

Abstract
Autosomal Dominant Polycystic kidney disease (ADPKD) is a common monogenic disorder marked by numerous progressively enlarging kidney cysts. Mettl3, a methyltransferase that catalyzes the abundant N6-methyladenosine (m6A) RNA modification, is implicated in development, but its role in most diseases is unknown. Here, we show that Mettl3 and m6A levels are increased in mouse and human ADPKD samples, and that kidney-specific transgenic Mettl3 expression produces tubular cysts. Conversely, Mettl3 deletion in three orthologous ADPKD mouse models slows cyst growth. Interestingly, methionine and S-adenosylmethionine (SAM) levels are also elevated in ADPKD models. Moreover, methionine and SAM induce Mettl3 expression and aggravate ex-vivo cyst growth, whereas dietary methionine restriction attenuates mouse ADPKD. Finally, Mettl3 activates the cyst-promoting c-Myc and cAMP pathways through enhanced c-Myc and Avpr2 mRNA m6A modification and translation. Thus, Mettl3 promotes ADPKD and links methionine utilization to epitranscriptomic activation of proliferation and cyst growth.
Keywords: Polycystic kidney disease, METTL3, m6A mRNA methylation, methionine, c-Myc, AVPR2
Graphical Abstract
eTOC Blurb
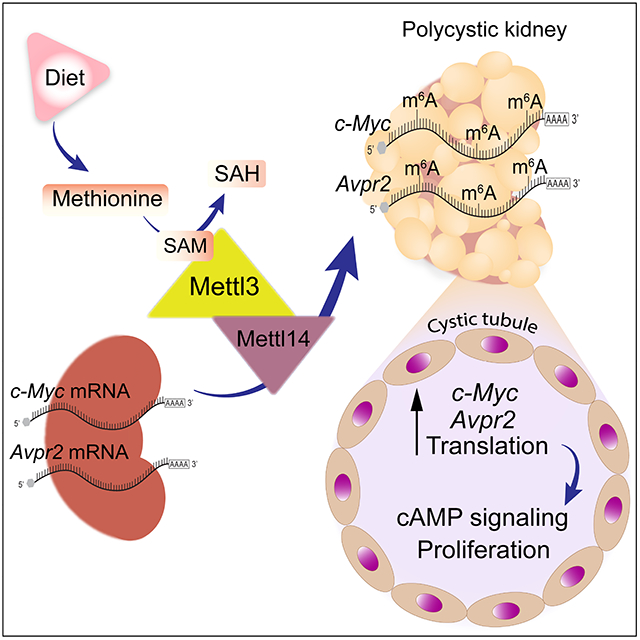
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic disorder with limited treatment options. Ramalingam et al. show that methionine dependency is a new ADPKD metabolic vulnerability as the RNA methyltransferase Mettl3 links methionine utilization to epitranscriptomic activation of pro-proliferative signaling and cyst growth. They further show that Mettl3 deletion or dietary methionine restriction improve ADPKD in mouse models.
Introduction
Like DNA, RNAs undergo dynamic chemical modifications, altering their processing, stability, or translational efficiency (Nachtergaele and He, 2018). The most common eukaryotic RNA modification is adenosine methylation at the 6th nitrogen position known as N6-methyladenosine (m6A). First discovered in the 1970s and thought to be a relic of bacterial and plant RNA, it is now clear that m6A is widely prevalent in mammalian RNAs. This revelation has sparked a rapidly evolving new field of 'epitranscriptomics.' A multi-protein m6A writer complex composed of Mettl3, Mettl14 and other adapter proteins mediates the m6A modification (Bokar et al., 1997), with Mettl3 acting as the core enzymatic subunit of this complex. Mettl3 binds S-adenosylmethionine (SAM), a derivative of methionine, and transfers an activated methyl group onto adenosine in RNAs, forming m6A (Jia et al., 2013). The biological impact of m6A is still not fully known, but initial studies point to its broad role in mammalian physiology. For example, Mettl3 deletion perturbs embryonic stem cell fate (Batista et al., 2014; Geula et al., 2015; Wang et al., 2014) and embryonic development of several organs (Zhang et al., 2020). The Mettl3-m6A axis is also implicated in multiple forms of cancers, where it promotes the translation of oncogenic mRNAs (Choe et al., 2018; Vu et al., 2017). However, with some exceptions, definitive studies linking Mettl3 to diseases are lacking. To our knowledge, the role of Mettl3 in any form of kidney disease has not been studied.
Autosomal dominant polycystic kidney disease (ADPKD), primarily caused by mutations of PKD1 or PKD2, is amongst the most common monogenetic human disorders (Patel et al., 2009; Ramalingam et al., 2020). The defining feature is the numerous renal tubule-derived cysts, which enlarge over time, causing massive bilateral kidney enlargement. Nearly 50% of patients develop end-stage renal disease. Excessive cyst epithelial proliferation and aberrant cAMP and c-Myc signaling are the key pathological hallmarks of ADPKD (Kurbegovic and Trudel, 2020; Sussman et al., 2020; Wang et al., 2018). Tolvaptan, the only FDA-approved drug for ADPKD, retards cyst growth via antagonizing AVPR2 (Vasopressin Receptor 2) and inhibiting cAMP signaling in cyst epithelia (Reif et al., 2011; Torres et al., 2012). Recent studies have also uncovered a substantial metabolic component in ADPKD (Hajarnis et al., 2017), raising the hope of targeted therapies (Lakhia et al., 2018; Lee et al., 2019) and dietary interventions to alleviate cyst growth (Torres et al., 2019; Warner et al., 2016). Activation of aerobic glycolysis and inhibition of fatty acid oxidative phosphorylation are considered the principal metabolic defects in PKD (Hajarnis et al., 2017; Menezes et al., 2016; Padovano et al., 2018; Rowe et al., 2013). This metabolic reprogramming may allow carbon usage for anabolic processes that support proliferating cyst epithelia. However, the impact of one-carbon and methionine metabolism, which generate the essential methyl groups for the various anabolic processes, is unknown. Moreover, the mechanism by which altered nutrient utilization facilitates a pro-proliferative transcriptomic output is also unclear.
Here, we report that the Mettl3-m6A pathway lies at the nexus of methionine metabolism and pro-proliferative signaling. We observed higher methionine-SAM levels and Mettl3 upregulation in multiple ADPKD mouse models. Methionine-SAM supplementation or Mettl3 overexpression induces cyst growth. Conversely, dietary methionine restriction or Mettl3 ablation attenuates PKD. Methionine and SAM promote Mettl3 expression, and the Pkd1-mutant cells are overtly reliant on the methionine-SAM-Mettl3 transmethylation axis for proliferation. Finally, Mettl3 activates cAMP and c-Myc signaling by enhancing Avpr2 and c-Myc mRNA methylation and translation. Thus, methionine is a new PKD metabolic vulnerability, with Mettl3 likely acting as a methionine sensing enzyme that links methionine dependence to the activation of pro-proliferative signaling.
Results
RNA methyltransferase Mettl3 is upregulated in mouse and human PKD.
We began by examining Mettl3 and Mettl14 expression in kidneys of Ksp/Cre; Pkd1F/RC (Pkd1F/RC-KO) and Ksp/Cre; Pkd1F/F(Pkd1F/F-KO) mice. Pkd1F/RC-KO is a compound mutant orthologous ADPKD model with a germline hypomorphic Pkd1 mutation in one allele and a somatic kidney tubule-specific null mutation in the other allele. The Pkd1F/F-KO model harbors kidney tubule-specific deletion of both Pkd1 alleles. By immunoblot analysis, we found marked Mettl3 upregulation in postnatal day (P)18 Pkd1F/RC-KO and P10 Pkd1F/F-KO kidneys compared to their respective, age-matched, control kidneys (Figure 1A). Moreover, Mettl14, another member of the m6A writer complex, was also upregulated in both ADPKD models (Figure 1A). Immunofluorescence staining further confirmed higher Mettl3 expression in P18 Pkd1F/RC-KO (Figure 1B) and P10 Pkd1F/F-KO cyst epithelia (Figure S1A) compared to their respective control kidneys.
Figure 1: Mettl3-m6A pathway is upregulated in mouse and human PKD.

(A) Immunoblots showing Mettl3 and Mettl14 expression in P18 Ksp-Cre;Pkd1F/RC (Pkd1F/RC-KO) and P10 Ksp-Cre;Pkd1F/F (Pkd1F/F-KO) compared to their respective age-matched control kidneys. Beta-actin acts as the loading control.
(B) Representative images of Mettl3 immunostaining in cysts (Cy) of P18 Pkd1F/RC-KO kidneys and control kidneys and in human ADPKD tissue and NHK. n = 10 images from 5 biological replicate mouse kidneys in mice and n = 8 images from 4 biological replicate human samples.
(C) ELISA-measured mRNA m6A level in P18 Pkd1F/RC-KO (blue circles) and P10 Pkd1-KO (red circles) kidneys compared to their respective age-matched control kidneys (grey circles). Error bars, SEM; Student’s t-test.
(D) Representative images of m6A immunostaining in P18 Pkd1F/RC-KO kidneys and control kidneys (n = 25 images from 5 biological replicate kidneys/group) and Human ADPKD tissue compared to NHK (n = 25 images from 4 biological replicate samples/group).
(E) H&E-staining of kidney sections from P10 Pkd1F/RC-KO and P50 i-Pkd1-KO mice. Scale bars, 0.5 mm and 80 um.
(F) Immunoblot showing Mettl3 expression in pre-cystic kidneys of P10 Pkd1F/RC-KO and P50 i-Pkd1-KO mice compared to kidneys of their respective age-matched control mice. Beta-actin acts as the loading control.
(G-H) Representative images of Mettl3 (red) immunostaining and Dolichos Biflorus Agglutinin (DBA, a collecting duct marker) (green) labeling in kidney sections from P10 Pkd1F/RC-KO (G) and P50 i-Pkd1-KO (H) mice and their respective age-matched control mice (n = 6 images from 3 biological replicate kidneys/group).
Scale bars, 20 um (B, D, G, and H); Error bars represent SEM; Student's t-test. See also Figure S1.
Next, we tested whether Mettl3 upregulation was associated with increased m6A mRNA modification. We found that ELISA-measured total mRNA m6A levels were 25.3% higher in Pkd1F/RC-KO kidneys and 26.1% higher in Pkd1F/F-KO kidneys compared to their respective age-matched control kidneys (Figure 1C). Moreover, staining with an anti-m6A antibody also demonstrated a higher m6A signal in P18 Pkd1F/RC-KO (Figure 1D) and P10 Pkd1F/F-KO kidneys (Figure S1A) compared to their respective control kidneys. Interestingly, our analysis of publicly available data from an independent unbiased metabolomic study (Podrini et al., 2018) showed higher m6A levels in P4 Ksp/Cre; Pkd1F/− compared to control kidneys (Figure S1B). To determine whether these findings are relevant to human ADPKD, we performed anti-METTL3 and anti-m6A immunofluorescence analysis using tissue sections from normal human kidney (NHK) samples and cystic kidney samples from individuals with ADPKD (n = 4, each group). We found that compared to renal tubules in NHK, METTL3 expression (Figure 1B & Figure S1C) and m6A levels (Figure 1D & Figure S1C) were higher in kidney cysts in human ADPKD samples. Thus, the Mettl3-m6A axis is upregulated in both mouse and human forms of ADPKD.
Next, we examined whether Mettl3 upregulation precedes cyst formation or if it is merely an epiphenomenon of cyst growth. At P10, Pkd1F/RC-KO kidneys are morphologically normal with only occasional cysts (Figure 1E). We noted robust Mettl3 upregulation, including in non-cystic Pkd1F/RC-KO collecting ducts (Figure 1F and 1G), even at this pre-cystic stage, indicating that Mettl3 activation is an early event in the cystogenesis process. We next turned to the doxycycline-inducible Pkd1-KO model to determine how soon after Pkd1 deletion Mettl3 becomes upregulated. First, we found that Mettl3 expression declines with kidney maturation in wild-type mice and is markedly lower at P28 than at younger time points (Figure S1D). Next, to inactivate Pkd1 in adult mice, we administered doxycycline to Ksp/rtTA; tetO-cre; Pkd1F/F (i-Pkd1-KO) and littermate control mice (Ksp/rtTA or tetO-cre negative) starting at P34. We harvested kidneys 16-days later at P50, allowing sufficient time for Pkd1 deletion but not enough for cysts to form (Figure 1E). Remarkably, we still observed greater Mettl3 expression in i-Pkd1-KO kidneys compared to control kidneys, including in collecting ducts (Figure 1F and 1H). Thus, Mettl3 upregulation occurs soon after Pkd1 deletion and before cyst formation.
