

Abstract
Background.
Oncogenic fusions involving the neuregulin 1 (NRG1) gene are found in approximately 0.2% of cancers of diverse histologies. The resulting chimeric NRG1 proteins bind predominantly to HER3, leading to HER3-HER2 dimerization and activation of downstream growth and survival pathways. HER3 is therefore a rational target for therapy in NRG1 fusion-driven cancers.
Methods.
We developed novel patient-derived and isogenic models of NRG1-rearranged cancers and examined the effect of the anti-HER3 antibody, seribantumab, on growth and activation of signaling networks in vitro and in vivo.
Results.
Seribantumab inhibited NRG1-stimulated growth of MCF-7 cells and growth of patient-derived breast (MDA-MB-175-VII, DOC4-NRG1) and lung (LUAD-0061AS3, SLC3A2-NRG1 fusion) cancer cells harboring NRG1 fusions or NRG1 amplification (HCC-95). In addition, seribantumab inhibited growth of isogenic HBEC cells expressing a CD74-NRG1 fusion (HBECp53-CD74-NRG1) and induced apoptosis in MDA-MB-175-VII and LUAD-0061AS3 cells. Induction of pro-apoptotic proteins and reduced expression of the cell cycle regulator cyclin D1 were observed in seribantumab-treated cells. Treatment of MDA-MB-175-VII, LUAD-0061AS3 and HBECp53-CD74-NRG1 cells with seribantumab, reduced phosphorylation of EGFR, HER2, HER3, HER4 and known downstream signaling molecules such as AKT and ERK1/2. Significantly, administration of seribantumab to mice bearing LUAD-0061AS3 PDX and OV-10–0050 (ovarian cancer with CUL-NRG1 fusion) PDX tumors induced regression of tumors by 50–100%. Afatinib was much less effective at blocking tumor growth.
Conclusion.
Seribantumab treatment blocked activation of the four ERBB family members and of downstream signaling, leading to inhibition of NRG1 fusion-dependent tumorigenesis in vitro and in vivo in breast, lung and ovarian patient-derived cancer models.
Keywords: NRG1 rearrangement, HER3 targeted therapy, seribantumab, preclinical NRG1 fusion models, afatinib
Introduction
Rearrangements of the neuregulin 1 gene (NRG1, also referred to as heregulin) are rare but recurrent clinically actionable genomic alterations identified in up to 0.2% of all solid tumors (1,2). A fusion involving NRG1 was first identified in the breast cancer cell line MDA-MB-175-VII in 1999 (3). Subsequently, fusions of the NRG1 gene with various upstream partners have been identified in multiple solid tumor subtypes by several groups (1,2,4). In the largest published study that looked at the distribution of NRG1 rearrangements among different cancer types, 41 of 21,858 tumors had a fusion including 0.5% gallbladder cancer, 0.5% renal clear cell carcinoma, 0.5% pancreatic cancer, 0.4% ovarian cancer and 0.2% sarcoma (1). The incidence in non-small cell lung cancer and breast cancer is approximately 0.2% (1). NRG1 fusions have also been identified in uterine and head and neck cancers (2). More recently, Jonna et al presented at the 2020 ASCO Annual Meeting a larger dataset with 82 of 44,570 tumors (0.2%) bearing an NRG1 fusion (5).
Nearly all NRG1 gene fusions contain NRG1 exon 6, which encodes the EGF-like domain of the protein (2) and is essential for oncogenic transformation by the NRG1 chimeric protein (6). Binding of the EGF-like domain of NRG1 to the catalytically inactive ERBB3 (HER3) in an autocrine or paracrine manner leads to heterodimer formation between HER3 and other ERBB family members, particularly ERBB2 (HER2). Dimerization results in autophosphorylation and activation of the HER3 partner, which is then able to trans-phosphorylate HER3, forming docking sites for downstream signaling molecules. Together, these active ERBB complexes activate known growth and survival pathways such as the PI3K, MTOR and MAPK pathways (2,7).
Given that HER3 is required for NRG1 fusion-driven tumor growth but does not have an active kinase domain to target with small molecule antagonists, targeting the extracellular domain of HER3 is an attractive therapeutic strategy. Our prior study has shown that shRNA-mediated down-regulation of HER3 leads to death of the DOC4-NRG1 fusion-positive breast cancer cell line MDA-MB-175-VII (2), providing evidence that this essential receptor tyrosine kinase (RTK) can be exploited for therapy. While there is still no FDA-approved therapy for NRG1 fusion-driven tumors, several monoclonal antibodies (mAb) targeting HER3 are in various stages of clinical development (8,9). It was previously demonstrated that therapy with a monoclonal antibody targeting HER3 (GSK2849330) achieved a durable response in a patient with invasive mucinous adenocarcinoma of the lung harboring a CD74-NRG1 fusion (2). This agent is currently being evaluated in a phase 1 trial (NCT01966445) in patients with advanced HER3-positive solid tumors.
Seribantumab (previously called MM-121) is a fully human IgG2 mAb that competes with NRG1 for binding to HER3 (10) and antagonizes receptor signaling (10,11). This antibody was developed to target HER3 after computational models revealed that this RTK was the most critical regulator of PI3K-AKT signaling (11). Treatment of mice bearing xenografts of prostate, ovarian and renal cancer cell lines with high levels of wildtype NRG1 and phospho-HER3 reduced tumor growth significantly (12,13). A phase 2 study evaluating the efficacy of seribantumab plus erlotinib in unselected non-small cell lung cancer (NSCLC) showed that patients with higher levels of NRG1 mRNA might benefit from seribantumab therapy (14). Taken together, these studies support the use of seribantumab to treat cancers that may be dependent on NRG1–HER3 signaling.
There have been no clinical or preclinical studies specifically testing the efficacy of seribantumab in tumors arising from NRG1 fusions. In this study, we developed novel cell line and patient-derived xenograft models with NRG1 fusions and examined the effect of seribantumab on growth, apoptosis and intracellular signaling in vitro and in vivo. Seribantumab was effective at blocking NRG1-stimulated growth of MCF7 cells. In cells with endogenous NRG1 fusions, blockade of HER3 with seribantumab reduced activation of other ERBB family members (HER2, HER4 and EGFR) and the PI3-kinase-AKT-MTOR, RAS-MAPK and STAT3 pathways. Importantly, seribantumab blocked growth and induced apoptosis in NRG1 fusion models derived from breast, lung and ovarian cancers in vitro and in vivo.
Materials and Methods
Additional experimental procedures and a list of materials used are provided in Supplementary Methods.
Patient-derived cell line and xenograft development.
Patient-derived cell lines and xenografts were developed under institutional review board-approved biospecimen protocols (06–107, 14–091) and written informed consent was obtained from patients for collection of tumor material in accordance with the Belmont report. Mice were cared for, and experiments conducted in accordance with a protocol approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee and Research Animal Resource Center. The LUAD-0061AS3 PDX model was generated from samples obtained from a patient with an SLC3A2-NRG1 fusion-driven lung cancer. The patient exhibited disease progression while on treatment with afatinib (40 mg/day) at the time of sample collection. A thoracentesis was performed and pleural effusion fluid sample was obtained. Heparin was added to a final concentration of 1 mg/L fluid. All cells were isolated by centrifugation (300 × g, 5 min, in a tabletop centrifuge) and red blood cells were removed by incubating for 5 min in ACK (ammonium-chloride-potassium) lysis buffer (ThermoFisher Scientific, Grand Island, NY, A1049201). Twenty million cells were then implanted into the subcutaneous flank of six-week-old female NSG (NOD/SCID gamma) mice (Envigo, Madison, WI). The LUAD-0061AS3 cell line was generated from LUAD-0061AS3 PDX tumor tissue obtained after seven serial passages. Briefly, fresh tumors were cut into small pieces and then digested in a cocktail of tumor dissociation enzymes obtained from Miltenyl Biotec (130–095-929) in 5 mL serum-free DME:F12 media for one hour at 37 oC, with vortexing every 5–10 min. Digested samples were resuspended in 45 mL complete growth media to inactivate the dissociation enzymes and then cells were pelleted by centrifugation. Finally, cells were plated in complete growth media and allowed to propagate over multiple generations in the absence of afatinib, trypsinized when necessary to subculture and eventually only single cells remained. The OV-10–0050 PDX model was established from a surgically resected clinical sample with a CLU-NRG1 fusion (CLU exon 8 fused to NRG1 exon 6) by WuXi AppTec (Kowloon, Hong Kong) (2). PDX tumors were serially transplanted three times before a model was considered established.
