

Conspectus

Carbohydrates (glycans, saccharides, and sugars) are essential molecules in all domains of life. Research on glycoscience spans from chemistry to biomedicine, including material science and biotechnology. Access to pure and well-defined complex glycans using synthetic methods depends on the success of the employed glycosylation reaction. In most cases, the mechanism of the glycosylation reaction is believed to involve the oxocarbenium ion. Understanding the structure, conformation, reactivity, and interactions of this glycosyl cation is essential to predict the outcome of the reaction. In this Account, building on our contributions on this topic, we discuss the theoretical and experimental approaches that have been employed to decipher the key features of glycosyl cations, from their structures to their interactions and reactivity.
We also highlight that, from a chemical perspective, the glycosylation reaction can be described as a continuum, from unimolecular SN1 with naked oxocarbenium cations as intermediates to bimolecular SN2-type mechanisms, which involve the key role of counterions and donors. All these factors should be considered and are discussed herein. The importance of dissociative mechanisms (involving contact ion pairs, solvent-separated ion pairs, solvent-equilibrated ion pairs) with bimolecular features in most reactions is also highlighted.
The role of theoretical calculations to predict the conformation, dynamics, and reactivity of the oxocarbenium ion is also discussed, highlighting the advances in this field that now allow access to the conformational preferences of a variety of oxocarbenium ions and their reactivities under SN1-like conditions.
Specifically, the ground-breaking use of superacids to generate these cations is emphasized, since it has permitted characterization of the structure and conformation of a variety of glycosyl oxocarbenium ions in superacid solution by NMR spectroscopy.
We also pay special attention to the reactivity of these glycosyl ions, which depends on the conditions, including the counterions, the possible intra- or intermolecular participation of functional groups that may stabilize the cation and the chemical nature of the acceptor, either weak or strong nucleophile. We discuss recent investigations from different experimental perspectives, which identified the involved ionic intermediates, estimating their lifetimes and reactivities and studying their interactions with other molecules. In this context, we also emphasize the relationship between the chemical methods that can be employed to modulate the sensitivity of glycosyl cations and the way in which glycosyl modifying enzymes (glycosyl hydrolases and transferases) build and cleave glycosidic linkages in nature. This comparison provides inspiration on the use of molecules that regulate the stability and reactivity of glycosyl cations.
Key References
Martin A.; Arda A.; Désiré J.; Martin-Mingot A.; Probst N.; Sinaÿ P.; Jiménez-Barbero J.; Thibaudeau S.; Blériot Y.. Catching Elusive Glycosyl Cations in a Condensed Phase with HF/SbF5 Superacid. Nat. Chem. 2016, 8, 186–191.1The first experimental demonstration of the existence of the glycosyl oxocarbenium ion, the intermediate in glycosylation reactions, together with the determination of its 3D structure by NMR.
Hansen T.; Lebedel L.; Remmerswaal W. A.; van der Vorm S.; Wander D. P. A.; Somers M.; Overkleeft H. S.; Filippov D. V.; Désiré J.; Mingot A.; Bleriot Y.; van der Marel G. A.; Thibaudeau S.; Codée J. D. C.. Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Cent. Sci. 2019, 5, 781–788.24A combined theoretical and computational approach is described to quantitatively predict the stereoselectivity of SN1-type glycosylation reactions based on the full ensemble of geometries that can be adopted by any particular oxocarbenium ion.
Montalvillo-Jimenez L.; Santana A. G.; Corzana F.; Jimenez-Oses G.; Jimenez-Barbero J.; Gomez A. M.; Asensio J. L.. Impact of Aromatic Stacking on Glycoside Reactivity: Balancing CH/π and Cation/π Interactions for the Stabilization of Glycosyl Oxocarbenium Ions. J. Am. Chem. Soc. 2019, 141, 13372–13384.79Experimental demonstration that different aromatic moieties can modulate the rate and outcome of SN1-like glycosylations, through cation/π interactions.
Santana A. G.; Montalvillo-Jiménez L.; Diaz-Casado L.; Corzana F.; Merino P.; Cañada F. J.; Jiménez-Osés G.; Jiménez-Barbero J.; Gómez A. M.; Asensio J. L.. Dissecting the Essential Role of Anomeric β-Triflates in Glycosylation Reactions. J. Am. Chem. Soc. 2020, 142, 12501–12514.4A combined approach using chemical, NMR, kinetic, and theoretical methods demonstrated that the prevalence of the Curtin–Hammett fast-exchange assumption may be ruled out for most glycosylations.
Introduction
While observation of simple oxocarbenium ions that resonance-stabilize carbenium ions was reported as early as 1937 by Meerwein,5 the characterization of glycosyl oxocarbenium ions remained elusive for years. It is obvious that, for any typical monosaccharide, the chemical environment within the pyranose ring, with several nearby C–O bonds, strongly degrades the stability and lifetime of the oxocarbenium ions. These species are extremely reactive, and for decades, their intrinsic fleeting nature has precluded the definition of clear structure–reactivity relationships. Their superelectrophilic nature, which forces even relatively weakly nucleophilic counterions, such as trifluoromethanesulfonate, to form covalently bound intermediates rather than ion pairs, is responsible for this situation together with their extremely short lifetimes (picoseconds).6 Therefore, in 2011, Crich stated that “the field of glycosyl oxocarbenium ions was therefore at a similar level of development as that of carbenium ions in general in the 1960s”.7 As we describe herein, recent methodological advances have now positioned this domain almost in line with that of carbenium ions, converting these elusive ionic species into tangible molecules.
