

Abstract

We report the synthesis and crystal structures of three new copper(II) Schiff-base complexes. The complexes have been characterized by elemental analysis and Fourier transform infrared (FT-IR) and UV–visible spectroscopies. The X-ray diffraction (XRD) analysis reveals that complexes 1 and 3 crystallize in a monoclinic space group C2/c and 2 in a triclinic space group P1̅, each adopting a square planar geometry around the metal center. We use a density functional theory method to explore the quantum chemical properties of these complexes. The calculation proceeds with the three-dimensional (3D) crystal structure characterization of the complexes in which the calculated IR and UV–vis values are comparable to the experimental results. Charge distribution and molecular orbital analyses enabled quantum chemical property prediction of these complexes. We study the drug-likeness properties and binding potentials of the synthesized complexes. The in silico outcome showed that they could serve as permeability-glycoprotein (P-gp) and different cytochrome P450 substrates. Our calculations showed that the complexes significantly bind to cytochrome P450 3A4.
1. Introduction
Schiff bases and their metal complexes have been studied extensively1−4 due to their vital roles in the main group and transition-metal coordination chemistry inherent from their simple method of preparation and structural variety.5 Their syntheses involve the condensation reaction of primary amines with active carbonyl in the presence of a suitable solvent.6 These metal complexes are prepared via the addition of the Schiff-base ligand to a metal precursor in an appropriate ratio together with suitable experimental conditions.7,8 Myriad applications of Schiff bases and their metal complexes have been reported.9 Researchers have employed them as chelating ligands in coordination chemistry,2,5,10 as a catalyst,11−14 as a dye,15,16 as an initiator in polymerization,17 and as luminescent compounds.18,19 Biologically, they have been tested as antibacterial,20−22 antifungal,23−26 antitumor,27−29 and antiviral30,31 agents including insecticides.32−34
With the emergence of cisplatin35 in the 1970s and auranofin36,37 in the 1990s, the exploration of metal complexes as drug candidates continued to attract scientists in drug design and discovery.38 As reviewed herein,39 studies showed copper (Cu) complexes’ use as anticancer compounds that could induce cancer cell death via various mechanisms such as proteosome inhibition, generation of reactive oxygen species (ROS), and DNA damage.
Earlier, Cu compounds used as therapeutic agents suffered some drawbacks,39,40 such as solubility issues, which could be solved as recently proposed by Wehbe et al.39 However, Cu-based complexes have resurfaced lately in inorganic synthesis investigations with rapt attention in which researchers have prepared Cu complexes for various medical indications.39,41−43 For instance, Cu(II)–indomethacin complexes have found application in veterinary science as anti-inflammatory medicine.44,45 This re-emergence is partly inherent from copper’s existence in many natural biological pathways that could control Cu levels and metabolize it appreciably.39
In this work, we report the synthesis and crystal structure of copper(II) Schiff-base complexes. We elucidate the complexes by single-crystal X-ray diffraction and characterize these by elemental analysis and Fourier transform infrared (FT-IR) and UV–visible spectroscopies. We calculate some quantum chemical parameters of these complexes using a density functional theory (DFT) approach. The application of the DFT methods in estimating the chemical reactivity concepts of bioactive metal complexes is relevant.46,47 Studies have shown its uses in calculating electron distribution to explore compounds’ reactivities.46−55 We predict the potentials of these complexes as drug candidates using web-based software. The prediction guided our selection of cytochrome P450 3A4 (CYP3A4) to study the complexes’ inhibitory potency against this enzyme using our own N-layered integrated molecular orbital and molecular mechanics (ONIOM) method.
Briefly, CYP3A4 is an enzyme responsible for over 50% of drug metabolism in humans; it functions as a xenobiotic expelling enzyme found in the liver. Studies have linked CYP3A4 overexpression to multidrug resistance in several diseased states such as cancer.56 Hence, its inhibition is a common practice in drug and pharmaceutical design. The natural substrates of CYP3A4 include erythromycin and protoporphyrin IX containing Fe (heme). A concise description of this enzyme’s functions, structural details, and biochemistry is available in the literature.57
2. Results and Discussion
Scheme 1 shows the synthesis route for the copper complex [Cu(L)2]. The complexes synthesized at ambient temperatures under the magnetic stirring of a methanolic solution containing Schiff-base ligands and copper acetates [Cu(OAc)2·H2O] in a 2:1 molar ratio gave an air-stable brown solid in good yield (84%). The precipitate formed was then collected by filtration and washed thoroughly with ethanol. The complex showed good solubility in chloroform, dichloromethane, acetonitrile, dimethylformamide (DMF), toluene, and dimethyl sulfoxide (DMSO). However, it is insoluble in ether, methanol, ethanol, and heptane.
Scheme 1. Synthesis of the Copper Complexes.

2.1. Spectroscopic Studies
2.1.1. UV–Vis Spectroscopy and Magnetic Susceptibilities
Figure 1 depicts the electronic absorption spectra of Cu(II) complexes 1–3 recorded in a dichloromethane solution. Generally, transition due to the ligands appeared in the UV region, while the d–d transition appeared in the visible domain.42 The spectra of 1–3 majorly showed two absorption bands in the UV region between 287 and 300 nm and 381 and 387 nm. Studies showed the assignment of the observed intense high-energy band of 287 and 300 nm to the π → π* intraligand charge transfer (ILCT) transition,9,58,59 while the band observed at around 381 and 387 nm was attributed to the ligand-to-metal charge transfer (LMCT) transition.60 In a high-concentration solution of the complex, a very weak broad band also appeared between 723 and 757 nm in the electronic spectra, and the authors assigned this to d–d transitions (2Eg → 2T2g) for Cu(II) complexes having a tetragonal distortion geometry due to the Jahn–Teller effect.61
Figure 1.
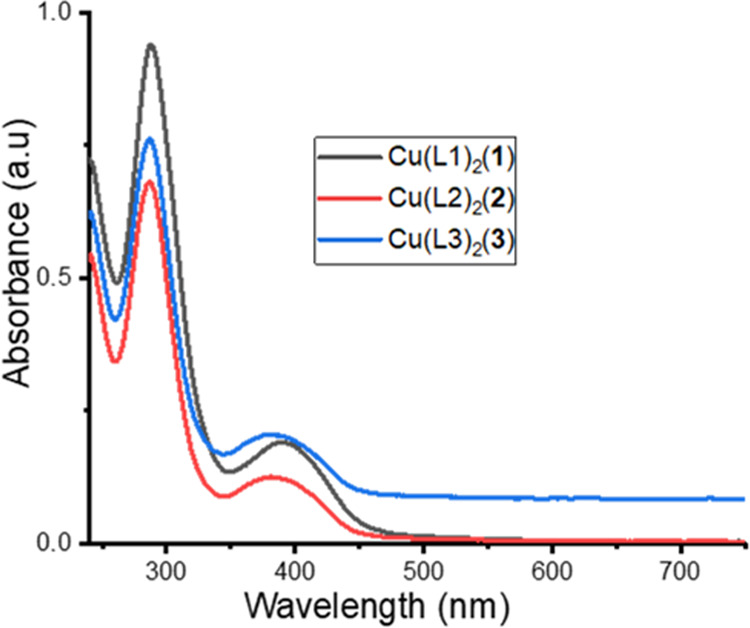
Electronic absorption spectra of complexes 1–3.
Calculated output comparison with experimental data enables the prediction of the applied theoretical model accuracy. Simulations of the UV–vis properties done at the TD-B3LYP/6-31+G(d) level of theory with dichloromethane as a solvent mimic the experimental procedure. Calculated absorption spectra were within 309.09 and 820.98 nm for the three complexes with prominent peaks at around 309.09–329.52 and 372.67–411.67 nm with weak bands at 722.50 and 820.98 nm. These values are slightly higher but seem to be qualitatively in relative order with the experimental data.
The magnetic moments for mononuclear copper(II) complexes are usually observed in the range of 1.7–2.2 B.M regardless of stereochemistry. The magnetic moments of the copper(II) complexes are in the range 1.79–1.91 B.M, which confirm the presence of mononuclear Cu(II) because those for Cu(I) are expected to be diamagnetic.
2.1.2. Fourier Transform Infrared Spectroscopy
Four major diagnostic bands appeared in the IR spectra (Figure 2) of the complexes 1–3, which are ν(C=N), ν(C–O), ν(M–O), and ν(M–N). The stretching band of ν(C=N) displayed a red shift in the complexes’ spectra compared to those for the ligands. It appeared between 1559 and 1552 cm–1 in the HL and shifted to 1550–1545 cm–1 in the complexes. This shift indicates the involvement of azomethine nitrogen during coordination.61 This lower shift might be inherent from a weakening in the C=N bond after complexation;62 we could explain this based on the donation of an electron from the imine nitrogen to the copper ion empty d-orbital.63,64 The ν(C–O) stretching vibrational bands in the complexes shifted to a region of higher wavenumber. They were observed in the wavelength 1227–1220 cm–1 and showed blue shifts of about 2–7 cm–1 relative to the parent ligands. The ν(C=C) vibrational bands for the aromatic ring appeared between 1605 and 1553 cm–1 in all three complexes. The bands at 477–444 cm–1 in the spectra assigned to ν(M–O) and ν(M–N) stretching bands, respectively, indicate the phenoxide (O−) and imine (C=N) groups of HL coordinated to Cu(II) ions. This coordination facilitates the formation of chelated complexes confirmed by the single-crystal structure of the complexes.
Figure 2.
