

Abstract
Disease models, including in vitro cell culture and animal models, have contributed significantly to developing diagnostics and treatments over the past several decades. The successes of traditional drug screening methods were generally hampered by not adequately mimicking critical in vivo features, such as a 3D microenvironment and dynamic drug diffusion through the extracellular matrix (ECM). To address these issues, we developed a 3D dynamic drug delivery system for cancer drug screening that mimicks drug dissemination through the tumor vasculature and the ECM by creating collagen-embedded microfluidic channels. Using this novel 3D ECM microsystem, we compared viability of tumor pieces to traditionally used 2D methods in response to 3 different drug combinations. Drug diffusion profiles were evaluated by simulation methods and tested in the 3D ECM microsystem and a 2D 96 well set up. Compared to the 2D control, the 3D ECM microsystem produced reliable data on viability, drug ratios and combination indeces. This novel approach enables higher throughput and sets the stage for future applications utilizing drug sensitivity predicting algorithms based on dynamic diffusion profiles requiring only minimal patient tissue. Our findings moved drug sensitivity screening closer to clinical implications with a focus on testing combinatorial drug effects – an option often limited by the amount of available patient tissues.
Keywords: tumor on a chip, combinatorial drug screening, bioengineering, microfluidics, extracellular matrix, cancer therapy
INTRODUCTION
Advancements in drug screening build off a substantial body of work with ex vivo cell culture models on 2D systems [96-well plates, gelatin sponge, etc. (1,2)]. While, these approaches are powerful they often are resembling physiological relevant environments (3). Therefore, building 3D microenvironments that better mimic in vivo conditions has been of great interest (4–6). Part of these advances is the development of microfluidic systems for vasculature (7–9). Advancements in drug circulatory diffusion were developed with microfluidic systems to replicate the surrounding tumor vasculature (7–9). Polydimethylsiloxane (PDMS) is often used as a primary material for microfluidic chip fabrication due to its flexibility and high gas permeability. However, PDMS does not form a biocompatible extracellular matrix for 3D cell culture, but rather serves as an inorganic substrate that lacks physiological features (10,11) of biomaterials of the ECM, such as collagen type I (12). In addition, PDMS has a strong hydrophobic nature that causes hydrogen bonding or pola-polar interactions negatively influencing drug diffusion (13).
Collagen makes up approximately 30% of protein in the body, while PDMS does not exist in the body. Thus, combining microfluidic collagen embedded channels with techniques such as sacrificial template molding (14) and 3D bioprinting (15,16) is providing advantages that close the gap between ex vivo cell culture models and animal models. Clearly, animal models provide much greater physiological relevance, but are less cost effective and can yield responses which are relevant to animals but are unintentionally misleading in humans (17,18). Importantly, animal models often lack immune system functionality (19) and species-specific drug response (19,20) thus, presenting inconsistent responses compared to clinical trials in human patients (21).
Several tumor-on-a-chip models have been developed in recent years to overcome limitations of exsiting techniques. For example, Chang et al (22) developed a PDMS-based drug screening device on 96-well plates to test xenograft mouse brain tissues. Although this device offered a high-throughput design compared to traditional 2D drug screening, the systems were made of non-physiological PDMS. Skardal et al. (23) developed a metastasis-on-a-chip model in which metastatic tumor cell migration was demonstrated in a circulating microfluidic system that connected a gut organoid with a liver organoid in two independent chambers. While this approach mimicked in vivo conditions with more complexity, it still utilized an external PDMS-based microfluidic system. Other material-based approaches such as hydrogels have been used but not with respect to drug screening. Kolesky et al. (24) created microfluidic channels in gelatin and fibrinogen using 3D bioprinting, and Nguyen et al. (25) fabricated channels in collagen type I with 400 μm needles and coated the channel with endothelial cells. These designs introduced ECM embedded channels toward drug screening applications, but the 3D printing method was limited by channel resolution (26), and the use of needles was not extendable to more complex channel designs.
Here, we tested a 3D ECM microsystem that provides spatial, temporal and biological controlled microenvironment that resembles in vivo circulatory diffusion dynamics, which can greatly enhance the ability of tumor drug screening approaches as it mimics the in vivo tumor environment (TME) (Fig. 1A). A small number (5) of 1mm3 in size tumor pieces were placed next to opposing channels (0.5mm) and aligned with the direction of the diffusion and drug gradient. This set up allows the implementation of diffusion simulation in a ECM-resembling scaffold, provides temporal and importantly, spatial control any 2D systems lacks. In addition, this 3D ECM microsystem allows for higher throughput, along with ease of manipulation for combinatorial drug dosing supported by simulation modelling to interpret responses to combinatorial dosing.
Figure 1. 3D Collagen vascular tumor-on-a-chip mimetics for dynamic combinatorial drug screening.

A. A microfluidic 3D extracellular matrix (ECM) based drug screening device could fill the gap between ex vivo models and animal models, with more physiological relevance compared to ex vivo models, and more direct controllability than animal models. B. Design of a 3D ECM based dynamic tumor-on-a-chip drug screening device. Tumors were positioned between two parallel channels. Microfluidic channels on both ends were available for drug perfusion. The 3D dynamic diffusion approach (left) was designed to mimic in vivo drug delivery from blood vessels to tumors (right). Drug molecules were perfused into ECM embedded microfluidic channels, and then the molecules physically diffused into ECM, generating a drug gradient in the scaffold.
RESULTS
3D ECM microsystem design and validation
Intravenous drug delivery from the vasculature into local tissue depends on diffusion profiles of the individual compounds. To replicate this process, we created a 3D collagen type I scaffolding in which 5 tumor tissues (1 mm3, derived from MDA-MB-231 human breast cancer cell line mouse xenografts) could be embedded (Fig. 1B). We previously developed a micromilling technique to fabricate microfluidic channels directly in a 3D ECM microsystem (14). Circular microfluidic channels were incorporated on both ends to establish drug gradients that perfuse across the device and the tumor samples (Fig. 1B). First, the clinically relevant breast cancer therapeutic drug adriamycin (doxorubicin) (27) was tested to assess if drug diffusion can be reliably predicted in the 3D-collage scaffold. Doxorubicin (30 μl) was injected into the left channel of the device, and allowed to diffuse into the collagen scaffold for 24 h. As doxorubicin molecules are auto-fluorescent (28), direct determination of the diffusion was determined through confocal microscopy imaging (Fig. 2A). Doxorubicin fluorescent signals were captured and intensity profiles were measured at 1 and 24 h. After 24 h (Fig. 2B), an approximately linear concentration profile was obtained from the left to the right channel. This allowed a one dimensional simulation using Matlab, by applying Fick’s second law in one dimension (1D), with the finite difference method as a simplified math model. The obtained diffusion profile was compared to a mathematically simulated diffusion profile predicted through diffusion equations as described (29,30). Notably, experimentally obtained values for the slope of the curve were not significantly different from mathematically modeled curves (Fig. 2C). To further examine the accuracy of the 1D simulation in the 3D collage scaffold the rhodamine 6G diffusion profile was predicted by using rhodamine 6G diffusion coefficients (31,32) and comparing these modeling results to experimentally obtained values (Fig. S1). Reassuringly, the 6G rhodamine diffucion profile matched the simulation as well.
Figure 2. Diffusion profiles and cell viability in our ECM based systems.
