

Key Points
Question
What is the association between pathogenic or likely pathogenic variants of inherited cardiomyopathies and arrhythmia syndromes and the risk of unexplained sudden cardiac death (SCD)?
Findings
In this genetic association study of 413 adults who had unexplained SCD, nearly one-fifth of individuals had pathogenic or likely pathogenic genetic variants consistent with inherited cardiomyopathies or arrhythmia syndromes, despite having normal cardiac findings. These clinically significant variants were predominantly associated with hypertrophic cardiomyopathy, dilated cardiomyopathy, and long QT syndrome.
Meaning
Further research is needed to assess the utility of integrating genetic testing into risk assessment for the prevention of unexplained sudden cardiac death.
This genetic association study investigates cases of unexplained sudden cardiac death to determine the frequency of pathogenic or likely pathogenic genetic variants of inherited cardiomyopathies and arrhythmia syndromes.
Abstract
Importance
Unexplained sudden cardiac death (SCD) describes SCD with no cause identified. Genetic testing helps to diagnose inherited cardiac diseases in unexplained SCD; however, the associations between pathogenic or likely pathogenic (P/LP) variants of inherited cardiomyopathies (CMs) and arrhythmia syndromes and the risk of unexplained SCD in both White and African American adults living the United States has never been systematically examined.
Objective
To investigate cases of unexplained SCD to determine the frequency of P/LP genetic variants of inherited CMs and arrhythmia syndromes.
Design, Setting, and Participants
This genetic association study included 683 African American and White adults who died of unexplained SCD and were included in an autopsy registry. Overall, 413 individuals had DNA of acceptable quality for genetic sequencing. Data were collected from January 1995 to December 2015. A total of 30 CM genes and 38 arrhythmia genes were sequenced, and variants in these genes, curated as P/LP, were examined to study their frequency. Data analysis was performed from June 2018 to March 2021.
Main Outcomes and Measures
The frequency of P/LP variants for CM or arrhythmia in individuals with unexplained SCD.
Results
The median (interquartile range) age at death of the 413 included individuals was 41 (29-48) years, 259 (62.7%) were men, and 208 (50.4%) were African American adults. A total of 76 patients (18.4%) with unexplained SCD carried variants considered P/LP for CM and arrhythmia genes. In total, 52 patients (12.6%) had 49 P/LP variants for CM, 22 (5.3%) carried 23 P/LP variants for arrhythmia, and 2 (0.5%) had P/LP variants for both CM and arrhythmia. Overall, 41 P/LP variants for hypertrophic CM were found in 45 patients (10.9%), 9 P/LP variants for dilated CM were found in 11 patients (2.7%), and 10 P/LP variants for long QT syndrome were found in 11 patients (2.7%). No significant difference was found in clinical and heart characteristics between individuals with or without P/LP variants. African American and White patients were equally likely to harbor P/LP variants.
Conclusions and Relevance
In this large genetic association study of community cases of unexplained SCD, nearly 20% of patients carried P/LP variants, suggesting that genetics may contribute to a significant number of cases of unexplained SCD. Our findings regarding both the association of unexplained SCD with CM genes and race-specific genetic variants suggest new avenues of study for this poorly understood entity.
Introduction
Estimates of the incidence of sudden cardiac death (SCD) in the United States range from 180 000 to 450 000 cases annually.1,2 Coronary heart disease (CHD) is the most common cause for SCD in the West, representing 50% to 75% of SCD. While autopsies may lead to a diagnosis of structural heart disease in many of the remaining cases (eg, individuals with SCD without CHD), the cause remains uncertain in 30% to 40% of cases, despite toxicologic and histopathologic analysis (so-called unexplained SCD).2,3 Because many of these cases occur in the community setting, most are referred to the medical examiner for evaluation, and during the past 20 years, an increased understanding of genetics is leading to improved evaluation of such cases.
Molecular autopsy or genetic testing can improve diagnostic accuracy in individuals with normal cardiac autopsy results. Two groups of familial diseases are responsible for SCD: cardiomyopathies (CMs) and arrhythmia syndromes. In the young (ie, aged <40 years), arrhythmia syndromes are well-recognized causes of SCD in the absence of structural heart disease.4 Several hypertrophic and dilated CMs (HCM and DCM, respectively) gene variants have also been reported to increase risk of SCD; however, whether these variants can predict future SCD risk, especially in those without overt structural heart disease, remains controversial.5,6
Most studies of unexplained SCD conducted to date have concentrated on small, homogenous populations of young people (ie, <35 years) from Australia, New Zealand, Denmark, the Netherlands, Spain, the United Kingdom, France, and South Korea.7,8,9,10,11,12,13 One study from New York14 included 296 cases involving individuals who belonged to multiple races; however, more than half were infants or children. None of these studies has addressed the potential association of African American race with genetic risk of SCD. It is well known that different racial and ethnic groups have diverse genetic signatures with different allelic frequencies. However, no systematic comparison of the genetics underlying cases of unexplained SCD between adult White and African American decedents has ever been conducted.15
Our institute has been receiving SCD cases for diagnostic evaluation from the State of Maryland medical examiner for more than 20 years. Using this database, we conducted among the largest systematic genetic examinations of adult African American and White individuals in the US who died suddenly and were referred for autopsy for suspected SCD but had normal cardiac pathologic findings. Our goal was to understand the association between clinically significant variants for inherited CMs and arrhythmia syndromes and risk of unexplained SCD in adults living in the United States.
