

Abstract
Mycoplasma gallisepticum causes chronic respiratory disease in chickens leading to large economic losses in the poultry industry, and the impacts remain to be a great challenge for a longer period. Among the other approaches, a vaccine targeting the adhesion proteins of M. gallisepticum would be a promising candidate in controlling the infection. Thus, the present study is aimed to design a multi-epitope vaccine candidate using cytoadhesion proteins of M. gallisepticum through an advanced immunoinformatics approach. As a result, the multi-epitope vaccine was constructed, which comprised potential T-cell and B-cell binding epitopes with appropriate adjuvants. The designed multi-epitope vaccine represented high antigenicity with viable physiochemical properties. The prospective three-dimensional structure of the epitope was predicted, refined, and validated. The molecular docking analysis of multi-epitope vaccine candidates with the chicken Toll-like receptor-5 predicted effective binding. Furthermore, codon optimization and in silico cloning ensured high expression. Thus, the present finding indicates that the engineered multi-epitope vaccine is structurally stable and can induce a strong immune response. Furthermore, the multi-epitope vaccine is suggested to be a suitable vaccine candidate for the M. gallisepticum infection due to its effective binding capacity and precise specificity.
Introduction
Mycoplasma gallisepticum is an avian pathogen causing chronic respiratory disease in chickens1−4 and infectious sinusitis in turkeys.5M. gallisepticum are cell wall-less bacteria belonging to the family Mycoplasmataceae and class Mollicutes(6) and are usually characterized by small cells and a genome with an absence of a peptidoglycan layer.7 The remarkable characteristic of M. gallisepticum is colonization of host cells by cytoadhesion, followed by cilial shedding and inflammation.8 Initial adhesion of M. gallisepticum to the host cells is a crucial step in infection and pathogenesis and along with a family of variable lipoproteins (vlhA) it plays an important role in pathogenesis by immune evasion.9 The M. gallisepticum cytoadherence is a multistep progression involving the primary cytoadherence proteins GapA and CrmA10 and cytoadherence accessory proteins Hlp3 and PlpA.11 The cytoadherence proteins and cytoadherence accessory proteins are virulence factors in M. gallisepticum infection, and prophylactic strategies targeting these proteins may be an important preventive measure against M. gallisepticum infection.
Avian mycoplasmosis has caused a substantial economic loss globally by significantly reducing egg production, meat quality, and hatchability rate.12 Control of M. gallisepticum by antibiotics is practiced in most poultry farms, and the widely used antibiotics are tetracyclines, macrolides (tylosin and tilmicosin), fluoroquinolones (enrofloxacin and difloxacin), and pleuromutilins (tiamulin).13,14 The widespread use of antibiotics has led to the rise and spread of resistant bacteria over time.15 The quinolone resistance in M. gallisepticum has become common, which hinders treatment and control efforts.16 The application of vaccinology might be a potential approach in the prevention and control of M. gallisepticum infections in layer and breeder flocks. The currently available vaccines are live-attenuated vaccines, live vaccines, and bacterins.17 Live-attenuated vaccines such as the F-strain, ts-11 strain, and 6/85 strain and bacterins are widely used, but a sudden onset of M. gallisepticum infections can pose a challenge for the suppression of infection.18,19 Live-attenuated vaccines may get reverted back and cause side effects, whereas bacterins are highly expensive and involve repetitive doses. Thus, the development of highly efficient vaccines at low cost is of immense importance, which can be effectively achieved through new recombinant strategies. Reports on in silico approaches for the vaccine development for mycoplasma species are not observed, except the identification of some virulence genes.20 Since a large population of birds is to be administered concurrently, effortless administration with a least amount of labor is a vital requisite to consider the vaccine desirable for poultry.21 To fix the current state of mycoplasma infections in chickens, an effective vaccine is therefore necessary.
In both the preclinical and clinical phases, the development of a vaccine candidate is expensive, complex, and time-consuming, requiring in vitro and in vivo protocols for assessing efficacy. The computational methods have facilitated the in silico evaluations for an intended vaccine candidate with the advancement of bioinformatics, significantly reducing the needed time for preclinical and clinical procedures.22,23 The recent advancement in the field of bioinformatics and immunoinformatics had led to the efficient screening and validation of proteins, which can be a potential vaccine target.24 Earlier studies reported the use of in silico assays to investigate the efficiency of the designed vaccine for human diseases.25 The advantage of this approach is the identification of a larger range of vaccine candidates without the need to propagate the pathogenic microorganism in a laboratory while delivering a multivalent, highly purified, and sustainable alternative to whole-cell vaccines.26 Furthermore, the molecular docking approach predicts the binding domains and molecular interactions between the receptor and vaccine candidate.27,28 To date, a very few in silico studies were carried out to design a vaccine in veterinary diseases and no studies have been reported for M. gallisepticum.29,30 The major limitations of the in silico approach is that it cannot predict polysaccharides or lipids which may be active components in proteins,31 the requirement to identify suitable antigen-processing sites for the predicted epitopes which were not addressed by many epitope prediction tools.32 In this study, we used robust immunoinformatics tools to predict MHC-I and MHC-II binding T-cell-specific epitopes and B-cell epitopes from four M. gallisepticum antigenic proteins and developed a base to design a candidate for a multi-epitope-based vaccine against M. gallisepticum infection in chickens (Figure 1).
Figure 1.

Flow chart summarizing the designed vaccine. Several phases are involved in this approach: potential epitope prediction from the antigenic proteins and construction, evaluation, and validation of the multi-epitope vaccine construct; disulfide engineering and molecular docking with chicken immune receptors; and finally, codon adaptation and in silico cloning.
Results and Discussion
Vaccines are critically responsible for providing host organisms with protection from a specific disease, helping to save lives worldwide. The process for the production of vaccines is typically highly painstaking and costly and takes a long period of time for their achievement. The design of vaccines can be accomplished by using different immunoinformatics methods in less time. They include the detection of a pathogen’s latent antigenic proteins, accompanied by the determination of different immune-dominant epitopes that are responsible for the development of immune responses against the pathogen, both humoral and cell-mediated. Therefore, multi-epitope-based vaccines can be engineered for the probable antigenic proteins of a pathogen by identifying the B- and T-cell epitope regions. Currently, numerous peptide vaccines are under development, and most of them are for human diseases.33 Limited research studies have been carried out in the field of in silico vaccines for poultry and other animals. Among those, few studies focused on the prediction of epitopes for animal diseases such as foot and mouth disease33 and animal trypanosomiasis34 in designing the multi-epitope vaccine, which offered effective immunity when compared with the available vaccines. The immunoinformatics approach is a valid method for designing multi-epitope vaccines against infectious diseases in poultry.35−38 The immunoinformatics analysis and evaluation in the present study show that the vaccine construct has all the factors within the most favorable range for it to be considered as a potential vaccine candidate, which further needs to be experimentally validated.
Antigenicity and Allergenicity Prediction
Initially, the GapA, PlpA, Hlp3, and CrmA proteins were subjected to antigenicity and allergenicity prediction. Verifying the antigenic and allergic nature of the selected proteins is crucial in the selection of epitopes for the further study.39 The VaxiJen v 2.0 server was used for the evaluation of the antigenicity of the proteins. The threshold score for antigenicity is 0.4. All protein sequences selected are nonallergens that are predicted using AllerTop v.2.0.0. Table 1 summarizes both the proteins’ antigenic score and allergenicity.