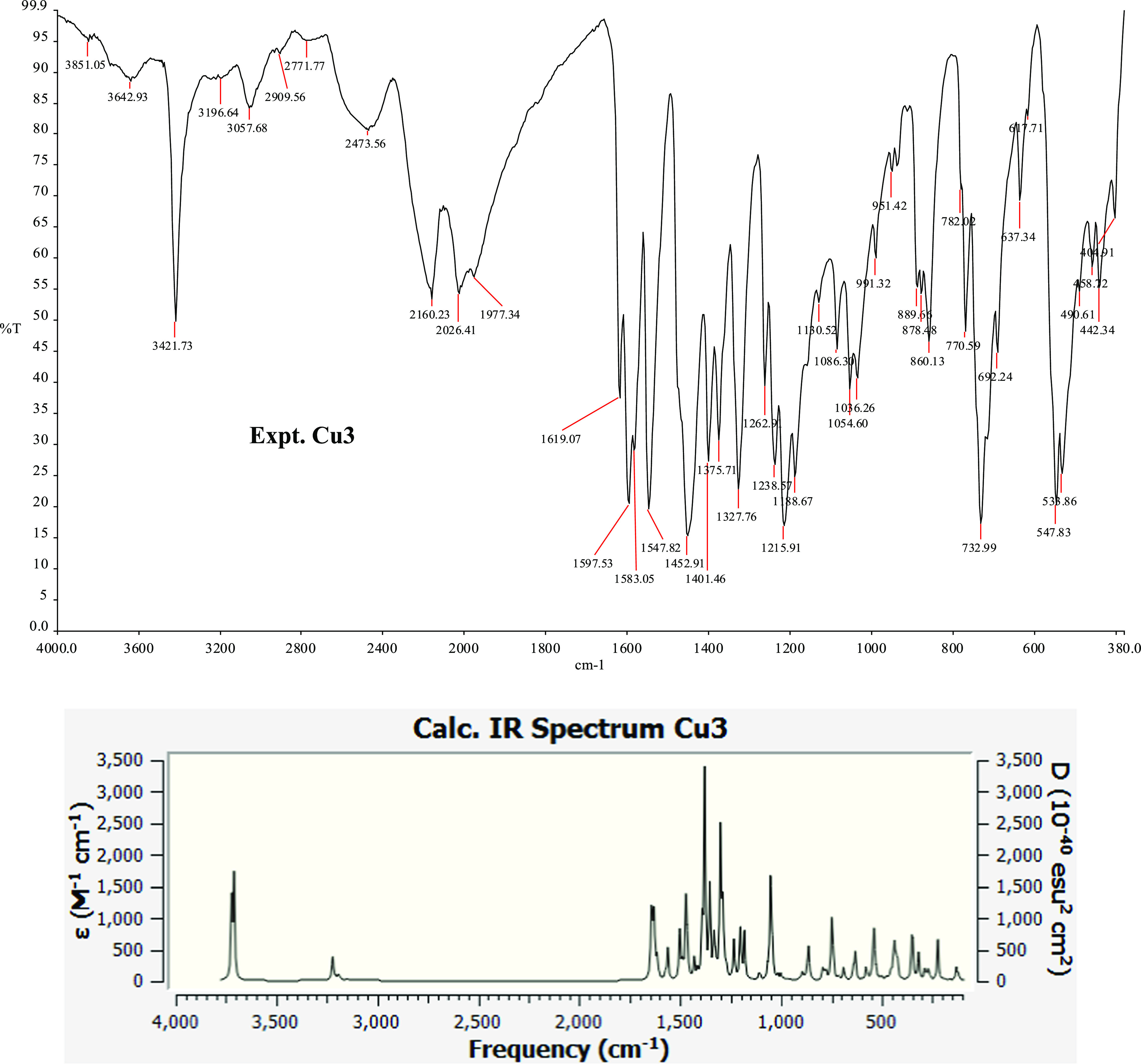
Experimental and calculated IR spectra of Cu3. Other IR spectra are available in the Supporting Information.
Post-simulation analysis of the DFT-calculated IR could provide a detailed view of the vibration associated with each spectrum. Presented in Table 1 is the output from the DFT calculation. The recorded ν(C–H) stretching bands are from the aromatic group, and the ν(O–H) vibrates at around 3722.35–3706.91 cm–1. The calculated values are close to the experimentally observed data. The tallest peak in each complex corresponds to a gentle vibration of the entire complex at 1384.13, 1392.46, and 1382.06 cm–1 for 1, 2, and 3, respectively. These calculated values correspond to experimental frequency values of 1370.91, 1374.00, and 1375.71 cm–1 for the respective complex.
Table 1. Calculated IR Values (in cm–1) for the Cu(II) Complexes in Methanol at the SMD/B3LYP/6-31+G(d) Level of Theory.
| ν(C=N) | ν(C–O) | ν(Cu–O) | ν(Cu–N) | ν(C=C) | ν(C–H) | ν(O–H) | |
|---|---|---|---|---|---|---|---|
| 1 | 1572.02–1552.34 | 1292.28–1283.72 | 455.01–436.16 | 408.47 | 1612.02–1589.71 | 3221.64–3217.07 | 3708.86–3708.35 |
| 2 | 1575.48–1562.37 | 1299.36–1296.15 | 442.35 | 430.36 | 1612.48–1579.71 | 3221.90–3213.60 | 3716.71–3706.91 |
| 3 | 1577.77–1565.65 | 1281.07–1275.39 | 468.68 | 411.82 | 1614.83–1588.56 | 3226.14–3221.38 | 3722.35–3710.83 |
2.2. Crystal Structure Description
We gave the pictorial representation of compounds 2 and 3 in Figure 3, with selected bond parameters around the metal centers in compounds 1–3 in Table 2. Unlike the previously reported crystal structure of 1, which had half of the Cu(II) complex in the asymmetric unit,4 the asymmetric unit of compounds 2 and 3 consists of complete Cu(II) complexes bearing two Schiff-base ligands HL2 and HL3, respectively. In all of the complexes, the ligands adopted a κ2N:O coordination mode via the imine nitrogen and phenolate oxygen atoms. Each of the metal centers in 2 and 3 adopted a slightly distorted square planar geometry with bond angles ranging from 87.782(4) to 173.222(4)° and 87.312(4) to 166.812(4)°, respectively.
Figure 3.

Crystal structures of Cu(II) complexes 2 and 3.
Table 2. Selected Bond Parameters for Compounds 1–3.
| compound |
|||
|---|---|---|---|
| bond parameter | 14 | 2 | 3 |
| Cu–O/Å | 1.89(1) | 1.884(9) | 1.89(1) |
| 1.880(9) | 1.90(1) | ||
| Cu–N/Å | 2.02(1) | 2.01(1) | 1.99(1) |
| 2.001(1) | 2.00(1) | ||
| O–Cu–O/deg | 180.000(2) | 173.222(4) | 166.812(4) |
| O–Cu–N/deg | 90.080(5) | 92.461(4) | 93.431(4) |
| 89.920(5) | 88.830(4) | 89.824(4) | |
| 90.081(5) | 91.870(4) | 91.851(4) | |
| 89.922(5) | 87.782(4) | 87.312(4) | |
| N–Cu–N/deg | 180.001(7) | 171.931(4) | 169.508(5) |
Replacing the methyl group in 2 with an electron-withdrawing ortho-chloro (in 3) substituent further constricts the O–Cu–O and N–Cu–N bond angles to 166.812(4) and 169.508(5)°, respectively. Also, we observe a gradual increase in the calculated root-mean-square deviation (RMSD) of the four donor atoms (N–O–N–O) around the compounds’ metal centers. As expected, the RMSD value in 1 is 0.000 Å and attributed to the ideal square planar geometry around the metal center. As the O–Cu–O and N–Cu–N angles become narrower in compounds 2 and 3, the RMSD increased to 0.126 and 0.188 Å, respectively. These values signify the degree of deformity of the square planar geometry in the Cu(II) complexes bearing ortho-substituted Schiff-base ligands. In all of the compounds, Cu–O bond distances were relatively shorter than Cu–N distances, and this observation is closely related to similar compounds in the literature.65−67
The identified plane that best describes the molecular conformation of compounds 1–3 depicted in Figure 4 is R1–4, while the dihedral angles between these planes appear in Table 3. The dihedral angle between the R1 and R1 planes gradually widens from 0.000(2)° (in 1) to 10.32(4)° (in 2) to 15.6(4)° (in 3), showing a similar trend to the square planar RMSD values. Each Cu(II) complex has two five-membered metallocycles resulting from the κ2N:O coordination mode of the Schiff-base ligands.
Figure 4.
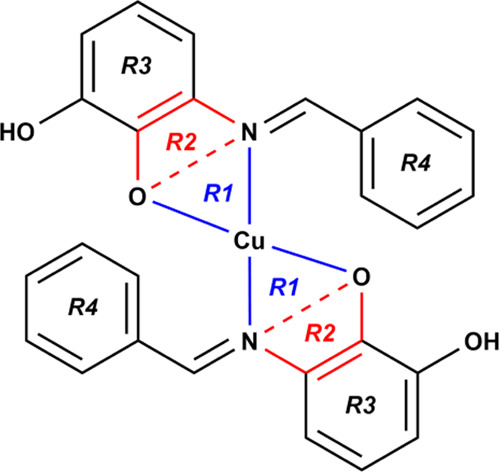
Planes identified in the Cu(II) complexes 1–3 denoted R1, R2, R3, and R4.
Table 3. Dihedral Angles between the Selected Planes.
| dihedral angle/deg |
|||
|---|---|---|---|
| compound | R1–R1 | R1–R2 | R3–R4 |
| 1(4) | 0.000(2) | 33.48(7) | 34.40(6) |
| 35.24(8) | 35.54(6) | ||
| 2 | 10.32(4) | 12.46(4) | 64.51(4) |
| 17.18(7) | 119.53(5) | ||
| 3 | 15.6(4) | 13.3(6) | 121.4(4) |
| 7.8(6) | 120.8(4) | ||
The spatial arrangement of these metallocycles shown in Figure 4 lies in planes R1 and R2. The widest dihedral angle between R1 and R2 planes in this series showed when the Cu(II) center adopted an ideal square planar geometry (in 1). The R1–R2 dihedral angle has a constriction along with the O–Cu–O and N–Cu–N bond angles as the degree of deformity of the square planar geometry increases. The geometric orientation of the coordinated ligands is best outlined by the phenyl rings and denoted R3 and R4. The ortho-substituted electron-donating group in 2 broadens the R3–R4 dihedral angle from 34.40(6) and 35.54(6)° (in 1) to 64.51(4) and 119.53(5)°. Interestingly, the R3–R4 dihedral angles in 2 indicate that the phenyl rings have gauche (64.51(4)°) and anticlinal (119.53(5)°) conformations. The presence of the ortho-chloro substituent causes the phenyl rings to have the same anticlinal (120.8(4) and 121.4(4)°) conformation.
Figure 5 shows the intra- and intermolecular interactions observed in the crystal packing of compounds 1–3, with the relevant parameters listed in Table 3. Complex 1 forms a distinct hydrogen-bonding pattern involving the hydroxyl group. The structure of 1 consists of a classical intermolecular O–H···O hydrogen bonds with a graph set descriptor between hydroxyl groups of neighboring molecules. On the contrary, hydroxyl groups in 2 and 3 form intramolecular O–H···π interactions with the ortho-methyl- and ortho-chloro-substituted phenyl ring, respectively. Interestingly, we observed intermolecular Cu···π contacts of 3.5540(7) and 3.502(6) Å in compounds 2 and 3, respectively. Moreover, compounds 2 and 3 both exhibited nonclassical C–H···O hydrogen-bonding patterns between one of the aromatic hydrogen atoms and the hydroxyl group’s oxygen atom. These unique interactions observed between these two complexes could indicate the spatial arrangement of the ligands and the slight distortion of the square planar geometry around the metal.
Figure 5.