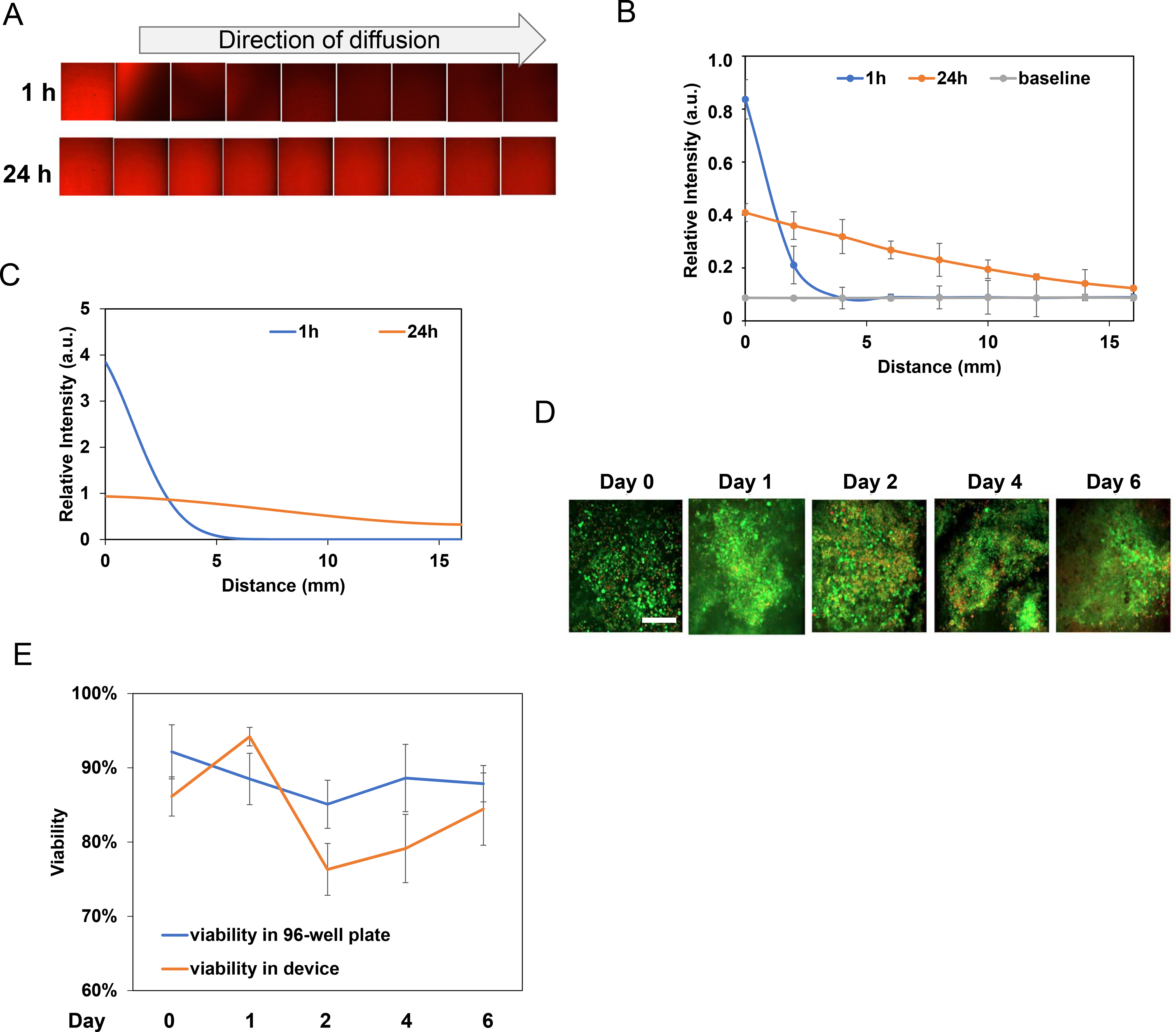
A. A doxorubicin diffusion profile was captured through confocal microscopy imaging of doxorubicin fluorescent intensity at 1 and 24 h. B. Quantification of doxorubicin diffusion for 1 and 24 h. The grey line was the baseline fluorescence intensity without the presence of doxorubicin. C. 1D simulation of doxorubicin diffusion profile generated by applying the diffusion coefficient of doxorubicin from literature data. The simulation was similar to experimental results for the 1 and 24 h diffusion profiles. D. Viability tests for tumor samples cultured in 96-well plates and in our ECM tumor-on-a-chip device. E. Live/dead staining of tumor samples from Day 0 to Day 6, in 96-well plates and in our device. Scale bar: 200μm.
To test next drug responses of actual tumor tissues in the device, human breast tumor xenograft fragments were created by injecting MDA-MB-231 cells into nude mice (33). Tumor fragments were were dissected into two pieces (1 mm3) and characterized for viability to exclude dead tissue samples (Fig. S2) prior to screening. 3D ECM microsystems were prepared with DMEM + 10%FBS to provide tumor samples with essential nutrients. To first examine tumor tissue survival over time, tumor tissue viability was examined up to six days with five tumor samples per device. Tumor samples were then analyzed with a live/dead stain (CalAM & EthD-1) and confocal microscopy (Figs. 2D and 2E). To avoid autofluorescence from tumor tissue, its ECM, and the collagen scaffold (34–36) to alter the analysis two methods where used to determine cell viability (Fig. S3). First, since tumor autofluorescence is rather uniform throughout the tumor, image processing software was used (ImageJ bundled with Java 1.8.0_172; https://imagej.nih.gov/ij/download.html) to subtract autofluorescence from tumor tissue images. Secondly, tumors were cut into smaller pieces, gently manually detached with tweezers and against the coverslip. That way, it was found that tumor samples maintained over 75% viability in the device compared to initial viability of the tumor being approximately 80% (Fig. 2D). To get a better sense if tumor viability in the 3D ECM microsystem differs from the one found in conventional 1D methods, tumor pieces were seeded in a 96 well plate, immersed in media and analyzed in parallel. As Fig. 2E shows, tissue viability varied from about 76%−95% in both methods, but was comparable over time (Fig. 2E).
Single drug screening
To better mimic dynamic in vivo diffusion profiles and to identify drug concentrations that would fit best with COMPUSYN algorithms (37,38), drug concentration simulation was used through Matlab coding to compare one-time dosing vs constant-flow dosing (replenish drugs every 12 hours) over three days (Fig. S4). The one-fill mode will cause a drop of initial high dose at the left channel, and the multiple-fills mode will cause a continuous increasing dose over the whole device. The constant-flow dosing mode suggested that compared to one-time dosing, higher max/min dose differences occur especially after 72 and 48 hours compared to 24 hours, which is preferable for more accurate simulation and estimation of a dose-effect curve using COMPUSYN.
To next correlate simulation with tumor viability, five tumor samples were placed in the 3D collage scaffold and 30 μl of media containing 100 μM of doxorubicin was introduced into the left channel. To achieve a constant channel drug concentration and match the simulation of constant-flow mode, drug containing media was replenished every 12 h. As a control, five tumor samples were placed individually in 96-well plates, and treated directly with 100 μl media (DMEM+10% FBS) with doxorubicin (every 12 hours) doses corresponding to a 48 h drug concentration simulated by Matlab for each tumor piece in the 3D device (Fig. 3A). The 48 h drug concentration was chosen to create a comparable dosing scheme between the 2 devices. After three days, all 10 samples (device and 96-well plate) were analyzed by live/dead staining and tissue viability was determined (Fig. 3B). As expected, a dose-dependent decrease in viability that correlated with the drug dose gradient was observed in both 2D controls as well as 3D ECM microsystem. Notably, the 3D ECM microsystem samples showed lower viability compared to 2D controls. As a reference, tumor samples from the 3D ECM microsystem were also analyzed for proliferation and apoptosis by immunofluorescence staining for Ki-67 and caspase 3, respectively (Fig. 3C). As expected, a dose-dependent increase in Ki-67 (proliferation) and decrease in caspase 3 (apoptosis) was observed, which was quantified and consistent with the live/dead staining results (Fig. 3D). Doxorubicin intrinsic fluorescence decreased as well across the spatially distributed samples, which correlated with the drug simulation analysis (Figs. 2A–2C).