Kidney-specific transgenic Mettl3 expression produces tubular cysts:
Whether Mettl3-m6A upregulation is functionally significant in PKD is unknown. As a first step towards addressing this question, we generated transgenic mice with a conditional, cre-inducible Mettl3 gain-of-function allele. We designed a transgene that contains (from 5′ to 3′ orientation) CAG promoter, loxP-flanked STOP cassette, cDNA fragment encoding HA-tagged mouse Mettl3, internal ribosomal entry site-enhanced green fluorescent protein (IRES-EGFP), and polyadenylation signal (Figure 2A, see STAR Methods for details). We began by validating this transgene in the mIMCD3 kidney epithelial cell line. We observed Mettl3 upregulation and de novo EGFP and HA expression only in the presence of cre recombinase (Figure S2A and S2B). Next, after pronuclear injection into fertilized eggs of C57BL6 female mice, we obtained nine F0 mice that carried the Mettl3 transgene. We confirmed germline transmission in four F0 mice (Figure S2C). We bred these four founder mice with Ksp-Cre mice to produce kidney tubule-specific Mettl3-overexpressing mice (Mettl3Tg). We phenotypically and molecularly characterized the F1 and F2 progeny from these crosses. By qRT-PCR and immunoblot analysis, we observed that compared to control (cre-negative; Mettl3Tg) kidneys, Mettl3Tg kidneys expressed approximately 2-fold higher Mettl3 (Figure 2B). We also noted EGFP and HA expression in Mettl3Tg kidneys but not in control kidneys (Figure 2B). H&E staining revealed normal histology in P60 control kidneys, whereas we observed tubular dilation and cysts in P60 Ksp-Cre;Mettl3Tg kidneys (Figure 2C). We found that these cysts were GFP-positive and expressed higher levels of Mettl3 and m6A and were derived from collecting ducts, the primary site of cre expression in this model (Figure 2C). Phenotypic characterization of additional Ksp-Cre;Mettl3Tg mice is shown in Figure S2D. Collectively, these findings demonstrate that transgenic Mettl3 expression autonomously induces kidney cyst formation in mice.
Figure 2: Kidney-specific transgenic Mettl3 expression produces renal tubule cysts.

(A) Schematic illustrating the genetic approach used for generating Mettl3Tg mice. Briefly, they harbor a transgene containing CAG promoter upstream of a loxP-STOP-loxP cassette and a cDNA sequence encoding HA-tagged Mettl3 and Ires-EGFP. Cre-mediated recombination deletes the STOP cassette leading to conditional Mettl3 and GFP expression. Mettl3F/F mice carry loxP sites flanking Mettl3 exon-4. Recombination results in the insertion of a premature stop codon and thus prevents Mettl3 expression.
(B) qRT-PCR analysis showing Mettl3 expression in P60 Ksp-Cre;Mettl3Tg (green circles; Mettl3Tg) compared to Cre-negative;Mettl3Tg kidneys (purple circles; control). Immunoblot analysis showing Mettl3 and de novo GFP and HA expression in Mettl3Tg kidneys compared to control kidneys. Beta-Actin serves as the loading control. qRT-PCR analysis showing Mettl3 expression in P21 Ksp-Cre;Mettl3F/F (brown circles, Mettl3-KO) compared to control kidneys (grey circles).
(C) Representative H&E-stained, Mettl3 immunostained, DBA labeled and GFP immunostained kidney sections from P60 control mice, P60 Mettl3Tg mice, and 20-week-old Mettl3-KO. n = 6 images from 3 biological replicate kidneys/group.
Scale bars, 50 um; Error bars represent SEM; Student's t-test. See also Figure S2.
Mettl3 deletion slows cyst growth in orthologous ADPKD mouse models:
We next asked whether Mettl3 inhibition ameliorates cyst growth in PKD models. To test this hypothesis, we first generated Mettl3F/F mice that contain loxP sites in the genomic DNA, flanking the fourth exon of the Mettl3 gene (Figure 2A, see STAR Methods for details). We used the Ksp-Cre driver to study the effects of Mettl3 deletion in normal renal tubules. Using qRT-PCR (Figure 2B) and immunofluorescence staining (Figure 2C), we confirmed reduced Mettl3 expression in Ksp-Cre;Mettl3F/F (Mettl3-KO) kidneys compared to control (cre-negative;Mettl3F/F) kidneys. Moreover, by RNA dot blot analysis, we observed reduced mRNA m6A levels in Mettl3-KO compared to control kidneys (Figure S2E). However, we found that P21 Mettl3-KO mice exhibit normal kidney histology (Figure S2F). We analyzed a second cohort of 20-week-old Mettl3-KO mice to examine the effects of prolonged Mettl3 deletion. Again, we noted normal kidney histology (Figure 2C), kidney-weight-to-body-weight ratio (KW/BW), and serum creatine levels (Figure S2G).
As Mettl3 inactivation did not affect renal tubule maturation or maintenance, we were able to study the effects of its deletion in ADPKD mouse models. We first inactivated Mettl3 in the compound mutant Pkd1F/RC-KO ADPKD model. Considering that this model harbors a missense mutation, we found that Pkd1 levels between control, Pkd1F/RC-KO, and Pkd1F/RC-Mettl3-DKO kidneys were unchanged (Figure S3A). However, qRT-PCR and immunoblot analysis confirmed Mettl3 downregulation in Ksp-Cre;Pkd1F/RC;Mettl3F/F (Pkd1F/RC-Mettl3-DKO) compared to Ksp-Cre;Pkd1F/RC (Pkd1F/RC-KO) kidneys (Figure S3B-E). Furthermore, by immunofluorescence staining, we noted no Mettl3 expression in cyst epithelial cells of Pkd1F/RC-Mettl3-DKO mice (Figure S3C). Histological analysis revealed smaller kidneys with fewer cysts after Mettl3 deletion (Figure 3A). We observed a 40% lower KW/BW (Figure 3B) and a 50% lower cyst index (Figure 3C) in Pkd1F/RC-Mettl3-DKO mice compared to Pkd1F/RC-KO mice. Moreover, as a sign of improved kidney function, we noted a 26% lower serum creatine level (Figure 3D) in Pkd1F/RC-Mettl3-DKO compared to Pkd1F/RC-KO mice. The expression of kidney injury markers Kim1 and Ngal was lower by 60% and 54%, respectively, in Pkd1F/RC-Mettl3-DKO compared to Pkd1F/RC-KO kidneys (Figure 3E).
Figure 3: Mettl3 deletion alleviates cyst growth in three ADPKD models.

(A) H&E-stained kidney sections from P18 Pkd1F/RC-KO and Pkd1F/RC-Mettl3-DKO mice.
(B-E) Kidney-weight-to-body-weight (KW/BW) ratio (B), cyst index (C), serum creatinine levels (D), and kidney Kim1 and Ngal mRNA expression (E) in P18 Pkd1F/RC-Mettl3-DKO mice (orange circles) compared to Pkd1F/RC-KO mice (blue circles).
(F) H&E-stained kidney sections from P10 Pkd1-KO and Pkd1-Mettl3-DKO mice.
(G-I) KW/BW ratio (G) ,kidney Kim1 and Ngal mRNA expression (H), and survival analysis (I) of P10 Pkd1-Mettl3-DKO mice (green circles/line) compared to Pkd1-KO mice (light pink circles/line). The blue line depicts survival of mice with kidney tubule-specific loss of one Mettl3 allele.
(J) Schematic illustrating the approach used to generate i-Pkd1-KO and littermate i-Pkd1-Mettl3-DKO mice. Representative immunofluorescence images showing Mettl3 and m6A immunostaining in cysts of i-Pkd1-KO and i-Pkd1-Mettl3-DKO cysts (n = 8 images from 3 biological replicate kidneys/group).
(K) H&E-stained kidney sections of P80 i-Pkd1-KO and i-Pkd1-Mettl3-DKO mice are shown.
(L-M) KW/BW ratio (L) and cyst index (M) of P80 i-Pkd1-Mettl3-DKO mice (light orange circles) compared to i-Pkd1-KO mice (dark pink circles). Control mice (Ksp/rtTA; tetO-cre) are shown as grey circles.
Scale bars, 0.5 mm (A, F, and K) and 20 um (J); Error bars represent SEM; Student's t-test (B, D, E, G, and M), Mann-Whitney (C, H), One-way ANOVA Tukey's multiple-comparisons test (F), and Mantel-Cox test (I). See also Figure S3.
Next, we inactivated Mettl3 in the developmental Pkd1-KO ADPKD model (Figure 3F). By qRT-PCR analysis, we confirmed that Pkd1 was downregulated equally in both Pkd1F/F-KO and Pkd1F/F-Mettl3-DKO kidneys compared to control kidneys (Figure S3A), but, as expected, Mettl3 expression was reduced only in the Pkd1F/F-Mettl3-DKO kidneys (Figure S3B). Using immunofluorescence and immunoblot analysis, we further confirmed Mettl3 deletion in cyst epithelia of Pkd1F/F-Mettl3-DKO mice compared to Pkd1F/F-KO mice (Figure S3C-E). Even in this rapidly fatal ADPKD model, we found that Mettl3 deletion resulted in a 21.9% lower KW/BW ratio Figure 3G), lower Kim1 (down by 37%) and Ngal (down by 46%) expression Figure 3H), and importantly, improved median survival (Figure 3I). Finally, we examined if Mettl3 also exerts pathogenic effects in the adult-onset, inducible Pkd1-KO model. We administered doxycycline to littermate control, Ksp-rtTA;tetO-cre;Pkd1F/F (i-Pkd1-KO), and Ksp-rtTA;tetO-cre;Pkd1F/F;Mettl3F/F (i-Pkd1-Mettl3-DKO) mice in their drinking water from P28 to P42 and sacrificed them at P80 (Figure 3J and Figure S3F). Using qRT-PCR, we found that Pkd1 expression was equally reduced in i-Pkd1-KO and i-Pkd1-Mettl3-DKO kidneys compared to control kidneys (Figure S3G) but Mettl3 expression was reduced only in i-Pkd1-Mettl3-DKO kidneys (Figure S3H). By immunofluorescence (Figure 3J) and immunoblot analysis (Figure S3D), we noted higher Mettl3 and m6A levels in i-Pkd1-KO than in control kidneys. As expected, we observed reduced Mettl3 and m6A levels in i-Pkd1-Mettl3-DKO compared to i-Pkd1-KO kidneys (Figure 3J and Figure S3D and S3H). By H&E staining, we observed fewer cysts in i-Pkd1-Mettl3-DKO than in i-Pkd1-KO kidneys (Figure 3K). We also noted a normal KW/BW ratio (Figure 3L) and a lower cyst index (Figure 3M) in i-Pkd1-Mettl3-DKO than in i-Pkd1-KO kidneys. Thus, our data collectively indicate that Mettl3 deletion attenuates cyst growth in ADPKD models irrespective of the type of Pkd1 mutation or dynamics of cyst growth.
c-Myc and Avpr2 are Mettl3 targets:
The canonical Mettl3 function is its m6A-forming catalytic activity. Therefore, to investigate how Mettl3 promotes cyst growth, we began by identifying and cross-comparing the m6A epitranscriptomic landscapes in control, Pkd1F/RC-KO, and Pkd1F/RC-Mettl3-KO kidneys. We immunoprecipitated m6A-modified mRNAs and performed high-throughput sequencing (MeRIP-seq) (Dominissini et al., 2012; Meyer et al., 2012). We used two biological replicate samples per group; each sample contained pooled kidney RNA from either three male or female mice. Using principal component analysis, we found that biological replicates clustered together, indicating similar m6A profiles between the male and female samples with the same genotype. We also found that Pkd1F/RC-KO samples segregated separately from Pkd1F/RC-Mettl3-KO samples suggesting that Mettl3 deletion alters the m6A landscape of cystic kidneys (Figure 4A). We observed that the m6A GGACU motif was enriched in Pkd1F/RC-KO but not in the Pkd1F/RC-Mettl3-KO samples (Figure S4A) and that the m6A peaks mostly mapped to 5' or 3' untranslated regions of mRNAs (Figure S4B). We next identified ADPKD-relevant targets of Mettl3 (P < 0.01, FDR < 0.1). We reasoned that due to increased Mettl3 activity in Pkd1F/RC-KO kidneys, m6A modification on its putative mRNA targets would also increase. Conversely and as a pertinent negative control, m6A modification on these same mRNAs will be lower in Pkd1F/RC-Mettl3-KO kidneys because of Mettl3 deletion. Applying these criteria, we identified c-Myc and Avpr2 amongst 133 putative Mettl3 targets (Figure 4B and 4C, and Table S1). We found that m6A levels on c-Myc and Avpr2 mRNAs were >4-fold higher in Pkd1F/RC-KO kidneys than in control kidneys. Both mRNAs exhibited markedly lower m6A modification in Pkd1F/RC-Mettl3-DKO kidneys (Figure 4D and Figure S4C).
