PHENIX software package (52) in an iterative manner. The same R-free test set was used throughout the refinement. The statistics for data collection and structural refinement of the covalent DRM2-DNA and DRM2C397R-DNA complexes are summarized in table S1.
In vitro methylation assay
In vitro methylation assay was performed in 20-μl reactions containing 1 μM DRM2 (wild type or mutants), 3 μM synthesized DNA duplexes, 0.56 μM S-adenosyl-l-[methyl-3H] methionine with a specific activity of 18 Ci/mmol (PerkinElmer), 1.96 μM nonradioactive SAM, 50 mM tris-HCl (pH 8.0), 0.05% β-mercaptoethanol, 5% glycerol, and bovine serum albumin (BSA; 200 μg/ml). The DNA substrates were synthesized either containing (GAC)12, (TAC)12, (AAC)12, or (TGC)12 sequences to serve as CG, CTA, CAA, or CTG substrates, respectively, or with a 30-bp ATATATTATAAATACXTATTATTATATAAT sequence harboring a single CG, CC, CT, or CA motif for CG, CC, CT, or CA substrates, respectively. For clarity of the CC substrate, the second cytosine in the CC site is replaced with a 5-methylcytosine. Reactions were incubated at 37°C for 20 min before being quenched by the addition of 5 μl of 10 mM nonradioactive SAM. The reaction mixtures (12.5 μl) were then loaded onto a DEAE membrane (PerkinElmer) and air dried. The membrane was washed with 0.2 M ammonium bicarbonate (pH 8.2) three times for 5 min each, deionized water once for 5 min, and 95% ethanol once for 5 min. After air drying, the membrane was transferred into vials containing 4 ml of scintillation buffer (Fisher) and subjected to tritium scintillation recording by a Beckman LS6500 counter. Each reaction was replicated three times. For control, all the methylation assays included samples containing enzymes and SAM only in the reaction buffer, which gave basal levels of radioactivity to be subtracted from the actual reaction readings for data analysis.
For the enzymatic analysis of abasic site–containing DNA, the 30-bp CG DNA described above was modified such that the CG guanine on the target strand is replaced with an abasic site and the cytosine on the complementary strand is replaced with a 5-methylcytosine. The CG DNA with a hemimethylated site was used as control. DRM2 (3 μM) and DNA substrates (10 μM) were used for the enzymatic assay with DRM2, and 0.3 μM DNMT3A/3L and 3 μM DNA substrates were used for the enzymatic assay with DNMT3A.
Sanger bisulfite sequencing
A 637-bp DNA substrate, containing multiple target sites (upper strand: 15 CG, 38 CA, 26 CT, and 29 CC sites; lower strand: 15 CG, 57 CA, 31 CT, and 26 CC sites), was derived from a fragment of pGEX-6P-1 vector (nucleotides 302 to 938) via polymerase chain reaction (PCR) amplification. The methylation assay was performed in vitro in 20-μl reaction mixtures containing 0.5 μM DRM2, 0.05 μM DNA substrate, 400 μM SAM (Sigma-Aldrich), 50 mM tris-HCl (pH 8.0), 0.05% β-mercaptoethanol, 5% glycerol, and BSA (200 μg/ml). The reaction was incubated at 37°C for 1 hour, followed by bisulfite conversion using EZ DNA Methylation-Gold Kit (Zymo Research). The bisulfite-converted DNA upper and lower strands were subsequently amplified by 2× Taq RED DNA Polymerase Master Mix (Apex) using respective sets of primers (for the upper stand, 5′-TTGAAGAAAAATATGAAGAGGATTTGTATGAG-3′ and 5′-CCCCTCCAACACAACTTCC-3′ were used as forward and reverse primers, respectively; for the lower strand, 5′-ACCCACTCCACTTCTTTTCCAATATC-3′ and 5′-AGGGTGTGAGGTGGGAGAT-3′ were used as forward and reverse primers, respectively). The PCR products were cloned into the pCR4-TOPO vector (Invitrogen) and subjected to sequencing analysis. In total, two biological replicates were assayed.