The Structure and Conformation of Glycosyl Oxocarbenium Ions: From Computational to Experimental Evidence
Computational Characterization
Chemical and physical sugar properties depend on their conformations. The substitution pattern of the pyranoses governs their conformational preferences. Therefore, the introduction of a positive charge associated with the O5–C1 double bond character has a dramatic impact on the pyranose.8
Glycosylations are experimentally carried out under kinetic control, with some exceptions.9 Therefore, the diastereoselectivity depends on the attack to the glycosyl oxocarbenium.10 For years, the fleeting nature of oxocarbenium ions required theoretical calculations to predict their geometries and their impact on glycosylation outcomes, especially for those following a SN1 mechanism. The relationship between short lifetime and reactivity may be explained by the fact that the lone pair of any nucleophile may assist the departure of the leaving group. The nucleophile approaches the donor [initially separated by 3 Å with an O5–C1–Nu angle of 90° (α) or 180° (β)], interacting with its LUMO orbital.11
Theoretical calculations predicted that the 4H3 conformer of the d-Glcp oxocarbenium ion has a C–O bond distance of 1.25 Å and a torsion angle τ5 (C5–O5–C1–C2) close to zero (τ5 < 20°), and its positive charge is located on the anomeric carbon.12 For monosaccharides, there are two or more oxocarbenium conformers computationally found for Galp (4H3 and 5S1),13 Glcp (4H3 and 5S1), and Manp (4H3, 3E, B2,5). Ab initio molecular dynamics suggest that these conformers are connected through a transition state (TS), 1S3 for Glcp.14 These geometries can be attacked by a nucleophile through the diastereotopic α/β faces to yield the product. Therefore, at least four possibilities are feasible (Figure 1). Pioneering works by Whitfield developed this field years ago. The relative stability of each conformation depends on the degree of flexibility15 and the presence of ion pairs.16 There are low energy barriers (ΔG°⧧ ca. 10–15 kcal/mol) for interconversion between conformers.17 Given the improved accuracy of current computational methodologies, we provide here the energies for the Whitfield geometries reoptimized at PBE0-D3/def2-TZVP level, using CH2Cl2 (PCM) as solvent and employing per-O-methyl derivatives as models to avoid the formation of hydrogen bonds.
Figure 1.

(a) Idealized energy profile for the SN1 mechanism without considering proton transfer reactions. The two-conformer hypothesis of per-O-methyl glycosyl cations is shown. The intermediate B0 geometry is most similar to that of the starting glycoside. The 2D projection shows the interconversion of selected C (chair), B (boat), H (half-chair), and S (skew-boat) conformations. (b) Main conformations and Gibbs free energies of mannopyranosyl cations (4H3, 3E, and B2,5). (c) Single point calculations upon rotation around the H2–C2–O2–CAc torsion. (d) Optimized 4H3 and 5S1 conformations and ΔG differences (kcal/mol) for 2-O-Me and 2-O-isopropyl Glcp. Calculations are carried out at the PBE0-D3/def2-TZVP level (CH2Cl2). Configurations are defined by a three-letter code (Gal, Glc, or Man). Ring size is denoted by p (pyranose).
The preferred geometry depends on the C-2 substituent, with additional stabilizations when it displays a pseudoequatorial orientation. Thus, the computations predict that the 3E conformation is more stable (ΔG = −2.0 kcal/mol) than the 4H3 for Manp oxocarbenium. However, the B2,5 conformer (Figure 1b) is more stable (ΔG = −2.6 kcal/mol), in agreement with the findings in glycosyl transferases (GTs) and glycosyl hydrolases (GHs, see below). Moreover, 4H3 is 15.2 kcal/mol more stable than 5S1 in the Glcp oxocarbenium.
Another situation arises when there is neighboring-group participation. For instance, a 2-O-acetyl moiety allows the formation of dioxolenium ions (Figure 1c). Indeed, these species are more stable than the oxocarbenium ones, providing key contributions to the outcome.18,19 Electron-withdrawing (deactivating) or electron-donating (activating) substituents at C-2 also affect the reactivity of glycosyl donors. The calculations suggest that activating substituents produce a syn-preference, ruled by the interaction of O2 lone pair and an empty p-orbital at the anomeric position. Deactivating substituents increase the anti-preference.20 The introduction of chiral protecting groups induces different populations of the glycosyl cation conformations, favoring the nucleophilic attack at one face.21 In addition, the starting anomeric orientation may also modulate the conformational preferences.22
The two possible TSs (α-TS, β-TS) with oxocarbenium-like character23 lead to different products. Knowledge of the energy differences and geometries of the intermediates and/or transitions states is essential to predict the final outcome of the reaction. The differences between transition state and intermediate are rather subtle, and both terms mainly arise from their lifetime (shorter for TSs) and their energy values (larger for TSs) along a reaction coordinate (see Figure 1a in which B0 and B1 are intermediates). Indeed, Codée and co-workers have described the full ensemble of conformations that these species can adopt.24 They employed conformational energy landscapes (CEL) to quantify the dynamics of different oxocarbenium ions and were able to connect the predicted conformational ensembles to the observed stereoselectivities in SN1-type reactions with triethylsilane or allyl-trimethyl silane.
Despite the relevance of the glycosyl cation properties for pure SN1 reactions, it should be noted that, in most cases, the departure of the activated leaving group and the nucleophilic attack of the acceptor alcohol to the anomeric center takes place in a concerted, asynchronous manner, determining an oxocarbenium-like transition state. Indeed, anomeric substitutions are known to proceed through a continuum of mechanisms between formal SN2 and SN1 reactions. As a final consideration, these processes can also be directed by preassociation of the acceptor with the leaving group or even more remote pyranose sites and SNi-like mechanisms have been described for some glycosylations.25−27
Experimental Characterization: The Cation Pool Method
Initial structural information on glycosylation intermediates was provided by electrochemical activation of glycosyl donors.28 The pioneer “cation pool” method, which generates carboxonium ions through the anodic oxidation of α-silyl- or α-arylthio-substituted ethers at low temperature in the absence of a nucleophile allowed accumulation and characterization of cyclic alkoxycarbenium ions by NMR (Figure 2a). The chemical shifts exhibited by the methine proton and carbon supported its cationic character. However, the switch to glycosyl donors showed the limits of this approach. While accumulation of the glycosyl cations was evidenced by the isolation of methyl glycosides in the presence of methanol (Figure 2b), their spectroscopic observation was not possible even under flow conditions. Nevertheless, the stability of glycosyl cations was mapped as a function of temperature (T) and reactor residence time (tR). Lifetimes are on the order of a second at −78 °C in CH2Cl2 for the perbenzylated Glc cation.29
Figure 2.

(a) Generation and accumulation of cyclic oxocarbenium ions as a cation pool. (b) Generation of the glycosyl cation or its equivalent by the cation pool method.
