

Abstract
SARS-CoV-2 infection of human airway epithelium activates genetic programs leading to progressive hyperinflammation in COVID-19 patients. Here, we report on transcriptomes activated in primary airway cells by interferons and their suppression by Janus kinase (JAK) inhibitors. Deciphering the regulation of the angiotensin-converting enzyme 2 (ACE2), the receptor for SARS-CoV-2, is paramount for understanding the cell tropism of SARS-CoV-2 infection. ChIP-seq for activating histone marks and Pol II loading identified candidate enhancer elements controlling the ACE2 locus, including the intronic dACE2 promoter. Employing RNA-seq, we demonstrate that interferons activate expression of dACE2 and, to a lesser extent, the genuine ACE2 gene. Interferon-induced gene expression was mitigated by the JAK inhibitors baricitinib and ruxolitinib, used therapeutically in COVID-19 patients. Through integrating RNA-seq and ChIP-seq data we provide an in-depth understanding of genetic programs activated by interferons, and our study highlights JAK inhibitors as suitable tools to suppress these in bronchial cells.
Subject terms: Gene regulation, High-throughput screening
Hye Kyung Lee et al. identify additional candidate enhancers linked to the ACE2 promoters producing biologically active ACE2, the receptor for SARS-CoV-2. They also demonstrate JAK inhibitors suppress the interferon-induced genetic programs, including ACE2 expression, in human airway epithelial cells.
Introduction
The angiotensin-converting enzyme 2 (ACE2) receptor is the gateway for SARS-CoV-2 to airway epithelium1,2 and the strong inflammatory response after viral infection is a hallmark in COVID-19 patients. Single cell RNA sequencing (scRNA-seq) studies have revealed that ACE2 expression in type II pneumocytes is induced by interferons3–7, suggesting that the presence of an autoregulatory loop could result in increased viral infection. Recent studies8–10 have identified a hitherto unknown short form of ACE2, called dACE2, that originates from an intronic promoter activated by interferons, calling into question the earlier studies that transcription of the full length native ACE2 is under interferon control. Onabajo and colleagues9 used ENCODE data for histone modification marks (H3K4me3, H3K4me1, and H3K27ac) as well as DNase I hypersensitive (DHS) sites in cell lines to mark putative regulatory elements at the newly identified exon (ex1c) located within intron 9 of the ACE2 gene. However, no regulatory elements were detected in the vicinity of the 5′ end of the full-length transcript encoding biologically active ACE2, and in sequences distal to the genuine promoter. Since these data sets were obtained from a wide range of cell lines and not from human primary airway cells, the principal target of SARS-CoV-2, they might not present a comprehensive picture of the regulatory regions controlling expression of the entire ACE2 locus, including dACE2, in bronchial tissue.
Janus kinase (JAK) inhibitors suppress signal transduction pathways leading to immune activation and inflammation and they are used therapeutically to harness cytokine storms in COVID-19 patients11,12. However, the genetic programs activated in airway cells by interferons and their response to JAK inhibitors remain to be elucidated. To better understand these programs, we investigated the transcriptomes activated by interferon α, β, γ, and λ as well as the cytokines growth hormone (GH), IL6, and IL7 in human primary airway cells. Using ChIP-seq for active histone marks and Pol II loading, we identified candidate enhancer elements controlling genes regulated by the different interferons. This also led to a better understanding of the structure and regulation of the ACE2 locus, including the intronic promoter encoding the dACE2 transcript. Our study also highlights that JAK inhibitors are suitable tools that efficiently blunt interferon-activated genetic programs.
Results
Interferons regulate the ACE2 locus in human airways cells
To comprehensively identify the genetic elements controlling the extended ACE2 locus, with an emphasis on its response to interferons, we focused on human primary Small Airway Epithelial Cells (SAECs) expressing a wide range of cytokine receptors and key mechanistic components of the executing JAK/STAT signal transduction pathway (Supplementary Data 1). We stimulated SAECs with interferon type I (IFNα and IFNβ), type II (IFNγ), and type III (IFNλ) as well as with growth hormone (GH), Interleukin 6 (IL6) and IL7, followed by RNA-seq transcriptome analyses (Supplementary Data 2–8). ACE2 expression increased ~16 to 18-fold upon stimulation with the four interferons but not with GH, IL6 and IL7 (Fig. 1a). This degree of induction was similar to some classical interferon stimulated genes (ISG), such as STAT1 and ISG20 (Fig. 1b–c). In contrast, expression of ISG15 was induced by approximately 500-fold (Fig. 1c).
Fig. 1. Expression of ACE2 isoforms is induced by interferons.

a–c ACE2, and STAT1 and ISGs mRNA levels from control cells and cells treated with different cytokines were measured by RNA-seq. Results are shown as the means ± s.e.m. of independent biological replicates (n = 3). One-way ANOVA followed by Dunnett’s multiple comparisons test was used to evaluate the statistical significance of differences. d RNA-seq reads were matched with each exon, including the new exon (ex1c), of the ACE2 gene. The CELF1 gene served as a control for the quality of the RNA-seq data. e SAECs were cultured in the absence and presence of interferon alpha (IFNα) and beta (IFNβ) followed by RNA-seq analyses. The reads covering key exons (1a, 1b, 9, 1c and 10) are shown. f mRNA levels of exon9 and exon1c were measured using qRT-PCR and normalized to GAPDH levels. Results are shown as the means ± s.e.m. of independent biological replicates (Control and IFNβ, n = 9; IFNα, n = 3). Two-way ANOVA with followed by Tukey’s multiple comparisons test was used to evaluate the statistical significance of differences between control and cytokine-treated cells.