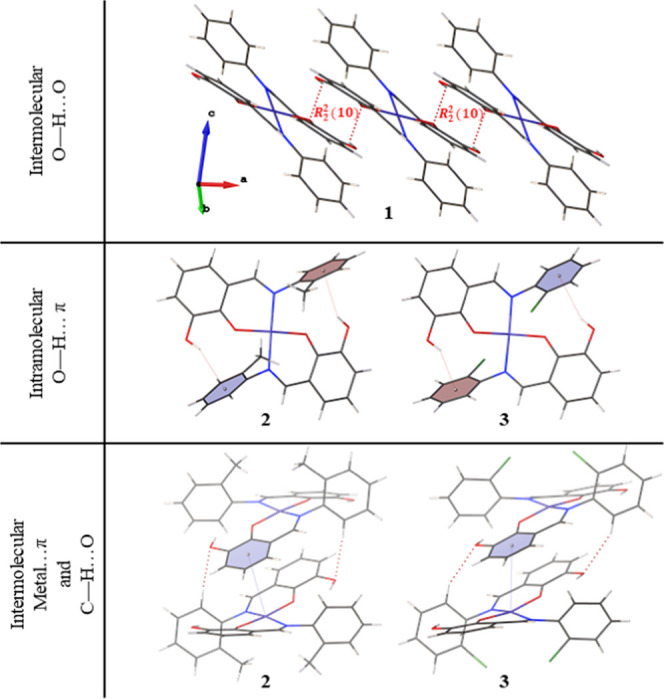
Intramolecular and intermolecular interactions observed in the crystal packing of compounds 1–3.
2.3. DFT Analysis of Structural Properties
We noticed full geometry optimization of the complexes in both gas and solvent media with no negative eigenvalue to indicate structural stability. As expected, there was a small change in the geometric arrangement of the structures in both phases. We observed an increase in the bond length distances around the coordinating atoms to the Cu atom of the complexes in the solvent phase; the bond length distances were about 0.025 Å longer in the solvent media than in the gas phase. Thus, the structures are modified in the solvent with bond and angle parameters in related order with the experimental geometries. As shown in Figure 6, the overlay of the optimized structures with the corresponding crystal structures yielded RMSD values of 0.94, 1.25, and 1.24 Å for 1, 2, and 3, respectively. This deviation is due to the orientations of the phenyl moieties and bond parameters around the metal centers after the optimization.
Figure 6.

Molecular overlays with RMSD values of the crystal structures (gray) and their respective optimized structures (orange) at the SMD/B3LYP/6-31+G(d) level of theory.
Interaction energy estimation through the counterpoise approach68 denotes the energy required to pull in the coordination of a metal atom with its ligands by partitioning the metal and its constituents separately. We fragment the fully optimized gas-phase structures into two to calculate their interaction energies. Calculated complexation energy values are −191.33, −194.25, and −197.28 kcal/mol for 1, 2, and 3, respectively. These values indicate that compound 3 is energetically favorable compared to the other two complexes. We can attribute the values in 3 to the presence of chlorine atoms in this compound (Scheme 1 and Figure 6). Bulk solvent contribution to each complex gave values of −65.88, −66.33, and −67.82 kcal/mol for 1, 2, and 3, respectively. These high energies show the importance of solvation in metal complexes; it redefines their electronic behavior and reshapes their geometries.46,47
2.3.1. NBO Study
Effective atomic charges are essential to a molecule’s reactivity prediction as a potentially active moiety.69 We provide in Figure 7 the pictorial representation charge distribution derived from orbital analysis calculated using the NBO 3.1 program70 implemented in the Gaussian 16 package. The green color indicates an atomic attraction to the positive nuclei, the black region is a neutral charge, and the red color denotes a negative nuclei attraction. Despite the positively charged state of the Cu(II) complexes, the atoms retained their default charges revealed from the analysis. The charge distribution from this NBO calculation is called natural atomic charges (NACs), and values obtained are within −0.727e and 1.171e.
Figure 7.

Charge distribution derived from natural bond orbital analysis calculated at the SMD/B3LYP/6-31+G(d) level of theory for the studied Cu(II) complex.
The copper atoms are the most positively charged, while the oxygen is the most negatively charged. All of the nitrogen atoms and the methyl (CH3) carbon atoms of the compound 2 side chain have negative charges to indicate the availability of electron pairs or unoccupied orbitals. The hydrogen atoms are neutral, while the benzene carbon atoms have mixtures of both partially negative and positive charges. The even distribution of all atomic charge types across the complexes could provide a basis for potentially appreciable molecular interaction if coupled with another system or a group of atoms.
2.3.2. Quantum Chemical Descriptors of the Studied Compounds
The lowest unoccupied molecular orbital–highest occupied molecular orbital (LUMO–HOMO) gap defines the properties of the electrons in a stable compound. Analysis of orbital distribution often reflects the ability of electrons to move from occupied orbitals to unoccupied orbitals, thus providing a fundamental basis for evaluating the chemical reactivity, selectivity, and stability of the compounds.71 In this study, energies of four molecular orbitals were reported: the highest and second-highest occupied molecular orbitals (EHOMO and EHOMO–1) as well as the lowest and second-lowest unoccupied molecular orbitals (ELUMO and ELUMO+1) (Table 4). We also present in Table 4 the quantum chemical properties derived from LUMO and HOMO energies (eqs 2–9), while we illustrate their occupancy in Figure 8.
Table 4. Quantum Chemical Descriptors for the Copper Complexes at the SMD/B3LYP/6-31+G(d) Level of Theory.
| 1 | 2 | 3 | |
|---|---|---|---|
| LUMO + 1 (eV) | –2.310 | –2.258 | –2.352 |
| LUMO (eV) | –4.565 | –4.530 | –4.590 |
| HOMO – 1 (eV) | –6.244 | –6.193 | –5.992 |
| HOMO (eV) | –5.927 | –5.933 | –6.076 |
| IP (eV) | 5.927 | 5.933 | 6.076 |
| EA (eV) | 4.565 | 4.530 | 4.590 |
| ΔE (eV) | 1.362 | 1.403 | 1.486 |
| μ (eV) | –5.246 | –5.232 | –5.333 |
| η (eV) | 0.681 | 0.702 | 0.743 |
| S (eV–1) | 1.469 | 1.425 | 1.346 |
| ω (eV) | 20.209 | 19.505 | 19.139 |
| χ | 5.246 | 5.232 | 5.333 |
Figure 8.

LUMO and HOMO plots for the studied copper complexes at the SMD/B3LYP/6-31+G(d) level of theory.
The energy difference between LUMO + 1 and LUMO is high, with LUMO having twice the value of LUMO + 1 to indicate a high-energy transition on moving from LUMO + 1 to LUMO. In contrast, HOMO – 1 and HOMO energy values are similar, denoting negligible differences in these orbital transitions (Table 4). As shown in Figure 8, unfilled orbitals are seen across most atoms, while phenoxy unit orbitals are highly filled. The chlorine atoms of compound 3 seem to have accurately filled orbitals, and this could be related to the charge distribution analysis in which this atom is neutral (Figure 7). Studies have shown that compounds with high EHOMO values easily donate their electrons compared to compounds with lower EHOMO values.72 The relative EHOMO value of −6 eV observed in this calculation reflects the plausibility of electron donation of the complexes coupled with another molecular system; we thus envisage favorable interaction. These results indicate the involvement of more energy during electron transition in the HOMO relative to LUMO.
The nucleophilicity and electron-withdrawing ability of a compound estimated through ionization potential (IP) and electron affinity (EA), respectively,71 are feasible. These quantities derived from EHOMO and ELUMO, respectively, reflect the nucleophilic properties and electron attraction power (Table 4). Accounting for the difference between EHOMO and ELUMO yields band gap (ΔE), a critical stability index.71 The calculated ΔE values are relatively low with 1.36, 1.40, and 1.49 eV for complexes 1, 2, and 3, respectively; low ΔE values have been akin to appreciable reactivity and stability.73,74
The global indices of reactivity presented in Table 4 also show that the values of the electrochemical potential, hardness, softness, electrophilicity, and electronegativity do not vary hugely from one compound to another due to their analogous nature. However, 1 produced the most favorable values in some of these parameters with the lowest ΔE and hardness index η and the highest global softness. The lowest hardness index and highest softness values denote the potential ease of reactivity in this compound. We can measure the electrophilic power of molecules by global electrophilicity indices (ω).75 Therefore, a molecule with a high electrophilicity index exhibits electrophilic behavior. The electrophilicity indices among the compounds appear to be relatively high. We noticed lower electronegativity (χ) power for the structures (Table 4), which is tenable due to the positively charged coordinating metal (Cu).
2.4. Analysis of Drug-Likeness and Pharmacokinetics of the Cu Complexes
Table 5 shows some of the results from the web-based analytical tool, SwissADME. The predictions showed that 1 obeyed the three drug-likeness theories due to its lower molecular weight (MW = 487.99 g/mol). The other two complexes have molecular weights above 500.00 g/mol. The complexes have a uniform bioavailability score of 0.55, and this is sufficient. However, they have low water-solubility indices typical of most metal complexes intended as lead compounds; possible solutions have been highlighted.39 Pharmacokinetics study of the complexes shows high gastrointestinal uptake and skin permeation rate (Table 5). The copper complexes could be applicable as a P-gp substrate and cytochrome P450 (CYP) 2C19, CYP2C9, and CYP3A4 inhibitor. Therefore, we study the binding potency of these complexes toward CYP3A4 using the hybrid ONIOM QM/MM method.
Table 5. Predicted Drug-Likeness and Pharmacokinetic Properties of the Cu(II) Complexesa.
| ADME properties | |
|---|---|
| 1 | physicochemical properties and lipophilicity |
| MW: 487.99 g/mol, H-bond donor: 2, H-bond acceptor: 4, log P: 2.75 | |
| drug-likeness theory | |
| Lipinski: yes, Veber: yes, Muegge: yes | |
| bioavailability score: 0.55 | |
| pharmacokinetics | |
| GI absorption: high, P-gp substrate: yes, CYP2C19 inhibitor: yes, CYP2C9: yes, CYP3A4 inhibitor: yes, log Kp (skin permeation): –5.56 cm/s | |
| 2 | physicochemical properties and lipophilicity |
| MW: 516.05 g/mol, H-bond donor: 2, H-bond acceptor: 4, log P: 3.31 | |
| drug-likeness theory | |
| Lipinski: no, Veber: yes, Muegge: yes | |
| bioavailability score: 0.55 | |
| pharmacokinetics | |
| GI absorption: high, P-gp substrate: yes, CYP2C19 inhibitor: yes, CYP2C9: yes, CYP3A4 inhibitor: yes, log Kp (skin permeation): –5.21 cm/s | |
| 3 | physicochemical properties and lipophilicity |
| MW: 558.90 g/mol, H-bond donor: 2, H-bond acceptor: 4, log P: 3.94 | |
| drug-likeness theory | |
| Lipinski: no, Veber: yes, Muegge: yes | |
| bioavailability score: 0.55 | |
| pharmacokinetics | |
| GI absorption: high, P-gp substrate: yes, CYP2C19 inhibitor: yes, CYP2C9: yes, CYP3A4 inhibitor: yes, log Kp (skin permeation): –4.38 cm/s |
MW = Molecular weight, H = hydrogen, GI = gastrointestinal.
