

Abstract
T cell-mediated immunity plays an important role in controlling SARS-CoV-2 infection, but the repertoire of naturally processed and presented viral epitopes on class I human leukocyte antigen (HLA-I) remains uncharacterized. Here, we report the first HLA-I immunopeptidome of SARS-CoV-2 in two cell lines at different times post infection using mass spectrometry. We found HLA-I peptides derived not only from canonical open reading frames (ORFs) but also from internal out-of-frame ORFs in spike and nucleocapsid not captured by current vaccines. Some peptides from out-of-frame ORFs elicited T cell responses in a humanized mouse model and individuals with COVID-19 that exceeded responses to canonical peptides, including some of the strongest epitopes reported to date. Whole-proteome analysis of infected cells revealed that early expressed viral proteins contribute more to HLA-I presentation and immunogenicity. These biological insights, as well as the discovery of out-of-frame ORF epitopes, will facilitate selection of peptides for immune monitoring and vaccine development.
Keywords: SARS-CoV-2, out-of-frame ORF immunopeptidomics, T Cell response, immunogenicity, HLA Class I, viral infection, coronavirus
Graphical abstract
Analysis of the HLA-I peptidome of SARS-CoV-2 infection identifies peptides derived from canonical and out-of-frame ORFs in viral S and N protein that are not captured by current vaccines and yield potent T cell responses in a mouse model as well as individuals with COVID-19.
Introduction
As efforts continue to develop effective vaccines and therapeutic agents against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus causing the ongoing coronavirus disease 2019 (COVID-19) pandemic (Lu et al., 2020), it is critical to decipher how infected host cells interact with the immune system. Previous insights from SARS-CoV and Middle East respiratory syndrome (MERS)-CoV as well as emerging evidence from SARS-CoV-2 imply that T cell responses play an essential role in SARS-CoV-2 immunity and viral clearance (Altmann and Boyton, 2020; Grifoni et al., 2020a; Le Bert et al., 2020; Rydyznski Moderbacher et al., 2020; Sekine et al., 2020). Growing concerns about emerging viral variants and potential resistance to antibody defenses have spurred renewed discussions about other immune responses and, in particular, cytotoxic T cells (Ledford, 2021). When viruses infect cells, their proteins are processed and presented on the host cell surface by class I human leukocyte antigen (HLA-I). Circulating cytotoxic T cells recognize the presented foreign antigens and initiate an immune response, resulting in clearance of infected cells. Investigating the repertoire of SARS-CoV-2-derived HLA-I peptides will enable identification of viral epitopes responsible for activation of cytotoxic T cells.
Most studies that have interrogated the interaction between T cells and SARS-CoV-2 antigens to date utilized overlapping peptide tiling approaches and/or bioinformatics predictions of HLA-I binding (Campbell et al., 2020; Ferretti et al., 2020; Grifoni et al., 2020b; Nguyen et al., 2020; Poran et al., 2020; Saini et al., 2020; Tarke et al., 2020). Although HLA-I binding prediction is undoubtedly a useful tool to identify putative antigens, it has limitations. First, antigen processing and presentation is a multi-step biological pathway that includes source protein degradation by the proteasome, peptide cleavage by aminopeptidases, translocation into the endoplasmic reticulum (ER), and HLA-I binding (Neefjes et al., 2011). Although many computational predictors now account for some of these steps, the average positive predictive value achieved across HLA alleles is still ∼64% (Sarkizova et al., 2020). Second, prediction models do not account for ways in which viruses may manipulate cellular processes that affect antigen presentation. For example, viruses can attenuate translation of host proteins, downregulate proteasome machinery, and interfere with HLA-I expression (Hansen and Bouvier, 2009; Sonenberg and Hinnebusch, 2009). These changes shape the collection of viral and human-derived HLA-I peptides presented to the immune system. Third, prediction models do not capture the dynamics of viral protein expression during the course of infection. Kinetics studies in vaccinia and influenza viruses have shown that HLA-I presentation of viral epitopes can peak 3.5–9.5 h post infection (hpi) (Croft et al., 2013; Wu et al., 2019). Moreover, because viruses can suppress HLA-I presentation, proteins that are expressed earlier in the virus life cycle may contribute more to the repertoire of viral epitopes. In light of these limitations, experimental measurements of naturally presented peptides upon infection can deepen our understanding of T cell responses to SARS-CoV-2.
Mass spectrometry (MS)-based HLA-I immunopeptidomics is a direct and untargeted method to discover endogenously presented peptides (Abelin et al., 2017; Bassani-Sternberg and Gfeller, 2016; Chong et al., 2018; Sarkizova et al., 2020). This technology has facilitated detection of virus-derived HLA-I peptides for West Nile virus, vaccinia virus, human immunodeficiency virus (HIV), human cytomegalovirus (HCMV), and measles virus (Croft et al., 2013; Erhard et al., 2018; McMurtrey et al., 2008; Rucevic et al., 2016; Schellens et al., 2015; Ternette et al., 2016). These infectious disease studies revealed new antigens, characterized the kinetics of presented peptides during infection, and identified viral peptide sequences that activate T cell responses.
Identifying viral protein sequences from MS data commonly relies on matching spectra against a database of known viral open reading frames (ORFs) and has largely focused on canonical ORFs. Over the past decade, genome-wide profiling of translated sequences has revealed a striking number of non-canonical ORFs in mammalian and viral genomes (Finkel et al., 2020a; Ingolia et al., 2009, 2011; Stern-Ginossar et al., 2012). Although the function of most of these non-canonical ORFs remains unknown, it is becoming clear that the translated polypeptides serve as fruitful substrates for the antigen presentation machinery in viral infection, uninfected cells, and cancer (Chen et al., 2020b; Hickman et al., 2018; Ingolia et al., 2014; Maness et al., 2010; Ouspenskaia et al., 2020; Ruiz Cuevas et al., 2021; Starck and Shastri, 2016; Yang et al., 2016). Importantly, a recent study identified 23 unannotated ORFs in the genome of SARS-CoV-2, some of which have higher expression levels than the canonical viral ORFs (Finkel et al., 2020b). Whether these non-canonical ORFs give rise to HLA-I-bound peptides remains unknown.
Here we present the first examination of the HLA-I immunopeptidome in two SARS-CoV-2-infected human cell lines and complement this analysis with RNA sequencing (RNA-seq) and global proteomics measurements. We identify viral HLA-I peptides that are derived from canonical and non-canonical ORFs and monitor the dynamics of viral protein expression and peptide presentation over multiple time points post infection. We show that peptides derived from out-of-frame ORFs elicit T cell responses in immunized mice and individuals with COVID-19 using ELISpot and multiplexed barcoded tetramer assays combined with single-cell sequencing. Whole-proteome measurements suggest that the time of viral protein expression correlates with HLA-I presentation and immunogenicity and that SARS-CoV-2 interferes with the cellular proteasomal pathway, potentially resulting in lower presentation of viral peptides. Computational predictions and biochemical binding assays demonstrate that the detected HLA-I peptides can be presented by additional HLA-I alleles beyond the nine alleles tested in our study. Our findings can inform future immune monitoring assays in affected individuals and aid in the design of efficacious vaccines.
Results
Profiling HLA-I peptides in SARS-CoV-2-infected cells by MS
To interrogate the repertoire of human and viral HLA-I peptides, we immunoprecipitated (IP) HLA-I proteins from SARS-CoV-2-infected human lung A549 cells and HEK293T cells that were transduced to stably express ACE2 and TMPRSS2, two known viral entry factors. We then analyzed their HLA-bound peptides by liquid chromatography-tandem MS (LC-MS/MS) (Figure 1 A). We also analyzed the whole proteome of the IP flowthrough by LC-MS/MS and performed RNA-seq to examine the effect of SARS-CoV-2 on human gene expression. To allow detection of peptides from the complete translatome of SARS-CoV-2, we combined the recently identified 23 ORFs (Finkel et al., 2020b) with the list of canonical ORFs and the human RefSeq database for LC-MS/MS data analysis.
Figure 1.

HLA-I peptidome and whole-proteome measurements in SARS-CoV-2-infected cells
(A) Schematic of the experiment and the antigen presentation pathway.
(B) Population frequency of the 9 endogenous HLA-I alleles expressed in A549 and HEK293T cells.
(C) Length distribution of HLA peptides in infected and naive cells.
(D) Motif of 9-mer sequences identified in infected and naive cells.
(E) Fraction of observed peptides assigned to alleles using HLAthena prediction (%rank cutoff < 0.5) for the immunopeptidome of infected and uninfected cells.
See also Figures S1 and S2 and Table S1.
When choosing cell types for this study, we focused on achieving biological relevance and high HLA-I allelic coverage. A549 cells are lung carcinoma cells and represent the key biological target of SARS-CoV-2; thus, they are commonly used in COVID-19 studies. HEK293T cells endogenously express HLA-A∗02:01 and B∗07:02, two high-frequency HLA-I alleles. Together, the nine HLA-I alleles expressed by HEK293T and A549 cells cover at least one allele in 63.8% of the human population (Figure 1B; STAR Methods). Using immunofluorescence staining of the nucleocapsid protein, we evaluate that ∼70% of the transduced cells were infected at the peak infection time (Figure S1 ).
Figure S1.

SARS-CoV-2 infection of HEK293T/ACE2/TMPRSS2 and A549/ACE2/TMPRSS2, related to Figure 1
(A) A549 cells expressing ACE2 and TMPRSS2 were infected with SARS-CoV-2 at MOI of 3 for 3, 6, 12, 18, and 24 hours. Fixed cells were incubated with a fluorescence antibody to the nucleocapsid and DAPI stain was used to label the nuclei. Immunofluorescent images were taken using an EVOS microscope with a 10x lens. Bars show mean ± SD (B) Similar to (A) for HEK293T cells. (C) Plaque assay confirming SARS-CoV-2 inactivation for HLA-IP experiments. A549 cells were infected with SARS-CoV-2 at MOI of 3 for 24 hours. 10-fold serial dilutions were prepared in Opti-MEM and used to infect Vero cells in a 24-wells plate. Comparing plaques in (left) cultured media of infected A549 cells; (middle) SARS-CoV-2 infected A549 cells treated with a lysis buffer containing 1.5% Triton-X and Benzonase for 3 hours; and (right) non-infected A549 cells. When adding the 1:10 dilution of the lysis buffer, infected and non-infected cells died immediately due to the relatively high Triton-X concentration.
We validated the technical performance of our assays by examining the overall characteristics of presented HLA peptides. We identified 5,837 and 6,372 HLA-bound 8- to 11-mer peptides in uninfected and infected (24 hpi) A549 cells and 4,281 and 1,336 unique peptides in HEK293T cells, respectively (Table S1). The reduction in the total number of peptides after infection in HEK293T cells is likely due to cell death (∼50% of cells 24 hpi). As expected, peptide length distribution was not influenced by infection, and the majority of HLA-I peptides were 9-mers (Figure 1C). Next we compared the binding motifs of all 9-mer peptides between uninfected and infected cells per cell line and per individual HLA allele (Figure 1D; Figures S2 A and S2B). We did not find major differences following infection, and the observed amino acids at the main anchor positions 2 and 9 were in line with the expected binding motifs of the alleles expressed in the two cell lines.
Figure S2.

Peptide logos and allele assignment for all experiments, related to Figure 1
(A) Logo plots for individual alleles of peptides identified and assigned to cell line specific alleles with HLAthena percentile rank < 0.5 for naive and 24h post Sars-CoV-2 infected A549 (left) and HEK293 (right) cells. (B) Peptide logo plots aggregated over all alleles for label free time course experiments in A549 and HEK293 samples. (C) Allele assignment for peptides identified in time course experiments using HLAthena with a percentile rank < 0.5 cutoff. (D) Expression level of HLA-A, -B, and -C alleles as measured by RNA-seq in A549 and HEK293T cell lines pre- and 24hr post-infection. (E) All 9-mer peptides tiled along human protein sequences were predicted for binding to the HLA alleles present in HEK293T (top) and A549 (bottom) cell lines. Per allele, the fraction of those 9-mers with predicted binding scores that are better than 50% of previously identified known binders in mono-allelic experiments for the allele is shown.
To evaluate whether the MS-detected peptides were indeed predicted to bind to the expressed HLA-I alleles, we inferred the most likely allele to which each peptide binds using HLAthena (Sarkizova et al., 2020). At a stringent cutoff of predicted percentile rank of 0.5 or less, 87% of A549 and 73% of HEK293T cell identified peptides post infection were assigned to at least one of the alleles in the corresponding cell line (Figure 1E; Figure S2C). Differences in the relative representation of HLA alleles on the cell surface are influenced by the expression level as well as the permissiveness of the binding motif of each allele (Figures S2D and S2E).
SARS-CoV-2 HLA-I peptides
Next we examined HLA-I peptides that are derived from the SARS-CoV-2 genome (Figure 2 A; Table S1). We identified 28 peptides from canonical proteins (non-structural protein 1 [nsp1], nsp2, nsp3, nsp5, nsp8, nsp10, nsp14, nsp15, spike (S), M, ORF7a, and nucleocapsid [N]). Strikingly, 9 peptides were derived from out-of-frame ORFs in S and N. Four peptides matched to an in-silico six-frame translation database of the SARS-CoV-2 genome. However, manual inspection of ribosome profiling data (Finkel et al., 2020b) did not support translated ORFs in these regions. Most of the HLA-I peptides were detected in more than one experiment and predicted as good binders by HLAthena (%rank < 2) to at least one of the expressed HLA alleles. We confirmed binding for 19 of the 20 HLA-I peptides predicted to be presented by four HLA alleles expressed in A549 and HEK293T cells (A∗02:01, B∗07:02, B∗18:01, and B∗44:03) using biochemical binding assays (IC50 < 500 nM; Figure 2B; Table S2). One peptide, HADQLTPTW, was also detected in non-infected A549 cells; thus, we removed it from all subsequent MS analyses.
Figure 2.

SARS-CoV-2 HLA-I immunopeptidome and whole proteome
(A) Summary of peptide location across the SARS-CoV-2 genome from the HLA-I immunopeptidome, whole proteome, and predictions.
(B) Biochemical binding of HLA-I peptides to purified major histocompatibility complexes (MHCs). Shown are the fractions of peptides that were confirmed to bind the assigned alleles (half maximal inhibitory concentration [IC50] < 500 nM; Table S2).
(C) SARS-CoV-2 protein abundance in A549 and HEK293T cells 24 hpi. iBAQ, intensity-based absolute quantification.
(D and E) Comparison of our protein abundance measurements 24 hpi and Ribo-seq (Finkel et al., 2020b) in A549 (D) and HEK293T (E) cells.
(F) HLA-I presentation potential of SARS-CoV-2 ORFs in A549 cells. ORFs were ranked according to the ratio between the number of peptides predicted to bind any of the six HLA-I alleles in A549 and the total number of 8- to 11-mers.
(G) Similar to (F) for HEK293T cells.
(H) Presentation potential across 92 HLA-I alleles, shown as boxplots (median ratio, whiskers reach to lowest and highest values no further than 1.5× interquartile range [IQR] of the ratio between the number of peptides predicted to bind each allele and total number of peptides). SARS-CoV-2 ORFs are ranked by the median across HLA-I alleles.
Surprisingly, we detected only one HLA-I peptide from N: a SARS-CoV-2 protein expected to be highly abundant based on previous RNA-seq and ribosome profiling (Ribo-seq) studies (Finkel et al., 2020b; Kim et al., 2020). To test whether this low representation could be explained by lower expression of N in our experiment, we examined the whole-proteome MS data. We found a strong correlation between the abundance of viral proteins in the proteome of the two cell lines (Pearson R = 0.91; Figure 2C) and with recently published translation measurements in infected Vero cells (Finkel et al., 2020b) (Pearson R = 0.86 and R = 0.78 for A549 and HEK293T, respectively; Figures 2D and 2E; Table S3A). The N protein remained the most abundant viral protein in both cell lines.
An alternative hypothesis for lower N representation could be that the protein harbors fewer peptides compatible with the HLA binding motifs. Therefore, for each SARS-CoV-2 ORF, we computed the ratio between the number of peptides that are predicted to be presented by at least one of the HLA-I alleles in each cell line and the number of total 8- to 11-mers. Notably, N had fewer than expected presentable peptides than most SARS-CoV-2 proteins in both cell lines (Figures 2F and 2G; Table S3B). We then expanded our analysis to 92 HLA-I alleles with high population coverage and with immunopeptidome-trained predictors (Sarkizova et al., 2020; Figure 2H; Table S3B). This analysis also categorized N among the least presentable canonical proteins of SARS-CoV-2. Our results hint that N might be less presented than expected, given its high expression level in infected cells (∼10-fold greater than the next most abundant viral protein; Figure 2C).