Our RNA-seq data sets (Supplementary Data 2–8) created a foundation to dig deeper and comprehensively investigate gene classes differentially activated by cytokines in SAECs. IFNα treatment resulted in a more than 2-fold activation of 1405 genes, 647 genes were induced by IFNβ, 1570 by IFNγ, and 1285 by IFNλ3. IL6 activated 1432 genes, IL7 1782 genes, and GH 2807 genes. A total of 229 genes (Supplementary Data 9) were activated by all four interferons, and four genes (IRF7, IFI27, IFI35, and PARP10) were induced by all seven cytokines used in this study. Gene Set Enrichment Analysis (GSEA) showed, depending on the interferon, that between 17% and 26% of the induced genes were linked to immune responses, TNFα signaling, inflammatory response, IL-JAK/STAT signaling, among others (Supplementary Fig. 1a and Supplementary Data 2–5). While the family of interferon induced genes (IFIs) was, in general, activated by all four interferons, the magnitude differed more than 10-fold between individual genes and gene families (Supplementary Fig. 2). Notably, genes encoding the family of Interferon-induced proteins with tetratricopeptide repeats (IFIT) were induced up to several thousand-fold. In contrast activation of the gene family of interferon-induced transmembrane proteins (IFITM) was at least one order of magnitude lower. Other studies used bulk RNA-seq of primary human alveolar epithelial type-2 cells (AT2s) infected with SARS-CoV-27 and scRNA-seq from human bronchial epithelium (BEAS-2B) stimulated with interferons5. There was an overlap between genes induced by interferons in SAECs and SARS-CoV-2 in AT2s (Supplementary Fig. 2). In contrast, scRNA-seq provides less read depth and a direct comparison with bulk RNA-seq is therefore not possible. As a positive control, we also measured the activation of STAT1, STAT2, SOCS1 SOCS3, and SIRF1 and IRF7 (Supplementary Fig. 3e–f). The cytokines IL6, IL7, and GH activated different gene sets (Supplementary Data 6–8). Our results not only identified gene sets activated by interferons but also demonstrated their differential response to individual interferons (Supplementary Fig. 3).
The ACE2 locus encodes two mRNAs, one encoding the full-length ACE2 and one encoding the N-terminally truncated form of ACE2 (dACE2). The dACE2 mRNA is initiated from an intronic promoter and in agreement with earlier studies8,9, we detected the dACE2 exon (ex1c) within intron 9 of the ACE2 gene (Fig. 1d). To obtain additional information on the interferon response of the dACE2 and ACE2 promoters, we used RNA-seq and determined the respective read counts over the three alternative first exons (Fig. 1d–e). While the increase of RNA-seq reads induced by IFNα/β was highest (~25-fold) over ex1c, a significant, ~2–10-fold increase was detected over ex1a and ex1b, supporting the notion that expression of the full-length ACE2 transcript is also under interferon control. The CELF1 gene served as a control (Fig. 1d). As an independent assay we used qRT-PCR and determined that IFN α/β stimulation led to an 8 to 15-fold increase of dACE2 (ex1c) and an approximately ~3-fold increase of ACE2 RNA (ex9) (Fig. 1f). In addition, both AEC2 isoforms were detected by Western blot (Supplementary Fig. 4). Previous studies in normal human bronchial epithelium (NHBE) did not reveal an interferon response of the native ACE2 promoter8,9 suggesting differences between cell types or culture conditions. The mouse Ace2 gene is also induced by cytokines through a JAK/STAT5-activated enhancer in the second intron13 and a DHS site is located in the equivalent location on the human ACE2 gene in SAECs and lung tissue. This suggests the presence of additional regulatory elements controlling expression of the full-length ACE2 mRNA.
Interferons activate enhancers in the ACE2 locus
To identify candidate regulatory elements controlling the extended ACE2 locus, including ACE2 and dACE2, in primary airway cells, we conducted ChIP-seq for the histone marks H3K27ac (activate loci), H3K4me1 (enhancers), and H3K4me3 (promoters). We also probed RNA Polymerase II (Pol II) loading in SAECs in the absence and presence of IFNβ (Fig. 2). DNase I hypersensitive (DHS) sites from human lung tissues14 and SAECs15 served as bona fide predictors of regulatory regions. In addition to ACE2, expression of the neighboring TMEM27 gene, an ACE2 homologue, as well as BMX were activated by interferons (Fig. 2a) suggesting the presence of shared regulatory elements. ACE2 and TMEM27 originated from a gene duplication and their response to interferon is equivalent. The positions of the chromatin boundary factor CTCF suggests that ACE2 and TMEM27 are located within a sub-TAD (Fig. 2a).
Fig. 2. Structure of the extended ACE2 locus and regulation of the ACE2 locus in primary airway epithelial cells.

a Regulatory marks in the 600 kb locus including ACE2 and neighboring genes in SAECs. mRNA levels in the absence and presence of IFNβ were measured by RNA-seq. ChIP-seq for the histone marks H3K4me1, H3K27ac, and H3K4me3, and Pol II was conducted in SAECs in the absence and presence of IFNβ. CTCF and STAT5 ChIP-seq data and DHS data are from ENCODE. b ChIP-seq experiments for the histone marks, H3K4me3 (promoter), H3K4me1 (enhancers), H3K27ac (active genes), and Pol II loading. The DHS data were obtained from ENCODE14,15. Yellow shade, candidate enhancers and blue shade, predicted promoter. The P/E region within intron 9 probably constitutes a combined promoter/enhancer unit. The BCL6 gene severed as ChIP-seq control. Solid arrows indicate the orientation of genes. c A putative STAT5 enhancer in the ACE2 gene was identified using ChIP-seq data from IFNβ-treated K562 cells16. d ChIP-seq data for STAT transcription factors, histone marks H3K4me3 (promoter), H3K4me1 (enhancers) and H3K27ac (active genes), and RNA Pol II at the STAT1 locus.