2.5. Binding Affinity Study of CYP3A4 with the Cu(II) Compounds
CYP3A4 has a wide-open active site region that enables natural substrate binding, inhibitor binding, or both. This spacious active domain might allow an inhibitor to shift from its exact position during the simulation. To confirm that all of the Cu complexes did not displace from the substrate-binding region of CYP3A4 as modeled, we re-docked the heme compound in the enzyme and superimposed it with all of the optimized systems (Chart 1A). Besides the completeness of the optimized enzyme–ligand complexes confirmed with frequency analysis, the RMSD values of these complexes (Chart 1B) analyzed in the Discovery Studio76 showed low deviations. The Cu compounds remained at the heme-binding region and showed appreciable interaction with CYP3A4 active site residues (Figure 9).
Chart 1. (A) Overlay of the Initial X-ray Structure (Blue) Containing the Heme Ligand with the Optimized CYP3A4–Cu(II) Complex 1 (Pink), 2 (Green), and 3 (Cyan); (B) Binding Energy (ΔEbind) and RMSD (Å) of Cu(II) Compounds and Heme with CYP3A4 at the B3LYP/6-31+G(d): AMBER ONIOM Level.

Figure 9.

Ligand-active residues’ nonbonded interaction network for CYP3A4 binding to (A) Cu(II) compounds 1, (B) 2, (C) 3, and (D) heme natural substrate.
The recorded RMSD values averaged to 0.670 Å indicate that the applied modeling approach finds the lowest energy structures, and the system converged. The RMS deviation decreased in the order of complex 3 > 1 > 2 during binding to CYP3A4, while the heme substrate showed an RMSD value of 0.691 Å (Chart 1B). The potency of these Cu(II) compounds as CYP3A4 substrates measured as a function of their interaction energy with the enzyme showed appreciable binding. The calculated values for CYP3A4 binding to the Cu(II) complexes 1, 2 and 3 are approximately −35.5, −15.7, and −58 kcal/mol, respectively. The compound 3 binding energy value is favorable over that of the CYP3A4–heme system with −21.7 kcal/mol to denote that this Cu(II) complex could displace natural substrate binding to inhibit CYP3A4 effectively. The binding energy in the CYP3A4–complex 1 system is similar to that in the heme, indicating that complex 1 would likely bind competitively to inhibit CYP3A4. We observe a correlation between the binding energy and RMSD values; the more favored the binding (more negative), the higher (more positive) the RMSD value (Chart 1B).
Analysis of the Cu(II) complex–CYP3A4 nonbonded interactions with the Discovery Studio76 at 4.0 Å ligand–neighbor residue interaction showed the binding orientation of these complexes at the active site of the enzyme (Figure 9). The contact maps showed that all of the compounds interacted through more than 25 active residues of the CYP3A4. These amino acids showed favorable interactions in addition to residues 402–406 placed at the QM region (Figure 1A) during the calculation. The residues formed nonbonded interactions like van der Waals (vdW), conventional hydrogen bonds (HBs), alkyl, and pi–alkyl (Figure 9). These residues, revealed through computational and experimental studies,57,77 are crucial for substrate binding.
For instance, Arg77/102/337/402, Trp98, and Cys404 actively interact with the heme propionate binding.57,77,78 The interaction profile showed at least three of these residues in the Cu(II) complexes’ (Figure 9A–C) binding, while all of the six residues formed various nonbonded links to the heme compound (Figure 9D) as expected. Cys404 is a pivotal residue involved with the heme central iron stability.57 Our results showed Cys404 cruciality in the ligand-binding with pi–sulfur and pi–alkyl nonbonded interactions (Figure 9A–C). Cys404 coordinating power is well-pronounced with the metal–acceptor interaction observed in the iron of the heme–CYP3A4 natural substrate system (Figure 9D). Met414 consistently formed a pi–sulfur interaction with all of the Cu(II) complexes through one of their N-benzyl substituents. Ala267 also formed hydrophobic interactions in all of the Cu complexes.
In compound 1/2–CYP3A4 systems, Thr272 formed a conventional HB (an interaction between H and N/O/Cl atoms of a nonbonded complex) with the ligands’ parent benzoyl bases (Figure 9A,B). In the compound 3–CYP3A4 system (Figure 9C), we observe two HBs through an Arg102 and Gly406 interaction with this complex. This HB interaction coupled with chlorine atoms’ presence in 3 could perhaps account for its favorable binding affinity over the remaining complexes. Overall, vdW forces drive the binding of all of these Cu(II) complexes, and all of the interacting residues are vital to CYP3A4 inhibitor/substrate binding.57
3. Conclusions
This study showed the successful preparation of Schiff bases and Cu(II) metal complexes characterized using UV–vis and FT-IR spectrometries. We confirmed their purity through elemental analysis. The structural properties of three new copper(II) complexes reported in this study showed the crystal structures adopting square planar geometries around the central metal atom. Classical intermolecular hydrogen bonds exist in complex 1 (Figure 5). Complexes 2 and 3 showed intramolecular O–H···π interactions with the ortho-methyl- and ortho-chloro-substituted phenyl rings. The three experimentally identified Cu(II) complexes were studied at the theoretical level to give more details on their structural and spectroscopic properties (Table 2). DFT calculations revealed the quantum chemical properties (Table 4) of the complexes with closely related values due to their analogous nature. They have high HOMO energy values and show interatomic charge transfer within the Schiff-base ligand (ligand-to-ligand charge transfer, LLCT). Possible metal-Cu(II)-to-ligand charge transfer (MLCT) is also obtainable. There also exists good coordination around the copper(II) atom and the ligand (Figure 6) to indicate favorable metal–ligand charge transfer (MLCT or LMCT).
Due to the structural and chemical properties of these complexes, we evaluated their potential as drug moieties and observed that they could potentially serve as P-gp substrates that might inhibit multidrug resistance enzymes, among other favorable properties (Table 5). A theoretical study of these complexes’ interaction as potential CYP3A4 substrates showed appreciable binding affinity (Chart 1). The synthesized Schiff-base Cu(II) metal complexes, particularly complex 3, are proposed as prospective inhibitors of CYP3A4. Complex 3 has two chlorine substituents that improved the chemical properties of the positively charged copper through electronic induction as indicated with its highest electronegative values (Table 4). It also has the most satisfactory binding to CYP3A4, which is better than that of its substrate. The predicted interaction energy between Schiff-base Cu(II) complexes and CYP3A4 using the ONIOM approach is likely the maiden; literature survey reveals no research in this direction. Hence, comparing with available experimental or theoretical studies that are not related might not be sufficient. Subsequently, we would provide further insight into the activities of these compounds using classical experiments.
4. Methodology
4.1. Materials and Methods
Cu(CH3COO)2·H2O, o-toluidine, 2-chloroaniline, aniline, and 2,3-dihydoxybenzaldehyde were purchased from Merck and used without further purification. All solvents were purchased from commercial sources and used without any purification. We determine melting points (mp) and infrared spectra of the compounds on an Electrothermal 9100 and an FT-IR Perkin Elmer Spectrometer, respectively. This spectrometer model 100 is equipped with a universal ATR sampling accessory. All characteristic peaks are reported in wavenumbers (cm–1) in the range 4000–400 cm–1. The electronic absorption spectra recorded on a Shimadzu UV-3600 UV–vis–NIR spectrophotometer using quartz cuvettes have a path length of 1 cm in ranges 200–400 nm for UV and 400–900 nm for visible regions. The room temperature magnetic moment measurements were performed on a Sherwood Scientific magnetic susceptibility balance (Model Mk1).
4.2. General Synthesis
4.2.1. Synthesis of a Schiff-Base Ligand
The ligands (E)-3-((phenylimino)methyl)benzene-1,2-diol (HL1), (E)-3-((o-tolylimino)methyl)benzene-1,2-diol (HL2), and (E)-3-(((2-chlorophenyl)imino)methyl)benzene-1,2-diol (HL3) were synthesized in excellent yields following our recently reported method.79 An equimolar ratio of the appropriate primary amine and the aldehyde was ground in a poly top vial for 5 min using a glass rod to afford yellow solids.
4.2.2. Synthesis of Complex [Cu(L1)2] (1)
The ligand (HL1) (0.204 g, 0.96 mmol) was dissolved in 20 mL of ethanol and added to a stirring methanolic solution of Cu(CH3COO)2·H2O (0.100 g, 0.48 mmol), and the mixture was stirred for 2 h. The resulting brown precipitate was filtered and washed with ether and dried under vacuum. A single crystal suitable for X-ray diffraction was obtained by vapor diffusion of hexane into acetone in 7 days. Yield = 0.22 g (94%); μeff = 1.92; mp = 215–217 °C. FT-IR ν(cm–1): 3351(s), 3047(w), 1604(m), 1545(s), 1451(s), 1323(m), 1241(s), 1323(m), 1241(s), 1078(m), 478(m), 439(w). UV–vis (CH2Cl2), λmax (ε): 289(93 000), 392 (22 000), and 757(11 000) nm (M–1 cm–1). C28H24CuN2O4 calculated: C, 63.99; H, 4.13; N, 5.74; found: C, 63.54; H, 4.01, N, 5.69.
4.2.3. Synthesis of Complex [Cu(L2)2] (2)
We dissolved ligand (HL2) in 50 mL of methanol, and a methanolic solution of Cu(CH3COO)2·H2O (0.100 g, 0.48 mmol) was added dropwise and stirred for 2 h. The resulting brown precipitate was filtered and washed with ether and dried under vacuum. Suitable specimens for single-crystal X-ray diffraction were obtained by vapor diffusion of hexane into a dichloromethane solution of 2 in 7 days. Yield = 0.21 g (84%); μeff = 1.83 B.M; mp = 232–234 °C. FT-IR ν(cm–1): 3382(s), 2911(w), 1593(m), 1545(m), 1456(s), 1402(m), 1374(m), 1323(s), 1227(s), 1178(m), 1111(m), 1042(m), 887(m), 859(m), 734(s), 569(s) and 476(m). UV–vis (CH2Cl2) λmax (ε): 287(83 000), 383(20 000), and 723(11 000) nm (M–1 cm–1). C28H24CuN2O4 calculated: C, 65.17; H, 4.69; N, 5.43; found: C, 65.17; H, 4.77; N, 5.39.