Figure 4: Mettl3 deletion inhibits c-Myc and Avpr2-cAMP signaling in cystic kidneys.

(A) Principle component analysis of MeRIP-Seq data from P18 control (Ctl), Pkd1F/RC-KO (SKO), and Pkd1F/RC-Mettl3-KO (DKO) kidneys.
(B) Comparative analysis (Ctl vs. SKO vs. DKO) used to identify Mettl3 mRNA targets in PKD.
(C) Heatmap showing m6A levels on the 133 Mettl3 mRNA targets in P18 Ctl, SKO, and DKO kidneys.
(D) IGV tracks showing m6A modification on c-Myc and Avpr2 mRNAs in Ctl, SKO, and DKO kidneys.
(E) qRT-PCR analysis showing c-Myc and Avpr2 mRNA expression in P18 Ctl (grey circles), SKO (blue circles), and DKO (orange circles) kidneys or P60 control (purple circles) and Mettl3Tg (green circles) kidneys.
(F) Immunoblot showing c-Myc and Avpr2 expression in P18 Ctl, SKO, and DKO kidneys, and P60 control and Mettl3Tg kidneys. Beta-actin acts as the loading control.
(G) Quantification of immunoblots in (F) is shown.
(H) Immunoblots showing pCreb1, PCNA, and Yap1 expression in P18 SKO and DKO kidneys, and P60 control and Mettl3Tg kidneys. Beta-actin acts as the loading control.
(I) Representative images showing pCreb1 and Yap1 immunostaining and O-propargyl puromycin (OPP) incorporation in P18 SKO and DKO kidneys. (n = 8 images from 4 biological replicate kidneys/group).
(J) Representative images showing pCreb1 and Yap1 immunostaining and OPP incorporation in P60 control (n = 10 images from 4 biological replicate kidneys) and Mettl3Tg kidneys (n = 11 images for pCreb1, 5 images for Yap1 and 15 images for OPP from 4 biological replicate kidneys).
Scale bars: 50 um; Error bars represent SEM; Student's t-test and One-way ANOVA, Tukey's multiple-comparisons test (E, G). See also Figures S4 & S5 and Table S1.
c-Myc, an oncogenic transcription factor, is upregulated in PKD and is required for cyst growth (Kurbegovic and Trudel, 2020; Trudel et al., 1998). Moreover, c-Myc is an established Mettl3 target in cancer cell lines (Li et al., 2019; Yang et al., 2020; Yuan et al., 2020; Zhao et al., 2020). AVPR2 is a G-protein-coupled receptor for vasopressin (Lolait et al., 1992). AVPR2 stimulation propagates the Adenylyl cyclase/cAMP pathway in collecting ducts, which is well-known to promote PKD (Sussman et al., 2020). These compelling pre-existing links prompted us to test whether Mettl3 promotes c-Myc and Avpr2 expression. Using RNA-seq (Figure S4D-E) and qRT-PCR analysis (Figure 4E), we noted a robust but equal upregulation of c-Myc and Avpr2 mRNAs Pkd1F/RC-KO and Pkd1F/RC-Mettl3-DKO kidneys compared to control kidneys. These findings indicated that Mettl3 deletion did not affect c-Myc and Avpr2 mRNA levels. In contrast, using immunoblot analysis, we found that c-Myc and Avpr2 protein levels were greater in Pkd1F/RC-KO compared to control kidneys but were lower by approximately 50% in Pkd1F/RC-Mettl3-DKO kidneys compared to the Pkd1F/RC-KO kidneys (Figure 4F-G). Similarly, we found that siRNA-mediated Mettl3 knockdown in normal (mIMCD3) or Pkd1 mutant (Pkd1RC/−) collecting duct cells also resulted in lower c-Myc and Avpr2 protein without affecting mRNA levels (Figure S4F). The fact that mRNA abundance was unaffected, but protein expression was still lower implied that Mettl3-m6A inhibition reduces c-Myc and Avpr2 mRNA translation. Conversely, but consistent with this idea, we found that Mettl3 overexpression promotes c-Myc and Avpr2 protein expression by >2-fold in Mettl3Tg compared to control kidneys (Figure 4F-G) but again, c-Myc and Avpr2 mRNA levels remained unchanged (Figure 4E). Consistently, transgenic Mettl3 overexpression in mIMCD3 and Pkd1RC/− cells also increased c-Myc and Avpr2 protein (Figure S4G). Finally, to test whether Mettl3 directly promotes Avpr2 translation in renal epithelial cells, we transfected Pkd1RC/+ and Pkd1RC/− cells with a luciferase reporter plasmid containing Avpr2 exon-2. Luciferase reporter assays revealed that Mettl3 knockdown reduced, whereas its overexpression enhanced Avpr2 expression (Figure S4H-I). Thus, collectively these studies identify and validate c-Myc and Avpr2 as Mettl3 targets in PKD.
Mettl3 promotes c-Myc and cAMP signaling, cyst proliferation, and protein synthesis:
We next tested whether Mettl3 independently affects downstream functions of Avpr2 and c-Myc. Avpr2 stimulation raises intracellular cAMP, which through protein kinase A catalytic activity phosphorylates CREB (cAMP-response element-binding protein) (Wang et al., 2010). pCREB binds to regulatory DNA regions and transactivates cAMP target genes. Yap1 (yes-associated protein 1), a pro-proliferative oncogenic transcription factor, is a critical c-Myc target gene in cystic kidneys (Cai et al., 2018; Happe et al., 2011). Thus, as readouts of cAMP and c-Myc signaling in vivo, we measured pCREB and Yap1 levels (Figure 4H). We noted that both pCREB and Yap1 were markedly upregulated in Pkd1F/RC-KO compared to control kidneys. Mettl3 deletion reduced pCREB and Yap1 expression by >50% in Pkd1F/RC-Mettl3-DKO compared to Pkd1F/RC-KO kidneys (Figure 4H). Similarly, by immunofluorescence analysis, we observed reduced pCREB and Yap1 expression in cyst epithelia of Pkd1F/RC-Mettl3-DKO compared to Pkd1F/RC-KO kidneys (Figure 4I). Consistently, from an unbiased analysis of the RNA-seq data, we noted reduced expression of gene networks controlled by AVP-cAMP-CREB1 and Yap1 in the Pkd1F/RC-Mettl3-DKO compared to the Pkd1F/RC-KO kidneys (Figure S4J). Conversely, we observed pCREB and Yap1 upregulation in the collecting ducts of Ksp-Cre;Mettl3Tg compared to control mice (Figure 4H and 4J). The physiological effect of the AVPR2-cAMP axis is to promote water reabsorption and concentrate urine. As an additional sign of activated cAMP signaling in vivo, we found that 24-hour urine output (relative to body weight) is lower by 35%, and urine osmolality is greater by 25% in Ksp-Cre;Mettl3Tg mice compared to control mice (Figure S5A).
The transcriptional output of c-Myc/YAP1 and AVPR2/cAMP pathways eventually converges on pro-proliferative and pro-growth processes. Accordingly, we found that expression of PCNA, a marker of proliferating cells, was elevated in Pkd1F/RC-KO compared to control kidneys, but Mettl3 deletion reduced PCNA levels by ~50% in the Pkd1F/RC-Mettl3-DKO kidneys (Figure 4H). Further indicating lower proliferation, we noted 52.4% fewer phosphohistone H3-positive cyst epithelia in the Pkd1F/RC-Mettl3-DKO kidneys than the Pkd1F/RC-KO kidneys (Figure S5B-C). Conversely, we found that Mettl3 overexpression raised proliferation, as evidenced by higher PCNA levels in Mettl3Tg kidneys than control kidneys (Figure 4H). To assess in vivo protein synthesis, we injected mice with puromycin or its alkyne analog o-propargyl-puromycin (OPP) and harvested kidneys 90-minutes later. Puromycin and OPP are both efficiently incorporated into nascent peptides (Liu et al., 2012). Using immunoblot (global puromycin incorporation) and click-chemistry-based fluorescence (OPP incorporation in cells), we found that the neo-protein synthesis rate was higher in cyst epithelia of Pkd1F/RC-KO mice compared to normal tubule epithelia of control mice (Figure S5D-F). We found that Mettl3 deletion lowered puromycin incorporation in whole kidney protein lysate by ~25% in Pkd1F/RC-Mettl3-DKO compared to Pkd1F/RC-KO mice (Figure S5E-F). We also observed a markedly lower OPP incorporation in the cyst epithelia of Pkd1F/RC-Mettl3-DKO kidneys than in Pkd1F/RC-KO kidneys (Figure 4I). Conversely, we found that cyst epithelia in kidneys of Mettl3Tg mice exhibited notable OPP incorporation, whereas there was minimal to no OPP incorporation in the renal tubules of control mice (Figure 4J). These results indicate that Mettl3 independently activates c-Myc and cAMP signaling and promotes cyst proliferation and neo-protein synthesis.
Methionine and SAM activate Mettl3 and promote ex-vivo cyst growth:
Methionine is converted to SAM, which binds to Mettl3 and provides the essential activated methyl group for the m6A reaction (Chiang et al., 1996; Finkelstein, 1990). As a first sign that the methionine-SAM axis maybe independently implicated in cyst growth, we noted that ELISA-measured SAM level was >5-fold higher in P18 Pkd1F/RC-KO and P10 Pkd1F/F-KO kidneys than their respective age-matched control kidneys (Figure 5A). Moreover, we found that SAM level was also higher in P10 pre-cystic Pkd1F/RC-KO kidneys, indicating that similar to Mettl3, SAM upregulation is an early event in the cystogenesis process (Figure 5B). Next, to causally link methionine-SAM to cystogenesis, we examined the effects of methionine or SAM supplementation in the ex-vivo kidney cystogenesis model. We cultured embryonic day (E) 12.5 wild-type kidneys for four days in media containing 100 uM 8-Bromo-cAMP and 0, 0.2, 0.8, or 2 mM methionine. We noted a concentration-dependent increase in cyst index, indicating that methionine fuels cyst formation in this model (Figure 5C). Similarly, we found that SAM supplementation also aggravated ex-vivo cyst growth, implicating the cellular methylation pathway rather than the function of methionine as an amino acid component of proteins in driving cystogenesis (Figure 5D).
Figure 5: Methionine upregulates Mettl3 and potentiates proliferation and ex-vivo cyst growth.

(A) ELISA-measured SAM levels in P18 Pkd1F/RC-KO and P10 Pkd1F/F-KO kidneys compared to their respective age-matched control kidneys.
(B) ELISA-measured SAM levels in precystic P10 Pkd1F/RC-KO kidneys and control kidneys.
(C) Images and cyst index quantification of E12.5 wild-type kidneys grown for four days in culture media containing 100 uM 8-Bromo-cAMP and 0, 0.2, 0.8, or 2-mM L-methionine.
(D) Images and cyst index quantification of E12.5 wild-type kidneys grown for four days in culture media containing 100 uM 8-Bromo-cAMP that was supplemented with either 250 uM S-adenosyl methionine (SAM) or a control vehicle.