Table 1. Antigenicity and Allergenicity Prediction of M. gallisepticum Cytoadherence Proteins.
| s. no. | protein | antigenicity score | allergen |
|---|---|---|---|
| 1 | GapA | 0.5284 | nonallergen |
| 2 | PlpA | 0.5424 | nonallergen |
| 3 | Hlp3 | 0.5010 | nonallergen |
| 4 | CrmA | 0.5579 | nonallergen |
T-Cell Epitope Prediction
Due to the lack of chicken MHC alleles, human MHC alleles were used to predict the epitopes. Earlier studies used human alleles because of the lack of chicken alleles in immunoinformatics online tools.30,40,41 Studies have shown that chicken B–F alleles can stimulate an immune response similar to human class I homologous alleles, particularly in antigen presentation.42,43 Due to the lack of chicken MHC-I and MHC-II alleles in the T-cell epitope prediction database, the closest similar human alleles (HLA*B 40:06, HLA*B 41:04, and HLA*B 41:03 for MHC I and DRB1:1482, DRB1:1366, DRB1:1310, and DRB1:1445 for MHC II) were used. The NetMHCcons 1.1 server predicted MHC-I binding epitopes. The epitopes with % rank <0.50 were preferred as strong binders for MHC I (Tables 2 and S1). The NetMHCpan server predicted MHC-II binding epitopes. The epitopes with % rank <2 were preferred as strong binders for MHC II (Tables 3 and S2).
Table 2. List of MHC-I Binding Epitopes Predicted by the NetMHCcons 1.1 Server with Threshold: Threshold for Strong Binding Peptide 0.50% (% Rank)a.
| s. no. | protein | position | allele | peptide | 1-log50k(aff)* | affinity (nM)# | % rank | class I immunogenicity score |
|---|---|---|---|---|---|---|---|---|
| 1. | GapA | 984 | HLA-B40:06 | QEFTGFDAL | 0.417 | 549.34 | 0.50 | 0.26184 |
| HLA-B41:04 | 0.582 | 92.19 | 0.17 | |||||
| HLA-B41:03 | 0.666 | 37.11 | 0.12 | |||||
| 2. | PlpA | 817 | HLA-B40:06 | YEYPEYEEI | 0.427 | 492.48 | 0.40 | 0.22466 |
| HLA-B41:03 | 0.601 | 74.57 | 0.50 | |||||
| 3. | Hlp3 | 907 | HLA-B41:03 | QELLRYNVI | 0.602 | 74.16 | 0.50 | 0.05082 |
| HLA-B41:04 | 0.588 | 86.16 | 0.17 | |||||
| 4. | CrmA | 769 | HLA-B40:06 | NEIGVILPL | 0.518 | 184.51 | 0.12 | 0.22694 |
| HLA-B41:03 | 0.693 | 27.86 | 0.05 | |||||
| HLA-B41:04 | 0.594 | 80.45 | 0.15 |
*Prediction score, #affinity as the IC50 value in nanometer.
Table 3. List of MHC-II Binding Epitopes Predicted by the NetMHCpan Servera.
| s. no. | protein | position | allele | peptide | core peptide | 1-log50k(aff)* | binding affinity (nM)# | % rank |
|---|---|---|---|---|---|---|---|---|
| 1 | GapA | 941 | DRB1_1482 | IRLRLLVIDRSRATN | LLVIDRSRA | 0.660 | 39.48 | 0.09 |
| DRB1_1445 | 0.560 | 117.07 | 0.12 | |||||
| DRB1_1366 | 0.780 | 10.84 | 0.40 | |||||
| DRB1_1310 | 0.731 | 18.29 | 0.70 | |||||
| 2 | PlpA | 666 | DRB1_1445 | KTFSLNKGLNKVIIR | LNKGLNKVI | 0.467 | 320.42 | 1.80 |
| DRB1_1310 | 0.700 | 25.58 | 1.60 | |||||
| 3 | Hlp3 | 5 | DRB1_1482 | KIHNKILKNLAKLKK | ILKNLAKLK | 0.601 | 75.28 | 0.60 |
| DRB1_1445 | 0.523 | 173.55 | 0.40 | |||||
| DRB1_1366 | 0.782 | 10.61 | 0.40 | |||||
| DRB1_1310 | 0.728 | 19.06 | 0.80 | |||||
| 4 | CrmA | 356 | DRB1_1445 | FSRLYLNSVNSLSFI | LYLNSVNSL | 0.477 | 286.75 | 1.40 |
| DRB1_1366 | 0.743 | 16.09 | 1.40 | |||||
| DRB1_1310 | 0.730 | 18.57 | 0.70 |
Threshold for strong binders (% rank): 2%. * Predicted binding affinity in a log scale, #affinity as the IC50 value in nanometer.
B-Cell Epitope Prediction
An important step in the vaccine design includes the prediction and identification of B-cell epitopes.39 The B-cell epitopes in the cytoadherence proteins were predicted using the BepiPred server. The threshold was 0.350, and the peptides above this threshold are considered as potential B-cell epitopes (Figure 2). The epitopes from all the selected proteins with length varying from 10 to 15 peptides are listed in Table 4.
Figure 2.

BepiPred B-cell epitope prediction of (a) GapA, (b) PlpA, (c) Hlp3, and (d) CrmA. The yellow color in the graph denotes the epitopic regions in the protein sequence.
Table 4. Predicted Linear B-Cell Epitopes from M. gallisepticum Cytoadherence Proteins.
| s. no. | protein | epitope | length |
|---|---|---|---|
| 1 | GapA | FDPGNTNDLT | 10 |
| VVEATQDQEDP | 11 | ||
| AVQQEQKTKDQ | 11 | ||
| GGVSSPRGAN | 10 | ||
| PAVIEDAPTT | 10 | ||
| WSLGTRKDSAWL | 12 | ||
| GAITTWPEVQVNYK | 14 | ||
| KRLETQTTTPLID | 13 | ||
| FSPYEHPEWYED | 12 | ||
| LSSTGDQQGWY | 11 | ||
| SFTPSSQGYTWQ | 12 | ||
| KAGYSIRPDDDTVF | 14 | ||
| RVSPDSSALA | 10 | ||
| TTEADGKEVL | 10 | ||
| RYTPPQNNPD | 10 | ||
| 2 | PlpA | YDANGNPVSDPSLA | 13 |
| TVNQPDQTPITPHLE | 14 | ||
| AVHHDAEDDV | 10 | ||
| TKEANDSLQNRVKE | 14 | ||
| 3 | Hlp3 | EQPVENPQVQETK | 13 |
| PIEVNQPVQQ | 10 | ||
| KEEAKKSNSN | 10 | ||
| TAALNKPEPSTVEL | 14 | ||
| EKVVQQPKEVVA | 12 | ||
| QFVPPQSLNQVETP | 14 | ||
| SLAPRGYNQQPRVRR | 15 | ||
| 4 | CrmA | DYTTSRNRFDQRQT | 14 |
| TNSNRIGNRNNN | 12 | ||
| DDGTKFNFTKQTQGE | 15 | ||
| PEWTGSEENKN | 11 | ||
| PGTPQVTLKE | 10 | ||
| YNGDQRPTGNF | 11 | ||
| LTEEGARSFSNT | 12 | ||
| YIRAQGDTPESRSI | 14 | ||
| NSNRPNPNGL | 10 | ||
| TSGLPNQQPFGT | 12 | ||
| DSSNPDASSSFS | 12 | ||
| DSSNPDASSSFS | 12 | ||
| SSNPGSPGSYTAV | 13 | ||
| FEGSGAKYTSD | 11 | ||
| VVDPDGNLTN | 10 |
Multi-epitope Vaccine Design
The initiation of adaptive immunity largely depends on the regulation of T cells in poultry.44 Avian β-defensin, an adjuvant, was added at the N-terminal to enhance the efficacy of the vaccine, followed by a sequence of T-cell and B-cell epitopes present on the cytoadhesion and cytoadhesion accessory proteins, viz., GapA, PlpA, Hlp3, and CrmA (Figure 3).45 These epitopes along with the adjuvant have been merged together with EAAAK, which enhances the bifunctional catalytic activity and firmness of the vaccine construct.46 The AAY and GPGPG linkers were inserted between the T-cell and B-cell epitopes, respectively, which allows dissociation and identification of individual epitopes.47,48
Figure 3.