In agreement with earlier studies9, we identified DHS sites at ex1c located in intron 9 and in intron 17 (Fig. 2b). In addition, we identified DHS sites in the vicinity of ex1a, likely marking the genuine ACE2 promoter, a distal site marking a possible enhancer and one in intron 15 coinciding with activate chromatin marks. The DHS site in intron 9 overlaps with strong H3K4me3 marks, identifying it as a genuine promoter region (Fig. 2b). This site is also decorated with H3K4me1 and H3K27ac marks and extensive Pol II loading, hallmarks of a complex promoter/enhancer. H3K27ac and Pol II loading was further induced by IFNβ, reflecting increased ACE2 expression. The cytokine regulated BCL6 gene served as a control. IFNβ activated the transcription factors STATs 1, 2, and 5 (Fig. 1b and Supplementary Data 3) and ChIP-seq experiments from K562 erythroid cells stimulated with IFNβ16 revealed preferential binding of STAT5 to the intronic promoter/enhancer (Fig. 2c) further supporting regulation through the JAK/STAT pathway. The STAT1 locus served as a control for the binding of STAT transcription factors (Fig. 2d). While the ACE2 promoter associated with ex1a is marked by a DHS site and H3K4me1 marks, there is little evidence of H3K4me3 and H3K27ac marks. However, it is well known that there is no direct relationship between gene activity and the presence of these marks.
Based on the presence of DHS sites, activating histone marks and Pol II loading, either in combination or by themselves, several candidate enhancers regulating the ACE2 locus were identified (Fig. 2b). In addition, activating histone marks and DHS sites14,15 marked candidate regulatory elements in other loci under interferon control, such as STAT1 and ISG15, in human lung tissues (Fig. 3)5,17. Members of the IFIT gene family were induced several thousand-fold by interferons (Supplementary Fig. 3) and ChIP-seq profiled revealed the regulatory landscape in this genetic locus (Supplementary Fig. 5). Candidate regulatory elements were also identified in IRF1 and 9, STAT1, IFI44 and members of the CXCL gene family. Our RNA-seq and ChIP-seq data should foster research aimed at understanding interferon-regulated genetic programs in airway cells.
Fig. 3. Activating histone marks at the ACE2 locus in lung tissue.
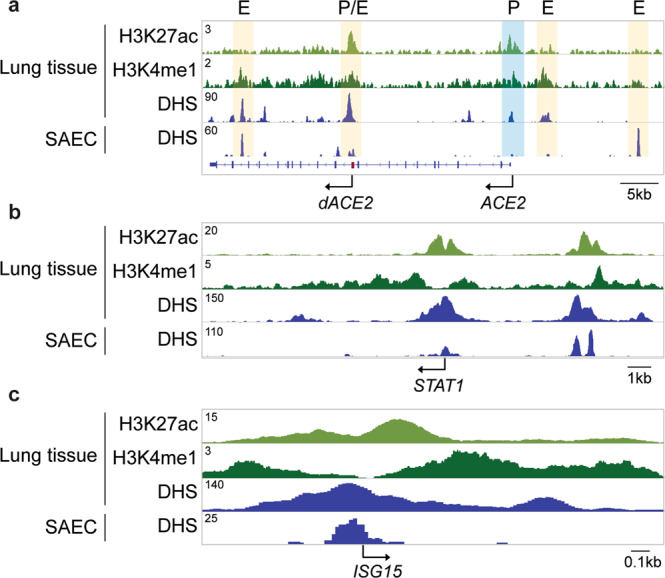
ChIP-seq for histone marks H3K4me1 (enhancers) and H3K27ac (active genes), and DHS from human lung tissues and DHS from SAECs display regulatory elements at the ACE2 (a), STAT1 (b), and ISG15 (c) loci.
JAK inhibitors suppress the IFNβ-sensing enhancers in the ACE2 locus
Interferons activate genetic programs through the JAK/STAT signaling pathway and JAK inhibitors are used clinically in COVID-19 patients in an effort to suppress the genomic consequences of cytokine storms11,18,19. To investigate if interferon-induced ACE2 and dACE2 expression is controlled by the JAK/STAT pathway, we cultured SAECs in the presence of IFNβ and the JAK inhibitors, baricitinib and ruxolitinib, followed by RNA-seq, qRT-PCR assays (Fig. 4a–c and Supplementary Data 10–11) and ChIP-seq analyses (Fig. 4e–h). Both JAK inhibitors blunted the IFNβ-induced increase of the full-length ACE2 (ex1a, ex1b, and ex9) and the dACE2 (ex1c) transcripts (Fig. 4a–b), supporting that their respective promoters are under JAK/STAT control. The efficacy of the two inhibitors extended to a range of genetic programs activated through the pan JAK/STAT pathway (Supplementary Fig. 1b) and induction of bona fide interferon stimulated genes (ISG), such as STAT1 and ISG15, was suppressed (Fig. 4c, d). In ACE2, as in other ISGs, ruxolitinib treatment mitigated the establishment of activating H3K27ac marks and Pol II loading over distal and intronic regulatory elements (Fig. 4e–h). IFNβ-induced expression of ISG, IFIT, and IFITM (Supplementary Fig. 3) was suppressed by baricitinib and ruxolitinib (Supplementary Data 9; Fig. 5). In addition to key transcription factors, primary RNA pattern recognition receptors (PRRs), including (RIG-I)-like receptors, are critical for the interferon production by lung epithelial cells upon infection by SARS-CoV-220. Specifically, LGP221 (DHX58) and MDA5 are receptors binding double-stranded viral RNA intermediates and they primarily regulate IFN induction in response to SARS-CoV-220. Notably, LGP2 itself was induced by interferons and this activation was mitigated by JAK inhibitors (Supplementary Figs. 3h and 5).