Cell Lines.
The breast cancer epithelial cell lines MDA-MB-175-VII (Cat# HTB-25, RRID: CVCL_1400) and MCF-7 (Cat# HTB-22, RRID: CVCL_0031) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). MDA-MB-175-VII cells express a DOC4-NRG1 fusion (2,15). MCF-7 cells were derived from pleura effusion isolated from a patient with breast cancer and are ER positive (16). This cell line has been profiled by the Broad Institute Depmap program and does not have any NRG1 rearrangement (17). Human bronchial epithelial cells were immortalized by overexpression of CDK4 and TERT (HBEC-3KT cell line) and were obtained from Dr. John Minna (UT South Western, TX, USA) (18). A p53 C-terminal mutant was introduced into HBEC-3KT (HBECp53) as described previously (19) and a CD74-NRG1 fusion was expressed in these cells by lentiviral-mediated transduction of the cDNA. Cells expressing the fusion were selected using 200 μg/mL hygromycin. The HBECp53-SLC3A2-NRG1 cells are an unselected population in which the SLC3A2-NRG1 fusion has been introduced by CRISPR-Cas9-mediated genome editing as we have described previously for ROS1 and BRAF fusions (20,21). HCC-95 cells were obtained from Dr. William Lockwood (BC Cancer Center, Vancouver, BC, Canada, RRID: CVCL_5137) and these cells were found to have NRG1 amplification by whole exome sequencing (2). Cell lines were tested for mycoplasma every six months (MycoAltert kit, Lonza) with the most recent testing conducted six months prior to completion of the studies in this manuscript. Authenticated cell lines purchased from ATCC one year prior to the studies were expanded and stocks were frozen. A new vial of cells were thawed and used for 10–15 passages (every 2 months) and the known oncogene verified by RT-PCR each time. The identity of cell lines that were created in our laboratory was routinely confirmed by testing for the known oncogene fusion.
Growth and propagation of cell lines.
The MDA-MB-175-VII cell line was maintained in DMEM: Ham’s F12 (1:1) medium supplemented with 20% FBS. For experiments, MDA-MB-175-VII cells were plated and grown in DMEM: Ham’s F12 medium containing 10% FBS. MCF-7 cells were grown in DMEM supplemented with 10% FBS. HBECp53 cells were grown in KSM supplemented with bovine pituitary extract and EGF. Isogenic HBECp53 cell lines expressing NRG1 fusions were grown in DMEM: Ham’s F12 (1:1) medium supplemented with 10% FBS. HCC-95 cells were grown in RPMI-1640 supplemented with 10% FBS. All growth media were supplemented with 1% antibiotic (penicillin/streptomycin mixture). Cells were sub-cultured using trypsin (0.25%)/EDTA (1 mM) when stock flasks reached 75% confluency and re-plated at a 1:3 dilution. Cells were kept in a humidified incubator infused with 5% CO2 and maintained at 37oC.
Growth assays.
For the time-course experiments, cells were plated at a density of 5,000 (HCC-95) or 10,000 (all others) cells per well in 12-well tissue culture plates and then treated 24 h later (time 0) with the respective agents. For the MCF-7 growth assay, cells were treated with 1 μM seribantumab for one hour prior to incubation with 10 ng/mL NRG1-β1. Cells were trypsinized and counted at the relevant time point shown on the graphs. For dose-response studies, cells were plated at a density of 7,500–10,000 cells in white clear-bottom 96-well plates in a volume of 90 μL complete growth media and 10 μL chemicals added at 10X concentration (to achieve 1X concentration) in a final volume of 100 μL. After 96 h incubation, 10 μL alamarBlue cell viability reagent was added to achieve a final concentration of 10%. AlamarBlue is a cell-permeable pH-sensitive dye that is reduced when it enters the mitochondria and emits fluorescence at a different wavelength (22). Fluorescence was measured (Ex: 530 nm, Em: 585 nm) using a Molecular Dynamics Spectramax M2 fluorescence plate reader as we have described previously (23). In each experiment background fluorescence was determined in cells treated with 1 μM of the 20S proteasome inhibitor carfilzomib, which is toxic to most cells at high concentrations, and was subtracted from all values. There were 3–4 replicates of each condition. Relative IC50 and 95% CI values were determined by non-linear regression analysis using GraphPad Prism 8 software using either a variable slope model or in cases where inhibition was only partial, a three-parameter fit was used. The curve fitting resulted in R2>0.8 for the data sets. Each condition was assayed in triplicate in 2–5 independent experiments.
Efficacy studies in animals.
Crushed PDX tumor samples were mixed with matrigel (50%) and injected into the subcutaneous flank of six-week-old female NSG (LUAD-0061AS3) or Balb/c nude (OV-10–0050) mice. When tumors reached approximately 100–150 mm3, mice were randomly assigned to groups of 5–8 and treatment commenced. There were two mice per group with bilateral flank tumors for the protein phosphorylation/expression study in the LUAD-0061AS3 PDX model. Drugs were administered once and then tumors were collected at 2, 24, and 168 h post-treatment. Afatinib was administered by oral gavage once daily as a suspension (in 0.5% methylcellulose-0.4% Tween-80) on a 5 days-on and 2 days-off schedule. Seribantumab was administered in phosphate buffered saline by injection into the peritoneal cavity once every three days for a biweekly dosing schedule. Mice were observed daily throughout the treatment period for signs of morbidity and mortality. Tumor length and width, and animal weights were measured twice weekly. Tumor volume was calculated using the empirical formula V = length × width2 × 0.52. The percent change in tumor volume of each tumor was calculated using the formula ((V2-V1)/V1))*100 where V1 is the starting tumor volume and V2 is the final tumor volume.
RT-PCR and qPCR.
For detection of the SLC3A2-NRG1 fusion transcript, RNA was extracted using a Qiagen RNA mini kit and cDNAs were synthesized using SuperScript IV VILO (ThermoFisher) according to the manufacturer’s instructions. The SLC3A2-NRG1 fusion was detected by RT-PCR using 5’-ATGCTTGCTGGTGCCGTGGTCA-3’ (forward, SLC3A2 exon 4) and 5’-GGTCTTTCACCATGAAGCACTCCCC-3’ (reverse, NRG1 exon 6) primers. For detection of the CD74-NRG1 fusion, forward primers targeting CD74 exon 6 (5’-AGAGCTGGATGCACCATTGG-3’) were used. For detection of the CLU-NRG1 fusion, forward primer targeting CLU (5’-TGAAGACTCTGCTGCTGTTTGTG-3’) and two reverse primers targeting NRG1 (R1: 5’-GTTTTCTCCTTCTCCGCACATTT; R2: 5’-TATCTCGAGGGGTTTGAAAGGTC-3’) were used. For expression of NRG1 splice variants by qPCR, TaqMan gene expression master mix was used (ThermoFisher 4369016) with the following expression assays: NRG1α (Hs01103794_m1), NRG1β (Hs00247624_m1) and GAPDH (Hs02786624_G1). NRG1 mRNA levels are expressed relative GAPDH mRNA level. All cell line values were normalized to the HBECp53 cells.
Histology and Immunohistochemistry.