Combining Superacids and Low Temperature NMR
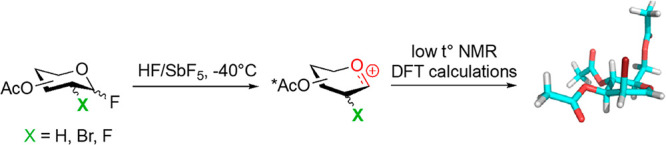
Superacids, defined as acids stronger than 100% anhydrous sulfuric acid, constitute a unique chemical tool popularized by Nobel laureate George Olah. His strategy of suppressing bases and nucleophiles, to avoid either cation deprotonation or quenching, was key for the successful generation and NMR-based observation of long-lived carbenium and oxocarbenium ions, then considered as elusive species.30 Extension of this methodology to carbohydrates was first reported by Paulsen, who observed isomerization reactions in HF.31 More recently, Akien and Subramanian presented NMR data for fructopyranosyl, per-O-methyl-fructofuranosyl, and 2-deoxy-glucopyranosyl oxocarbenium ions,32 after dissolving the corresponding methyl glycosides in fluorosulfonic acid. However, details of these experiments have not yet appeared in the scientific literature. Concomitantly, Blériot, Thibaudeau, and Jiménez-Barbero successfully reported 1H/13C NMR spectra for peracetylated Glc oxocarbenium ions generated from dissolving the corresponding peracetylated Glc fluorides in superacidic HF/SbF5 medium (Figure 3).1 Bromination or deoxygenation at C2 position was needed to generate glycosyl cations with extended lifetimes, up to 4 h at −40 °C, suitable for extensive low temperature NMR analysis, supported by DFT calculations. Protonation of the acetoxy groups and through space stabilization of the anomeric carbocation by the bromine atom, which was forced to adopt a pseudoaxial orientation, was demonstrated. This methodology was successfully extended to d-Gal, d-Man,1,24l-Fuc, and l-Rha33 and Ferrier cation34 (Table 1). Introduction of fluorine at the C2 position gave strikingly different results, as the corresponding glycosyl cations could not be detected by NMR. It was hypothesized that a fast equilibrium between a contact ion pair, observed by NMR, and a transient solvent separated ion pair, confirmed by its trapping with deuterium, was operative suggesting that, in the absence of nucleophiles in the reaction medium, the superelectrophilic 2-fluoroglycosyl cation is stabilized by noncoordinating SbnF5n+1– species.33 In any case, given the intrinsic nature of the employed chemical conditions, which generate protonated oxygens all around the ring, the use of the described conformations to predict the outcome of general glycosylation reactions should be taken with caution.
Figure 3.

1H NMR (a) and 13C NMR (b) spectra of the 2-deoxy-Glc cation in HF/SbF5. Data from ref (1)
Table 1. Set of Glycosyl Cations Observed in HF/SbF5 and Their Conformational Characterization by Low Temperature NMR Data Assisted by DFT Calculations.
| glycosyl
cation |
|||||||
|---|---|---|---|---|---|---|---|
| 2-deoxy-d-Glc | 2-deoxy-d-Gal | 2-bromo-d-Glc | 2-bromo-d-Gal | 2-bromo-d-Man | 2-deoxy-l-Fuc | 2-deoxy-l-Rha | |
| δ H-1 (ppm) | 8.89 | 9.18 | 8.36 | 8.63 | 8.50 | 8.74 | 8.84 |
| δ C-1 (ppm) | 227.0 | 227.9 | 197.4 | a | 195.6 | 224.2 | 224.0 |
| conformer | 4E | 4E | 4H5 | 4H5 | 4H3 | 3H4 | b |
Not observed.
Not determined.
Combining Mass Spectrometry and IR Ion Spectroscopy
Mass spectrometry (MS) has also been used to study glycosyl cations. Collision-induced fragmentation–electrospray ionization (CID-ESI) allowed examination of the influence of the stereochemistry and nature of the protecting groups on their stability.35 Tandem MS/MS was used to generate glycosyl cations and study their gas-phase fragmentation but did not provide structural information at the atomic level. The combination of MS with infrared (IR) ion spectroscopy has emerged as a new tool to determine the gas-phase structures of glycosyl cations.3,35 Electrospray ionization of a glycosyl donor produced a parent [M + NH4]+ ion that was fragmented using CID. The fragment ion corresponding to the glycosyl cation was isolated in a quadrupole ion trap and characterized by IR ion spectroscopy using a laser operating in the 700–1850 cm–1 range. The IR region above 1450 cm–1, where C=O stretch vibrations of the acetyl group and oxocarbenium ion are found, provides characteristic bands for unambiguous assignment of the cation. In oxocarbenium structures, strong absorptions above 1600 cm–1 indicate a free or weakly interacting carbonyl group, while the C1-bridged acetyl group in dioxolenium ions yields absorption bands below 1600 cm–1. Thus, the oxocarbenium ions are assigned through their characteristic C1=O5+ stretch (∼1600 cm–1) and preservation of the acetyl C=O stretch near 1800 cm–1, while formation of a dioxolenium leads to the absence of acetyl C=O and C1=O5+ stretches and appearance of a dioxolenium O–C=O+ stretch (∼1550 cm–1) and bending mode (∼1500 cm–1) (Figure 4a). To support the IR data and obtain deeper structural information on the formed ions, IR ion spectroscopy was combined with ab initio and DFT calculations. Experimental IR spectra were superimposed on the DFT-predicted IR spectra for the lowest-energy conformers and demonstrated good overlap.3,36 This led to the characterization of the mannosyl oxocarbenium ion derived from a permethylated donor adopting the 3E conformation (Figure 4b). This work also highlighted the possibility to characterize a mixture of isomers using IR ion spectroscopy thereby providing crucial insights into the dynamics of glycosyl cations in the gas phase. In parallel, cryogenic ion infrared (IR) spectroscopy was also used, exploiting helium nanodroplets as a useful cryogenic matrix that resembles the environment of low dielectric constant solvents commonly used during glycosylations.33,36
Figure 4.
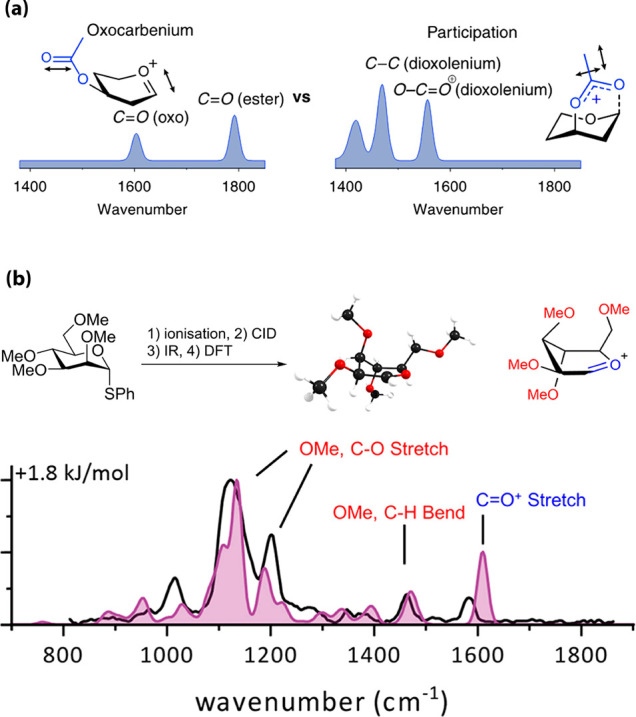
(a) Infrared ion spectroscopy of glycosyl cations: oxocarbenium and dioxolenium ions give different diagnostic peaks (blue). (b) Comparison of the calculated spectrum (filled) for 3E Man oxocarbenium conformer with the measured IR ion spectrum (black lines). Reproduced with permission from ref (3). Copyright 2018 American Chemical Society.