4.2.4. Synthesis of Complex [Cu(L3)2] (3)
We dissolved ligand (HL3) in 50 mL of methanol, and a methanolic solution of Cu(CH3COO)2·H2O (0.10 g, 0.48 mmol) was added dropwise and stirred for 2 h. The resulting brown precipitate was filtered and washed with ether and dried under vacuum. Suitable specimens for single-crystal X-ray diffraction were obtained by vapor diffusion of hexane into a dichloromethane solution of 3 in 7 days. Yield = 0.32 g (90%); mp = 260–262 °C; μeff = 1.79. FT-IR ν(cm–1): 3421.73(s), 2909(w), 1597(m), 1547(m), 1452(s), 1401(m), 1374(m), 1215(s), 1188(m), 1086(m), 1036(m), 889(m), 860(m), 733(s), and 469(m). UV–vis (CH2Cl2) λmax (ε): 287(72 000), 383(16 000), and 723(9000) nm (M–1 cm–1). C28H24CuN2O4 calculated: C, 56.08; H, 3.26; N, 5.03; found: C, 56.01; H, 3.12; N, 5.00.
4.3. Single-Crystal X-ray Diffraction
Crystal evaluation and data collection of all complexes were done on a Bruker Smart APEXII diffractometer at Mo Kα radiation (λ = 0.71073 Å), which was equipped with an Oxford Cryostream low-temperature apparatus operating at 100 K for the sample. We collected reflections at different starting angles using the APEXII program suite to index these reflections.80 Data reduction involved using the SAINT81 software usage. The scaling and absorption corrections were applied using the SADABS82 multiscan technique. We solved and refined the structure by the direct method using the SHELXS and SHELXL programs, respectively.83 We drew the graphics of the crystal structures with the Mercury software.84 For the first nonhydrogen atoms’ refinement, we did it isotropically. Then, for the second, anisotropic refinement with the full-matrix least square method based on F2 using SHELXL was used. Geometrical positioning of all of the hydrogen atoms allowed them to ride on their parent atoms and be refined isotropically. The data set for compound 3 consists of a two-component, nonmerohedral twin. We did an HKLF5 refinement and recorded a volume ratio of 0.579(1):0.421(1) using the twin law [1̅ 0 0; 0 1̅ 0; 0.788 0 1]. Table 6 shows the crystallographic data and structure refinement parameters for the complex.
Table 6. Crystal Data and Structure Refinement for 2 and 3.
| parameters | 2 | 3 |
|---|---|---|
| empirical formula | C28H24CuN2O4 | C26H18Cl2CuN2O4 |
| formula weight | 516.03 | 556.86 |
| temperature/K | 100.02 | 100.33 |
| crystal system | monoclinic | monoclinic |
| space group | C2/c | P21/n |
| a/Å | 24.8417(6) | 12.0280(9) |
| b/Å | 14.2633(4) | 14.800(1) |
| c/Å | 17.1697(4) | 13.524(1) |
| β/deg | 128.9150(10) | 110.518(4) |
| volume/Å3 | 4733.6(2) | 2254.6(3) |
| Z | 8 | 4 |
| ρcalc g/cm3 | 1.448 | 1.641 |
| μ/mm–1 | 0.961 | 1.245 |
| F(000) | 2136.0 | 1132.0 |
| crystal size/mm3 | 0.35 × 0.31 × 0.18 | 0.23 × 0.14 × 0.08 |
| 2θ range for data collection/deg | 3.548–56.884 | 3.906–52.992 |
| index ranges | –33 ≤ h ≤ 32 −19 ≤ k ≤ 18-22 ≤ l ≤ 22 | –15 ≤ h ≤ 16, −18 ≤ k ≤ 18, −16 ≤ l ≤ 16 |
| reflections collected | 31 813 | 29 792 |
| independent reflections | 5938 [Rint = 0.0188, Rsigma =0.0142] | 29 792 [Rint = 0, Rsigma = 0.0720] |
| data/restraints/parameters | 5938/2/320 | 29 792/0/163 |
| goodness of fit on F2 | 1.065 | 1.182 |
| final R indices [I > =2σ (I)] | R1 = 0.0251, wR2 = 0.0697 | R1 = 0.1036, wR2 = 0.3217 |
| final R indices [all data] | R1 = 0.0291, wR2 = 0.0722 | R1 = 0.1133, wR2 = 0.3257 |
| largest diff. peak/hole/e Å–3 | 0.45/–0.42 | 3.22/–3.65 |
4.4. Computational Protocol
We performed all calculations within the Gaussian 16 Rev. B01 (G16) program package85 in vacuum and using methanol as the solvent. The choice of methanol as the solvent premised on the synthetic pathway of these complexes (Scheme 1). For the metal complex, we executed their electronic calculations at the DFT level of theory using the combination of Becke 3 Lee Yang Parr86,87 hybrid exchange functional and 6-31+G(d)88,89 basis set. We applied the density-based solvent model (SMD)90 to account for solvent contribution implicitly. Other studies include drug-likeness and binding affinity predictions with SwissADME91 and Gaussian 16, respectively.
4.4.1. DFT Study of the Structural Properties and Compound Characterization
We set up the structures for calculations in the GaussView 6.0.1692 suite of programs specifying minimum geometry optimization and the vibrational frequency at the B3LYP/6-31+G(d) level of theory. From the frequency calculation, we evaluated the compounds’ stability and obtained infrared (IR) properties. We calculate the UV–vis spectra using the time-dependent (TD) keyword implemented in the Gaussian package and dichloromethane as the solvent (similar to the experiment). The energy required to complex the copper(II) with the ligand atoms was estimated using the counterpoise approach.68 We calculated the bulk solvent energy contribution to the complexes, thus providing a better understanding of their behavior in a solvent. Solute–solvent interactions are required to modify the structure, energy, and total behavior of systems. As displayed in equation (eq 1), we calculated the solvation energy (ΔGsolv*) by subtracting the total gas-phase energy from the solvent phase energy.
| 1 |
4.4.2. Charge Distribution and Molecular Orbital Analyses
We used the natural bond orbital (NBO)93 approach to calculate the atomic charge distribution on each complex atom. From the NBO analysis, we can predict electron delocalization, atomic charge distribution, and intra-atomic interaction of atoms within a compound.93 The energies of the frontier molecular orbitals (FMOs),72 the highest and second-highest occupied molecular orbitals (EHOMO and EHOMO–1) and the lowest and second-lowest unoccupied molecular orbitals (ELUMO and ELUMO+1), were calculated. Some quantum chemical descriptors including ionization potential (IP),71 electron affinity (EA),71 band gap (ΔE), chemical hardness (η),94 global softness (S),94 electrochemical potential (μ),95 electrophilicity index (ω),75 and electronegativity (χ)95 were estimated as outlined in eqs 2–9.
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
4.5. Drug-Likeness and Pharmacokinetics Calculations
To establish the probability of these crystallized copper structures as potential drug moieties, we evaluated the three complexes utilizing SwissADME.91 Through this software, we predicted the drug-likeness and pharmacokinetic properties of these compounds. SwissADME is an in-depth analytical absorption, distribution, metabolism, and excretion (ADME) software developed to estimate pharmacokinetic and the drug-likeness of compounds. The protocol entails 3D structures’ upload and canonical smiles’ generation within the software interface. Subsequently, we ran the generated canonical smiles to predict the physicochemical and drug-likeness properties of the complexes.
4.6. ONIOM Calculations
The 3D coordinates of CYP3A4 are available in complex with several molecules in the RCSB Protein Data Bank (PDB),96 and we chose CYP3A4 with code 4D7D containing a natural-substrate-based inhibitor.78 The simulation protocol entails the addition of missing residues of 4D7D in MODELLER 9.19,97 separation of the fused inhibitor from the natural heme substrate, replacement of the natural substrate with our Schiff-base complexes, charge determination with the H++ server,98 and the calculation setup in GaussView 6.0.16. We calculated the binding affinity of these potent complexes toward CYP3A4 using the hybrid ONIOM quantum mechanics/molecular mechanism (QM/MM) method. Further analyses included system stability estimation and ligand–enzyme interaction profiling.
Previous literature49,99,100 showed that the hybrid B3LYP approach is sufficient with relative energies in good trend with experimental values. Therefore, we selected the B3LYP/6-31+G(d):AMBER ONIOM level to optimize the metal complexes, CYP3A4, and Cu complex–CYP3A4 systems. An illustration of this calculation setup (Figure 10A) indicates residues 402–406 and the Cu complex at the QM level with +2 charge. The MM layer has a +5 revealed through the H++ server charge distribution analysis of the enzyme. CYP3A4 residues 402–406 showed consistent interaction with all of the complexes after coupling them with the protein (Figure 10B) before the simulation. Although there are other interacting residues with these compounds, the number of atoms DFT can handle accurately and within the available resource limits our selection. We estimated the stability of the fully optimized geometries by calculating their vibrational modes to ensure no negative eigenstate values. For comparison, we applied the calculation procedure to the CYP3A4–heme natural substrate system.
Figure 10.