Figure 3. Tumor-on-a-chip response to single drug exposure.
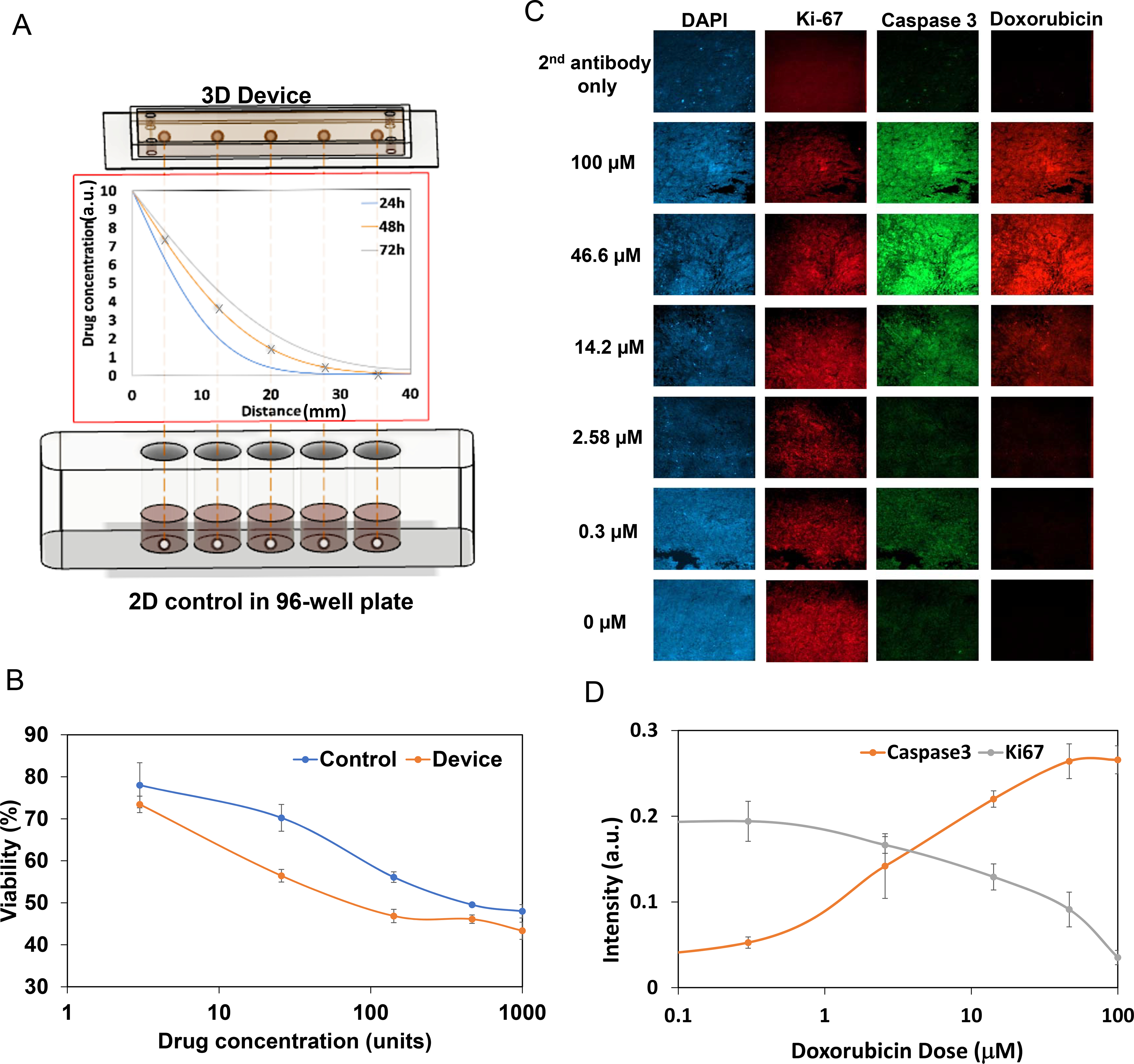
A. 3D devices vs. 2D controls setup. Top: Five tumors were inserted in series into the system. Middle: Simulation of drug diffusion profile for 24, 48, and 72 h. Bottom: In a 2D control, tumors were cultured in 96-well plates and treated directly with constant drug doses equal to the drug dose in the 3D device at 48 h. B. Doxorubicin drug screening results for the device and control. Note that both presented similar trends, yet the absolute drug effect was different. For doxorubicin, 1000 units=100 μM. Experiments were done in triplicates. C. Immunofluorescent staining for DAPI, Ki-67, and caspase 3, and autofluorescence of doxorubicin, in tumor samples treated with doxorubicin. Expression of caspase 3 increased and Ki-67 decreased with higher dosing, which agreed with the live/dead staining results. All images are 700×700μm D. Quantification of immunofluorescent staining intensity of caspase 3 and Ki-67. The lines are polynomial interpolations.
Tumor viability during combination treatment
Drug synergism is an essential determinant in evaluating drug combination therapies. The advantage of syngergistic drug effects, when compared to additive ones, is that a specific drug ratio generates higher effects than the combined individual drug responses (additive). Thus, drug synergism is a desired factor in evaluating combinatorial drug schemes. The Chou-Talalay method (39) is frequently used to determine if two drugs act syngergistic, additive or antagonistic. This method uses the combination index (CI) as a readout. Here, CI values, where CI<1 marks synergistic, CI=1 additive and CI>1 antagonistic drug effects, were calculated using COMPUSYN software that is based on the Chou-Talalay method (39). The power of this method lies in determining optimal (less toxic) and efficacious drug ratios for combination treatments. In general, CI analyses are done in vitro using cell lines which allow testing of many different drug concentrations, but often lack in vivo reproducibility.
To test the feasibility of the 3D ECM microsystem, a drug combination was chosen that is commonly used to treat breast cancer: doxorubicin and cyclophosphamide act synergistically to kill tumor cells through different mechanisms (40). Doxorubicin kills cells by intercalation of DNA, topoisomerase II inhibition and free radical formation (41,42). Cyclophosphamide metabolites (e.g. 4-hydroxycyclophosphamide) mediates cell death by alkylating and crosslinking DNA in cancer cells (41,43–45). Four sample setups were used as illustrated in Figs. S5 to analyze the known synergistic effect of both drugs in the 3D ECM microsystem: doxorubicin alone (denoted as doxo), 4-hydroxycyclophosphamide alone (denoted as cyclo), doxorubicin and 4-hydroxycyclophosphamide in opposite directions (denoted as oppo), and both drugs diffusing in the same direction (denoted as para for parallel). For oppo, doxorubicin and 4-hydroxycyclophosphamide were injected from the left and right channels, respectively; and for para, both drugs were injected from the left channel. Similar to the one drug screening approach, 96-well controls were prepared, with drug doses matching 48 h dosing in the 3D ECM microsystem (Figs. S5). In the 3D device, the concentration of the drugs at each tumor sample location was again determined through 1D simulation as before (Fig. S6A). The simulations revealed that compared to doxorubicin, 4-hydroxycyclophosphamide (277g/mol) diffused faster than doxorubicin (543 g/mol), as expected with its lower molecular weight (Fig. S6B). Based on this and the median effect dose (Dm) of doxorubicin being previously reported as much lower than 4-hydroxycyclophosphamide for MDA-MB-231 cell treatment (46), we selected 100 μM of doxorubicin and 100 mM of 4-hydroxycyclophosphamide as the initial injection dose for each channel. Single drug treatments showed reduced tumor viability (1-effect) (Figs. S7A and S7B, where viability is 1-effect) as the drug dose increased in the 96 well control and 3D ECM microsystem (Figs. 4C and 4D, blue and red lines).