Our deep coverage of the viral proteins in the whole-proteome analysis (24 proteins) allowed us to observe several interesting findings. Although the translation of ORF1a and 1ab, the source polyproteins of nsps1–nsp16, is 10- to 1,000-fold lower than the structural ORFs (Finkel et al., 2020b), we found that the abundance of some nsps was comparable with that of structural proteins (e.g., nsp1 and nsp8; Figure 2C). Interestingly, although nsp1–nsp11 were cleaved post-translationally from the same polyproteins, their expression levels were variable. This finding is consistent with two additional proteomics studies of SARS-CoV-2-infected cells utilizing different detergents in their lysis buffers (Schmidt et al., 2020; Stukalov et al., 2020), suggesting that the observed differences in expression are not due to detergent solubility. Moreover, nsp12–nsp15, which originate from polyprotein 1ab downstream to the frameshift signal, are, as expected, expressed at lower levels. Another observation is that the S protein appeared as an outlier in both cell lines with higher expression in the proteome data compared with Ribo-seq measurements, suggesting that it may undergo positive post-translational regulation (Figures 2D and 2E; computed Pearson R when omitting S increased from 0.86 to 0.99 and 0.78 to 0.92 in A549 and HEK293T cells, respectively).
Kinetics of SARS-CoV-2 protein expression and HLA-I peptide presentation
To investigate the dynamics of HLA-I presentation during infection, we compared the relative abundance of HLA-I peptides in A549 and HEK293T cells at 3, 6, 12, 18 and 24 hpi. For technical reasons, we split the infection time course analysis into two batches (3, 6, and 24 hpi and 12, 18, and 24 hpi) and normalized to the 24-hpi time point.
Labeling with tandem mass tags (TMT) enabled detection of 10 viral HLA-I peptides in A549 cells; four of these peptides were quantified across all time points, two were only detected in the 12|18|24-h plex, and four were only detected in the 3|6|24-h plex (Figure 3 A; Table S1). It is likely that peptides that were detected only in the 3|6|24-h plex were also presented on HLA-I at 12 and 18 hpi, however, because of separate cell culture experiments and data acquisition, they were not detected in the 12|18|24-h plex. HLA-I presentation of most detected viral peptides peaked at 6 hpi, similar to previous reports for vaccinia virus (Croft et al., 2013) and influenza virus (Wu et al., 2019). Although some human-derived HLA-I peptides changed over time, the majority were fairly stable. In HEK293T cells, we detected 13 peptides from SARS-CoV-2, with the caveat of observing some peptides only in the 3|6|24-h plex as described above (Figure 3B; Table S1). Examining the dynamics of HLA-I peptides observed across all time points, we found that the abundance of some viral peptides peaked at 6 hpi; however, we also observed maximal presentation at 12, 18 and 24 hpi for others.
Figure 3.

HLA-I peptides dynamics in SARS-CoV-2-infected cells
(A and B) Dynamics of TMT-labeled HLA-I peptides 3, 6, 12, 18, and 24 hpi in A549 (A) and HEK293T cells (B). TMT intensity values of peptides detected in two independent experiments (3, 6, and 24 hpi and 12, 18, and 24 hpi) were normalized to the respective abundance at 24 h present in both experiments. Dashed lines indicate detection in the 3|6|24-h plex only.
(C) Dynamics of SARS-CoV-2 protein expression according to whole-proteome analysis.
(D) Venn diagram showing SARS-CoV-2 proteins according to their earliest expression time and the source proteins for HLA-I-presented peptides in A549 and HEK293T cells. The hypergeometric p value represents the enrichment of early-expressed proteins (3 hpi) in the group of proteins presented on HLA-I.
(E) CD8+ responses to early/late-expressed SARS-CoV-2 proteins in convalescent COVID-19 individuals according to a recent study. The box shows the quartiles, the bar indicates median, and the whiskers show the distribution (see Table S3 in Tarke et al., 2020).
To assess the relationship between HLA peptide presentation and the time of viral protein expression, we performed fractionated whole-proteome MS analysis across the 3, 6, and 24 hpi time points from the same cell lysates. Although the majority of viral proteins were expressed in cells at 6 hpi, only eight and nine proteins were detected at 3 hpi in A549 and HEK293T cells, respectively (Figure 3C). We found that viral proteins detected as early as 3 hpi contributed to HLA-I presentation more than viral proteins expressed at 6 hpi or later (hypergeometric p < 0.0375; Figure 3D) and elicited stronger CD8+ T cell responses in COVID-19 convalescent individuals (Tarke et al., 2020) (Wilcoxon rank-sum p < 0.0181; Figure 3E). This observation may explain a recent surprising finding that nsp3 is among the four most immunogenic proteins of SARS-CoV-2 (Tarke et al., 2020). Although nsp3 is not expressed at high levels, its early expression in infected cells may contribute to presentation of nsp3-derived HLA-I peptides.
SARS-CoV-2 infection interferes with cellular pathways that may affect antigen processing
To investigate how the levels of viral source proteins affect their ability to be processed and presented, we ranked the individual SARS-CoV-2 proteins and HLA-I peptides according to their abundance in comparison with human proteins. Although the overall abundance of viral proteins in the infected cells proteome at 24 hpi was relatively low (HEK293T, 2.6%; A549, 3%; Figure S3 A), individual viral proteins were highly expressed and exceeded most of the host proteins (Wilcoxon rank-sum test; A549, p < 10−4; HEK293T, p < 10−6; Figure 4 A; Figure S3B; Table S4). In contrast to the high expression of their source proteins, the intensities of viral HLA-I peptides are similar to peptides from the host proteome, indicating that viral peptides are not presented preferentially (Wilcoxon rank-sum test; A549, p > 0.8; HEK293T, p > 0.4; Figure 4B, Figure S3C; Table S1). Moreover, as shown recently for influenza virus (Wu et al., 2019), we found that the intensities of the viral HLA-I peptides do not directly correspond to their source protein abundances (Figures 4A and 4B).
Figure S3.

SARS-CoV-2 peptide abundance and antigen presentation pathway proteins in infected cells, related to Figure 4
(A) Table showing the percentage of the total whole proteome abundance represented by SARS-CoV-2 derived proteins at 0, 3, 6, 12, 18, 24hpi identified in singleshot whole proteome LC-MS/MS analyses. (B) Rank plot of the protein abundances represented by log10 protein iBAQ values for each human (gray), canonical SARS-CoV-2 (blue), and noncanonical SARS-CoV-2 proteins detected in the whole proteome analysis of HEK293T cells 24hpi. SARS-CoV2 proteins are annotated with their respective gene names. (C) Similar rank plot to (A) but for observed HLA-I peptides and their abundances represented by log2 peptide intensities in HEK293T cells 24hpi. Peptides mapping to SARS-CoV-2 are annotated with their respective amino acid sequence and source protein name. (D) Heatmap of log10 iBAQ values for antigen presentation pathway proteins observed across uninfected and 24hpi in A549 and HEK293T cells. (E) Volcano plot comparing protein levels across uninfected and infected A549/ACE2 cells 6hpi reported in publically available whole proteome data (PXD020019). Proteins from SARS-CoV-2 (red), ubiquitination pathways (teal), proteasomal function (purple), antigen processing (pink), and IFN pathways (orange) are colored accordingly. Significantly changing proteins are shown above the dashed line (p value < 0.01) along with annotations of specific proteins involved in the above pathways.
Figure 4.

The effect of SARS-CoV-2 infection on antigen presentation in host cells
(A) Rank plot of protein abundances (log10 protein iBAQ) from human and SARS-CoV-2 detected in the whole-proteome analysis.
(B) Similar rank plot as in (A) but for observed HLA-I peptide abundance (log2 intensity).
(C) Venn diagram showing the overlap between total HLA-I peptides in uninfected and infected A549 cells.
(D) Expression heatmap of central antigen presentation pathway proteins in uninfected and infected cells (24 hpi).
(E) Volcano plot comparing protein levels in uninfected and infected A549 and HEK293T cells 24 hpi (dashed line, p < 0.01, moderated t test).
(F) Similar to (E); a volcano plot representing whole-proteome data from A549/ACE2 cells 24 hpi (Stukalov et al., 2020).
To assess whether there are global changes in HLA-I antigen presentation upon infection, we compared the overlap between HLA-I peptidomes of uninfected and infected (24 hpi) A549 cells. The overlap among peptides detected in both experiments (62%; Figure 4C) was similar to what was observed in biological replicates of the same sample (Abelin et al., 2017; Demmers et al., 2019). This high overlap and the relatively low HLA-I peptide representation from viral proteins that are expressed at 6 hpi or later (Figure 3D) led us to interrogate the whole-proteome data for evidence of viral interference with the antigen presentation pathway. Because we analyzed the whole proteome from the cell lysate post HLA immunopurification, the levels of HLA-A, HLA-B, and HLA-C could not be evaluated. However, all other host proteins should remain intact and enable proteomic analyses of host responses to infection.
First, we compared the expression of central HLA-I presentation pathway proteins (e.g., B2M, ERAP1/2, TAP1/2, and proteasome subunits) between uninfected and infected cells using our fractionated proteome data (∼7,000 quantified proteins; Figure 4D; Figure S3D; Table S4). Although some antigen presentation proteins had cell-type-specific expression patterns, we observed no significant differences in these proteins upon infection. Of note, HLA-F, which interacts with KIR3DS1 on natural killer (NK) cells during viral infection (Lunemann et al., 2018), had increased expression in infected cells.
Next we compared all proteins detected in uninfected and infected cells to determine whether proteins involved in ubiquitination, proteasomal function, antigen processing, and interferon (IFN) signaling were altered (Figure 4E; Table S4). We observed a general decrease in ubiquitination pathway proteins, with several of them depleted significantly in response to SARS-CoV-2 infection, including RNF181, UBE2B, and TRIM11. POMP, a chaperone critical for assembly of 20S proteasomes and immunoproteasomes, was the most significantly depleted proteasomal protein in infected cell lines (p < 0.0095). POMP has been reported recently to affect ORF9c stability, which has been implicated in suppressing the antiviral response (Dominguez Andres et al., 2020). As reported across multiple cell lines infected with SARS-CoV-2 (Chen et al., 2020a), the tyrosine kinase JAK1, critical for IFN signaling, was depleted in A549 and HEK293T cells upon infection (Figure 4E). We confirmed the observed depletion of POMP and ubiquitination pathway proteins in an independent proteome study (Stukalov et al., 2020) that profiled uninfected and infected A549/ACE2 cells at 6 hpi (Figure S3E) and 24 hpi (Figure 4F). These data suggest that SARS-CoV-2 may interfere with IFN signaling proteins and the HLA-I pathway through POMP depletion and by altering ubiquitination pathway proteins, that in turn, may prevent abundant SARS proteins expressed later in infection from being effectively processed and presented.
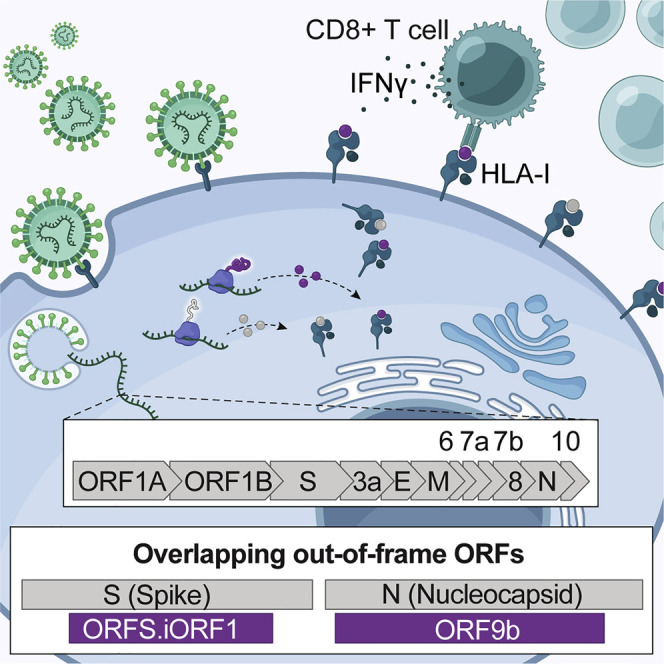
HLA-I peptides are derived from internal out-of-frame ORFs in S and N
Remarkably, we detected nine HLA-I peptides processed from internal out-of-frame ORFs in the coding region of S and N, termed S.iORF1 (also known as ORF2b; Jungreis et al., 2021) and ORF9b. From S.iORF1/2, we detected three HLA-I peptides (GPMVLRGLIT, GLITLSYHL, and MLLGSMLYM) in HEK293T cells (Figure 5 A). In addition, we detected six HLA-I peptides from ORF9b in A549 cells (LEDKAFQL and DEFVVVTV) and HEK293T cells (SLEDKAFQL, KAFQLTPIAV, ELPDEFVVV, and ELPDEFVVVTV) (Figure 5B). These HLA-I peptides cover overlapping protein sequences and contain binding motifs compatible with the expressed HLA-I alleles. To validate the amino acid sequences of these non-canonical peptides, we compared the tandem mass spectra of synthetic peptides with the experimental spectra and observed high correlation between fragment ions and retention times (±2 min; Figure 5C).
Figure 5.

SARS-CoV-2 HLA-I peptides from S.iORF1/2 and ORF9b
(A) HLA-I peptides derived from S.iORF1/2. Underscored methionines (M) represent the start codons of S.iORF1 and S.iORF2.
(B) HLA-I peptides derived from ORF9b (N.iORF1) and N.iORF2.
(C) Mirror plots with fragment ion mass spectra confirming the sequences of four HLA-I peptides that were identified in S.iORF1/2 and ORF9b (positive y axis, HLA IP samples; negative y axis, synthetic peptide).
(D) Biochemical HLA-A∗02:01/peptide binding measurements. The concentration of peptide yielding 50% inhibition of the binding of the radiolabeled A∗02:01 ligand (IC50) was used to calculate peptide affinity.
(E) The effect of human codon optimization on HLA-I peptides derived from S.iORF1/2. Shown is Needleman-Wunsch pairwise global alignment between the SASR-CoV-2 sequence (NC_045512.2) and the human optimized S from the Krogan library (Gordon et al., 2020) in the S.iORF1/2 coding region. Purple boxes indicate the position of the HLA-I peptides in the out-of-frame ORFs.
(F) similar to (E) but for N in the ORF9b coding region.
Six of the peptides from out-of-frame ORFs were predicted to bind HLA-A∗02:01 in HEK293T cells, suggesting potential for widespread presentation of these non-canonical HLA-I peptides in the population. We confirmed binding for all six peptides using biochemical measurements in the presence of a high-affinity radiolabeled A∗02:01 ligand (IC50 < 500 nM; Figure 5D; Table S2). Interestingly, the three peptides with highest affinity among all tested HLA-I peptides originated from out-of-frame ORFs: two from S.iORF1/2 (MLLGSMLYM and GLITLSYHL, IC50 < 0.5 nM) and one from ORF9b (ELPDEFVVVTV, IC50 = 1.6 nM).
In the context of T cell immunity and vaccine development, it is crucial to understand the effect of optimizing RNA sequences on the endogenously processed and presented HLA-I peptides derived from internal out-of-frame ORFs. Exogenous expression of viral proteins in vaccines often involve manipulating the native nucleotide sequences (e.g., via codon optimization) to enhance expression. These techniques maintain the amino acid sequence of the canonical ORF but may alter the sequence of proteins encoded in alternative reading frames. In addition to the two current mRNA vaccines targeting the S glycoprotein (Callaway, 2020; Jackson et al., 2020; Mulligan et al., 2020), the N protein is also considered for vaccine development (Dutta et al., 2020; Zhu et al., 2004).
To investigate the effect of codon optimization on HLA-I peptides derived from S.iORF1/2 and ORF9b, we compared the native viral sequence with synthetic S and N from a SARS-CoV-2 human optimized ORF library (Gordon et al., 2020). As expected, there was no change in the main ORFs; however, the amino acid sequences in the +1 frame encoding S.iORF1/2 and ORF9b were significantly different (Figures 5E and 5F). In the case of S.iORF1, it is possible that this ORF is expressed in the human optimized construct because the methionine driving its translation is preserved, however, the sequence of potential HLA-I peptides would be different (Figure 5E). In the case of ORF9b, the start codon was mutated, few stop codons were introduced along the ORF, and the sequence of the detected HLA-I peptides was altered (Figure 5F). These data suggest that human codon optimization of the main ORF may preclude the HLA-I presentation of peptides encoded from alternative ORFs.