Histology and immunohistochemistry (IHC) were performed as previously described (24). Briefly, xenograft tissues were collected, fixed in 4% buffered formalin-saline at room temperature for 24 h, embedded in paraffin blocks and then sections of 4 μM thickness were mounted on glass slides. After deparaffinization, the tissue sections were subjected to hematoxylin and eosin (H&E) staining, or antigen retrieval for IHC staining. For IHC assays, slides were immersed in 3% H2O2 for 5 min, washed, then blocked for 15 min in 5% bovine serum albumin. Slides were incubated in primary antibodies overnight at 4 oC, washed and then incubated with biotinylated anti-rabbit secondary antibody using a DAB kit (Dako) for 30 min at 37 oC. The positive signals from IHC staining were detected using a diaminobenzidine (DAB) detection kit (Dako) according to the manufacturer’s instructions. Slides were stained with antibodies against WT1 (6F-H12, Dako), PR (PgR 636, Dako), p53 (318–6-11, Dako) and napsin A (MRQ-60, Roche) and counterstained with hematoxylin.
Statistical Analysis.
Tumor data sets were compared by two-way ANOVA, with Dunnett’s or Tukey’s multiple comparison test to determine significance. P<0.05 was considered a statistically significant difference between two values or data sets. All statistical analysis was conducted using GraphPad Prism 8 software (RRID: SCR_002798). The area under curve was calculated by the trapezoid rule (25) and groups were compared using one-way ANOVA. Caspase 3/7 activity was compared using Student’s t-test. All experiments consisted of 2–3 replicates per condition and data is expressed as mean ± SD or SEM.
Results
Expression of NRG1 alpha and beta isoforms in patient-derived cell lines with NRG1 alterations.
Oncogenic NRG1 fusions retain only a small part of NRG1 and this portion invariably includes the EGF-like domain. This domain in NRG1 exists in two forms, namely the alpha and beta isoforms. To comparatively assess the expression level of NRG1 in the different cell lines, we focused on the EGF-like domain as this is required for transformation and used isoform-specific qPCR assays. Cancer cell lines with NRG1 fusion or NRG1 amplification were compared to cells without an NRG1 alteration. This was achieved by qPCR analysis using TaqMan assays that were specific for each of the alpha and beta splice variants of NRG1. The breast cancer cell line MDA-MB-175-VII harbors a chromosomal translocation between NRG1 and DOC4 and the lung cancer cell line LUAD-0061AS3 harbors a translocation between NRG1 and SLC3A2. Expression of the DOC4-NRG1 and SLC3A2-NRG1 fusions in the cell lines was confirmed by RT-PCR (Figure 1A and B). The HCC-95 cell line is a lung cancer cell line that has amplification of NRG1 (2). For comparison we used the MCF-7 breast cancer cell line and HBECp53 cell line (untransformed immortalized human bronchiolar epithelial cells); neither cell lines are known to harbor any NRG1 alteration. All cell lines expressed NRG1α and NRG1β mRNAs at varying levels (Supplementary Figure S1A). The mRNA level in each cell line is expressed relative to corresponding mRNA in HBECp53 cells. The MCF-7 cells were found to have the lowest expression of NRG1 isoforms. HCC-95 expressed very high levels of NRG1α and NRG1β mRNA, likely due to the NRG1 amplification. Whereas HCC-95 cells had the highest level of NRG1α mRNA expression compared to cell lines with NRG1 fusions and the control cells (Supplementary Figure S1B), the LUAD-0061AS3 had the highest level of NRG1β mRNA. HCC-95 cells had 14-fold more NRG1β mRNA than the MDA-MB-175-VII cells. These results suggest that cell lines with NRG1 alterations express both NRG1 isoforms. However, the limited number of cell lines analyzed suggests that caution should be exercised in interpreting these results.
Figure 1. Seribantumab inhibits growth of cells harboring NRG1 alterations.
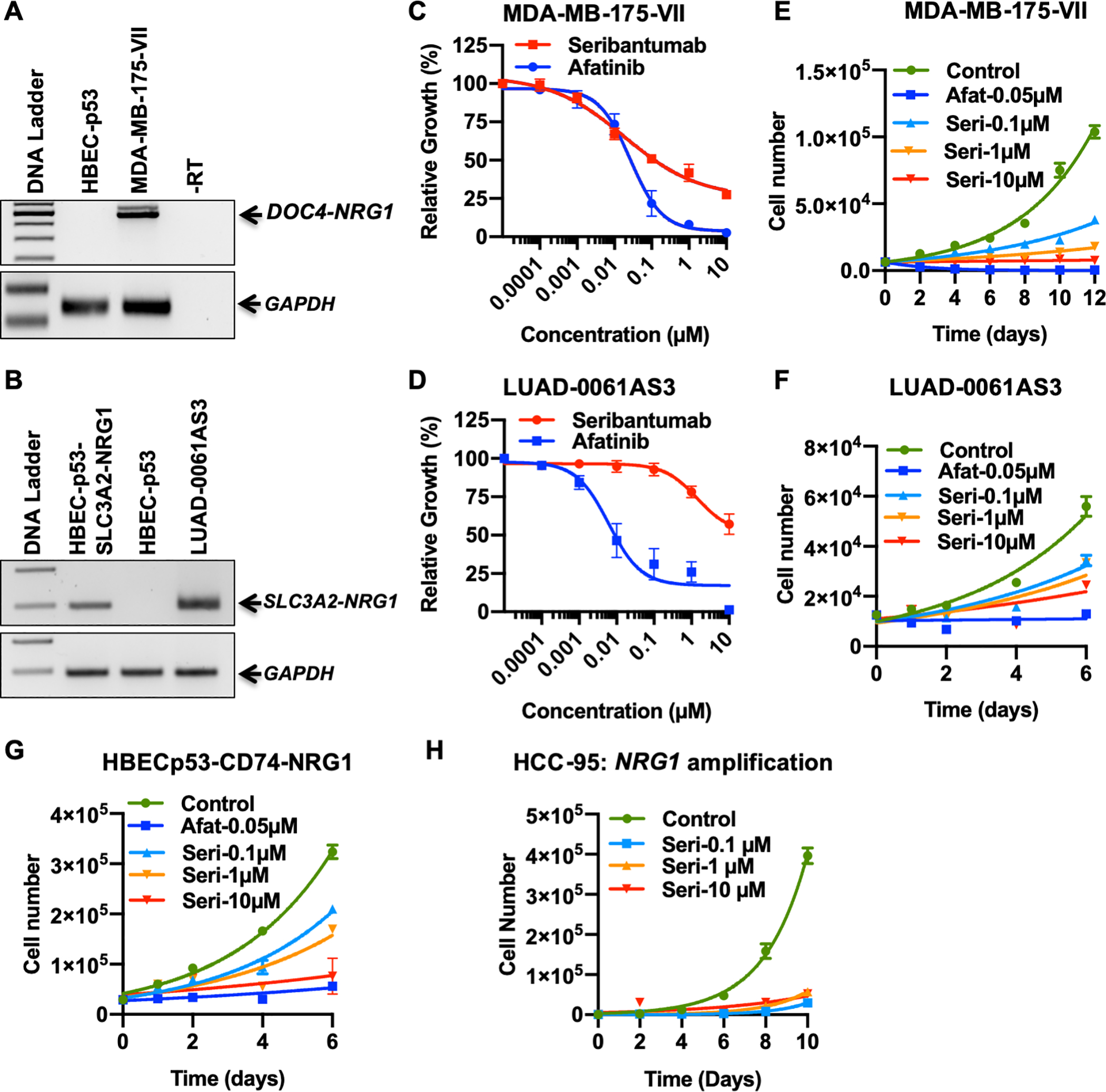
Expression of DOC4-NRG1 (A) or SLC3A2-NRG1 (B) fusions in MDA-MB-175-VII and LUAD-0061AS3 cells, respectively, was determined by RT-PCR. The HBECp53 (NRG1 fusion-negative) and HBECp53-SLC3A2-NRG1 cells were used as negative and positive controls, respectively. (C-D) Cells were treated with the indicated concentrations of seribantumab or afatinib for 96 h and then the relative number of cells estimated with alamarBlue viability dye. Viability results represent the mean ± SEM of 2–5 independent experiments in which each condition was assayed in triplicate determinations. Viability data was analyzed by non-linear regression and the IC50 values for growth inhibition and the 95% confidence interval were determined with GraphPad Prism 8 and are given in Supplementary Figure S2C. (E-H). Cells were treated as indicated with afatinib or seribantumab and then counted every 24–48 h. Results represent the mean ± SD for one experiment in which each condition was assayed in duplicate. Afat: afatinib. Seri: seribantumab.