(E) Proliferation rate of control (Pkd1RC/+) and Pkd1RC/− cells grown in normal (purple circles) or methionine-depleted (orange circles) culture media.
(F) Proliferation rate of control and Pkd1RC/− cells grown in normal (vehicle) or SAM-supplemented culture media.
(G) Proliferation rate of Pkd1RC/+ and Pkd1RC/− cells grown in normal (vehicle) or SAH-supplemented culture media.
(H) Proliferation rate of Pkd1RC/− cells that were transfected with either control siRNA (circles) or Mettl3 siRNA (triangles) and then grown in normal (purple) or high methionine (2 mM, green) culture media.
(I) Immunoblots showing Mettl3, c-Myc, and Avpr2 expression in Pkd1RC/− cells treated with rising concentration of methionine or SAM. Beta-actin acts as the loading control.
(J) Immunoblots showing Mettl3, c-Myc and Avpr2 expression in Pkd1RC− cells that were transfected with control or Mettl3 siRNA and then grown in culture media containing 2 mM methionine or 100 uM SAM. Beta-actin acts as the loading control.
Scale bars: 50 um; Error bars represent SEM; Student's t-test (A and B) and One-way ANOVA, Tukey's multiple-comparisons test (C-H). See also Figures S6.
These unexpected observations prompted us to test whether methionine-SAM is a metabolic necessity for Pkd1 mutant cells. We found that the proliferation rate – assessed using the alamarBlue assay – was nearly 2-fold higher in Pkd1RC/− cells compared to control (Pkd1RCI+) cells (Figure 5E). A 12-hour methionine depletion eliminated the proliferative advantage of Pkd1RC/− cells but had no impact on control cells (Figure 5E). Conversely, we found that SAM supplementation increased the already elevated proliferation of Pkd1RC/− cells by ~30% but again did not affect control cells (Figure 5F). Importantly, we noted that S-adenosylhomocysteine (SAH), the immediate byproduct of SAM-mediated methylation reactions, did not promote the proliferation of Pkd1RC/− or control cells (Figure 5G). These results suggest that Pkd1 mutant cells are overtly reliant on the methionine-SAM transmethylation axis for their proliferation.
We next investigated if the methionine-SAM axis impinges on the Mettl3-m6A pathway. We cultured Pkd1RC/− cells in growth media containing normal (0.2 mM) or high (2 mM) methionine. We then transfected these cells with control or Mettl3 siRNAs. We observed that methionine enhanced the proliferation of Pkd1RC/− cells transfected with control siRNA. However, this stimulatory effect was lost in Pkd1RC/− cells transfected with Mettl3 siRNA (Figure 5H). Moreover, we found that both methionine and SAM upregulated Mettl3 expression in Pkd1RC/− cells in a concentration-dependent manner (Figure 5I). Conversely, we noted that methionine depletion reduced Mettl3 expression in Pkd1RC/− cells. SAM, but not SAH, was sufficient to rescue this downregulation (Figure S6A-B). Moreover, we noted higher c-Myc and Avpr2 expression in the Pkd1RC/− cells exposed to rising methionine or SAM concentrations (Figure 5I). Mettl3 inhibition prevented methionine or SAM-mediated c-Myc and Avpr2 upregulation (Figure 5J). Thus, these results reveal that methionine/SAM promotes Mettl3 expression and that Mettl3 facilitates methionine/SAM-stimulated proliferation of Pkd1RC/− cells.
Dietary methionine restriction reduces Mettl3 expression and ameliorates PKD:
Finally, we addressed the intriguing possibility that the methionine-Mettl3 axis is a targetable new metabolic necessity for in vivo cyst growth. First, we analyzed the metabolomic profile of wild-type and cystic Pkd1RC/RC kidneys. The Pkd1RC/RC mice harbor homozygous germline Pkd1 R3277C (RC) mutations and develop slowly progressive PKD, mimicking the human phenotype (Hopp et al., 2012). An additional benefit is that this model allows for dietary intervention studies. We performed untargeted mass spectrometry on kidney samples from 30-week-old wild-type and Pkd1RC/RC mice (n = 6/group). We analyzed each biological kidney sample twice. PCA revealed a robust correlation between the technical replicates (Figure 6A). We found 109 altered metabolites in Pkd1RC/RC compared to wild-type kidneys (Figure 6B and Table S2). Notably, we observed that methionine levels were 1.83-fold higher in cystic Pkd1RC/RC kidneys than in wild-type kidneys (Figure 6C). Moreover, we found that ELISA-measured SAM levels were also higher in Pkd1RC/RC kidneys than in control kidneys (Figure 6D). Concomitantly, we noted increased Mettl3 and m6A levels in cyst epithelia of Pkd1RC/RC mice compared to normal tubules of control mice (Figure 6E-F), indicating that the RNA methylation pathway is activated in Pkd1RC/RC kidneys.
Figure 6: Low methionine diet inhibits Mettl3 expression and ameliorates in vivo cyst growth.

(A) Principle component analysis of the kidney metabolomics data of 30-week-old control and Pkd1RC/RC mice.
(B) Heatmap showing the 109 differentially present kidney metabolites in 30-week-old control and Pkd1RC/RC mice. Arrow indicates methionine.
(C) Mass spectrometry-measured kidney methionine levels in 30-week-old control mice (n = 6, grey) and Pkd1RC/RC mice (n = 6, purple).
(D) ELISA-measured SAM levels in the kidneys of 48-week-oldcontrol (n = 3, grey) and Pkd1RC/RC mice (n = 3, purple).
(E) Immunoblot showing Mettl3 and Mettl14 expression in kidneys of 48-week-old control and Pkd1RC/RC mice. Beta-actin acts as the loading control.
(F) Representative images showing Mettl3 and m6A immunostaining and DBA labeling in kidneys of 48-week-old control (n = 9 images from 4 biological replicate kidneys) and Pkd1RC/RC mice (n = 8 images from 4 biological replicate kidneys). Anti-Mettl3 or anti-m6A antibodies (red) and DBA (green).
(G) Whole kidney images and H&E-stained kidney sections from 5-month-old Pkd1RC/RC mice on standard (purple circles) or methionine-restricted (MR) diet (orange circles).
(H) ELISA-measured kidney SAM levels in 5-month-old Pkd1RC/RC mice on a standard or MR diet.
(I-K) KW/BW ratio, cyst index, and kidney Ngal mRNA expression in 5-month-old Pkd1RC/RC mice on a standard or MR diet.
(L) qRT-PCR analysis showing c-Myc and Avpr2 expression in kidneys of 5-month-old Pkd1RC/RC mice on standard or MR diet.
(M-N) Immunoblot analysis showing Mettl3, c-Myc, and Avpr2 protein expression in kidneys from 5-month-old Pkd1RC/RC mice on standard or MR diet. Beta-actin acts as the loading control.
Scale bars: (E) 100 uM (Mettl3 images) and 50 uM (m6A images), (F) 1 cm (top) and 2 mm (bottom); Error bars represent SEM; Student’s t-test (all graphs). See also Figure S6 & Table S2.
To provide a direct link to in vivo PKD progression, we conducted a 15-week preclinical dietary methionine intervention study in the Pkd1RC/RC model. We fed five-month-old Pkd1RC/RC mice either a standard diet containing 0.62% methionine or a methionine-restricted (MR) diet containing 0.16% methionine. Mice on the MR diet consumed more food but still had lower body-weight and fat mass by the end of the 15-week trial (Figure S6C-F). Importantly, by morphological and histological analysis, we found that the MR diet reduced kidney size and cysts (Figure 6G). Moreover, we observed lower kidney SAM and Mettl3 levels in Pkd1RC/RC mice on MR compared to standard diet (Figure 6H). We also observed a lower KW/BW ratio (down by 25%) and cyst index (down by 37.9%), and reduced kidney Ngal (down by 64.7%) (Figure 6I-K and S6G) and PCNA expression in Pkd1RC/RC mice on MR compared to the standard diet (Figure S6H-I). Finally, we found that while the MR diet did not affect c-Myc and Avpr2 mRNA abundance (Figure 6L), it still lowered their protein levels in Pkd1RC/RC kidneys compared to mice on the replete diet (Figure 6M and 6N). Thus, systemic methionine restriction lowers kidney SAM and Mettl3 levels and ameliorates PKD progression.
Discussion
We show that methionine and Mettl3-m6A are novel interlinked pathogenic axes in ADPKD. First, Mettl3 is consistently and robustly upregulated across multiple ADPKD mouse models and human samples. This upregulation occurs soon after Pkd1 deletion and precedes cystogenesis in murine models. Importantly, levels of m6A, the end-product of Mettl3 catalytic activity, and methionine-SAM, the precursors for all methylation reactions, are also higher in cystic kidneys. Next, in a series of complementary experiments, we demonstrate that Mettl3 and methionine are causally linked to cyst growth. Using gain and loss-of-function alleles, we show that Mettl3 is partially necessary and sufficient to promote in vivo cyst growth. Cyst epithelia in Mettl3Tg mice were positive for the Mettl3-GFP transgene indicating that Mettl3 overexpression cell-autonomously produces cysts. Conversely, Mettl3 deletion attenuated cyst growth in multiple ADPKD models irrespective of the type of Pkd1 mutation (null or hypomorphic) or dynamics of cyst growth (rapidly fatal, aggressive, or slowly progressive). Similarly, methionine supplementation aggravated ex-vivo cyst growth, whereas dietary methionine restriction ameliorated murine PKD. Methionine’s role in RNA methylation can partially explain its cyst-promoting effects because (i) enhancing the transmethylation potential of kidney cells via SAM is also aggravates ex-vivo cyst growth, (ii) methionine and SAM (but not SAH) promote Mettl3 expression in kidney cells, and (iii) the pro-proliferative effect of methionine in Pkd1-mutant cells is lost in the absence of Mettl3.
c-Myc and Avpr2/cAMP are among the classical pathways linked to PKD progression. However, how c-Myc is upregulated or whether Avpr2 expression is even altered in PKD is unclear. Our work suggests a novel, Mettl3-regulated, epitranscriptomic mechanism for c-Myc and cAMP signaling activation in PKD. First, we report that Mettl3 enhances c-Myc mRNA translation in PKD models and thereby promotes Yap1 expression, in vivo neo-protein synthesis, and cyst epithelial proliferation. Second, in somewhat of a surprising finding, we found that Avpr2 is upregulated in cystic kidneys, in part due to Mettl3-enhanced translation of Avpr2 mRNA, suggesting a new mechanism for the high cAMP levels observed in PKD. In further support of this idea, we noted that Mettl3 deletion decreases, whereas its overexpression increases pCREB expression. Moreover, kidney-specific Mettl3 overexpression produces an antidiuretic effect indicative of elevated Avpr2-cAMP signaling. In keeping with the notion of a linear methionine-SAM-Mettl3 pathogenic axis, methionine and SAM also independently promote c-Myc and Avpr2 expression. Conversely, dietary methionine restriction lowers c-Myc and Avpr2 expression. Interestingly, methionine restriction has been reported to induce polyuria in mice (Cooke et al., 2018), further suggesting an inhibited Avpr2-cAMP axis.
An important insight from our work is that methionine utilization is a previously unrecognized ADPKD metabolic vulnerability. We report that Pkd1-mutant cells, but not control cells, depend on transmethylation for their proliferation, a finding that is reminiscent of methionine dependence observed in numerous cancer cell lines (Cellarier et al., 2003; Wang et al., 2019). Interestingly, methionine reliance may connect to aerobic glycolysis, another major metabolic adaptation of cyst epithelium (Padovano et al., 2018; Rowe et al., 2013). Methionine, an essential amino acid, cannot be synthesized de novo in humans or mice. However, it can be regenerated via homocysteine remethylation, which is sustained by converting serine to glycine. In turn, serine is synthesized via the glycolysis intermediate 3-phosphoglycerate, providing a path for aerobic glycolysis to feed into methionine metabolism (Maddocks et al., 2016). Indeed, diversion of glycolytic flux into serine biosynthesis has been shown to promote oncogenesis (Locasale et al., 2011; Possemato et al., 2011) and contribute to the cellular SAM pool (Yu et al., 2019). A recent study found that glucose restriction leads to decreased intracellular methionine levels in yeast cells (Zou et al., 2020). The life span extension by glucose restriction was abrogated by external methionine supplementation, further suggesting that glucose and methionine metabolism are intertwined.