Fig. 4. JAK inhibitors mitigate activation of IFNβ-stimulated genes.

a–b SAECs were cultured in the presence of IFNβ and JAK inhibitors, either baricitinib or ruxolitinib followed by RNA-seq analyses (a) and qRT-PCR (b). Reads covering key exons are displayed. qRT-PCR results were normalized to GAPDH levels and are shown as the means ± s.e.m. of independent biological replicates (Control and IFNβ, n = 9; IFNβ+JAK inhibitors, n = 3). Two-way ANOVA with followed by Tukey’s multiple comparisons test was used to evaluate the statistical significance of differences between control and cytokine/Jak inhibitor-treated cells. c–d STAT1 and ISG15 mRNA levels from control and experimental cells were measured by RNA-seq. Results are shown as the means ± s.e.m. of independent biological replicates (n = 3). One-way ANOVA with followed by Dunnett’s multiple comparisons test was used to evaluate the statistical significance of differences between control and cytokine/JAK inhibitor-treated cells. e–h H3K27ac marks, and Pol II loading at the ACE2, STAT1, and ISG15 loci in SAECs in the absence and presence of IFNβ and the JAK inhibitor, ruxolitinib. The FAR1 locus was used as a ChIP-seq control.
Fig. 5. JAK inhibitors suppress distinct interferon-regulated immune pathways.
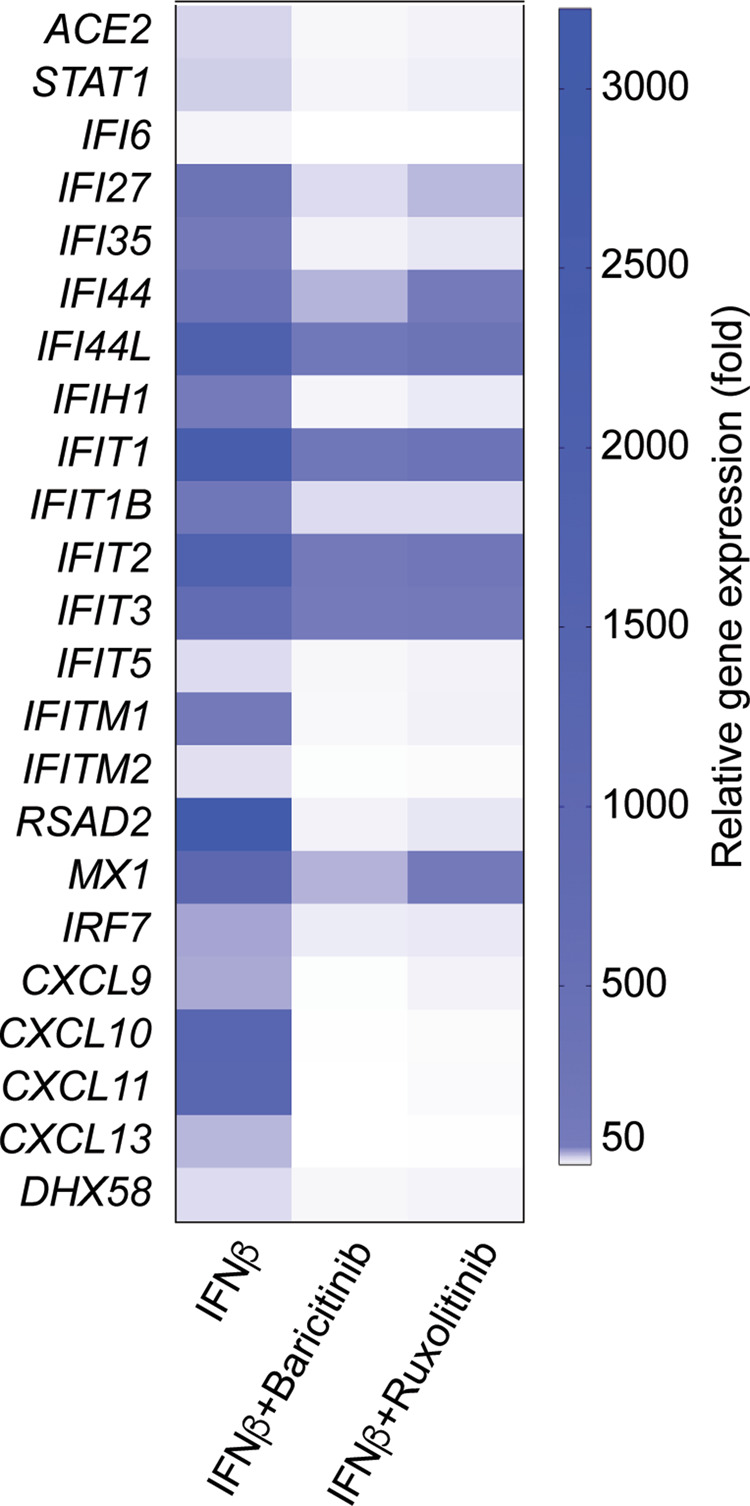
Heat map comparison of relative expression levels of representative interferon-responsive genes in SAECs stimulated with IFNβ (n = 3) alone or with IFNβ and JAK inhibitors (n = 3).
