

SUMMARY
Secondary bile acids (SBAs) are derived from primary bile acids (PBAs) in a process reliant on biosynthetic capabilities possessed by few microbes. To evaluate the role of BAs in intestinal inflammation, we performed metabolomic, microbiome, metagenomic, and transcriptomic profiling of stool from ileal pouches (surgically created resevoirs) in colectomy-treated patients with ulcerative colitis (UC) versus controls (familial adenomatous polyposis, FAP). We show relative to FAP, UC pouches have reduced levels of lithocholic acid and deoxycholic acid (normally the most abundant gut SBAs), genes required to convert PBAs to SBAs, and Ruminococcaceae (one of few taxa known to include SBA-producing bacteria). In three murine colitis models, SBA supplementation reduces intestinal inflammation. This anti-inflammatory effect is in part dependent on the TGR5 bile acid receptor. These data suggest that dysbiosis induces SBA deficiency in inflammatory-prone UC patients, which promotes a pro-inflammatory state within the intestine that may be treated by SBA restoration.
Keywords: Pouchitis, bile acids, Inflammatory bowel disease, colitis, metabolomics, dysbiosis, ulcerative colitis
Graphical Abstract
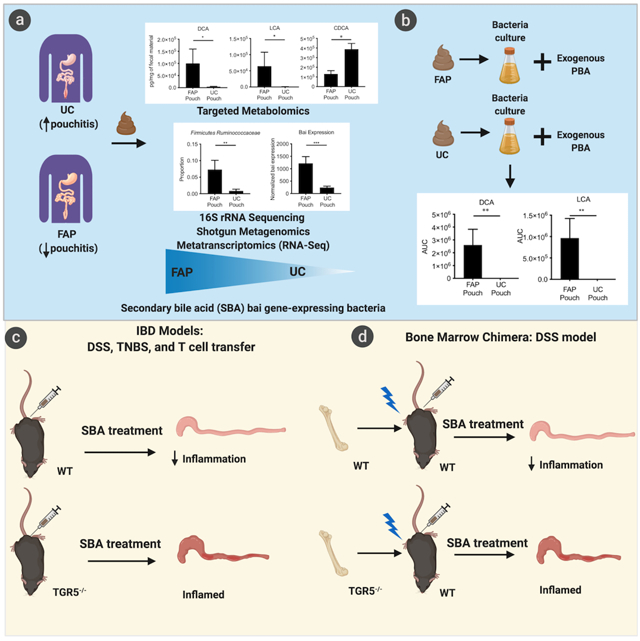
eTOC:
Secondary bile acids (SBAs) are some of the most concentrated bacterially-derived gut metabolites. Sinha et al. find UC pouch patients have reduced SBAs and Ruminococcaceae (one of few SBA-producing taxa) compared to FAP-control patients. In colitis models, SBAs ameliorate disease in a process reliant on the TGR5 bile acid receptor.
INTRODUCTION
Ulcerative colitis (UC) is a chronic, relapsing illness affecting millions of patients worldwide. Prevalence in western countries exceeds 0.3% and incidence is rising in newly industrialized countries in Asia, Africa, and South America (Ng et al., 2018). Patients often suffer many debilitating symptoms starting at a young age (Ramos and Papadakis, 2019). UC has no medical cure and can only be surgically cured with colectomy, the removal of the entire colon, often with the creation of an ileal-anal “pouch” to mimic the function of the removed rectum. Unfortunately, the pro-inflammatory state in UC often persists even after colectomy, as studies have shown about 50% of patients who undergo surgery continue to have intestinal inflammation in the pouch, known as pouchitis (Navaneethan and Shen, 2009, Hata et al., 2017). Remarkably, pouchitis is rarely seen in patients who undergo the same surgical procedure due to a relatively rare non-inflammatory disease known as familial adenomatous polyposis syndrome (FAP) (Nyam et al., 1997, Hata et al., 2017). This observation further supports the notion that pouchitis is strongly associated with UC and may arise through mechanisms similar to those involved in UC (Tyler et al., 2013). Treatments for this disorder are limited, consisting most commonly of multiple courses of antibiotics, which carry their own risks, such as the development of dysbiosis, secondary infections, and bacterial resistance (Navaneethan and Shen, 2009). The fact that antibiotics treat pouchitis clearly underscores the role of microbiota in this disease.
Microbes are well-known to possess the biosynthetic capability to modify and produce molecules affecting intestinal inflammation (Rooks and Garrett, 2016, Postler and Ghosh, 2017). The enzymatic action of intestinal bacteria, for example, creates a full complement of bile acids, which have many physiologic roles, including glucose regulation and intestinal motility (Rao et al., 2010, Joyce and Gahan, 2017, Macpherson et al., 2016) The process by which bile acids exert their many effects is complex and relies on both the host and intestinal bacteria (Pols et al., 2011, Schaap et al., 2014) to convert the liver-produced primary bile acids (PBAs), cholic acid (CA) and chenodeoxycholic acid (CDCA), into secondary bile acids (SBAs), known to be potent signaling molecules (Wang et al., 2019). Two key bacteria-reliant processes are bile acid deconjugation and 7-alpha hydroxylation; these two distinct metabolic activities influence the physiologic response in the intestine differently. PBAs are conjugated with glycine or taurine to increase water solubility before being excreted into the biliary tree. The enzyme, bile salt hydrolase, produced by many common commensal genera, such as Bacteroides, Clostridium, and Enterococcus, deconjugates PBAs as the initial step. However, only a few known bacteria—all from the Lachnospiraceae and Ruminococcaceae families—perform the subsequent 7α-dehydroxylation of CA and CDCA to respectively generate deoxycholic acid (DCA) and lithocholic acid (LCA), the two most common SBAs (Stellwag and Hylemon, 1978, Stellwag and Hylemon, 1979) and known influencers of the inflammatory response (Duboc et al., 2013).
While the vast majority of bile acids are reabsorbed by the distal small intestine, the remaining enter the colon at concentrations that range from about 200 μM to 1000 μM (Hamilton et al., 2007, Devlin and Fischbach, 2015). In fact, SBAs are some of the most highly concentrated metabolites in the normal gut microenvironment (Devlin and Fischbach, 2015).
The role of SBAs, however, in intestinal inflammation is only beginning to be deciphered. Previous studies have largely focused on dysbiosis and in some cases, bile acid and other metabolomic changes in UC patients with intact colons compared to healthy controls (Duboc et al., 2013, Matsuoka and Kanai, 2015, Zella et al., 2011). Although the similarities of pre-colectomy versus pouch microbial profiles is unknown, by investigating both the microbiota and metabolite differences of pouches in patients with history of UC or the non-inflammatory disease FAP, we hypothesized being able to identify signals that may be lost in this otherwise heterogeneous population. Our data indicate that there are important differences in the stool SBA profiles from UC patients treated with colectomy (UC pouch) compared to FAP pouch patients. In our 16S rRNA gene sequencing studies, we have found that only one particular family of bacteria, the Ruminococcaceae, is present in significantly lower amounts in stool from UC compared to FAP pouch patients. Importantly, the Ruminococcaceae are known to contain members capable of SBA generation (Stellwag and Hylemon, 1979, Duboc et al., 2012). Our metagenomics analysis confirms this finding. Furthermore, using metatranscriptomics, we show that the bile acid-inducible (bai) genes, which are needed for the critical 7α-dehydroxylation step that converts PBAs to SBAs are expressed in significantly lower amounts in UC pouches compared to FAP pouches. Based on these SBA differences, we then show that the SBAs LCA and DCA mitigate inflammation in both acute and chronic murine colitis models and reduce expression of key cytokines and chemokines involved in inflammation. We also utilize transgenic mice and bone marrow chimera models to show the critical role of the TGR5 bile acid receptor in mitigating the inflammation (STAR methods).
RESULTS
Secondary bile acids, DCA and LCA, are reduced in inflammation-prone UC pouches
To better understand the potential role of bile acids in intestinal inflammation, we first conducted a targeted metabolomics study using liquid chromatography–mass spectrometry (LC-MS) to assess the differences in bile acid composition of stool from UC versus FAP (control) pouch patients. While UC and FAP are clinically quite different, apart from UC pouch patients having significantly more history of pouchitis than FAP pouch patients, the two populations compared were otherwise similar (Table S1).
The most striking difference in bile acid profiles was that two SBAs, LCA and DCA, were significantly reduced in UC compared to FAP pouches. Conversely, the PBA CDCA was significantly higher in UC compared to FAP pouches (Figure 1A). Cholic acid levels were also higher in UC compared to FAP pouches, but this was not significant. There were no other significant differences in the bile acid profiles between UC compared to FAP pouches (Table S2). We also compared bile acid profiles of UC pouch stool from patients with a history of pouchitis and those with quiescent disease and found that there were no notable differences in the bile acid profiles. Prior investigations in the literature have also shown remarkably consistent species richness, diversity, and general composition when comparing UC pouches with or without pouchitis (Johnson et al., 2009). Additional untargeted metabolite testing was performed. No clear differences of other metabolite groups thought to be involved in intestinal inflammation, such as short chain fatty acids, tryptophan/indoles, and arginine-derived polyamines were identified in this untargeted analysis (data not shown).
Figure 1. UC pouch stool has diminished secondary bile acids and altered microbial composition, which is associated with lower expression of bai genes, and less efficiency to convert primary bile acid to secondary bile acid in vitro than FAP pouch.