This MS/IR approach also deciphered the impact of leaving group anomericity on the glycosyl cation structure3,37 and allowed the study of neighboring and remote participation,38 which plays a decisive role in shaping the stereochemical outcome in glycosylations.39 Regarding the Ferrier cation, this approach led to the identification of a dioxalenium ion,40 distinct from the glycosyl cation obtained in superacid. As described for the superacid approach,1 the employed conditions are far from those employed in saccharide synthesis as no nucleophiles or nucleophilic counterions, such as triflate, are present under these conditions, which requires one to be very cautious when using these conformations to predict the outcome of glycosylations.
Reactivity from Naked Glycosyl Oxocarbenium Ions to Bimolecular Processes: Glycosyl Triflates as Reservoirs of Glycosyl Cations
Despite the indubitable relevance of glycosyl cations, pure SN1 processes with naked oxocarbenium ions only represent an extreme case among all possible pathways involving dissociative TSs.2,41−46 Upon activation, glycosyl donors generate a plethora of highly reactive species in chemical exchange, many of which have been characterized by NMR.42 The subsequent attack of the acceptor alcohol to the anomeric center of these intermediates proceeds through a continuum of substitution mechanisms spanning the gap between formal SN2 and SN1 processes (Scheme 1) to furnish the glycosylation products (SNi mechanisms involving a preassociation of the alcohol to the donor species prior to this step have also been reported and might be operative in some cases).25−27
Scheme 1. Schematic Representation of the SN2/SN1 Mechanistic Continuum in Anomeric Substitutions.

Glycosylations promoted by triflate agents provide a paradigmatic example of this situation.47−54 Pioneering work by Crich showed that under the low dielectric conditions employed in glycosylations, weakly nucleophilic anions, such as triflate, react with the activated donor to yield highly reactive intermediates.43 In order for this transformation to occur, the pKa of the conjugate acids of leaving groups should be close to that of triflic acid. The resulting glycosyl triflates exist as a mixture of α- and β-anomers, presumably in exchange with more reactive glycosyl oxocarbenium-like species, in the form of either close or solvent-separated ion pairs (CIPs or SSIPs). It should be noted that the ionization of both species might be assisted by the ring oxygen, which for β-anomer would require a conformational adjustment, as supported by the antiperiplanar lone pair hypothesis (APLPH theory).8 In any case, α- and β-glycosylations can proceed through alternative reaction pathways (Figure 5), whose mechanistic relevance depends on the experimental conditions and the donor/acceptor structural and electronic properties. Importantly, most of the species involved are barely populated in solution and therefore virtually undetectable under conventional conditions. This applies to glycosyl cations and, in most cases, also to β-triflates, which are severely destabilized by the anomeric effect. Nevertheless, since they still play a key role in glycosylations, Curtin–Hammett schemes, which imply a rapid interconversion among species in the reaction time scale, have been traditionally invoked for explanatory purposes.48−51
Figure 5.

(a) Alternative SN2 and SN1 reaction pathways leading to formation of α- and β-glycosides from glycosyl triflate intermediates. CIP, SSIP, and SEIP stand for contact ion pair, solvent-separated ion pair, and solvent-equilibrated ion pair, respectively. (b) Calculated free energy differences between α- and β-glycosyl triflates for d-Man, d-Glc and d-All in the gas phase. (c) NMR detection of β-glycosyl triflate signals (red) in reaction mixtures from Glc (left) and All (right) donors. Estimated equilibrium constants for the anomerization process are given in green.
The first evidence for the existence of alternative reaction pathways involving α- and β-glycosyl triflates were provided by Crich using kinetic isotopic measurements.48,49 The reaction of α- and β-Glc triflates with isopropanol proceeds through a loosely associative TS to form the corresponding β- and α-products, respectively. Similarly, α-Man triflates participate in SN2-like substitutions to yield the β product. In contrast, the α-product results from a highly dissociative process, which probably involves the participation of a CIP, transiently formed from the highly unstable β-Man triflate. These findings were supported by kinetic experiments with customized cation-clock reactions.50,51 Accordingly, the triflate anomerization equilibrium is central to the glycosylation mechanism and, together with the individual anomeric substitution reactions, eventually determines the stereochemical outcome of the reaction, which is opposite for Glc and Man. In agreement with this view, the β-selectivity displayed by Man52 would reflect the reduced significance of those pathways involving the corresponding β-triflate, greatly destabilized by the axial substituent at C2. In contrast, these paths must dominate the Glc case, explaining its increased tendency to provide α-products.
A simple strategy to interrogate the reactivity of the most unstable species within activated reaction mixtures consists of attenuating the nucleophilic character of the acceptor, pushing the reaction mechanism toward the SN1 limit. This invariably translates into an increased α-selectivity as demonstrated by Codée and colleagues.53,54 For Glc donors, glycosylation with poor nucleophiles, such as trifluoroethanol, proceeds with total stereoselectivity to yield a unique α-product. This behavior reflects the key role played by β-glycosyl triflates or glycosyl oxocarbenium-like species under these circumstances.
Despite the significant knowledge accumulated on these processes, key aspects of the mechanism leading to the formation of α-products from dominant α-glycosyl triflates remained undetermined until recently. Indeed, the detection of the minor, yet relevant, β-glycosyl triflate intermediates in activated reaction mixtures had proven challenging, with very few examples identified by NMR.55,56 Also, the glycosyl-triflate anomerization exchange rate was basically unknown. A recent NMR-based study addressed these key questions.4
Thus, by using a 13C-labeled Glc donor, the previously undetected β-Glc triflates were identified in the reaction mixtures. Interestingly, the anomerization equilibrium was extremely sensitive to the orientation of the substituent at C3, whose inversion from equatorial to axial, from Glc to All, led to a large fraction of β-triflate (Figure 4b,c). Both the anomerization equilibrium and exchange rate constants were determined under diverse experimental conditions. Importantly, the exchange kinetics increased linearly with the concentration of added triflate, showing a slope fully consistent with a bimolecular anomerization mechanism.