Figure 4. Double drug screening with doxorubicin (doxo) and cyclophosphamide (cyclo).
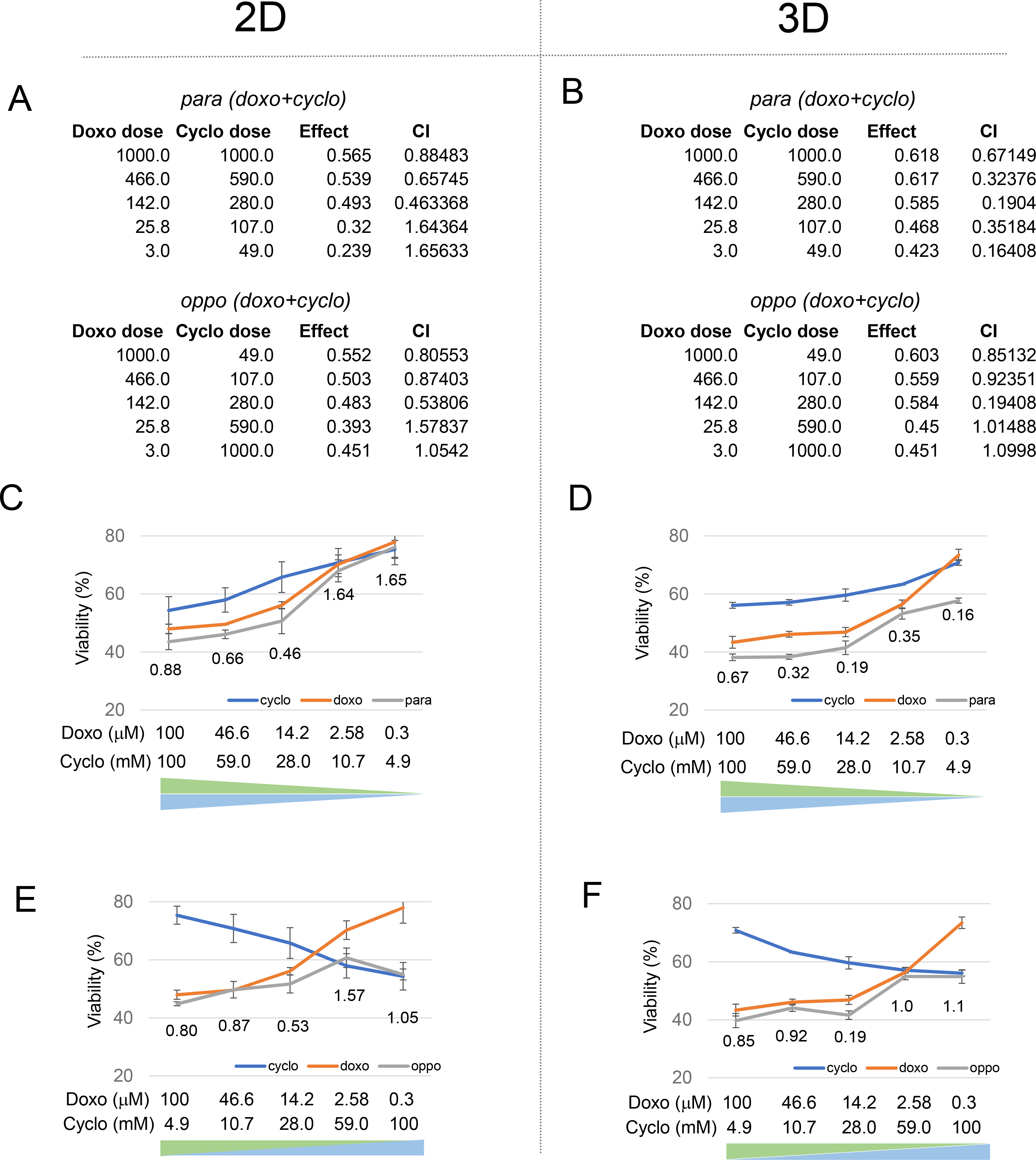
A. COMPUSYN analysis of 2D 96 well-plate control. B. COMPUSYN analysis of 3D ECM microsystem. C. Drug screening results for cyclo & doxo in para dosing in 2D control. The blue and orange curves indicate single drug treatments, and the grey curves indicate parallel direction drug combination results. Combination index values are labeled at each data point. D. Drug screening results for cyclo & doxo in para dosing in 3D ECM microsystem. E. Drug screening results for cyclo & doxo in oppo dosing in 2D control. F. Drug screening results for cyclo & doxo in oppo dosing in 3D ECM microsystem. Experiments were done in triplicates.
As expected, treatment with both drugs at the same time in para showed lower tumor tissue viability than either of the single drugs alone, which indicated that the addition of either drug increased the overall effect (Figs. 4A upper part, 4C and 4D, grey lines). In both oppo treatment schemes, 2D and 3D, the tissue viability after drug combination was dominated by the highest dose of doxo (Figs. 4A lower part, 4E and 4F, grey lines). For the 3D ECM microsystem this suggests that the initial increase in tissue viability was due to a rapid drop of the dominating doxo effect, and that the subsequent decrease was mainly caused by synergistic combination effect of doxo and cyclo. The tissue viability curve (for both 2D control and 3D device) fluctuated from one side to the other, and reached a local minimum at double median dosing in the 3D ECM microsystem.
Comparison of synergistic and antagonistic combination indeces in 2D and 3D
Analyzing the CI values (Figs. 4A and 4B, and placed below each combination data point Figs. 4C to 4F), it was noticed that while CI values of 2D and 3D analyses were mostly comparable in para and oppo, some subtle differences stood out. For example, the 2D midpoint CI value for para treatment (doxo & cyclo) started at 0.88 for high doses of doxo and cyclo and decreased to 0.46 for median doses, and then increased to 1.65 for low doses, suggesting better synergism, at midpoint (Fig. 4C). Similar trends with a 2.4-fold lower CI value of 0.19 was observed in the para 3D ECM microsystem (Fig. 4D). Importantly, for oppo combinations, maximum synergy occurred at “double-median” dosing in both the 2D control with a CI value of 0.53 and more convincingly in the 3D ECM microsystem with a CI value of 0.19 (Figs. 4E and 4F). This suggests that the dynamic drug delivery in the 3D ECM microsystem provides a more robust synergy effect compared to the 2D analysis.
To examine a broader applicability of the 3D ECM microsystem, another drug combination common in breast cancer treatment was used: cisplatin and paclitaxel (cis & pac). Unlike the doxo & cyclo combination, the clinical outcomes using this drug combination are mixed for drug synergy and effectiveness (47,48). While one study reported a 85% response rate with the cis and pac combination treatment (49), an only 21% response rate was found in another similar study (50). As before, drug diffusion profiles were determined for cis and pac individually (Fig. S8), before viability and CI were examined. As shown in Fig. S9, similar trends although less distinct were observed as for the doxo & cyclo combination, with a dominance of high cis doses in viability and lower midpoint CIs in the 3D ECM microsystem compared to the 2D control. As expected, given the clinical outcomes mentioned above, CI values were overall higher than found with the doxo & cyclo combination.