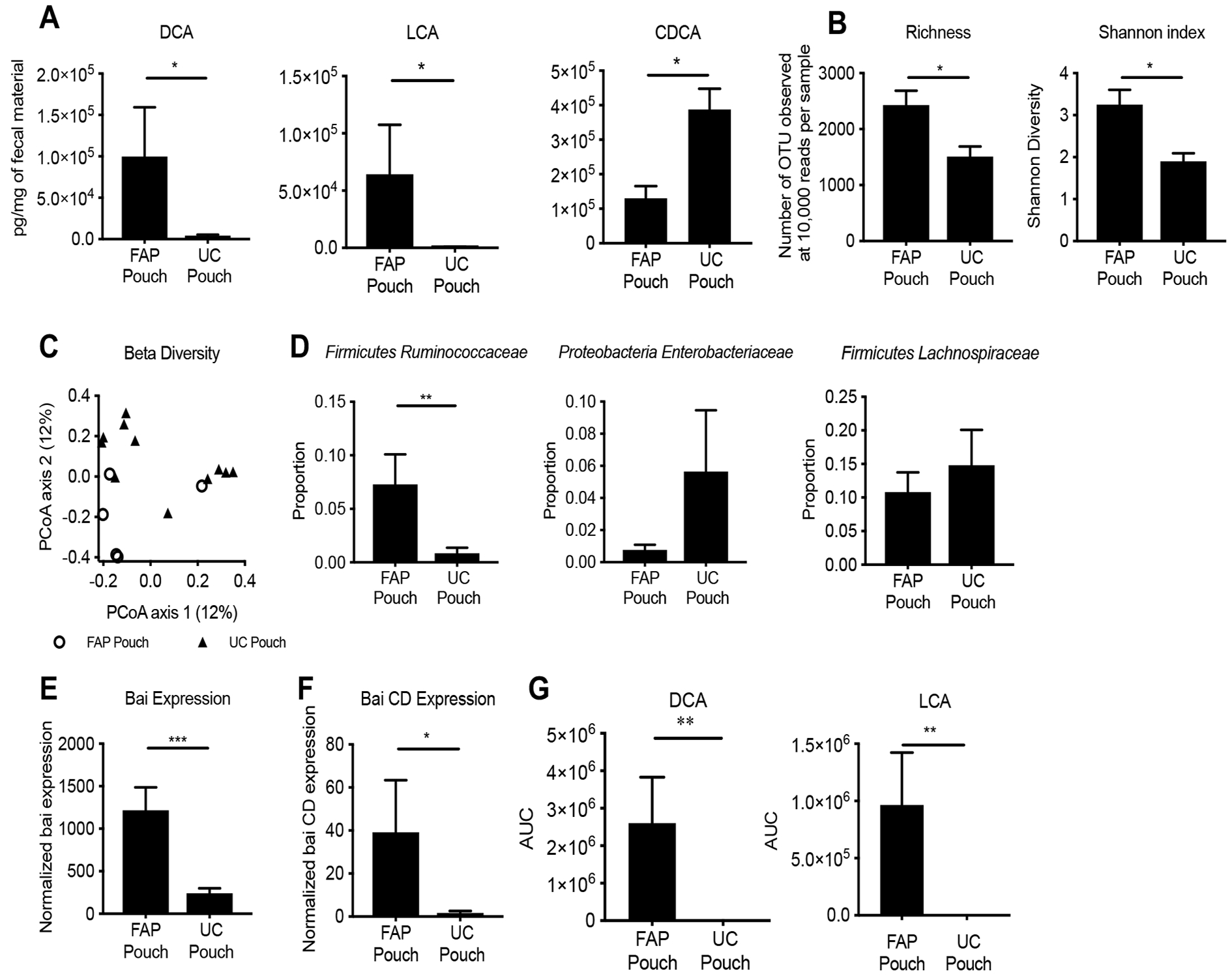
(A) Levels of DCA and LCA are significantly lower in UC versus FAP pouches, whereas CDCA is significantly higher measured by LC-MS. (B) Total bacterial species present (at 10,000 reads per sample) show reduced richness and Shannon diversity in UC pouch microbiota. (C) Bray-Curtis distances to assess β-diversity at community level differences between UC pouch and FAP pouch, as measured by PERMANOVA test. (D) Relative proportions of most abundant species revealed reduced taxon abundance of Firmicutes Ruminococcaceae in UC compared to FAP pouches. (E) Bai A-K gene transcription (established by RNA-Seq) is lower in UC pouches compared to FAP pouches. (F) Bai CD gene expression, a key enzyme in SBA biosynthesis, is also lower in UC pouches compared to FAP pouches. (G) The stool from UC pouch was less efficient in converting exogenous CA and CDCA to DCA and LCA respectively than FAP pouch. Data represented as mean ± SEM. (A) UC pouch n=17, FAP pouch n=7, * P<.05, two-tailed t-test, post multiple t-test with Benjamini and Hochberg posttest correction, (B) UC pouch n=11, FAP pouch n=5; * P<.05, two-tailed t-test, (C) UC pouch n=11, FAP pouch n=5, Permanova test, R2=.093, (D) UC pouch n=11, FAP pouch n=5, ** P<.01, two-tailed t-test, (E) UC pouch n=7, FAP pouch n=3, *** P<0.001, two-tailed t-test, (F) UC pouch n=7, FAP pouch n=3, * P<0.05, two-tailed t-test, (G) UC pouch n=17, FAP pouch n=7, ** P<.01, two-tailed t-test.
Our finding showing the essential absence of LCA and DCA in UC pouches suggests a potential differential ability to convert PBAs to SBAs in UC versus FAP pouch microenvironments. Based on the lower levels of LCA and DCA in UC than in FAP pouch stool samples, we postulate that these SBAs may be important in maintaining immune homeostasis in the pouch. In addition, we found the ratio of DCA:CA is highest in healthy stool, followed by FAP pouch and lowest in UC pouch (Figure S1A). This suggests increased DCA (which is derived from CA) is seen in healthier intestines. The conversion of PBA to SBA is notably a very efficient reaction and can be carried to equilibrium by a small number of bacteria and not necessarily reflect microbial density (Ridlon et al., 2006). However, the total reads per sample in both our 16S and shotgun metagenomic studies described below were not found to be different in UC compared to FAP pouch (Figure S1B).
UC pouches have less bacterial diversity than control FAP pouches
We then sought to determine the bacterial composition of the pouch samples and elucidate any compositional differences between UC and FAP pouch stool. Stool samples were processed identically for 16S rRNA marker gene sequencing as described in the methods section. We calculated the α-diversity using richness and Shannon indices, as a measure of species diversity across the samples. We found that UC pouches demonstrated decreased α-diversity compared to FAP pouches (Figure 1B). This is consistent with findings that patients with UC (and also Crohn’s disease) have lower bacterial diversity (Lane et al., 2017). UC and FAP pouch samples also showed significant differences in β-diversity, a measure of compositional similarity, as measured by unweighted Unifrac distances (Figure 1C). While it is known that the microbial composition of stool from various parts of the human intestine vary considerably, our findings, taken together, indicate UC and FAP pouch samples showed significantly different bacteria composition (Behr et al., 2018, Smith et al., 2005).
Ruminococcaceae are reduced in UC pouches
Other studies have shown a reduction in Firmicutes and an increase in Proteobacteria phyla is a frequent finding in inflammatory bowel disease (IBD)-associated dysbiosis (Lloyd-Price et al., 2019, Frank et al., 2007, Gevers et al., 2014). Indeed, we also found a reduction in Firmicutes families that contain the most common gut microbes in our UC pouch samples. We also noted a nonsignificant increase in Proteobacteria that is consistent with IBD-associated dysbiosis (Matsuoka and Kanai, 2015, Sartor and Wu, 2017). Interestingly, of all the taxa identified, we observed only a significant reduction in the Ruminococcaceae family (Figure 1D, Table S3), indicating that this particular microbial family might play a role in maintaining intestinal immune homeostasis. In alignment with this notion is the fact that reduced Ruminococcaceae have been shown to significantly correlate with lower SBA levels in other pathologic states such as Clostridium difficile colonization (Kakiyama et al., 2013, Theriot et al., 2016). Only members of the Ruminococcaceae and Lachnospiraceae (both in the Clostridium Cluster XIVa) are known to possess the relatively rare 7α-dehydroxylation capability necessary to convert PBAs to SBAs (Mullish et al., 2019, Ridlon et al., 2013). Notably, in our 16S rRNA investigation showing the reduction of Ruminococcaceae in UC pouches, we also found reduction in bacterial strains showing higher homology to Clostridium (C.) leptum, a member of the Ruminooccaceae family known to possess 7α-dehydroxylation capability, than any other named species (Figure S1C). In our metagenomic analysis, we confirmed significantly reduced Ruminococcaceae in UC versus FAP pouch stool (Figure S1D). In addition, our metagenomic analysis identified reads with high homology to gene sequences in the bai operon (genes needed to convert PBAs to SBAs) to be less in the UC compared to FAP pouch stool, potentially indicating a decreased capability to generate SBAs in the UC compared to FAP pouch (Figure S1E).
Bai expression is reduced in UC pouches
The bai operon contains genes that encode enzymes necessary to transform PBAs to SBAs. Only specific gut bacteria are known to possess the capability to perform this transformation (Kitahara et al., 2001, Solbach et al., 2018). Of the several proteins required in SBA biosynthesis, one key enzyme is a stereo-specific NAD(H)-dependent 3-dehydro-4-bile acid oxidoreductase, encoded by the baiCD gene cluster, which is part of the bai operon (Kang et al., 2008). To assess whether the lower concentrations of DCA and LCA in UC compared to FAP pouch stool samples corresponded with differences in the expression of bai genes, we performed metatranscriptomics analysis (using whole transcriptome shotgun sequencing (RNA-Seq) of stool microbial genomes). We found that there was significantly lower expression of bai genes in UC compared to FAP pouches (Figure 1E and 1F).
Stool from FAP pouches are able to produce significantly more SBAs in vitro compared to UC pouches
The process of transforming PBAs to SBAs is complex and requires microbes to perform critical steps. Reproducing this gut microbial ecosystem in vitro by selecting specific bacteria is challenging, particularly when it is known that very low concentrations of select bacteria can be critical in performing such a conversion of PBAs to SBAs (Ericsson and Franklin, 2015, Solbach et al., 2018).
To confirm the differential production of SBAs in the UC compared to FAP pouches, we cultured stool from UC and FAP pouch with PBAs in anaerobic chambers. We then assessed LCA and DCA concentrations in the supernatant using LC-MS. We again found significantly reduced LCA and DCA production from microbes in the stool from UC compared to FAP pouches (Figure 1G), confirming our initial direct findings from human samples (Figure 1A).
DCA and LCA mitigate inflammation in dextran sodium sulfate (DSS) murine colitis model
Given that LCA and DCA were nearly undetectable in UC pouch stool (Figure 1A), we next sought to investigate whether restoration of these SBAs could reduce intestinal inflammation in a murine model of colitis. To test this hypothesis, colitis was induced by giving 2.5% DSS in drinking water over a 10 day period. SBAs were administered per rectum on days 4, 6 and 8 (Figure 2A). We also noted that LCA and DCA levels decreased in response to DSS, a finding that provides further credibility for the use of a DSS model as it does seem to at least partially mimic what occurs in the pro-inflammatory UC pouch (Figure S2A). We did not, however, identify meaningful changes in the bacterial composition in response to DSS (Figure S2B). No additional bile acids were found to be significant (Table S4). To assess for a dose-dependent effect, mice were treated with increasing concentrations of SBAs. Treatments with 1 mg (1X) and 5 mg (5X) suspension of LCA or DCA maintained body weight with increasing efficacy compared to vehicle control. At 10 mg (10X) dose, LCA ameliorated disease while DCA did not (Figure 2B and C). Similarly, all doses of LCA, and the 1X and 5X dose of DCA showed reduced colon histopathology compared to control (Figure 2D).
Figure 2. LCA and DCA ameliorate DSS-induced colitis.