Out-of-frame HLA-I peptides elicit T cell responses in humanized HLA-A2 mice and individuals with COVID-19
To evaluate the immunogenicity of the HLA-I peptides detected by MS, we conducted three assays probing T cell responses in a humanized mouse model, individuals with COVID-19, and unexposed humans. First, we immunized five transgenic HLA-A2 mice with a pool of 9 A∗02:01 peptides for 10 days and tested the T cell responses to individual peptides using an INFγ ELISpot assay. We found positive response to three non-canonical peptides from out-of-frame ORFs, two from S.iORF1/2 (GLITLSYHL and MLLGSMLYM), and one from ORF9b (ELPDEFVVVTV), as well as a canonical peptide from nsp3 (YLNSTNVTI) (Figures 6A and 6B).
Figure 6.

T cell responses to SARS-CoV-2 HLA-I peptides
(A) Five HLA-A2 transgenic mice were immunized with a pool of nine HLA-I peptides detected on A∗02:01 in HEK293T cells for 10 days. Splenocytes were incubated with individual peptides and monitored for IFNγ secretion. HLA-A∗02:01 restricted HIV-Gag peptide and non-stimulated wells were used as negative controls. Anti-CD3 and phytohemagglutinin (PHA) were used as positive controls. The dashed line represents the threshold for positive responses (3× the median of the HIV-Gag). The box shows the quartiles, the bar indicates median, and the whiskers show the distribution.
(B) ELISpot images from one of the five vaccinated mice. Numbers indicate the spot count.
(C) PBMCs from convalescent COVID-19 individuals expressing A∗02:01 alleles were incubated with a pool of HLA-I peptides from canonical or out-of-frame ORFs. A pool of 102 peptides tiling the entire nucleocapsid (N) protein that was evaluated in the same samples (Gallagher et al., 2021) served as positive control. Bars show the mean of duplicates.
(D) ELISpot images of individuals #1 and #3. Numbers indicate the spot count.
(E) Illustration of the multiplexed tetramer assay and T cell single-cell profiling.
(F) CD8+ T cell reactivity detected in convalescent COVID-19 individuals and unexposed subjects expressing A∗02:01 to individual HLA-I peptides. The score in the heatmap indicates the fraction of peptide-specific reacting T cells from total CD8+ cells in the sample.
(G) Single-cell transcriptomics of reactive T cells. Top panel: uniform manifold approximation and projection (UMAP) embedding of all tetramer-positive cells colored by unsupervised clustering. Center panel: expression levels of 15 genes associated with different states of T cells, as characterized previously (Su et al., 2020). Bottom panel: expression level of these 15 genes in individual T cells reactive to ELPDEFVVVTV peptide from ORF9b.
Next we investigated the immunogenicity of the HLA-I peptides in the context of COVID-19. We performed ELISpot assays with peripheral blood mononuclear cells (PBMCs) from six convalescent individuals expressing HLA-A∗02:01 and monitored IFNγ secretion in response to a pool of 15 HLA-I peptides from canonical ORFs and 7 peptides from the out-of-frame ORFs. As a positive control, we compared the T cell responses with a pool of 102 peptides tiling the N protein measured in the same individuals as part of another study (Gallagher et al., 2021). We observed positive responses to the non-canonical pool in two of the six samples (Figures 6C and 6D). Notably, in one individual, the T cell responses to the non-canonical pool exceeded the responses to the N pool, although the number of tested peptides was 14-fold lower (7 versus 102 peptides in the non-canonical and N pools, respectively).
To delineate the T cell responses against individual HLA-I peptides in humans, we utilized a multiplexed technology combining a barcoded tetramer assay and single-cell sequencing of epitope-reactive CD8+ T cells (Figure 6E; Francis et al., 2021). Using this method, we obtained information about (1) the ex vivo frequency of CD8+ T cells reactive to each peptide in each sample; (2) the sequences of the T cell receptors (TCRs; paired a/b chains) recognizing each peptide; and (3) gene expression profiles of individual reactive CD8+ T cells. Testing nine HLA-A∗02:01 samples (seven COVID-19 convalescent and two unexposed), we found reactivity to positive control peptides from influenza and SARS-CoV-2 (Figure 6F; Table S5A). As expected, HLA-I peptides that bind A∗02:01 according to our affinity measurements (Table S2) elicited stronger CD8+ responses than peptides that were detected on other HLA alleles (Wilcoxon rank-sum p < 10−6; Figure S4 A). Two non-canonical peptides from ORF9b, ELPDEFVVVTV and SLEDKAFQL, were in the top five reactive peptides (Table S5A). Strikingly, ELPDEFVVVTV invoked the strongest CD8+ response among all tested HLA-I peptides, with the frequency of detected T cells similar to that observed for the influenza epitope and above those for three commonly recognized SARS-CoV-2 epitopes: YLQPRTFLL, KLWAQCVQL, and LLYDANYFL (Ferretti et al., 2020). Of note, YLQPRTFLL has been considered the most reactive SARS-CoV-2 epitope in a few independent studies (Ferretti et al., 2020; Habel et al., 2020; Shomuradova et al., 2020).
Figure S4.

CD8+ responses to HLA-I peptides in individuals with COVID-19 and TCR homology in ELPDEFVVVTV-reactive T cells, related to Figure 6
(A) The number of unique CD8+ T cell clones reacting to HLA-I peptides that were found to bind HLA-A∗02:01 in biochemical binding measurement and HLA-I peptides that did not bind HLA-A∗02:01. Wilcoxon rank-sum p value is indicated. The box shows the quartiles, bar indicates median and the whiskers show the distribution. (B) Network plot showing the relationship of unique clonotypes within and across subjects. Clonotypes, shown as nodes, are connected to other clonotypes with similar alpha or beta CDR3 with edges (scirpy v0.6.0). (C) CDR3 size distributions for alpha and beta TCR chains. (D) TCR α/β-paired sequence logo for related clonotypes represented in the interconnected cluster at the bottom of the network shown in (B). (E) CD8+ T cell reactivity detected in convalescent COVID-19 patients and unexposed subjects expressing B∗07:02 to individual peptides that bind HLA-B∗07:02 or other alleles. The score in the heatmap indicates the fraction of peptide-specific reacting T cells from total CD8+ T cells in the sample. (F) Similar to (A) for HLA-B∗07:02.
Examining the gene expression profile and the TCR sequence of the reacting T cells provided additional supporting evidence for the functional relevance of the ELPDEFVVVTV epitope during the course of COVID-19. Most cells reactive to ELPDEFVVVTV showed high expression of effector markers and moderate to high expression of memory markers based on gene sets described in a recent COVID-19 CD8+ subpopulation profiling study (Figure 6G; Su et al., 2020). In addition, the TCR sequences of CD8+ T cells reactive to ELPDEFVVVTV revealed significant CDR3 homology across affected individuals (Figures S4B–S4D).
Although our T cell data provide evidence of CD8+ responses to peptides from ORF9b in individuals with COVID-19, we did not detect significant responses to HLA-I peptides from S.iORF1/2, GLITLSYHL and MLLGSMLYM, in the seven tested COVID-19 samples. To evaluate the immunogenicity of the third HLA-I peptide from S.iORF1/2, GPMVLRGLIT, we performed an additional barcoded tetramer assay with PBMCs from individuals with COVID-19 expressing HLA-B∗07:02. We observed the expected positive reactivity to control peptides from EBV (RPPIFIRRL) and SARS-CoV-2 (SPRWYFYYL) as well as overall greater CD8+ responses to HLA-I peptides that bind B∗07:02 (Wilcoxon rank-sum p < 10−10; Figures S4E and S4F; Table S5B). However, we found no significant responses to GPMVLRGLIT in affected individuals, although we detected this peptide multiple times in our MS experiments (Table S1). It is possible that our assay was not sensitive enough to capture T cell responses to the three non-canonical peptides from S.iORF1/2 because we also observed weak responses to KLWAQCVQL, a commonly recognized A∗02:01 epitope in individuals with COVID-19 (Ferretti et al., 2020; Takagi and Matsui, 2020), exhibiting similar reactivity as GLITLSYHL from S.iORF1/2.
SARS-CoV-2 HLA-I peptides can be presented by additional alleles in the population
Increasingly accurate HLA-I presentation prediction tools are applied routinely to the full transcriptome or proteome of an organism to computationally nominate presentable epitopes. However, these tools are trained on data that are agnostic to virus-specific processes that may interfere with the presentation pathway. Thus, the sensitivity and specificity of in silico predictions for any particular virus are characterized insufficiently. To assess how well computational tools would recover the MS-identified HLA-I peptides, we used HLAthena (Abelin et al., 2017; Sarkizova et al., 2020) to retrospectively predict all 8- to 11-mer peptides tiling SARS-CoV-2 proteins against the complement of HLA-I alleles expressed by A549 and HEK293T cells (Figure 7 A; Table S6A). Of the 36 MS-identified peptides, 23 had a predicted percentile rank (%rank) of less than 0.5, and 31 had a %rank of less than 2.
Figure 7.

Presentation prediction and population coverage estimates of MS-identified SARS-CoV-2 HLA-I peptides
(A) Summary of LC-MS/MS-identified SARS-CoV-2 epitopes with corresponding HLAthena predictions for the 6 HLA-I alleles expressed by A549 cells and the 3 HLA-I alleles in HEK293T cells.
(B) HLAthena predictions for 92 HLA-I alleles. Left: the number of unique HLA-I alleles predicted as strong binders. Right: estimated population coverage. Alleles are colored and ordered according to loci and world population frequency (high to low color intensity).
(C) Biochemical binding measurements of HLA-I peptides and five HLA alleles that were not profiled in our cell lines. Shown are the fractions of peptides that were confirmed to bind the predicted alleles (IC50 < 500 nM; Table S2).
(D) IC50 nM affinity measurements of HLA-I peptides for nine alleles separated by predicted binders (%rank < 2) and predicted non-binders (%rank ≥ 2) (Welch Two Sample t test, data are presented as median, whiskers reach to lowest and highest values no further than 1.5× IQR).
Within 39,875 possible SARS-CoV-2 8- to 11-mers, 14 of 18 A549 HLA-I peptides and 11 of 18 HEK293T peptides had %rank scores within the top 1,000 viral peptides (top 1.5% and 1.7% for A549 and HEK293T cells, respectively). To account for variability in viral protein expression levels, we repeated this analysis within the source protein of each peptide. We found that 16 of 36 peptides scored within the top 10 among all 8- to 11-mers of the source protein, and 21 scored within the top 20. These observations suggest that, although an in silico epitope prediction scheme that nominates the top 10–20 peptides of each viral protein would recover ∼50% (16–21 of 36) of observed epitopes with very high priority, this list would still only encompass ∼5%–10% true LC-MS/MS positives (16–21 of 10 × #proteins).
Next we estimated the HLA allele coverage achieved by the observed endogenously processed and presented viral epitopes among African Americans (AFA), Asian Pacific Islanders (API), European (EUR), Hispanic (HIS), United States, and world populations at different %rank cutoffs based on HLAthena predictions across 92 HLA-I alleles (Figure 7B; Figure S5 ; Tables S6B and S6C). At the second most stringent cutoff, %rank of 0.5 or less, 31 of the 36 individual peptides were predicted to bind at least one allele (range, 1–21; median, 4.9; mean, 4.5). Combined, the MS-identified peptide pool was estimated to cover at least one HLA-A, HLA-B, or HLA-C allele for 99% of the population with at least one peptide.
Figure S5.

Population coverage estimates of LC-MS/MS-identified SARS-CoV-2 HLA-I peptides, related to Figure 7
HLAthena predictions for 92 HLA-I alleles using percentile rank cutoff values of 0.1, 0.5, 1, and 2% were used to show the number of alleles and estimated coverage for each LC-MS/MS-observed SARS-CoV-2 peptides across (A) AFA, (B) API, (C) EUR, (D) HIS, and (E) USA populations. Alleles are colored and ordered according to loci and the corresponding population frequency (high to low color intensity). Peptides are ordered according to their estimated coverage at %rank cutoff of 0.5.
To validate the predicted binding of the HLA-I peptides, we performed biochemical binding measurement with 30 synthetic peptides and 5 HLA alleles not present in the two profiled cell lines. We confirmed binding for 5 of 9 (56%) HLA-I peptides predicted at a 0.5 %rank threshold and 12 of 29 (41%) peptides predicted at a %rank threshold of 2 (Figure 7C), with significantly higher measured affinities for predicted binders versus non-binders (Figure 7D; Table S2). Moreover, two peptides with predicted presentation on HLA alleles not profiled in our cell lines have been found recently to elicit T cell responses in convalescent COVID-19 individuals expressing the predicted alleles (EILDITPCSF and QLTPTWRVY, detected on A∗25:01 and C∗16:01, were predicted to bind A∗26:01 and A∗30:02 at a %rank of 0.5 or less, respectively; Table S7; Tarke et al., 2020). These results indicate that HLA-I immunopeptidomics on only two cell lines, combined with epitope prediction tools, can help prioritize CD8+ T cell epitopes with high population coverage.
Discussion
We provide the first view of SARS-CoV-2 HLA-I peptides that are endogenously processed and presented by infected cells. Although our study profiled two cell lines, it uncovers insights into SARS-CoV-2 antigen presentation that extend beyond the nine HLA alleles tested here. (1) A substantial fraction, 9 of 36 (25%), of viral peptides detected are derived from internal out-of-frame ORFs in S (S.iORF1/2) and N (ORF9b). Remarkably, HLA-I peptides from non-canonical ORFs were strongly immunogenic in immunized mice and convalescent COVID-19 individuals, as shown by pooled ELISpot and multiplexed tetramer assays. These observations imply that current interrogations of T cell responses in individuals with COVID-19, which focus on the canonical viral ORFs (Grifoni et al., 2020a; Weiskopf et al., 2020), exclude an important source of virus-derived HLA-I epitopes. (2) A large fraction of detected HLA-I peptides were from nsps. Although earlier studies focused mostly on T cell responses to structural proteins, this finding, together with recent studies that expanded their epitope pools to include nsps (Dan et al., 2020; Kared et al., 2021; Tarke et al., 2020), portray nsps as an integral part of the T cell response to SARS-CoV-2. (3) The timing of SARS-CoV-2 protein expression appears to be a key determinant for antigen presentation and immunogenicity. Proteins expressed earlier in infection (3 hpi) were more likely to be presented on the HLA-I complex and elicit a T cell response in individuals with COVID-19.
Recent findings highlight the need to look beyond antibodies for strategies to achieve long-lasting protection against COVID-19 (Ledford, 2021). Several newly emerged SARS-CoV-2 variants are poorly neutralized by antibodies raised against the parental isolates used in the current vaccines (Chen et al., 2021; Wu et al., 2021). Importantly, recent studies have shown that CD8+ T cell responses are not substantially affected by mutations found in prominent SARS-CoV-2 variants (Redd et al., 2021; Tarke et al., 2021). Thus, integrating T cell epitopes into the design of next-generation vaccines has the potential to provide prolonged protection in the face of emerging variants. Our work reveals that ORF9b is an important source of T cell epitopes that remains largely unexplored in the context of T cell immunity. Although relatively short (97 amino acids [aa]), ORF9b yielded six HLA-I peptides (16% of total detected peptides) in A549 and HEK293T cells that bind at least four different alleles (A∗02:01, B∗18:01, B∗44:03, and A∗26:01). We identified two A∗02:01 peptides, ELPDEFVVVTV and SLEDKAFQL, that elicit CD8+ T cell responses in convalescent individuals, demonstrating that ORF9b is translated and presented on HLA-I in-vivo during the course of COVID-19. Moreover, ORF9b is highly expressed and among the few viral proteins that are detected early in infection, two traits that correlate with HLA-I presentation and immunogenicity. Specifically, our study highlights ELPDEFVVVTV as a promising T cell epitope. It binds A∗02:01 and A∗26:01, elicits strong T cell responses in immunized mice and individuals with COVID-19, and is recognized by TCRs from different affected individuals sharing a mutual CDR3 motif. Importantly, ELPDEFVVVTV elicits stronger T cell responses (in five of seven individuals studied here) than the three most commonly recognized A∗02:01 SARS-CoV-2 epitopes (Ferretti et al., 2020), including YLQPRTFLL, which was recorded as the most potent SARS-CoV-2 epitope in three independent studies (Ferretti et al., 2020; Habel et al., 2020; Shomuradova et al., 2020) and is the target of commercial monomer and tetramer assays.