Another implication of our findings is that Mettl3 may be a methionine-sensing enzyme that facilitates altered methionine utilization and links this amino acid to a pro-proliferative transcriptomic output. A paradoxical observation is that even though transmethylation is essential for cancer cell growth, their DNA is globally hypomethylated (Ehrlich, 2009). Similarly, ADPKD tissues also exhibit DNA hypomethylation (Bowden et al., 2018). Our work suggests that one reason for methionine dependence is that, rather than DNA, methionine is used in the RNA methylation pathway.
Finally, and perhaps most importantly, our work suggests two new treatment strategies for PKD. Firstly, dietary methionine restriction, such as a vegan diet, is a potential novel therapeutic intervention to slow cyst growth. We have previously shown that reduced food and caloric intake ameliorates cyst growth (Warner et al., 2016), an effect that is recapitulated simply by methionine restriction.
Therefore, the beneficial effects of lower food intake may be partly due to reduced cellular methionine levels. A recent study also demonstrated the feasibility of methionine restriction in humans suggesting a potential translational path (Gao et al., 2019). Second, our work has uncovered Mettl3 as a new drug target for PKD. Indeed, several biotech companies and academic laboratories have already developed small molecule inhibitors of the METTL3-METTL14 complex (Cully, 2019 Bedi et al., 2020). Two of these inhibitors are poised to enter phase I clinical trials with the eventual goal of treating AML. Considering that the methionine-Mettl3 axis promotes the AVPR2-cAMP pathway, dietary methionine restriction or a potential Mettl3 inhibitor may act in synergy with tolvaptan. However, these are early days, and there are many open questions, including if these drugs will have off-target effects or be safely tolerated.
Limitations of Study
First, while we show that activation of the SAM-Mettl3 axis is an early event in the murine cystogenesis process, how Pkd1 deletion leads to elevated SAM levels is unknown. It is tempting to speculate that aerobic glycolysis, which can fuel the methionine cycle via serine synthesis, contributes to the high SAM levels (Yu et al., 2019). Moreover, SAM-independent mechanisms for Mettl3 activation may exist, which were not investigated. Second, we have validated only two Mettl3 targets. We also did not test if combined heterozygous deletions of c-Myc and Avpr2 recapitulates the effects of Mettl3 deletion. Finally, the cyst-reducing effect may not be unique to methionine restriction, and whether the dietary restriction of other essential amino acids produces similar benefits remains unstudied.
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Vishal D. Patel (vishald.patel@utsouthwestern.edu).
Materials Availability
The Mettl3F/F and Mettl3Tg mouse lines were generated in this study and are available at the University of Texas Southwestern Medical Center.
Data and Code Availability
The MeRIP-seq and RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE165956.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All experiments involving animals were approved by the UT Southwestern Institutional Animal Care and Research Advisory Committee or the Institutional Animal Care and Use Committee at the Mayo Clinic (Protocol no. A52112 and A00003864). The studies were conducted in adherence to the NIH Guide for the Care and Use of Laboratory Animals. We utilized the Ksp-Cre, Ksp-rtTA, tetO-Cre (Stock No: 006234, Jackson Laboratories), Pkd1F/F and Pkd1RC/RC(Pkd1 p.R3277C) mouse lines in our studies (Hopp et al., 2012; Pan et al., 2013; Shibazaki et al., 2008). Additionally, we generated new Mettl3F/F and Mettl3Tg mouse lines for these studies (described below). Mice were housed in the UT Southwestern Animal Facility room that is always maintained at 18-24 °C and at 30-50% humidity. Mice used for the methionine diet study were housed at the Mayo Clinic Animal Facility. These mice were maintained at regulated temperature (20-22 °C) and humidity (30-70%). All the mice were always maintained on a 24-h light/dark cycle of 6:00 AM - 6:00 PM ‘light on’ schedule. Ad libitum access to food and water was provided to mice at all times. All mice were maintained in the C57BL/6 background and were used as the result of in-house matings. Similar numbers of male and female mice were analyzed for each experiment. Mice were generally healthy at the time of sacrifice. Age of the mice used for different experiments are described in the figure legends. Cre-negative, age-matched littermates were used as controls. For the ex vivo embryonic organ culture studies, the corresponding contralateral kidneys were used as controls.
Generation of Mettl3 Mouse models
Conditional Mettl3 knockout mouse
As global Mettl3 knockout is embryonically lethal, we generated a conditional null mouse line. As the first step, we created a Mettl3 conditional knockout allele in C57BL/6 ES cells using a vector modeled from L1L2_Bact_P (#45359, KOMP repository). This vector contains the L1L2_Bact_P cassette inserted at position 52299195 of Chromosome 14 (mMettl3 genomic region). The cassette contains an FRT site followed by the lacZ sequence and a loxP site. The loxP site is followed by the neomycin resistance gene under the control of the human beta-actin promoter, SV40 polyA, a second FRT site, and a second loxP site. A third loxP site is inserted downstream of exon four at position 52298473. The vector has two homology arms to the mMettl3 gene. One is upstream of position 52299195, and the second is downstream of position 52298473 of Chromosome 14. Through homologous recombination, the vector was inserted into the C57BL/6 ES cells at the homology arms. ES cells were screened to identify a correctly targeted clone. The clone, with one wild-type Mettl3 allele and one L1L2_Bact_P cassette inserted allele, was injected into C57BL/6 blastocysts. Mettl3-targeted mouse line was established from a germline-transmitting chimera. The chimeric mouse was crossed to C57BL/6 Flp mice to excise the neomycin resistance system. The IRES-LacZ is also removed in the process. The resultant heterozygous Mettl3F/+ mouse contains two LoxP sites flanking exon 4 of Mettl3. This mouse was bred to homozygosity and maintained in the C57BL/6 background. Ksp/Cre mouse line was used to ablate Mettl3 in the kidney tubules. Cre-mediated recombination of the floxed allele causes a frameshift, which inserts a premature stop codon. Litters were genotyped by Dreamtaq PCR utilizing two primers: Forward: GTT TCG AGG AAG TCC TTA TTT ACA GAA TCT GG 5′; Reverse: TGT AGC AGA GTT GTT ATA GGC TCA GTC CTC C, 5′ (95°C, 3 min; 95°C, 30 s; 65°C, 30 s; 72°C, 1 min; 30 times to step two). We generated this mouse in collaboration with Genoway S.A.
Conditional Mettl3 overexpression mouse
We generated the Mettl3 overexpression construct from the VectorBuilder’s mammalian gene expression vector that contains a pUC origin of replication, an ampicillin resistance gene, and an SV40 late polyadenylation site. A CAG promoter, an LSL sequence, and the Mettl3 cDNA (NM_019721.2) fused with HA/HA/HA tag were cloned into the plasmid. We also included an IRES-GFP reporter downstream of the Mettl3 cDNA and upstream of the pA site. The resulting Mettl3-Tg construct (8226 bps) was transfected into IMCD3 cells and validated whether Mettl3/HA and GFP expression were Cre-dependent.
We then linearized this construct with SapI and AscI restriction enzymes and removed the pUC and Ampicillin gene sequences. The resulting 6228 bp Mettl3-Tg fragment: CAG-LSL-Mettl3/HA/HA/HA - IRES-GFP- pA was injected into fertilized embryos. The embryos were subsequently transferred to pseudo-pregnant c57BL6 females. The G0 founders (29 mice) were PCR genotyped using primers specific to the Mettl3-Tg transgene. 9/29 animals were positive for the transgene. We bred each of these nine founders to determine germline transmission of Mettl3-Tg transgene. Four founder mice (two males and two females) passed on the transgene at Mendelian ratios to their offsprings. These F0 founders were bred with Ksp-Cre mice to assess transgene activity and phenotypic characterization. F1 and F2 progeny were also evaluated for transgene activity and phenotype. The vector was generated with VectorBuilder, and the transgenic Core performed the pronuclear injection at UT Southwestern Medical Center. Litters were genotyped by DreamTaq PCR using the primers: forward - 5' ATATGGAGTTCCGCGTTACA, reverse – 5' TGGCGTTACTATGGGAACAT.
Human specimens
The PKD Biomarkers and Biomaterials Core in the Kansas PKD Center at the University of Kansas Medical Center (KUMC) provided the frozen ADPKD and NHK specimens, which have we have used in previous studies (Lakhia et al., 2020). The OCT blocks were sectioned at 5 um and stained with an anti-Mettl3 or anti-m6A antibody using the protocol described in the immunofluorescence staining section of Methods. The protocol for the use of surgically discarded kidney tissues complied with federal regulations and was approved by the Institutional Review Board at KUMC.
Low-Methionine Diet Studies
The low-methionine diet study was performed on 5-month-old Pkd1RC/RC mice on a mixed background. Male and female mice were randomized into two groups: (i) control diet (0.62% methionine, 0.36% cysteine) and (ii) low-methionine diet (0.16% methionine, 0.00% cysteine). The mice had ad libitum access to food and water. One week before the study, the mice were placed in individual metabolic cages for acclimation. Daily food intake was monitored throughout the study. After 15 weeks of dietary intervention, mice were euthanized according to approved protocols. Blood was collected, and the kidneys were weighed. Portions of kidney tissue were placed in formalin and processed for histological studies or snap-frozen in liquid nitrogen and stored at −80 °C for further analysis. The low-methionine diet was purchased from Envigo Teklad Diets (Madison, WI). Glutamic acid was increased to keep the diet isonitrogenous.
Cell Lines
mIMCD3, Pkd1RC/−, and Pkd1RC/+ cell lines were used in the current study. The Pkd1RC/− and Pkd1RC/+ are immortalized, male, murine collecting duct cells. The mIMCD3 cells were grown in DMEM media supplemented with 10% FBS at 37 °C. Pkd1RC/− and Pkd1RC/+ cells were grown in DMEM:F-12 medium (Gibco, #10565-018) supplemented with 2% FBS, 5 μg/ml insulin, 5g/ml transferrin, and 5 ng/ml sodium selenite (Lonza, #17-838Z).
METHOD DETAILS:
Mouse kidney tissue harvest
Mice were euthanized in accordance with approved protocols. Blood was obtained via cardiac puncture when animals were under anesthesia. The right kidney was extracted, weighed, flash-frozen, and stored at −80 °C for molecular analysis. The left kidney was fixed with perfusion of 4% (wt/vol) paraformaldehyde for histological and immunofluorescence analysis. The fixed kidneys were sagittally bisected, and one half was treated with sucrose overnight and then embedded in OCT. The other half of the left kidney was embedded in paraffin.
Histology and cyst index analysis
Tissue embedding in paraffin and subsequent sectioning was performed using standard protocols by the Histology core at UT Southwestern Medical Center (Mettl3 studies) or the Mayo Clinic Histology Core Laboratory (methionine diet restriction studies). The tissues were cut into 5 μm sections and stained with hematoxylin-eosin (H&E) to analyze the cyst index. The stained sections were imaged using the slide scanner and analyzed for the cyst index (cyst area proportional to cross-sectional kidney area) using the Image J software described earlier (Kashyap et al., 2020; Warner et al., 2016; Yheskel et al., 2019).
Serum creatinine measurement
Serum creatinine was measured using capillary electrophoresis. Serum was separated via centrifugation from blood obtained through cardiac puncture of anesthetized mice and then frozen at −80 °C until needed.