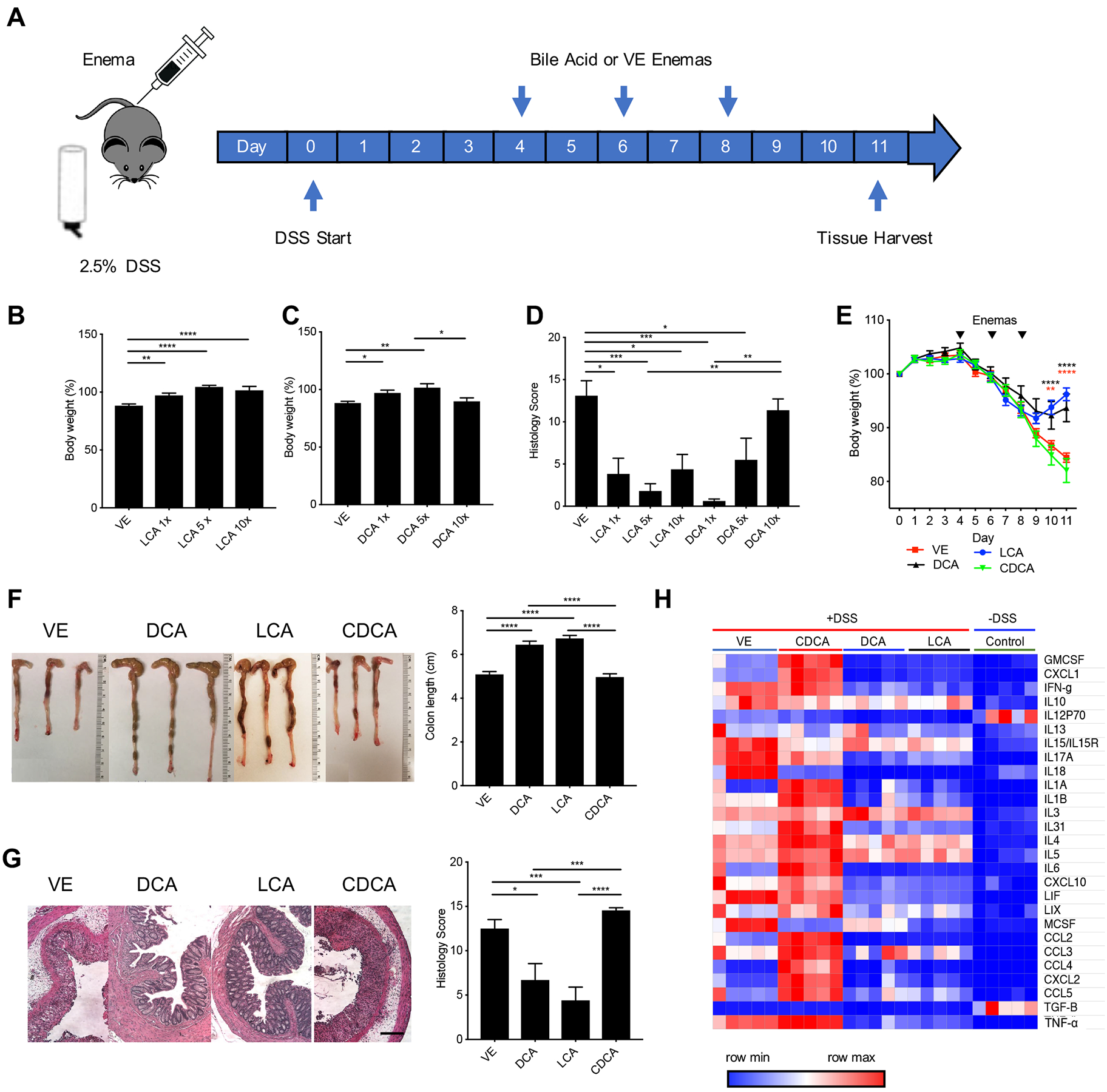
(A) C57BL/6 mice were given water containing 2.5% DSS (w/v) for 11 days and treated with suspension of bile acid or vehicle control (VE) per rectum on days 4, 6 and 8 with 150 μL of either 1 mg (1X), 5 mg (5X), or 10 mg (10X) of bile acid. LCA and DCA treatment result in dose-dependent protection, evident by an improvement in (B, C) body weight and (D) histopathology. LCA and DCA-treated mice maintained (E) higher body weights, (F) healthier gross colon morphology and longer colon lengths compared to VE or CDCA-treated mice. (G) LCA and DCA reduced distal colon inflammation and histopathology score. Scale bar = 20 m. (H) Luminex heat map data shows a decrease of pro-inflammatory associated cytokines in colon homogenate isolated from LCA and DCA-treated mice. Colon homogenate from mice untreated with DSS was used as a control. Data represented as mean ± SEM, analyzed by one-way ANOVA with Tukey’s post-hoc (B, C, D, F, G) or two-way ANOVA with Bonferroni’s post-hoc (E), and significance reported as * P<.05, ** P<.01, *** P<.001, and **** P<0.0001 (B) VE n=23, LCA 1X n=20, LCA 5x n=10, LCA 10x n=9, (C) VE n=23, DCA 1X n=17, DCA 5x n=9, DCA 10x n=9 (D) VE n=5, LCA 1X n=3, LCA 5x n=5, LCA 10x n=4, DCA 1X n=4, DCA 5x n=4, DCA 10x n=4, (E-F) VE, LCA, DCA n=14, CDCA n=13, (G) All groups n=10, (H) All groups n=5.
Given our findings of reduced SBAs and increased CDCA in UC compared to FAP pouches (Figure 1A), we next compared the protective effect of LCA and DCA to CDCA and vehicle control in mice with DSS-induced colitis. LCA, DCA, CDCA, or vehicle control was administered 3 times per rectum to mice with DSS-induced colitis (Figure 2A). LCA and DCA reproducibly reduced colitis disease parameters as shown by weight loss, colon length, and gross colon morphology (Figure 2E and F). These SBAs also significantly reduced the leukocyte infiltration and histologic inflammation of the colon (Figure 2G). Unlike LCA and DCA, CDCA treatment did not ameliorate disease and was indistinguishable from or slightly worse than the vehicle control. These studies demonstrate that LCA and DCA can reduce experimental colitis.
Treatment with DCA and LCA promote an anti-inflammatory profile
To gain initial insight into the mechanism by which SBAs influence intestinal inflammation, we next conducted Luminex multiplex immunoassay on distal colon tissue from colitis-inflamed mice treated with LCA, DCA, CDCA, or vehicle control as described above. Our results show that LCA and DCA treatments caused a remarkable and significant decrease in multiple chemokines and cytokines associated with inflammation, including those often increased in intestinal inflammation such as CCL5, CXCL10, IL17A, and TNFα (Figure 2H and Figure S3) (Banks et al., 2003). This finding suggests that restoration of SBAs, either directly or potentially through restoration of SBA-producing bacteria, may reduce intestinal inflammation seen in pouchitis and IBD.
In addition, quantitative PCR was performed on RNA extracted from colon tissue which confirmed a generalized reduction in inflammatory cytokines, including TNFα and IL-17, in mice with DSS-induced colitis treated with LCA in both WT as well as TGR5−/− mice compared to control (Figure S4A); this dampening in the expression of pro-inflammatory cytokines was also seen in mice with TNBS-induced colitis (Figure S4B). This finding is consistent with the literature, which shows reasonable evidence supporting that the anti-inflammatory results are at least in part due to the effect of SBAs on inflammatory gene expression (Fiorucci et al., 2018). Taken together these data suggest a direct effect of SBAs on inflammatory gene expression.
DCA and LCA also mitigate inflammation in both trinitrobenzenesulfonic acid (TNBS) and CD45RBhi T cell transfer murine colitis models
To confirm the findings from the DSS model, we also conducted experiments assessing if SBAs could reduce intestinal inflammation in two additional murine models of colitis. We first tested the ability of SBAs to mitigate colitis in the acute TNBS model. Mice were pre-sensitized with skin application of TNBS acid followed by rectal administration of TNBS in 50% ethanol. We next administered LCA or vehicle control 3 times per rectum to mice with TNBS-induced colitis (Figure 3A). LCA reproducibly reduced colitis disease parameters as shown by weight loss, colon length, and gross colon morphology (Figure 3B and C). LCA also significantly reduced the leukocyte infiltration and histologic inflammation of the colon (Figure 3D). Unlike LCA, vehicle control administration did not ameliorate disease. Additionally, mice treated with LCA had lower disease activity and levels of fecal lipocalin 2 (a secreted protein that is upregulated in models of colitis) compared to controls (Figure S5A and B) (Chassaing et al., 2012).
Figure 3. LCA ameliorates TNBS-induced colitis and CD45RBhi CD4+ T cell transfer colitis.
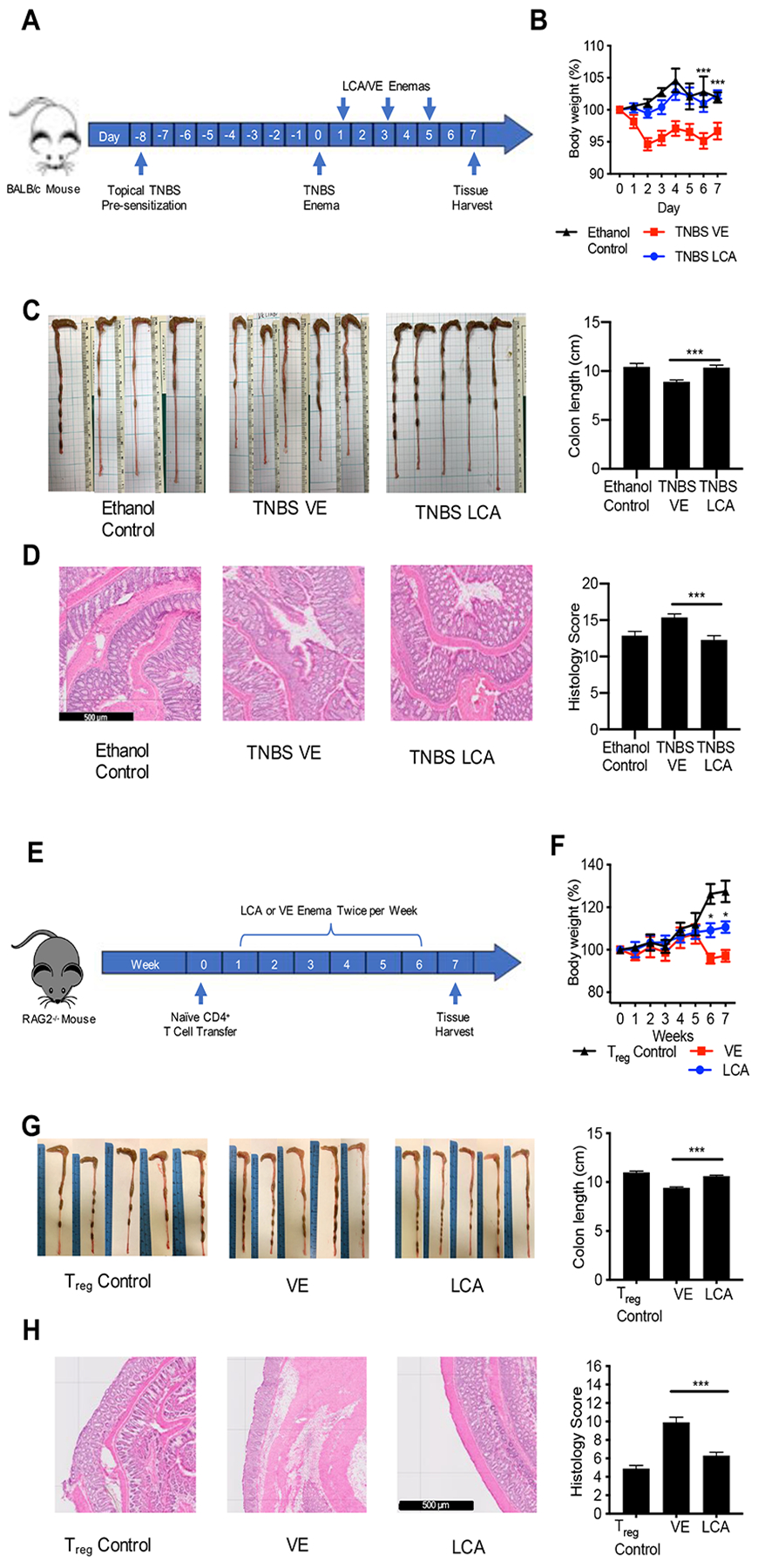
(A) Mice were pre-sensitized with topical TNBS, and further sensitized with recto-transfer of TNBS. Suppository treatment of TNBS mice with LCA on day 1, 3 and 5 resulted in (B) higher body weight, (C) longer colon length and healthier gross colon morphology, and (D) reduced distal colon inflammation and histopathology score. (E) Naïve T cells were transferred into RAG2−/− mice. The control group received Treg cells intravenous injection. Beginning at week 1, the naïve T cell mice were treated with LCA or VE suppository twice per week for 6 weeks. LCA treatment maintained (F) higher body weight, (G) longer colon lengths, and healthier gross colon morphology. (H) LCA reduced distal colon inflammation and histopathology score. Scale bar = 500 m. Data represented as mean ± SEM, analyzed by two-way ANOVA with Bonferroni’s post-hoc (B, F) or one-way ANOVA with Tukey’s post-hoc (C, D, G, H). and significance reported as * P<.05, ** P<.01, *** P<.001, and **** P<0.0001. (B) (B) TNBS VE n=19, TNBS LCA n=19 Ethanol Control n= 8, (C) TNBS VE n=18, TNBS LCA n=17 Ethanol Control n= 8, (D) TNBS VE n=15, TNBS LCA n= 14 Ethanol Control n= 8, (F-H) VE n=5, LCA n=5, Treg control n=5.