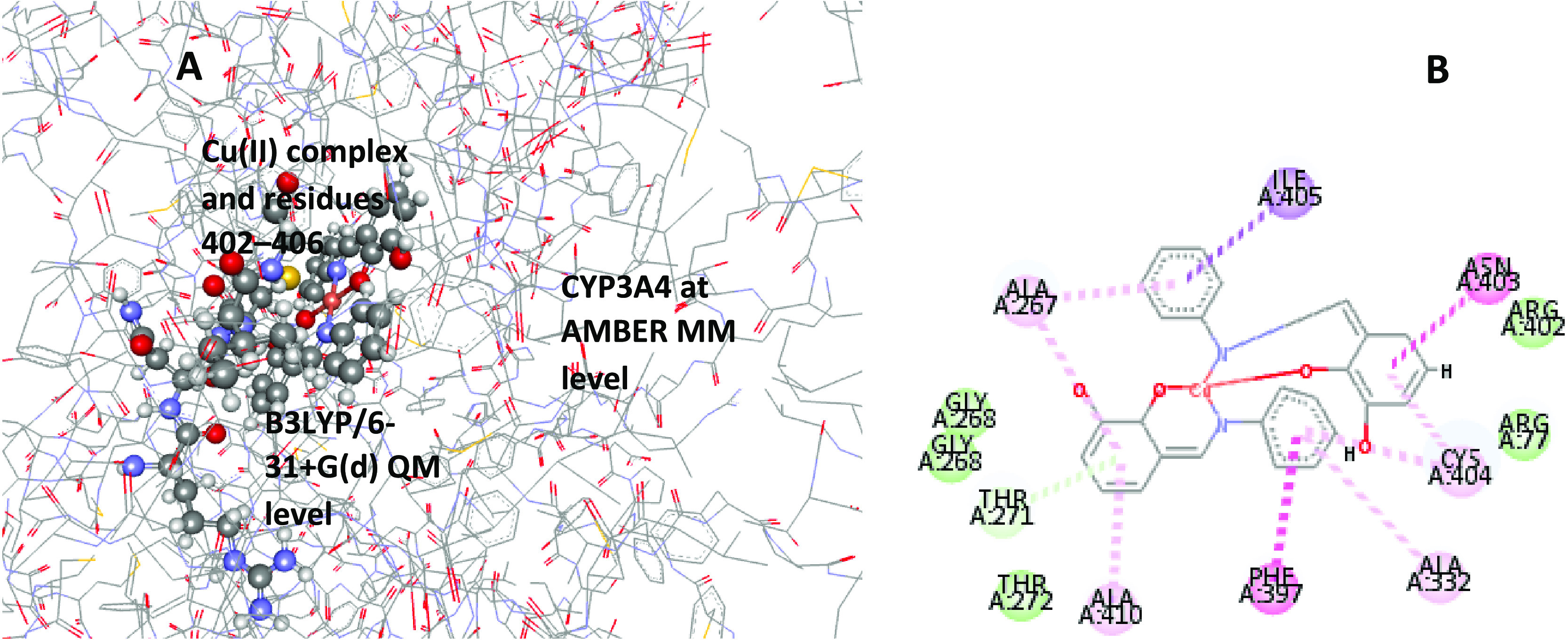
Schematic representation of (A) the two-layered ONIOM [B3LYP/6-31+G(d):AMBER] model for the CYP3A4–Cu(II) complex simulation and (B) interaction of the Cu complex with the enzyme before the calculation.
The calculated total energy change using eq 10 depicts the binding potency (ΔEbind) of these Cu complexes toward CYP3A4. ΔEbind represents the difference in the energy components existing between the CYP3A4 and the complexes, ΔEmodel is the energy of the model system calculated at the high (QM) and low level (MM), and ΔEreal represents the energy of the entire (real) system at the MM level. We performed the ONIOM calculation in the gas phase due to the large memory required for implicit or explicit modeling. Note that the ONIOM approach often comes with some limitations like bond and angle parameters’ definition for a system. Thus, we use input parameters from reports in the literature.101,102
| 10 |
Acknowledgments
The authors acknowledge the Department of Chemical Sciences, University of Johannesburg, School of Chemistry and Physics, University of KwaZulu-Natal, and the Centre for High-Performance Computing (www.chpc.ac.za) in South Africa for financial and technical support. Dr. T.L.Y. is grateful to the Faculty of Science (FRC/URC) postdoctoral support, the University of Johannesburg, for the awarded Postdoctoral research fellowship. Dr. M.M.L. is grateful to the South Africa National Research Foundation (NRF) financial support for awarding Grant 120707 DSI/NRF Innovation Postdoctoral Fellowship 2020.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c00906.
The authors declare no competing financial interest.
Supplementary Material
References
- Prakash A.; Adhikari D. Application of Schiff bases and their metal complexes-A Review. Int. J. ChemTech Res. 2011, 3, 1891–1896. [Google Scholar]
- Di Bernardo P.; Zanonato P.; Tamburini S.; Tomasin P.; Vigato P. Complexation behaviour and stability of Schiff bases in aqueous solution. The case of an acyclic diimino (amino) diphenol and its reduced triamine derivative. Dalton Trans. 2006, 4711–4721. 10.1039/b604211b. [DOI] [PubMed] [Google Scholar]
- Yusuf T. L.; Oladipo S. D.; Olagboye S. A.; Zamisa S. J.; Tolufashe G. F. Solvent-free synthesis of nitrobenzyl Schiff bases: Characterization, antibacterial studies, density functional theory and molecular docking studies. J. Mol. Struct. 2020, 1222, 128857 10.1016/j.molstruc.2020.128857. [DOI] [Google Scholar]
- Olagboye S. A.; Yusuf T. L.; Oladipo S. D.; Zamisa S. J. Crystal structure of bis (2-hydroxy-6-((phenylimino) methyl) phenolato-κ2N, O) copper (II), C26H20CuN2O4. Z. Kristallogr. - New Cryst. Struct. 2020, 235, 689–692. 10.1515/ncrs-2019-0900. [DOI] [Google Scholar]
- Keypour H.; Rezaeivala M.; Valencia L.; Pérez-Lourido P.; Khavasi H. R. Synthesis and characterization of some new Co(II) and Cd(II) macroacyclic Schiff-base complexes containing piperazine moiety. Polyhedron 2009, 28, 3755–3758. 10.1016/j.poly.2009.08.021. [DOI] [Google Scholar]
- da Silva C. M.; da Silva D. L.; Modolo L. V.; Alves R. B.; de Resende M. A.; Martins C. V. B.; de Fátima Â. Schiff bases: A short review of their antimicrobial activities. J. Adv. Res. 2011, 2, 1–8. 10.1016/j.jare.2010.05.004. [DOI] [Google Scholar]
- Wesley Jeevadason A.; Kalidasa Murugavel K.; Neelakantan M. A. Review on Schiff bases and their metal complexes as organic photovoltaic materials. Renewable Sustainable Energy Rev. 2014, 36, 220–227. 10.1016/j.rser.2014.04.060. [DOI] [Google Scholar]
- Jeewoth T.; Li Kam Wah H.; Bhowon M. G.; Ghoorohoo D.; Babooram K. Synthesis and anti-bacterial/catalytic properties of Schiff bases and Schiff base metal complexes derived from 2, 3-diaminopyridine. Synth. React. Inorg. Met.-Org. Chem. 2000, 30, 1023–1038. 10.1080/00945710009351817. [DOI] [Google Scholar]
- Salehi M.; Faghani F.; Kubicki M.; Bayat M. New complexes of Ni(II) and Cu(II) with tridentate ONO Schiff base ligand: synthesis, crystal structures, electrochemical and theoretical investigation. J. Iran. Chem. Soc. 2018, 15, 2229–2240. 10.1007/s13738-018-1412-1. [DOI] [Google Scholar]
- Temel H.; Ziyadanoğullari B.; Aydin I.; Aydin F. Synthesis, spectroscopic and thermodynamic studies of new transition metal complexes with N,N′-bis(2-hydroxynaphthalin-1-carbaldehydene)-1,2-bis(m-aminophenoxy)ethane and their determination by spectrophotometric methods. J. Coord. Chem. 2005, 58, 1177–1185. 10.1080/00958970500078890. [DOI] [Google Scholar]
- Tümer M.; Akgün E.; Toroğlu S.; Kayraldiz A.; Dönbak L. Synthesis and characterization of Schiff base metal complexes: their antimicrobial, genotoxicity and electrochemical properties. J. Coord. Chem. 2008, 61, 2935–2949. 10.1080/00958970801989902. [DOI] [Google Scholar]
- Champouret Y. D.; Fawcett J.; Nodes W. J.; Singh K.; Solan G. A. Spacially confined M2 centers (M = Fe, Co, Ni, Zn) on a sterically bulky binucleating support: Synthesis, structures and ethylene oligomerization studies. Inorg. Chem. 2006, 45, 9890–9900. 10.1021/ic061286x. [DOI] [PubMed] [Google Scholar]
- Liu X.; Manzur C.; Novoa N.; Celedón S.; Carrillo D.; Hamon J.-R. Multidentate unsymmetrically-substituted Schiff bases and their metal complexes: Synthesis, functional materials properties, and applications to catalysis. Coord. Chem. Rev. 2018, 357, 144–172. 10.1016/j.ccr.2017.11.030. [DOI] [Google Scholar]
- Laidler D. A.; Milner D. J. Asymmetric synthesis of cyclopropane carboxylates: Catalysis of diazoacetate reactions by copper(II) Schiff base complexes derived from α-amino acids. J. Organomet. Chem. 1984, 270, 121–129. 10.1016/0022-328X(84)80341-1. [DOI] [Google Scholar]
- Befta U.1: 2 Chromium Complex Dyes. EP Patent EP0150676A31985, p 120.
- Mennicke W.Westphal, Mixtures of 1: 2 chromium complex dyes, Ger Offen3, 409,082 (to Bayer AG)19 Sep. 1985, DE Appl, 13 Mar 1984 Chemical Abstracts 1986, 111359.