To generate the WebLogos, 56 clones were analyzed to calculate the average methylation level of CG, CA, CT, and CC sites. Of the total 237 cytosine sites, 21 most methylated sites (methylation efficiency of 57.1% or higher) and 43 least methylated sites (methylation frequency of 5.0% or lower) were selected to generate WebLogos using the server (http://weblogo.threeplusone.com/create.cgi) (53), spanning from −7- to +8-flanking sites.
For Sanger bisulfite sequencing in plants, genomic DNA was extracted from 10-day-old seedlings using the cetyltrimethylammonium bromide (CTAB) method. DNA (500 ng) was bisulfite-treated using the EZ DNA Methylation-Gold Kit (Zymo Research, D5006) and amplified for SDC using the following primers: forward, 5′-GAAAAAGTTGGAATGGGTTTGGAGAGTTTAA-3′ and reverse, 5′-CAACAAACCCTAATATATTTTATATTAAAAC-3′. The PCR product was analyzed by gel electrophoresis, extracted, and purified using QIAEX II Gel Extraction kit (QIAGEN, 20021). Purified samples were then cloned into pCR2.1-TOPO TA vector (Invitrogen, 450641) and transformed into E. coli DH5α competent cells. The positive colonies were selected for plasmid DNA extraction followed by sequencing analysis.
Calculation of DNA shape parameters
The DNA shape parameters were calculated using the web server (http://web.x3dna.org/) (54), with the structures of DRM2-bound DNA molecules as input.
Plant materials and growth conditions
All A. thaliana transgenic lines were derived from ecotype Columbia-0. The triple mutant drm1 drm2 cmt3 (ddc) is a gift from S. Jacobsen (University of California, Los Angeles). Seeds were sown on ½ Murashige and Skoog (MS) plates containing 1% sucrose and kept at 4°C for 2 days before being transferred to long-day conditions (16-hour light/8-hour dark) at 22°C. After 10 days of growing on plates, the seedlings were transferred to soil and grown under long-day conditions at 22°C.
Construction of plasmids and generation of transgenic plants
Genomic DNA sequences of full-length DRM2 with the endogenous 1.3-kb promoter were amplified, and point mutations to residues C397 and R595 were made by site-directed mutagenesis using overlapping PCR. Wild-type and mutant DRM2 constructs were further cloned into the pCAMBIA1306 vector with a C-terminal 3xFLAG tag by ligation or ClonExpress II One Step Cloning Kit (Vazyme, C112). These constructs were then transformed into ddc plants via Agrobacterium-mediated floral dip method (55). Homozygous T3 generation plants were used for Western blotting and reverse transcription quantitative PCR (RT-qPCR) experiments. The primer sequences used for this study are summarized in table S5.
RNA extraction and RT-qPCR
Total RNA was extracted from 10-day-old seedlings grown on ½ MS plates using PureLink RNA Mini Kit (Thermo Fisher Scientific, 12183025). One microgram of total RNA was reverse-transcribed into cDNA with ProtoScript II (New England Biolabs, M0368L), followed by qPCR with SYBR Green Master Mix (Bio-Rad, 1725124) using CFX96 Real-Time System 690 (Bio-Rad). Relative SDC transcript level to ACTIN7 was calculated via the ∆Ct method.
Protein extraction and Western blotting
Total proteins were extracted from 10-day-old seedlings grown on ½ MS plates using 5% SDS and boiled for 10 min at 95°C before running on SDS–polyacrylamide gel electrophoresis gel. FLAG-tagged proteins were detected with horseradish peroxidase–conjugated anti-FLAG antibody (Sigma-Aldrich, A8592-1MG). Actin protein detected by an anti-actin antibody (Proteintech, 60008-1-Ig) was used as a loading control. All Western blots were developed using the ECL Plus Western Blotting Detection System (GE Healthcare, RPN2132) and chemiluminescent imaging using an ImageQuant LAS 4000 (GE Healthcare).