Seribantumab inhibits growth of cells harboring NRG1 alterations
Cells expressing NRG1 fusions rely on activation of HER3 for growth and survival (2). Here we evaluated the ability of seribantumab to inhibit the growth of two cell lines that harbor NRG1 rearrangements (MDA-MB-175-VII, DOC4-NRG1 fusion and LUAD-0061AS3, SLC3A2-NRG1 fusion) in comparison to tumor and non-tumor cell lines without an NRG1 fusion (MCF-7 and HBECp53, respectively). Treatment of the two NRG1 fusion-positive cell lines with seribantumab or afatinib reduced growth in a dose-dependent manner (Figure 1C and D). Both seribantumab and afatinib had minimal effect on the growth of MCF-7 breast cancer cells (Supplementary Figure S2A) or HBECp53 cells (Supplementary Figure S2B). The estimated IC50 values obtained for growth inhibition are given in Supplementary Figure S2C. MDA-MB-175-VII (IC50 = 0.02 μM) and LUAD-0061AS3 (IC50 = 1.4 μM) cells were approximately 2260- and 32.3-fold more sensitive to seribantumab than MCF-7 cells (IC50 = 45.2 μM), respectively. Similarly, MDA-MB-175-VII and LUAD-0061AS3 cells were approximately 10,000- and 145-fold more sensitive to seribantumab than the non-tumor HBECp53 cells (IC50 = 203 μM).
To further explore the temporal nature of cell growth inhibition by seribantumab, cells were treated for up to 12 days with vehicle, seribantumab (0.1, 1, 10 μM) or afatinib (0.05 μM) and then proliferation was estimated. In these experiments, we used the MDA-MB-175 and LUAD-0061AS3 cell lines, isogenic HBECp53 cells ectopically expressing a CD74-NRG1 fusion, and the NRG1-amplified lung cancer cell line HCC-95 (2) (Figure 1E–H). RT-PCR confirmed the presence of the CD74-NRG1 fusion in the HBECp53-CD74-NRG1 cells (Supplementary Figure S2D). The doubling time is given in Supplementary Figure S3. Seribantumab slowed the growth of the MDA-MB-175-VII cells as early as 24 h after treatment was initiated, and growth was blocked for the entire 12-day period of the experiment by the 1 and 10 μM concentrations (Figure 1E). Similar results were obtained with the LUAD-0061AS3 cells (Figure 1F). Although the HBECp53-CD74-NRG1 cells were less sensitive to seribantumab than LUAD-0061AS3 and MDA-MB-171-VII cells, we nevertheless observed an almost complete inhibition of growth at the highest concentration of seribantumab (Figure 1G). The HCC-95 cells were the most sensitive to seribantumab, with growth completely inhibited even at the lowest antibody concentration (Figure 1H). Afatinib treatment (0.05 μM) was also effective at inhibiting growth of the three cell lines with NRG1 rearrangements (Figure 1C–G). We did not examine afatinib sensitivity of HCC-95 cells in this study. These results suggest that seribantumab effectively inhibits growth of tumor cell lines that harbor NRG1 fusions or NRG1 amplification.
Seribantumab specifically inhibits NRG1-dependent cell growth.
First, we sought to confirm that NRG1 could activate known mitogen activated pathways in MCF-7 cells. To this end, cells were treated with increasing concentrations of NRG1-β1 (the EGF-like domain) for 10 min and then protein phosphorylation was determined by Western blotting (Figure 2A). Treatment of MCF-7 cells with NRG1-β1 caused a dose-dependent increase in phosphorylation of EGFR, HER3 and HER4. Increased phosphorylation of the three receptors was observed with as little as 10 ng/mL NRG1-β1, with phosphorylation of EGFR being the least sensitive. This was accompanied by an increase in phosphorylation of AKT, ERK1/2, and elements of the MTOR pathway including ribosomal protein S6 (Figure 2A). Next, we examined the ability of seribantumab to block NRG1-stimulated growth of MCF-7 cells. Cells were simultaneously treated with varying concentrations of NRG1-β1 (0–5 ng/mL) and seribantumab (0–0.5 μM) for 96 h and then viability was determined. Treatment of MCF-7 cells with NRG1-β1 resulted in a significant increase in cell viability likely due to enhanced proliferation (Figure 2B). The lowest concentration of seribantumab used (0.125 μM) largely suppressed growth of NRG1-β1-stimulated MCF-7 cells. This was further explored in temporal studies in which MCF-7-cells were pretreated with 2 μM seribantumab for one hour prior to addition of 10 ng/mL NRG1-β1 for up to 10 days and growth assessed. Seribantumab pretreatment prevented NRG1-β1-stimulated growth completely (Figure 2C). The results demonstrate that inhibiting HER3 with seribantumab effectively blocks NRG1-dependent cell proliferation.
Figure 2. Seribantumab specifically blocks NRG1-dependent growth and induces apoptosis.

(A) MCF-7 cells were treated with the indicated concentrations of NRG1-β1 for 10 min and then cell extracts were prepared and subjected to Western blotting for the indicated phosphorylated (p) or total (t) proteins. (B) MCF-7 cells were treated with escalating doses of NRG1-β1 and seribantumab for 96 h and then growth determined using alamarBlue viability dye. Data was analyzed by non-linear regression using GraphPad Prism 8. There were four replicates of each condition and data represent the mean ± SD. (C) MCF-7 cells were pretreated with 2 μM seribantumab for 1 h prior to stimulation with 10 ng/mL NRG1-β1. Cells were counted on the days indicated on the graph. Results represent the mean ± SD of duplicates of each condition in one representative experiment. MDA-MB-175-VII (D) and LUAD-0061AS3 (E) cells were treated with the indicated concentrations of inhibitors for 48 h and then caspase 3/7 enzymatic activity measured in cell homogenates. Carfilzomib (20S proteasome inhibitor) was used as a positive control for apoptosis. Results are the mean ± SEM of three independent experiments in which each condition was assayed in three replicate determinations. The absence of an error bar for any data point indicates an error value too small to be represented on the scale used.
Seribantumab induces apoptosis in cells harboring NRG1 rearrangements.
To examine if seribantumab can induce cell death, we measured caspase 3/7 enzymatic activity in cell homogenates as a surrogate for apoptosis. MDA-MB-175-VII and LUAD-0061AS3 cells were treated with 0–10 μM seribantumab or afatinib for 48 h. As a positive control for activation of caspase 3/7, 1 μM carfilzomib was used. A dose-dependent increase in caspase 3/7 activity in cells treated with afatinib or seribantumab was observed (Figure 2D). Afatinib was more effective at activating caspase 3/7 than seribantumab at lower concentrations in MDA-MB-175-VII. However, at the 10 μM concentration, afatinib and seribantumab were equally effective at activating caspase 3/7 (afatinib: 14.1 ± 3.6-fold above control, seribantumab: 12.7 ± 4.2-fold above control) and comparable to the level of caspase 3/7 activity stimulated by carfilzomib (16.6 ± 1.9-fold above control). Although afatinib and seribantumab stimulated caspase 3/7 activity to a similar extent at the highest concentration used in LUAD-0061AS3 (Figure 2E), the magnitude of the response was much less than that observed in MDA-MB-175-VII cells (afatinib: 3.3 ± 0.1-fold above control, seribantumab: 4.0 ± 0.3-fold above control). This may reflect a less active apoptosis pathway in LUAD-0061AS3 cells since carfilzomib also stimulated caspase 3/7 less in this cell line (5.8 ± 1.3-fold above control) compared to MDA-MB-175-VII cells. These results suggest that seribantumab can induce apoptosis in a dose-dependent manner in NRG1 fusion-positive breast and lung cancer cell lines.