Schematic representation of the final multi-epitope vaccine construct. The 196-amino acid long peptide containing the avian β-defensin adjuvant at the N-terminal followed T-cell epitopes and B-cell epitopes with EAAAK, AAY, and GPGPG linkers, respectively.
mRNA Structure Prediction
Prediction of the mRNA secondary structure is essential, which is a key factor in translation initiation, elongation, and mRNA biogenesis.49,50 Free energy related to the whole mRNA structure was obtained using Mfold online service. The minimum free energy of the secondary RNA structure was ΔG = −156.50 kcal/mol (Figure 4). This score indicates the stability and translation efficiency of protein in hosts. The increase in mRNA stability is directly related to the increases in the expression rate.
Figure 4.

(A) Prediction of the RNA secondary structure of the multi-epitope vaccine construct gene by the Mfold server. (B) Circle graph structure, which displays the base pairs of the structure.
Antigenicity, Allergenicity, and Solubility Profile Prediction of the Multi-epitope Vaccine
A creditable vaccine candidate should be capable of initiating an immune response without causing allergic reactions, thus the antigenicity and allergenicity of the vaccine construct were predicted.39,41 The main criteria that have to be ensured while designing a vaccine is the antigenicity to induce a humoral and/or cell-mediated immune response against the targeted microorganism and allergenicity of the constructed vaccine. The antigenic scores for the multi-epitope vaccine are 0.5669 and 0.5532 as predicted by VaxiJen v2.0 and ANTIGENpro servers, respectively. The vaccine was found to be nonallergenic using AllerTOP v2.0. The predicted solubility upon over expression by the SOLpro server showed the vaccine construct as soluble with probability 0.636137. The designed vaccine construct is antigenic and non-allergenic and can be endorsed as a suitable candidate.
Physiochemical Characterization of the Designed Vaccine
The assessment of physiochemical properties of the designed multi-epitope vaccine is essential for validating the safety and efficacy of the vaccine.51 Thus, various physical and chemical properties were analyzed. The designed multi-epitope vaccine is composed of 196 amino acids with a molecular weight of∼ 21.40 kDa. The theoretical pI was calculated as 9.41, which indicate that the multi-epitope vaccine is significantly basic in nature. The instability index was estimated to be 35.78, implying a stable protein. The computed aliphatic index and grand average of hydropathicity (GRAVY) were found to be 92.14 and −0.094, respectively, signifying that the vaccine is thermostable and hydrophilic (Table 5).
Table 5. Physiochemical Property Assessment of the Primary Sequence of the Multi-epitope Vaccine Construct.
| parameters | result |
|---|---|
| number of amino acids | 196 |
| molecular weight | 21.40 kDa |
| theoretical pI | 9.41 |
| positively charged residues (Asp + Glu) | 12 |
| negatively charged residues (Arg + Lys) | 22 |
| extinction coefficient (at 280 nm in H2O) | 16,765 |
| estimated half-life (mammalian reticulocytes, in vitro) | 30 h |
| estimated half-life (yeast cells, in vivo) | >20 h |
| Estimated half-life (Escherichia coli, in vivo) | >10 h |
| instability index | 35.78 |
| aliphatic index | 92.14 |
| grand average of hydropathicity (GRAVY) | –0.094 |
Secondary and Tertiary Structure Prediction
Evaluating the secondary structure of the multi-epitope vaccine revealed that the percentage of random coils [57.14% (112 aa)] was higher than those of α-helices [20.92% (41 aa)] and β-sheets [21.94% (43 aa)]. Figure 5 shows the graphical descriptions of the GOR IV server and the performance of the secondary structure from the PSIPRED server. I-TASSER servers have been used to model the tertiary structure of the vaccine. Based on 10 threading templates, the I-TASSER server predicted five tertiary structure models of the designed chimeric protein. All the 10 templates chosen showed good alignment according to their Z-score values (ranging from 1.71 to 2.25). The C-score value of the selected model is −4.83. The C-score value is directly proportional to the quality of the structure. With an estimated root-mean-square deviation (rmsd) of 17.3 ± 2.7 Å, this model had an estimated TM-score of 0.22 ± 0.06. As a scale for evaluating the structural similarity between two structures, the TM-score has been proposed.52 A TM-score >0.5 reveals a correct topology model, and a TM-score <0.17 implies random similarity. These cut-off values are protein length-independent.
Figure 5.

Predicted secondary structure of the multi-epitope vaccine construct. (A) GOR IV server results: α helix (blue), random coil (yellow), and extended strand (red). (B) Graphical illustration of the secondary structure by the PSIPRED server.
Structure Refinement and Validation
I-TASSER-modeled three-dimensional (3D) structure was subjected to the PROCHECK server, where the Ramachandran plot was generated and the output revealed 37.1% residues that were present in the favored region. The Ramachandran plot allows one to visualize energetically allowed and disallowed dihedral angles psi (ψ) and phi (φ). Hence, the 3D model was subjected to further refinement by the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE), and the output provided five refined models for the vaccine construct. Model number 4 was the best refined structure with qualifying parameters such as a global distance test-high accuracy (GDT-HA) of 0.7908, rmsd of 0.828, MolProbity of 4.127, clash score of 117.2, poor rotamer of 8.2, and Rama favored of 62.4% (Table S3). Thus, this model (Figure 6) was finally selected for further investigations.
Figure 6.

Tertiary structure model of multi-epitope vaccine construction and validation. (A) 3D structural alignment of the modeled multi-epitope vaccine construct before (orange) and after (green) refinement. (B) Ramachandran plot for the multi-epitope vaccine construct.
Immunological Property Assessment
The presence of B-cell epitopes in the multi-epitope vaccine construct plays an important role in triggering humoral immune responses.53 The BcePred server was used for the prediction of continuous B-cell epitopes in the chimeric multi-epitope vaccine with default parameters. Important properties of epitopes such as hydrophilicity, antigenicity, flexibility, accessibility, polarity, and exposed surfaces were predicted (Table 6). Ellipro was used to predict the conformational B-cell epitopes in the multi-epitope vaccine. Only one conformational epitope region was predicted (Table 7 and Figure 7). The analysis of continuous and discontinuous B-cell epitopes in the designed vaccine construct revealed that they can interact with antibodies and are flexible.