Next, we tested the ability of LCA to mitigate colitis in the chronic CD45RBhi T cell transfer colitis model. Splenic CD4+ T cells were sorted to naïve CD4+ T cells and Treg cells and injected into Rag2−/− mice. Starting at week 1, the mice were given either LCA or vehicle control enema twice a week for 6 weeks (Figure 3E). LCA reproducibly reduced colitis disease parameters as shown by weight loss, colon length, and gross colon morphology (Figure 3F and G). LCA also significantly reduced the leukocyte infiltration and histologic inflammation of the colon (Figure 3H). Unlike LCA, vehicle control administration did not ameliorate disease.
The protective effect of LCA on DSS-induced colitis is lost in mice with TGR5 deficient immune cells
Bile acids are known to act on multiple receptors, including at least three nuclear receptors, the farnesoid X receptor (FXR), the pregnane X receptor (PXR), and the vitamin D receptor (VDR) as well as transmembrane receptors such as TGR5 (Long et al., 2017). These receptors are expressed on various cell types known to influence inflammatory processes, including macrophages, enteric neurons, and epithelial cells (Copple and Li, 2016, Poole et al., 2010). Given that DCA and LCA are the most potent natural bile acid (BA) ligands for the widely-expressed G protein-coupled receptor, TGR5, we sought to investigate its role in intestinal inflammation using knockout and congenic bone marrow chimeric mice deficient in TGR5 (Duboc et al., 2014, Yuan and Bambha, 2015).
We first tested the ability of LCA to mitigate intestinal inflammation in TGR5−/− compared to wildtype (WT) mice in the DSS-induced colitis model. The protective effect of LCA was lost in TGR5−/− mice, which was reflected by worsened colitis disease parameters as shown by weight loss, disease activity, colon length, and gross colon morphology compared to WT mice treated with LCA (Figure 4A–C). TGR5−/− mice treated with LCA also had significantly increased histologic inflammation of the colon compared to WT mice treated with LCA (Figure 4D). Unlike TGR5−/− mice, WT mice treated with LCA again continued to show amelioration of disease induced by DSS.
Figure 4. The protective effect of LCA on DSS-induced colitis is lost in mice with TGR5 deficient immune cells.
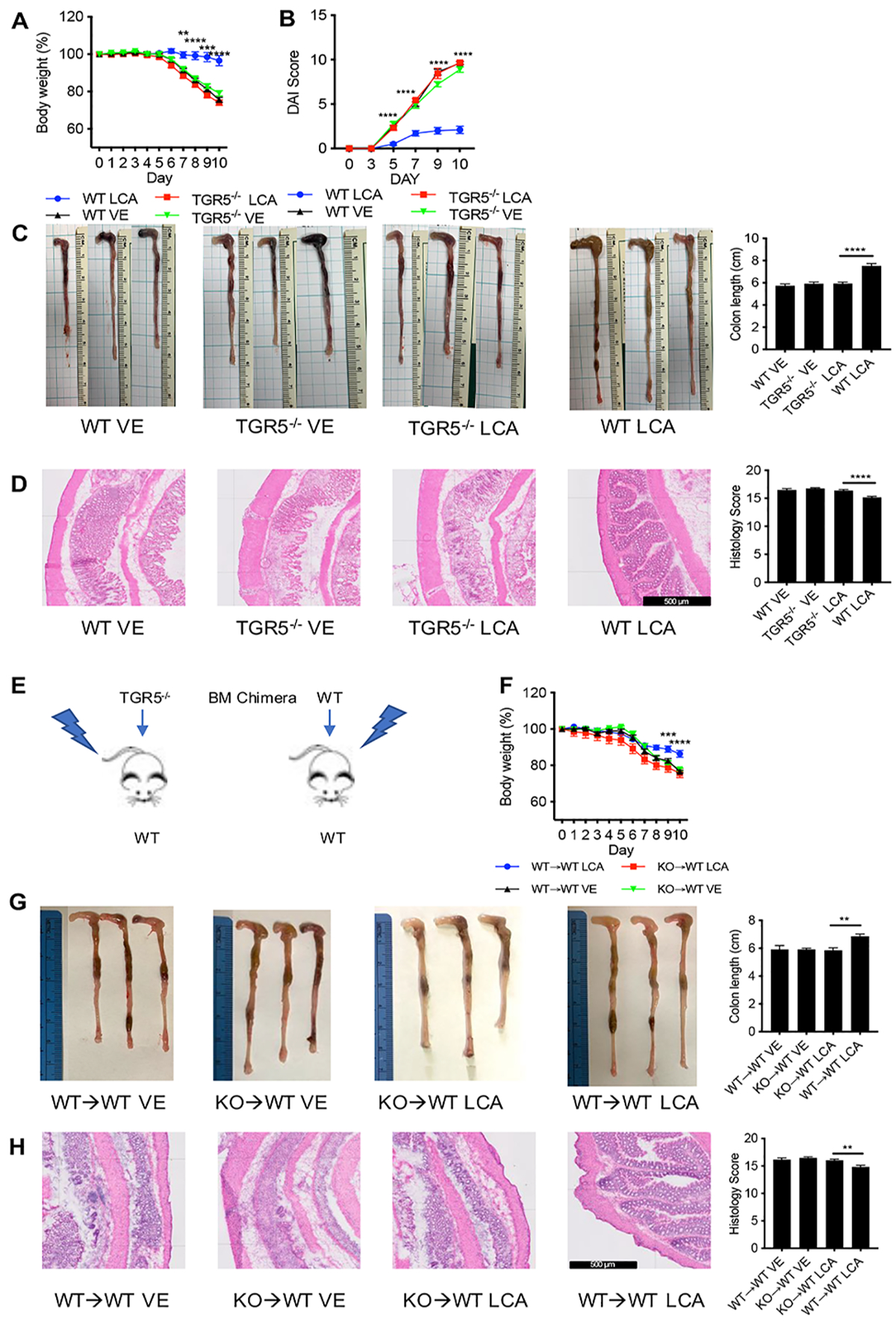
Treatment of TGR5−/− with LCA was not protective against (A) loss of body weight, (B) disease activity reflected by disease activity index (DAI), (C) shortening of colon length, and (D) inflammation and histopathology in DSS-induced colitis. (E) In chimeric mice generated by transplanting TGR5−/− (KO) bone marrow into lethally irradiate WT mice, 2.5% DSS resulted in (F) decreased body weight (G) shorter colon lengths, and (H) lower histology scores compared to mice receiving WT bone marrow grafts. Scale bar = 500 m. Data represented as mean ± SEM, analyzed by two-way ANOVA with Bonferroni’s post-hoc (A, B, and F) or one-way ANOVA with Tukey’s post-hoc (C, D, G, H). and significance reported as * P<.05, ** P<.01, *** P<.001, and **** P<0.0001. (A, C) WT VE n=15, TGR5−/− VE n=14, TGR5−/− LCA n=19, WT LCA n=19, (B) WT VE n=10, TGR5−/− VE n=9, TGR5−/− LCA n=9, WT LCA n=10, (D) WT VE n=10, TGR5−/− VE n=10, TGR5−/− LCA n=15, WT LCA n=15, (F-H) KO→WT VE n=6, WT→WT VE n=6, KO→WT LCA n=10, WT→WT LCA n=10.
To further characterize the role of TGR5 activation on immune versus non-immune cells (such as epithelial cells), we generated congenic bone marrow (BM) chimeric mice from lethally irradiated WT mice receiving BM transplantation from either TGR5−/− or WT mice (Figure 4E). Again, in DSS-induced colitis, the protective effect of LCA was lost in TGR5−/−→WT chimera, which was reflected by worsened colitis disease parameters as shown by weight loss, colon length, and gross colon morphology compared to WT→WT chimera treated with LCA (Figure 4F and G). TGR5−/−→WT chimera treated with LCA had significantly increased histologic inflammation of the colon compared to WT→WT chimera treated with LCA (Figure 4H). Furthermore, intracellular TNFα+ and IL17+ colonic leukocytes were higher in TGR5−/−→WT chimera mice treated with LCA compared to WT→WT LCA mice (Figure S6). These experiments highlight the contribution of immune cells expressing TGR5 in LCA/SBA mediated protection in DSS-induced colitis.
DISCUSSION
The most common long-term complication of the preferred surgery to cure UC—colectomy with pouch creation—is pouchitis (Shen and Lashner, 2008). While the same surgical procedure is used to treat FAP, pouchitis is rarely seen in this non-inflammatory condition. The remarkable difference in pouchitis propensity of UC versus FAP suggests constitutive differences of these two populations. We began our study with targeted metabolomics analysis to determine differences in bile acid profiles between these groups. The surgically-altered anatomy with pouch creation results in fecal stasis and colon-like transformation of ileal pouch mucosa that in itself is thought to create an environment that is more prone to inflammation (Shen and Lashner, 2008). To control for these anatomical changes we chose to investigate differences between pouches created for UC versus the non-inflammatory condition FAP. Remarkably, our data identify LCA and DCA to be almost undetectable in UC pouch patients. This striking finding in patients who underwent colectomy suggests that SBAs may play a role in dysregulated metabolism-induced intestinal inflammation. While this dramatic difference has not been previously reported in pouch patients, our findings are in line with the higher levels of SBAs in feces that has been observed in healthy subjects compared to patients with IBD (Duboc et al., 2013). These similar findings in pouchitis and IBD further suggest shared mechanisms for intestinal inflammation; in fact, pouchitis has been considered a “reactivation of UC” in the pouch ileal mucosa (McLaughlin et al., 2010, Shen, 2012).
Dysbiosis has been known to play a role in intestinal inflammation seen in IBD and pouchitis. It is also known that both bile acids and antibiotics, potentially through their alteration of PBA transformation to SBAs, alter the intestinal flora (Theriot et al., 2016, Buffie et al., 2015). Our findings confirm that significant changes in bacterial diversity and composition occur in UC versus FAP pouches. Notably, our finding of decreased Ruminococcaceae in UC compared to FAP pouch stool requires further exploration. As members of the Ruminococcaceae family, particularly C. leptum, have been shown to be significantly reduced in feces from IBD compared to healthy subjects, the therapeutic potential of Ruminococcaceae and other SBA producing-species in pouchitis and other intestinal inflammation warrants additional investigations (Gionchetti et al., 2003, Sokol et al., 2006). The therapeutic implications of correcting dysbiosis through restoration of key bacterial species (or metabolites) holds clinical promise as studies have indicated certain probiotics to be of some benefit in pouchitis (Singh et al., 2015).
While members of the Clostridiales order are known to possess the requisite 7α-dehydroxylating capability to generate SBAs, the family Ruminococcaceae, to our knowledge, has not been identified as a significant contributor to production of LCA or DCA from PBAs. In fact, while C. leptum members in the Ruminococcaceae family have been noted to produce SBAs, other non-Ruminococcaceae members such as C. scindens have been shown to perform this transformation to SBAs much more efficiently, at least by an order of magnitude (Kitahara et al., 2001). Mucosal samples could have bacterial profiles different from stool as reported in prior studies (Tang et al., 2015). While we did not evaluate microbiota changes in mucosal samples, other literature suggests high similarity of microbiota between stool and mucosa samples derived from pouches (Zella et al., 2011). In addition, there is also literature in IBD and other diseases showing high levels of microbial profile similarity between mucosal and fecal samples (Maharshak et al., 2018, Chen et al., 2014). In addition, there is potential for confounding effects caused by inflammation on both the microbiota community structure as well as on their metabolites. As we have shown, inflammation in the DSS-induced colitis model is associated with significant decrease in SBAs and these SBAs can alter bacterial communities (McKenney et al., 2019). Exactly how this inflammation affects the microbiota and metabolome in both human pouchitis and experimental colitis is currently unknown.