Lastly, a known antagonistic drug combination (paclitaxel and vincristine) was examined (38,51). Indeed, both combination curves showed significantly different profiles. For the para combination in 2D and 3D (grey lines in Figs. 5A and 5B), the tumor viability was relatively high and stable even with an increasing dose of both drugs, with CI values. In oppo combinations, tissue viability at both ends (high pac or high vin) was lower and only one of the two drugs affected the response as the other drug, as observed in the diffusion simulation (Fig. S10), could not effectively diffuse to the far end. This finding is an important distinction compared to the 2D model as in in vivo diffusion through the ECM defines drug efficacies and thus the 3D ECM microsystem provides a more accurate resemblance of drug effects.
Figure 5. Comparison of a known antagonistic drug interaction in 2D and the 3D ECM microsystem.
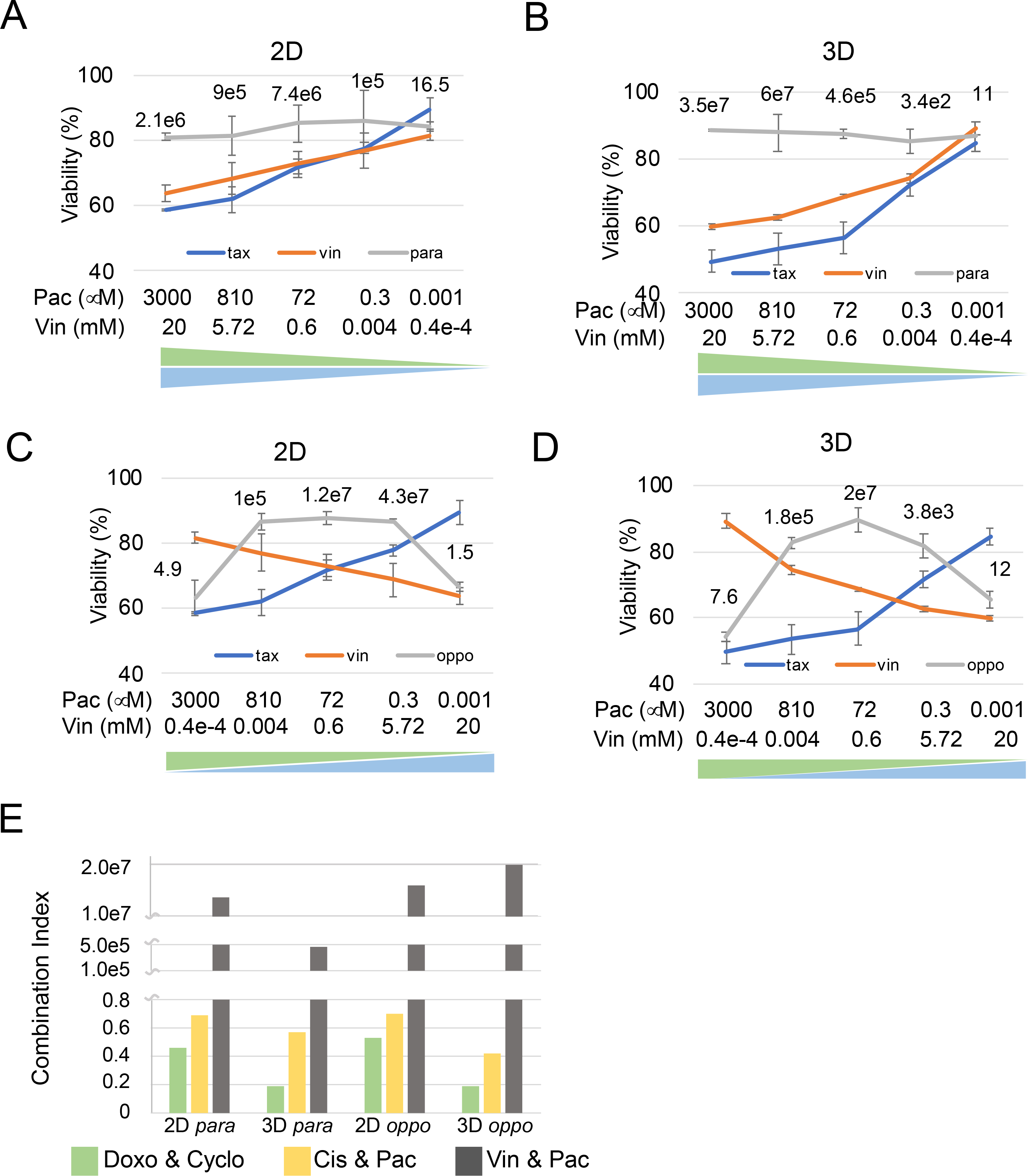
A. Drug screening results of paclitaxel (pac) and vincristine (vin) in para dosing in 2D 96 well plate control. The blue and orange curves indicate single drug treatments, and the grey curves indicate parallel direction drug combination results. Combination index values are labeled at each data point. B. Drug screening results for pac & vin in para dosing in 3D ECM microsystem. C. Drug screening results for pac & vin in oppo dosing in 2D control. D. Drug screening results for pac & vin in oppo dosing in 3D ECM microsystem. E. CI comparison between all 3 drug combinations (doxo & cyclo, cis & pac, and pac & vin) at double-median dosing.
Comparing then the midpoint para and oppo CI values for these 3 drug combinations (Fig. 5E) showed a stronger synergistic effect for the doxo & cyclo compared to cis & pac, while pac & vin presented an antagonistic effect. The CIs identified for the 3D ECM microsystem were comparable to CIs found in the 2D system. However, 3D CIs demonstrated lower CI values for the syngerstic drug combinations, where the dox & cyclo CI was distinctively lower than the cis & pac CI, compared to the 2D analysis where both CIs appeared similar, thus better reflecting reported success of both combinations in human trials (52).
DISCUSSION
Here, we introduce a novel 3D collagen tumor-on-a-chip approach with microfluidic channels applicable to combinatorial drug screening. The device permits 3D collagen-embedded tumor samples to be rapidly evaluated for drug sensitivy employing drug diffusion profiles from vessel-mimicking channels, which simulate in vivo dynamic drug delivery. Based on mathematical simulations of drug diffusion profiles, local drug concentrations for each tumor sample in the 3D ECM microsystem were determined in a time dependent manner and applied to the 2D controls. Comparison between the 3D ECM microsystem and the commonly used 2D 96-well approach confirmed the appropriateness of the 3D ECM microsystem that provides a much needed physiological-relevant experimental environment, in determining tissue viability, useful drug ratios and combination indeces. Within a tissue, concentration gradients exist not only for oxygen, pH, nutrients and effector molecules, but also for drugs. Therefore, proximity of a blood vessel, ECM compositions are important factors determining drug concentrations in tumor tissues. As 2D approaches fail to build meaningful gradients, it has long been realized that drug screening in 3D is far superior in high throughput drug screening, however, cost and limited tissue availability often restrict drug screens in 3D (53). The here presented 3D ECM microsystem helps to overcome these hurdles by offering blood vessel like structures (channels) embedded in collagen and diffusion gradients. Besides single drug gradient generation, we specifically designed and examined parallel dosing and opposite dosing strategies. Thus, the parallel setup mimicked intravenous chemotherapy delivery as a simplified version. As the opposite setup does not represent any in vivo application specifically, it offers an efficient way to test drug combinations in different dose ratios, which can only be achieved by manually adding different doses in parallel. This would require many more devices and tumor tissue pieces. In addition, we presented evidence here that the placement of 5 tumor pieces in series between two channels enables reliable testing of drug combinations to derive relevant drug ratios and thus, combination indeces.