In contrast to ORF9b, S.iORF1/2-derived peptides did not elicit significant T cell responses in convalescent COVID-19 individuals. This finding is surprising, given that GLITLSYHL and MLLGSMLYM had the highest affinity to HLA-A∗02:01 among all HLA-I peptides tested and were immunogenic in a humanized mouse model, demonstrating that they can elicit T cell responses in vivo. Moreover, GLITLSYHL immunogenicity in mice was 10-fold higher than ELPDEFVVVTV, the most potent SARS-CoV-2 epitope detected in individuals with COVID-19, with responses comparable only with an Influenza epitope. The discrepancy between the immunogenicity of S.iORF1/2-derived peptides in mice and individuals with COVID-19 could suggest an immune evasion mechanism to attenuate the translation and/or antigen processing of these non-canonical ORFs in affected individuals. Testing T cell responses in convalescent samples, as done in our study, is biased toward symptomatic individuals, and perhaps T cell reactivity to these peptides is associated with asymptomatic infection. Interestingly, although the sequence encoding the canonical and ORF9b-derived HLA-I peptides remained unchanged in the recent emerging SARS-CoV-2 variants B.1.1.7, P.1, and B.1.351 (originally detected in the United Kingdom, Brazil, and South Africa, respectively; Rambaut et al., 2020), the three HLA-I peptides derived from S.iORF1/2 were mutated (Figure S6 ; Table S8): S 69–70 deletion in the B.1.1.7 variant results in deletion of the last two amino acids of the S.iORF1-derived MLLGSMLYM epitope, whereas D80A substitution in canonical S of the B.1.351 variant results in I-to-L substitution in GPMVLRGLIT and GLITLSYHL. This could be due to relaxed selective pressure, allowing mutations to emerge, or may point to potential positive selective pressure on these T cell epitopes encoded from the alternative out-of-frame ORFs. Further studies, including testing T cell responses in asymptomatic subjects, are needed to elucidate the potential role of S.iORF1/2-derived peptides in COVID-19.
Figure S6.

HLA-I peptide sequences in B.1.1.7, P.1, and B.1.351 SARS-CoV-2 variants, related to Figure 6
(A) The sequence of the HLA-I peptides detected in our study were used as tblastn queries against a database containing early representative genomes of SARS-CoV-2 lineages with the pango designations B.1.1.7 (29 genomes), P.1 (14 genomes), and B.1.351 (23 genomes); see GISAID acknowledgment table for accessions (Table S8). Identity scores for each peptide in each variant are shown in the heatmap. (B,C) Mutations in the S.iORF1/2 region of B.1.351 (B) and B.1.1.7 (C) variants in comparison to the SARS-CoV-2 RefSeq sequence NC_045512.2 isolated from Wuhan. The position of the three HLA-I peptides is indicated.
Our analyses demonstrate that synthetic approaches aiming to enhance the expression of canonical ORFs, some of which are utilized in current vaccine strategies, can inadvertently eliminate or alter production of HLA-I peptides derived from overlapping reading frames. Researchers may need to carefully examine the effect of sequence manipulation and codon optimization on internal overlapping ORFs, especially those encoding HLA-I peptides. In broader terms, many viral genomes have evolved to increase their coding capacity by utilizing overlapping ORFs and programmed frameshifting (Ketteler, 2012). Thus, our findings suggest a more general principle in vaccine design according to which optimizing expression of desired antigens using codon optimization can be at the expense of CD8+ response when the same region encodes a source protein for T cell epitopes in an alternative frame. Combining insights from ribosome profiling and HLA-I immunopeptidomics can uncover the presence of non-canonical peptides that will enable more informed decisions in vaccine design.
Proteomics analyses of infected cells show that SARS-CoV-2 may interfere with presentation of HLA-I peptides and expression of ubiquitination and immune signaling pathway proteins. We found that SARS-CoV-2 infection leads to a significant decrease in expression of POMP and ubiquitination pathway proteins. By affecting ubiquitin-mediated proteasomal degradation and immune signaling proteins, SARS-CoV-2 may reduce the precursors for downstream processing and HLA-I presentation and alter the immune response. The effects of SARS-CoV-2 on HLA-I presentation may be influenced by additional factors. such as translation inhibition by nsp1 (Schubert et al., 2020) and degradation of host transcripts (Finkel et al., 2020c), that can diminish antigen presentation by attenuating the expression of HLA-I molecules. Moreover, a recent study reports that ORF8 protein disrupts HLA-I antigen presentation and reduces recognition and the elimination of virus-infected cells by cytotoxic T lymphocytes (CTLs) (Park, 2020; Zhang et al., 2020). Further research is needed to directly probe the various effects of SARS-CoV-2 on HLA-I antigen presentation.
Our work uncovers previously uncharacterized SARS-CoV-2 HLA-I peptides from out-of-frame ORFs in the SARS-CoV-2 genome and highlights the contribution of these viral epitopes to the immune response in a mouse model and convalescent COVID-19 individuals. These new CD8+ T cell targets and the insights into HLA-I presentation in infected cells will enable more precise selection of peptides for COVID-19 immune monitoring and vaccine development.
Limitations of the study
The results of this study should be interpreted within the context of its technical limitations. First, immunopeptidome profiling was performed in infected cell lines and may not capture the in vivo conditions in a faithful manner. Nevertheless, T cell responses in affected individuals to HLA-I peptides, including non-canonical epitopes, support the in vivo presentation of at least some of the peptides reported in this study. Second, our study spans nine HLA alleles expressed endogenously expressed in two cell lines. Further studies of SARS-CoV-2-infected cell lines from diverse lineages and primary tissues expressing different HLA alleles will likely facilitate identification of additional epitopes. Third, LC-MS/MS based assays can suffer from false negatives when peptide abundance is below the limit of detection or the sequence does not ionize well.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HLA Class I antibody W6/32 for HLA-IP | Santa Cruz Biotechnology | Cat # sc-32235; RRID:AB_627934 |
| HLA Class I antibody W6/32 for in-vitro binding assay | ATCC | Cat # HB-95; RRID_CVCL_7872 |
| SARS-CoV Nucleocapsid (N) Protein (RABBIT) polyclonal antibody | Rockland | Cat # 200-401-A50; RRID:AB_828403 |
| Alexa Fluor 568 goat anti rabbit antibody | Invitrogen | Cat # A11011; RRID:AB_143187 |
| anti mouse IFNy mAB AN18 purified 250 ug (1mg/mL) | Mabtech | Cat # 3321-3-250; RRID:AB_907279 |
| anti-mouse IFNy mAb R4-6A2, biotinylated 250ug (1mg/mL) | Mabtech | Cat # 3321-6-250; RRID:AB_907271 |
| MS CD3E pure mAB 0.5 mg/mL | BD Biosciences | Cat # 553058, RRID:AB_394591 |
| Bacterial and virus strains | ||
| SARS-CoV-2 | Centers for Disease Control and Prevention and BEI Resources | 2019-nCoV/USA-WA1/2020 isolate (NCBI accession number: MN985325.1) |
| Biological samples | ||
| Convalescent donor blood sample | Precision for Medicine (USA) | https://www.precisionformedicine.com/ |
| Convalescent donor blood sample | Sanguine (USA) | https://sanguinebio.com/ |
| Convalescent donor blood sample | CTL (USA) | http://www.immunospot.com/ImmunoSpot-ePBMC |
| Convalescent donor blood sample | The Immune Monitoring Laboratory, MGH | https://www.massgeneral.org/cancer-center/clinical-trials-and-research/immunotherapy/cellular-immunotherapy-program/immune-monitoring-laboratory |
| Chemicals, peptides, and recombinant proteins | ||
| 1 M Tris, pH 8.0 | Invitrogen | Cat # AM9855G |
| EDTA | Sigma Aldrich | Cat #7789 |
| Sodium chloride | Sigma Aldrich | Cat #71376 |
| Triton-X | Sigma Aldrich | Cat #T9284 |
| Octyl β-d-glucopyranoside | Sigma Aldrich | Cat # 08001 |
| Phenylmethanesulfonyl fluoride | Sigma Aldrich | Cat # 78830 |
| C0mplete Protease Inhibitor Tablet-EDTA free | Sigma Aldrich | Cat # 5056489001 |
| Aprotinin | Sigma Aldrich | Cat: # A6103 |
| Leupeptin | Roche | Cat: # 11017101001 |
| Sodium fluoride | Sigma Aldrich | Cat: #S7920 |
| Phosphatase inhibitor cocktail 2 | Sigma Aldrich | Cat: #P5726 |
| Phosphatase inhibitor cocktail 3 | Sigma Aldrich | Cat: #P0044 |
| DNase I | Sigma Aldrich | Cat # 4716728001 |
| Gammabind Plus Sepharose beads | Millipore Sigma | Cat #17-0886-01 |
| Dithiothretiol, No-Weigh Format | Fisher Scientific | Cat # 20291 |
| Iodoacetamide | Sigma Aldrich | Cat: # A3221 |
| Lysyl endopeptidase (LysC), | Wako Chemicals | Cat # 129-02541 |
| Trypsin, Mass Spec Grade | Promega | Cat # V511X |
| Formic Acid | Sigma Aldrich | Cat # F0507 |
| Acetonitrile | Honeywell | Cat # 34967 |
| Trifluoretic acid | Sigma Aldrich | Cat # 302031 |
| TMT sixplex Isobaric Label Reagent Set | ThermoFisher | Cat # 90061 |
| 0.5M HEPES, pH 8.5 | Alfa Aesar | Cat # J63218 |
| Hydroxylamine solution, 50% (vol/vol) in H2O | Sigma Aldrich | Cat #467804 |
| Methanol | Honeywell | Cat # 34966 |
| Ammonium hydroxide solution, 28% (wt/vol) in H2O | Sigma Aldrich | Cat # 338818 |
| Acetic acid, glacial | Sigma Aldrich | Cat # AX0073 |
| Avicel | DuPont | Cat # RC-581 |
| Benzonase | Thomas Scientific | Cat # E1014-25KU |
| Synthetic peptides, 5mg > 90% purity | Genscript | Customized quote |
| SARS-CoV-2 Nucleocapsid peptides pool | JPT | PM-WCPV-NCAP |
| Human IFNγ ELISpot | CTL | Cat #hIFNgp-2M/2 |
| Illumina TruSeq Stranded mRNA (LT) | Illumina | Cat # FC-122-2101 |
| Agilent 2200 TapeStation D1000 ScreenTape | Agilent | Cat # 5067-5582 |
| NextSeq V2.5 High Output 75 cycle kit | Illumina | Cat # 20024906 |
| NextSeq V2.5 High Output 150 cycles kit | Illumina | Cat # 20024907 |
| PolyIC/LC, Hiltonol (2mg/ml) | Oncovir | |
| Streptavidin HRP | Mabtech | Cat # 3310-9-1000 |
| Substrate for Elispot: TMB | Mabtech | Cat # 3651-10 |
| M. Tuberculosis H37 RA | DIFCO Laboratories | Cat # 231141 |
| Adjuvant complete freund | Becton, Dickinson and Co | Cat # 263810 |
| GIBCO Phytohemagglutinin, M form (PHA-M) | GIBCO | Cat #10576015 |
| Multiscreen IP WHT STRL | Thermo Fisher Scientific | Cat #MSIPS4W10 |
| SDB-XC disk, 47mm | Empore 3M | Cat # 2240 |
| Critical commercial assays | ||
| HLA-A,B,C typing | Histogenetics | Customized quote |
| Deposited data | ||
| RNA sequencing data | GEO | GSE159191 |
| original mass spectra, peptide spectrum matches and databases | MassIVE | MSV000087225 |
| Experimental models: Cell lines | ||
| A549 | ATCC | CCL-185 |
| HEK293T | ATCC | CRL-3216 |
| VERO E6 | ATCC | CRL-1586 |
| Experimental models: Organisms/strains | ||
| B6.Cg-Immp2lTg(HLA-A/H2-D)2Enge/J | The Jackson Laboratory | Stock # 004191 |
| Software and algorithms | ||
| Spectrum Mill software package v7.1 pre-Release | Broad Institute | https://proteomics.broadinstitute.org/ |
| Bowtie 1.1.2 | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| HLAthena binding prediction | Broad Institute | http://hlathena.tools/ |
| Other | ||
| Easy-nLC 1200 System | Thermo Fisher Scientific | LC140 |
| Orbitrap Exploris 480 | Thermo Fisher Scientific | BRE725532 |
| FAIMS Pro Interface | Thermo Fisher Scientific | FNS02-10001 |
| Sequences, analyses, and barcoded tetramer pools used to determine peripheral T cell specificity | This paper | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Shira Weingarten-Gabbay (shirawg@broadinstitute.org).
Materials availability
Cell lines transduced with ACE2 and TMPRSS2 are available upon request.
Data and code availability
The raw RNA sequencing data generated in this study have been submitted to the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE159191. The original mass spectra, peptide spectrum matches, and the protein sequence databases used for searches have been deposited in the public proteomics repository MassIVE (https://massive.ucsd.edu) and are accessible at ftp://MSV000087225@massive.ucsd.edu.
Experimental model and subject details
Human subjects
Peripheral blood samples for pooled ELISpot assays were collected from COVID-19 convalescent patients (2 male and 4 female) under informed consent. Ethical review for the collection of peripheral blood samples and the secondary use of the PBMCs was conducted by Partners Healthcare Services (PHS) Institutional Review Board (IRB protocol IDs: 2020P000804 and 2020P001446). Peripheral blood samples for multiplexed tetramer assays (9 male and 8 female) were purchased from Precision 4 Medicine (USA), Sanguine (USA), or CTL (USA). These companies ethically collected samples under informed consent or as clinical excess specimens under a waiver of consent.
Cell culture
Human embryonic kidney HEK293T cells (female), human lung A549 cells (male), and African green monkey kidney Vero E6 cells (female) were maintained at 37°C and 5% CO2 in DMEM containing 10% FBS. We generated stable HEK293T and A549 cells expressing human ACE2 and TMPRSS2 by transducing them with lentivirus particles carrying these two cDNAs. A549 cells express A∗25:01/30:01, B∗18:01/44:03 and C∗12:03/16:01, while HEK293T cells express A∗02:01, B∗07:02 and C∗07:02 (determined by HLA typing, Histogenetics, USA).
SARS-CoV-2 virus stock preparation
The 2019-nCoV/USA-WA1/2020 isolate (NCBI accession number: MN985325) of SARS-CoV-2 was obtained from the Centers for Disease Control and Prevention and BEI Resources. To generate the virus P1 stock, we infected Vero E6 cells with this isolate for 1h at 37°C, removed the virus inoculum, rinsed the cell monolayer with 1X PBS, and added DMEM supplemented with 2% FBS. Three days later, when the cytopathic effect of the virus became visible, we harvested the culture medium, passed through a 0.2μ filter, and stored it at −80°C. To generate the virus P2 stock, we infected Vero E6 cells with the P1 stock at a multiplicity of infection (MOI) of 0.1 plaque forming units (PFU)/cell and harvested the culture medium three days later using the same protocol as for the P1 stock. All experiments in this study were performed using the P2 stock.
HLA-A02 transgenic mice
6-8 week old, female HLA-A2 transgenic AAD mice were used in the experiments (B6.Cg-Immp2l Tg(HLA-A/H2-D)2Enge/J, The Jackson Laboratory). These animals express a chimeric molecule, which contains peptide-binding α1 and α2 domains of the HLA-A2.1 molecule and the α3 domain of the mouse H-2 Dd molecule, under the control of the HLA-A2.1 promoter in addition to mouse MHC H-2b. These animals allow the testing of human T cell immune responses to HLA-A2 presented antigens. Animals were maintained and bred in the animal facility at Dana Farber Cancer Institute in compliance with the Institutional Animal Care and Use Committee.