Our metagenomic investigation of the bai genes, which included Ruminococcaceae, confirmed reads with high homology to gene sequences in the bai operon associated with DCA and LCA production and our transcriptomics analysis showed higher expression of bai mRNA in FAP compared to UC pouch stool samples. While identifying the exact species responsible for these decreased SBAs in UC compared to FAP is beyond the scope of this investigation, strains with highest homology to C. leptum may be determinant contributors, as they were shown to be reduced in UC compared to FAP pouches. Taken together, we propose that the reduction in 7α-dehydroxylating bacteria, such as in Ruminococcaceae, results in decreased SBAs seen in UC pouch stool, which in turn promotes intestinal inflammation.
This conclusion is consistent with published findings showing a role for SBAs in intestinal inflammation (Sun et al., 2018, Duboc et al., 2013). DCA, for example, has been shown to promote the maintenance of mucosal integrity by increasing intestinal epithelial cell migration (Strauch et al., 2003). LCA and DCA have also been shown to suppress in vitro pro-inflammatory cytokine production from human peripheral blood-derived macrophages, key mediators of intestinal inflammation in IBD, through activation of the TGR5 receptor (Yoneno et al., 2013). LCA has also been recently shown to impair Th1 activation, as evidenced by reduced TNFα and IFNγ. This was shown to be mediated through the VDR, a known bile acid receptor, at physiologic concentrations (Pols et al., 2017). Clearly, additional experiments using colitis models would further our understanding of the mechanism by which intestinal homeostasis is disrupted in patients with intestinal inflammation.
We confirmed the ability of LCA and DCA to mitigate the effects of colitis in three murine models. Previous studies have shown that the bile acid FXR agonist obeticholic acid and ursodeoxycholic acid reduce gut inflammation in experimental murine colitis models at very high doses (Gadaleta et al., 2011, Martinez-Moya et al., 2013). However, we are not aware of previous studies showing LCA or DCA to alleviate murine colitis at concentrations that are physiologically relevant. The concentrations used in our per rectum treatments were roughly in line with bile acids in the intestine (mM concentrations), particularly when accounting for additional dilution that occurs when administered into rectal contents and the fact that animal retention of liquid enemas is incomplete (Raufman et al., 2003). Our Luminex analysis on colon tissue from mice with DSS-induced colitis suggests a broad anti-inflammatory role for these SBAs, and the mechanism by which they exert their effect in part appears to be through TGR5 expression by immune cells. Further investigation into the role of various bile acid receptors on immune and non-immune cells is needed to better understand their ability to mitigate intestinal inflammation and in so doing, will potentially identify promising means to treat patients with UC. In this regard, we have initiated a clinical study (ClinicalTrials.gov Identifier: NCT03724175) to investigate the role of SBAs in patients with antibiotic refractory pouchitis, a population for which there are no approved therapies. Insights from this study will further inform our understanding of the role of SBAs in intestinal inflammation and hold promise to provide an effective treatment.
STAR METHODS
Lead Contact and Materials Availability
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Aida Habtezion. aidah@stanford.edu.
Experimental Model and Subject Details
Human Studies
All studies were approved by the Stanford University Institutional Review Board (IRB). Fecal samples were collected from patients with confirmed ulcerative colitis (UC) or familial adenomatous polyposis (FAP) diagnoses who had colectomies with creation of ileal pouch. All subjects had undergone colectomy with the creation of an ileo-anal pouch. Patient recruitment was done in two cohorts. The first cohort included 10 UC and 4 FAP patients. Subsequently, a second cohort was added with 7 UC and 3 FAP patients. During the study, none of the patients were receiving bile acid binders (such as choleystyramine) or bile acid medication (such as ursodeoxycholic acid). During the study, 35.3% and 29.4% of the UC pouch were using antibiotics and probiotics, respectively as shown in Table S1. Stool samples from the 17 UC and 7 FAP were used for metabolomic studies. Stool samples were snap frozen with dry ice and stored at −80 °C until use.
Mouse Studies
C57BL/6 and Rag2−/− and, Balb/c mice were purchased from Taconic (Hudson, NY) and Jackson Laboratory respectively and housed at the Stanford research animal facility for 1 week until used in experiments. TGR5−/− mice were kindly provided to us by Merck & Co., Inc., (Kenilworth, NJ USA). TGR5−/+ male mice were mated with TGR5−/+ female mice. The resultant pups were separated based on their sex, after weaning. After genotyping, the TGR5−/− mice and the littermate wild type were housed in separate cages (5 mice per cage) during DSS treatment. All animal use and handling was performed in compliance with National Institute of Health guidelines and with approval of the Stanford University Institute of Animal Care and Use Committees.
Methods Details
Targeted Metabolomics
The targeted bile acid metabolomics analysis from both human and mouse stool was performed at the NIH West Coast Metabolomics Center (University of California, Davis). The extraction was performed according to the La Frano protocol (La Frano et al., 2017). 1 mL cold methanol containing 10 μL anti-oxidant solution, (0.2 mg/mL solution BHT/EDTA in 1:1 MeOH:water), followed by adding each surrogate (SSTD, 250 nM (deuterated bile acids for recovery calculations during data processing)). The homogenized solution was then centrifuged, dried, resuspended and filtered using 0.2um PVDF membrane (Agilent 203980–100) for LC-MS analysis. LC-MS analysis was performed on Thermo Scientific Vaquish Horizon UPLC / AB Sciex 6500+ Qtrap with targeted MRM method (developed by Newman lab, optimized for Fiehn lab setup) (La Merrill et al., 2014).
In vitro Assessment of PBA to SBA Conversion by UC and FAP Pouch Stool Cultures
Frozen stool samples (~1 mg) from FAP or UC pouch were inoculated into a 5 mL of mega medium supplemented with 100 M cholic acid-2,2,4,4-d4 (d4-CA, Sigma-614149) and chenodeoxycholic acid-2,2,4,4-d4 (d4-CDCA, Sigma- 614122) as described in the literature (Goodman et al., 2011). After incubation under strict anaerobic conditions in a Coy chamber at 37 °C for 2 days, 1 mL of aliquots were taken an d frozen at −80 °C until use. To extract the metabolites, 100 μL of bacterial cultures were mixed with 400 μL of methanol, and then centrifuged at 13,000g for 10 min at 4 °C. The cell-free supernata nts were subsequently filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), and 5 μL of the filtrate was injected into LC-MS. The compounds were separated and analyzed using Agilent UPLC Q-TOF platform as described in the literature (Studer et al., 2016). The MassHunter Qualitative Analysis Software (Agilent, version 7.0) was used for targeted feature quantification, including standard parameters.
16S rRNA Sequencing and Shotgun Metagenomics Analysis:
The Children’s Hospital of Pennsylvania (CHOP) Microbiome Center conducted DNA sequencing studies to identify bacterial and metagenomic differences between UC and FAP pouch stool. DNA was extracted using the Qiagen DNEasy PowerSoil kit (Germantown, MD). For 16S rRNA marker gene sequencing, barcoded PCR primers targeting the V1–V2 region of the 16S rRNA gene were used (forward: 5’-AGAGTTTGATCCTGGCTCAG-3’, reverse: 5’-TGCTGCCTCCCGTAGGAGT-3’). PCR reactions were carried out in quadruplicate and pooled for sequencing on the Illumina MiSeq instrument, yielding 250bp paired-end sequence reads. For shotgun metagenomic sequencing, DNA libraries were generated using the NexteraXT kit (Illumina, San Diego, CA, USA) and sequenced on the Illumina HiSeq 2500 instrument.
Bioinformatics analysis of 16S marker gene sequencing data was carried out using QIIME v1.9.1. Read pairs were joined and filtered to remove low quality sequence, using a threshold of Q19. Operational taxonomic units were selected at a 97% sequence similarity threshold using UCLUST v1.2.22. Taxonomic assignments were generated using the Greengenes reference database v13_8. Representative sequences were aligned by PyNAST and a phylogenetic tree was constructed using FastTree2. The estimated tree was used to compute UniFrac distances between each pair of samples (Lozupone and Knight, 2005). Subsequent statistical analysis was carried out in the R programming language.
Bioinformatics analysis of shotgun metagenomic sequencing data was carried out using a custom pipeline (https://github.com/PennChopMicrobiomeProgram/ShotgunPipeline). Quality filtering was carried out using Trimmomatic v0.33. Reads matching to the human genome were removed by alignment with BWA v0.7.12. Reads aligning to genes in the bai operon were identified by BLAST search, and alignment scores were inspected to ensure valid alignments. Subsequent statistical analysis was carried out in the R programming language.
Transcriptomics Analysis
RNA was extracted from stool using the RNeasy PowerMicrobiome Kit (Qiagen) according to manufacturer’s protocol. Ribosomal RNA was removed using a Ribo-Zero Epidemiology kit (Illumina). A sequencing library was prepared using a standard HT Illumina Stranded mRNA kit (Illumina). Libraries were pooled according to the manufacturer’s protocol and submitted for paired-end sequencing on an Illumina HiSeq at the Mayo Clinic in Rochester, MN. For DNA and RNA sequencing, raw reads were assessed for nucleotide quality and adapter contamination with FastQC v. 0.11.5. Contaminating sequencing adapters, terminal bases below PHRED quality score 30, and trimmed reads shorter than 80 bp were removed with Cutadapt v. 1.15. Reads identified as originating from ribosomal RNA were removed using SortMeRNA version 2.0. Reads identified as originating from the host were removed by alignment to the human genome GRCh38.p11 and with BWA v0.7.1. Remaining reads were mapped to the UniRef50 functionally annotated gene family database (accessed 12/24/17) at the protein level using the Diamond v0.9.15.116 aligner. Gene counts - obtained were imported into MATLAB. Genes with ≤1 read at all time points were excluded from further analysis. Transcriptional data was normalized with the standard DEseq2 normalization (Anders and Huber, 2010).
Dextran Sodium Sulfate Colitis Model
DSS (36,000–50,000 MW) was purchased from MP Biomedicals (Santa Ana, CA) and dissolved in drinking water to 2.5% (w/v) and given ad libitum to 8 week-old female C57BL/6 beginning on day 0 for 10 days. Body weights were recorded daily. In the control group, the mice were given regular drinking water. Solutions of deoxycholic (DCA), lithocholic (LCA) and chenodeoxycholic acid (CDCA) (Sigma-Aldrich, St. Louis, MN) were prepared by mixing with water and sonicating. Volume for all enemas was 150 μL with either 1 mg (100D7), 5 mg (5X), or 10 mg (10X) of bile acid. Enema suspensions were instilled via the rectum using a 1-mL Luer Lock syringe attached to a 23G needle and lubricated polyethylene tubing (0.048 O.D. in) on day 4, 6 and 8. Lubricated tubing was inserted 1 cm into the rectum of isoflurane-anesthetized mice. Colon tissues were harvested and colon length measured. Distal segments of the colon were used for Luminex and H&E staining. Stool was collected from the DSS and control treated mouse colons at the time of sacrifice. The stool samples were snap frozen and kept at −80 °C for metabolomic and 16S marker gene sequencing analysis. The metabolomic and 16S marker gene sequencing analysis was performed as described above.
Trinitrobenzenesulfonic Acid Colitis Model
8 week-old female Balb/c mice were pre-sensitized with 1% TNBS acid solution (Sigma-Aldrich; St. Louis, MO) in acetone by skin application. After 8 days mice were anesthetized and rectally treated with either 100 μL of 50 % ethanol (control group) or 2.5% TNBS in 50% ethanol, as previously described (Wirtz et al., 2007). On day 1, 3 and 5 after TNBS treatment, the mice received 150 μL of water (VE) or 150 μL of LCA (1 mg per rectum) dissolved in water. The body weight of the mice was measured daily. On day 7, the mice were sacrificed. Colon tissues were harvested for colon length measurement and for H&E staining as in the DSS model above.