Methods
Study Design and Oversight
Between January 1995 and December 2015, CVPath Institute was referred 5262 hearts from cases of unexpected sudden death from the Office of the Chief Medical Examiner of the State of Maryland (OCME-MD). Race (ie, White or African American) was determined through the OCME-MD report through inquiry of family members. Analysis of racial differences in genetic variants associated with unexplained SCD will help researchers better understand the role of racial genetics in this phenomenon. Although there was a desire to include different racial and ethnic groups in this analysis, the number of individuals belonging to these groups with unexplained SCD was too few to make the results reliable.
For every case, the heart was systematically evaluated with detailed histopathological analyses performed by an experienced cardiac pathologist at CVPath Institute. The protocol for the study was approved by the institutional review board of the CVPath Institute. A waiver of consent was granted by the institutional review board because this used only autopsy material. This study followed the Strengthening the Reporting of Genetic Association Studies (STREGA) reporting guideline. Figure 1A presents the study flowchart, and the eMethods in the Supplement contain further details.
Figure 1. Study Flow and Variant Filtering Schematic Diagrams.
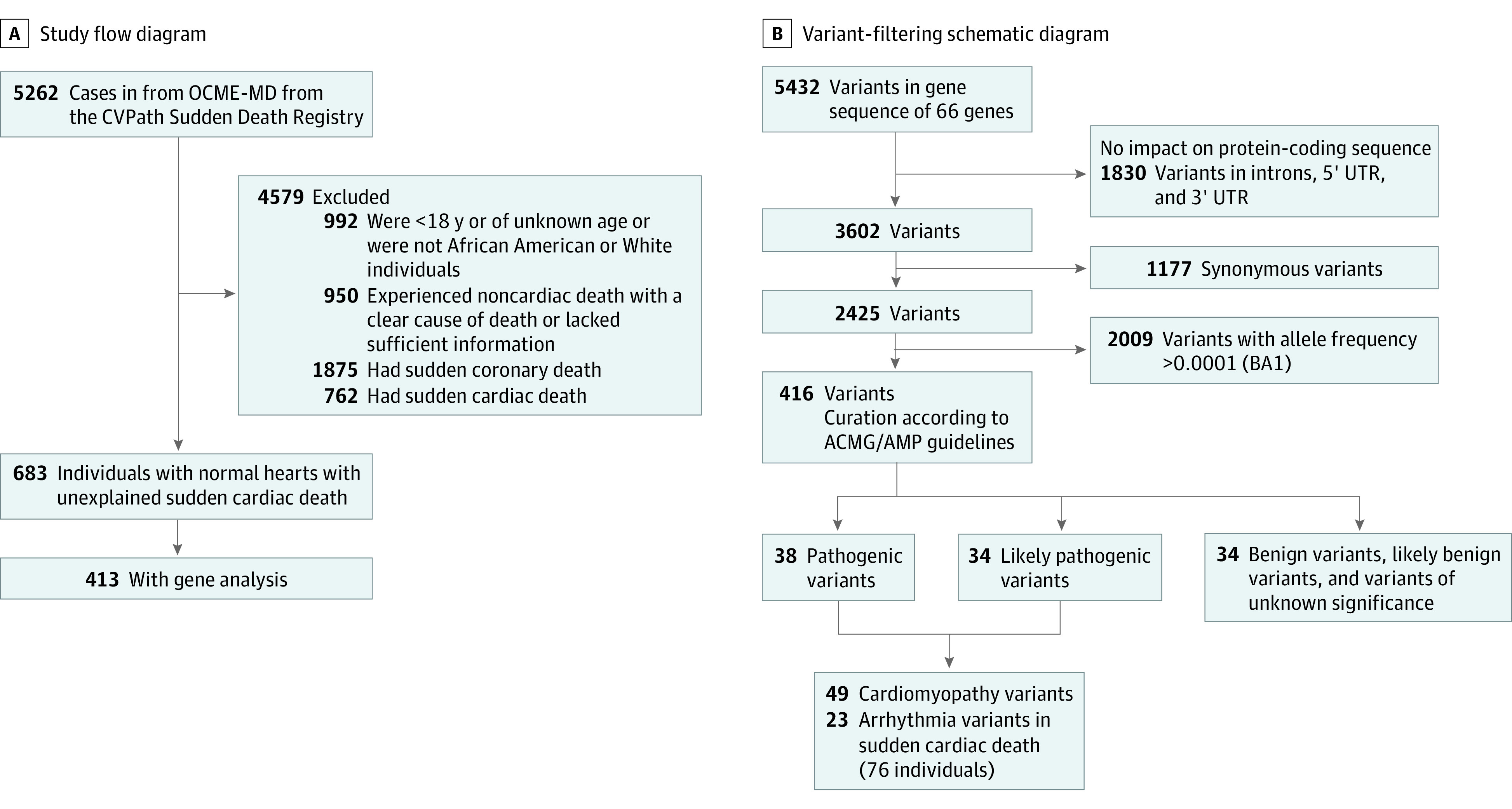
A, Noncardiac death included intracranial hemorrhage or drug overdose. Lack of sufficient information indicates missing data (eg, cause of death, heart dimensions); sudden coronary death, acute coronary syndrome, 75% or greater stenosis in any epicardial coronary arteries, or poststent implantation or bypass graft; sudden cardiac death, cardiomyopathy, valvular heart disease, congenital heart disease, myocarditis, pericarditis, or infective carditis, severe hypertrophy in myocardium, and severe fibrosis in myocardium. ACMG/AMP indicates American College of Medical Genetics and Genomics and the Association for Molecular Pathology; OCME-MD, Office of the Chief Medical Examiner of the State of Maryland; and UTR, untranslated region.