Method details
Quantification of virus infectivity
A549 and 293T cells expressing ACE2 and TMPRSS2 were infected with SARS-CoV-2 (Washington isolate) at an MOI of 3 for indicated times (3, 6, 12, 18, and 24 hpi). After infection, supernatants were removed, and cells were fixed with 4% paraformaldehyde for 30 minutes at room temperature. Cells were then permeabilized with 0.1% of Triton X-100 in PBS for 10 minutes and hybridized with Anti-SARS-CoV Nucleocapsid (N) Protein (RABBIT) polyclonal antibody (1:2000, Rockland, #200-401-A50) at 4°C overnight. Alexa Fluor 568 goat anti rabbit antibody (Invitrogen, A11011) were used as the secondary antibody for labeling virus infected cells. Finally, DAPI was added to label the nuclei. Immunofluorescent images were taken using an EVOS microscope with 10x lens and infection rates were calculated with ImageJ.
Plaque assay
Vero E6 cells were used to determine the titer of our virus stock and to evaluate SARS-CoV-2 inactivation following lysis of infected cells in our HLA-IP buffer. Briefly, we seeded Vero E6 cells into a 12-well plate at a density of 2.5 × 105 cells per well, and the next day, infected them with serial 10-fold dilutions of the virus stock (for titration) or the A549 lysates (for the inactivation assay) for 1h at 37°C. We then added 1 mL per well of the overlay medium containing 2X DMEM (GIBCO: #12800017) supplemented with 4% FBS and mixed at a 1:1 ratio with 1.2% Avicel (DuPont; RC-581) to obtain the final concentrations of 2% and 0.6% for FBS and Avicel, respectively. Three days later, we removed the overlay medium, rinsed the cell monolayer with 1X PBS and fixed the cells with 4% paraformaldehyde for 30 minutes at room temperature. 0.1% crystal violet was used to visualize the plaques.
Immunoprecipitation of HLA-I complexes
Cells engineered to express SARS-CoV-2 entry factors were seeded into nine 15 cm dishes (three dishes per time point) at a density of 15 million cells per dish for A549 cells and 20 million cells per dish for HEK293T cells. The next day, the cells were infected with SARS-CoV-2 at a multiplicity of infection (MOI) of 3. To synchronize infection, the virus was bound to target cells in a small volume of opti-MEM on ice for one hour, followed by addition of DMEM/2% FBS and switching to 37°C. At 3, 6, 12, 18, and 24h post-infection, the cells from three dishes were scraped into 2.5ml/dish of cold lysis buffer (20mM Tris, pH 8.0, 100mM NaCl, 6mM MgCl2, 1mM EDTA, 60mM Octyl β-d-glucopyranoside, 0.2mM Iodoacetamide, 1.5% Triton X-100, 50xC0mplete Protease Inhibitor Tablet-EDTA free and PMSF) obtaining a total of 9 mL lysate. This lysate was split into 6 eppendorf tubes, with each tube receiving 1.5 mL volume, and incubated on ice for 15 min with 1ul of Benzonase (Thomas Scientific, E1014-25KU) to degrade nucleic acid. The lysates were then centrifuged at 4,000 rpm for 22min at 4°C and the supernatants were transferred to another set of six eppendorf tubes containing a mixture of pre-washed beads (Millipore Sigma, GE17-0886-01) and 50 ul of an MHC class I antibody (W6/32) (Santa Cruz Biotechnology, sc-32235). The immune complexes were captured on the beads by incubating on a rotor at 4°C for 3hr in the BSL3 lab. Virus inactivation was confirmed before subsequent samples processing outside the BSL3 using plaque assay (Figure S1C). The unbound lysates were kept for whole proteomics analysis while the beads were washed to remove non-specifically bound material. In total, nine washing steps were performed; one wash with 1mL of cold lysis wash buffer (20mM Tris, pH 8.0, 100mM NaCl, 6mM MgCl2, 1mM EDTA, 60mM Octyl β-d-glucopyranoside, 0.2mM Iodoacetamide, 1.5% Triton X-100), four washes with 1mL of cold complete wash buffer (20mM Tris, pH 8.0, 100mM NaCl, 1mM EDTA, 60mM Octyl β-d-glucopyranoside, 0.2mM Iodoacetamide), and four washes with 20mM Tris pH 8.0 buffer. Dry beads were stored at −80°C until mass-spectrometry analysis was performed.
HLA-I peptidome LC-MS/MS data generation
HLA peptides were eluted and desalted from beads as described previously (Sarkizova et al., 2020). After the primary elution step, HLA peptides were reconstituted in 3% ACN/5% FA and subjected to microscaled basic reverse phase separation. Briefly, peptides were loaded on Stage-tips with 2 punches of SDB-XC material (Empore 3M) and eluted in three fractions with increasing concentrations of ACN (5%, 10% and 30% in 0.1% NH4OH, pH 10). For the time course experiment, one third of a pool of 6 IPs (for 12|18|24h) or a pool of 2 IPs (for 3|6|24hpi) was also labeled with TMT6 (Thermo Fisher Scientific, lot # UC280588, A549: 12h:126, 3h:127, 18h:128, 129: 6h, 24h:130, HEK293T: 3h: 126, 12h:127, 6h:128, 18h:129, 24h:131) (Thompson et al., 2003), combined and desalted on a C18 Stage-tip, and then eluted into three fractions using basic reversed phase fractionation with increasing concentrations of ACN (10%, 15% and 50%) in 5 mM ammonium formate (pH 10). Peptides were reconstituted in 3% ACN/5%FA prior to loading onto an analytical column (25-30cm, 1.9 μm C18 (Dr. Maisch HPLC GmbH), packed in-house PicoFrit 75 μm inner diameter, 10 μm emitter (New Objective)). Peptides were eluted with a linear gradient (EasyNanoLC 1200, Thermo Fisher Scientific) ranging from 6%–30% Solvent B (0.1%FA in 90% ACN) over 84 min, 30%–90% B over 9 min and held at 90% B for 5 min at 200 nl/min. MS/MS were acquired on a Thermo Orbitrap Exploris 480 equipped with FAIMS (Thermo Fisher Scientific) in data dependent acquisition. FAIMS CVs were set to −50 and −70 with a cycle time of 1.5 s per FAIMS experiment. MS2 fill time was set to 100ms, collision energy was 29CE or 32CE for TMT respectively.
Whole proteome LC-MS/MS data generation
200 uL aliquot of HLA IP supernatants were reduced for 30 minutes with 5mM DTT (Pierce DTT: A39255) and alkylated with 10mM IAA (Sigma IAA: A3221-10VL) for 45 minutes both at 25°C on a shaker (1000 rpm). Protein precipitation using methanol/chloroform was then performed. Briefly, methanol was added at a volume of 4x that of HLA IP supernant aliquot. This was followed by a 1x volume of chloroform and 3x volume of water. The sample was mixed by vortexing and incubated at −20°C for 1.5 hours. Samples were then centrifuged at 14,000 rpm for 10 minutes and the upper liquid layer was removed leaving a protein pellet. The pellet was rinsed with 3x volume of methanol, vortexed lightly, and centrifuged at 14,000 rpm for 10 minutes. Supernatant was removed and discarded without disturbing the pellet. Pellets were resuspended in 100 mM triethylammonium bicarbonate (pH 8.5) (TEAB). Samples were digested with LysC (1:50) for 2h on a shaker (1000 rpm) at 25°C, followed by trypsin (1:50) overnight. Samples were acidified by 1% formic acid final concentration and dried. Samples were reconstituted in 4.5 mM ammonium formate (pH 10) in 2% (vol/vol) acetonitrile and separated into four fractions using basic reversed phase fractionation on a C-18 Stage-tip. Fractions were eluted at 5%, 12.5%, 15%, and 50% ACN/4.5 mM ammonium formate (pH 10) and dried. Fractions were reconstituted in 3%ACN/5%FA, and 1 ug was used for LC-MS/MS analysis. MS/MS were acquired on a Thermo Orbitrap Exploris 480 (Thermo Fisher Scientific) in data dependent acquisition (MS2 isolation width 0.7 m/z, top20 scans, collision energy 30%) (Figures 2, 3, 4, and S3B–S3D). Uninfected 1 ug single shot samples were analyzed similarly. For the time course experiment, the samples (12h, 18h, 24h) were not fractionated and 1 ug was used for LC-MS/MS analysis, as described above except that FAIMS with −50, −65, and −85 CV was applied and cycle time was 0.8 s for each CV (Figure S3A).
LC-MS/MS data interpretation
Peptide sequences were interpreted from MS/MS spectra using Spectrum Mill (v 7.1 pre-release) to search against a RefSeq-based sequence database containing 41,457 proteins mapped to the human reference genome (hg38) obtained via the UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables) on June 29, 2018, with the addition of 13 proteins encoded in the human mitochondrial genome, 264 common laboratory contaminant proteins, 553 human non-canonical small open reading frames, 28 SARS-CoV2 proteins obtained from RefSeq derived from the original Wuhan-Hu-1 China isolate NC_045512.2 (https://www.ncbi.nlm.nih.gov/nuccore/1798174254; Wu et al., 2020), and 23 novel unannotated virus ORFs whose translation is supported by Ribo-seq (Finkel et al., 2020b) for a total of 42,337 proteins. Among the 28 annotated SARS-CoV2 proteins we opted to omit the full-length polyproteins ORF1a and ORF1ab, to simplify peptide-to-protein assignment, and instead represented ORF1ab as the mature 16 individual non-structural proteins that result from proteolytic processing of the 1a and 1ab polyproteins. We added the D614G variant of the SARS-Cov2 Spike protein that is commonly observed in European and American virus isolates. For additional searches, we also added 2036 entries from 6-frame translation of the SARS-Cov2 genome for all possible ORFs longer than 6 amino acids (Table S1).
For immunopeptidome data MS/MS spectra were excluded from searching if they did not have a precursor MH+ in the range of 600-4000, had a precursor charge > 5, or had a minimum of < 5 detected peaks. Merging of similar spectra with the same precursor m/z acquired in the same chromatographic peak was disabled. Prior to searches, all MS/MS spectra had to pass the spectral quality filter with a sequence tag length > 1 (i.e., minimum of 3 masses separated by the in-chain masses of 2 amino acids) based on HLA v3 peak detection. MS/MS search parameters included: ESI-QEXACTIVE-HCD-HLA-v3 scoring parameters; no-enzyme specificity; fixed modification: carbamidomethylation of cysteine; variable modifications: cysteinylation of cysteine, oxidation of methionine, deamidation of asparagine, acetylation of protein N-termini, and pyroglutamic acid at peptide N-terminal glutamine; precursor mass shift range of −18 to 81 Da; precursor mass tolerance of ± 10 ppm; product mass tolerance of ± 10 ppm, and a minimum matched peak intensity of 30%. Peptide spectrum matches (PSMs) for individual spectra were automatically designated as confidently assigned using the Spectrum Mill auto-validation module to apply target-decoy based FDR estimation at the PSM level of < 1.5% FDR. For the TMT-labeled time course experiments, two parameters were revised: the MH+ range filter was 800-6000, and TMT labeling was required at lysine, but peptide N-termini were allowed to be either labeled or unlabeled. Relative abundances of peptides in the time-course experiments were determined in Spectrum Mill using TMT reporter ion intensity ratios from each PSM. TMT reporter ion intensities for the 3 time points split across two plexes were not corrected for isotopic impurities because the respective adjacent intervening labels were not included. Each peptide-level TMT ratio was calculated as the median of all PSMs contributing to that peptide. PSMs were excluded from the calculation that lacked a TMT label, or had a negative delta forward-reverse identification score (half of all false-positive identifications). Intensity values for each time point were normalized to the 24h time point to compare between the 12|18|24h and 3|6|24h plex.
For whole proteome data MS/MS spectra were excluded from searching if they did not have a precursor MH+ in the range of 600-6000, had a precursor charge > 5, had a minimum of < 5 detected peaks, or failed the spectral quality filter with a sequence tag length > 0 (i.e., minimum of 2 masses separated by the in-chain masses of 1 amino acid) based on ESI-QEXACTIVE-HCD-v4-30-20 peak detection. Similar spectra with the same precursor m/z acquired in the same chromatographic peak were merged. MS/MS search parameters included: ESI-QEXACTIVE-HCD-v4-30-20 scoring parameters; Trypsin allow P specificity with a maximum of 4 missed cleavages; fixed modification: carbamidomethylation of cysteine and seleno-cysteine; variable modifications: oxidation of methionine, deamidation of asparagine, acetylation of protein N-termini, pyroglutamic acid at peptide N-terminal glutamine, and pyro-carbamidomethylation at peptide N-terminal cysteine; precursor mass shift range of −18 to 64 Da; precursor mass tolerance of ± 20 ppm; product mass tolerance of ± 20 ppm, and a minimum matched peak intensity of 30%. Peptide spectrum matches (PSMs) for individual spectra were automatically designated as confidently assigned using the Spectrum Mill auto-validation module to apply target-decoy based FDR estimation at the PSM level of < 1.0% FDR. Protein level data was summarized by top uses shared (SGT) peptide grouping and non-human contaminants were removed. SARS-CoV-2 derived proteins were manually filtered to include identifications with > 6% sequence coverage and at least 2 or more unique peptides.
Validation of peptide identifications
Peptide identifications were validated using synthetic peptides. Synthetic peptides were obtained from Genscript, at purity > 90% purity and dissolved to 10 mM in DMSO. For LC-MS/MS measurements, peptides were pooled and further diluted with 0.1% FA/3% ACN to load 120 fmol/μl on column. One aliquot of synthetic peptides was also TMT labeled as described above. LC-MS/MS measurements were performed as described above. For plots, peak intensities in the experimental and the synthetic spectrum were normalized to the highest peak.
RNA-Seq of SARS-CoV-2 infected cells
A549 and HEK293T cells were seeded into 6-well plates at a density of 5 × 105 cells per well (one well per condition). After 11-24h, the cells were infected with SARS-CoV-2 at an MOI of 3. At 12, 18 and 24h post-infection, the cells were lysed in Trizol (Thermo, 15596026), and the total RNA was isolated using standard phenol chloroform extraction. Standard Illumina TruSeq Stranded mRNA (LT) was performed using 500 ng of total RNA (illumina, FC-122-2101). Oligo-dT beads were used to capture polyA-tailed RNA, followed by fragmentation and priming of the captured RNA (8 minutes at 94°C). Immediately first strand cDNA synthesis was performed. Second strand cDNA synthesis was performed using second strand marking master and DNA polymerase 1 and RNase H. cDNA was adenylated at the 3′ ends followed immediately by RNA end ligation single-index adapters (AR001-AR012). Library amplification was performed for 12-15 cycles under standard illumina library PCR conditions. Library quantitation was performed using Agilent 2200 TapeStation D1000 ScreenTape (Agilent, 5067-5582). RNA sequencing was performed on the NextSeq 550 System using a NextSeq V2.5 High Output 75 cycle kit (illumina, 20024906) or 150 cycles kit (illumina, 20024907) for paired-end sequencing (70nt of each end).
In-vitro MHC-peptide binding assay
Classical competition assays, based on the inhibition of binding of a high affinity radiolabeled ligand to purified MHC molecules, were utilized to quantitatively measure peptide binding to HLA-A and -B class I MHC molecules. The assays were performed, and MHC purified, as detailed previously (Sidney et al., 2013). Briefly, 0.1-1 nM of radiolabeled peptide was co-incubated at room temperature with 1 μM to 1 nM of purified MHC in the presence of a cocktail of protease inhibitors and 1 μM B2-microglobulin. MHC bound radioactivity was determined following a two-day incubation by capturing MHC/peptide complexes on W6/32 (anti-class I) antibody coated Lumitrac 600 plates (Greiner Bio-one, Frickenhausen, Germany), and measuring bound cpm on the TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter. The concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide was calculated. Under the conditions utilized, where [label] < [MHC] and IC50 ≥ [MHC], the measured IC50 values are reasonable approximations of the true Kd values (Cheng and Prusoff, 1973; Gulukota et al., 1997). Each competitor peptide was tested at six different concentrations covering a 100,000-fold dose range, and in three or more independent experiments. As a positive control, the unlabeled version of the radiolabeled probe was also tested in each experiment.
Vaccinations of HLA-A2 transgenic mice
Five mice were immunized subcutaneously in the flank with a vaccine. The vaccine contained nine A∗02:01 peptides (50ug each peptide per mice) emulsified in Complete Freunds Adjuvant (CFA BD Bioscience/Difco) supplemented with 20ug PolyIC/LC (Hiltonol/Oncovir). 10 days post-vaccination, animals were euthanized using CO2, and Spleens were removed for ELISpot assays.