Adressing tumor heterogeneity through the 3D ECM microsystem
While 3D cell culture models, such as organoids, allow cell-to-ECM contact, they fail to reflect intratumoral tumor heterogeneity. For example, fibroblasts, adipocytes, immune cells a part of the tumor microenvironment (TME) and modulate the response of cancer cells to chemotherapies and targeted therapies through production of secreted factors (52). Therfore, analyzing tumor tissue that contains all TME cell types is necessary for valuable drug screening. We provide evidence that tumor tissues show decent viability up to 6 days. However, while that can be useful for some research purposes, in drug sensitivity testing such long time periods are not advisable, as prolonged interactions of tumor cells with the collagen scaffold induces cells to migrate out of the tumor that way resulting in different drug responses found in tumors with intact architecture. Another important factor requiring terse analysis time comes from the relative short viability of immune cells ex vivo, compared to cancer cells (54). As we show here overall tumor viability, future studies are needed to examine individual cell type viabilities in 3D ECM microsystem. Intratumoral heterogeneity is also a result of clonal heterogeneity that influences drug responsiveness and has been addressed by emerging computational prediction models used to optimize cancer therapies (55). Intertumoral heterogeneity which describes genomic differences of the same cancer between two or more patients as well as clonal differences of metastases within one patient, is an equally pressing issue difficult to address in conventional drug screening aproaches (56). Clearly, the 3D ECM microsystem presented here offers a personalized way to screen for effective drug combinations of the tested tissues.
Comparisons of time-dependent effects or dosing frequencies offer important pharmacological insight into how dosing regimens influence diffusion profiles, drug dosing range, and can lead to variations in drug treatment outcomes. For example, comparisons between the 2D and the 3D ECM microsystem showed that all four different drug application methods generated similar trends with dose-effect patterns in 2D and the 3D ECM microsystem that mostly agreed with each other: fluctuations and curve inflections. However, absolute cell death amounts differed between 2D and 3D, probably due to differences in the actual drug delivery profile over time: constant dosing in 2D vs. dynamic dosing 3D. In the control (96-well plates), tumor fragments were soaked in drugs, and thus experienced constant drug dosing, with 12h replenishing over a three day period. In the 3D device, the tumor fragments were exposed to drugs through diffusion in the 3D ECM microsystem and experienced a dynamic and nonlinearly increasing dose profile, which is physiological more relevant as drug delivery in 2D. Therefore, in vitro drug screening by constant dosing is an overly simplified model that provides an inaccurate reference to in vivo drug treatment. With the development of computational models and accuracy in simulations, a more reliable in vitro reference can be provided. Our study is merely a first step in dynamic diffusion simulation, but already presents different drug effects especially in combination index values. For example, our Matlab simulation in 1D applying Fick’s second law was a simplified tool to calculate spatiatemperal distribution of all drugs in the device, while a 3D simulation optimized by experimental data are preferrable for more accurate determination in the future.
Other important advantages of the 3D ECM microsystem are: 1) Ease of experimental manipulation: single dosing for the channels for each five-tumors in the device is easier than diluting and combining drug doses multiple times as requied for the traditional 2D approach. 2) Low drug consumption: the micrometer scaled fluidic system requires only small volumes of reagents. As some drugs (especially new drugs in development) are expensive or initially only synthesized in small batches, our approach provides useful insight early in the drug development process.
In conclusion, we developed an effective and convenient new 3D collagen tumor-on-a-chip approach that offers microfluidic channels to mimic tumor vasculature in vivo for drug screening and a cancer appropriate ECM. Our device enables 3D embedded tumor samples to be examined with single/double drug combinations in a physiologically meaningful way as it enables drug diffusion through the ECM, which simulates in vivo dynamic drug delivery. The 3D ECM microsystem can be adapted depending on individual ECM requirements, availability of tumor tissues, etc. to provide personalized patient treatment.
METHODS
Tumor preparation
All animal experiments were approved by the University of Pittsburgh IACUC. MDA-MB-231 cells were injected into the mammary fat pad of 3–4 -week-old female nude immune-compromised SCID-Beige mice and the tumors were harvested 8 weeks after implantation. The tumors were stored in liquid nitrogen (33). Before drug testing, tumor samples were thawed and immediately sliced with a scalpel into roughly 2 mm3 (1 mm*1 mm*2 mm) fragments. Tumor sections close to the necrotic core and the surface were removed. Each fragment was divided into two 1 mm3 fragments, one for live/dead staining to ensure the fragment had over 70% viability. Fragments that were viable were embedded into the 3D ECM microsystems for drug screening.
Tumor device fabrication
Building off our previous work (57), the device chamber was fabricated by cutting PMMA boards (TAP Plastic, 2.4 cm in thickness) into chamber walls (inner: 40*16 mm, outer: 44*20 mm) through a laser cutting system (Epilog, CO). Then the chamber was attached to a 50*22 coverslip with optical adhesive (NOA 81, Norland products Inc., NJ). The device was cured with UV for 2 min and rinsed with 70% ethanol under UV light for 2 h, then rinsed twice with 1xPBS to remove residual ethanol. The gelatin template was prepared as described in our previous work (57), then positioned in the chamber. Three or five tumor samples were placed between the parallel channels, with equal distance between each sample, or any other locations for desired local dosing, based on our 1D simulation. Collagen with 3 mg/ml concentration (10% 10xPBS, high concentration collagen type I with corresponding concentration ratio, 1N NaOH = 0.023*collagen, and DMEM+10%FBS) was injected into the chamber and covered the tumor samples. Then the device was maintained at room temperature for 30 min, followed by incubation at 37oC for 30 min. The gelatin template was then removed with a syringe.
Doxorubicin drug gradient characterization
Doxorubicin (Selleckchem, in DMSO) was first diluted by DMEM+10%FBS to 100 μM, then injected into the left channel of one blank device (without the presence of tumor samples). The device was next imaged using confocal microscopy. Doxorubicin had an excitation/emission wavelength of approximately 480/560 nm. Images were captured moving spatially across the device from the left channel to right channel, at 1 mm intervals. The average intensity of the images (intensity of doxorubicin fluorescence) was determined through image analysis of the confocal microscope images (Axio Observer Z1 Microscope System, Zeiss). The sample was incubated at 37oC for 24 h, then removed and another set of images was capture. A baseline intensity was also determined by imaging a blank device without the doxorubicin injection.
Viability test for tumor samples
Tumors embedded in the device were stained with 100 μl of CalAM + EthD-1 solution (adding droplets directly on top of tumors in the device) for 1 h, then rinsed with 1XPBS twice (10 min each). Next the device was imaged with a confocal microscope. This test though is destructive to tumor samples, so multiple samples were needed for Day 0 up to Day 6 for viability tests. For tumors cultured in 96-well plates, the media was removed, and 100 μl of CalAM + EthD-1 solution was added per well for 30 min staining followed by 1XPBS rinse twice (10 min each). Then the tumors were imaged with confocal microscopy. Images were then analyzed with ImageJ (ImageJ.nih.gov) to count green (live) and red (dead) cells. Viability was determined by their ratio.