While elevated interferon levels following the initial virus infection modulate the host’s immune system, persistent upregulation can lead to cytokine storms and more severe symptoms22,23. To address the efficacy of JAK inhibitors in the suppression of ACE2 expression, we performed additional kinetic experiments. The expression of ACE2 and STAT1 significantly increased within four hours after IFNβ stimulation and peaked at 12 h (Fig. 6a). Increased expression from the dACE2 promoter accounted for most of elevated ACE2 levels. This IFNβ-induced gene expression was suppressed by the JAK inhibitor ruxolitinib and persisted for 12 h (Fig. 6b). Lastly, pretreatment of cells with ruxolitinib for two hours abrogated the interferon response of ACE2 and STAT1 (Fig. 6c). JAK inhibitors efficiently suppress IFN-induced gene expression; these results mechanistically clarify that JAK inhibitors are therapeutically beneficial in COVID-19 patients, including those experiencing cytokine storms.
Fig. 6. JAK inhibitor ruxolitinib suppresses IFNβ-stimulation.

a SAECs were stimulated with IFNβ for 12 h (0, 1, 2, 4, 6, 8, and 12 h) and analyzed by qRT-PCR. b SAECs were cultured for two hours in the presence of IFNβ followed by the addition of ruxolitinib and qRT-PCR after 0–12 h (0, 1, 2, 4, 6, 8, and 12 h). c SAECs were pretreated with ruxolitinib for two hours before stimulating the cells with IFNβ, followed by qRT-PCR for 0–12 h (0, 1, 2, 4, 6, 8, and 12 h). qRT-PCR results were normalized to GAPDH levels and are shown as the means ± s.e.m. of independent biological replicates (all conditions, n = 4). Two-way ANOVA with followed by Tukey’s multiple comparisons test was used to evaluate the statistical significance of differences between control and cytokine/Jak inhibitor-treated cells.
Discussion
SARS-CoV-2 entry into nasal epithelia and lung tissue is facilitated by ACE2 and deciphering the regulation of the ACE2 gene will aid our understanding of the viral tropism and also non-pulmonary pathology. Here, we have identified candidate regulatory elements in the extended ACE2 locus and propose roles in controlling gene expression induced by interferons and other, not yet defined, stimuli. The intronic region associated with the dACE2 transcript8–10 bears promoter and enhancer marks and binds STAT transcription factors, suggesting a complex regulatory element controlling interferon-induced expression. Additional candidate enhancers were identified distal to the genuine ACE2 promoter and within introns3,4. Our high-resolution bulk RNA-seq data sets on differentiated human primary small airway epithelial cells (SAECs) revealed the presence of the genuine full-length ACE2 transcript and the short form of ACE2, named dACE2, which had been identified in other studies using bulk RNA-seq8–10. Our transcriptome studies and qRT-PCR demonstrate that interferons strongly activate expression of dACE2 but only marginally ACE2. dACE2 encodes a non-functional protein, which does not bind SARS-CoV-29,10, strongly suggesting that the interferon response does not result in increased viral infection. However, further studies are needed to investigate whether dACE2 could be a decoy and thus impede cellular uptake of SARS-CoV-2 upon interferon treatment. Single cell RNA-seq (scRNA-seq) studies3–7 have shown elevated ACE2 expression in primary human airway cells stimulated with type I interferons. Based on these findings a feedback loop hypothesis was formulated suggesting that increased ACE2 levels induced by the cytokine storm would result in an increased infectivity. However, current evidence suggests interferons largely activate expression of the non-functional dACE2 isoform. In general, the lower sequencing depth of scRNA-seq makes it more challenging to identify alternative transcripts. Whether ACE2 RNA levels correlate with protein levels remains an open question24. Since previous scRNA-seq studies did not detect the more abundant dACE2 transcript3–7,24, which would encode a protein lacking the 356 N-terminal amino acids, additional studies are needed.
Our ChIP-seq data also point to several additional candidate enhancers controlling the entire ACE2 locus, including the neighboring TMEM27 gene. Further studies are warranted to investigate ACE2 regulation in other SARS-CoV-2 target cells, such as pancreas25,26, gut27, and mammary epithelium. While the regulation of the human and mouse ACE213 loci displays distinct differences, they share their response to cytokines and the JAK/STAT pathway, with the mouse Ace2 gene being activated in mammary tissue by cytokines through an intronic enhancer13. Further studies in tissues and primary cells are needed to better understand the complex, possibly cell-specific, regulation of the human ACE2 locus in organs with extrapulmonary manifestations of SARS-CoV-2 infection.