The time evolution of the Glc and All triflate mixtures was also analyzed at increasing concentration of trifluoroethanol, allowing the estimation of the kinetic constants governing the triflate anomerization and alcohol substitution steps. The obtained results ruled out the prevalence of Curtin–Hammett conditions, showing that, under certain circumstances, the α → β triflate exchange becomes the rate-limiting step for α-glycosylation. Finally, kinetic isotopic measurements, assisted by theoretical analyses, indicated that the α-product forms through a dissociative process involving the participation of a β-CIP transiently formed upon the β-glycosyl triflate. According to this, the latter intermediate acts as a reservoir of extremely reactive oxocarbenium-like species, whose lifetime is too short to permit diffusional separation of the triflate anion, and reacts with trifluoroethanol in a stereoselective manner to yield the α-glycoside. These results confirmed the key role played by β-glycosyl triflates in glycosylations involving poorly nucleophilic acceptors, in agreement with Crich and Codée.41,53−55
While glycosyl-sulfoxide donors are usually employed under preactivation conditions, an alternative single-step protocol, in which activation is performed in the presence of the acceptor alcohol is also feasible. Remarkably, the NMR study of these processes employing 13C-labeled donors revealed that even under these circumstances, glycosyl triflate intermediates play a key role. Of note, a competing reaction, which involves the formation of cationic donor/acceptor sulfonium adducts, and consequently the nonproductive consumption of the acceptor, was identified as the main yield-limiting problem.57
Interactions of Glycosyl Cations: From Enzymes to Model Systems
Glycosyl Cations As Central Entities in Glycosylation Biomachinery
Carbohydrate-active enzymes (CAZymes) sharply fine-tune the different architectures of complex glycans in nature.58 GTs and GHs synthesize and cleave glycosidic bonds with exquisite specificity. For GTs, the selective assembly of the glycosyl-bond donor and acceptor proceeds through glycosyl oxocarbenium species. The consideration of these ions as intermediates or transition states remains a matter of discussion, related to the way these enzymes achieve catalysis. Most inverting GTs (and GHs) employ a SN2 mechanism, and therefore, the glycosyl oxocarbenium ion might merely be considered as a transition state. In contrast, glycosyl oxocarbenium-like intermediates might be plausible for those enzymes that operate through SN1 and SNi (retaining) mechanisms (Scheme 2).
Scheme 2. Schematic Representation of the Inverting and Retaining Mechanisms for GHs and GTs.

From top to bottom: particular case of inverting GT through the SN2 mechanism; typical double displacement mechanism for GHs showing substrate distortion; inverting SN1 mechanism through single displacement; SNi-like retaining mechanism involving a front-side attack of the nucleophile.
Besides suitable residues acting as proton shuttles (often overlooked from a chemical perspective), any efficient catalytic site of these CAZymes requires two main features: (a) appropriate shape; (b) increasing the lifetime of the oxocarbenium-like intermediates. The first feature is covered since enzymes are highly flexible.59 Substrate flexibility is also important, and each pyranose ring displays a specific conformational itinerary that allows it to provide the best structural and electronic complementarity in the CAZyme active sites. Itineraries in GHs are deduced through free energy landscapes (FELs) by QM/MM metadynamics.60,61 Despite the existing 38 canonical conformations of pyranoses, only a few are found in these itineraries, depending on the particular enzyme. For instance, retaining α-mannosidases follow a oS2 → [B2,5]⧧ → 1S5 itinerary, whereas retaining β-mannosidases engage a 1S5 → [B2,5]⧧ → oS2 path.62 Alternatively, retaining β-glucosidases operate through the 1S3 → [4H3]⧧ → 1C4 itinerary, with the opposite for α-glucosidases.63
Upon binding, the substrate should be preorganized toward a distorted conformation (ES complex) closely related to the oxocarbenium TS. This preorganization also exploits a key point in carbohydrates, the hydroxymethyl rotamers, for providing through-space electrostatic transition state stabilizations. Thus, glucosidases and β-mannosidases bind their ligands as gg rotamers (Figure 6), whereas α-galactosidases and N-acetyl galactosidases proceed through gt conformations.64 Then, the bound molecule evolves toward the glycosyl oxocarbenium. The structure of this cation implies a limited conformational space of oxocarbenium-like transition states. In fact, they are restricted to B2,5 (or enantiomeric 2,5B), 4H3 (3H4), 3E (E3), and 4E (E4). In general, they are conserved within a specific family (Table 2).
Figure 6.

Side chain conformational preferences: (a) (PDB ID 6J34) with α-Glcp in gg conformation, (b) α-Manp in gg (PDB ID 5A7V), and (c) β-Galp in gt (PDB ID 1T0O). In all cases, there is a H-bond between O6 and an Asp residue. Conformational distributions for (d) α- and β-glucosidases, (e) α- and β-mannosidases, and (f) α- and β-galactosidases. Each population in the bound state (gg, gt, or tg compared to those in free solution (crossed bars). Data from ref (64). Relative energies for torsion angles calculated for the (g) Glcp oxocarbenium ion, (h) mannosyl oxocarbenium, and (i) galactosyl oxocarbenium ions. Scans were computed at the PBE0-D3/def2-SVP level in gas phase.
Table 2. Some Examples of Catalytic Itineraries of GH and G Families.
| enzyme | family | substrate | oxocarbenium | product | ref |
|---|---|---|---|---|---|
| α-mannosidases | inverting GH125 | oS2 | B2,5 | 1S5 | (65) |
| β-mannosidases | retaining GHs | 1S5 | B2,5 | oS2 | (66) |
| 1,3-1,4-β-glucanase | retaining GH16 | 1,4B/1S3 | 4E/4H3 | 4C1 | (67) |
| β-galactocerebroside | retaining GH59 | 1S3/4C1 | 4H3 | 4C1 | (68) |
| MGAT5 | inverting GT | 4C1 | 4H3 | 1,4B | (69) |
| LgtC | retaining GT | 4C1 | 4E/4H5 | 4C1 | (70) |
Regarding the second feature, noncovalent interactions stabilize glycosyl oxocarbenium species. GTs have a particular functional profile71 and display multiple noncovalent interactions with the ligands in their binding sites. For instance, a common feature of SNi GTs is a hydrogen bond between the acceptor and the leaving group.72 One crucial point is how the positive charge of glycosyl cations is stabilized by these enzymes, which often rely on intermolecular polar contacts, forming ion-pairs, typically involving carboxylate side chains or even phosphate modifications.73 It has also been shown that GHs may stabilize transition states even from remote positions to the anomeric center.74
The Influence of Aromatic Interactions on the Stability of Glycosyl Cations. From Enzymes to Model Systems
As described above, chemical modifications of the glycosyl donors that facilitate the development of positive charge at the anomeric center provide enhanced reactivities.43
Interestingly, aromatic platforms also assist cleavage and formation of glycosidic linkages by establishing favorable interactions with the transition states. Pioneering ideas on a feasible role for π-systems in the context of glycosylation reactions were first put forward by Krepinsky and co-workers.75 Later on, a data-mining comparison of protein sequences corresponding to glycosyl hydrolases allowed the identification of a conserved phenylalanine presumably involved in catalysis. Such an aromatic residue was described as a “mechanistically relevant transition-state stabilization factor” (Figure 7a).76 Similarly, it has been hypothesized that ADP-ribosyl cyclase uses a tryptophan ring to assist the process through cation/π interactions in the transition state.77 A novel mechanism for a “SNi synthase”, engineered from a retaining “double-SN2” hydrolase, has also been described, involving a tyrosine to stabilize the oxocarbenium transition state generated during the front-face attack of the glycosidic acceptor (Figure 7b).78
Figure 7.