RNA isolation & quantitative RT-PCR
Total RNA was isolated from mouse kidney tissues or cells using the miRNeasy Mini kit from Qiagen (catalog #217004). The RNA was used for cDNA synthesis or mRNA purification, as described below. The first-strand cDNA synthesis was performed using the iScript cDNA synthesis kit (catalog #170-8891, Bio-Rad) and the Superscript III first-strand synthesis system (catalog#18080051, Invitrogen) or QuantiTect Reverse Transcription Kit (Qiagen). quantitative RT-PCR (qRT-PCR) was performed using the iQ SYBR Green Supermix (catalog 170-8882, from Bio-Rad) or TaqMan Gene Expression Master Mix (Applied Biosystems). The samples were loaded in duplicates or triplicates on a CFX ConnectTM Real-time PCR detection system. 18S RNA or Gapdh was used to normalize gene expression. The raw data were analyzed using the Bio-Rad CFX software. qRT-PCR primers are listed below:
| Mettl3 F: GTC AGC GTC ACT GGC TTT CTT | R: GTC AGT CAG GAG ATC CTA GAG CTA |
| Pkd1 F: CTA GAC CTG TCC CAC AAC CTA | R: GCA AAC ACG CCT TCT TCT AAT GT |
| Ngal F: CTC TTG TAG CTC ATA GAT GGT GC | R: GCA GGT GGT ACG TTG TGG G |
| c-Myc F: CTG AGC CCC TAG TGC TGC ATG A | R: GGG GTT TGC CTC TTC TCC ACA G |
| Avpr2 F: TGA CCG AGA CCC GCT GTT A | R: CGA CCC CGT CGT ATT AGG G |
m6A ELISA
mRNA was isolated from 40 ug of total RNA using the Dynabeads mRNA purification kit (catalog number: 61006). m6A modification on mRNAs was measured using the EpiQuik m6A RNA Methylation Quantification Kit (catalog #P-9005-96, Epigentek). We performed ELISA on all samples at the same time.
S-adenosylmethionine ELISA
SAM levels in kidney tissues of age-matched control and ADPKD mice were measured using the ELISA kit from Cell Biolabs (# STA-671-C) following the manufacturer's protocol. The ELISA was performed using protein lysate prepared from snap-frozen kidney tissues. The lysis buffer was freshly prepared with 1X PBS (#10010049, Invitrogen) and a protease inhibitor cocktail (cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail, Catalog #11836170001 from Roche) at a ratio of 1 tablet per 7 ml of 1X PBS. The homogenization was performed using either the microtube hand-held homogenizer (Bel-Art™ F65000-0000, Fisher) or the Bead Mill 24 (Fisher Scientific), kept on ice and maintained at 4 degrees at all times. The homogenates were centrifuged for 15 minutes at 10,000 g in 4 degrees. The lysates were diluted to approximately one ug/ul concentration and immediately proceeded with ELISA. The 450 nm OD readings of the samples were plotted on the standard curve obtained using the kit’s SAM-BSA standards to derive SAM levels in the test samples. Further, the SAM levels were normalized to the exact protein amount loaded for comparative analysis. Protein concentration was measured using the Bradford Assay reagent on the same day.
Immunofluorescence staining
Following PFA fixation, the kidney tissues were washed three times with 1X PBS and then treated with 30% sucrose overnight. The samples were embedded in OCT and stored at −80 °C degrees until further use. The OCT blocks were cryo-sectioned at 5 um thickness and kept at −20 °C degrees. Before immunofluorescence staining, the slides were left at room temperature for 30 minutes. The slides were then washed in 1X PBS for 5 minutes. The slides were antigen retrieved with sodium citrate for anti-m6A, anti-pCreb, anti-pHH3, and anti-Yap1 antibodies. For anti-Mettl3, anti-GFP, and Biotinylated-DBA antibodies, the slides were antigen retrieved with 0.25% TritonX 100 at room temperature for 40 minutes. Subsequently, the slides were treated with sodium borohydride to quench autofluorescence for 40 minutes and with 1X PBS+10% goat serum+0.1% BSA (antibody block) for 1-2 hours at room temperature. Sections were incubated with primary antibodies (1:500 dilution) overnight. The next day, slides were washed with PBS, incubated with appropriate Alexa Fluor secondary antibodies (1:500 dilution) for 1 hour, and mounted using Vecta Shield containing Dapi. We imaged the slides using a Zeiss Compound Light microscope.
Western blot analysis
Protein was extracted from cells or tissues using T-per tissue protein extraction reagent (catalog #78510) lysis buffer. Concentration was measured using the Bradford Assay reagent. 10-40 ug of protein was loaded on SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. The membrane was blotted with anti-Mettl3 (#ab195352 Abcam, #MA5-27527 Invitrogen), anti-Mettl14 (#HPA038002, Sigma), anti-GFP (#GFP-1020 Aves Lab), anti-HA (#3724, Cell Signaling), anti-c-Myc (#ab185656 Abcam, #OP10-200ug Sigma), anti-Avpr2 (#AB1797P, EMD Millipore), anti-Yap1 (#4912, Cell Signaling), anti-pCREB (#9198S, pCreb1), anti-PCNA (#sc-7907, Santa Cruz), or anti-puromycin (#PMY-2A4, Developmental Studies Hybridoma Bank) antibodies. All the primary antibodies were used at 1:1000 dilution, except for anti-puromycin, which was used at 1:50 dilution. Goat anti-rabbit-HRP conjugate (#7074S, Cell Signaling) or anti-mouse HRP-conjugated IgG (#7076S, Cell Signaling) was used as the secondary antibody. HRP-conjugated Actin antibody (#A3865, Sigma) was used at 1:40,000 dilution for measuring total protein. The blots were developed using the X-ray film developer or the Bio-rad digital imager. The protein bands were quantified using the Imagelab software from Bio-rad. Each western blot was repeated at least three times.
MeRIP-Seq and RNA-seq analysis
m6A RNA immunoprecipitation was performed using the Magna MeRIP™ m6A Kit (EMD Millipore, catalog #17-10499). Briefly, we chemically fragmented RNA into 100 nucleotides or smaller fragments. Next, we performed magnetic immunoprecipitation using mouse monoclonal anti-m6A (ab151230; Abcam) and rabbit IgG (sc-2027; Santa Cruz Biotechnology) antibody as a negative control. We used kidney RNA from 18-day-old control, Pkd1F/RC-KO, and Pkd1F/RC-Mettl3-KO mice. Two biological replicate samples per group were analyzed, each sample containing pooled fragmented RNA from either three male or three female mice.
We prepared RNA libraries for Illumina sequencing using the NEBNext® Ultra™ RNA Library Prep Kit (catalog E7530S, New England Biolabs). We used the immunoprecipitated (anti-m6A and anti-IgG) and input RNA to prepare libraries for sequencing. Libraries were sequenced on Illumina NextSeq 500 at Next Generation Sequencing Core at Eugene McDermott Center for Human Growth and Development at UT Southwestern Medical Center. 35-45 million single-end reads with a read length of 76 bp were generated for each library. FASTQ files were checked for quality using fastqc (v0.11.2) found at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, fastq_screen (v0.4.4) found at http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen. The reads from FASTQ files were aligned to the mouse genome (mm10) using STAR, a splice-aware aligner for RNA-seq data (Dobin et al., 2013). Duplicate alignments were marked using Picard-tools (v1.127 https://broadinstitute.github.io/picard/). The peaks were called using MACS2 (v2.1.0) (Zhang et al., 2008), with a q-value threshold of 0.05 and using the RNA input of MeRIP samples as controls. The fragment size of each library was used to extend reads at their 3′ ends to a fixed length with "–extsize" parameter in MACS2. The peaks were annotated using ChIPseeker (Yu et al., 2015). The motif analysis was performed using HOMER (v4.7.2) (Heinz et al., 2010). The differential expression analysis of input RNA samples of control, Pkd1F/RC-KO and Pkd1F/RC; Mettl3F/F-KO was performed using edgeR (Robinson et al., 2010). The BAM files were input normalized using the bamCompare module of deepTools (v3.1.1) (Ramirez et al., 2016). The input normalized bigwig files were used to visualize the coverage tracks in the IGV browser app (http://software.broadinstitute.org/software/igv/). The differentially bound m6A sites between groups of samples (control, Pkd1F/RC-KO and Pkd1F/RC;Mettl3F/F-KO) were analyzed using Diffbind (Ross-Innes et al., 2012). The normalized read count of all binding sites was used to cluster samples. The Heat map was plotted using the Morpheus software from MIT Broad Institute. The MeRIP sequencing data were deposited to the Gene Expression Omnibus GSE165956.
Proliferation studies in mouse kidneys
Sagittal kidney sections of control, Pkd1F/RC-KO, and Pkd1F/RC-Mettl3-DKO mice were stained with anti-pHH3 antibody (#H0412, Sigma) and anti-Rabbit IgG Alexa Fluor 594 conjugate secondary antibody and counterstained with Dapi. We scanned the entire tissue section on the slides using the digital slide scanner microscope. The images were analyzed using the Zeiss software, Zen Blue edition. We counted all the pHH3 positive cells in the kidney section utilizing the software's custom draw tool.
In vivo Protein synthesis Measuremen
Puromycin
We injected mice with 40 ug/g body weight puromycin (Invitrogen, #A1113803) intraperitoneally 90 minutes before kidney harvest (Hato et al., 2019). The kidneys were flash-frozen and stored at −80 °C. Protein extraction, sample preparation, and western blots were performed as described above. Newly synthesized peptides were detected using an anti-puromycin antibody (1:50 dilution) from Hybridoma Studies. The blots were stained with Ponceau for total protein measurement. The blots were imaged using the Bio-Rad imager and quantified using the ImageLab software.
O-propargyl-puromycin
We adapted the protocol to detect OPP-tagged neo-peptides from previous studies (Jao and Salic, 2008; Liu et al., 2012). OPP was purchased from JenaBioScience (catalog # NU-931-5) and reconstituted in PBS at ten ug/ul. OPP or PBS was injected into mice at 40 ug/g body weight intraperitoneally 90 minutes before kidney harvest. Kidney section slides were washed three times for 10 minutes each with 1X TBS containing 0.1% of TritonX100. We prepared the Click Chemistry mix by adding the following reagents in this order: 0.1X Tris Base (pH7.5) 100 mM, 0.2% TritonX100, one mM Copper Sulphate solution, 100 uM AlexaFluor Azide555, and 100 mM Ascorbic Acid. The mix was freshly prepared right before performing the Click Chemistry reaction. The slides were incubated with the Click Chemistry Mix for 30 minutes at room temperature in the dark. The slides were washed five times with 1X TBS containing 1% TritonX100 for 30 minutes each before mounting with coverslips. We obtained images using the digital slide scanner and the Zeiss compound microscope.
Mettl3 Knockdown and Overexpression in cells
We used mIMCD3, Pkd1RC/+, and Pkd1RC/− cells for the Mettl3 knockdown and overexpression studies. Cells were transfected siRNAs or the Mettl3Tg plasmid using Lipofectamine 3000 (catalog #L3000008, Invitrogen). The cells were lysed 72 hours after siRNA treatment to extract RNA or protein. We obtained scrambled and Mettl3 siRNAs from Dharmacon. For the overexpression studies, infection with the Ad-CMV-iCre virus (Vector Biolabs, #1045) was performed 24 hours after transfection with the Mettl3Tg plasmid. We obtained protein and RNA four days after Cre infection.
Luciferase Assay in Cells
Avpr2 (exon2) was PCR-amplified using forward and reverse primers that introduced XbaI and XhoI restriction sites at the 5′- and 3′- ends, respectively. The forward primer sequence was 5' -TAG AAT TCT AGA CAG TGC CTG GGG CCC TTT CGT CCC CTA-3' whereas the reverse primer was 5'-TAG AAT CTC GAG TTT CCA GAG GAG CTT CTG GAT CCC ACG-3'. This PCR product and the pLightSwitch Empty (pLS) 3′-UTR plasmid were digested with XbaI and XhoI restriction enzymes and were ligated to generate the Avpr2-pLS plasmid. 0.2 ug of Avpr2-pLS plasmid or the empty pLS plasmid was transfected in control or Pkd1RC/− cells (seeded at 2.5 x 104 density/well, 24-well plates). The cells were also transfected with 0.02 ug pGL3 vector to control for transfection efficiency. For Mettl3 knockdown studies, the cells were co-transfected with scrambled or Mettl3 siRNAs at 100 nM concentration. For Mettl3 overexpression studies, the cells were co-transfected with 0.75 ug of constitutively-active Mettl3Tg plasmid. Seventy-two hours later, the cells were lysed using 100 ul of 1x passive lysis buffer, and 10 μl of the cell lysate was added to 96-well plates. Photinus and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega Corp) according to the manufacturer's directions.