Type I interferon (IFNα and IFNβ) secreted by SARS-CoV-2-infected airway epithelium controls autocrine and paracrine genetic programs. Here, we used small airway epithelial cells (SAECs) that, like human bronchial epithelial cells (HBEs), differentiate in air liquid interphase (ALI) culture systems and form tight junctions and displaying cilia. Other studies have focused on primary tonsil epithelial cells9, primary normal human bronchial epithelial cells (NHBE)8–10, nasal epithelial cells (NECs)10, BEAS-2B cells (bronchial epithelium)5, primary human air way epithelial cells from human donors5 as well as HEK293T, A549, SCC-4, SCC-25, Vero, CV-1, MDCK, R9ab, and MCA-38 cell lines8. While any of these cells respond to cytokines, the magnitude of the transcriptome response likely differs between experimental systems and is dependent on the presence and abundance of various components of the JAK/STAT signaling pathway. Since some studies used bulk RNA-seq and others scRNA-seq a definitive comparison of the genetic programs induced by interferons is difficult to achieve. Bulk RNA-seq is an appropriate approach to gauge interferon-activated genetic programs as indicated by the up to 50,000-fold induction of the IFIT gene family encoding antiviral proteins. Integrating the comprehensive RNA-seq and ChIP-seq data sets also permits the genome-wide identification of candidate enhancers responsive to interferons and other cytokines.
JAK inhibitors28–30 are used therapeutically to treat COVID-19 patients11,12,31 and our studies have identified IFN-induced genetic pathways that are suppressed by ruxolitinib and baricitinib. Both JAK inhibitors had an equivalent capacity to effectively mitigate IFNβ-induced gene activation and blunt immune signaling pathways. Autosomal-recessive and autosomal-dominant mutations in interferon pathway genes have been identified in some patients with life-threatening COVID-1932 and high TYK2 expression was linked to COVID-19 disease severity33. The promising clinical results with JAK inhibitors, which dampen the inflammation, and molecular studies, including ours, on the efficacy of JAK inhibitors in suppressing interferon-induced genetic programs has practical implications for their use in treating COVID-19 patients.
Methods
Cell culture
Human small airway epithelial cells (SAECs) obtained from Lifeline Technology (FC-0016) were expanded using the complete BronchiaLifeTM media kit (Lifeline Technology, LL-0023). All culture wares were pre-coated in 30 μg/ml of Fibronectin (ThermoFisher Scientific, 33016015) for at least 1 h at room temperature. Calu-3 line (ATCC, HTB-55TM) were cultured using Eagle’s Minimum Essential Medium (ATCC, 30-2003TM) containing 10% fetal bovine serum (Cytiva, SH3007103) in 5% CO2 atmosphere at 37 °C.
Cytokines (10 ng/ml; Human IFNβ, 300-02BC; Human IFNγ, 300-02; Human IL6, 200-06; Human IL7, 200-07; Human Growth hormone, 100-40, Peprotech; Human IFNα2b, 78077.1, Stem Cell Technologies; Human IFNλ3, 5259-IL-025, R&D systems) were added in respective culture media after serum starvation for 2 h, and the cells were incubated for 12 h in 5% CO2 atmosphere at 37 °C. The cells were washed with PBS (Gibco, 14190144) twice and harvested.
Jak inhibitors, 10 μM of either Baricitinib (HY-15315A, MedChemExpress) or Ruxolitinib (HY-50856A, MedChemExpress), were added to BronchiLiafeTM media with or without IFNβ. SAECs and incubated for 12 h and then washed with PBS twice and harvested.
RNA isolation and quantitative real-time PCR (qRT–PCR)
Total RNA was extracted from the collected cells and purified using the PureLink RNA Mini Kit (Invitrogen) according to the manufacturer’s instructions. cDNA was synthesized from total RNA using Superscript II (Invitrogen). Quantitative real-time PCR (qRT-PCR) was performed using TaqMan probes (ACE2, Hs01085333_m1; STAT1, Hs01013996_m1; GAPDH, Hs02786624_g1, Thermo Fisher scientific) on the CFX384 Real-Time PCR Detection System (Bio-Rad) according to the manufacturer’s instructions. Exon9 and exon1c mRNA were measured with the following primers, that were used by Onabajo et al.9, using TaqMan expression assay: Forward, 5′-GGGCGACTTCAGGATCCTTAT-3′, Reverse, 5′-GGATATGCCCCATCTCATGATGG-3′, Probe, 5′-ATGGACGACTTCCTGACAG-3′; Forward, 5′-GGAAGCAGGCTGGGACAAA-3′, Reverse, 5′-AGCTGTCAGGAAGTCGTCCATTG-3′, Probe, 5′-AGGGAGGATCCTTATGTG-3′. PCR conditions were 95 °C for 30 s, 95 °C for 15 s, and 60 °C for 30 s for 40 cycles. All reactions were done in triplicate and normalized to the housekeeping gene GAPDH. Relative differences in PCR results were calculated using the comparative cycle threshold (CT) method.
Total RNA sequencing (Total RNA-seq) and data analysis
Total RNA was extracted from the collected cells and purified using the PureLink RNA Mini Kit (Invitrogen) according to the manufacturer’s instructions. Ribosomal RNA was removed from 1 μg of total RNAs and cDNA was synthesized using SuperScript III (Invitrogen). Libraries for sequencing were prepared according to the manufacturer’s instructions with TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold (Illumina, RS-122-2301) and paired-end sequencing was done with a HiSeq 3000 instrument (Illumina).