Table 6. Prediction of Linear B-Cell Epitopes in the Multi-epitope Vaccine by Hydrophilicity, Flexibility, Accessibility, Exposed Surface, Polarity, and Antigenic Propensity Based on the BcePred Server.
| prediction parameters | epitope position |
|---|---|
| hydrophilicity | 28–31, 33, 69–70, 133–136, 138–139, 158, 180 |
| flexibility | 130–132, 137, 153, 155–156, 177–178, 187 |
| accessibility | 25–30, 65, 68–70, 85–90, 96–102, 132–136, 143, 147, 152–153, 155–158, 185, 189–190 |
| exposed surface | 152–153, 155–156 |
| polarity | 42–48, 50, 70, 143–147, 152–153 |
| antigenic propensity | 4–5, 9, 13–17, 40–41, 43–46, 65–63, 112–115, 125–129, 167–168, 173 |
Table 7. List of Predicted Conformational B-Cell Epitopes from the Multi-epitope Vaccine Construct by the Ellipro Server.
| s. no. | epitope position | number of residues | score |
|---|---|---|---|
| 1 | Q16, S17, G20, F21, M22, R23, V24, P25, N26, N27, E28, A29, Q30, C31, E32, Q33, A34, I37, C38, S39, K40, D41, H42, C43, H47, T48, R49, A50, F51, G52, H53, C54, Q55, R56, G57, V58, P59, C60, C61, V64, Y65, D66, E67, A68, A69, A70, K71, Q72, E73, F74, T75, G76, F77, D78, A79, A82, Y83, E85, N86, Q87, Y88, Y89, P90, P91, A92, A93, A94, Y95, Q96, E97, Y98, D99, Y100, Y101, P102, P103, A104, A105, Y107, N108, E109, L116, P118, G119, P120, G121, I122, D130, R131, S132, R133, A134, T135, N136, G137, P138, G139, P140, G141, K142, I143, H144, N145, K146, L148, K149, N150, L151, A152, K153, L154, K155, K156, G157, P158, G159, P160, G161, F162, L165, Y166, L167, N168, S169, N171, S172, L173, S174, F175, I176, G177, P178, G179, P180, G181, K182, T183, F184, S185, L186, N187, K188, G189, L190, N191, K192, V193, I194 | 148 | 0.589 |
Figure 7.
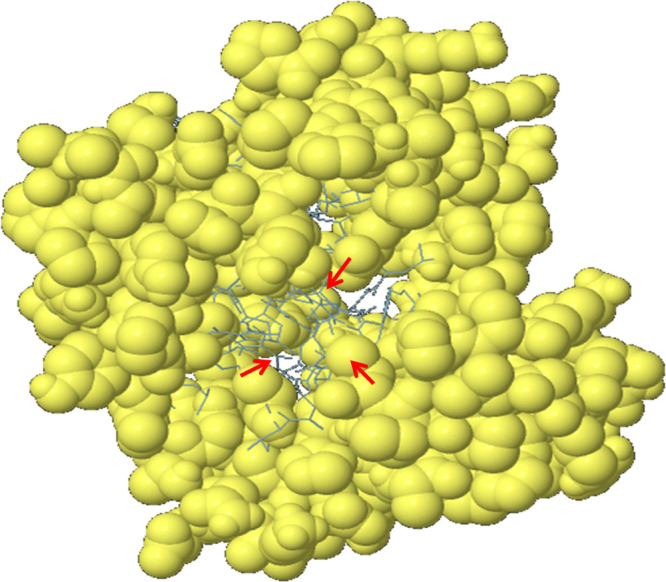
Predicted conformational B-cell epitopes by the ElliPro tool. The immunogenic epitopes are depicted as yellow globules on the ball and stick (red arrow) representation of the multi-epitope vaccine construct structure.
Vaccine Protein Disulfide Engineering
Disulfide engineering was performed to stabilize the vaccine construct by particular geometric confirmations.36,54 It was predicted that a total number of 60 pairs of amino acid residues may form a disulfide bond through the DbD2 server. Among them, only three residues, including LYS 2- TYR 5, ARG 49-ASP 66, and TYR 88-TYR 95, which were replaced by cysteine residues, were able to create disulfide bond formation following the residue assessment by chi3 and B-factor energy parameters (Figure 8). On the basis of −87 to +97 chi3 values and <2.5 energy values, residue screening was performed.
Figure 8.

Disulfide engineering of the vaccine construct. (A) Initial model without disulfide bonds, (B) mutant model, the yellow stick, within the circle represents the disulfide bond formation.
Molecular Docking
Molecular docking was carried out between the multi-epitope vaccine construct and chicken Toll-like receptor-5 (TLR5). Among the chicken TLRs, TLR5 has a propensity toward the recognition of bacterial components present on the extracellular surfaces,71 thus chicken TLR5 was selected for docking. HADDOCK uses biochemical and biophysical interaction data to perform docking.55 HADDOCK clustered 104 structures in 13 clusters, representing 52% of the models produced by water-refined HADDOCK. The most accurate cluster of all is the top cluster with the smallest HADDOCK ranking. Using the HADDOCK refinement server, a representative model of the top cluster was subjected to further refinement, where 20 structures were grouped into one cluster, resulting in 100% of the HADDOCK water-refined version. The results of the refined model are presented in Table 8. A strong binding affinity between the vaccine and the receptor is indicated by a Haddock score of 297.1 ± 3.1; a negative score suggests better docking (Figure 9). A total of 19 hydrogen bonds, 1 salt bridge, and 182 nonbonded contacts were analyzed (Table 9). The binding affinity of a complex, or in terms of thermodynamics, the Gibbs free energy (ΔG), is a critical quantity for deciding whether or not an interaction can actually occur under particular conditions in the cell. Using PRODIGY web servers, the binding affinity of the docked complex was analyzed. The G-values were found to be −14.4 kcal/mol for the vaccine construct and chicken TLR5, and the dissociation constant was 3.0 × 10–11. The results show that, as indicated by negative Gibbs free energy (ΔG), the docked complex is energetically viable.
Table 8. Docking Statistics of Best Refined Docked Chicken TLR5 and Vaccine Construct.
| parameters | result |
|---|---|
| HADDOCK score | –297.1 ± 3.1 |
| cluster size | 20 |
| rmsd from the overall lowest-energy structure | 0.8 ± 0.4 |
| van der Waals energy | –154.8 ± 5.3 |
| electrostatic energy | –454.1 ± 36.8 |
| desolvation energy | –51.5 ± 5.2 |
| restraints violation energy | 0.0 ± 0.0 |
| buried surface area | 5197.8 ± 122.0 |
| Z-score | 0.0 |
Figure 9.

Molecular docking of the multi-epitope vaccine with chicken TLR5. (A) Chicken TLR5 and the vaccine construct-docked complex. The vaccine construct is shown in red while chicken TLR5 in blue. (B) Interacting residues between docked TLR5 (chain A) and vaccine (chain B).
Table 9. List of Hydrogen Bond Interactions between TLR5 and Multi-epitope Vaccine Construct.
| TLR5 |
multi-epitope vaccine |
||
|---|---|---|---|
| position | residue | position | residue |
| 21 | ALA | 174 | SER |
| 26 | TYR | 160 | PRO |
| 27 | SER | 163 | SER |
| 27 | SER | 168 | ASN |
| 28 | GLU | 164 | ARG |
| 28 | GLU | 164 | ARG |
| 48 | ASP | 92 | ALA |
| 48 | ASP | 96 | GLN |
| 605 | ALA | 149 | LYS |
| 606 | GLY | 149 | LYS |
| 621 | GLY | 142 | LYS |
| 666 | CYS | 108 | ASN |
| 667 | ARG | 108 | ASN |
| 679 | LYS | 108 | ASN |
| 683 | GLY | 104 | ALA |
| 724 | ASP | 101 | TYR |
| 725 | LYS | 101 | TYR |
| 725 | LYS | 155 | LYS |
| 726 | ASN | 97 | GLU |
Codon Optimization and In Silico Cloning
The initial step in validating a vaccine candidate is to carry out serological analysis,56 which requires the expression of the vaccine in a suitable expression system. A similar approach has been used in earlier studies for in silico designed vaccines prior to in vitro expression.57 We chose E. coli as the expression system. The Java codon adaptation tool (JCAT) aids in optimized cloning and expression in the E. coli K12 strain as a host. The codon adaptation index (CAI) in the adapted sequence was 0.92, and the GC content of the improved sequence was estimated to be 54.08% and the GC content of E. coli was 50.7. The graphical representation of codon usage in the optimized multi-epitope vaccine gene is shown in Figure 10. In order to perform in silico cloning, the codon-optimized vaccine construct sequence was inspected for restriction enzyme sites; HindIII and BamHI enzymes were not found in the vaccine sequence, so these enzymes were used for in silico cloning purpose. Finally, a successful clone of 5938 bp was obtained following the insertion of the vaccine construct into the pET28a(+) vector (Figure 11).