Assessment of Hearts
All specimens had been fixed in 10% buffered formalin, and any significant pathology was ruled out (eMethods in the Supplement). Normal hearts were defined as described in the eMethods in the Supplement. Evaluation for myocardial fibrosis is described in the eMethods in the Supplement, as are the details of DNA extraction, next-generation sequencing, and data analysis.
Variant Filtering and Curation
The 30 target CM genes and 38 target arrhythmia genes are shown in eTable 1 in the Supplement. Intronic variants more than 10 bases from the exon, variants in the 5′ untranslated region (UTR) and 3′UTR, and synonymous variants were filtered according to their estimated impact on protein-coding sequence. All variants with a minor allele frequency (MAF) of greater than 0.01% (according to BA1 criteria in American College of Medical Genetics and Genomics and the Association for Molecular Pathology [ACMG/AMP] guidelines) in the Exome Aggregation Consortium (ExAC), TOPMED, and GnomAD were excluded from the analysis. Overall, 416 variants were subject to curation according to their estimated risk of SCD, using the ClinGen variant curation interface.16 Variants were curated according to the ACMG/AMP guidelines,17 with minor modifications for the SCD phenotype. A genetic counselor (K.A.M.) and a clinical molecular geneticist (L.J.B.J.) experienced in variant curation reviewed the variant interpretation procedure, independently annotated a subset of variants, and provided feedback to further refine the modifications and interpretation procedure. More details of the curation criteria appear in the eMethods in the Supplement.
Statistical Analysis
Results for continuous variables with normal distribution were expressed as means with SDs. Normality of distribution was tested by the Shapiro-Wilk test. Variables with nonnormal distribution were expressed as medians with interquartile ranges (IQRs). Categorical data were expressed as numbers with percentages and analyzed by χ2 test or Fisher exact tests. The t test was used to analyze the significance of differences for continuous variables with normal distribution, whereas comparisons of variables with nonparametric distribution were performed by the Kruskal-Wallis test. For analysis of percentage dying by age range, a Fisher exact test was used. A P < .05 was considered statistically significant, and all tests were 2-tailed. JMP software version 13.0 (SAS Institute), SPSS software version 22 (IBM Corp), and Prism version 8.0 (GraphPad Software) were used for statistical analyses. Data analysis was performed from June 2018 to March 2021.
Results
Demographic Characteristics and Heart Measurements of the Study Cohort
The study design is shown in Figure 1. Of the 5262 hearts collected, 992 decedents were excluded because they did not meet study demographic criteria (569, <18 years; 423, belonged to racial/ethnic group other than African American or White). Another 950 decedents were excluded because the cause of death (CoD) was noncardiac or there was a lack of detailed information about final diagnosis or heart dimensions. An additional 2637 decedents were excluded because the CoD was a known cardiac cause, such as obstructive coronary artery disease (including, but not limited, to acute coronary syndromes), valvular disease, CM, or myocarditis. Overall, 683 decedents with normal cardiac autopsies were adjudicated to have had unexplained SCD (Figure 1A). Among these, 413 cases with acceptable DNA quality for sequencing were chosen. These decedents approximately matched the racial makeup of the original 683 decedents. The demographic and clinical characteristics of the 413 included patients are shown in eTable 1 in the Supplement. Overall, 259 patients (62.7%) were men, the median (IQR) age at death was 41 (29-48) years, with 208 (50.4%) were African American adults.
Variant Filtration and Classification
Analysis of exome sequencing data in 30 CM and 38 arrhythmia genes identified a total of 5432 variants present in the 413 study participants. Overall, 1177 variants were identified and annotated as synonymous variants, which are commonly used as negative controls in genetic studies. We used bioinformatic filtering to remove intronic variants more than 10 bases from the exon, variants in the 5′UTR and 3′UTR, synonymous variants, and variants with an allele frequency of less than 0.0001. This filtering narrowed the list to 416 variants, which were then subject to curation evaluation according to ACMG/AMP criteria.17 Among these, a total of 72 variants were classified as pathogenic (P) or likely pathogenic (LP) (Figure 1B).