- Young R.; Cooper G. Dissociation of intermolecular linkages of the sperm head and tail by primary amines, aldehydes, sulphydryl reagents and detergents. Reproduction 1983, 69, 1–10. 10.1530/jrf.0.0690001. [DOI] [PubMed] [Google Scholar]
- Hamada Y.; Sano T.; Fujii H.; Nishio Y.; Takahashi H.; Shibata K. White-light-emitting material for organic electroluminescent devices. Jpn. J. Appl. Phys. 1996, 35, L1339. 10.1143/JJAP.35.L1339. [DOI] [Google Scholar]
- Kawamoto T.; Nishiwaki M.; Tsunekawa Y.; Nozaki K.; Konno T. Synthesis and characterization of luminescent zinc (II) and cadmium (II) complexes with N, S-chelating Schiff base ligands. Inorg. Chem. 2008, 47, 3095–3104. 10.1021/ic7020758. [DOI] [PubMed] [Google Scholar]
- Salvat A.; Antonnacci L.; Fortunato R. H.; Suárez E. Y.; Godoy H. Screening of some plants from Northern Argentina for their antimicrobial activity. Lett. Appl. Microbiol. 2001, 32, 293–297. 10.1046/j.1472-765X.2001.00923.x. [DOI] [PubMed] [Google Scholar]
- Yousif E.; Majeed A.; Al-Sammarrae K.; Salih N.; Salimon J.; Abdullah B. Metal complexes of Schiff base: preparation, characterization and antibacterial activity. Arabian J. Chem. 2017, 10, S1639–S1644. 10.1016/j.arabjc.2013.06.006. [DOI] [Google Scholar]
- Al Zoubi W.; Al-Hamdani A. A. S.; Ahmed S. D.; Ko Y. G. Synthesis, characterization, and biological activity of Schiff bases metal complexes. J. Phys. Org. Chem. 2018, 31, e3752 10.1002/poc.3752. [DOI] [Google Scholar]
- Gudasi K. B.; Patil M. S.; Vadavi R. S.; Shenoy R. V.; Patil S. A.; Nethaji M. X-ray crystal structure of the N-(2-hydroxy-1-naphthalidene) phenylglycine Schiff base. Synthesis and characterization of its transition metal complexes. Transition Met. Chem. 2006, 31, 580–585. 10.1007/s11243-006-0031-3. [DOI] [Google Scholar]
- Cukurovali A.; Yilmaz İ.; Kirbag S. Spectroscopic characterization and biological activity of salicylaldehyde thiazolyl hydrazone ligands and their metal complexes. Transition Met. Chem. 2006, 31, 207–213. 10.1007/s11243-005-6353-8. [DOI] [Google Scholar]
- Iftikhar B.; Javed K.; Khan M. S. U.; Akhter Z.; Mirza B.; McKee V. Synthesis, characterization and biological assay of Salicylaldehyde Schiff base Cu(II) complexes and their precursors. J. Mol. Struct. 2018, 1155, 337–348. 10.1016/j.molstruc.2017.11.022. [DOI] [Google Scholar]
- Tadavi S. K.; Yadav A. A.; Bendre R. S. Synthesis and characterization of a novel schiff base of 1,2-diaminopropane with substituted salicyaldehyde and its transition metal complexes: Single crystal structures and biological activities. J. Mol. Struct. 2018, 1152, 223–231. 10.1016/j.molstruc.2017.09.112. [DOI] [Google Scholar]
- Malik M. A.; Dar O. A.; Gull P.; Wani M. Y.; Hashmi A. A. Heterocyclic Schiff base transition metal complexes in antimicrobial and anticancer chemotherapy. MedChemComm 2018, 9, 409–436. 10.1039/C7MD00526A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andiappan K.; Sanmugam A.; Deivanayagam E.; Karuppasamy K.; Kim H.-S.; Vikraman D. In vitro cytotoxicity activity of novel Schiff base ligand–lanthanide complexes. Sci. Rep. 2018, 8, 3054 10.1038/s41598-018-21366-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanimurugan A.; Kulandaisamy A. DNA, in vitro antimicrobial/anticancer activities and biocidal based statistical analysis of Schiff base metal complexes derived from salicylalidene-4-imino-2,3-dimethyl-1-phenyl-3-pyrazolin-5-one and 2-aminothiazole. J. Organomet. Chem. 2018, 861, 263–274. 10.1016/j.jorganchem.2018.02.051. [DOI] [Google Scholar]
- Meng F.; Zhao Q.; Li M.; Xin Y.. Yingyong Huaxue, 19 (2002) 1183-1185 Chemical Abstracts, 2003, 330746.
- Kumar K. S.; Ganguly S.; Veerasamy R.; De Clercq E. Synthesis, antiviral activity and cytotoxicity evaluation of Schiff bases of some 2-phenyl quinazoline-4(3)H-ones. Eur. J. Med. Chem. 2010, 45, 5474–5479. 10.1016/j.ejmech.2010.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi K.; Kureshy R.; Khan N.; Tabassum S.; Zaidi S. Schiff base derived from sulfane thoxazole and salicylaldehyde or thiophene-2-aldehydes. Inorg. Chim. Acta 1988, 151, 95–100. 10.1016/S0020-1693(00)91888-7. [DOI] [Google Scholar]
- Zhu X.; Wang C.; Dang Y.; Zhou H.; Wu Z.; Liu Z.; Ye D.; Zhou Q. The Schiff base N-salicylidene-O, S-dimethylthiophosphorylimine and its metal complexes: synthesis, characterization and insecticidal activity studies. Synth. React. Inorg. Met.-Org. Chem. 2000, 30, 625–636. 10.1080/00945710009351787. [DOI] [Google Scholar]
- Nath M.; Goyal S. Spectral studies and bactericidal, fungicidal, insecticidal and parasitological activities of organotin (IV) complexes of thio schiff bases having no donor atoms. Met.-Based Drugs 1995, 2, 297–309. 10.1155/MBD.1995.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loehrer P. J.; Einhorn L. H. Cisplatin. Ann. Intern. Med. 1984, 100, 704–713. 10.7326/0003-4819-100-5-704. [DOI] [PubMed] [Google Scholar]
- Hill D. T.; Lantos I.; Sutton B. M.. Process and Intermediate for Preparing Auranofin. U.S. Patent US4133952A1979.
- Nardon C.; Pettenuzzo N.; Fregona D. Gold Complexes for Therapeutic Purposes: an Updated Patent Review (2010–2015). Curr. Med. Chem. 2016, 23, 3374–3403. 10.2174/0929867323666160504103843. [DOI] [PubMed] [Google Scholar]
- Rigamonti L.; Reginato F.; Ferrari E.; Pigani L.; Gigli L.; Demitri N.; Kopel P.; Tesarova B.; Heger Z. From solid state to in vitro anticancer activity of copper(ii) compounds with electronically-modulated NNO Schiff base ligands. Dalton Trans. 2020, 49, 14626–14639. 10.1039/D0DT03038D. [DOI] [PubMed] [Google Scholar]
- Wehbe M.; Leung A. W.; Abrams M. J.; Orvig C.; Bally M. B. A Perspective–can copper complexes be developed as a novel class of therapeutics?. Dalton Trans. 2017, 46, 10758–10773. 10.1039/C7DT01955F. [DOI] [PubMed] [Google Scholar]
- Weiss R. B.; Christian M. C. New cisplatin analogues in development. Drugs 1993, 46, 360–377. 10.2165/00003495-199346030-00003. [DOI] [PubMed] [Google Scholar]
- Wang T.; Guo Z. Copper in medicine: homeostasis, chelation therapy and antitumor drug design. Curr. Med. Chem. 2006, 13, 525–537. 10.2174/092986706776055742. [DOI] [PubMed] [Google Scholar]
- Oladipo S. D.; Omondi B.; Mocktar C. Co(III) N,N′-diarylformamidine dithiocarbamate complexes: Synthesis, characterization, crystal structures and biological studies. Appl. Organomet. Chem. 2020, 34, e5610 10.1002/aoc.5610. [DOI] [Google Scholar]
- Oladipo S. D.; Mocktar C.; Omondi B. In vitro biological studies of heteroleptic Ag (I) and Cu(I) unsymmetrical N, N′-diarylformamidine dithiocarbamate phosphine complexes; the effect of the metal center. Arabian J. Chem. 2020, 13, 6379–6394. 10.1016/j.arabjc.2020.05.039. [DOI] [Google Scholar]
- Weder J. E.; Dillon C. T.; Hambley T. W.; Kennedy B. J.; Lay P. A.; Biffin J. R.; Regtop H. L.; Davies N. M. Copper complexes of non-steroidal anti-inflammatory drugs: an opportunity yet to be realized. Coord. Chem. Rev. 2002, 232, 95–126. 10.1016/S0010-8545(02)00086-3. [DOI] [Google Scholar]
- Weder J. E.; Hambley T. W.; Kennedy B. J.; Lay P. A.; MacLachlan D.; Bramley R.; Delfs C. D.; Murray K. S.; Moubaraki B.; Warwick B.; et al. Anti-inflammatory dinuclear copper (ii) complexes with indomethacin. synthesis, magnetism and EPR spectroscopy. Crystal structure of the N, N-dimethylformamide adduct. Inorg. Chem. 1999, 38, 1736–1744. 10.1021/ic981100x. [DOI] [PubMed] [Google Scholar]
- Ndagi U.; Lawal M. M.; Soliman M. E. DFT Study of the Structural and Electronic Properties of Selected Organogold (III) Compounds with Characteristic Anticancer Activity. Russ. J. Phys. Chem. A 2019, 93, 1543–1558. 10.1134/S0036024419080302. [DOI] [Google Scholar]
- Lawal M. M.; Lawal I. A.; Klink M. J.; Tolufashe G. F.; Ndagi U.; Kumalo H. M. Density functional theory study of gold (III)-dithiocarbamate complexes with characteristic anticancer potentials. J. Inorg. Biochem. 2020, 206, 111044 10.1016/j.jinorgbio.2020.111044. [DOI] [PubMed] [Google Scholar]
- Galvan M.; Vela A.; Gazquez J. L. Chemical reactivity in spin-polarized density functional theory. J. Phys. Chem. A. 1988, 92, 6470–6474. 10.1021/j100333a056. [DOI] [Google Scholar]
- Ugbaja S. C.; Sanusi Z. K.; Appiah-Kubi P.; Lawal M. M.; Kumalo H. M. Computational modelling of potent β-secretase (BACE1) inhibitors towards Alzheimer’s disease treatment. Biophys. Chem. 2021, 270, 106536 10.1016/j.bpc.2020.106536. [DOI] [PubMed] [Google Scholar]
- Magwenyane A. M.; Mhlongo N. N.; Lawal M. M.; Amoako D. G.; Somboro A. M.; Sosibo S. C.; Shunmugam L.; Khan R. B.; Kumalo H. M. Understanding the Hsp90 N-Terminal Dynamics: Structural and Molecular Insights into the Therapeutic Activities of Anticancer Inhibitors Radicicol (RD) and Radicicol Derivative (NVP-YUA922). Molecules 2020, 25, 1785 10.3390/molecules25081785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejalonibu M. A.; Elrashedy A. A.; Lawal M. M.; Soliman M. E.; Sosibo S. C.; Kumalo H. M.; Mhlongo N. N. Dual targeting approach for Mycobacterium tuberculosis drug discovery: insights from DFT calculations and molecular dynamics simulations. Struct. Chem. 2020, 31, 557–571. 10.1007/s11224-019-01422-w. [DOI] [Google Scholar]
- Akinpelu O. I.; Lawal M. M.; Kumalo H. M.; Mhlongo N. N. Computational studies of the properties and activities of selected trisubstituted benzimidazoles as potential antitubercular drugs inhibiting MTB-FtsZ polymerization. J. Biomol. Struct. Dyn. 2020, 1–13. 10.1080/07391102.2020.1830176. [DOI] [PubMed] [Google Scholar]
- Akinpelu O. I.; Lawal M. M.; Kumalo H. M.; Mhlongo N. N. Drug repurposing: Fusidic acid as a potential inhibitor of M. tuberculosis FtsZ polymerization–Insight from DFT calculations, molecular docking and molecular dynamics simulations. Tuberculosis 2020, 121, 101920 10.1016/j.tube.2020.101920. [DOI] [PubMed] [Google Scholar]
- Adeowo F. Y.; Ejalonibu M. A.; Elrashedy A. A.; Lawal M. M.; Kumalo H. M. Multi-target approach for Alzheimer’s disease treatment: computational biomolecular modeling of cholinesterase enzymes with a novel 4-N-phenylaminoquinoline derivative reveal promising potentials. J. Biomol. Struct. Dyn. 2020, 121, 1–17. 10.1080/07391102.2020.1826129. [DOI] [PubMed] [Google Scholar]
- Ibeji C. U.; Lawal M. M.; Tolufashe G. F.; Govender T.; Naicker T.; Maguire G. E.; Lamichhane G.; Kruger H. G.; Honarparvar B. The Driving Force for the Acylation of β-Lactam Antibiotics by l, d-Transpeptidase 2: Quantum Mechanics/Molecular Mechanics (QM/MM) Study. ChemPhysChem 2019, 20, 1126–1134. 10.1002/cphc.201900173. [DOI] [PubMed] [Google Scholar]
- Sun X.; Tang S.; Hou B.; Duan Z.; Liu Z.; Li Y.; He S.; Wang Q.; Chang Q. Overexpression of P-glycoprotein, MRP2, and CYP3A4 impairs intestinal absorption of octreotide in rats with portal hypertension. BMC Gastroenterol. 2021, 21, 2 10.1186/s12876-020-01532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyenhuis D.Cytochrome P450 3A4, https://collab.its.virginia.edu/access/content/group/f85bed6c-45d2-4b18-b868-6a2353586804/2/Ch00_Nyenhuis_D_Cytochrome_P450_3A4/Ch00_Nyenhuis_D_Cytochrome_P450_3A4_CYP3A4.html.