Figure 10.

Graphical view of codon usage in the optimized vaccine construct gene. The red line indicates relative adaptiveness, and the blue line represents mean codon usage.
Figure 11.

In silico restriction cloning of the gene sequence of the vaccine construct into the pET28a(+) expression vector. The red part indicates the gene coding the multi-epitope vaccine construct, and the black part indicates the vector backbone.
Conclusions
A multi-epitope-based vaccine was developed against M. gallisepticum through an immunoinformatics approach, which is advantageous over traditional methods in many aspects. The application of computational methods can be used to design and develop an effective vaccine in a short time and low cost. The amino acid sequences of cytoadherence and cytoadherence accessory proteins were retrieved, their potential B-cell and T-cell binding epitopes were predicted, and a vaccine was constructed, including epitopic sequences with an immunogenic adjuvant merged together with the suitable linkers that can trigger strong immunogenic responses. The constructed multi-epitope vaccine was found to be nonallergenic and -antigenic. Disulfide engineering further increased the stability of the multi-epitope vaccine construct. A stable and strong interaction of the vaccine with the chicken immune receptor was confirmed by molecular docking analysis. The multi-epitope cDNA in silico cloning experiments were also subsequently conducted to verify its probable efficacy of being expressed in the expression vector. Nevertheless, these findings provide novel and valuable epitope candidates and also showed a novel way to develop a vaccine against M. gallisepticum.
Materials and Methods
Sequence Retrieval
The Uniprot Knowledgebase data was used to retrieve the amino acid sequences of antigenic proteins from M. gallisepticum strain R low in the FASTA format. The proteins GapA (accession no: Q9REM8), PlpA (accession no: Q7NBF9), Hlp3 (accession no: Q7NBT3), and CrmA (accession no: F8WJY4) were retrieved and used for further analysis.
Evaluation of Protein Antigenicity and Allergenicity
The selected protein sequences were submitted to the VaXiJen v 2.0 server to identify antigenicity, assisting in the prediction of potential antigens with a threshold score of 0.4. The models were tested using leave-one-out cross-sectional models; the validations were with 70–89% accuracy.58 Similarly, the prediction of allergenicity was made using AllerTop v. 2.0.59 Therefore, allergens and nonallergens with possible sensitivity and protein specificity have been predicted.
Prediction of T-Cell Epitopes
Human MHC-I and MHC-II alleles were used as alternates for chicken MHC alleles due to the nonavailability of chicken alleles for peptide-binding predictions in immunoinformatics tools.40 Hence, the best suitable human alleles for chicken were chosen from a previous study.41 Three alleles for MHC class I (HLA*B 40:06, HLA*B 41:04, and HLA*B 41:03) and four DRB1 alleles for MHC class II (DRB1:1482, DRB1:1366, DRB1:1310, and DRB1:1445) were selected from this study.
MHC-I and -II Binding Prediction
The NetMHCcons 1.1 server was used to predict MHC-I binding affinity (http://www.cbs.dtu.dk/services/NetMHCcons);60 the thresholds were selected for strong binding peptides with IC50 values of 50 nM for strong binders and 500 nM for weak binders. Thus, IC50 < 50 nM peptides were categorized as strong binders and 50 < IC50 < 500 nM peptides were considered weak binders. Similarly, in terms of the percentage (percent) rank, the thresholds for strong binding peptides were set at 0.5 while those for weak binders were maintained at 2. The protein sequence was set to a length of nine amino acids. Using the IEDB analysis resource (http://tools.iedb.org/immunogenicity),61 the class I immunogenicity score was predicted. The NetMHCIIpan 3.1 server was used to predict the MHC-II binding epitope (http://www.cbs.dtu.dk/services/NetMHCIIpan-3.1/);62 the peptide length was set to 15 amino acids. The server then predicted a number of peptides binding to MHC alleles, from which only peptides which were recognized as strong binders (% rank <2) for all the four MHC alleles were selected.
B-Cell Epitope Prediction
BepiPred was used to predict linear B-cell epitopes (http://tools.iedb.org/bcell/) from the IEDB research resource.63 By combining two residue properties with the hidden Markov model, BepiPred predicts continuous epitopes. BepiPred was assessed on the data set of epitopes extracted from the literature, AntiJen, and HIV databases.
Construction of the Multi-epitope Vaccine
The multi-epitope vaccine against M. gallisepticum was designed using the MHC-I and MHC-II binding epitopes from the GapA, PlpA, Hlp3, and CrmA proteins. Furthermore, to improve the immunogenicity of the multi-epitope vaccine, the TLR5 agonist, avian β defensin 8, was selected as an adjuvant. The EAAAK linker was used to link the adjuvant with CTL epitopes. The intra-CTL and intra-HTL epitopes were linked using AAY and GPGPG linkers, respectively.