Seribantumab inhibits phosphorylation of downstream mediators in cells with NRG1 alterations
To investigate the cellular signaling networks affected by seribantumab, the phosphorylation states of EGFR, HER2, HER3, HER4, and elements of the PI3K, MTOR and MAPK pathways were examined by Western blotting following treatment of serum-starved LUAD-0061AS3, HBECp53-CD74-NRG1 and MDA-MB-175-VII cells with the indicated concentrations of seribantumab (Figures 3 and 4A). Treatment of LUAD-0061AS3 cells with seribantumab resulted in almost complete inhibition of phosphorylation of EGFR, HER2, HER3, HER4, AKT and STAT3 (Figure 3A). Phosphorylation of ERK1/2 was less sensitive to seribantumab treatment. The inhibitory effect of seribantumab on protein phosphorylation was similar to that obtained with afatinib in most instances (Figure 3A). In HBECp53-CD74-NRG1 cells, seribantumab treatment completely inhibited HER3 phosphorylation and reduced phosphorylation of HER2, EGFR and HER4 to a lesser extent (Figure 3B). Similar to observations in LUAD-0061AS3 cells, phosphorylation of AKT, p70S6K and STAT3 were almost completely inhibited by seribantumab treatment (Figure 3B). In MDA-MB-175-VII cells, seribantumab fully inhibited phosphorylation of HER3, HER2, EGFR and HER4 and reduced phosphorylation of AKT, ERK1/2 and STAT3 to a large extent (Figure 4A). Neither seribantumab nor afatinib had any effect on expression of any protein after the treatment, suggesting that loss of phosphorylation observed in response to seribantumab treatment was due entirely to a block in signal transduction. In HCC-95 cells, seribantumab treatment also inhibited phosphorylation of HER2, HER3 and downstream effectors with little effect on EGFR phosphorylation (Supplementary Figure S4). Taken together, these results suggest that treatment with seribantumab can disrupt HER3-dependent signaling, block phosphorylation of ERBB receptors and downstream signaling, reduce expression of cell cycle proteins, and induce expression of pro-apoptotic proteins. These events likely culminate in inhibition of growth and impaired survival.
Figure 3. Seribantumab inhibits intracellular signaling in lung cancer cell lines with NRG1 fusion.

Serum-depleted LUAD-0061AS3 (A) and HBECp53-CD74-NRG1 (B) cells were treated with the indicated concentrations of seribantumab or afatinib for 1 h. Whole-cell extracts were prepared after all treatments and subjected to SDS-PAGE followed by immunoblotting for the phosphorylated (p) or total (t) proteins shown in each panel. All Western blotting studies were conducted at least two times and representative immunoblots of phosphorylated (p) and total (t) proteins are shown.
Figure 4. Seribantumab inhibits intracellular signaling in breast cancer cells with NRG1 fusion.

(A) Serum-depleted MDA-MB-175-VII cells were treated with the indicated concentrations of seribantumab for 3 h. (B-D) Serum-depleted MDA-MB-175-VII cells were treated with 2 μM seribantumab for up to 24 h. Whole-cell extracts were prepared after all treatments and subjected to SDS-PAGE followed by immunoblotting for the phosphorylated (p) or total (t) proteins shown in each panel. All Western blotting studies were conducted at least two times and representative immunoblots of phosphorylated (p) and total (t) proteins are shown.
To obtain a more comprehensive understanding of the mechanistic action of seribantumab, the temporal relationship between seribantumab treatment and phosphorylation of signaling proteins or expression of proteins that regulate apoptosis and the cell cycle were evaluated. Serum-deprived MDA-MB-175-VII cells were treated with 2 μM seribantumab for up to 24 h and then whole-cell extracts were prepared and subjected to Western blotting. Seribantumab treatment rapidly reduced phosphorylation of HER3, HER4, and downstream signaling, with full inhibition observed 30 min after treatment was initiated (Figure 4B). Phosphorylation of AKT remained completely inhibited for the entire 24 h treatment period even though there appears to be a slight increase in HER3, HER4, and p70S6 kinase phosphorylation at the 12 and 24 h time points. Phosphorylation of MEK1/2 and ERK1/2 was inhibited rapidly but reactivation was seen earlier than was observed for HER3 and HER4 (Figure 4B). Expression of the pro-apoptotic proteins cleaved-PARP and PUMA was elevated by seribantumab treatment in a time-dependent manner (Figure 4C) and remained elevated from 6–24 h. This is in agreement with observations shown in Figure 2D highlighting that seribantumab induced activation of caspase 3/7 by 48 h of treatment. The level of cyclin D1, a protein that permits transit through the G1 phase of the cell cycle (26), was reduced in seribantumab-treated cells by 1 h and was undetectable by 6 h (Figure 4D). As observed with phosphorylation of some proteins, cyclin D1 level began to be restored by 12 h (Figure 4D).
Seribantumab treatment induces tumor regression in an NSCLC PDX model with an SLC3A2-NRG1 rearrangement.
The inhibition of growth of cell lines with NRG1 fusions and NRG1-stimulated MCF-7 cells by seribantumab supported the evaluation of seribantumab efficacy in vivo. We generated an NSCLC PDX model from a patient with invasive mucinous adenocarcinoma harboring an SLC3A2-NRG1 fusion. Histological characterization of the PDX tumors is shown in Figure 5A. As expected, the tumor was positive for TTF-1 (lung adenocarcinoma marker) and showed membranous phospho-HER3 staining as demonstrated previously (27).
Figure 5. Efficacy of seribantumab in an NSCLC PDX model with NRG1 fusion.
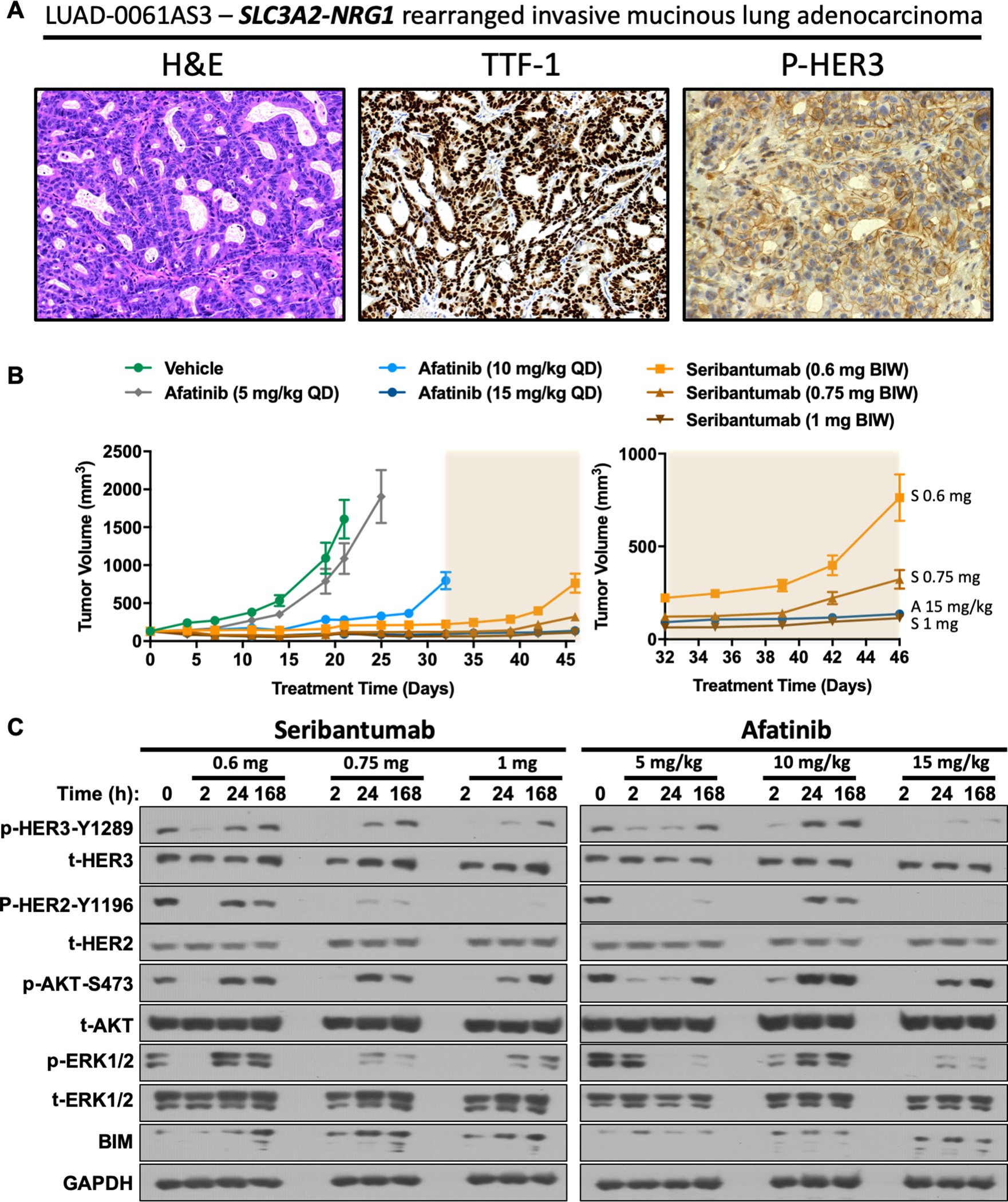
(A) Characterization of the LUAD-0063AS1 PDX model. Hematoxylin and eosin staining, TTF-1 and phospho-HER3 IHC (left to right). (B) Mice bearing LUAD-0061AS3 PDX tumors (7 animals per group) were treated with the indicated doses of afatinib (QD) or seribantumab (BIW). Tumor volumes were measured twice weekly and plotted over time. Results represent the mean ± SEM. The right panel shows a zoom-in on the last 14 days of treatment (shaded area) to show that 1 mg seribantumab is as effective as the highest dose of afatinib. A: afatinib; S: seribantumab. Individual tumor measurements showing the percent change in tumor volume is shown in Supplementary Figure S5B. (C) Mice bearing LUAD-0061AS3 PDX tumors were treated with a single administration of vehicle, afatinib or seribantumab and then tumors collected at 2, 24, or 168 h. Western blotting was performed twice for each protein and representative immunoblots of phosphorylated (p) and total (t) proteins are shown.