Frequency of P/LP Genetic Variants for CM and Arrhythmia Disorders in the Study Cohort
The 413 decedents with suspected unexplained SCD were analyzed for 30 CM and 38 primary arrhythmia genes for P/LP variants (eTable 1 in the Supplement). In total, 76 decedents (18.4%) had variants considered P/LP for HCM and DCM or arrhythmia (Brugada syndrome, long QT syndrome [LQTS], arrhythmogenic right ventricular CM, and catecholaminergic polymorphic ventricular tachycardia). Of these, 52 decedents (12.6%) had 49 P/LP variants for CMs and 22 decedents (5.3%) carried 23 P/LP variants for arrhythmia. Two decedents (0.5%) carried variants for both conditions (Figure 2A and 2B). There were 41 P/LP variants for HCM in 45 decedents (10.9%); 9 P/LP variants for DCM in 11 decedents (2.7%); and 10 P/LP variants for LQTS in 11 decedents (2.7%). Among those carrying P/LP variants, combinations of conditions were found, including 2 (0.5%) having both HCM and LQTS variants, and 2 (0.5%) with HCM and DCM variants (Figure 2A and 2B). P/LP variants in MYBPC3 (OMIM 600958; 20 of 61 variants [32.8%]) and MYH7 (OMIM 160760; 18 [29.5%]) were the most frequent CM genes (Figure 2C), while variants in SCN5A (OMIM 600163; 6 of 24 variants [25.0%]), KCNH2 (OMIM 152427; 5 [20.8%]), and KCNQ1 (OMIM 607542; 5 [20.8%]) were the most frequently found arrhythmia genes consistent with Brugada syndrome and LQTS (Figure 2D). More than 90% of P/LP variants in MYH7, TNNT2 (OMIM 191045), TNNI3 (OMIM 191044), SCN5A, KCNQ1, and KCNH2 were missense variants. By contrast, almost all P/LP variants in MYBPC3 and DSP (OMIM 125647) (21 of 22 variants [95.5%]) were truncating variants, including nonsense and canonical splicing variants (eFigure 1 in the Supplement). The demographic characteristics, CoD, and relevant genetic information for every decedent with P/LP variants are provided in eTable 3 in the Supplement.
Figure 2. Genetic Testing for Cardiomyopathy and Arrhythmia Genes in Decedents With Unexplained Sudden Cardiac Death.

ARVC indicates arrhythmogenic right ventricular cardiomyopathy; BS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LQTS, long QT syndrome; P/LP, pathogenic or likely pathogenic.
When the MAF for benign and P/LP variants examined in the current study were compared with the general population, MAF for benign variants showed a linear association between our SCD study cohort and the general population, as expected (R2 = 0.86; P < .001) (eFigure 2A in the Supplement). In contrast, MAF for P/LP variants showed a nonlinear association between the SCD study cohort and the general population (R2 = 0.002; P = .99) and was highly enriched in the study cohort (eFigure 2B in the Supplement). These data suggest the association of these variants with unexplained SCD. Among the genes analyzed, only the MYH7 variants have been reviewed and classified by ClinGen expert panel.18 By comparing MAF of MYH7 P/LP variants in our SCD study cohort to the MAF of all 39 panel-reviewed MYH7 P/LP variants in the general population, a 105-fold enrichment was identified in our unexplained SCD study cohort compared with the general population (mean [SD] enrichment, 0.003 [0.001] vs 2.948 × 10−5 [2.139 × 10−5]) (Figure 3A), suggesting a strong association with unexplained SCD. When stratified by age range, a significantly greater percentage of individuals carrying P variants died between the ages of 18 and 30 years (16 of 40 [40.0%]) compared with similar-aged individuals not carrying P/LP (75 of 337 [22.3%]) or carrying only LP variants (8 of 36 [22.2%]) (Figure 3B). The other age ranges examined did not show any differences between groups.
Figure 3. Frequency and Characteristics of Decedents With Unexplained Sudden Cardiac Death Carrying Pathogenic or Likely Pathogenic (P/LP) Gene Variants.

HCM indicates hypertrophic cardiomyopathy; MAF, minor allele frequency.
Demographic Characteristics and Heart Dimensions in Decedents With and Without P/LP Gene Variants
No significant difference was found in demographic characteristics and heart dimensions between decedents with and without P/LP variants of either male or female sex (Table). Overall, among 76 decedents carrying P/LP variants, 32 (42.1%) were African American individuals, and 44 (57.9%) were White individuals; 29 (38.2%) were women, and 47 (61.8%) were men.