mRNA Secondary Structure Prediction
The Mfold online server was used to predict the RNA secondary structure of the multi-epitope vaccine (http://unafold.rna.albany.edu/?q=mfold). The Mfold server provides the prediction of true positive base pairs and structures within minimum ΔG thermodynamically.64
Antigenicity, Allergenicity, and Solubility Profile Prediction
The antigenicity of the multi-epitope vaccine was predicted using the VaxiJen server (http://www.ddgpharmfac.net/vaxijen/VaxiJen/VaxiJen.html) and ANTIGENpro (http://scratch.proteomics.ics.uci.edu/);65 the allergenicity of the vaccine was verified using AllerTOP v2.0 (http://www.ddg-pharmfac.net/AllerTOP/). The AllerTOP v2.0 server utilizes machine learning techniques to sort the allergens, such as nearest neighbors, auto- and cross-variance transformation, and amino acid descriptors. The accuracy of this server is 85.3% at fivefold cross validation.59 In addition, the SOLpro online tool was employed for the prediction of solubility upon overexpression of the protein construct in the E. coli host (http://scratch.proteomics.ics.uci.edu/), with 74% accurate prediction and a 10-fold cross-validation method.66
Determination of Physiochemical Properties
The ExPASy-ProtParam server, accessible at (http://web.expasy.org/protparam/), has calculated the physiochemical properties of the vaccine construct. Multiple parameters, aliphatic index, instability index, half-life, isoelectric point (pI), molecular weight, and atomic composition, including grand average hydropathicity (GRAVY), were analyzed. The half-life of the protein represents the time that the molecule disappears after its synthesis in the cell. The instability index indicates the in vitro stability of a protein molecule.67
Secondary Structure Prediction
The secondary structure of the multi-epitope vaccine construct was predicted using the Garnier-Osguthorpe-Robson (GOR IV) online server (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_gor4.html) with a mean accuracy of 64.4%68 and position-specific iterated prediction (PSIPRED) analysis on outputs from PSI-BLAST (http://bioinf.cs.ucl.ac.uk/psipred/).69
Tertiary Structure Prediction, Refinement, and Validation
The tertiary structure was modeled using the I-TASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/).70 The 3D structure modeled by I-TASSER was subjected to refinement by the GalaxyRefine server (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE).71 This server replaces amino acids with high-probability rotamers and applies molecular dynamic simulation for overall structural relaxation. The output usually includes five refined models, with different parameter scores, comprising GDT-HA, rmsd, MolProbity, Clash score, Poor rotamers, and Rama favored.72 The refined structure was validated by PROCHECK (https://servicesn.mbi.ucla.edu/PROCHECK/) which analyzes the stereochemical quality of a protein structure by analyzing the residue-by-residue geometry and overall structure geometry.73
Immunological Analysis for the Chimeric Multi-epitope Vaccine
The multi-epitope vaccine was analyzed for linear (continuous) and conformational (discontinuous) B-cell epitopes. Continuous B-cell epitopes were predicted by BcePred (https://webs.iiitd.edu.in/raghava/bcepred/index.html).74 Discontinuous B-cell epitopes were predicted by ElliPro, which gives insights about linear and discontinuous antibody epitopes based on a protein antigen’s 3D structure (http://tools.iedb.org/ellipro/).75
Multi-epitope Vaccine Protein Disulfide Engineering
Disulfide engineering is an important biotechnological tool to design new disulfide bonds in the target protein through cysteine mutation of residues in the highly mobile region of protein. Disulfide bonds provide substantial stability and reinforce the protein geometric conformation. For this purpose, the DbD2 online server was used (http://cptweb.cpt.wayne.edu/DbD2/index.php). The pair of residues capable of forming a disulfide bond can be detected by this web server if each amino acid residue is mutated to cysteine.76
Molecular Docking
To generate a stable immune response, it is necessary for the vaccine to interact with the target immune cell receptor. In this study, interactions between the vaccine construct and chicken TLR5 were studied as they are bacterial-sensing TLR.77 CPORT78 was used in predicting the active and passive residues for the interactions. The docking of the vaccine with chicken TLR5 was performed by HADDOCK 2.4 (https://www.bonvinlab.org/software/haddock2.4/).79 To refine the chosen cluster, HADDOCK Refinement Interface was used. The best structure after refinement from the docked complex was chosen and its binding affinity was calculated using the PRODIGY web server.80,81 Last, the interacting residues between the vaccine construct and chicken TLR5 were mapped using PDBsum (https://www.ebi.ac.uk/thorntonsrv/databases/pdbsum/Generate.html).82
Codon Optimization and In Silico Cloning of the Vaccine Construct
The JCAT server (http://www.jcat.de) was used to execute codon optimization according to E. coli codon usage.83 Parameters that are significant to the effectiveness of gene expression including CAI and GC content adjustment were optimized. The assessment of cloning and expression of the vaccine construct in an appropriate expression vector was carried out using the SnapGene restriction cloning tool (GSL Biotech LLC 2015).
Acknowledgments
The authors are thankful to the Thiruvalluvar University for providing infrastructural support to carry out the research work. The authors received no financial support for the research, authorship, and/or publication of this article. The authors are thankful to Dr. D. Vasanth (ex-IIT Madras) for his English language corrections.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01032.
List of predicted MHC-I binding epitopes, list of MHC-II binding epitopes, and (five refined models from the Galaxy refine server (PDF)
Author Contributions
S.P.M. and M.C.H. designed and performed the experimental studies. S.P.M. carried out the in silico experiments. The manuscript was written by S.P.M. and M.C.H.
The authors declare no competing financial interest.
Supplementary Material
References
- Jordan F. T. W.Avian mycoplasmas, The mycoplasmas. In Human and Animal Mycoplasmas; Tully J. G., Whitcomb R. F., Eds.; Academic Press: New York, NY, 1979; Vol. II, pp 1–48. [Google Scholar]
- Levisohn S.; Kleven S. H. Avian mycoplasmosis ( Mycoplasma gallisepticum). Rev. Sci. Technol. 2000, 19, 425–442. 10.20506/rst.19.2.1232. [DOI] [PubMed] [Google Scholar]
- Johansson K.-E.; Pettersson B.. Taxonomy of Mollicutes. In Molecular Biology and Pathogenicity of Mycoplasma; Razin S., Hermann R., Eds.; Kluwer Academic/Plenum Publishers: New York, 2002; pp 1–29. [Google Scholar]
- Mycoplasma gallisepticum Infection; Yoder H. W. J., Ed.; Iowa State University Press: Ames, IA, 1991. [Google Scholar]
- Davidson W. R.; Nettles V. F.; Couvillion C. E.; Yoder H. W. Jr. Infectious sinusitis in wild turkeys. Avian Dis. 1982, 26, 402–405. 10.2307/1590112. [DOI] [PubMed] [Google Scholar]
- Razin S.; Yogev D.; Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol. Mol. Biol. Rev. 1998, 62, 1094–1156. 10.1128/mmbr.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin S.; Hayflick L. Highlights of mycoplasma research: an historical perspective. Biologicals 2010, 38, 183–190. 10.1016/j.biologicals.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Muto A.; Ushida C.. Transcription and Translation. In Molecular Biology and Pathogenicity of Mycoplasmas; Razin S., Herrmann R., Eds.; Springer: Boston, MA, 2002. [Google Scholar]
- Noormohammadi A. H. Role of phenotypic diversity in pathogenesis of avian mycoplasmosis. Avian Pathol. 2007, 36, 439–444. 10.1080/03079450701687078. [DOI] [PubMed] [Google Scholar]
- Indiková I.; Much P.; Stipkovits L.; Siebert-Gulle K.; Szostak M. P.; Rosengarten R.; Citti C. Role of the GapA and CrmA cytadhesins ofMycoplasma gallisepticum in promoting virulence and host colonization. Infect. Immun. 2013, 81, 1618–1624. 10.1128/IAI.00112-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May M.; Papazisi L.; Gorton T. S.; Geary S. J. Identification offibronectin-binding proteins in Mycoplasma gallisepticum strain R. Infect. Immun. 2006, 74, 1777–1785. 10.1128/iai.74.3.1777-1785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromati J.; Mavromati E.; Gjeta Z. The Effect of a macrolid antibiotic on the control of mycoplasmas And production efficiency in broilers. Biotechnol. Animal Husb. 2011, 27, 721–731. 10.2298/bah1103721m. [DOI] [Google Scholar]
- El Gazzar M.; Laibinis V. A.; Ferguson-Noel N. Characterization of a ts-1-like Mycoplasma galisepticum isolate from commercial broiler chickens. Avian Dis. 2011, 55, 569–574. 10.1637/9689-021711-reg.1. [DOI] [PubMed] [Google Scholar]
- Kleven S. H. Control of avian mycoplasma infections in commercial poultry. Avian Dis. 2008, 52, 367–374. 10.1637/8323-041808-review.1. [DOI] [PubMed] [Google Scholar]
- Chernova O. A.; Medvedeva E. S.; Mouzykantov A. A.; Baranova N. B.; Chernov V. M. Mycoplasmas and Their Antibiotic Resistance: The Problems and Prospects in Controlling Infections. Acta Nat. 2016, 8, 24–34. 10.32607/20758251-2016-8-2-24-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumbe N.; Louie A.; Leary R.; Liu W.; Deziel M. R.; Tam V. H.; Bachhawat R.; Freeman C.; Kahn J. B.; Bush K.; Dudley M. N.; Miller M. H.; Drusano G. L. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Invest. 2003, 112, 275–285. 10.1172/jci200316814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance A. M.; Branton S. L.; Collier S. D.; Gerard P. D.; Peebles E. D. Effects of time-specific F-strain Mycoplasma gallisepticum inoculation overlays on prelay ts11-strain Mycoplasma gallisepticum inoculation on performance characteristics of commercial laying hens. Poult. Sci. 2008, 87, 655–660. 10.3382/ps.2007-00492. [DOI] [PubMed] [Google Scholar]
- Farran M. T.; Ellakany H. F.; Shaib H. A.; Majed H. M. Evaluation of Antibiotics to Control Mycoplasma gallisepticum in Broiler Breeder Chickens. Poult. Fish Wildl. Sci. 2018, 06, 191. 10.4172/2375-446X.1000191. [DOI] [Google Scholar]
- Gingerich E.Vaccine and vaccination for Mycoplasma gallisepticum (MG). Proceedings in Cornell Poultry Conference, Ithaca, NY, 2002; pp 2–5.