LUAD-0061AS3 PDX tumors were implanted into the subcutaneous flank of immunocompromised mice (7 animals per group) and treatment was initiated two weeks later with seribantumab (0.6 mg, 0.75 mg, or 1 mg per dose, twice weekly) or afatinib (5 mg/kg, 10 mg/kg, or 15 mg/kg, once daily). The 5 mg/kg daily dose of afatinib is equivalent to the human dose of 50 mg daily, which is the maximum approved dose for patients. Seribantumab is being evaluated at 3000 mg weekly in a new Phase 2 clinical study (CRESTONE, NCT04383210), which is equivalent to a dose of 11.5 mg two times per week in mice.
The tumor volume as a function of time is illustrated in Figure 5B, and the area under curve was computed for each group to facilitate comparison of tumor volume between groups at the last date all groups had surviving animals (day 35) is shown in Supplementary Figure S5A. The 5 mg/kg afatinib dose caused a small but significant reduction in tumor growth (Figure 5B and Supplementary S5A). However, higher doses of afatinib and all doses of seribantumab tested caused a bigger decrease in tumor volume (Figure 5B and Supplementary S5A). Treatment with 0.75 mg or 1 mg seribantumab resulted in regression of 4/7 and 6/7 tumors, respectively (Supplementary Figure S5B). Tumors in the 1 mg seribantumab group continued to shrink, resulting in a maximum tumor reduction of 57.2 ± 2.6% by day 42 and this was maintained for another two weeks. The highest dose of afatinib (15 mg/kg) was also effective at causing regression of 6/7 tumors in the group (Supplementary Figure S5B). However, the maximum response to the 15 mg/kg afatinib dose was not sustained for more than a few days and a slow tumor regrowth was observed while the animals were still receiving treatment. The 1 mg BIW seribantumab dosage, which is lower than the human dosage, was as effective as the 15 mg/kg QD dose of afatinib (Figure 5B, right). No treatment caused any statistically significant reduction in animal weight (Supplementary Figure S5C) or negatively influenced animal health in any noticeable way. Taken together, these results suggest that seribantumab is more effective at reducing tumor growth than afatinib.
Seribantumab treatment blocks phosphorylation of known growth modulators and induces expression of apoptosis markers in vivo.
The data presented above indicate that seribantumab effectively reduces growth and blocks activation of growth-promoting pathways in cancer cell lines with NRG1 fusions and abrogated growth of NRG1 fusion-positive cell lines and LUAD-0061AS3 PDX tumors, irrespective of tissue of origin or fusion partner. Phosphorylation of HER2, HER3, AKT, and ERK1/2 was examined to evaluate the ability of seribantumab and afatinib to interfere with NRG1-dependent signaling. Animals bearing LUAD-0061AS3 PDX tumors were given a single administration of seribantumab (0.6 mg, 0.75 mg or 1 mg) or afatinib (5 mg/kg, 10 mg/kg or 15 mg/kg) and then tumors removed at 2, 24, or 168 hours post-drug administration. Protein phosphorylation was then detected by Western blotting of PDX tumor lysates.
As shown in Figure 5C (left panel) all doses of seribantumab resulted in reduced phosphorylation of HER2, HER3, AKT and ERK1/2 by the 2 h time point, with higher doses being more effective at the longer time points. Although there was some reactivation of phosphorylation of HER3, AKT, and ERK1/2 at the later time points, HER2 phosphorylation remained inhibited even after 168 h of treatment at higher doses. Similarly, afatinib reduced phosphorylation of HER2, HER3, AKT, and ERK1/2 with the best effect seen with the highest dose studied (15 mg/kg). With the exception of HER2, reactivation of protein phosphorylation was observed at the later time points (Figure 5C, right panel). At 5 mg/kg in mice, a dose that is equivalent to that used clinically, afatinib was able to inhibit HER2 phosphorylation completely by 2 h and caused a major loss in HER3 phosphorylation.
The ability of seribantumab and afatinib to induce expression of the pro-apoptotic protein BIM in the same tumor lysates probed above for protein phosphorylation was next examined. Induction of BIM expression was clearly seen after 24 h of treatment with all doses of seribantumab. Tumors isolated from mice treated with 0.75 and 1 mg seribantumab had higher levels of BIM than vehicle-treated tumors by two hours after drug administration. Elevated BIM was present at the 168 h time point following administration of all doses of seribantumab studied. Only the higher doses of afatinib were able to induce sustained BIM expression, but to a lesser degree than that elicited by seribantumab (Figure 5C, right panel). Importantly, the clinically relevant dose of afatinib (5 mg/kg once daily in mice) caused a small but transient increase in BIM level.
Seribantumab treatment induces complete tumor regression in an HGSOC PDX model with a CLU-NRG1 rearrangement.
High-grade serous ovarian cancer (HGSOC) accounts for 70–80% of ovarian cancer deaths, and overall survival has not changed significantly for several decades (28). Seribantumab was previously shown to block growth of xenograft tumors generated from OVCAR8 cells (12), which exhibit HGSOC histology (29). Here we examined the efficacy of seribantumab in an ovarian PDX model (OV-10–0050), which was derived from a surgically resected ovarian tumor and harbors a CLU-NRG1 fusion (2). RT-PCR confirmed the presence of the CLU-NRG1 fusion (Supplementary Figure S6A). Xenograft tissue morphology and immunohistochemistry markers (positive for WT1 and strong nuclear staining for TP53) are consistent with HGSOC histology (30) (Figure 6A). Mice bearing OV-10–0050 PDX tumors (5–8 animals per group) were treated with 1, 2.5, 5, or 10 mg seribantumab (twice weekly) or 5 mg/kg afatinib (once daily) and tumor growth assessed. As with the above experiments in the NSCLC PDX model, the doses of seribantumab used here are lower than the dose that is used in patients. Treatment was terminated at day 27 and tumor growth was monitored for an additional 63 days (90 days after initiation of treatment or once tumors reached the maximum allowable size). The tumor volume as a function of time is illustrated in Figure 6B, and the area under curve was computed for each group to compare tumor volumes between groups at the last date of treatment (Supplementary Figure S6B). Afatinib treatment caused a small but significant decrease in tumor volume (p=0.003) (Figure 6B and Supplementary Figure S6B). Seribantumab administration rapidly inhibited growth of OV-10–0050 PDX tumors leading to significant tumor shrinkage at all doses tested (Figure 6B and Supplementary Figure S6B). The average tumor volumes at the time of the last treatment was: 983.7 ± 254.5 (vehicle); 786.4 ± 190.5 (afatinib); 1.3 ± 0.3 (1 mg seribantumab); 1.9 ± 0.6 (2.5 mg); 17.9 ± 14.5 (5 mg seribantumab); 2.1 ± 0.6 (10 mg seribantumab) and is illustrated in Figure 6C as the percent change in tumor size. Following treatment cessation, tumors that were previously treated with seribantumab continued to shrink, while tumors in the vehicle and afatinib-treated groups continued to grow (Figure 6B, right). Forty-one days after treatment began, mice bearing vehicle- and afatinib-treated tumors were sacrificed due to the high tumor burden. By day 73 (46 days after treatment termination), tumors started to regrow in the 1, 2.5 and 5 mg groups. However, only 1 of 8 tumors started to regrow in the two highest dose groups at the end of the study, suggesting that seribantumab likely eliminated the vast majority of tumor cells. No treatment caused any significant change in overall animal health or weight (Supplementary Figure S6C).