McrBC digestion
Genomic DNA was extracted from 100 mg of rosette leaf tissue of 3-week-old plants using PureLink Plant Total DNA Purification Kit (Thermo Fisher Scientific, 45-7004). Genomic DNA (100 ng) was treated with McrBC enzyme (New England Biolabs, M0272L) at 37°C for 7 hours and then for 20 min at 65°C to deactivate the enzyme. Digested DNA and undigested DNA were amplified using genomic locus–specific primers.
Bisulfite sequencing library construction and data analysis
Bisulfite treatment and sequencing library construction were conducted as previously described (56). Briefly, genomic DNA was extracted from 3-week-old plants using DNeasy Plant Mini Kit (QIAGEN, 69104), and ~1 μg of DNA was sheared to 300 to 400 bp using the Covaris S220 (Covaris) using the Covaris SonoLab 7.5. Sheared DNA was used to construct the library using the Illumina TruSeq DNA PCR-Free Low Throughput Library Prep Kit (Illumina, 20015962). After adapter ligation, samples were bisulfite-treated using EZ DNA Methylation-Lightning Kit (Zymo Research, 11-338) and then amplified for 10 cycles using Kapa HiFi HotStart Uracil ReadyMix (Kapa Biosystems, KK 2801) before sequencing on a HiSeq 4000 (Illumina) with 50-bp single-end reads. Sequencing reads were trimmed using FASTP (57) and aligned to the Arabidopsis TAIR10 genome using bsmap version 2.9 (58), allowing for 8% mismatches, trimming anything with a quality score of 33 or less, and removing any reads with more than five N’s. Methylation at every cytosine was called using bsmap’s methratio.py script, processing only unique reads and removing duplicate reads. Differentially methylate cytosines were identified using both methylKit (59) and bsmap’s methdiff.py script with the following cutoffs for each sequence context: 40% difference for CG, 20% difference for CHG, and 10% difference for CHH. Differentially methylated cytosines called by methylKit and methdiff.py were compared using BEDtools intersectBed (60), and only overlapped differentially methylated cytosines were used for subsequent analysis. Flanking sequence surrounding each differentially methylated cytosine was found using BEDtools getfasta, and the resulting fastas were compiled into a motif using WebLogo (53). TE metaplots were created by deepTools computeMatrix (61) using bsmap methylation file and a list of all TEs from TAIR10.
Statistics
Two-tailed Student’s t tests were performed to compare distributions between different groups, and a P value lower than 0.05 was considered to be statistically significant.
Acknowledgments
We would like to thank J. Liu and D. Sanders for assistance of BS-seq analysis. We also thank staff members at the Advanced Light Source (DE-AC02-05CH11231), Lawrence Berkeley National Laboratory and the NE-CAT beamlines (GM124165), Advanced Photon Source (DE-AC02-06CH11357), Argonne National Laboratory for access to x-ray beamlines and the Northwestern University Sequencing Core Facility for high-throughput sequencing. Funding: This work was supported by an NIH grant (R35GM119721) to J.S., a University of California Cancer Research Coordinating Committee (UC CRCC) grant (CRR-20-634140) to J.S., and NIH (1R35GM124806) and NSF CAREER (1552455) to X.Z. S.M.L. is supported by NIH T32 (GM008349). The group of J.Z. is supported by the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2016ZT06S172). Author Contributions: J.F., S.M.L., J.J., M.B., J.L., Z-M.Z., and W.R. performed the experiments. J.Z. provided technical support. Q.C. performed computational analysis. X.Z. and J.S. conceived and organized the study. J.F., S.M.L., J.J., X.Z., and J.S. prepared the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Coordinates and structure factors for the DRM2-CTT, DRM2-CAT, DRM2-CCT, DRM2-CTG, DRM2-CCG, and DRM2C397R-CCG complexes have been deposited in the Protein Data Bank under accession codes 7L4C, 7L4F, 7L4M, 7L4H, 7L4K, and 7L4N, respectively. The bisulfite sequencing data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE146700. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/23/eabd9224/DC1
REFERENCES AND NOTES
- 1.Bergman Y., Cedar H., DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 20, 274–281 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Jones P. A., Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Law J. A., Jacobsen S. E., Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenberg M. V. C., Bourc’his D., The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Ronemus M. J., Galbiati M., Ticknor C., Chen J., Dellaporta S. L., Demethylation-induced developmental pleiotropy in Arabidopsis. Science 273, 654–657 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Zhang H., Lang Z., Zhu J.-K., Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506 (2018). [DOI] [PubMed] [Google Scholar]