Ex vivo Organ Culture
Embryonic day (E) 12.5 kidneys were cultured on the Whatman membrane (Sigma, WHA110409) in an air-medium interface as described previously in (Ramalingam et al., 2018). We used regular DMEM media (Thermo fisher #12500) containing 10% Fetal Bovine Serum (FBS) and 2% PenStrep (Invitrogen #1514022). After 24 hours, the media was replaced with the DMEM media containing the following cyst growth ingredients: 100 uM of 8-Br-cAMP (Catalog #B7880, Sigma), 10% FBS, ten mM HEPES, 5 μg/ml insulin, 5 μg/ml transferrin, 2.8 nM selenium, 25 ng/ml prostaglandin E, 32 pg/ml T3, 250 U/ml penicillin, and 2% PenStrep (Magenheimer et al., 2006), for another 24 hours to induce cyst formation. These were further cultured for an additional 48 hours in three different ways: (i) for the SAM supplementation experiments, we treated one kidney from each embryo either with 250 uM SAM (Cayman Chemicals #13956) (Albrecht et al., 2019) or with a vehicle (20mM HCl). (ii) For methionine depletion experiments, we cultured one kidney from each embryo in the special DMEM growth media without methionine (Invitrogen #21013024). The other kidney was cultured in the equivalent media that contains 0.2 mM methionine (Invitrogen #11965092). In addition to the cyst growth ingredients listed above, 110 mg/L sodium pyruvate (Gibco, #11360070) was included to maintain a consistent media formulation. Additionally, four mM L-glutamine (Sigma, #G7513) was supplemented to match the formulations of methionine-depletion media and control media. (iii) For the methionine supplementation assays, the kidneys were cultured in DMEM media containing the cyst growth ingredients and supplemented with a freshly prepared L-Methionine (Sigma, # M5308) to reach a final concentration of 0.8 mM or 2 mM methionine. The cultures were imaged using the AmScope SE306R-PZ Forward Binocular Stereo Microscope at 3.75x magnification. The cysts were measured and analyzed using the ImageJ software.
AlamarBlue Proliferation Assay in Cells
SAM and SAH treatment
Pkd1RC/+ and Pkd1RC/− cells (1x104 density) were seeded on 24-well plates. One day later, these cells were treated with DMEM culture media containing 1X alamarBlue reagent (Invitrogen, DAL1025) and 100 uM of vehicle, freshly reconstituted SAM or 100 uM of SAH (Cayman Chemical, #13603). Calorimetric readings were taken at 570 nm and 600 nm in a microplate reader after 12 hours. The REDOX reaction of alamarBlue was used to assess cell proliferation quantitatively. A higher reduction in alamarBlue reagent implies a higher proliferation rate.
Methionine Depletion
Pkd1RC/+ and Pkd1RC/− cells (1x104 density) were seeded on 24-well plates. One day later, these cells were treated with DMEM culture media containing 0.2 mM or 0 mM methionine and 1X alamarBlue reagent (Invitrogen, DAL1025). Calorimetric readings were taken at 570 nm and 600 nm in a microplate reader after 12 hours.
Mettl3 knockdown experiments
Pkd1RC/− cells were seeded at an initial density of 0.8x104 per well in 24-well plates. siRNA transfection was performed 12 hours later. After 48 hours of siRNA treatment, these cells were treated with DMEM culture media containing alamarBlue reagent and 0 uM or 2 uM methionine. Twelve hours later, the calorimetric readings were taken at 570 nm and 600 nm in a microplate reader.
Methionine/SAM treatment in Mettl3 KD cells
Pkd1RC/− cells were seeded at an initial density of 0.5x105 per well in 6-well plates. siRNA transfection was performed 12 hours later. After 48 hours of siRNA treatment, these cells were treated with DMEM culture media containing 0 uM or 2 uM methionine or 100 uM vehicle or freshly reconstituted SAM. Twelve hours later, the protein lysates were prepared from cells and proceeded to western blot analysis.
Mettl3Tg Urine Analysis
Mice were placed in the metabolic cages and were allowed to acclimatize for three days. Following acclimatization, we collected urine every 24 hours for three days. Osmolality was measured using the Advanced Instruments OsmoPro osmometer from the Bone and Mineral Metabolism department at UT Southwestern (Aboudehen et al., 2017). Throughout the experiment, mice were maintained in a controlled 12-hour day-night cycle in separate cages and provided water and a regular diet. Sex-matched littermate Cre- Mettl3Tg were used as controls.
Untargeted Metabolomic Analysis of Kidneys
Untargeted metabolomics in kidneys was carried out at The Mayo Clinic Metabolomics Resource Core facility. We used kidney tissues from 7.5 months old wild-type and Pkd1RC/RC mice. Samples were deproteinized with six times volume of cold acetonitrile: methanol (1:1 ratio), kept on ice with intermittent vortexing for 30 minutes at 4°C, followed by centrifugation at 18000xg. Three μl of an internal standard solution of 13C6-phenylalanine (250ng/μl) was added as an internal standard to each sample before deproteinization. The supernatants were divided into two aliquots and dried down for analysis on a Quadrupole Time-of-Flight Mass Spectrometer (Agilent Technologies 6550 Q-TOF) coupled with an Ultra High-Pressure Liquid Chromatograph (1290 Infinity UHPLC Agilent Technologies). We acquired profiling data both under positive and negative electrospray ionization conditions over a mass range of 100 - 1200 m/z at a resolution of 10,000-35,000. Metabolite separation was achieved using two columns of differing polarity, a hydrophilic interaction column (HILIC, ethylene-bridged hybrid 2.1 x 150 mm, 1.7 mm; Waters) and a reversed-phase C18 column (high-strength silica 2.1 x 150 mm, 1.8 mm; Waters). For each column, the run time was 20 min using a flow rate of 400 μl/min. A total of four runs per sample were performed to give maximum coverage of metabolites. Samples were injected in duplicate, and a quality control sample consisting of a subset of samples from the study was injected several times during a run. All raw data files obtained were converted to a compound exchange file format using Masshunter DA reprocessor software (Agilent). Mass Profiler Professional (Agilent) was used for data alignment and to convert each metabolite feature (m/z x intensity x time) into a matrix of detected peaks for compound identification. An unsupervised principal component analysis, ANOVA, 3D plot and heat map, and a Partial Least Square discrimination analysis (PLS-DA) comparison between groups were obtained for analysis giving a list of accurate mass molecular weights of differentially expressed components that were run against the Metlin database to provide putative identification (IDs). IDs were further examined by comparison to a purchased reference standard of the proposed compound. Mass accuracy of the Q-TOF method is <5ppm with retention time precision better than 0.2%. A 1.2x fold change can be detected with a precision of 4%.
Data analysis was performed using Mass profiler Professional (MPP, Agilent Inc.). Each sample was normalized to the median of the baseline, and log 2 transformed. Metabolites in the completed data matrix were normalized and scaled to account for technical variations. Metabolites detected in at least ≥50% of the samples in the study groups were selected for multivariate and differential expression analyses. Normalized data were analyzed by a multivariate approach such as Principal Component Analysis (PCA) to reveal data homogeneity, groupings, outliers, and trends. Univariate statistical analysis, Students’ unpaired t-test was performed with multiple testing correction (q-value/FDR≤0.05) using the bootstrap analysis to identify the differentially expressed metabolites between two groups with statistical significance (P≤0.05). Missing values are excluded from the analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
The graphs depict mean and SEM. Student's t-test or Mann-Whitney was used for pairwise comparisons. Analysis of variance (ANOVA) followed by Tukey's post hoc test was used for multiple comparisons. Mantel-Cox test was used for survival analysis. P < 0.05 was considered statistically significant. A priori power analysis was not performed to determine sample sizes. We chose the sample sizes based on our experience of working with PKD mouse model and dietary intervention studies. The sample size, the type of statistical analysis used, and P values are mentioned in the figure graphs, the figure legends, and the results section. The data showed a continuous normal distribution. Mice were randomly assigned to the standard diet or MR diet group. Investigators were not blinded to the treatment or the genotypes of animals. Exclusion criteria were based on mouse well-being. No mice were excluded from this study.
Supplementary Material
Table S1. Putative Mettl3 targets in control, Pkd1F/RC-KO, and Pkd1F/RC-Mettl3-DKO kidneys, related to Figure 4. The table lists all Mettl3 mRNAs targets identified from MeRIP-seq that exhibit higher m6A levels in Pkd1F/RC-KO but lower m6A levels in Pkd1F/RC-Mettl3-DKO compared to control kidneys. Refer to STAR Methods section for how MeRIP-Seq analysis was performed.
Table S2. Untargeted metabolomic analysis between wild-type and Pkd1RC/RC kidneys, related to Figure 6. The table lists metabolites detected from Mass Spectrometry analysis. Refer to STAR Methods section on Untargeted Metabolomic analysis of Kidneys.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-METTL3 | Abcam | ab195352 |
| Anti-METTL3 | Invitrogen | MA5-27527 |
| Anti-m6A | Abcam | ab151230 |
| Anti-c-Myc | Abcam | ab185656 |
| Anti-c-Myc | Sigma | OP10-200UG |
| Anti-Avpr2 | EMD | AB1797P |
| Anti-Puromycin | Developmental Studies Hybridoma Bank | PMY-2A4 |
| Anti-HA | Cell Signaling | #3724 |
| Anti-PCNA | Santa Cruz | sc-7907 |
| Anti-pHH3 | Sigma | H0412 |
| Anti-pCreb1 | Cell Signaling | 9198S |
| Anti-Yap | Cell Signaling | #4912 |
| Anti-GFP | Aves | GFP-1020 |
| Anti-DBA | Vector Laboratories | B-1035 |
| Anti-Mettl14 | Sigma | HPA038002 |
| Bacterial and Virus Strains | ||
| Ad-CMV-iCre virus | Vector Biolabs | #1045 |
| Biological Samples | ||
| Normal human and ADPKD kidney samples | PKD Biomarkers and Biomaterials Core in the Kansas PKD Center at the Kansas University Medical Center (KUMC) | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin | Invitrogen | A1113803 |
| O-Propargyl-Puromycin | JenaBioScience | NU-931-5 |
| 8-Br-cAMP | Sigma | B7880 |
| alamarBlue | Invitrogen | DAL1025 |
| methionine | Sigma | M5308 |
| s-adenosylmethionine | Cayman Chemicals | 13956 |
| s-adenosylhomocysteine | Cayman Chemicals | 13603 |
| Critical Commercial Assays | ||
| m6A elisa kit | Epigentek | P-9005-96 |
| SAM elisa kit | Cell Biolabs | STA-671-C |
| Magna MeRIP™ m6A Kit | EMD Millipore | 17-10499 |
| Submitted data | ||
| MeRIP-seq | NCBI Gene Expression Omnibus | GSE165956 |
| RNA-seq | NCBI Gene Expression Omnibus | GSE165956 |
| Experimental Models: Organisms/Strains | ||
| Ksp/Cre | Shibazaki et al., 2008 | N/A |
| Ksp/rtTA | Pan et al., 2013 | N/A |
| tetO-Cre | Jackson Laboratories | Stock No: 006234 |
| Pkd1F/F | Jackson Laboratories | Stock No: 010671 |
| Pkd1RC/RC(PKD1 p.R3277C) | Hopp et al., 2012 | N/A |
| Mettl3F/F | UT Southwestern Med Ctr | N/A |
| Mettl3Tg | UT Southwestern Med Ctr | N/A |
| mIMCD3 cells | ATCC | CRL 2123 |
| Oligonucleotides | ||
| siRNA against Mettl3 (UGGUUUACAUGUCGACUAA, UGGUUUACAUGUUGUGUGA, UGGUUUACAUGUUUUCUGA, UGGUUUACAUGUUUUCCUA) | Dharmacon | L-049446-01-0020 |
| Scrambled siRNAs (ACAGCAGGCACAGACAGGCAGU) | Dharmacon | D-001810-10-20 |
| Recombinant DNA | ||
| Plasmid - CAG-LSL-Mettl3cDNA-HA/HA/HA-IRES-GFP | This paper | N/A |
| Plasmid - pLightSwitch Empty (pLS) 3′-UTR | Switchgear Genomics | S890005 |
| Plasmid - pGL3 | Promega | E1751 |
| Software and Algorithms | ||
| Imagej | NIH | https://imagej.nih.gov/ij/ |
| Zen blue software | Zen | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| ImageLab | Bio-rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| Ingenuity Pathway Analysis (IPA) | Qiagen | https://analysis.ingenuity.com/pa/installer/select |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| Integrative Genomics Viewer (IGV) | Broad Institute | http://software.broadinstitute.org/software/igv/ |
Highlights.