Table. Patient Characteristics in Participants With and Without P/LP Gene Variants.
| Characteristic | Median (IQR) | P value | ||||
|---|---|---|---|---|---|---|
| With P/LP variant (n = 76) | Without P/LP variant (n = 337) | |||||
| Age, y | 39.0 (28.0-48.0) | 41.2 (31.0-48.0) | .22 | |||
| Sex, No. (%) | ||||||
| Women | 29 (38.2) | 125 (37.1) | .52 | |||
| Men | 47 (61.8) | 212 (62.9) | ||||
| African American, No. (%) | 32 (42.1) | 176 (52.2) | .47 | |||
| Body/heart dimensions | Women (n = 29) | Men (n = 47) | Women (n = 125) | Men (n = 212) | P value for women | P value for men |
| Body height, cm | 165.1 (162.6-174.0) | 177.8 (170.2-180.3) | 167.6 (162.6-172.1) | 177.8 (172.7-184.8) | .82 | .37 |
| Body weight, kg | 74.4 (62.8-108.4) | 81.7 (72.1-91.2) | 80.3 (62.6-96.6) | 83.0 (71.2-97.8) | .96 | .49 |
| BMI | 27.0 (22.0-36.2) | 26.2 (23.0-29.0) | 29.0 (23.3-34.6) | 26.5 (22.6-30.2) | .98 | .70 |
| Body surface area, m2 | 1.72 (1.56-2.11) | 1.87 (1.74-1.98) | 1.80 (1.60-1.96) | 1.90 (1.73-2.07) | .87 | .48 |
| Heart weight, g | 350.0 (300.0-420.0) | 400.0 (372.0-450) | 350.0 (300.0-420.0) | 430.0 (380.0-466) | .81 | .10 |
| LV diameter, mm | 35.0 (15.0-39.5) | 30.0 (25.0-38.0) | 35.0 (28.5-40.0) | 35.0 (30.0-40.0) | .99 | .39 |
| LV free wall thickness, mm | 13.0 (12.0-15.0) | 14.0 (12.0-15.0) | 12.0 (10.0-14.0) | 14.0 (12.0-15.0) | .06 | .18 |
| Septum thickness, mm | 13.0 (12.0-15.0) | 14.0 (11.0-16.0) | 14.0 (12.0-15.0) | 15.0 (14.0-17.0) | .26 | .73 |
| RV thickness, mm | 4.0 (4.0-5.0) | 5.0 (4.0-5.5) | 4.0 (4.0-5.0) | 5.0 (4.0-5.0) | .56 | .30 |
| Septum to free wall ratio | 1.08 (1.00-1.18) | 1.09 (1.06-1.18) | 1.13 (1.10-1.20) | 1.08 (1.00-1.19) | .16 | .19 |
Abbreviations: BMI, body mass index (calculated as weight in kilograms divided by height in meters squared); IQR, interquartile range; LV, left ventricle; P/LP, pathogenic/likely pathogenic; RV, right ventricle.
Because of the high overall frequency of decedents with unexplained SCD carrying HCM P/LP variants (45 [10.9%]), we conducted a subanalysis in this group. We compared heart and body dimensions of individuals with HCM P/LP variants in the study cohort with 49 hearts with a pathologic diagnosis of HCM from the CVPath Institute registry and found significant differences in most heart measurements (eTable 2B in the Supplement). When we examined the distribution of HCM P/LP variants by septal thickness across all cases of unexplained SCD, we found an equal distribution of HCM P/LP across all septal thicknesses (Figure 3C). Sections of left ventricular myocardium from 21 individuals with P/LP HCM gene variants and 22 consecutive individuals without P/LP gene variants (median [IQR] age, 39 [25-47] years vs 31 [22-44] years; P = .19) were evaluated for evidence of myocardial fibrosis. The median (IQR) percentage area myocardial fibrosis in participants with and without P/LP HCM gene variants was similar (1.02% [0.76%-1.55%] vs 0.93% [0.70%-1.40%); P = .52) (Figure 3D; eTable 4 in the Supplement). Thus, in participants with unexplained SCD, there was no association between heart dimensions, myocardial fibrosis, and the presence of P/LP HCM variants.
Racial Differences in P/LP Gene Variants Between African American and White Individuals
African American individuals carried a similar number of P/LP gene variants compared with White individuals (32 of 208 [15.4%] vs 44 of 205 [21.5%]; P = .14). Full demographic details of African American and White decedents are shown in eTable 5 in the Supplement. P/LP gene variant carriers by race showed a significantly higher prevalence of CM variants in White individuals compared with African American individuals (39 of 205 [19.0%] vs 22 of 208 [10.6%]; P = .02) (Figure 4A). The largest numbers of P/LP variants in CM genes were in MYBPC3 and MYH7 in both groups. The total numbers of P/LP gene variant carriers for arrhythmia genes were not different between African American and White decedents; however, the most frequent P/LP variants were different: KCNQ1 and RYR2 were predominant in African American individuals, while KCNH2 and DSP were more prevalent in White individuals, with SCN5A being similar between the 2 races (Figure 4B).
Figure 4. Differences in Pathogenic or Likely Pathogenic (P/LP) Gene Variants Between African American and White Individuals.

Further variant analyses of racial distribution showed that there was a tendency for P/LP variants to be more prevalent in 1 race (African American vs White, CM genes: 16 of 61 [26.2%] vs 34 of 61 [55.7%]; arrhythmia genes: 14 of 24 [58.3%] vs 10 of 24 [41.7%]). Only 11 of 61 P/LP variants (18.0%) were found in both races for CM (eFigures 3A and 3B in the Supplement). The MAF for benign variants in African American vs White individuals was linear (R2 = 0.6607; P < .001) (eFigure 3C in the Supplement). There was no difference in prevalence for variants with unknown significance (VUS) (R2 = 0.0018; P = .12) (eFigure 3D in the Supplement). We identified 2 VUS (1 in MYH7 and 1 in JUP [OMIM 173325]) only in African American individuals, for which we found multiple carriers, but none or only a few reported in the general population, suggesting they could be associated with risk of unexplained SCD in African American individuals (eTable 6 in the Supplement).