The raw data were subjected to QC analyses using the FastQC tool (version 0.11.9) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Total RNA-seq read quality control was done using Trimmomatic34 (version 0.36) and STAR RNA-seq35 (version STAR 2.5.4a) using 50 bp paired-end mode was used to align the reads (hg19). HTSeq36 (version 0.9.1) was to retrieve the raw counts and subsequently, R (https://www.R-project.org/), Bioconductor37, and DESeq238 were used. Additionally, the RUVSeq39 package was applied to remove confounding factors. The data were pre-filtered keeping only those genes, which have at least ten reads in total. The visualization was done using dplyr (https://CRAN.R-project.org/package=dplyr) and ggplot240. Genes were categorized as significantly differentially expressed with an adjusted p-value (pAdj) below 0.05 and a fold change > 2 for upregulated genes and a fold change of < −2 for downregulated ones. Gene Set Enrichment Analysis (GSEA, https://www.gsea-msigdb.org/gsea/msigdb) were performed. Sequence read numbers were calculated using Samtools41 software with sorted bam files.
Chromatin immunoprecipitation sequencing (ChIP-seq) and data analysis
Chromatin was fixed with formaldehyde (1% final concentration) for 15 min at room temperature, and then quenched with glycine (0.125 M final concentration). Chromatin washed with PBS was homogenized using a tissue grinder in Farnham’s lysis buffer and fragmented by sonication. Fragmentation efficiency in the range of 250–500 bp was checked using gel electrophoresis and ChIP was performed with Dynabeads Protein A (Invitrogen) and the following antibodies (5–10 μg) were used for ChIP-seq: H3K27ac (Abcam, ab4729), RNA polymerase II (Abcam, ab5408), H3K4me1 (Active Motif, 39297), and H3K4me3 (Millipore, 07-473). Libraries for next-generation sequencing were prepared and sequenced with a HiSeq 3000 instrument (Illumina).
The raw data were subjected to QC analyses using the FastQC tool (version 0.11.9) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Quality filtering and alignment of the raw reads was done using Trimmomatic34 (version 0.36), Bowtie42 (version 1.2.2), and Samtools41 (version 1.8), with the parameter ‘-m 1’ to keep only uniquely mapped reads, using the reference genome hg19. Picard tools (Broad Institute. Picard, http://broadinstitute.github.io/picard/. 2016) was used to remove duplicates. Homer43 (version 4.8.2) and DeepTools44 (version 3.1.3) software was applied to generate bedGraph files, separately. Integrative Genomics Viewer45 (version 2.5.3) was used for visualization. Each ChIP-seq experiment was conducted for more than two replicates. DeepTools was used to obtain the Pearson and Spearman correlation between the replicates.
Western blot
Proteins (100 μg) from SAECs extracted using lysis buffer (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 0.5% Na-DOC, 1% NP-40, 0.1% SDS, 5 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail) were separated on a 4–12% NuPage gradient gel (Invitrogen) and transferred to a PVDF membrane (Invitrogen). Membranes were blocked for 1 h with 5% nonfat dry milk in PBS-T buffer (PBS containing 0.1% Tween 20) and incubated for 1.5 h at 4 °C with the primary antibody (1:1000) against ACE2 (Proteintech, 21115-1-AP) and GAPDH (Cell signaling, #5174). After washing, membranes were incubated for 1 h with HRP-conjugated secondary antibodies (Cell signaling). Labeled protein bands were detected using an enhanced chemiluminescence system (Thermo scientific) and Amersham Imager 600 (GE healthcare). Band density was analyzed using this imager.
Statistics and reproducibility
Data were presented as the means ± s.e.m. (standard error of the mean) of all experiments with n = number of biological replicates. For comparison of samples, data were presented as standard deviation in each group and were evaluated with a two-way ANOVA followed by Tukey’s multiple comparisons test or a one-way ANOVA with Dunnett’s multiple comparisons test using PRISM 8 GraphPad (version 8.2.0). Statistical significance was obtained by comparing the measures from control group and each treated group. A value of *P < 0.05, **P < 0.001, ***P < 0.0001, ****P < 0.00001 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by the Intramural Research Programs (IRPs) of National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and National Center for Advancing Translational Sciences (NCATS). We thank Ilhan Akan, Sijung Yun, and Harold Smith from the NIDDK genomics core for NGS and NCATS Chemical Genomics Center Team for the JAK inhibitors. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Author contributions
H.K.L.: project conception, experimental design and execution, data analysis, preparation of figures, writing manuscript; O.J.: experimental design and execution; L.H.: project conception, experimental design, data analysis, preparation of figures, writing manuscript.
Data availability
The data-sets generated here were uploaded under the accession GSE161665 (ChIP-seq in GSE161663 and RNA-seq in GSE161664). In addition, we analyzed the ChIP-seq for STATs from GSE31477, CTCF from GSE101051, H3K27ac and H3K4me1 from human lung tissues from GSE143115 and GSE142958, and DNase I hypersensitive (DHS) data from human lung tissues and SAECs from GSE90364 and GSE29692, respectively. RNA-seq data from other types of lung cells, BEAS-2B cells and AT2s, was obtained from GSE148829 and GSE152586, respectively.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hye Kyung Lee, Email: hyekyung.lee@nih.gov.
Lothar Hennighausen, Email: lotharh@niddk.nih.gov.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-021-02167-1.