CD45RBhi T cell Transfer Model of Colitis
Using the CD4+ T cell isolation Kit II (Miltenyi Biotec), CD4+ T cells were first enriched from spleen cells of 8 week-old WT female mice. The enriched CD4+ T cells were further sorted to naïve CD4+ T cells (CD4+, CD45RBhi, CD25lo) and Tregs (CD4+, CD45RBlo, CD25hi), as previously described (Nguyen et al., 2015). Cells were washed with sterile PBS before being injected intravenously (I.V.) into 8 week-old Rag2−/− female mice. The Rag2−/− mice received 0.5×106 naïve CD4+ T cells with or without 0.1 ×106 Tregs. 1 week following adoptive CD4+ T transfer, the mice were given 150 μL of either LCA (1 mg per rectum) or VE enema twice a week until sacrifice (week 7). The mice were weighed weekly until sacrificed. Colon tissues were harvested for assessment of colon length and histology as in the DSS and TNBS models.
TGR5 Chimeric Mice DSS-Induced Colitis
To generate congenic bone marrow chimeric mice, 8 week-old C57BL/6 WT mice were lethally irradiated with two doses 9.5-Gy radiation ~4h apart. Bone marrow cells (2×106), isolated from TGR5−/− mice, were transferred into gender matched 8 week-old irradiated mice via retro-orbital injection under anesthesia (TGR5−/−→WT). The control group received bone marrow cells from WT mice (WT→WT). Following engraftment period (8 weeks) as previously described (Nguyen et al., 2015), the chimeric mice were subjected to DSS colitis. The mice were then treated with either LCA or VE enema as in the DSS model above.
Histology
The colon was fixed in 10% buffered formalin and processed for haematoxylin and eosin (H&E) analysis (Histo-Tec Laboratory, Inc., Hayward, CA). The slides were blindly scored for intestinal inflammation on a scale of 1–15 as previously described in the literature (Nguyen et al., 2015).
DAI Score
To assess the severity of the DSS and TNBS colitis, disease activity index (DAI) was recorded by scoring the body weight loss, stool consistency, and blood in the stool, as described in the literature (Rangan et al., 2019). The loss of the body weight was scored: score 0, no body weight loss; score 1, body weight loss 1%−5%; score 2, body weight loss 5%−10%; score 3, body weight loss 10%−20%; score 4, more than 20% body weight loss. Stool consistency was measured: score 0, solid pellets; score 1, soft but pellet shaped; score 2, loose stool with some solidity; score 3, loose stool with some liquidity; score 4, diarrhea. The blood in the stool was detected using Sure-Vue Fecal Occult Blood test (Fisher, Houston, TX). Scoring was as follows: score 0, no blood detected; score 1, occult blood positive; score 2, occult blood positive with visual blood in the stool; score 3, occult blood positive with visual blood in the stool and rectal bleeding; score 4, occult blood positive with overall visual blood in the stool and rectal bleeding. The total sum from body weight, stool consistency and blood in the stool was calculated as the overall DAI score.
Luminex immunoassay
Luminex was performed at the Stanford Human Immune Monitoring Core. Briefly, mouse 38 plex kits were purchased from eBiosciences/Affymetrix (Waltham, MA) and used according to the manufacturer’s recommendations with modifications as described below. Beads were added to a 96 well plate and washed in a Biotek (Winooski, VT) ELx405 washer. Samples were added to the plate containing the mixed antibody-linked beads and incubated at room temperature for 1 hour followed by overnight incubation at 4°C with shaking. Cold and room temperature incubation steps were performed on an orbital shaker at 500–600 rpm. Following the overnight incubation, plates were washed in a Biotek ELx405 washer and then biotinylated detection antibody was added for 75 minutes at room temperature with shaking. The plate was washed as above and streptavidin-PE was added. After incubation for 30 minutes at room temperature, wash was performed and reading buffer was added to the wells. Each sample was measured in duplicate. Plates were read using a Luminex 200 instrument or Flex3D with a lower bound of 50 beads per sample per cytokine. Custom assay control beads by Radix Biosolutions (Georgetown, TX) were added to all wells.
Lipocalin 2 ELISA
Fecal samples were collected on the last day of the TNBS experiment. The fecal samples were reconstituted in PBS containing 0.1% TWEEN®−20 at a concentration of 100 mg feces/mL and vortexed for 20 min to yield a homogenous suspension (Chassaing et al., 2012). Samples were then centrifuged at 12,000 RPM for 10 minutes. Supernatant was collected and stored at −20°C until analysis. Lipocalin 2 (Lcn-2) levels were quantified using Mouse Lipocalin-2/NGAL ELISA kit according to the manufacturer’s recommendation (R&D Systems, Minneapolis, MN). The optical density was measured Spectramax iD3 (Molecular Devices Corp., Sunnyvale, CA, USA) at 450 nm. Bellow detection limit and above detection limit value was set 6000 pg/g feces and 600,000 pg/g feces, respectively.
Colon Leukocytes Isolation and Flow Cytometry
Leukocytes were isolated from the colon as described in the literature (Nguyen et al., 2015). The colons were cleansed in HBSS containing 2% BCS without Ca2+/Mg2+. The tissues were first incubated twice with HBSS containing 2% BCS 2 mM EDTA at 37°C for 20 minutes. The EDTA was then rinsed with HBSS containing 2% BCS without EDTA. The colon tissues were cut into ~0.5 cm pieces and, incubated twice with collagenase IV (Sigma Aldrich, St Louis, Mo) in 5% BCS RPMI at 37 °C (30 minutes each). The cell suspension was then collected and washed. Density gradient centrifugation with 40/80% percoll (GE Healthcare Bio Science AB, Uppsala, Sweden) was used to enrich the leukocytes at the interface. The leukocytes were then washed and stimulated with a mixture of LPS (1μg/mL) + PMA (50ng/mL) + Ionomycin (1 μg/mL) + Brefeldin A for 3 hours, before staining for flow cytometry analysis. The following dyes and antibodies were used to stain the cells: Zombie Aqua (Biolegend, San Diego, CA), CD45.2-PE/Dazzle 594 (Biolegend), CD45.1-Qdot 605 (Biolegend), IL17-Alexa 700 (Biolegend) and TNFα-Alexa 488 (Biolegend). Intracellular staining of cytokines was performed using Foxp3 fixation and permeabilization solutions per manufacturer recommendations (eBioscience). Data was acquired and analyzed on an LSRII (BD Biosciences) and FlowJo software (FlowJo, LLC), respectively.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction
RNA was extracted from the colon tissue using miRNeasy kit (Qiagen, Hilden, Germany). The RNA was converted to complementary DNA using the High-Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA). Quantitative reverse-transcription polymerase chain reaction of the cDNA was conducted on an ABI StepOne Plus real-time instrument (Thermo Fisher Scientific, Waltham, MA) using the following Applied Biosystems Powerup SYBR Green Master Mix and primers: IL17: forward, 5’-CCCTGGACTCTCCACCGCAA-3’; reverse, 5’-TCCCTCCGCATTGACACAGC-3’; IL12: forward, 5’-GGTGTCCAGGCACATCAGACC-3’; reverse, 5’-TTCTCCAGCTCCCACATGGC-3’; TNFα: forward, 5’-ATGAGCACAGAAAGCATGA-3’; reverse, 5’-AGTAGACAGAAGAGCGTGGT-3’ (Liu et al., 2016); IL6: forward, 5’-CCTCTGGTCTTCTGGAGTACC-3’; reverse, 5’-ACTCCTTCTGTGACTCCAGC-3’ (Liu et al., 2016); IL1β: forward, 5’-GCAACTGTTCCTGAACTCAACT-3’; reverse, 5’-ATCTTTTGGGGTCCGTCAACT-3’ (Tomita et al., 2016). The IL17 and IL12 primers were designed using Genscript online platform. All primers were synthesized in protein and nucleic acid facility (PAN) at Stanford. For each gene, the housekeeping gene 18s rRNA was used to normalize; the alteration in gene expression between groups was analyzed using the Pfaffl method (Pfaffl, 2001).
Quantification and Statistical analysis
Statistical analyses for the targeted metabolomics was performed using Prism 7 (GraphPad Software, Inc., La Jolla, CA) and multiple t-test with Benjamini and Hochberg posttest correction. Two-tailed t-test or Mann Whitney’s test were used in a single variable with two group comparison, one-way ANOVA with Tukey posttest or Kruskal-Wallis test were used in single-variable comparisons with more than two groups, and two-way ANOVA with Bonferroni posttest for multi-variable analyses. Differences with P < .05 were regarded as statistically significant. For analysis of 16S rRNA marker gene sequencing data, the Wilcoxon rank-sum test was used to assess differences in richness and Shannon index. The PERMANOVA test was used to assess differences in UniFrac distance between groups (Anderson, 2001). For differences in taxonomic abundance, we used a t-test of log-transformed abundance values, and applied a false discovery rate correction to account for multiple comparisons.
Data and Code Availability
The datasets for 16S rRNA sequence, the shotgun metagenomics and the transcriptomics generated during this study are available at Sequence Read Archive (SRA) with the respective accession numbers: PRJNA599442, PRJNA600008 and PRJNA600269.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| IL17-Alexa 700 | Biolegend | 560820 | |
| TNFα-Alexa 488 | Biolegend | 506313 | |
| CD45.2-PE/Dazzle 594 | Biolegend | 565390 | |
| CD45.1-Qdot 605 | Biolegend | 110737 | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| Cholic acid-2,2,4,4-d4 | Sigma-Aldrich | 614149 | |
| Chenodeoxycholic acid-2,2,4,4-d4 | Sigma-Aldrich | 614122 | |
| Dextran Sodium Sulfate | MP Biomedicals | 0216011080 | |
| Deoxycholic acid | Sigma-Aldrich | D2510 | |
| Lithocholic acid | Sigma-Aldrich | L6250 | |
| Chenodeoxycholic acid | Sigma-Aldrich | C9377 | |
| Picrylsulfonic acid solution, 5 % (w/v) in H2O | Sigma-Aldrich | P2297 | |
| Collagenase IV | Sigma-Aldrich | C5138 | |
| Zombie Aqua | Biolegend | 423102 | |
| Critical Commercial Assays | |||
| Mouse Lipocalin-2/NGAL ELISA kit | R&D Systems | DY1857 | |
| Foxp3 fixation and permeabilization solutions | Life Technologies | 00-5521-00 | |
| miRNeasy kit | Qiagen | 217084 | |
| High-Capacity RNA-to-cDNA kit | Applied Biosystems | 4387406 | |
| Permibilization Buffer (10X) | Affymetrix, Inc. | 00-8333-56 | |
| Applied Biosystems Powerup SYBR Green Master Mix | Applied Biosystems | A25742 | |
| Experimental Models: Organisms/Strains | |||
| C57BL/6 | Taconic | B6 | |
| Balb/CJ | Jackson Laboratory | 651 | |
| TGR5−/− | Merck (Gift) | Model 11695 | |
| RAG2−/− | Taconic | RAGN12 | |
| Deposited Data | |||
| 16S rRNA sequencing | SRA | PRJNA599442 | |
| Transcriptomics data | SRA | PRJNA600008 | |
| Shotgun Metagenomics | SRA | PRJNA600269 | |
| Oligonucleotides | |||
| TNFα | Forward, 5’-ATGAGCACAGAAAGCATGA-3’ | Stanford Protein and Nucleic Acid Facility (PAN) |
(Liu et al., 2016) |
| reverse, 5’-AGTAGACAGAAGAGCGTGGT-3’ | |||
| IL17 | forward, 5’-CCCTGGACTCTCCACCGCAA-3’; | Stanford Protein and Nucleic Acid Facility (PAN) |
This manuscript |
| reverse, 5’-TCCCTCCGCATTGACACAGC-3’ | |||
| IL12 | forward, 5’-GGTGTCCAGGCACATCAGACC-3’; | Stanford Protein and Nucleic Acid Facility (PAN) |
This manuscript |
| reverse, 5’-TTCTCCAGCTCCCACATGGC-3’ | |||
| IL6 | forward, 5’-CCTCTGGTCTTCTGGAGTACC-3’ | Stanford Protein and Nucleic Acid Facility (PAN) |
(Liu et al., 2016) |
| reverse, 5’-ACTCCTTCTGTGACTCCAGC-3’ | |||
| IL1β | forward, 5’-GCAACTGTTCCTGAACTCAACT-3’ | Stanford Protein and Nucleic Acid Facility (PAN) |
(Tomita et al., 2016) |
| reverse, 5’- ATCTTTTGGGGTCCGTCAACT-3’ | |||
| Software and Algorithms | |||
| Flow Jo | FlowJo LLC | https://www.flowjo.com | |
| Prism | Graphpad software Inc. | https://www-graphpad-com.laneproxy.stanford.edu/scientific-software/prism/ | |
Highlights.