Age at death was examined in the CVPath Registry, in both the excluded group of autopsy-determined SCD (sudden coronary death and sudden cardiac death) as well as in the unexplained SCD group. Overall, the percentage of individuals dying of SCD aged 18 to 30 years was significantly greater for African American than for White individuals, while the opposite was seen in individuals older than 60 years (eFigure 4 in the Supplement). The percentage of individuals dying aged 18 to 30 years was greater in both White and African American decedents with unexplained SCD compared with their counterparts dying of SCD, while the opposite tendency was seen in later age ranges (ie, 51-60 and >60 years) (eFigure 4 in the Supplement). Overall, there was no differences in age at death between White and African American individuals carrying P/LP variants (Figure 4C).
Discussion
The current study examined the association of CM and arrhythmia genetic variants with unexplained SCD and assessed whether race-based differences in these genetic variants exist. To our knowledge, this is the largest and most comprehensive genetic autopsy study of unexplained SCD ever to be performed in the United States. In our autopsy registry of more than 5000 cases of suspected SCD referred from OCME-MD, we identified 683 hearts from White and African American decedents with unexplained SCD. Genetic analyses of 30 CM and 38 primary arrhythmia genes were performed in 413 decedents and showed that slightly less than one-fifth of individuals dying of unexplained SCD carried P/LP variants for CM or arrhythmia genes, representing significantly greater enrichment in the frequency of these variants vs the general population. Although our results were not combined with screening of surviving family members, largely because such practices were not considered routine during most of the 20 years these data were collected. Nonetheless, our data suggest that genetic causes may contribute to a greater number of unexplained SCD cases in adults than previously appreciated. One surprising finding was the large number of decedents with unexplained SCD carrying genetic variants for HCM (10.9%), which suggests an association of these genes with risk of SCD in the absence of structural heart disease. We found that 96% of P/LP variants in MYBPC3 and DSP were truncating variants, including nonsense and canonical splicing variants, which is consistent with previous reports describing the type of variants seen in these disorders.19,20 In addition, our study is the first to examine the frequency of P/LP variants in African American and White decedents of unexplained SCD. Overall, African American individuals had a similar frequency of arrhythmia P/LP gene variants vs White individuals but carried fewer P/LP variants for CM genes. Although individuals dying of unexplained SCD had an earlier risk of death (regardless of race) than those dying of known causes of SCD (eg, coronary artery disease, CM), there was no difference in survival among carriers of P/LP variants according to race.
Autopsy studies have demonstrated the utility of genetic analyses in decedents with unexplained SCD, yielding potential diagnoses in as many as one-third of young SCD cases. Although the definition of P/LP variants was different in studies published before the ACMG/AMP guidelines were released, they all demonstrated the well-known association between arrhythmia gene variants and unexplained SCD8,21,22 and between CM variants and unexplained SCD.7,8
Of the CM genes studied, most variants detected were considered P/LP for HCM. Furthermore, the finding of 2 individuals (0.5%) with both HCM and LQTS variants and 2 (0.5%) with HCM and DCM variants has been reported in other unexplained SCD studies.8 Although HCM is a common genetic heart disorder and a well-recognized cause of SCD in those with overt heart disease, the role of genetics in risk prediction remains uncertain in those without clinically detectable disease.5,6 The prevailing clinical perception is that risk of sudden death in HCM is related to the presence of ventricular hypertrophy. However, case reports have documented the occurrence of SCD in those carrying genetic HCM variants without overt clinical disease.23 There is currently disagreement among consensus expert panels about the significance of HCM P/LP variants in those without overt cardiac disease.5,6 Studies of single families with specific variants are limited by numbers, length of follow-up, and the possibility that shared genetic and as-yet unknown environmental modifiers may affect overall risk. Indeed, large-scale studies of SCD in individuals with genotype-positive, phenotype-negative individuals have not been performed. Further work is needed to understand whether carrying certain CM variants without overt clinical disease increases risk for unexplained SCD.
The inclusion of individuals who belong to an underrepresented race, ie, African American adults, is another unique aspect of our study. Overall, African American decedents with unexplained SCD harbored a similar frequency of P/LP variants as White decedents, although White decedents were more likely to carry P/LP variants for CM genes and African American decedents had a higher frequency of VUS. While the most frequent P/LP variants in CM genes were MYBPC3 and MYH7 for both races, the most frequent P/LP variants in arrhythmia genes were different: KCNQ1 and RYR2 in African American individuals and KCNH2 in White individuals, with SCN5A being equally frequent in both groups. Moreover, the prevalence of VUS was higher in African American individuals than White individuals, which is consistent with the previous study.24 Although African American decedents with SCD tended to die at an earlier age than White decedents in our registry, supporting previous clinical observations that African American individuals have higher risk of SCD at an earlier age,25 no overall differences in survival by race in carriers of P/LP variants were found.