(a) Aromatic interactions in GH complexes with a functional role in transition-state stabilization. (b) Aromatic assistance promotes catalysis in an engineered SNi-synthase. (c) Model I, evolution of the reference (blue, left) and model compounds (red, right) upon treatment with TfOH and methanol in CDCl3 at 25 °C. Reaction profiles for the consumption of both derivatives (triangles) and product formation (circles). (d) Model II, evolution of reference (blue, top) and model compounds (red, bottom) upon treatment with Tf2O and 8 equiv of TFE in CDCl3 at −65 °C. Reaction curves for formation of β (top) and α (bottom) products.
The first systematic study on the role of aromatic residues in stabilizing glycosyl oxocarbenium-like transition states79 used designed model systems, which featured a glycosyl donor or a labile glycoside involved in CH/π stacking with one or two aromatic platforms. In a second step, the models, classified as type I and II (Figures 7c,d), were assayed in methanolysis and glycosylation reactions, respectively. The results showed a complex scenario in which aromatic platforms can exert opposite effects on the reaction rate, yield, and stereoselectivity, depending on the specific mechanism, the geometry and strength of the CH/π interactions, and the conformational flexibility of the reacting glycoside (Figures 7c,d). Thus, the formation of stable CH/π complexes with a defined geometry, in conjunction with the absence of conformational constraints in the reactive unit (type I, Figure 7c) led, in all cases, to a significant inhibition of reactivity in methanolysis reactions, accompanied by an increase in the stereoselectivity (Figure 7c). In fact, there is a balance between two opposing contributions of the carbohydrate/aromatic stacking in the activation energy: electrostatic and van der Waals. The degradation of the latter, promoted by the conformational distortions required by the glycosyl cation, is not compensated by strengthening the former. In agreement with this, the formation of dynamic and adaptable complexes, along with the presence of conformational restrictions in the reactive pyranose, which limits conformational distortions in the transition state, promoted a steady increase in the glycosylation rates, especially for the more dissociative processes involving 2-deoxy donors (type II, Figure 7d). Interestingly, the rate augmentations due to cation/π interactions were not accompanied by improvements in stereoselectivity, which remained the same or slightly decreased: the oxocarbenium/aromatic interactions are highly dynamic, signaling a mismatched shape complementarity. Fittingly, aromatic residues present in the active sites of glycosidases tend to participate in unusual parallel-shifted or lateral stacking geometries with the substrate (Figure 7a), a feature that likely reflects the need to maintain the sugar ring relatively unconstrained by the aromatic residue, thus allowing the penalty-free pyranose distortions required for the reaction to proceed.
Summary and Perspectives
Recent investigations on the structure, conformation, reactivity and interactions of glycosyl oxocarbenium ions have been reported in the last years. Given the current importance of carbohydrates and their interactions in different scientific domains and the urgent necessity of accessing well-defined complex glycan structures for these studies, further advances in this field are expected. The combination of advanced theoretical protocols with state-of-the-art structural and synthetic chemistry methods will expand our knowledge of these key species, allowing us in the near future to predict the outcome of most glycosylation reactions, independently of the involved mechanism, from SN1 to SN2-like processes, always considering the reaction kinetics, the counterions, the possible intra- or intermolecular participation of functional groups, and the donors. Long live these short-lived intermediates!
Biographies
Antonio Franconetti (Sevilla, Spain) received his Ph.D. in Chemistry (2016). After postdoctoral appointments at Barcelona and Balearic Islands, in 2019 he moved to CIC bioGUNE (Bilbao) as Juan de la Cierva postdoctoral researcher. His research interests are focused on supramolecular chemistry, carbohydrates, and modeling.
Ana Ardá (La Coruña, Spain) received her Ph.D. in Chemistry (2006). After a postdoctoral appointment at Utrecht, she joined the Center for Biological Research (CSIC) in Madrid in 2008. In 2014, she moved to CIC bioGUNE (Bilbao) where she is Associated Principal Investigator as Ikerbasque Research Associate. Her research interests include carbohydrate chemistry and glycan recognition by protein receptors.
Juan Luis Asensio (Barcelona) received his Ph.D. in Chemistry (1995) at Madrid. After a postdoctoral stay at the National Institute for Medical Research at London, he moved back to Madrid (IQOG-CSIC) in 1998, becoming a tenured scientist in 2000. He was promoted to senior research scientist in 2008 and has focused his research on glycochemistry and molecular recognition.
Yves Blériot (Niort, France) completed his Ph.D. in Chemistry at Nantes (1994). After a postdoctoral stay at Oxford, he was appointed Associate Professor in 1997 at Ecole Normale Supérieure (Paris). In 2009, he moved to Poitiers University (France) as Full Professor. His research activity deals with glycosciences, from the design of therapeutically relevant carbohydrate derivatives to the study of chemical and enzymatic glycosyl transfer.
Sébastien Thibaudeau (France) received his Ph.D. in Chemistry (2002) at Poitiers. After a Marie-Curie postdoctoral appointment at Oxford, he joined the Department of Chemistry at the University of Poitiers in 2004, where he is currently Full Professor. His research interests include organic synthesis, organofluorine chemistry, glycosciences, superacids, and carbocation chemistry.
Jesús Jiménez-Barbero (Madrid) received his Ph.D. in Chemistry (1987) at Madrid. After postdoctoral stays at Zurich, Mill Hill, and Pittsburgh, he returned to Madrid (CSIC). In 2002, he was promoted to CSIC Research Professor and was appointed Ikerbasque Research Professor and Scientific Director of CIC bioGUNE (Bilbao) in 2014. His scientific interest is focused on molecular recognition and chemical glycobiology, employing a multidisciplinary approach that combines synthesis, biochemistry, molecular biology, biophysics, molecular modeling, and NMR.