- Zolezzi S.; Decinti A.; Spodine E. Syntheses and characterization of copper(II) complexes with Schiff-base ligands derived from ethylenediamine, diphenylethylenediamine and nitro, bromo and methoxy salicylaldehyde. Polyhedron 1999, 18, 897–904. 10.1016/S0277-5387(98)00376-3. [DOI] [Google Scholar]
- Satheesh C. E.; Raghavendra Kumar P.; Shivakumar N.; Lingaraju K.; Murali Krishna P.; Rajanaika H.; Hosamani A. Synthesis, structural characterization, antimicrobial and DNA binding studies of homoleptic zinc and copper complexes of NO Schiff bases derived from homoveratrylamine. Inorg. Chim. Acta 2019, 495, 118929 10.1016/j.ica.2019.05.028. [DOI] [Google Scholar]
- Satheesh C. E.; Raghavendra Kumar P.; Sharma P.; Lingaraju K.; Palakshamurthy B. S.; Raja Naika H. Synthesis, characterisation and antimicrobial activity of new palladium and nickel complexes containing Schiff bases. Inorg. Chim. Acta 2016, 442, 1–9. 10.1016/j.ica.2015.11.017. [DOI] [Google Scholar]
- Hazra M.; Dolai T.; Pandey A.; Dey S. K.; Patra A. Synthesis and Characterisation of Copper(II) Complexes with Tridentate NNO Functionalized Ligand: Density Function Theory Study, DNA Binding Mechanism, Optical Properties, and Biological Application. Bioinorg. Chem. Appl. 2014, 2014, 104046 10.1155/2014/104046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şenol C.; Hayvali Z.; Dal H.; Hökelek T. Syntheses, characterizations and structures of NO donor Schiff base ligands and nickel(II) and copper(II) complexes. J. Mol. Struct. 2011, 997, 53–59. 10.1016/j.molstruc.2011.04.037. [DOI] [Google Scholar]
- Signorini O.; Dockal E. R.; Castellano G.; Oliva G. Synthesis and characterization of aquo[N,N′-ethylenebis(3-ethoxysalicylideneaminato)]dioxouranium(VI). Polyhedron 1996, 15, 245–255. 10.1016/0277-5387(95)00243-L. [DOI] [Google Scholar]
- Rezvani Z.; Abbasi A. R.; Nejati K.; Seyedahmadian M. Syntheses, characterization and glass-forming properties of new bis[5-((4-ndodecyloxyphenyl)azo)-N-(4-nalkoxyphenyl)-salicylaldiminato]nickel (II) complex homologues. Polyhedron 2005, 24, 1461–1470. 10.1016/j.poly.2005.03.096. [DOI] [Google Scholar]
- Sixt T.; Kaim W. Copper (I) complexes with N, O-donor Schiff base ligands related to intermediate forms of the TPQ cofactor in amine oxidases. Inorg. Chim. Acta 2000, 300–302, 762–768. 10.1016/S0020-1693(99)00612-X. [DOI] [Google Scholar]
- Mansilla-Koblavi F.; Tenon J. A.; Toure S.; Ebby N.; Lapasset J.; Carles M. Une série de N-(2, 3-dihydroxybenzilidene) amines: manifestation d’équilibres tautomères. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1995, 51, 1595–1602. 10.1107/S0108270194002945. [DOI] [Google Scholar]
- Lavaee P.; Eshtiagh-Hosseini H.; Housaindokht M. R.; Mague J. T.; Esmaeili A. A.; Abnous K. Synthesis, characterization and fluorescence properties of Zn(II) and Cu(II) complexes: DNA binding study of Zn(II) complex. J. Fluoresc. 2016, 26, 333–344. 10.1007/s10895-015-1719-6. [DOI] [PubMed] [Google Scholar]
- Boys S. F.; Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors (Reprinted from Molecular Physics, vol 19, pg 553-566, 1970). Mol. Phys. 2002, 100, 65–73. 10.1080/00268970110088901. [DOI] [Google Scholar]
- Frezza M.; Hindo S.; Chen D.; Davenport A.; Schmitt S.; Tomco D.; Dou Q. P. Novel Metals and Metal Complexes as Platforms for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. 10.2174/138161210791209009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glendening E. D.; Landis C. R.; Weinhold F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. 10.1002/jcc.23266. [DOI] [PubMed] [Google Scholar]
- Zhan C.-G.; Nichols J. A.; Dixon D. A. Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J. Phys. Chem. A 2003, 107, 4184–4195. 10.1021/jp0225774. [DOI] [Google Scholar]
- Bradley J. D.; Gerrans G. C. Frontier molecular orbitals. A link between kinetics and bonding theory. J. Chem. Educ. 1973, 50, 463 10.1021/ed050p463. [DOI] [Google Scholar]
- Lawal M. M.; Govender T.; Maguire G. E.; Kruger H. G.; Honarparvar B. DFT study of the acid-catalyzed esterification reaction mechanism of methanol with carboxylic acid and its halide derivatives. Int. J. Quantum Chem. 2018, 118, e25497 10.1002/qua.25497. [DOI] [Google Scholar]
- Lawal M. M.; Govender T.; Maguire G. E.; Honarparvar B.; Kruger H. G. Mechanistic investigation of the uncatalyzed esterification reaction of acetic acid and acid halides with methanol: a DFT study. J. Mol. Model. 2016, 22, 235 10.1007/s00894-016-3084-z. [DOI] [PubMed] [Google Scholar]
- Liu S. B. Dynamic behavior of chemical reactivity indices in density functional theory: A Bohn-Oppenheimer quantum molecular dynamics study. J. Chem. Sci. 2005, 117, 477–483. 10.1007/BF02708352. [DOI] [Google Scholar]
- Biovia D. S.Discovery Studio Modeling Environment, Release 2017, San Diego, CA, USA, 2016.
- Ekroos M.; Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13682–13687. 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P.; Chamberlin A. R.; Poulos T. L.; Sevrioukova I. F. Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J. Med. Chem. 2016, 59, 4210–4220. 10.1021/acs.jmedchem.5b01146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olagboye S. A.; Yusuf T. L.; Oladipo S. D.; Zamisa S. J. Crystal structure of (E)-1-(2-nitrophenyl)-N-(o-tolyl) methanimine, C14H12N2O2. Z. Kristallogr. - New Cryst. Struct. 2020, 235, 833–836. 10.1515/ncrs-2020-0034. [DOI] [Google Scholar]
- Bruker,APEXII; APEXII Bruker AXS Inc: Madison, Wisconsin, USA, 2009.
- Bruker,SAINT; SAINT Bruker AXS InC: Madison, Wisconsin, USA, 2009.
- Bruker,SADABS; SADABS Bruker AXS Inc: Madison, Wisconsin, USA, 2009.
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Macrae C. F.; et al. Mercury CSD 2.0 - New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.. et al. Gaussian 16, rev. B.01, Wallingford, CT, 2016.
- Becke A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Lee C.; Yang W.; P R. G. Development of Colle-Salvetti correlation-energy formula into a functional of electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Rassolov V. A.; Ratner M. A.; Pople J. A.; Redfern P. C.; Curtiss L. A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. 10.1002/jcc.1058. [DOI] [Google Scholar]
- Hehre W. J.; Stewart R. F.; Pople J. A. self-consistent molecular-orbital methods. i. use of gaussian expansions of Slater-type atomic orbitals. J. Chem. Phys. 1969, 51, 2657–2664. 10.1063/1.1672392. [DOI] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennington R.; Keith T.; Millam J.; Eppinnett K.; Hovell W.; Gilliland R.. GaussView, version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016.
- Weinhold F.; Landis C. R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. 2001, 2, 91–104. 10.1039/B1RP90011K. [DOI] [Google Scholar]
- Yang W. T.; Parr R. G. Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 6723–6726. 10.1073/pnas.82.20.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz M. Density functional approach to frontier controlled reactions. J. Am. Chem. Soc. 1987, 109, 4823–4825. 10.1021/ja00250a012. [DOI] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B.; Sali A.. Protein structure modeling with MODELLER. In Methods in Molecular Biology; Springer: 2014; Vol. 1137, pp 1–15. [DOI] [PubMed] [Google Scholar]
- Anandakrishnan R.; Aguilar B.; Onufriev A. V. H++ 3.0: automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. 10.1093/nar/gks375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanusi Z. K.; Govender T.; Maguire G. E.; Maseko S. B.; Lin J.; Kruger H. G.; Honarparvar B. An insight to the molecular interactions of the FDA approved HIV PR drugs against L38L↑ N↑ L PR mutant. J. Comput.-Aided Mol. Des. 2018, 32, 459–471. 10.1007/s10822-018-0099-9. [DOI] [PubMed] [Google Scholar]
- Sanusi Z.; Govender T.; Maguire G.; Maseko S.; Lin J.; Kruger H.; Honarparvar B. Investigation of the binding free energies of FDA approved drugs against subtype B and C-SA HIV PR: ONIOM approach. J. Mol. Graphics Modell. 2017, 76, 77–85. 10.1016/j.jmgm.2017.06.026. [DOI] [PubMed] [Google Scholar]
- Cornell W. D.; Cieplak P.; Bayly C. I.; Gould I. R.; Merz K. M.; Ferguson D. M.; Spellmeyer D. C.; Fox T.; Caldwell J. W.; Kollman P. A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. 10.1021/ja00124a002. [DOI] [Google Scholar]
- Rappe A.; Colwell K.; Casewit C. Application of a universal force field to metal complexes. Inorg. Chem. 1993, 32, 3438–3450. 10.1021/ic00068a012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.