- Gaurivaud P.; Baranowski E.; Pau-Roblot C.; Sagné E.; Citti C.; Tardy F. Mycoplasma agalactiae Secretion of β-(1→6)-Glucan, a Rare Polysaccharide in Prokaryotes, Is Governed by High-Frequency Phase Variation. Appl. Environ. Microbial. 2016, 82, 3370–3383. 10.1128/AEM.00274-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma J. M. Introduction to poultry vaccines and immunity. Adv. Vet. Med. 1999, 41, 481–494. 10.1016/s0065-3519(99)80036-6. [DOI] [PubMed] [Google Scholar]
- Buckland B. C. The process development challenge for a new vaccine. Nat. Med. 2005, 11, S16–S19. 10.1038/nm1218. [DOI] [PubMed] [Google Scholar]
- Di Pasquale A.; Bonanni P.; Garçon N.; Stanberry L. R.; El-Hodhod M.; Tavares Da Silva F. Vaccine safety evaluation: practical aspects in assessing benefits and risks. Vaccine 2016, 34, 6672–6680. 10.1016/j.vaccine.2016.10.039. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; van der Spek P. J.; Sharma H. S.; Chandra R. Molecular Binding Mechanism and Pharmacology Comparative Analysis of Noscapine for Repurposing against SARS-CoV-2 Protease. J. Proteome Res. 2020, 19, 4678–4689. 10.1021/acs.jproteome.0c00367. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; Sharma N.; Chandra R. Multiepitope Subunit Vaccine to Evoke Immune Response against Acute Encephalitis. J. Chem. Inf. Model. 2020, 60, 421–433. 10.1021/acs.jcim.9b01051. [DOI] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; Chandra R. Vaccine Formulation and Optimization for Human Herpes Virus-5 through an Immunoinformatics Framework. ACS Pharmacol. Transl. Sci. 2020, 3, 1318–1329. 10.1021/acsptsci.0c00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; Tomar R.; Chandra R. Antimicrobial Peptide Designing and Optimization Employing Large-Scale Flexibility Analysis of Protein-Peptide Fragments. ACS Omega 2019, 4, 21370–21380. 10.1021/acsomega.9b03035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Sood D.; Singh S.; Kumar S.; Chandra R. High bio-recognizing aptamer designing and optimization against human herpes virus-5. Eur. J. Pharm. Sci. 2021, 156, 105572. 10.1016/j.ejps.2020.105572. [DOI] [PubMed] [Google Scholar]
- Idris S.; Salih S.; Basheir M.; Elhadi A.; Kamel S.; Abd-elrahman K.; Hamdi A.; Hassan M. A. In silico Prediction of Peptide based Vaccine against Fowlpox Virus (FPV). Immunome Res. 2018, 14, 1. 10.4172/1745-7580.1000154. [DOI] [Google Scholar]
- Ali S. A.; Almofti Y. A.; Abd-Elrahman K. A. Immunoinformatics Approach for Multiepitopes Vaccine Prediction against Glycoprotein B of Avian Infectious Laryngotracheitis Virus. Adv. Bioinf. 2019, 2019, 1270485. 10.1155/2019/1270485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge S.; Dellagostin O. A. The development of veterinary vaccines: a review of traditional methods and modern biotechnology approaches. Biotechnol. Res. Innov. 2017, 1, 6–13. 10.1016/j.biori.2017.10.001. [DOI] [Google Scholar]
- Silva-Arrieta S.; Goulder P. J. R.; Brander C. In silico veritas? Potential limitations for SARS-CoV-2 vaccine development based on T-cell epitope prediction. PLoS Pathog. 2020, 16, e1008607 10.1371/journal.ppat.1008607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.-F.; Lin C.-Y.; Hong H.-M. In silico Design, Synthesis and Potency of an Epitope-based Vaccine Against Foot-and-mouth Disease Virus. Int. J. Pharmacol. 2017, 13, 122–133. 10.3923/ijp.2017.122.133. [DOI] [Google Scholar]
- Michel-Todó L.; Bigey P.; Reche P. A.; Pinazo M.-J.; Gascón J.; Alonso-Padilla J. Design of an Epitope-Based Vaccine Ensemble for Animal Trypanosomiasis by Computational Methods. Vaccines 2020, 8, 130. 10.3390/vaccines8010130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman M. M.; ElAmin E. E.; Al-Nour M. Y.; Alam S. S.; Adam R. S.; Ahmed A. A.; Ahmed A. E.; Abdalla M. H.; Salih M. A. In Silico Design of Epitope Based Peptide Vaccine against Virulent Strains of (HN)-Newcastle Disease Virus (NDV) in Poultry Species. Int. J. Multidiscip. Curr. Res. 2016, 4, 868–878. [Google Scholar]
- Hasan M.; Ghosh P. P.; Azim K. F.; Mukta S.; Abir R. A.; Nahar J.; Hasan Khan M. M. Reverse vaccinology approach to design a novel multi-epitope subunit vaccine against avian influenza A (H7N9) virus. Microb. Pathog. 2019, 130, 19–37. 10.1016/j.micpath.2019.02.023. [DOI] [PubMed] [Google Scholar]
- Ingale A. G.; Goto S. Prediction of CTL epitope, in silico modeling and functional analysis of cytolethal distending toxin (CDT) protein of Campylobacter jejuni. BMC Res. Notes 2014, 7, 92. 10.1186/1756-0500-7-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise L.; Biron B. M.; Boyle C. M.; Kurt Yilmaz N.; Jang H.; Schiffer C.; Ross T. M.; Martin W. D.; De Groot A. S. T cell epitope engineering: an avian H7N9 influenza vaccine strategy for pandemic preparedness and response. Hum Vaccin Immunother. 2018, 14, 2203–2207. 10.1080/21645515.2018.1495303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni P. A.; Ali A. M. M. T.; Rout M.; Thabitha A.; Vino S.; Lulu S. S. Designing of an epitope-based peptide vaccine against walking pneumonia: an immunoinformatics approach. Mol. Biol. Rep. 2019, 46, 511–527. 10.1007/s11033-018-4505-0. [DOI] [PubMed] [Google Scholar]
- Aziz F.; Tufail S.; Shah M. A.; Salahuddin Shah M.; Habib M.; Mirza O.; Iqbal M.; Rahman M. In silico epitope prediction and immunogenic analysis for penton base epitope-focused vaccine against hydropericardium syndrome in chicken. Virus Res. 2019, 273, 197750. 10.1016/j.virusres.2019.197750. [DOI] [PubMed] [Google Scholar]
- Valdivia-Olarte H.; Requena D.; Requena D.; Ramirez M.; Saravia L. E.; Izquierdo R.; Falconi-Agapito F.; Zavaleta M.; Best I.; Fernández-Díaz M.; Zimic M. Design of a predicted MHC restricted short peptide immunodiagnostic and vaccine candidate for Fowl adenovirus C in chicken infection. Bioinformation 2015, 11, 460–465. 10.6026/97320630011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainio O.; Koch C.; Toivanen A. B-L antigens (class II) of the chicken major histocompatibility complex control T-B cell interaction. Immunogenetics 1984, 19, 131–140. 10.1007/BF00387856. [DOI] [PubMed] [Google Scholar]
- Hala K.; Boyd R.; Wick G. Chicken major histocompatibility complex and disease. Scand. J. Immunol. 1981, 14, 607–616. 10.1111/j.1365-3083.1981.tb00602.x. [DOI] [PubMed] [Google Scholar]