Figure 6. Efficacy of seribantumab in an ovarian cancer PDX model with NRG1 fusion.

(A) Immunohistochemical characterization of the OV-10–0050 PDX model. Hematoxylin and eosin staining, WT1 and TP53 IHC (left to right). (B) Mice bearing OV-10–0050 PDX tumors (5–8 animals per group) were treated with vehicle, afatinib (5 mg/kg QD) or the doses of seribantumab shown (BIW). Treatment was terminated on day 27 and animals were monitored for tumor regrowth until tumors reached maximum allowable size or until 90 days after treatment initiation. Results represent the mean tumor volume ± SEM. The right panel shows a zoom-in view on tumor volumes during the last 40 days of monitoring of seribantumab-treated groups. The highest dose of seribantumab blocked tumor regrowth after cessation of treatment. (C) Change in the volume of individual tumors (day 27 vs. volume at start of treatment).
Discussion
The NRG1 fusion gene encodes a chimeric protein that engages HER3 to drive tumorigenesis irrespective of histology, and therefore, targeting HER3 for therapy of NRG1 fusion-positive cancers constitutes a rational therapeutic strategy that can be exploited. Currently no FDA approved therapy for patients with NRG1 fusion-driven cancers exits. The only HER3-specific targeted agent in clinical trials for this group of malignancies is the monoclonal anti-HER3 antibody seribantumab (13).
By utilizing novel disease models that represent NRG1 fusion-driven lung, breast, and ovarian cancers (each with a different NRG1 fusion partner), we analyzed the effect of seribantumab on growth, apoptosis and activation state of signaling molecules that regulate proliferation, cell cycle progression and survival. We report that an anti-HER3 antibody (seribantumab) is able to block activation of all ERBB family members in NRG1 fusion-positive cell lines, similar to observations with afatinib. This prominent blockade of the ERBB family resulted in loss of downstream activation of the PI3K-AKT, MTOR, and ERK pathways, culminating in a significant reduction of proliferation and induction of apoptosis. In cells in culture, afatinib was more effective at inhibiting growth than seribantumab. The reason for this difference is unclear. It is possible that the presence of other growth factors in culture media may dampen the in vitro efficacy of seribantumab.
In two in vivo PDX models, seribantumab administration, at a dosage lower than that used in human trials, led to substantial tumor regression of more than 50% in a NSCLC PDX model and 100% regression in the PDX model of high-grade serous ovarian cancer (HGSOC) harboring a CLU-NRG1 fusion. HGSOC accounts for 70–80% of ovarian cancer deaths (28). In the HGSOC model, tumor growth was largely repressed for the 63 days after treatment was stopped and animals were monitored for tumor regrowth. In contrast to the effectiveness of seribantumab, a 5 mg/kg afatinib daily dose (human equivalent dose of 50 mg daily), was a poor antagonist of tumor growth, despite full inhibition of HER2 phosphorylation (indicating that tumor penetration of afatinib was not an issue). These observations were contrary to the in vitro results where afatinib were more effective at blocking cell growth. It is unlikely that the higher efficacy of seribantumab observed in our in vivo studies is due to any ADCC activity since the NSG strain of mice lacks mature T-cells, B-cells and NK-killer cells (31), precluding any ADCC-mediated effects. Instead, the higher in vivo potency of seribantumab could be partially attributed to the sustained increase in expression of pro-apoptotic proteins such as BIM in PDX tumors, compared to afatinib. Afatinib (5 mg/kg QD) administration did not induce BIM expression in PDX tumors. The lack of cell death may contribute to the poor response to afatinib seen in the two PDX models in our study and in clinical reports where there have been stable disease, short duration of response, or no response, in the majority of cases reported (2,32). These results suggest that specific inhibition of HER3 with the monoclonal antibody seribantumab in a tumor-agnostic fashion should be explored as a therapy for NRG1 fusion-dependent cancers.
One major drawback of preclinical studies attempting to develop therapies for NRG1 fusion-driven cancers is a lack of patient-derived models. Most studies employ either murine NIH-3T3 cells or cancer cell lines in which NRG1 fusions are artificially expressed. In our study we used patient-derived breast, lung, and ovarian cancer cell lines and PDX models to assess the efficacy of seribantumab. The preclinical results presented here demonstrate that seribantumab is an effective therapeutic agent for cancers arising from NRG1 rearrangements, perhaps aided by its ability to block activation of all ERBB family members, thereby inhibiting the cell cycle, and inducing apoptosis. Importantly, we also show that seribantumab can block growth of a lung cancer cell line with NRG1 amplification. It is possible that NRG1 amplification may emerge as a molecularly-defined cancer subset as more diagnostic platforms begin to profile for NRG1 alterations. We believe that these novel models can be used to thoroughly compare other potential therapies for cancers with NRG1 fusions such as the HER2-HER3 bispecific antibody MCLA-128 (33) and other HER3 antibodies to be able to better recognize the best-in-class drugs.
In summary, we find that the anti-HER3 mAb seribantumab reduces growth and induces apoptosis in disease models derived from three different histological cancer subtypes with NRG1 rearrangements at dosages that are clinically achievable and lower than the human dosage. These results provide a clear preclinical rationale for a tumor-agnostic trial of seribantumab to treat NRG1 gene fusion-positive solid tumors. A Phase 2 trial of seribantumab in this setting is currently open and accruing patients (CRESTONE, NCT04383210).
Supplementary Material
Statement of translational relevance.
Oncogenic fusions involving the neuregulin 1 (NRG1) gene are found in approximately 0.2% of cancers of diverse histologies. Previous studies have shown that activation of HER3 is essential for NRG1-fusion-dependent tumorigenesis. Using patient-derived lung, breast and ovarian cancer models with NRG1 fusions, we show the anti-HER3 monoclonal antibody seribantumab to be effective at blocking activation of HER3 and its downstream growth-promoting signaling. Importantly, seribantumab administration to mice bearing patient-derived xenograft tumors resulted in 50–100% tumor regression. Our results provide a rationale for the clinical evaluation of seribantumab in patients with advanced NRG1 fusion-positive cancers in a tumor type-agnostic fashion.
Acknowledgments
The authors are grateful to Drs. Rajeev Chillakuru, Doug Plessinger and Amy C. Cavers for critical reading of the manuscript. Financial support for this study was provided by a grant from Cycle for Survival to M. Ladanyi, U54 OD020355 grant from the National Institute of Health to E. de Stanchina, a Memorial Sloan Kettering Cancer Center Support Grant (P30 CA008748) and Elevation Oncology Inc. to R. Somwar and M. Ladanyi.
Conflicts of interest disclosures
Igor Odintsov, Allan J.W. Lui, Eric Gladstone, Morana Vojnic, Lukas Delasos, Zebing Liu, Whitney J. Sisso, Exequiel M. Sisso, Inna Khodos, Marissa S. Mattar and Elisa de Stanchina report no potential conflict of interest.