The authors declare no competing financial interest.
References
- Martin A.; Arda A.; Désiré J.; Martin-Mingot A.; Probst N.; Sinaÿ P.; Jiménez-Barbero J.; Thibaudeau S.; Blériot Y. Catching Elusive Glycosyl Cations in a Condensed Phase with HF/SbF5 Superacid. Nat. Chem. 2016, 8, 186–191. 10.1038/nchem.2399. [DOI] [PubMed] [Google Scholar]
- Crich D. En Route to the Transformation of Glycoscience: A Chemist’s Perspective on Internal and External Crossroads in Glycochemistry. J. Am. Chem. Soc. 2021, 143, 17–34. 10.1021/jacs.0c11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elferink H.; Severijnen M. E.; Martens J.; Mensink R. A.; Berden G.; Oomens J.; Rutjes F. P. J. T.; Rijs A. M.; Boltje T. J. Direct Experimental Characterization of Glycosyl Cations by Infrared Ion Spectroscopy. J. Am. Chem. Soc. 2018, 140, 6034–6038. 10.1021/jacs.8b01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana A. G.; Montalvillo-Jimenez L.; Diaz-Casado L.; Corzana F.; Merino P.; Canada F. J.; Jimenez-Oses G.; Jimenez-Barbero J.; Gomez A. M.; Asensio J. L. Dissecting the Essential Role of Anomeric β-Triflates in Glycosylation Reactions. J. Am. Chem. Soc. 2020, 142, 12501–12514. 10.1021/jacs.0c05525. [DOI] [PubMed] [Google Scholar]
- Meerwein H.; Hinz G.; Hofmann P.; Kroning E.; Pfeil E. Über Tertiäre Oxoniumsalze, I. J. Prakt. Chem. 1937, 147, 257–285. 10.1002/prac.19371471001. [DOI] [Google Scholar]
- Amyes T. L.; Jencks W. P. Lifetimes of oxocarbenium ions in aqueous solution from common ion inhibition of the solvolysis of .alpha.-azido ethers by added azide ion. J. Am. Chem. Soc. 1989, 111, 7888–7900. 10.1021/ja00202a033. [DOI] [Google Scholar]
- Bohé L.; Crich D. A Propos of Glycosyl Cations and the Mechanism of Chemical Glycosylation. C. R. Chim. 2011, 14, 3–16. 10.1016/j.crci.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott M.Carbohydrate Chemistry and Biochemistry: Structure and Mechanism; RSC Publisher, 2007, ISBN: 978-0-85404-256-2. [Google Scholar]
- Satoh H.; Manabe S.; Ito Y.; Lüthi H. P.; Laino T.; Hutter J. Endocyclic Cleavage in Glycosides with 2,3-trans Cyclic Protecting Groups. J. Am. Chem. Soc. 2011, 133, 5610–5619. 10.1021/ja201024a. [DOI] [PubMed] [Google Scholar]
- Cumpstey I. On a so-called “kinetic anomeric effect” in chemical glycosylation. Org. Biomol. Chem. 2012, 10, 2503–2508. 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]
- Whitfield D. M. In a glycosylation reaction how does a hydroxylic nucleophile find the activated anomeric carbon?. Carbohydr. Res. 2015, 403, 69–89. 10.1016/j.carres.2014.05.021. [DOI] [PubMed] [Google Scholar]
- Franconetti A.; Jiménez-Barbero J.; Cabrera-Escribano F. The Stabilization of Glycosyl Cations Through Cooperative Noncovalent Interactions: A Theoretical Perspective. ChemPhysChem 2018, 19, 659–665. 10.1002/cphc.201700988. [DOI] [PubMed] [Google Scholar]
- Whitfield D. M. In Advances in Carbohydrate Chemistry and Biochemistry; Derek H., Ed.; Academic Press: New York, 2009; Vol. 62, p 83. [DOI] [PubMed] [Google Scholar]
- Ionescu A. R.; Whitfield D. M.; Zgierski M. Z.; Nukada T. Investigations into the role of oxacarbenium ions in glycosylation reactions by ab initio molecular dynamics. Carbohydr. Res. 2006, 341, 2912–2920. 10.1016/j.carres.2006.09.027. [DOI] [PubMed] [Google Scholar]
- Nukada T.; Bérces A.; Wang L.; Zgierski M. Z.; Whitfield D. M. The two-conformer hypothesis: 2,3,4,6-tetra-O-methyl-mannopyranosyl and -glucopyranosyl oxacarbenium ions. Carbohydr. Res. 2005, 340, 841–852. 10.1016/j.carres.2004.12.021. [DOI] [PubMed] [Google Scholar]
- Hosoya T.; Kosma P.; Rosenau T. Contact ion pairs and solvent-separated ion pairs from D-mannopyranosyl and D-glucopyranosyl triflates. Carbohydr. Res. 2015, 401, 127–131. 10.1016/j.carres.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Hosoya T.; Takano T.; Kosma P.; Rosenau T. Theoretical Foundation for the Presence of Oxacarbenium Ions in Chemical Glycoside Synthesis. J. Org. Chem. 2014, 79, 7889–7894. 10.1021/jo501012s. [DOI] [PubMed] [Google Scholar]
- Bérces A.; Enright G.; Nukada T.; Whitfield D. M. The Conformational Origin of the Barrier to the Formation of Neighboring Group Assistance in Glycosylation Reactions: A Dynamical Density Functional Theory Study. J. Am. Chem. Soc. 2001, 123, 5460–5464. 10.1021/ja001194l. [DOI] [PubMed] [Google Scholar]
- Whitfield D. M.; Nukada T. DFT studies of the role of C-2-O-2 bond rotation in neighboring-group glycosylation reactions. Carbohydr. Res. 2007, 342, 1291–1304. 10.1016/j.carres.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Ionescu A. R.; Whitfield D. M.; Zgierski M. Z. O-2 Substituted pyranosyl oxacarbenium ions are C-2-O-2 2-fold rotors with a strong syn preference. Carbohydr. Res. 2007, 342, 2793–2800. 10.1016/j.carres.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Whitfield D. M. Could Diastereoselectivity in the Presence of O-2 Chiral Nonparticipating Groups Be an Indicator of Glycopyranosyl Oxacarbenium Ions in Glycosylation Reactions?. J. Org. Chem. 2012, 77, 3724–3739. 10.1021/jo202563f. [DOI] [PubMed] [Google Scholar]