Secondary bile acids (SBAs) are reduced in UC pouch patients, relative to FAP patients
Reduced Ruminococcaceae in UC pouches is associates with SBA deficiency
SBA supplementation ameliorates inflammation in animal models of colitis
The protective effect of SBAs is in part dependent on the TGR5 bile acid receptor
ACKNOWLEDGMENTS
We thank Yujun Yang and Yi Wei for their technical assistance. We thank Cuiping Li and Gotzone Garay for their assistance in recruiting patient samples. We are also thankful for the metabolomic services provided by the NIH West Coast Metabolomics Center at the University of California, Davis, the 16S and metagenomic sequencing services provided by the Children’s Hospital of Pennsylvania Microbiome Center, Flow analysis service was done on instruments in the Stanford Shared FACS Facility; tissue sectioning was performed by Stanford Animal Histology Service, and Luminex immunoassay service provided by Stanford’s Human Immune Monitoring Core. SS was supported by the Crohn’s and Colitis Foundation Career Development Award and KL2 Mentored Career Development Award of the Stanford Clinical and Translational Science Award to Spectrum (NIH KL2 TR 001083 AND NIH UL1 TR 001085). AH was supported by National Institute of Health (R01 DK101119), the Ann and Bill Swindells Charitable Trust, and Leslie and Douglas Ballinger. SS, JS, and AH were supported by the Kenneth Rainin Foundation Synergy Award. Graphical abstract was created using BioRender.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
All authors declare no competing interests.
REFERENCES
- ANDERS S & HUBER W 2010. Differential expression analysis for sequence count data. Genome Biol, 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDERSON MJ 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecology, 26, 32–46. [Google Scholar]
- BANKS C, BATEMAN A, PAYNE R, JOHNSON P & SHERON N 2003. Chemokine expression in IBD. Mucosal chemokine expression is unselectively increased in both ulcerative colitis and Crohn’s disease. J Pathol, 199, 28–35. [DOI] [PubMed] [Google Scholar]
- BEHR C, SPERBER S, JIANG X, STRAUSS V, KAMP H, WALK T, HEROLD M, BEEKMANN K, RIETJENS I & VAN RAVENZWAAY B 2018. Microbiome-related metabolite changes in gut tissue, cecum content and feces of rats treated with antibiotics. Toxicol Appl Pharmacol, 355, 198–210. [DOI] [PubMed] [Google Scholar]
- BUFFIE CG, BUCCI V, STEIN RR, MCKENNEY PT, LING L, GOBOURNE A, NO D, LIU H, KINNEBREW M, VIALE A, LITTMANN E, VAN DEN BRINK MR, JENQ RR, TAUR Y, SANDER C, CROSS JR, TOUSSAINT NC, XAVIER JB & PAMER EG 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature, 517, 205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHASSAING B, SRINIVASAN G, DELGADO MA, YOUNG AN, GEWIRTZ AT & VIJAY-KUMAR M 2012. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One, 7, e44328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN L, WANG W, ZHOU R, NG SC, LI J, HUANG M, ZHOU F, WANG X, SHEN B, K. M,A, WU K & XIA B 2014. Characteristics of fecal and mucosa-associated microbiota in Chinese patients with inflammatory bowel disease. Medicine (Baltimore), 93, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COPPLE BL & LI T 2016. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res, 104, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVLIN AS & FISCHBACH MA 2015. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nature chemical biology, 11, 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBOC H, RAINTEAU D, RAJCA S, HUMBERT L, FARABOS D, MAUBERT M, GRONDIN V, JOUET P, BOUHASSIRA D, SEKSIK P, SOKOL H, COFFIN B & SABATE JM 2012. Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil, 24, 513–20, e246–7. [DOI] [PubMed] [Google Scholar]
- DUBOC H, RAJCA S, RAINTEAU D, BENAROUS D, MAUBERT MA, QUERVAIN E, THOMAS G, BARBU V, HUMBERT L, DESPRAS G, BRIDONNEAU C, DUMETZ F, GRILL JP, MASLIAH J, BEAUGERIE L, COSNES J, CHAZOUILLERES O, POUPON R, WOLF C, MALLET JM, LANGELLA P, TRUGNAN G, SOKOL H & SEKSIK P 2013. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut, 62, 531–9. [DOI] [PubMed] [Google Scholar]
- DUBOC H, TACHE Y & HOFMANN AF 2014. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis, 46, 302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERICSSON AC & FRANKLIN CL 2015. Manipulating the Gut Microbiota: Methods and Challenges. Ilar j, 56, 205–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIORUCCI S, BIAGIOLI M, ZAMPELLA A & DISTRUTTI E 2018. Bile Acids Activated Receptors Regulate Innate Immunity. Frontiers in Immunology, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRANK DN, ST AMAND AL, FELDMAN RA, BOEDEKER EC, HARPAZ N & PACE NR 2007. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A, 104, 13780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GADALETA RM, VAN ERPECUM KJ, OLDENBURG B, WILLEMSEN EC, RENOOIJ W, MURZILLI S, KLOMP LW, SIERSEMA PD, SCHIPPER ME, DANESE S, PENNA G, LAVERNY G, ADORINI L, MOSCHETTA A & VAN MIL SW 2011. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut, 60, 463–72. [DOI] [PubMed] [Google Scholar]
- GEVERS D, KUGATHASAN S, DENSON LA, VAZQUEZ-BAEZA Y, VAN TREUREN W, REN B, SCHWAGER E, KNIGHTS D, SONG SJ, YASSOUR M, MORGAN XC, KOSTIC AD, LUO C, GONZALEZ A, MCDONALD D, HABERMAN Y, WALTERS T, BAKER S, ROSH J, STEPHENS M, HEYMAN M, MARKOWITZ J, BALDASSANO R, GRIFFITHS A, SYLVESTER F, MACK D, KIM S, CRANDALL W, HYAMS J, HUTTENHOWER C, KNIGHT R & XAVIER RJ 2014. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe, 15, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIONCHETTI P, RIZZELLO F, HELWIG U, VENTURI A, LAMMERS KM, BRIGIDI P, VITALI B, POGGIOLI G, MIGLIOLI M & CAMPIERI M 2003. Prophylaxis of pouchitis onset with probiotic therapy: a double-blind, placebo-controlled trial. Gastroenterology, 124, 1202–9. [DOI] [PubMed] [Google Scholar]
- GOODMAN AL, KALLSTROM G, FAITH JJ, REYES A, MOORE A, DANTAS G & GORDON JI 2011. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A, 108, 6252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILTON JP, XIE G, RAUFMAN JP, HOGAN S, GRIFFIN TL, PACKARD CA, CHATFIELD DA, HAGEY LR, STEINBACH JH & HOFMANN AF 2007. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol, 293, G256–63. [DOI] [PubMed] [Google Scholar]
- HATA K, ISHIHARA S, NOZAWA H, KAWAI K, KIYOMATSU T, TANAKA T, KISHIKAWA J, ANZAI H & WATANABE T 2017. Pouchitis after ileal pouch-anal anastomosis in ulcerative colitis: Diagnosis, management, risk factors, and incidence. Digestive Endoscopy, 29, 26–34. [DOI] [PubMed] [Google Scholar]
- JOHNSON MW, ROGERS GB, BRUCE KD, LILLEY AK, VON HERBAY A, FORBES A, CICLITIRA PJ & NICHOLLS RJ 2009. Bacterial community diversity in cultures derived from healthy and inflamed ileal pouches after restorative proctocolectomy. Inflamm Bowel Dis, 15, 1803–11. [DOI] [PubMed] [Google Scholar]
- JOYCE SA & GAHAN CG 2017. Disease-Associated Changes in Bile Acid Profiles and Links to Altered Gut Microbiota. Dig Dis, 35, 169–177. [DOI] [PubMed] [Google Scholar]
- KAKIYAMA G, PANDAK WM, GILLEVET PM, HYLEMON PB, HEUMAN DM, DAITA K, TAKEI H, MUTO A, NITTONO H, RIDLON JM, WHITE MB, NOBLE NA, MONTEITH P, FUCHS M, THACKER LR, SIKAROODI M & BAJAJ JS 2013. Modulation of the Fecal Bile Acid Profile by Gut Microbiota in Cirrhosis. Journal of hepatology, 58, 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG DJ, RIDLON JM, MOORE DR 2ND, BARNES S & HYLEMON PB 2008. Clostridium scindens baiCD and baiH genes encode stereo-specific 7alpha/7beta-hydroxy-3-oxo-delta4-cholenoic acid oxidoreductases. Biochim Biophys Acta, 1781, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITAHARA M, TAKAMINE F, IMAMURA T & BENNO Y 2001. Clostridium hiranonis sp. nov., a human intestinal bacterium with bile acid 7alpha-dehydroxylating activity. Int J Syst Evol Microbiol, 51, 39–44. [DOI] [PubMed] [Google Scholar]
- LA FRANO MR, HERNANDEZ-CARRETERO A, WEBER N, BORKOWSKI K, PEDERSEN TL, OSBORN O & NEWMAN JW 2017. Diet-induced obesity and weight loss alter bile acid concentrations and bile acid-sensitive gene expression in insulin target tissues of C57BL/6J mice. Nutr Res, 46, 11–21. [DOI] [PubMed] [Google Scholar]
- LA MERRILL M, KAREY E, MOSHIER E, LINDTNER C, LA FRANO MR, NEWMAN JW & BUETTNER C 2014. Perinatal exposure of mice to the pesticide DDT impairs energy expenditure and metabolism in adult female offspring. PLoS One, 9, e103337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANE ER, ZISMAN TL & SUSKIND DL 2017. The microbiota in inflammatory bowel disease: current and therapeutic insights. J Inflamm Res, 10, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU T, SHI Y, DU J, GE X, TENG X, LIU L, WANG E & ZHAO Q 2016. Vitamin D treatment attenuates 2,4,6-trinitrobenzene sulphonic acid (TNBS)-induced colitis but not oxazolone-induced colitis. Sci Rep, 6, 32889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LLOYD-PRICE J, ARZE C, ANANTHAKRISHNAN AN, SCHIRMER M, AVILA-PACHECO J, POON TW, ANDREWS E, AJAMI NJ, BONHAM KS, BRISLAWN CJ, CASERO D, COURTNEY H, GONZALEZ A, GRAEBER TG, HALL AB, LAKE K, LANDERS CJ, MALLICK H, PLICHTA DR, PRASAD M, RAHNAVARD G, SAUK J, SHUNGIN D, VAZQUEZ-BAEZA Y, WHITE RA 3RD, BRAUN J, DENSON LA, JANSSON JK, KNIGHT R, KUGATHASAN S, MCGOVERN DPB, PETROSINO JF, STAPPENBECK TS, WINTER HS, CLISH CB, FRANZOSA EA, VLAMAKIS H, XAVIER RJ & HUTTENHOWER C 2019. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature, 569, 655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LONG SL, GAHAN CGM & JOYCE SA 2017. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med, 56, 54–65. [DOI] [PubMed] [Google Scholar]
- LOZUPONE C & KNIGHT R 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol, 71, 8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MACPHERSON AJ, HEIKENWALDER M & GANAL-VONARBURG SC 2016. The Liver at the Nexus of Host-Microbial Interactions. Cell Host Microbe, 20, 561–571. [DOI] [PubMed] [Google Scholar]
- MAHARSHAK N, RINGEL Y, KATIBIAN D, LUNDQVIST A, SARTOR RB, CARROLL IM & RINGEL-KULKA T 2018. Fecal and Mucosa-Associated Intestinal Microbiota in Patients with Diarrhea-Predominant Irritable Bowel Syndrome. Dig Dis Sci, 63, 1890–1899. [DOI] [PubMed] [Google Scholar]