Mouse ELISpot assay
ELISpot was performed using red blood cell-depleted mouse splenocytes (200,000 cells/well) co-incubated with the individual peptides (10 μg/ml) in triplicate in ELISpot plates (Millipore, Billerica, MA) for 18h. Interferon-γ (IFNγ) secretion was detected using capture and detection antibodies as described (Mabtech AB, Nacka Strand, Sweden) and imaged using an ImmunoSpot Series Analyzer (Cellular Technology, Ltd, Cleveland, OH). HLA-A∗02:01 restricted HIV-GAG peptide and non-stimulated wells were used as negative controls. Spot numbers were normalized by removing the average background spot numbers calculated from negative control wells. AntiCD3 (2C11 BD BioScience) and PHA was used as a positive control. 55 spot-forming units/106 cells and a ≥ 3-fold increase over baseline is used as a threshold for positive responses. Methods were described in detail previously (Keskin et al., 2015).
ELISpot assay with COVID-19 PBMCs
Peripheral blood samples were collected from COVID-19 convalescent patients and PBMC were isolated using ficoll density gradient centrifugation. PBMC were plated out in serum free T cell assay media at 2.5e5 cells per well in a Human IFNγ single color ELISpot plate (Cellular Technology Limited [CTL], Cat# hIFNgp-2M/2). The canonical and non canonical pools (canonical pool: APHGHVMVEL, EEFEPSTQYEY, EIKESVQTF, EILDITPCSF, EILDITPCSFG, FASEAARVV, FAVDAAKAY, IRQEEVQEL, KNIDGYFKIY, KRVDWTIEY, NATNVVIKV, QLTPTWRVY, SEFSSLPSY, VGYLQPRTF, and YLNSTNVTI; non-canonical pool: DEFVVVTV, ELPDEFVVVTV, GLITLSYHL, GPMVLRGLIT, LEDKAFQL, MLLGSMLYM, and SLEDKAFQL) and a commercial nucleocapsid overlapping peptide pool (JPT peptide Technologies) were added to duplicate wells at a concentration of 2ug/ml of each peptide. A negative control well (to which just the equivalent concentration of DMSO was added) was used to assess background IFNg secretion. Cells were incubated for 16-20 hours at 37oC before developing according to manufacturer’s instructions. Spots were counted using an ImmunoSpot CoreS6 ELISpot counter (ImmunoSpot). The negative control background was subtracted from the antigen wells and the results are shown as spot forming units (SFU) per 2.5e5 PBMC. A spot cut off of 8 after background subtraction is used here to denote a positive response.
Multiplexed tetramer assay
HLA-A∗02:01, and HLA-B∗07:02 extracellular domains were expressed in E. coli and refolded along with beta-2-microglobulin and UV-labile place-holder peptides KILGFVFJV, and AARGJTLAM, respectively (Altman and Davis, 2016). The MHC monomer was then purified by size exclusion chromatography (SEC). MHC tetramers were produced by mixing alkylated MHC monomers and azidylated streptavidin in 0.5 mM copper sulfate, 2.5 mM BTTAA and 5 mM ascorbic acid for up to 4 h on ice, followed by purification of highly multimeric fractions by SEC. Individual peptide exchange reactions containing 500 nM MHC tetramer and 60 uM peptide were exposed to long-wave UV (366 nm) at a distance of 2-5 cm for 30 min at 4°C, followed by 30 min incubation at 30°C. A biotinylated oligonucleotide barcode (Integrated DNA Technologies) was added to each individual reaction followed by 30 minute incubation at 4°C. Individual tetramer reactions were then pooled and concentrated using 30 kDa molecular weight cut-off centrifugal filter units (Amicon).
De-identified peripheral blood mononuclear cells (PBMCs) from convalescent COVID-19 positive donors or unexposed donors were purchased from Precision 4 Medicine (USA), Sanguine (USA), or CTL (USA). These companies ethically collected samples under informed consent or as clinical excess specimens under a waiver of consent. PBMCs were thawed, and CD8+ T cells were enriched by magnetic-activated cell sorting (MACS) using a CD8+ T Cell Isolation Kit (Miltenyi) following the manufacturer’s protocol. The CD8+ T cells were then stained with 1 nM final concentration tetramer library in the presence of 2 mg/mL salmon sperm DNA in PBS with 0.5% BSA solution for 20 minutes. Cells were then labeled with anti-TCR ADT (IP26, Biolegend) for 15 minutes followed by washing. Tetramer bound cells were then labeled with PE conjugated anti-DKDDDDK-Flag antibody (BioLegend) followed by dead cell discrimination using 7-amino-actinomycin D (7-AAD). The live, tetramer positive cells were sorted using a Sony MA900 Sorter (Sony).
Single-cell sequencing
Tetramer positive cells were counted by Nexcelom Cellometer (Lawrence, MA, USA) using AOPI stain following manufacturer’s recommended conditions. Single-cell encapsulations were generated utilizing 5′ v1 Gem beads from 10x Genomics (Pleasanton, CA, USA) on a 10x Chromium controller and downstream TCR, and Surface marker libraries were made following manufacturer recommended conditions. All libraries were quantified on a BioRad CFX 384 (Hercules, CA, USA) using Kapa Biosystems (Wilmington, MA, USA) library quantified kits and pooled at an equimolar ratio. TCRs, surface markers, and tetramer generated libraries were sequenced on Illumina (San Diego, CA, USA) NextSeq550 instruments.
HLA-I antigen presentation prediction
HLAthena, a prediction tool trained on endogenous LC-MS/MS-identified epitope data, was used to predict HLA class I presentation for all unique 8-11-mer SARS-Cov-2 peptides across 31 HLA-A, 40 HLA-B and 21 HLA-C alleles (Sarkizova et al., 2020).
HLA allele frequencies and coverage estimates
World frequencies of HLA-A, -B, and -C allele in Table S6B are based on a meta-analysis of high-resolution HLA allele frequency data describing 497 population samples representing approximately 66,800 individuals from throughout the world (Solberg et al., 2008), downloaded from http://pypop.org/popdata/2008/byfreq-A.php.html, http://pypop.org/popdata/2008/byfreq-B.php.html, http://pypop.org/popdata/2008/byfreq-C.php. Subpopulation frequencies for AFA, API, EUR, HIS, and USA were obtained from supplemental data in Poran et al. (2020).
The cumulative phenotypic frequency (CPF) of peptides was calculated using , assuming Hardy-Weinberg proportions for the HLA genotypes (Dawson et al., 2001), where p i is the population frequency of the i th alleles within a subset of HLA-A, -B, or C alleles, denoted C. Coverage across HLA-A, -B, and -C alleles was calculated similarly: , where A, B, and C denote a subset of HLA-A, -B, and/or -C alleles for which the coverage is computed, as recently done in (Poran et al., 2020).
Quantification and statistical analysis
Whole proteome analysis
Postfiltering, intensity-based absolute quantification (iBAQ) was performed on the whole proteome LC-MS/MS as described in Schwanhäusser et al. (2011). Briefly, iBAQ values were calculated as follows: log10(totalIntensity/numObservableTrypticPeptides), the total precursor ion intensity for each protein was calculated in Spectrum Mill as the sum of the precursor ion chromatographic peak areas (in MS1 spectra) for each precursor ion with a peptide spectrum match (MS/MS spectrum) to the protein, and the numObservableTrypticPeptides for each protein was calculated using the Spectrum Mill Protein Database utility as the number of tryptic peptides with length 8 - 40 amino acids, with no missed cleavages allowed. Of note, S coverage was 55% in the HEK293T and 44% in the A549 post 24 hour fractionated data, which may be due to the high levels of glycosylation. Lower peptide coverage may lead to underestimation of S protein in our data. Both log10 transformed total intensity and iBAQ values were median normalized by subtracting sample specific medians and adding global medians for each abundance metrics and reported in Table S4.
To evaluate post infection protein level changes for a large set of proteins across cell lines and time points (Figures 4E and 4F; Figure S3E), all proteins detected in at least 6 out of 8 samples (A549 and HEK293T, each profiled at the 0, 3, 6, and 24 hpi) were retained in the analysis, with missing values imputed to the lowest 10th percentile of the log10iBAQ value distribution (Tyanova et al., 2016). A similar approach was used for the reanalysis of PXD020019 with values of proteins observed in eight of the twelve samples considered for imputation, and zero values replaced with a value below the minimum LFQ value reported.
Infected cells RNA-seq reads alignment
Sequencing reads were mapped to SARS-CoV-2 genome (RefSeq NC_045512.2) and human transcriptome (Gencode v32). Alignment was performed using Bowtie version 1.2.2 (Langmead et al., 2009) with a maximum of two mismatches per read. The fraction of human and viral reads was determined based on the total number of reads aligned to either SARS-CoV-2 or human transcripts.
Scoring pMHC-TCR interactions
Tetramer data analysis was performed using Python 3.7.3. For each single-cell encapsulation, tetramer UMI counts (columns) were matrixed by cell (rows) and log-transformed. The matrix was then Z-score transformed row-wise and subsequently, median-centered by column. Means were calculated by clonotype, and those with a value greater than 4 were characterized as positive interactions.
T cells transcriptomics analysis
Hydrogel-based RNA-seq data were analyzed using the Cell Ranger package from 10X Genomics (v3.1.0) with the GRCh38 human expression reference (v3.0.0). Except where noted, Scanpy (v1.6.0; Wolf et al., 2018) was used to perform the subsequent single-cell analyses. Any exogenous control cells identified by TCR clonotype were removed before further gene expression processing. Hydrogels that contain UMIs for less than 300 genes were excluded. Genes that were detected in less than 3 cells were also excluded from further analysis. The following additional quality control thresholds were also enforced. To remove data generated from cells likely to be damaged, upper thresholds were set for percent UMIs arising from mitochondrial genes (13%). To exclude data likely arising from multiple cells captured in a single drop, upper thresholds were set for total UMI counts based on individual distributions from each encapsulation (from 1500 to 3000 UMIs). A lower threshold of 10% was set for UMIs arising from ribosomal protein genes. Finally, an upper threshold of 5% of UMIs was set for the MALAT1 gene. Any hydrogel outside of any of the thresholds was omitted from further analysis. A total of 15,683 hydrogels were carried forward. Gene expression data were normalized to counts per 10,000 UMIs per cell (CP10K) followed by log1p transformation: ln(CP10K + 1).
Clustering and annotation of single cell data
Highly variable genes were identified (1,567) and scaled to have a mean of zero and unit variance. They were then provided to scanorama (v1.7; Hie et al., 2019) to perform batch integration and dimension reduction. These data were used to generate the nearest neighbor graph which was in turn used to generate a UMAP representation that was used for Leiden clustering. The hydrogel data (not scaled to mean zero, unit variance, and before extraction of highly variable genes) were labeled with cluster membership and provided to SingleR (v1.4.0; Aran et al., 2019) using the following references from Celldex (v1.0.0; Aran et al., 2019): MonacoImmuneData, DatabaseImmuneCellExpressionData, and BlueprintEncodeData. SingleR was used to annotate the clusters with their best-fit match from the cell types in the references. Clusters that yielded cell types other than types of the T Cell lineage were removed from consideration and the process was repeated starting from the batch integration step. The best-fit annotations from SingleR after the second round of clustering and annotation were assigned as putative labels for each Leiden cluster.
In order to provide corroboration for the SingleR best-fit annotations and further evidence as to the phenotype of the clusters, gene panels representing functional categories (Naive, Effector, Memory, Exhaustion, Proliferation) were used to score each hydrogel’s expression profiles using scanpy’s “score_genes” function (Wolf et al., 2018) which compares the mean expression values of the target gene set against a larger set of randomly chosen genes that represent background expression levels. The gene panels for each class were (Su et al., 2020): Naive - TCF7, LEF1, CCR7; Effector - GZMB, PRF1, GNLY; Memory - AQP3, CD69, GZMK; Exhaustion - PDCD1, TIGIT, LAG3, HAVCR2 (TIM3); Proliferation - MKI67, TYMS. The gene expression matrix for all hydrogels were first imputed using the MAGIC algorithm (v2.0.4; van Dijk et al., 2018). These functional scores were the only data generated from imputed expression values.
Consortia
The members of the MGH COVID-19 Collection & Processing Team are Kendall Lavin-Parsons, Blair Parry, Brendan Lilley, Carl Lodenstein, Brenna McKaig, Nicole Charland, Hargun Khanna, Justin Margolin, Anna Gonye, Irena Gushterova, Tom Lasalle, Nihaarika Sharma, Brian C. Russo, Maricarmen Rojas-Lopez, Moshe Sade-Feldman, Kasidet Manakongtreecheep, Jessica Tantivit, and Molly Fisher Thomas.
Acknowledgments
This project was started in April 2020 while SARS-CoV-2 was spreading fast in the United States and the people of Boston were sheltering in place. We are grateful to the tremendous efforts of the supporting teams, both at the Broad Institute and the NEIDL, who facilitated our research under those challenging circumstances. We thank J. Leon, O. Mizrahi, and E. Normandin for technical assistance; D.R. Mani and H. Metsky for computational assistance; and T. Ouspenskaia and P.M. Jean-Beltran for insightful discussions. We thank the Maus Lab volunteers who collected samples: E. Silva, K. Grauwet, and M. Jan. This study was supported in part by grants from the National Institute of Allergy and Infectious Diseases (U19AI110818 to P.C.S. and U19AI082630 to N.H.) and National Cancer Institute Clinical Proteomic Tumor Analysis Consortium grants (NIH/NCI U24-CA210986 and NIH/NCI U01 CA214125 to S.A.C. and NIH/NCI U24CA210979 to D.R.M.). S.W.-G. is the recipient of a Human Frontier Science Program fellowship (LT-000396/2018), EMBO non-stipendiary long-term fellowship (ALTF 883-2017), the Gruss-Lipper postdoctoral fellowship, the Zuckerman STEM Leadership Program fellowship, and the Rothschild postdoctoral fellowship. S.K. is a Cancer Research Institute/Hearst Foundation fellow. C.H.T.T. was supported by a National Science Foundation graduate research fellowship (1745303). M.G. is the recipient of an EMBO long-term fellowship (ALTF 486-2018) and a Cancer Research Institute/Bristol-Myers Squibb fellow (CRI2993). N.B. is an extramural member of the Parker Institute for Cancer Immunotherapy. C.C.B is a Parker Institute for Cancer Immunotherapy Bridge Fellow. D.B.K. acknowledges funding support from Emerson Collective and NIH/NCI R21 CA216772-01A1. C.M.R is supported by the G. Harold and Leila Y. Mathers Charitable Foundation, the Bawd Foundation, and NIH NIAID 3 R01-AI091707-10S1. M.S. is supported by Boston University startup funds. The MGH/MassCPR COVID biorepository was supported by a gift from Ms. Enid Schwartz, by the Mark and Lisa Schwartz Foundation, the Massachusetts Consortium for Pathogen Readiness, and the Ragon Institute of MGH, MIT and Harvard. This project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research under contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Laboratory, Center for Cancer Research.
Author contributions
S.W.-G., S.K., S.S., and J.G.A. conceptualized the study. S.W.-G., S.K., J.G.A., C.M.R., and M.S. designed the experiments. S.K., S.S., L.R.P., D.-Y.C., K.M.E.G., M.R.B., H.B.T., C.T., J.S., S.R., H.L.C., K.K., Y.W., D.L.-E., M.R.D., K.D.R., I.P.C., V.A.C., A.C., C.C.B., M.C., D.B.K., J.G.A., and M.S. performed experiments. S.W.-G., S.K., S.S., M.R.B., W.A.D., D.C.P., C.H.T.-T., Y.F., A.N., M.G., K.R.C., and J.G.A. performed data analysis. N.B., D.H.B., A.S., M.V.M., D.B.K., D.C.P., N.H., S.A.C., M.S., and P.C.S. supervised experiments. S.W.-G., S.K., S.S., K.R.C., N.H., S.A.C., J.G.A., M.S., and P.C.S. wrote the manuscript with comments from all authors.