S-adenosylmethoinine (SAM), Mettl3 and m6A levels are higher in ADPKD.
Methionine and SAM promote Mettl3 expression.
Mettl3 post-transcriptionally activates cyst-promoting c-Myc and cAMP signaling.
Mettl3 deletion or methionine dietary restriction improve ADPKD in mice.
Acknowledgments
The work is supported by the National Institutes of Health (R01DK102572) to VP and a PKD Foundation Research Grant Award to (E.N.C and S.K.). HR is supported by the PKD Foundation Research Fellowship. We thank the O'Brien Kidney Research Core Center (P30DK079328) at UT Southwestern Medical Center (UTSW), the Eugene McDermott Center for Human Growth and Development Sequencing Core and Bioinformatics Lab at UTSW, the UTSW Molecular Pathology Core, and the Whole Brain Microscopy Facility at UTSW for providing critical reagents and services. Human ADPKD and normal human kidney cells and tissues were provided by the PKD Biomarkers, Biomaterials and Cellular Models Core, located at the University of Kansas Medical Center. The Core is part of the PKD Research Resource Consortium, supported by the National Institutes of Health/NIDDK (U54 DK126126). We thank Dr. Ronak Lakhia and Dr. Abheepsa Mishra for their critical review and valuable inputs on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
E.N.C has a patent on the use of PAPP-A inhibitors in ADPKD. E.N.C. is a consultant for TeneoBio, Calico, Mitobridge, and Cytokinetics. E.N.C. is on the advisory board of Eolo Pharma. E.N.C. owns stocks in Teneobio. Unrelated to this work, V.P has patents involving the use of anti-miR-17 for the treatment of ADPKD (16/466,752 and 15/753,865). V.P. has previously held investment interests in Regulus Therapeutics and serves as a consultant for Otsuka Pharmaceuticals. V.P. lab has a sponsored research agreement with Regulus Therapeutics (unrelated to this work).
References
- Aboudehen K, Noureddine L, Cobo-Stark P, Avdulov S, Farahani S, Gearhart MD, Bichet DG, Pontoglio M, Patel V, and Igarashi P (2017). Hepatocyte Nuclear Factor-1beta Regulates Urinary Concentration and Response to Hypertonicity. J Am Soc Nephrol 28, 2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht LV, Bui MH, and De Robertis EM (2019). Canonical Wnt is inhibited by targeting one-carbon metabolism through methotrexate or methionine deprivation. Proc Natl Acad Sci U S A 116, 2987–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, and Caflisch A (2020). Small-Molecule Inhibitors of METTL3, the Major Human Epitranscriptomic Writer. ChemMedChem 15, 744–748. [DOI] [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, and Rottman FM (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Bowden SA, Rodger EJ, Bates M, Chatterjee A, Eccles MR, and Stayner C (2018). Genome-Scale Single Nucleotide Resolution Analysis of DNA Methylation in Human Autosomal Dominant Polycystic Kidney Disease. Am J Nephrol 48, 415–424. [DOI] [PubMed] [Google Scholar]
- Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, and Pan D (2018). A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev 32, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellarier E, Durando X, Vasson MP, Farges MC, Demiden A, Maurizis JC, Madelmont JC, and Chollet P (2003). Methionine dependency and cancer treatment. Cancer Treat Rev 29, 489–499. [DOI] [PubMed] [Google Scholar]
- Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, and McCann PP (1996). S-Adenosylmethionine and methylation. FASEB J 10, 471–480. [PubMed] [Google Scholar]
- Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al. (2018). mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke D, Ouattara A, and Ables GP (2018). Dietary methionine restriction modulates renal response and attenuates kidney injury in mice. FASEB J 32, 693–702. [DOI] [PubMed] [Google Scholar]
- Cully M (2019). Chemical inhibitors make their RNA epigenetic mark. Nat Rev Drug Discov 18, 892–894. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Ehrlich M (2009). DNA hypomethylation in cancer cells. Epigenomics 1, 239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein JD (1990). Methionine metabolism in mammals. J Nutr Biochem 1, 228–237. [DOI] [PubMed] [Google Scholar]
- Gao X, Sanderson SM, Dai Z, Reid MA, Cooper DE, Lu M, Richie JP Jr., Ciccarella A, Calcagnotto A, Mikhael PG, et al. (2019). Dietary methionine influences therapy in mouse cancer models and alters human metabolism. Nature 572, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, Aboudehen K, Zhang S, Kersjes K, Galasso R, et al. (2017). microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun 8, 14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happe H, van der Wal AM, Leonhard WN, Kunnen SJ, Breuning MH, de Heer E, and Peters DJ (2011). Altered Hippo signalling in polycystic kidney disease. J Pathol 224, 133–142. [DOI] [PubMed] [Google Scholar]
- Hato T, Maier B, Syed F, Myslinski J, Zollman A, Plotkin Z, Eadon MT, and Dagher PC (2019). Bacterial sepsis triggers an antiviral response that causes translation shutdown. J Clin Invest 129, 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, Rossetti S, Torres VE, and Harris PC (2012). Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 122, 4257–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CY, and Salic A (2008). Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 105, 15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, and He C (2013). Reversible RNA adenosine methylation in biological regulation. Trends Genet 29, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap S, Hein KZ, Chini CC, Lika J, Warner GM, Bale LK, Torres VE, Harris PC, Oxvig C, Conover CA, et al. (2020). Metalloproteinase PAPP-A regulation of IGF-1 contributes to polycystic kidney disease pathogenesis. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurbegovic A, and Trudel M (2020). The master regulators Myc and p53 cellular signaling and functions in polycystic kidney disease. Cell Signal 71, 109594. [DOI] [PubMed] [Google Scholar]
- Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, and Patel V (2018). PPARalpha agonist fenofibrate enhances fatty acid beta-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol 314, F122–F131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhia R, Yheskel M, Flaten A, Ramalingam H, Aboudehen K, Ferre S, Biggers L, Mishra A, Chaney C, Wallace DP, et al. (2020). Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EC, Valencia T, Allerson C, Schairer A, Flaten A, Yheskel M, Kersjes K, Li J, Gatto S, Takhar M, et al. (2019). Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat Commun 10, 4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, et al. (2019). METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer 18, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu Y, Stoleru D, and Salic A (2012). Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc Natl Acad Sci U S A 109, 413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolait SJ, O'Carroll AM, McBride OW, Konig M, Morel A, and Brownstein MJ (1992). Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 357, 336–339. [DOI] [PubMed] [Google Scholar]
- Maddocks OD, Labuschagne CF, Adams PD, and Vousden KH (2016). Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell 61, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, and Calvet JP (2006). Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(−) Co-transporter-dependent cystic dilation. J Am Soc Nephrol 17, 3424–3437. [DOI] [PubMed] [Google Scholar]
- Menezes LF, Lin CC, Zhou F, and Germino GG (2016). Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine 5, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachtergaele S, and He C (2018). Chemical Modifications in the Life of an mRNA Transcript. Annu Rev Genet 52, 349–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovano V, Podrini C, Boletta A, and Caplan MJ (2018). Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat Rev Nephrol 14, 678–687. [DOI] [PubMed] [Google Scholar]
- Pan X, Small EV, Igarashi P, and Carroll TJ (2013). Generation and characterization of KsprtTA and KsptTA transgenic mice. Genesis 51, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Chowdhury R, and Igarashi P (2009). Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens 18, 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podrini C, Rowe I, Pagliarini R, Costa ASH, Chiaravalli M, Di Meo I, Kim H, Distefano G, Tiranti V, Qian F, et al. (2018). Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun Biol 1, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam H, Fessler AR, Das A, Valerius MT, Basta J, Robbins L, Brown AC, Oxburgh L, McMahon AP, Rauchman M, et al. (2018). Disparate levels of beta-catenin activity determine nephron progenitor cell fate. Dev Biol 440, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam H, Yheskel M, and Patel V (2020). Modulation of polycystic kidney disease by non-coding RNAs. Cell Signal 71, 109548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, and Wallace DP (2011). Tolvaptan inhibits ERK-dependent cell proliferation, Cl(−) secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol 301, F1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, et al. (2013). Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19, 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, et al. (2008). Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 17, 1505–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman CR, Wang X, Chebib FT, and Torres VE (2020). Modulation of polycystic kidney disease by G-protein coupled receptors and cyclic AMP signaling. Cell Signal 72, 109649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres JA, Kruger SL, Broderick C, Amarlkhagva T, Agrawal S, Dodam JR, Mrug M, Lyons LA, and Weimbs T (2019). Ketosis Ameliorates Renal Cyst Growth in Polycystic Kidney Disease. Cell Metab 30, 1007–1023 e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, et al. (2012). Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367, 2407–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudel M, Barisoni L, Lanoix J, and D'Agati V (1998). Polycystic kidney disease in SBM transgenic mice: role of c-myc in disease induction and progression. Am J Pathol 152, 219–229. [PMC free article] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Cobo-Stark P, Patel V, Somlo S, Han PL, and Igarashi P (2018). Adenylyl cyclase 5 deficiency reduces renal cyclic AMP and cyst growth in an orthologous mouse model of polycystic kidney disease. Kidney Int 93, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ward CJ, Harris PC, and Torres VE (2010). Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int 77, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, Teo CC, Ang HY, Peh KLE, Yuan J, et al. (2019). Methionine is a metabolic dependency of tumor-initiating cells. Nat Med 25, 825–837. [DOI] [PubMed] [Google Scholar]
- Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, Harris PC, Torres VE, and Chini EN (2016). Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J Am Soc Nephrol 27, 1437–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Jiang X, Li D, and Jiang X (2020). HBXIP promotes gastric cancer via METTL3-mediated MYC mRNA m6A modification. Aging (Albany NY) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yheskel M, Lakhia R, Cobo-Stark P, Flaten A, and Patel V (2019). Anti-microRNA screen uncovers miR-17 family within miR-17~92 cluster as the primary driver of kidney cyst growth. Sci Rep 9, 1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yu W, Wang Z, Zhang K, Chi Z, Xu T, Jiang D, Chen S, Li W, Yang X, Zhang X, et al. (2019). One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol Cell 75, 1147–1160 e1145. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Du Y, Wang L, and Liu X (2020). The M6A methyltransferase METTL3 promotes the development and progression of prostate carcinoma via mediating MYC methylation. J Cancer 11, 3588–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhai Y, Zhang S, Dai X, and Li Z (2020). Roles of N6-Methyladenosine (m(6)A) in Stem Cell Fate Decisions and Early Embryonic Development in Mammals. Front Cell Dev Biol 8, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y, Liu Z, Ma S, Liu J, and Wu J (2020). METTL3 Facilitates Oral Squamous Cell Carcinoma Tumorigenesis by Enhancing c-Myc Stability via YTHDF1-Mediated m(6)A Modification. Mol Ther Nucleic Acids 20, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou K, Rouskin S, Dervishi K, McCormick MA, Sasikumar A, Deng C, Chen Z, Kaeberlein M, Brem RB, Polymenis M, et al. (2020). Life span extension by glucose restriction is abrogated by methionine supplementation: Cross-talk between glucose and methionine and implication of methionine as a key regulator of life span. Sci Adv 6, eaba1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Putative Mettl3 targets in control, Pkd1F/RC-KO, and Pkd1F/RC-Mettl3-DKO kidneys, related to Figure 4. The table lists all Mettl3 mRNAs targets identified from MeRIP-seq that exhibit higher m6A levels in Pkd1F/RC-KO but lower m6A levels in Pkd1F/RC-Mettl3-DKO compared to control kidneys. Refer to STAR Methods section for how MeRIP-Seq analysis was performed.
Table S2. Untargeted metabolomic analysis between wild-type and Pkd1RC/RC kidneys, related to Figure 6. The table lists metabolites detected from Mass Spectrometry analysis. Refer to STAR Methods section on Untargeted Metabolomic analysis of Kidneys.
Data Availability Statement
The MeRIP-seq and RNA-seq data have been deposited in the NCBI Gene Expression Omnibus under accession number GSE165956.