- 7.He X.-J., Chen T., Zhu J.-K., Regulation and function of DNA methylation in plants and animals. Cell Res. 21, 442–465 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okano M., Bell D. W., Haber D. A., Li E., DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., Nery J. R., Lee L., Ye Z., Ngo Q.-M., Edsall L., Antosiewicz-Bourget J., Stewart R., Ruotti V., Millar A. H., Thomson J. A., Ren B., Ecker J. R., Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Y., Ecker J. R., Non-CG methylation in the human genome. Annu. Rev. Genomics Hum. Genet. 16, 55–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goll M. G., Bestor T. H., Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Finnegan E. J., Dennis E. S., Isolation and identification by sequence homology of a putative cytosine methyltransferase from Arabidopsis thaliana. Nucleic Acids Res. 21, 2383–2388 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindroth A. M., Cao X., Jackson J. P., Zilberman D., McCallum C., Henikoff S., Jacobsen S. E., Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292, 2077–2080 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Cao X., Springer N. M., Muszynski M. G., Phillips R. L., Kaeppler S., Jacobsen S. E., Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc. Natl. Acad. Sci. U.S.A. 97, 4979–4984 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson I. R., Jacobsen S. E., Epigenetic inheritance in plants. Nature 447, 418–424 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Matzke M. A., Mosher R. A., RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Johnson L. M., Du J., Hale C. J., Bischof S., Feng S., Chodavarapu R. K., Zhong X., Marson G., Pellegrini M., Segal D. J., Patel D. J., Jacobsen S. E., SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 507, 124–128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wierzbicki A. T., Ream T. S., Haag J. R., Pikaard C. S., RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat. Genet. 41, 630–634 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhong X., du J., Hale C. J., Gallego-Bartolome J., Feng S., Vashisht A. A., Chory J., Wohlschlegel J. A., Patel D. J., Jacobsen S. E., Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 157, 1050–1060 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong X., Hale C. J., Law J. A., Johnson L. M., Feng S., Tu A., Jacobsen S. E., DDR complex facilitates global association of RNA polymerase V to promoters and evolutionarily young transposons. Nat. Struct. Mol. Biol. 19, 870–875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cokus S. J., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C. D., Pradhan S., Nelson S. F., Pellegrini M., Jacobsen S. E., Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gouil Q., Baulcombe D. C., DNA methylation signatures of the plant chromomethyltransferases. PLOS Genet. 12, e1006526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao L., Emperle M., Guo Y., Grimm S. A., Ren W., Adam S., Uryu H., Zhang Z.-M., Chen D., Yin J., Dukatz M., Anteneh H., Jurkowska R. Z., Lu J., Wang Y., Bashtrykov P., Wade P. A., Wang G. G., Jeltsch A., Song J., Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun. 11, 3355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klimasauskas S., Kumar S., Roberts R. J., Cheng X., HhaI methyltransferase flips its target base out of the DNA helix. Cell 76, 357–369 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Lin C.-C., Chen Y.-P., Yang W.-Z., Shen J. C. K., Yuan H. S., Structural insights into CpG-specific DNA methylation by human DNA methyltransferase 3B. Nucleic Acids Res. 48, 3949–3961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinisch K. M., Chen L., Verdine G. L., Lipscomb W. N., The crystal structure of HaeIII methyltransferase convalently complexed to DNA: An extrahelical cytosine and rearranged base pairing. Cell 82, 143–153 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Song J., Teplova M., Ishibe-Murakami S., Patel D. J., Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 335, 709–712 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z.