Declaration of interests
S.W.-G., S.K., S.S., K.R.C, N.H., S.A.C., J.G.A., M.S., and P.C.S. are named co-inventors on a patent application related to immunogenic compositions of this manuscript filed by The Broad Institute that is being made available in accordance with the COVID-19 technology licensing framework to maximize access to university innovations. D.L.-E., C.T., Y.W., M.R.D., W.A.D., and D.C.P. are employees and stockholders of Repertoire Immune Medicines. N.B. is an extramural member of the Parker Institute for Cancer Immunotherapy; receives research funds from Regeneron, Harbor Biomedical, DC Prime, and Dragonfly Therapeutics; and is on the advisory boards of Neon Therapeutics, Novartis, Avidea, Boehringer Ingelheim, Rome Therapeutics, Roswell Park Comprehensive Cancer Center, BreakBio, Carisma Therapeutics, CureVac, Genotwin, BioNTech, Gilead Therapeutics, Tempest Therapeutics, and the Cancer Research Institute. A.S. is a consultant for Gritstone, Flow Pharma, CellCarta, OxfordImmunotech, Immunoscape, and Avalia. La Jolla Institute for Immunology has filed for patent protection for various aspects of T cell epitope and vaccine design work. D.B.K. has previously advised Neon Therapeutics and has received consulting fees from Neon Therapeutics. D.B.K. owns equity in AduroBiotech, Agenus Inc., Armata Pharmaceuticals, Breakbio Corp., Biomarin Pharmaceutical Inc., Bristol Myers Squibb Com., Celldex Therapeutics Inc., Editas Medicine Inc., Exelixis Inc., Gilead Sciences Inc., IMV Inc., Lexicon Pharmaceuticals Inc., Moderna Inc., and Regeneron Pharmaceuticals. D.B.K. receives SARS-CoV-2 research support from BeiGene for a project unrelated to this publication. N.H. is a founder of Neon Therapeutics, Inc. (now BioNTech US), was a member of its scientific advisory board, and holds shares. N.H. is also an advisor for IFM Therapeutics. S.A.C. is a member of the scientific advisory boards of Kymera, PTM BioLabs, and Seer and a scientific advisor to Pfizer and Biogen. J.G.A. is a past employee and shareholder of Neon Therapeutics, Inc. (now BioNTech US). P.C.S. is a co-founder and shareholder of Sherlock Biosciences and a non-executive board member and shareholder of Danaher Corporation.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper self-identifies as living with a disability.
Published: June 3, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.05.046.
Contributor Information
MGH COVID-19 Collection & Processing Team:
Kendall Lavin-Parsons, Blair Parry, Brendan Lilley, Carl Lodenstein, Brenna McKaig, Nicole Charland, Hargun Khanna, Justin Margolin, Anna Gonye, Irena Gushterova, Tom Lasalle, Nihaarika Sharma, Brian C. Russo, Maricarmen Rojas-Lopez, Moshe Sade-Feldman, Kasidet Manakongtreecheep, Jessica Tantivit, and Molly Fisher Thomas
Supplemental information
Sheet 1: Legend for Table, Sheet 2: HLA-I bound peptide sequences from SARS-CoV-2 proteins identified in both A549 and HEK293T cells. Cysteines were detected in their carbamidomethylated or cysteinylated modification state, methionines were oxidized. “x” denotes identification in individual experiments, “T” in TMT experiments, “-” indicates that the peptide was detected but did not pass FDR correction in that sample, confidence level in identification was evaluated based on Spectrum Mill Score, after manual inspection of MS/MS spectra and comparison to synthetic peptide spectra for available peptides (Y: validated by synthetic peptide, N/A: synthetic peptide not available). For binding prediction using HLAthena, residues were considered unmodified; columns show the most likely HLA allele a peptide is bound to in the respective cell line as well as the percentile binding rank. Sheet 3-11: Peptide identifications from HLA-I immunoprecipitation experiments in A549 and HEK293T cells ± SARS-CoV-2 after filtering for contaminants and length 8-11. For TMT time course experiments, peptides were filtered for quantitative values and TMT ratios to the 24h time point were median normalized. Sheet 12: Coverage estimates for the alleles in the two profiled cell lines (A549 and HEK293T) per HLA locus and per HLA).
Binding affinities of 30 synthetic HLA-I peptides and nine purified MHC complexes matching four HLA alleles expressed in A549 and HEK293T cells (A∗02:01, B∗07:02, B∗18:01 and B∗44:03) and five alleles for which we predicted presentation based on HLAthena (A∗01:01, A∗26:01, B∗15:01, B∗44:02 and B∗51:01). Affinity scores were computed for each allele based on the concentration of synthetic peptide required to inhibit the binding of a high affinity radiolabeled ligand by 50% (IC50). IC50 < 500 nM is considered positive binding (highlighted by bold font); +++ indicates IC50 < 0.5 nM (below detection levels); dash line indicates IC50 > 70000 nM.
(A) SARS-CoV-2 protein expression values as determined by whole proteome measurements in A549 and HEK293T cells and Ribo-seq translation measurements that were previously published for Vero cells (Finkel et al., 2020b). iBAQ - intensity-based absolute quantification. (B) HLA-I presentability estimates of SARS-CoV-2 ORFs based on HLAthena predictions.
Data associated with whole proteome analyses of A549 and HEK293T cells ± SARS-CoV-2 at 0, 3, 6, and 24hpi. The “gene_anno” sheet contains the protein groups used to plot the results from the two-sample t test results (Figures 4E and 4F). These annotations are a combination of manual curation, KEGG, and GO annotations of genes from proteasomal pathway, antigen processing, ubiquitination pathway, IFN signaling, and SARS-CoV-2 genes. The “all proteins” sheet contains t test results from uninfected versus 3,6, and 24 hpi. The “sig01_sortFC” tab contains all proteins that are statistically enriched or depleted in response to SARS-CoV-2 infection in HEK293T and A549 cells. The sheet “Input_WholeProteome_data” contains the whole proteome data from the Spectrum Mill database search from uninfected, 3,6, and 24hpi fractionated whole proteome samples. The sheet “PXD020019_data” represents the MaxQuant LFQ results reported by this study used for comparison in Figures 4F and S3E.
(A) A table of tested peptides in a multiplexed tetramer assay and the summary statistics of the T cell responses in seven convalescent COVID-19 patients and two unexposed subjects expressing HLA-A∗02:01. clonotype - the total number of unique reacting T cell clones in positive samples based on the CDR3 region of the TCR; nCells - the total number of reacting T cells in positive samples; subjects - the number of patients in which positive T cells were obtained. Peptides were ranked according to the number of detected reacting T cell clones. GILGFVFTL from Influenza virus and YLQPRTFLL, KLWAQCVQL, LLYDANYFL from SARS-CoV-2 served as positive controls. (B) Similar to (A) but for B∗07:02 tetramer library. Summary statistics of the T cell responses in five convalescent COVID-19 patients and three unexposed subjects expressing HLA-B∗07:02. RPPIFIRRL from EBV and SPRWYFYYL from SARS-CoV-2 served as positive controls.
(A) HLA-I presentation prediction of SARS-CoV-2 epitopes per cell line. (B) HLA-I alleles frequencies. (C) Estimated population coverage of LC-MS/MS-detected SARS-CoV-2 epitopes based on predictions across 92 HLA-I alleles.
HLA-I presented peptides identified in our study were compared against epitopes tested for CD8+ responses in Tarke et al. (2020) in patient samples with known HLA restriction. We looked for exact peptide matched (left table) and partial peptide matches (defined as our peptide with up to two N- or C-terminal residues missing being a subsequence within a Tarke et al., 2020 peptide; right table) and tabulated the number of positive and negative responses seen against patients with the most likely HLA allele in our cell lines (“Assigned allele”; “Same allele tested”; “Same allele positive”) as well as other HLA alleles (“Other allele positive”).
Accessions and references for SARS-CoV-2 variants B.1.1.7, P.1, and B.1.351 analyzed in Figure S6.
References
- Abelin J.G., Keskin D.B., Sarkizova S., Hartigan C.R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G.L., Eisenhaure T.M., et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J.D., Davis M.M. MHC-Peptide Tetramers to Visualize Antigen-Specific T Cells. Curr. Protoc. Immunol. 2016;115:17.3.1–17.3.44. doi: 10.1002/cpim.14. [DOI] [PubMed] [Google Scholar]
- Altmann D.M., Boyton R.J. SARS-CoV-2 T cell immunity: Specificity, function, durability, and role in protection. Sci. Immunol. 2020;5:eabd6160. doi: 10.1126/sciimmunol.abd6160. [DOI] [PubMed] [Google Scholar]
- Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Gfeller D. Unsupervised HLA Peptidome Deconvolution Improves Ligand Prediction Accuracy and Predicts Cooperative Effects in Peptide-HLA Interactions. J. Immunol. 2016;197:2492–2499. doi: 10.4049/jimmunol.1600808. [DOI] [PubMed] [Google Scholar]
- Callaway E. The race for coronavirus vaccines: a graphical guide. Nature. 2020;580:576–577. doi: 10.1038/d41586-020-01221-y. [DOI] [PubMed] [Google Scholar]
- Campbell K.M., Steiner G., Wells D.K., Ribas A., Kalbasi A. Prediction of SARS-CoV-2 epitopes across 9360 HLA class I alleles. bioRxiv. 2020 doi: 10.1101/2020.03.30.016931. [DOI] [Google Scholar]
- Chen D.-Y., Khan N., Close B.J., Goel R.K., Blum B., Tavares A.H., Kenney D., Conway H.L., Ewoldt J.K., Kapell S., et al. SARS-CoV-2 desensitizes host cells to interferon through inhibition of the JAK-STAT pathway. bioRxiv. 2020 doi: 10.1101/2020.10.27.358259. [DOI] [Google Scholar]
- Chen J., Brunner A.-D., Cogan J.Z., Nuñez J.K., Fields A.P., Adamson B., Itzhak D.N., Li J.Y., Mann M., Leonetti M.D., Weissman J.S. Pervasive functional translation of noncanonical human open reading frames. Science. 2020;367:1140–1146. doi: 10.1126/science.aay0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.E., Zhang X., Case J.B., Winkler E.S., Liu Y., VanBlargan L.A., Liu J., Errico J.M., Xie X., Suryadevara N., et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021;27:717–726. doi: 10.1038/s41591-021-01294-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Prusoff W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chong C., Marino F., Pak H., Racle J., Daniel R.T., Müller M., Gfeller D., Coukos G., Bassani-Sternberg M. High-throughput and Sensitive Immunopeptidomics Platform Reveals Profound Interferonγ-Mediated Remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Mol. Cell. Proteomics. 2018;17:533–548. doi: 10.1074/mcp.TIR117.000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft N.P., Smith S.A., Wong Y.C., Tan C.T., Dudek N.L., Flesch I.E.A., Lin L.C.W., Tscharke D.C., Purcell A.W. Kinetics of antigen expression and epitope presentation during virus infection. PLoS Pathog. 2013;9:e1003129. doi: 10.1371/journal.ppat.1003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan J.M., Mateus J., Kato Y., Hastie K.M., Yu E.D., Faliti C.E., Grifoni A., Ramirez S.I., Haupt S., Frazier A., et al. Immunological memory to SARS-CoV-2 assessed for up to eight months after infection. bioRxiv. 2020 doi: 10.1101/2020.11.15.383323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson D.V., Ozgur M., Sari K., Ghanayem M., Kostyu D.D. Ramifications of HLA class I polymorphism and population genetics for vaccine development. Genet. Epidemiol. 2001;20:87–106. doi: 10.1002/1098-2272(200101)20:1<87::AID-GEPI8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Demmers L.C., Heck A.J.R., Wu W. Pre-fractionation Extends but also Creates a Bias in the Detectable HLA Class Ι Ligandome. J. Proteome Res. 2019;18:1634–1643. doi: 10.1021/acs.jproteome.8b00821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez Andres A., Feng Y., Campos A.R., Yin J., Yang C.-C., James B., Murad R., Kim H., Deshpande A.J., Gordon D.E., et al. SARS-CoV-2 ORF9c Is a Membrane-Associated Protein that Suppresses Antiviral Responses in Cells. bioRxiv. 2020 doi: 10.1101/2020.08.18.256776. [DOI] [Google Scholar]
- Dutta N.K., Mazumdar K., Gordy J.T. The Nucleocapsid Protein of SARS-CoV-2: a Target for Vaccine Development. J. Virol. 2020;94:e00647. doi: 10.1128/JVI.00647-20. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhard F., Halenius A., Zimmermann C., L’Hernault A., Kowalewski D.J., Weekes M.P., Stevanovic S., Zimmer R., Dölken L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods. 2018;15:363–366. doi: 10.1038/nmeth.4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti A.P., Kula T., Wang Y., Nguyen D.M.V., Weinheimer A., Dunlap G.S., Xu Q., Nabilsi N., Perullo C.R., Cristofaro A.W., et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity. 2020;53:1095–1107.e3. doi: 10.1016/j.immuni.2020.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel Y., Schmiedel D., Tai-Schmiedel J., Nachshon A., Winkler R., Dobesova M., Schwartz M., Mandelboim O., Stern-Ginossar N. Comprehensive annotations of human herpesvirus 6A and 6B genomes reveal novel and conserved genomic features. eLife. 2020;9:e50960. doi: 10.7554/eLife.50960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel Y., Mizrahi O., Nachshon A. The coding capacity of SARS-CoV-2. Nature. 2020;589:125–130. doi: 10.1038/s41586-020-2739-1. [DOI] [PubMed] [Google Scholar]
- Finkel Y., Gluck A., Winkler R., Nachshon A., Mizrahi O. SARS-CoV-2 utilizes a multipronged strategy to suppress host protein synthesis. Cell. 2020;183:1325–1339.e21. [Google Scholar]
- Francis J.M., Leistritz-Edwards D., Dunn A., Tarr C., Lehman J., Dempsey C., Hamel A., Rayon V., Liu G., Wang Y., et al. Allelic variation in Class I HLA determines pre-existing memory responses to SARS-CoV-2 that shape the CD8+ T cell repertoire upon viral exposure. bioRxiv. 2021 doi: 10.1101/2021.04.29.441258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher K.M.E., Leick M.B., Larson R.C., Berger T.R., Katsis K., Yam J.Y., Brini G., Grauwet K., MGH COVID-19 Collection & Processing Team. Maus M.V. SARS -CoV-2 T-cell immunity to variants of concern following vaccination. bioRxiv. 2021 doi: 10.1101/2021.05.03.442455. [DOI] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Sidney J., Zhang Y., Scheuermann R.H., Peters B., Sette A. A Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe. 2020;27:671–680.e2. doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulukota K., Sidney J., Sette A., DeLisi C. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J. Mol. Biol. 1997;267:1258–1267. doi: 10.1006/jmbi.1997.0937. [DOI] [PubMed] [Google Scholar]
- Habel J.R., Nguyen T.H.O., van de Sandt C.E., Juno J.A., Chaurasia P., Wragg K., Koutsakos M., Hensen L., Jia X., Chua B., et al. Suboptimal SARS-CoV-2-specific CD8+ T cell response associated with the prominent HLA-A∗02:01 phenotype. Proc. Natl. Acad. Sci. USA. 2020;117:24384–24391. doi: 10.1073/pnas.2015486117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.H., Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- Hickman H.D., Mays J.W., Gibbs J., Kosik I., Magadán J.G., Takeda K., Das S., Reynoso G.V., Ngudiankama B.F., Wei J., et al. Influenza A Virus Negative Strand RNA Is Translated for CD8+ T Cell Immunosurveillance. J. Immunol. 2018;201:1222–1228. doi: 10.4049/jimmunol.1800586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hie B., Bryson B., Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 2019;37:685–691. doi: 10.1038/s41587-019-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Brar G.A., Stern-Ginossar N., Harris M.S., Talhouarne G.J.S., Jackson S.E., Wills M.R., Weissman J.S. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 2014;8:1365–1379. doi: 10.1016/j.celrep.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L.A., Anderson E.J., Rouphael N.G., Roberts P.C., Makhene M., Coler R.N., McCullough M.P., Chappell J.D., Denison M.R., Stevens L.J., et al. An mRNA vaccine against SARS-CoV-2—preliminary report. N. Engl. J. Med. 2020;383:1920–1931. doi: 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungreis I., Nelson C.W., Ardern Z., Finkel Y., Krogan N.J., Sato K., Ziebuhr J., Stern-Ginossar N., Pavesi A., Firth A.E., et al. Conflicting and ambiguous names of overlapping ORFs in the SARS-CoV-2 genome: A homology-based resolution. Virology. 2021;558:145–151. doi: 10.1016/j.virol.2021.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kared H., Redd A.D., Bloch E.M., Bonny T.S., Sumatoh H., Kairi F., Carbajo D., Abel B., Newell E.W., Bettinotti M.P., et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J. Clin. Invest. 2021;131:145476. doi: 10.1172/JCI145476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin D.B., Reinhold B.B., Zhang G.L., Ivanov A.R., Karger B.L., Reinherz E.L. Physical detection of influenza A epitopes identifies a stealth subset on human lung epithelium evading natural CD8 immunity. Proc. Natl. Acad. Sci. USA. 2015;112:2151–2156. doi: 10.1073/pnas.1423482112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketteler R. On programmed ribosomal frameshifting: the alternative proteomes. Front. Genet. 2012;3:242. doi: 10.3389/fgene.2012.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bert N., Tan A.T., Kunasegaran K., Tham C.Y.L., Hafezi M., Chia A., Chng M.H.Y., Lin M., Tan N., Linster M., et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020;584:457–462. doi: 10.1038/s41586-020-2550-z. [DOI] [PubMed] [Google Scholar]