Limitations
Our study has several limitations. First, due to the nature of unexplained SCD, which mainly occurs in the community, detailed clinical and family history were often lacking. Thus, the data collected relied only on the association of the presence of a variant and the occurrence of death in a single individual, rather than within a familial cohort. Second, our study provides evidence for association only, although we did curate variants according to the ACMG/AMP guidelines to assign evidence of pathogenicity. Third, the study findings may not represent the frequency of genetic variants in decedents from SCD in the community. Fourth, genetic ancestry was defined by medical examiners in this study. However, a full genetic assessment of participants’ ancestry by genotyping would have further strengthened the race stratification performed in this study. Nonetheless, for all genes examined, the combined frequency of P/LP variants was orders of magnitude higher in SCD cases of both groups compared with the general population (eTable 7 in the Supplement), suggesting the results are applicable to both White and Black individuals. Lastly, the sequencing panels used in this study did not include CM genes TTN and ALPK3. Given that TTN truncating variants are a common cause of DCM, future genetic sequencing studies including these genes would be helpful in understanding their role in SCD. It should also be noted that the median age of SCD victims in this study was 41, which attests to the importance of understanding this entity better so that we can begin to improve mortality rates from unexplained SCD.
Conclusions
This study is among the largest examinations of the association of causative CM and arrhythmia genetic variants with unexplained SCD. Overall, nearly 20% of those with unexplained SCD were carriers of P/LP variants. Our findings regarding both the association of unexplained SCD with CM genes and race-specific genetic variants suggest new avenues of study for this poorly understood entity.
eMethods. Supplementary Methods
eFigure 1. Types of P/LP Variants in Arrhythmia and Cardiomyopathy Genes in Individuals With Unexplained SCD
eFigure 2. Comparison of Allele Frequency in Cardiomyopathy and Arrhythmia Gene Variants in Individuals With Unexplained SCD vs the General Population
eFigure 3. Racial Differences in P/LP, Benign and Variants of Unknown Significance (VUS) in Arrhythmia and Cardiomyopathy Genes in Unexplained SCD
eFigure 4. Racial Differences in Survival SCD Between African American and White Individuals
eTable 1. List of Genes in the Cardiomyopathy Panel and Arrhythmia Panel
eTable 2. Patient Characteristics of Participants With Unexplained SCD and HCM
eTable 3. Participants With Pathogenic or Likely Pathogenic Variants
eTable 4. Patient Characteristics and Percentage Area Myocardial Fibrosis in Unexplained Sudden Cardiac Death cases with P/LP in HCM Genes and Detailed Septal Evaluations
eTable 5. Patient Characteristics in African American and White Individuals With and Without P/LP Gene Variants
eTable 6. Variants With Unknown Significance in African American Individuals Only, With Suspicion for a Pathogenic Role in Unexplained SCD
eTable 7. Allele Frequencies of P/LP Variants in Unexplained SCD Cohort and in gnomAD
eReferences.
References
- 1.Benjamin EJ, Virani SS, Callaway CW, et al. ; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018;137(12):e67-e492. doi: 10.1161/CIR.0000000000000558 [DOI] [PubMed] [Google Scholar]
- 2.Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125(4):620-637. doi: 10.1161/CIRCULATIONAHA.111.023838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckart RE, Shry EA, Burke AP, et al. ; Department of Defense Cardiovascular Death Registry Group . Sudden death in young adults: an autopsy-based series of a population undergoing active surveillance. J Am Coll Cardiol. 2011;58(12):1254-1261. doi: 10.1016/j.jacc.2011.01.049 [DOI] [PubMed] [Google Scholar]
- 4.Wever EF, Robles de Medina EO. Sudden death in patients without structural heart disease. J Am Coll Cardiol. 2004;43(7):1137-1144. doi: 10.1016/j.jacc.2003.10.053 [DOI] [PubMed] [Google Scholar]
- 5.Elliott PM, Anastasakis A, Borger MA, et al. ; Authors/Task Force members . 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733-2779. doi: 10.1093/eurheartj/ehu284 [DOI] [PubMed] [Google Scholar]
- 6.Gersh BJ, Maron BJ, Bonow RO, et al. ; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons . 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124(24):2761-2796. doi: 10.1161/CIR.0b013e318223e230 [DOI] [PubMed] [Google Scholar]
- 7.Bagnall RD, Weintraub RG, Ingles J, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. 2016;374(25):2441-2452. doi: 10.1056/NEJMoa1510687 [DOI] [PubMed] [Google Scholar]