-M., Lu R., Wang P., Yu Y., Chen D., Gao L., Liu S., Ji D., Rothbart S. B., Wang Y., Wang G. G., Song J., Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng X., Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct. 24, 293–318 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Ren W., Gao L., Song J., Structural basis of DNMT1 and DNMT3A-Mediated DNA Methylation. Genes 9, 620 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stroud H., Greenberg M. V. C., Feng S., Bernatavichute Y. V., Jacobsen S. E., Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wada Y., Ohya H., Yamaguchi Y., Koizumi N., Sano H., Preferential de novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. J. Biol. Chem. 278, 42386–42393 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Cao X., Jacobsen S. E., Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. U.S.A. 99 ( suppl. 4), 16491–16498 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng X., Kumar S., Posfai J., Pflugrath J. W., Roberts R. J., Crystal structure of the HhaI DNA methyltransferase complexed with S-adenosyl-L-methionine. Cell 74, 299–307 (1993). [DOI] [PubMed] [Google Scholar]
- 35.Cao X., Jacobsen S. E., Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12, 1138–1144 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Henderson I. R., Jacobsen S. E., Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 22, 1597–1606 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ornstein R. L., Rein R., An optimized potential function for the calculation of nucleic acid interaction energies I. Base stacking. Biopolymers 17, 2341–2360 (1978). [DOI] [PubMed] [Google Scholar]
- 38.Yakovchuk P., Protozanova E., Frank-Kamenetskii M. D., Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 34, 564–574 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coman D., Russu I. M., Base pair opening in three DNA-unwinding elements. J. Biol. Chem. 280, 20216–20221 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Lebrun A., Lavery R., Modelling extreme stretching of DNA. Nucleic Acids Res. 24, 2260–2267 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohs R., Jin X., West S. M., Joshi R., Honig B., Mann R. S., Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 79, 233–269 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams L. D., Maher L. J. III, Electrostatic mechanisms of DNA deformation. Annu. Rev. Biophys. Biomol. Struct. 29, 497–521 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Anteneh H., Fang J., Song J., Structural basis for impairment of DNA methylation by the DNMT3A R882H mutation. Nat. Commun. 11, 2294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gowher H., Jeltsch A., Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpG sites. J. Mol. Biol. 309, 1201–1208 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Chan S. W.-L., Zilberman D., Xie Z., Johansen L. K., Carrington J. C., Jacobsen S. E., RNA silencing genes control de novo DNA methylation. Science 303, 1336 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Saredi G., Huang H., Hammond C. M., Alabert C., Bekker-Jensen S., Forne I., Reverón-Gómez N., Foster B. M., Mlejnkova L., Bartke T., Cejka P., Mailand N., Imhof A., Patel D. J., Groth A., H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714–718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Werner M. H., Gronenborn A. M., Clore G. M., Intercalation, DNA kinking, and the control of transcription. Science 271, 778–784 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Widom J., Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 34, 269–324 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Otwinowski Z., Minor W., [20] Processing of x-ray diffraction data collected in oscillation mode. Method Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 50.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sec. D Biol. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C., PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. Sec. D Biol. 58, 1948–1954 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E., WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S., Olson W. K., Lu X.-J., Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures. Nucleic Acids Res. 47, W26–W34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clough S. J., Bent A. F., Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998). [DOI] [PubMed] [Google Scholar]
- 56.Pignatta D., Erdmann R. M., Scheer E., Picard C. L., Bell G. W., Gehring M., Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. eLife 3, e03198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S., Zhou Y., Chen Y., Gu J., fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xi Y., Li W., BSMAP: Whole genome bisulfite sequence MAPping program. BMC bioinformatics 10, 232 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akalin A., Kormaksson M., Li S., Garrett-Bakelman F. E., Figueroa M. E., Melnick A., Mason C. E., methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramírez F., Dündar F., Diehl S., Grüning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/23/eabd9224/DC1