Shawn M. Leland is an Elevation Oncology Inc. employee and shareholder, and is on the Board of Directors.
Marc Ladanyi has received advisory board compensation from Merck, Bristol-Myers Squibb, Takeda, Bayer, Lilly Oncology, Janssen, and Paige.AI. In addition, he has received research support from LOXO Oncology, Helsinn Healthcare, Elevation Oncology Inc. and Merus.
Romel Somwar has received research support from Merus, Helsinn Healthcare, LOXO Oncology and Elevation Oncology Inc.
References
- 1.Jonna S, Feldman RA, Swensen J, Gatalica Z, Korn WM, Borghaei H, et al. Detection of NRG1 Gene Fusions in Solid Tumors. Clin Cancer Res 2019. doi 10.1158/1078-0432.CCR-19-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drilon A, Somwar R, Mangatt BP, Edgren H, Desmeules P, Ruusulehto A, et al. Response to ERBB3-Directed Targeted Therapy in NRG1-Rearranged Cancers. Cancer Discov 2018;8(6):686–95 doi 10.1158/2159-8290.CD-17-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaefer G, Fitzpatrick VD, Sliwkowski MX. Gamma-heregulin: a novel heregulin isoform that is an autocrine growth factor for the human breast cancer cell line, MDA-MB-175. Oncogene 1997;15(12):1385–94. [DOI] [PubMed] [Google Scholar]
- 4.Duruisseaux M, McLeer-Florin A, Antoine M, Alavizadeh S, Poulot V, Lacave R, et al. NRG1 fusion in a French cohort of invasive mucinous lung adenocarcinoma. Cancer Med 2016;5(12):3579–85 doi 10.1002/cam4.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonna S, Feldman RA, Ou SH, Brien A, Nagasaka M, Swensen J, et al. Characterization of NRG1 gene fusion events in solid tumors. Journal of Clinical Oncology 2020;38(15 suppl):3113. [Google Scholar]
- 6.Shin DH, Lee D, Hong DW, Hong SH, Hwang JA, Lee BI, et al. Oncogenic function and clinical implications of SLC3A2-NRG1 fusion in invasive mucinous adenocarcinoma of the lung. Oncotarget 2016;7(43):69450–65 doi 10.18632/oncotarget.11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F, et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov 2014;4(4):415–22 doi 10.1158/2159-8290.CD-13-0633. [DOI] [PubMed] [Google Scholar]
- 8.Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res 2014;20(6):1410–6 doi 10.1158/1078-0432.CCR-13-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Cuesta L, Thomas RK. Molecular Pathways: Targeting NRG1 Fusions in Lung Cancer. Clin Cancer Res 2015;21(9):1989–94 doi 10.1158/1078-0432.CCR-14-0854. [DOI] [PubMed] [Google Scholar]
- 10.Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, et al. Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal 2009;2(77):ra31 doi 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 11.Schoeberl B, Kudla A, Masson K, Kalra A, Curley M, Finn G, et al. Systems biology driving drug development: from design to the clinical testing of the anti-ErbB3 antibody seribantumab (MM-121). NPJ Syst Biol Appl 2017;3:16034 doi 10.1038/npjsba.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell 2010;17(3):298–310 doi 10.1016/j.ccr.2009.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res 2010;70(6):2485–94 doi 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sequist LV, Gray JE, Harb WA, Lopez-Chavez A, Doebele RC, Modiano MR, et al. Randomized Phase II Trial of Seribantumab in Combination with Erlotinib in Patients with EGFR Wild-Type Non-Small Cell Lung Cancer. Oncologist 2019;24(8):1095–102 doi 10.1634/theoncologist.2018-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang XZ, Jolicoeur EM, Conte N, Chaffanet M, Zhang Y, Mozziconacci MJ, et al. gamma-heregulin is the product of a chromosomal translocation fusing the DOC4 and HGL/NRG1 genes in the MDA-MB-175 breast cancer cell line. Oncogene 1999;18(41):5718–21 doi 10.1038/sj.onc.1202950. [DOI] [PubMed] [Google Scholar]
- 16.Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst 1973;51(5):1409–16 doi 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- 17.Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 2017;23(4):405–8 doi 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res 2006;66(4):2116–28. [DOI] [PubMed] [Google Scholar]
- 19.Li GG, Somwar R, Joseph J, Smith RS, Hayashi T, Martin L, et al. Antitumor Activity of RXDX-105 in Multiple Cancer Types with RET Rearrangements or Mutations. Clin Cancer Res 2017;23(12):2981–90 doi 10.1158/1078-0432.CCR-16-1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vojnic M, Kubota D, Kurzatkowski C, Offin M, Suzawa K, Benayed R, et al. Acquired BRAF rearrangements induce secondary resistance to EGFR therapy in EGFR-mutated lung cancers. J Thorac Oncol 2019. doi 10.1016/j.jtho.2018.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato H, Schoenfeld AJ, Siau E, Lu YC, Tai H, Suzawa K, et al. MAPK Pathway Alterations Correlate with Poor Survival and Drive Resistance to Therapy in Patients with Lung Cancers Driven by ROS1 Fusions. Clin Cancer Res 2020. doi 10.1158/1078-0432.CCR-19-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gloeckner H, Jonuleit T, Lemke HD. Monitoring of cell viability and cell growth in a hollow-fiber bioreactor by use of the dye Alamar Blue. J Immunol Methods 2001;252(1–2):131–8 doi 10.1016/s0022-1759(01)00347-7. [DOI] [PubMed] [Google Scholar]
- 23.Somwar R, Shum D, Djaballah H, Varmus H. Identification and preliminary characterization of novel small molecules that inhibit growth of human lung adenocarcinoma cells. J Biomol Screen 2009;14(10):1176–84 doi 10.1177/1087057109350919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Wei P, Yang Y, Cui W, Cao B, Tan C, et al. BATF2 Deficiency Promotes Progression in Human Colorectal Cancer via Activation of HGF/MET Signaling: A Potential Rationale for Combining MET Inhibitors with IFNs. Clin Cancer Res 2015;21(7):1752–63 doi 10.1158/1078-0432.CCR-14-1564. [DOI] [PubMed] [Google Scholar]
- 25.Gagnon RC, Peterson JJ. Estimation of confidence intervals for area under the curve from destructively obtained pharmacokinetic data. J Pharmacokinet Biopharm 1998;26(1):87–102 doi 10.1023/a:1023228925137. [DOI] [PubMed] [Google Scholar]
- 26.Tchakarska G, Sola B. The double dealing of cyclin D1. Cell Cycle 2020;19(2):163–78 doi 10.1080/15384101.2019.1706903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trombetta D, Graziano P, Scarpa A, Sparaneo A, Rossi G, Rossi A, et al. Frequent NRG1 fusions in Caucasian pulmonary mucinous adenocarcinoma predicted by Phospho-ErbB3 expression. Oncotarget 2018;9(11):9661–71 doi 10.18632/oncotarget.23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015;15(11):668–79 doi 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol Oncol 2015;138(2):372–7 doi 10.1016/j.ygyno.2015.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobel M, Rahimi K, Rambau PF, Naugler C, Le Page C, Meunier L, et al. An Immunohistochemical Algorithm for Ovarian Carcinoma Typing. Int J Gynecol Pathol 2016;35(5):430–41 doi 10.1097/PGP.0000000000000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 2005;106(5):1565–73 doi 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cadranel J, Liu SV, Duruisseaux M, Branden E, Goto Y, Weinberg BA, et al. Therapeutic Potential of Afatinib in NRG1 Fusion-Driven Solid Tumors: A Case Series. Oncologist 2020. doi 10.1634/theoncologist.2020-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geuijen CAW, De Nardis C, Maussang D, Rovers E, Gallenne T, Hendriks LJA, et al. Unbiased Combinatorial Screening Identifies a Bispecific IgG1 that Potently Inhibits HER3 Signaling via HER2-Guided Ligand Blockade. Cancer Cell 2018;33(5):922–36 e10 doi 10.1016/j.ccell.2018.04.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.