References
- 1.Hoffmann M, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280 e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lukassen, S. et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J.39, e105114 (2020). [DOI] [PMC free article] [PubMed]
- 3.Youk J, et al. Three-dimensional human alveolar stem cell culture models reveal infection response to SARS-CoV-2. Cell Stem Cell. 2020;27:905–919 e10. doi: 10.1016/j.stem.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sajuthi SP, et al. Type 2 and interferon inflammation regulate SARS-CoV-2 entry factor expression in the airway epithelium. Nat. Commun. 2020;11:5139. doi: 10.1038/s41467-020-18781-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziegler, C. G. K. et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell181, 1016–1035 (2020). [DOI] [PMC free article] [PubMed]
- 6.Chua RL, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 7.Katsura H, et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell. 2020;27:890–904.e8. doi: 10.1016/j.stem.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng, K. W. et al. Tissue-specific and interferon-inducible expression of nonfunctional ACE2 through endogenous retroelement co-option. Nat. Genet.52, 1294–1302 (2020). [DOI] [PMC free article] [PubMed]
- 9.Onabajo, O. O. et al. Interferons and viruses induce a novel truncated ACE2 isoform and not the full-length SARS-CoV-2 receptor. Nat. Genet.52, 1283–1293 (2020). [DOI] [PMC free article] [PubMed]
- 10.Blume C, et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet. 2021;53:205–214. doi: 10.1038/s41588-020-00759-x. [DOI] [PubMed] [Google Scholar]
- 11.La Rosee F, et al. The Janus kinase 1/2 inhibitor ruxolitinib in COVID-19 with severe systemic hyperinflammation. Leukemia. 2020;34:1805–1815. doi: 10.1038/s41375-020-0891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spinelli, F. R., Conti, F. & Gadina, M. HiJAKing SARS-CoV-2? The potential role of JAK inhibitors in the management of COVID-19. Sci. Immunol.5, eabc5367 (2020). [DOI] [PubMed]
- 13.Hennighausen, L. & Lee, H. K. Activation of the SARS-CoV-2 receptor Ace2 through JAK/STAT-dependent enhancers during pregnancy. Cell Rep. 32, 108199 (2020). [DOI] [PMC free article] [PubMed]
- 14.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurano MT, et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat. Genet. 2015;47:1393–1401. doi: 10.1038/ng.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Au-Yeung N, Mandhana R, Horvath CM. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAKSTAT. 2013;2:e23931. doi: 10.4161/jkst.23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai N, et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020;11:6319. doi: 10.1038/s41467-020-20139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeleswaram S, et al. Inhibition of cytokine signaling by ruxolitinib and implications for COVID-19 treatment. Clin. Immunol. 2020;218:108517. doi: 10.1016/j.clim.2020.108517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kinase Inhibitors: Bruton’s Tyrosine Kinase Inhibitors and Janus Kinase Inhibitors. (https://www.covid19treatmentguidelines.nih.gov/immune-based-therapy/immunomodulators/kinase-inhibitors/).
- 20.Yin X, et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021;34:108628. doi: 10.1016/j.celrep.2020.108628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui Y, et al. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics. 2001;78:129–134. doi: 10.1006/geno.2001.6661. [DOI] [PubMed] [Google Scholar]
- 22.Bost P, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell. 2020;181:1475–1488 e12. doi: 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao M, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 24.Ortiz ME, et al. Heterogeneous expression of the SARS-Coronavirus-2 receptor ACE2 in the human respiratory tract. EBioMedicine. 2020;60:102976. doi: 10.1016/j.ebiom.2020.102976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coate KC, et al. SARS-CoV-2 cell entry factors ACE2 and TMPRSS2 are expressed in the microvasculature and ducts of human pancreas but are not enriched in beta cells. Cell Metab. 2020;32:1028–1040 e4. doi: 10.1016/j.cmet.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller JA, et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021;3:149–165. doi: 10.1038/s42255-021-00347-1. [DOI] [PubMed] [Google Scholar]
- 27.Lamers, M. M. et al. SARS-CoV-2 productively infects human gut enterocytes. Science369, 50–54 (2020). [DOI] [PMC free article] [PubMed]
- 28.Luo W, et al. Targeting JAK-STAT signaling to control cytokine release syndrome in COVID-19. Trends Pharm. Sci. 2020;41:531–543. doi: 10.1016/j.tips.2020.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez GAM, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Investig. 2018;128:3041–3052. doi: 10.1172/JCI98814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi J, et al. Baricitinib-induced blockade of interferon gamma receptor and interleukin-6 receptor for the prevention and treatment of graft-versus-host disease. Leukemia. 2018;32:2483–2494. doi: 10.1038/s41375-018-0123-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goletti D, Cantini F. Baricitinib therapy in Covid-19 pneumonia - an unmet need fulfilled. N. Engl. J. Med. 2021;384:867–869. doi: 10.1056/NEJMe2034982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science370, eabd4570 (2020). [DOI] [PMC free article] [PubMed]
- 33.Pairo-Castineira E, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–98. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 34.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber W, et al. Orchestrating high-throughput genomic analysis with bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014;32:896–902. doi: 10.1038/nbt.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickham, H. Ggplot2: elegant graphics for data analysis, viii, 212 p. (Springer, 2009).
- 41.Masella AP, et al. BAMQL: a query language for extracting reads from BAM files. BMC Bioinform. 2016;17:305. doi: 10.1186/s12859-016-1162-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramirez F, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data-sets generated here were uploaded under the accession GSE161665 (ChIP-seq in GSE161663 and RNA-seq in GSE161664). In addition, we analyzed the ChIP-seq for STATs from GSE31477, CTCF from GSE101051, H3K27ac and H3K4me1 from human lung tissues from GSE143115 and GSE142958, and DNase I hypersensitive (DHS) data from human lung tissues and SAECs from GSE90364 and GSE29692, respectively. RNA-seq data from other types of lung cells, BEAS-2B cells and AT2s, was obtained from GSE148829 and GSE152586, respectively.