- Ledford H. How ‘killer’ T cells could boost COVID immunity in face of new variants. Nature. 2021;590:374–375. doi: 10.1038/d41586-021-00367-7. [DOI] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunemann S., Schöbel A., Kah J., Fittje P., Hölzemer A., Langeneckert A.E., Hess L.U., Poch T., Martrus G., Garcia-Beltran W.F., et al. Interactions Between KIR3DS1 and HLA-F Activate Natural Killer Cells to Control HCV Replication in Cell Culture. Gastroenterology. 2018;155:1366–1371.e3. doi: 10.1053/j.gastro.2018.07.019. [DOI] [PubMed] [Google Scholar]
- Maness N.J., Walsh A.D., Piaskowski S.M., Furlott J., Kolar H.L., Bean A.T., Wilson N.A., Watkins D.I. CD8+ T cell recognition of cryptic epitopes is a ubiquitous feature of AIDS virus infection. J. Virol. 2010;84:11569–11574. doi: 10.1128/JVI.01419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurtrey C.P., Lelic A., Piazza P., Chakrabarti A.K., Yablonsky E.J., Wahl A., Bardet W., Eckerd A., Cook R.L., Hess R., et al. Epitope discovery in West Nile virus infection: Identification and immune recognition of viral epitopes. Proc. Natl. Acad. Sci. USA. 2008;105:2981–2986. doi: 10.1073/pnas.0711874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan M.J., Lyke K.E., Kitchin N., Absalon J., Gurtman A., Lockhart S., Neuzil K., Raabe V., Bailey R., Swanson K.A., et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature. 2020;586:589–593. doi: 10.1038/s41586-020-2639-4. [DOI] [PubMed] [Google Scholar]
- Neefjes J., Jongsma M.L.M., Paul P., Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- Nguyen A., David J.K., Maden S.K., Wood M.A., Weeder B.R., Nellore A., Thompson R.F. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. 2020;94:e00510. doi: 10.1128/JVI.00510-20. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouspenskaia T., Law T., Clauser K.R., Klaeger S., Sarkizova S., Aguet F., Li B., Christian E., Knisbacher B.A., Le P.M., et al. Thousands of novel unannotated proteins expand the MHC I immunopeptidome in cancer. bioRxiv. 2020 doi: 10.1101/2020.02.12.945840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M.D. Immune evasion via SARS-CoV-2 ORF8 protein? Nat. Rev. Immunol. 2020;20:408. doi: 10.1038/s41577-020-0360-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poran A., Harjanto D., Malloy M., Arieta C.M., Rothenberg D.A., Lenkala D., van Buuren M.M., Addona T.A., Rooney M.S., Srinivasan L., Gaynor R.B. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Med. 2020;12:70. doi: 10.1186/s13073-020-00767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., du Plessis L., Pybus O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redd A.D., Nardin A., Kared H., Bloch E.M., Pekosz A., Laeyendecker O., Abel B., Fehlings M., Quinn T.C., Tobian A.A. CD8+ T cell responses in COVID-19 convalescent individuals target conserved epitopes from multiple prominent SARS-CoV-2 circulating variants. medRxiv. 2021 doi: 10.1101/2021.02.11.21251585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucevic M., Kourjian G., Boucau J., Blatnik R., Garcia Bertran W., Berberich M.J., Walker B.D., Riemer A.B., Le Gall S. Analysis of Major Histocompatibility Complex-Bound HIV Peptides Identified from Various Cell Types Reveals Common Nested Peptides and Novel T Cell Responses. J. Virol. 2016;90:8605–8620. doi: 10.1128/JVI.00599-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz Cuevas M.V., Hardy M.-P., Hollý J., Bonneil É., Durette C., Courcelles M., Lanoix J., Côté C., Staudt L.M., Lemieux S., et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021;34:108815. doi: 10.1016/j.celrep.2021.108815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydyznski Moderbacher C., Ramirez S.I., Dan J.M., Grifoni A., Hastie K.M., Weiskopf D., Belanger S., Abbott R.K., Kim C., Choi J., et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell. 2020;183:996–1012.e19. doi: 10.1016/j.cell.2020.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini S.K., Hersby D.S., Tamhane T., Povlsen H.R., Amaya Hernandez S.P., Nielsen M., Gang A.O., Hadrup S.R. SARS-CoV-2 genome-wide mapping of CD8 T cell recognition reveals strong immunodominance and substantial CD8 T cell activation in COVID-19 patients. Sci. Immunol. 2020;6:eabf7550. doi: 10.1126/sciimmunol.abf7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L., et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellens I.M., Meiring H.D., Hoof I., Spijkers S.N., Poelen M.C.M., van Gaans-van den Brink J.A.M., Costa A.I., Vennema H., Keşmir C., van Baarle D., van Els C.A. Measles Virus Epitope Presentation by HLA: Novel Insights into Epitope Selection, Dominance, and Microvariation. Front. Immunol. 2015;6:546. doi: 10.3389/fimmu.2015.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N., Lareau C.A., Keshishian H., Ganskih S., Schneider C., Hennig T., Melanson R., Werner S., Wei Y., Zimmer M., et al. The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat Microbiol. 2020;6:339–353. doi: 10.1038/s41564-020-00846-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert K., Karousis E.D., Jomaa A., Scaiola A., Echeverria B., Gurzeler L.-A., Leibundgut M., Thiel V., Mühlemann O., Ban N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020;27:959–966. doi: 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Sekine T., Perez-Potti A., Rivera-Ballesteros O., Strålin K., Gorin J.-B., Olsson A., Llewellyn-Lacey S., Kamal H., Bogdanovic G., Muschiol S., et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. 2020;183:158–168.e14. doi: 10.1016/j.cell.2020.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomuradova A.S., Vagida M.S., Sheetikov S.A., Zornikova K.V., Kiryukhin D., Titov A., Peshkova I.O., Khmelevskaya A., Dianov D.V., Malasheva M., et al. SARS-CoV-2 Epitopes Are Recognized by a Public and Diverse Repertoire of Human T Cell Receptors. Immunity. 2020;53:1245–1257.e5. doi: 10.1016/j.immuni.2020.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidney J., Southwood S., Moore C., Oseroff C., Pinilla C., Grey H.M., Sette A. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr. Protoc. Immunol. 2013 doi: 10.1002/0471142735.im1803s100. Chapter 18, Unit 18.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solberg O.D., Mack S.J., Lancaster A.K., Single R.M., Tsai Y., Sanchez-Mazas A., Thomson G. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: a meta-analytic review of 497 population studies. Hum. Immunol. 2008;69:443–464. doi: 10.1016/j.humimm.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N., Hinnebusch A.G. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck S.R., Shastri N. Nowhere to hide: unconventional translation yields cryptic peptides for immune surveillance. Immunol. Rev. 2016;272:8–16. doi: 10.1111/imr.12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N., Weisburd B., Michalski A., Le V.T.K., Hein M.Y., Huang S.-X., Ma M., Shen B., Qian S.-B., Hengel H., et al. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukalov A., Girault V., Grass V., Bergant V., Karayel O., Urban C., Haas D.A., Huang Y., Oubraham L., Wang A., et al. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv. 2020 doi: 10.1101/2020.06.17.156455. [DOI] [PubMed] [Google Scholar]
- Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., et al. ISB-Swedish COVID19 Biobanking Unit Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/j.cell.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi A., Matsui M. Identification of HLA-A∗02:01-restricted candidate epitopes derived from the non-structural polyprotein 1a of SARS-CoV-2 that may be natural targets of CD8+ T cell recognition in vivo. J. Virol. 2020;95:e01837. doi: 10.1128/JVI.01837-20. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarke A., Sidney J., Kidd C.K., Dan J.M., Ramirez S.I., Yu E.D., Mateus J., da Silva Antunes R., Moore E., Rubiro P., et al. Comprehensive analysis of T cell immunodominance and immunoprevalence of SARS-CoV-2 epitopes in COVID-19 cases. bioRxiv. 2020 doi: 10.1101/2020.12.08.416750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarke A., Sidney J., Methot N., Zhang Y., Dan J.M., Goodwin B., Rubiro P., Sutherland A., da Silva Antunes R., Frazier A., et al. Negligible impact of SARS-CoV-2 variants on CD4+ and CD8+ T cell reactivity in COVID-19 exposed donors and vaccinees. bioRxiv. 2021 doi: 10.1101/2021.02.27.433180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternette N., Yang H., Partridge T., Llano A., Cedeño S., Fischer R., Charles P.D., Dudek N.L., Mothe B., Crespo M., et al. Defining the HLA class I-associated viral antigen repertoire from HIV-1-infected human cells. Eur. J. Immunol. 2016;46:60–69. doi: 10.1002/eji.201545890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A.K.A., Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A.J., Burdziak C., Moon K.R., Chaffer C.L., Pattabiraman D., et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 2018;174:716–729.e27. doi: 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf D., Schmitz K.S., Raadsen M.P., Grifoni A., Okba N.M.A., Endeman H., van den Akker J.P.C., Molenkamp R., Koopmans M.P.G., van Gorp E.C.M., et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 2020;5:eabd2071. doi: 10.1126/sciimmunol.abd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Guan J., Handel A., Tscharke D.C., Sidney J., Sette A., Wakim L.M., Sng X.Y.X., Thomas P.G., Croft N.P., et al. Quantification of epitope abundance reveals the effect of direct and cross-presentation on influenza CTL responses. Nat. Commun. 2019;10:2846. doi: 10.1038/s41467-019-10661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Werner A.P., Koch M., Choi A., Narayanan E., Stewart-Jones G.B.E., Colpitts T., Bennett H., Boyoglu-Barnum S., Shi W., et al. Serum Neutralizing Activity Elicited by mRNA-1273 Vaccine - Preliminary Report. N. Engl. J. Med. 2021;384:1468–1470. doi: 10.1056/NEJMc2102179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Gibbs J.S., Hickman H.D., Reynoso G.V., Ghosh A.K., Bennink J.R., Yewdell J.W. Defining Viral Defective Ribosomal Products: Standard and Alternative Translation Initiation Events Generate a Common Peptide from Influenza A Virus M2 and M1 mRNAs. J. Immunol. 2016;196:3608–3617. doi: 10.4049/jimmunol.1502303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhang J., Chen Y., Luo B., Yuan Y., Huang F., Yang T., Yu F., Liu J., Liu B., et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. bioRxiv. 2020 doi: 10.1101/2020.05.24.111823. [DOI] [Google Scholar]
- Zhu M.-S., Pan Y., Chen H.-Q., Shen Y., Wang X.-C., Sun Y.-J., Tao K.-H. Induction of SARS-nucleoprotein-specific immune response by use of DNA vaccine. Immunol. Lett. 2004;92:237–243. doi: 10.1016/j.imlet.2004.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sheet 1: Legend for Table, Sheet 2: HLA-I bound peptide sequences from SARS-CoV-2 proteins identified in both A549 and HEK293T cells. Cysteines were detected in their carbamidomethylated or cysteinylated modification state, methionines were oxidized. “x” denotes identification in individual experiments, “T” in TMT experiments, “-” indicates that the peptide was detected but did not pass FDR correction in that sample, confidence level in identification was evaluated based on Spectrum Mill Score, after manual inspection of MS/MS spectra and comparison to synthetic peptide spectra for available peptides (Y: validated by synthetic peptide, N/A: synthetic peptide not available). For binding prediction using HLAthena, residues were considered unmodified; columns show the most likely HLA allele a peptide is bound to in the respective cell line as well as the percentile binding rank. Sheet 3-11: Peptide identifications from HLA-I immunoprecipitation experiments in A549 and HEK293T cells ± SARS-CoV-2 after filtering for contaminants and length 8-11. For TMT time course experiments, peptides were filtered for quantitative values and TMT ratios to the 24h time point were median normalized. Sheet 12: Coverage estimates for the alleles in the two profiled cell lines (A549 and HEK293T) per HLA locus and per HLA).
Binding affinities of 30 synthetic HLA-I peptides and nine purified MHC complexes matching four HLA alleles expressed in A549 and HEK293T cells (A∗02:01, B∗07:02, B∗18:01 and B∗44:03) and five alleles for which we predicted presentation based on HLAthena (A∗01:01, A∗26:01, B∗15:01, B∗44:02 and B∗51:01). Affinity scores were computed for each allele based on the concentration of synthetic peptide required to inhibit the binding of a high affinity radiolabeled ligand by 50% (IC50). IC50 < 500 nM is considered positive binding (highlighted by bold font); +++ indicates IC50 < 0.5 nM (below detection levels); dash line indicates IC50 > 70000 nM.
(A) SARS-CoV-2 protein expression values as determined by whole proteome measurements in A549 and HEK293T cells and Ribo-seq translation measurements that were previously published for Vero cells (Finkel et al., 2020b). iBAQ - intensity-based absolute quantification. (B) HLA-I presentability estimates of SARS-CoV-2 ORFs based on HLAthena predictions.
Data associated with whole proteome analyses of A549 and HEK293T cells ± SARS-CoV-2 at 0, 3, 6, and 24hpi. The “gene_anno” sheet contains the protein groups used to plot the results from the two-sample t test results (Figures 4E and 4F). These annotations are a combination of manual curation, KEGG, and GO annotations of genes from proteasomal pathway, antigen processing, ubiquitination pathway, IFN signaling, and SARS-CoV-2 genes. The “all proteins” sheet contains t test results from uninfected versus 3,6, and 24 hpi. The “sig01_sortFC” tab contains all proteins that are statistically enriched or depleted in response to SARS-CoV-2 infection in HEK293T and A549 cells. The sheet “Input_WholeProteome_data” contains the whole proteome data from the Spectrum Mill database search from uninfected, 3,6, and 24hpi fractionated whole proteome samples. The sheet “PXD020019_data” represents the MaxQuant LFQ results reported by this study used for comparison in Figures 4F and S3E.
(A) A table of tested peptides in a multiplexed tetramer assay and the summary statistics of the T cell responses in seven convalescent COVID-19 patients and two unexposed subjects expressing HLA-A∗02:01. clonotype - the total number of unique reacting T cell clones in positive samples based on the CDR3 region of the TCR; nCells - the total number of reacting T cells in positive samples; subjects - the number of patients in which positive T cells were obtained. Peptides were ranked according to the number of detected reacting T cell clones. GILGFVFTL from Influenza virus and YLQPRTFLL, KLWAQCVQL, LLYDANYFL from SARS-CoV-2 served as positive controls. (B) Similar to (A) but for B∗07:02 tetramer library. Summary statistics of the T cell responses in five convalescent COVID-19 patients and three unexposed subjects expressing HLA-B∗07:02. RPPIFIRRL from EBV and SPRWYFYYL from SARS-CoV-2 served as positive controls.
(A) HLA-I presentation prediction of SARS-CoV-2 epitopes per cell line. (B) HLA-I alleles frequencies. (C) Estimated population coverage of LC-MS/MS-detected SARS-CoV-2 epitopes based on predictions across 92 HLA-I alleles.
HLA-I presented peptides identified in our study were compared against epitopes tested for CD8+ responses in Tarke et al. (2020) in patient samples with known HLA restriction. We looked for exact peptide matched (left table) and partial peptide matches (defined as our peptide with up to two N- or C-terminal residues missing being a subsequence within a Tarke et al., 2020 peptide; right table) and tabulated the number of positive and negative responses seen against patients with the most likely HLA allele in our cell lines (“Assigned allele”; “Same allele tested”; “Same allele positive”) as well as other HLA alleles (“Other allele positive”).
Accessions and references for SARS-CoV-2 variants B.1.1.7, P.1, and B.1.351 analyzed in Figure S6.
Data Availability Statement
The raw RNA sequencing data generated in this study have been submitted to the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE159191. The original mass spectra, peptide spectrum matches, and the protein sequence databases used for searches have been deposited in the public proteomics repository MassIVE (https://massive.ucsd.edu) and are accessible at ftp://MSV000087225@massive.ucsd.edu.