1D-simulation of the drug profile
To demonstrate the feasibility of the model-based data-fitting approach, we simulated and predicted the diffusion profile of drugs through a simplified 1D diffusion equation (29). Since the double channels in our current design were parallel to each other, the diffusion profile along with the device was approximately parallel as well. We simulated the model by integrating the 1D diffusion equation:
using a fixed time step forward Euler method, and 2nd order center difference approaximation to the Laplacian. The simulation used Matlab (ver. R2018b; www.mathworks.com).
code:
clear all; range=40;% length between two parallel channels dx=0.1;% distance interval pi=3.1415; C=zeros(1,range/dx);%vector for drug concentration dt=0.001;%time interval D=1.635;%Diffusion coefficient of drug coe=D*dt/(dx*dx);% diffusion coefficient tumorp=[1,100,200,300,400];%tumor positions C(1)=10;%initial concentration value T=1/dt; P=[48];%time range, 48 indicates 48 hours m=1; for i=1:T*P(m) C(1)=10; %iteration for initial point, constant injection; for one time injection, C(1)=C(1)-D*dt*(C(1)-C(2))/dx; for j=2:range/dx-1 C(j)=C(j)+coe*(C(j+1)-2*C(j)+C(j-1)); %general iteration, Forward Euler C(j)=C(j)-0.000001; if C(j)<0 C(j)=0; %ensure concentration stays positive. end end C(range/dx)=C(range/dx)+D*dt*(C(range/dx-1)-C(range/dx))/dx; %iteration for endpoint end for l=1:5 tumorC(l)=C(tumorp(l)); %concentration at each tumor position end plot(C);%concentration profile
Tumor drug testing
Single drug testing:
In the device, 30 μl of doxorubicin (100 μM) or 4-hydroxycyclophosphamide (Toronto Research Chemicals, dissolved in DMSO,100 mM) diluted in DMEM+10%FBS was injected into one channel and the device was stored in a 5% CO2 37oC incubator for up to three days. Every 12 h the device was taken out, the drug was removed from channel, and the media in the channels was replenished with fresh drug solutions. In the 2D 96-well control, 100 μl of doxorubicin/4-hydroxycyclophosphamide with a designated concentration was added per well. The samples were stored in the incubator for three days. The solutions were refreshed every 12 h.
Double drug testing:
Opposite drug administration in the device: 30 μl of doxorubicin (100 μM) solution was injected into left channel, then 30 μl of 4-hydroxycyclophosphamide (100 mM) solution was injected into right channel. The solutions were refreshed every 12 h. Parallel drug administration in device: Mixture of doxorubicin (100 μM, final concentration) and 4-hydroxycyclophosphamide (100 mM, final concentration) was injected into left channel. The solutions were refreshed every 12 hours. Opposite/parallel drug administration in 96-well plates: Mixtures of doxorubicin & 4-hydroxycyclophosphamide with designed concentrations were added per well.
All samples were incubated for three days, removed, washed twice with PBS, and stained with CalAM and EthD-1 for viability imaging. The same approach was applied to cisplatin+paclitaxel, and paclitaxel+vincristine drug combinations.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by the Air Force Office of Scientific Research (FA9550–18-1–0262) P.R. LeDuc, Office of Naval Research (N00014–17-1–2566) P.R. LeDuc, Pennsylvania Department of Health (SAP4100077084) P.R. LeDuc, National Institute of Health (R01AG06100501A1), National Institute of Health (R56 CA233817) C.A. Neumann and National Institute of Health (NIH/NCI P30 CA047904) C.A. Neumann support from UPMC Hillman Cancer Center.
Footnotes
The authors declare no potential conflicts of interest.
DECLARATION OF COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Kumar S, Bajaj S, Bodla RB. Preclinical screening methods in cancer. Indian journal of pharmacology 2016;48(5):481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohie S, Udagawa Y, Aoki D, Nozawa S. Histoculture drug response assay to monitor chemoresponse. Chemosensitivity: Springer; 2005. p 79–86. [DOI] [PubMed] [Google Scholar]
- 3.Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnology and bioengineering 2009;103(4):655–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huh D, Hamilton GA, Ingber DE. From 3D cell culture to organs-on-chips. Trends in cell biology 2011;21(12):745–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbott A Cell culture: biology’s new dimension. Nature Publishing Group; 2003. [DOI] [PubMed] [Google Scholar]
- 6.Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nature reviews Molecular cell biology 2007;8(10):839. [DOI] [PubMed] [Google Scholar]
- 7.Ma H, Xu H, Qin J. Biomimetic tumor microenvironment on a microfluidic platform. Biomicrofluidics 2013;7(1):011501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loessner D, Stok KS, Lutolf MP, Hutmacher DW, Clements JA, Rizzi SC. Bioengineered 3D platform to explore cell–ECM interactions and drug resistance of epithelial ovarian cancer cells. Biomaterials 2010;31(32):8494–506. [DOI] [PubMed] [Google Scholar]
- 9.Szot CS, Buchanan CF, Freeman JW, Rylander MN. 3D in vitro bioengineered tumors based on collagen I hydrogels. Biomaterials 2011;32(31):7905–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hosseini Y, Verbridge SS, Agah M. Bio‐inspired microstructures in collagen type I hydrogel. Journal of Biomedical Materials Research Part A 2015;103(6):2193–7. [DOI] [PubMed] [Google Scholar]
- 11.Wan L, Neumann C, LeDuc P. Tumor-on-a-chip for integrating a 3D tumor microenvironment: chemical and mechanical factors. Lab on a Chip 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sokol ES, Miller DH, Breggia A, Spencer KC, Arendt LM, Gupta PB. Growth of human breast tissues from patient cells in 3D hydrogel scaffolds. Breast Cancer Research 2016;18(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halldorsson S, Lucumi E, Gomez-Sjoberg R, Fleming RMT. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens Bioelectron 2015;63:218–31 doi 10.1016/j.bios.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 14.Wan L, Skoko J, Yu J, LeDuc P, Neumann C. Mimicking Embedded Vasculature Structure for 3D Cancer on a Chip Approaches through Micromilling. Scientific reports 2017;7(1):16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy SV, Atala A. 3D bioprinting of tissues and organs. Nature biotechnology 2014;32(8):773. [DOI] [PubMed] [Google Scholar]
- 16.Kolesky DB, Truby RL, Gladman A, Busbee TA, Homan KA, Lewis JA. 3D bioprinting of vascularized, heterogeneous cell‐laden tissue constructs. Advanced materials 2014;26(19):3124–30. [DOI] [PubMed] [Google Scholar]
- 17.Esch EW, Bahinski A, Huh D. Organs-on-chips at the frontiers of drug discovery. Nature reviews Drug discovery 2015;14(4):248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huh D, Torisawa Y-s, Hamilton GA, Kim HJ, Ingber DE. Microengineered physiological biomimicry: organs-on-chips. Lab on a Chip 2012;12(12):2156–64. [DOI] [PubMed] [Google Scholar]
- 19.Asghar W, El Assal R, Shafiee H, Pitteri S, Paulmurugan R, Demirci U. Engineering cancer microenvironments for in vitro 3-D tumor models. Materials Today 2015;18(10):539–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hachey SJ, Hughes CC. Applications of tumor chip technology. Lab on a Chip 2018;18(19):2893–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature 2012;483(7391):531. [DOI] [PubMed] [Google Scholar]