- MARTINEZ-MOYA P, ROMERO-CALVO I, REQUENA P, HERNANDEZ-CHIRLAQUE C, ARANDA CJ, GONZALEZ R, ZARZUELO A, SUAREZ MD, MARTINEZ-AUGUSTIN O, MARIN JJ & DE MEDINA FS 2013. Dose-dependent antiinflammatory effect of ursodeoxycholic acid in experimental colitis. Int Immunopharmacol, 15, 372–80. [DOI] [PubMed] [Google Scholar]
- MATSUOKA K & KANAI T 2015. The gut microbiota and inflammatory bowel disease. Semin Immunopathol, 37, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKENNEY PT, YAN J, VAUBOURGEIX J, BECATTINI S, LAMPEN N, MOTZER A, LARSON PJ, DANNAOUI D, FUJISAWA S, XAVIER JB & PAMER EG 2019. Intestinal Bile Acids Induce a Morphotype Switch in Vancomycin-Resistant Enterococcus that Facilitates Intestinal Colonization. Cell Host Microbe, 25, 695–705.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLAUGHLIN SD, CLARK SK, TEKKIS PP, NICHOLLS RJ & CICLITIRA PJ 2010. The bacterial pathogenesis and treatment of pouchitis. Therap Adv Gastroenterol, 3, 335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLISH BH, MCDONALD JAK, PECHLIVANIS A, ALLEGRETTI JR, KAO D, BARKER GF, KAPILA D, PETROF EO, JOYCE SA, GAHAN CGM, GLEGOLA-MADEJSKA I, WILLIAMS HRT, HOLMES E, CLARKE TB, THURSZ MR & MARCHESI JR 2019. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut, 68, 1791–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAVANEETHAN U & SHEN B 2009. Pros and cons of antibiotic therapy for pouchitis. Expert Rev Gastroenterol Hepatol, 3, 547–59. [DOI] [PubMed] [Google Scholar]
- NG SC, SHI HY, HAMIDI N, UNDERWOOD FE, TANG W, BENCHIMOL EI, PANACCIONE R, GHOSH S, WU JCY, CHAN FKL, SUNG JJY & KAPLAN GG 2018. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet, 390, 2769–2778. [DOI] [PubMed] [Google Scholar]
- NGUYEN LP, PAN J, DINH TT, HADEIBA H, O’HARA E 3RD, EBTIKAR A, HERTWECK A, GOKMEN MR, LORD GM, JENNER RG, BUTCHER EC & HABTEZION A 2015. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol, 16, 207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NYAM DC, BRILLANT PT, DOZOIS RR, KELLY KA, PEMBERTON JH & WOLFF BG 1997. Ileal pouch-anal canal anastomosis for familial adenomatous polyposis: early and late results. Annals of Surgery, 226, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PFAFFL MW 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res, 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLS TW, NOMURA M, HARACH T, LO SASSO G, OOSTERVEER MH, THOMAS C, RIZZO G, GIOIELLO A, ADORINI L, PELLICCIARI R, AUWERX J & SCHOONJANS K 2011. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab, 14, 747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLS TWH, PUCHNER T, KORKMAZ HI, VOS M, SOETERS MR & DE VRIES CJM 2017. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS One, 12, e0176715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POOLE DP, GODFREY C, CATTARUZZA F, COTTRELL GS, KIRKLAND JG, PELAYO JC, BUNNETT NW & CORVERA CU 2010. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil, 22, 814–25, e227–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POSTLER TS & GHOSH S 2017. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab, 26, 110–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMOS GP & PAPADAKIS KA 2019. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin Proc, 94, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANGAN P, CHOI I, WEI M, NAVARRETE G, GUEN E, BRANDHORST S, ENYATI N, PASIA G, MAESINCEE D, OCON V, ABDULRIDHA M & LONGO VD 2019. Fasting-Mimicking Diet Modulates Microbiota and Promotes Intestinal Regeneration to Reduce Inflammatory Bowel Disease Pathology. Cell Rep, 26, 2704–2719.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO AS, WONG BS, CAMILLERI M, ODUNSI-SHIYANBADE ST, MCKINZIE S, RYKS M, BURTON D, CARLSON P, LAMSAM J, SINGH R & ZINSMEISTER AR 2010. Chenodeoxycholate in females with irritable bowel syndrome-constipation: a pharmacodynamic and pharmacogenetic analysis. Gastroenterology, 139, 1549–58, 1558.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAUFMAN JP, CHENG K & ZIMNIAK P 2003. Activation of muscarinic receptor signaling by bile acids: physiological and medical implications. Dig Dis Sci, 48, 1431–44. [DOI] [PubMed] [Google Scholar]
- RIDLON JM, ALVES JM, HYLEMON PB & BAJAJ JS 2013. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes, 4, 382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIDLON JM, KANG DJ & HYLEMON PB 2006. Bile salt biotransformations by human intestinal bacteria. J Lipid Res, 47, 241–59. [DOI] [PubMed] [Google Scholar]
- ROOKS MG & GARRETT WS 2016. Gut microbiota, metabolites and host immunity. Nat Rev Immunol, 16, 341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARTOR RB & WU GD 2017. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology, 152, 327–339.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAAP FG, TRAUNER M & JANSEN PL 2014. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol, 11, 55–67. [DOI] [PubMed] [Google Scholar]
- SHEN B 2012. Bacteriology in the etiopathogenesis of pouchitis. Dig Dis, 30, 351–7. [DOI] [PubMed] [Google Scholar]
- SHEN B & LASHNER BA 2008. Diagnosis and Treatment of Pouchitis. Gastroenterology & Hepatology, 4, 355–361. [PMC free article] [PubMed] [Google Scholar]
- SINGH S, STROUD AM, HOLUBAR SD, SANDBORN WJ & PARDI DS 2015. Treatment and prevention of pouchitis after ileal pouch-anal anastomosis for chronic ulcerative colitis. Cochrane Database Syst Rev, Cd001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH FM, COFFEY JC, KELL MR, O’SULLIVAN M, REDMOND HP & KIRWAN WO 2005. A characterization of anaerobic colonization and associated mucosal adaptations in the undiseased ileal pouch. Colorectal Disease, 7, 563–570. [DOI] [PubMed] [Google Scholar]
- SOKOL H, SEKSIK P, RIGOTTIER-GOIS L, LAY C, LEPAGE P, PODGLAJEN I, MARTEAU P & DORE J 2006. Specificities of the fecal microbiota in inflammatory bowel disease. Inflamm Bowel Dis, 12, 106–11. [DOI] [PubMed] [Google Scholar]
- SOLBACH P, CHHATWAL P, WOLTEMATE S, TACCONELLI E, BUHL M, GERHARD M, THOERINGER CK, VEHRESCHILD M, JAZMATI N, RUPP J, MANNS MP, BACHMANN O & SUERBAUM S 2018. BaiCD gene cluster abundance is negatively correlated with Clostridium difficile infection. PLoS One, 13, e0196977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STELLWAG EJ & HYLEMON PB 1978. Characterization of 7-alpha-dehydroxylase in Clostridium leptum. Am J Clin Nutr, 31, S243–s247. [DOI] [PubMed] [Google Scholar]
- STELLWAG EJ & HYLEMON PB 1979. 7alpha-Dehydroxylation of cholic acid and chenodeoxycholic acid by Clostridium leptum. J Lipid Res, 20, 325–33. [PubMed] [Google Scholar]
- STRAUCH ED, YAMAGUCHI J, BASS BL & WANG J-Y 2003. Bile salts regulate intestinal epithelial cell migration by nuclear factor–κB–induced expression of transforming growth factor–β. Journal of the American College of Surgeons, 197, 974–984. [DOI] [PubMed] [Google Scholar]
- STUDER N, DESHARNAIS L, BEUTLER M, BRUGIROUX S, TERRAZOS MA, MENIN L, SCHURCH CM, MCCOY KD, KUEHNE SA, MINTON NP, STECHER B, BERNIER-LATMANI R & HAPFELMEIER S 2016. Functional Intestinal Bile Acid 7alpha-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front Cell Infect Microbiol, 6, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN X, WINGLEE K, GHARAIBEH RZ, GAUTHIER J, HE Z, TRIPATHI P, AVRAM D, BRUNER S, FODOR A & JOBIN C 2018. Microbiota-Derived Metabolic Factors Reduce Campylobacteriosis in Mice. Gastroenterology, 154, 1751–1763.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG MS, POLES J, LEUNG JM, WOLFF MJ, DAVENPORT M, LEE SC, LIM YA, CHUA KH, LOKE P & CHO I 2015. Inferred metagenomic comparison of mucosal and fecal microbiota from individuals undergoing routine screening colonoscopy reveals similar differences observed during active inflammation. Gut Microbes, 6, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THERIOT CM, BOWMAN AA & YOUNG VB 2016. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOMITA K, FREEMAN BL, BRONK SF, LEBRASSEUR NK, WHITE TA, HIRSOVA P & IBRAHIM SH 2016. CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Scientific reports, 6, 28786–28786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TYLER AD, KNOX N, KABAKCHIEV B, MILGROM R, KIRSCH R, COHEN Z, MCLEOD RS, GUTTMAN DS, KRAUSE DO & SILVERBERG MS 2013. Characterization of the gut-associated microbiome in inflammatory pouch complications following ileal pouch-anal anastomosis. PLoS One, 8, e66934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S, MARTINS R, SULLIVAN MC, FRIEDMAN ES, MISIC AM, EL-FAHMAWI A, DE MARTINIS ECP, O’BRIEN K, CHEN Y, BRADLEY C, ZHANG G, BERRY ASF, HUNTER CA, BALDASSANO RN, RONDEAU MP & BEITING DP 2019. Diet-induced remission in chronic enteropathy is associated with altered microbial community structure and synthesis of secondary bile acids. Microbiome, 7, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WIRTZ S, NEUFERT C, WEIGMANN B & NEURATH MF 2007. Chemically induced mouse models of intestinal inflammation. Nat Protoc, 2, 541–6. [DOI] [PubMed] [Google Scholar]
- YONENO K, HISAMATSU T, SHIMAMURA K, KAMADA N, ICHIKAWA R, KITAZUME MT, MORI M, UO M, NAMIKAWA Y, MATSUOKA K, SATO T, KOGANEI K, SUGITA A, KANAI T & HIBI T 2013. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology, 139, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUAN L & BAMBHA K 2015. Bile acid receptors and nonalcoholic fatty liver disease. World J Hepatol, 7, 2811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZELLA GC, HAIT EJ, GLAVAN T, GEVERS D, WARD DV, KITTS CL & KORZENIK JR 2011. Distinct microbiome in pouchitis compared to healthy pouches in ulcerative colitis and familial adenomatous polyposis. Inflamm Bowel Dis, 17, 1092–100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets for 16S rRNA sequence, the shotgun metagenomics and the transcriptomics generated during this study are available at Sequence Read Archive (SRA) with the respective accession numbers: PRJNA599442, PRJNA600008 and PRJNA600269.