- 8.Lahrouchi N, Raju H, Lodder EM, et al. Utility of post-mortem genetic testing in cases of sudden arrhythmic death syndrome. J Am Coll Cardiol. 2017;69(17):2134-2145. doi: 10.1016/j.jacc.2017.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez O, Campuzano O, Fernández-Falgueras A, et al. Natural and undetermined sudden death: value of post-mortem genetic investigation. PLoS One. 2016;11(12):e0167358. doi: 10.1371/journal.pone.0167358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christiansen SL, Hertz CL, Ferrero-Miliani L, et al. Genetic investigation of 100 heart genes in sudden unexplained death victims in a forensic setting. Eur J Hum Genet. 2016;24(12):1797-1802. doi: 10.1038/ejhg.2016.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nunn LM, Lopes LR, Syrris P, et al. Diagnostic yield of molecular autopsy in patients with sudden arrhythmic death syndrome using targeted exome sequencing. Europace. 2016;18(6):888-896. doi: 10.1093/europace/euv285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song JS, Kang JS, Kim YE, et al. Identification of pathogenic variants in genes related to channelopathy and cardiomyopathy in Korean sudden cardiac arrest survivors. J Hum Genet. 2017;62(6):615-620. doi: 10.1038/jhg.2017.8 [DOI] [PubMed] [Google Scholar]
- 13.Mellor G, Raju H, de Noronha SV, et al. Clinical characteristics and circumstances of death in the sudden arrhythmic death syndrome. Circ Arrhythm Electrophysiol. 2014;7(6):1078-1083. doi: 10.1161/CIRCEP.114.001854 [DOI] [PubMed] [Google Scholar]
- 14.Lin Y, Williams N, Wang D, et al. Applying high-resolution variant classification to cardiac arrhythmogenic gene testing in a demographically diverse cohort of sudden unexplained deaths. Circ Cardiovasc Genet. 2017;10(6):e001839. doi: 10.1161/CIRCGENETICS.117.001839 [DOI] [PubMed] [Google Scholar]
- 15.Kannel WB, Wilson PW, D’Agostino RB, Cobb J. Sudden coronary death in women. Am Heart J. 1998;136(2):205-212. doi: 10.1053/hj.1998.v136.90226 [DOI] [PubMed] [Google Scholar]
- 16.ClinGen . ClinGen variant and gene curation. Accessed April 27, 2021. https://curation.clinicalgenome.org
- 17.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly MA, Caleshu C, Morales A, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018;20(3):351-359. doi: 10.1038/gim.2017.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viswanathan SK, Sanders HK, McNamara JW, et al. Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS One. 2017;12(11):e0187948. doi: 10.1371/journal.pone.0187948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh R, Thomson KL, Ware JS, et al. ; Exome Aggregation Consortium . Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19(2):192-203. doi: 10.1038/gim.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcondes L, Crawford J, Earle N, et al. ; Cardiac Inherited Disease Group New Zealand . Long QT molecular autopsy in sudden unexplained death in the young (1-40 years old): lessons learnt from an eight year experience in New Zealand. PLoS One. 2018;13(4):e0196078. doi: 10.1371/journal.pone.0196078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anastasakis A, Papatheodorou E, Ritsatos K, et al. Sudden unexplained death in the young: epidemiology, aetiology and value of the clinically guided genetic screening. Europace. 2018;20(3):472-480. doi: 10.1093/europace/euw362 [DOI] [PubMed] [Google Scholar]
- 23.Christiaans I, Lekanne dit Deprez RH, van Langen IM, Wilde AA. Ventricular fibrillation in MYH7-related hypertrophic cardiomyopathy before onset of ventricular hypertrophy. Heart Rhythm. 2009;6(9):1366-1369. doi: 10.1016/j.hrthm.2009.04.029 [DOI] [PubMed] [Google Scholar]
- 24.Manrai AK, Funke BH, Rehm HL, et al. Genetic misdiagnoses and the potential for health disparities. N Engl J Med. 2016;375(7):655-665. doi: 10.1056/NEJMsa1507092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao D, Post WS, Blasco-Colmenares E, et al. Racial differences in sudden cardiac death. Circulation. 2019;139(14):1688-1697. doi: 10.1161/CIRCULATIONAHA.118.036553 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods. Supplementary Methods
eFigure 1. Types of P/LP Variants in Arrhythmia and Cardiomyopathy Genes in Individuals With Unexplained SCD
eFigure 2. Comparison of Allele Frequency in Cardiomyopathy and Arrhythmia Gene Variants in Individuals With Unexplained SCD vs the General Population
eFigure 3. Racial Differences in P/LP, Benign and Variants of Unknown Significance (VUS) in Arrhythmia and Cardiomyopathy Genes in Unexplained SCD
eFigure 4. Racial Differences in Survival SCD Between African American and White Individuals
eTable 1. List of Genes in the Cardiomyopathy Panel and Arrhythmia Panel
eTable 2. Patient Characteristics of Participants With Unexplained SCD and HCM
eTable 3. Participants With Pathogenic or Likely Pathogenic Variants
eTable 4. Patient Characteristics and Percentage Area Myocardial Fibrosis in Unexplained Sudden Cardiac Death cases with P/LP in HCM Genes and Detailed Septal Evaluations
eTable 5. Patient Characteristics in African American and White Individuals With and Without P/LP Gene Variants
eTable 6. Variants With Unknown Significance in African American Individuals Only, With Suspicion for a Pathogenic Role in Unexplained SCD
eTable 7. Allele Frequencies of P/LP Variants in Unexplained SCD Cohort and in gnomAD
eReferences.