- Erf G. F. Cell-mediated immunity in poultry. Poult. Sci. 2004, 83, 580–590. 10.1093/ps/83.4.580. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Lee J.; Vu T. H.; Lee S.; Lillehoj H. S.; Hong Y. H. Chicken avian β-defensin 8 modulates immune response via the mitogen-activated protein kinase signaling pathways in a chicken macrophage cell line. Poult. Sci. 2020, 99, 4174–4182. 10.1016/j.psj.2020.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nain Z.; Karim M. M.; Sen M. K.; Adhikari U. K. Structural basis and designing of peptide vaccine using PE-PGRS family protein of Mycobacterium ulcerans—An integrated vaccinomics approach. Mol. Immunol. 2020, 120, 146–163. 10.1016/j.molimm.2020.02.009. [DOI] [PubMed] [Google Scholar]
- Ali M.; Pandey R. K.; Khatoon N.; Narula A.; Mishra A.; Prajapati V. K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. 10.1038/s41598-017-09199-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon N.; Pandey R. K.; Prajapati V. K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. 10.1038/s41598-017-08842-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Validi M.; Karkhah A.; Prajapati V. K.; Nouri H. R. Immuno-informatics based approaches to design a novel multi epitope-based vaccine for immune response reinforcement against Leptospirosis. Mol. Immunol. 2018, 104, 128–138. 10.1016/j.molimm.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Pourseif M. M.; Yousefpour M.; Aminianfar M.; Moghaddam G.; Nematollahi A. A multi-method and structure-based in silico vaccine designing against Echinococcus granulosus through investigating enolase protein. Bioimpacts 2019, 9, 131–144. 10.15171/bi.2019.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar T.; Narsaria U.; Basak S.; et al. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020, 10, 10895. 10.1038/s41598-020-67749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- Getzoff E. D.; Tainer J. A.; Lerner R. A.; Geysen H. M.. Advances in Immunology; Elsevier: Amsterdam, 1998; pp 1–98. [DOI] [PubMed] [Google Scholar]
- Pandey R. K.; Bhatt T. K.; Prajapati V. K. Novel Immunoinformatics Approaches to Design Multi-epitope Subunit Vaccine for Malaria by Investigating Anopheles Salivary Protein. Sci. Rep. 2018, 8, 1125. 10.1038/s41598-018-19456-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. J. J. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Gori A.; Longhi R.; Peri C.; Colombo G. Peptides for immunological purposes: design, strategies and applications. Amino Acids 2013, 45, 257–268. 10.1007/s00726-013-1526-9. [DOI] [PubMed] [Google Scholar]
- Foroutan M.; Ghaffarifar F.; Sharifi Z.; Dalimi A. Vaccination with a novel multi-epitope ROP8 DNA vaccine against acute Toxoplasma gondii infection induces strong B and T cell responses in mice. Comp. Immunol. Microbiol. Infect. Dis. 2020, 69, 101413. 10.1016/j.cimid.2020.101413. [DOI] [PubMed] [Google Scholar]
- Doytchinova I. A.; Flower D. R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. 2007, 8, 4. 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I.; Flower D. R.; Doytchinova I. AllerTOP--a server for in silico prediction of allergens. BMC Bioinf. 2013, 14, S4. 10.1186/1471-2105-14-S6-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karosiene E.; Lundegaard C.; Lund O.; Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012, 64, 177–186. 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- Calis J. J. A.; Maybeno M.; Greenbaum J. A.; Weiskopf D.; De Silva A. D.; Sette A.; Keşmir C.; Peters B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta M.; Karosiene E.; Rasmussen M.; Stryhn A.; Buus S.; Nielsen M. Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 2015, 67, 641–650. 10.1007/s00251-015-0873-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J.; Lund O.; Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J.; Randall A. Z.; Sweredoski M. J.; Baldi P. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnan C. N.; Zeller M.; Kayala M. A.; Vigil A.; Randall A.; Felgner P. L.; Baldi P. Highthroughput prediction of protein antigenicity using protein microarray data. Bioinform 2010, 26, 2936–2943. 10.1093/bioinformatics/btq551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S. e.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein identification and analysis tools on the ExPASy server. The Proteomics Protocols Handbook; Springer, 2005; pp 571–607. [Google Scholar]
- Garnier J.; Gibrat J.-F.; Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996, 266, 540–553. 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- Buchan D. W. A.; Jones D. T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. 10.1093/nar/gkz297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Yan R.; Roy A.; Xu D.; Poisson J.; Zhang Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo L.; Park H.; Seok C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J.; Park H.; Heo L.; Seok C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. 10.1093/nar/gks493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/s0021889892009944. [DOI] [Google Scholar]
- Saha S.; Raghava G. P. S.. BcePred: Prediction of Continuous B-Cell Epitopes in Antigenic Sequences Using Physico-chemical Properties; Nicosia G., Cutello V., Bentley P. J., Timis J., Eds.; Springer, 2004; pp 197–204, ICARIS 2004, LNCS 3239. [Google Scholar]
- Ponomarenko J.; Bui H.-H.; Li W.; Fusseder N.; Bourne P. E.; Sette A.; Peters B. ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf. 2008, 9, 514. 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig D. B.; Dombkowski A. A. Disulfide by Design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinf. 2013, 14, 346. 10.1186/1471-2105-14-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkler M.; Bainová H.; Bryja J. Protein evolution of Toll-like receptors 4, 5 and 7 within Galloanserae birds. Genet., Sel., Evol. 2014, 46, 72. 10.1186/s12711-014-0072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S. J.; Bonvin A. M. J. J. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One 2011, 6, e17695 10.1371/journal.pone.0017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zundert G. C. P.; Rodrigues J. P. G. L. M.; Trellet M.; Schmitz C.; Kastritis P. L.; Karaca E.; Melquiond A. S. J.; van Dijk M.; de Vries S. J.; Bonvin A. M. J. J. The HADDOCK2.2 web server: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- Vangone A.; Bonvin A. M. Contacts-based prediction of binding affinity in protein–protein complexes. Elife 2015, 4, e07454 10.7554/elife.07454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L. C.; Rodrigues J. P.; Kastritis P. L.; Bonvin A. M.; Vangone A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Jabłońska J.; Pravda L.; Vařeková R. S.; Thornton J. M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. 10.1002/pro.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote A.; Hiller K.; Scheer M.; Munch R.; Nortemann B.; Hempel D. C.; Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.