- 22.Chang TC, Mikheev AM, Huynh W, Monnat RJ, Rostomily RC, Folch A. Parallel microfluidic chemosensitivity testing on individual slice cultures. Lab on a Chip 2014;14(23):4540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skardal A, Devarasetty M, Forsythe S, Atala A, Soker S. A reductionist metastasis‐on‐a‐chip platform for in vitro tumor progression modeling and drug screening. Biotechnology and bioengineering 2016;113(9):2020–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kolesky DB, Homan KA, Skylar-Scott MA, Lewis JA. Three-dimensional bioprinting of thick vascularized tissues. Proceedings of the national academy of sciences 2016;113(12):3179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen D-HT, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, et al. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proceedings of the National Academy of Sciences 2013;110(17):6712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattacharjee N, Urrios A, Kang S, Folch A. The upcoming 3D-printing revolution in microfluidics. Lab on a Chip 2016;16(10):1720–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher B, Anderson S, Wickerham DL, DeCillis A, Dimitrov N, Mamounas E, et al. Increased intensification and total dose of cyclophosphamide in a doxorubicin-cyclophosphamide regimen for the treatment of primary breast cancer: findings from National Surgical Adjuvant Breast and Bowel Project B-22. Journal of Clinical Oncology 1997;15(5):1858–69. [DOI] [PubMed] [Google Scholar]
- 28.Motlagh NSH, Parvin P, Ghasemi F, Atyabi F. Fluorescence properties of several chemotherapy drugs: doxorubicin, paclitaxel and bleomycin. Biomedical optics express 2016;7(6):2400–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathews J, Walker RL. Mathematical methods of physics. WA Benjamin; New York; 1970. [Google Scholar]
- 30.Zhao S, Zhao H, Zhang X, Li Y, Du Y. Off-the-shelf microsponge arrays for facile and efficient construction of miniaturized 3D cellular microenvironments for versatile cell-based assays. Lab on a Chip 2013;13(12):2350–8. [DOI] [PubMed] [Google Scholar]
- 31.Müller C, Loman A, Pacheco V, Koberling F, Willbold D, Richtering W, et al. Precise measurement of diffusion by multi-color dual-focus fluorescence correlation spectroscopy. EPL (Europhysics Letters) 2008;83(4):46001. [Google Scholar]
- 32.Culbertson CT, Jacobson SC, Ramsey JM. Diffusion coefficient measurements in microfluidic devices. Talanta 2002;56(2):365–73. [DOI] [PubMed] [Google Scholar]
- 33.Lawson DA, Werb Z, Zong Y, Goldstein AS. The cleared mammary fat pad transplantation assay for mammary epithelial organogenesis. Cold Spring Harbor Protocols 2015;2015(12):pdb. prot078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuanlong Y, Yanming Y, Fuming L, Yufen L, Paozhong M. Characteristic autofluorescence for cancer diagnosis and its origin. Lasers in surgery and medicine 1987;7(6):528–32. [DOI] [PubMed] [Google Scholar]
- 35.Croce A, Bottiroli G. Autofluorescence spectroscopy and imaging: a tool for biomedical research and diagnosis. European journal of histochemistry: EJH 2014;58(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abujamra AL. Diagnostic techniques and surgical management of brain tumors. BoD–Books on Demand; 2011. [Google Scholar]
- 37.Zhang N, Fu J-N, Chou T-C. Synergistic combination of microtubule targeting anticancer fludelone with cytoprotective panaxytriol derived from panax ginseng against MX-1 cells in vitro: experimental design and data analysis using the combination index method. American journal of cancer research 2016;6(1):97. [PMC free article] [PubMed] [Google Scholar]
- 38.Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews 2006;58(3):621–81. [DOI] [PubMed] [Google Scholar]
- 39.Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 2010;70(2):440–6. [DOI] [PubMed] [Google Scholar]
- 40.Ganz PA, Romond EH, Cecchini RS, Rastogi P, Geyer CE, Swain SM, et al. Long-term follow-up of cardiac function and quality of life for patients in NSABP protocol B-31/NRG oncology: A randomized trial comparing the safety and efficacy of doxorubicin and cyclophosphamide (AC) followed by paclitaxel with ac followed by paclitaxel and trastuzumab in patients with node-positive breast cancer with tumors overexpressing human epidermal growth factor receptor 2. Journal of Clinical Oncology 2017;35(35):3942–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lori J, Stein T, Thamm D. Doxorubicin and cyclophosphamide for the treatment of canine lymphoma: a randomized, placebo‐controlled study. Veterinary and comparative oncology 2010;8(3):188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cappetta D, Rossi F, Piegari E, Quaini F, Berrino L, Urbanek K, et al. Doxorubicin targets multiple players: a new view of an old problem. Pharmacological research 2018;127:4–14. [DOI] [PubMed] [Google Scholar]
- 43.Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nature reviews Clinical oncology 2009;6(11):638. [DOI] [PubMed] [Google Scholar]
- 44.Bastert G, Voelcker G, Peter G, Schmidt-Matthiesen H, Hohorst H. In vitro assay for cyclophosphamide-sensitivity of human tumours: the effect of 4-hydro-peroxy-cyclophosphamide on the incorporation of 3H-uridine into the nucleic acids of human tumour cells (author’s transl). Zeitschrift fur Krebsforschung und Klinische Onkologie Cancer Research and Clinical Oncology 1976;85(3):299–307. [DOI] [PubMed] [Google Scholar]
- 45.Alberts DS, Einspahr JG, Struck R, Bignami G, Young L, Surwit EA, et al. Comparative in vitro cytotoxicity of cyclophosphamide, its major active metabolites and the new oxazaphosphorine ASTA Z 7557 (INN mafosfamide). Investigational new drugs 1984;2(2):141–8. [DOI] [PubMed] [Google Scholar]
- 46.Kirson ED, Schneiderman RS, Dbalý V, Tovaryš F, Vymazal J, Itzhaki A, et al. Chemotherapeutic treatment efficacy and sensitivity are increased by adjuvant alternating electric fields (TTFields). BMC medical physics 2009;9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sohn JH, Kim YT, Rha SY, Yoo NC, Roh JK, Kim BS, et al. Paclitaxel and Cisplatin Combination Chemotherapy in Pretreated Breast Cancer. Cancer Research and Treatment 2003;35(3):267–73. [DOI] [PubMed] [Google Scholar]
- 48.Rosati G, Riccardi F, Tucci A, De Rosa P, Pacilio G. A phase II study of paclitaxel/cisplatin combination in patients with metastatic breast cancer refractory to anthracycline-based chemotherapy. Tumori Journal 2000;86(3):207–10. [DOI] [PubMed] [Google Scholar]
- 49.Gelmon K, O’reilly S, Tolcher A, Campbell C, Bryce C, Ragaz J, et al. Phase I/II trial of biweekly paclitaxel and cisplatin in the treatment of metastatic breast cancer. Journal of clinical oncology 1996;14(4):1185–91. [DOI] [PubMed] [Google Scholar]
- 50.Sparano JA, Neuberg D, Glick JH, Robert NJ, Goldstein LJ, Sledge GW, et al. Phase II trial of biweekly paclitaxel and cisplatin in advanced breast carcinoma: an Eastern Cooperative Oncology Group study. Journal of clinical oncology 1997;15(5):1880–4. [DOI] [PubMed] [Google Scholar]
- 51.Chou T-C, Motzer RJ, Tong Y, Bosl GJ. Computerized quantitation of synergism and antagonism of taxol, topotecan, and cisplatin against human teratocarcinoma cell growth: a rational approach to clinical protocol design. JNCI: Journal of the National Cancer Institute 1994;86(20):1517–24. [DOI] [PubMed] [Google Scholar]
- 52.Fisusi FA, Akala EO. Drug Combinations in Breast Cancer Therapy. Pharm Nanotechnol 2019;7(1):3–23 doi 10.2174/2211738507666190122111224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langhans SA. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol 2018;9:6 doi 10.3389/fphar.2018.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018;18(20):3129–43 doi 10.1039/c8lc00322j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Metzcar J, Wang Y, Heiland R, Macklin P. A Review of Cell-Based Computational Modeling in Cancer Biology. JCO Clin Cancer Inform 2019;3:1–13 doi 10.1200/CCI.18.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skaga E, Kulesskiy E, Fayzullin A, Sandberg CJ, Potdar S, Kyttala A, et al. Intertumoral heterogeneity in patient-specific drug sensitivities in treatment-naive glioblastoma. BMC Cancer 2019;19(1):628 doi 10.1186/s12885-019-5861-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wan L, Skoko J, Yu J, Ozdoganlar O, LeDuc P, Neumann C. Mimicking embedded vasculature structure for 3D cancer on a chip approaches through micromilling. Scientific reports 2017;7(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.