- Whitfield D. M. Complications of modeling glycosylation reactions: Can the anomeric conformation of a donor determine the glycopyranosyl oxacarbenium ring conformation?. Carbohydr. Res. 2012, 356, 191–195. 10.1016/j.carres.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Whitfield D. M.; Guo J. Proton transfer and hydrogen bonding in glycosylation reactions. J. Carbohydr. Chem. 2017, 36, 59–99. 10.1080/07328303.2017.1365369. [DOI] [Google Scholar]
- Hansen T.; Lebedel L.; Remmerswaal W. A.; van der Vorm S.; Wander D. P. A.; Somers M.; Overkleeft H. S.; Filippov D. V.; Désiré J.; Mingot A.; Bleriot Y.; van der Marel G. A.; Thibaudeau S.; Codée J. D. C. Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Cent. Sci. 2019, 5, 781–788. 10.1021/acscentsci.9b00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.; Tang A.; Bennet A. J. A Stepwise Solvent-Promoted SNi Reaction of α-D-Glucopyranosyl Fluoride: Mechanistic Implications for Retaining Glycosyltransferases. J. Am. Chem. Soc. 2012, 134, 1212–1220. 10.1021/ja209339j. [DOI] [PubMed] [Google Scholar]
- Zeng J.; Wang R.; Zhang S.; Fang J.; Liu S.; Sun G.; Xu B.; Xiao Y.; Fu D.; Zhang W.; Hu Y.; Wan Q. Hydrogen-Bonding-Assisted Exogenous Nucleophilic Reagent Effect for β-Selective Glycosylation of Rare 3-Amino Sugars. J. Am. Chem. Soc. 2019, 141 (21), 8509–8515. 10.1021/jacs.9b01862. [DOI] [PubMed] [Google Scholar]
- Nielsen M. M.; Mala P.; Baldursson E. P.; Pedersen C. M. Self-promoted and stereospecific formation of N-glycosides. Chem. Sci. 2019, 10, 5299–5307. 10.1039/C9SC00857H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S.; Matsumoto K.; Kawamura K.; Suga S.; Yoshida J. Generation of Alkoxycarbenium Ion Pools from Thioacetals and Applications to Glycosylation Chemistry. Org. Lett. 2004, 6, 3755–3758. 10.1021/ol048524h. [DOI] [PubMed] [Google Scholar]
- Saito K.; Ueoka K.; Matsumoto K.; Suga S.; Nokami T.; Yoshida J. Indirect Cation-Flow Method: Flash Generation of Alkoxycarbenium Ions and Studies on the Stability of Glycosyl Cations. Angew. Chem., Int. Ed. 2011, 50, 5153–5156. 10.1002/anie.201100854. [DOI] [PubMed] [Google Scholar]
- Olah G. A.; Bollinger J. M. Stable Carbonium Ions. XXXIII. Primary Alkoxycarbonium Ions. J. Am. Chem. Soc. 1967, 89, 2993–2996. 10.1021/ja00988a034. [DOI] [PubMed] [Google Scholar]
- Paulsen H. Cyclic acyloxonium ions in carbohydrate chemistry. Adv. Carbohydr. Chem. Biochem. 1971, 26, 127–195. 10.1016/S0065-2318(08)60367-4. [DOI] [Google Scholar]
- Akien G. R.; Subramaniam B.. Direct observation of fructosyl oxocarbenium ions: key intermediates in biology and biofuels. Presented at the 246th ACS National Meeting & Exhibition, 2013.
- Lebedel L.; Ardá A.; Martin A.; Désiré J.; Mingot A.; Aufiero M.; Aiguabella Font N.; Gilmour R.; Jiménez-Barbero J.; Blériot Y.; Thibaudeau S. Structural and Computational Analysis of 2-Halogeno-Glycosyl Cations in the Presence of a Superacid: An Expansive Platform. Angew. Chem., Int. Ed. 2019, 58, 13758–13762. 10.1002/anie.201907001. [DOI] [PubMed] [Google Scholar]
- Bhuma N.; Lebedel L.; Yamashita H.; Shimizu Y.; Abada Z.; Arda A.; Desire J.; Michelet B.; Martin-Mingot A.; Abou-Hassan A.; Takumi M.; Marrot J.; Jimenez-Barbero J.; Nagaki A.; Bleriot Y.; Thibaudeau S. Insight into the Ferrier rearrangement by combining flash chemistry and superacids. Angew. Chem., Int. Ed. 2021, 60, 2036–2041. 10.1002/anie.202010175. [DOI] [PubMed] [Google Scholar]
- Denekamp C.; Sandlers Y. Formation and Stability of Oxocarbenium Ions from Glycosides. J. Mass Spectrom. 2005, 40, 1055–1063. 10.1002/jms.880. [DOI] [PubMed] [Google Scholar]
- Mucha E.; Marianski M.; Xu F.-F.; Thomas D. A.; Meijer G.; von Helden G.; Seeberger P. H.; Pagel K. Unravelling the Structure of Glycosyl Cations via Cold-Ion Infrared Spectroscopy. Nat. Commun. 2018, 9, 4174. 10.1038/s41467-018-06764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greis K.; Mucha E.; Lettow M.; Thomas D. A.; Kirschbaum C.; Moon S.; Pardo-Vargas A.; Helden G.; Meijer G.; Gilmore K.; Seeberger P. H.; Pagel K. The Impact of Leaving Group Anomericity on the Structure of Glycosyl Cations of Protected Galactosides. ChemPhysChem 2020, 21, 1905–1907. 10.1002/cphc.202000473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marianski M.; Mucha E.; Greis K.; Moon S.; Pardo A.; Kirschbaum C.; Thomas D. A.; Meijer G.; Helden G.; Gilmore K.; Seeberger P. H.; Pagel K. Remote Participation during Glycosylation Reactions of Galactose Building Blocks: Direct Evidence from Cryogenic Vibrational Spectroscopy. Angew. Chem., Int. Ed. 2020, 59, 6166–6171. 10.1002/anie.201916245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen T.; Elferink H.; van Hengst J. M. A.; Houthuijs K. J.; Remmerswaal W. A.; Kromm A.; Berden G.; van der Vorm S.; Rijs A. M.; Overkleeft H. S.; Filippov D. V.; Rutjes F. P. J. T.; van der Marel G. A.; Martens J.; Oomens J.; Codée J. D. C.; Boltje T. J. Characterization of Glycosyl Dioxolenium Ions and Their Role in Glycosylation Reactions. Nat. Commun. 2020, 11, 2664. 10.1038/s41467-020-16362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greis K.; Kirschbaum C.; Leichnitz S.; Gewinner S.; Schöllkopf W.; von Helden G.; Meijer G.; Seeberger P. H.; Pagel P. Direct Experimental Characterization of the Ferrier Glycosyl Cation in the Gas Phase. Org. Lett. 2020, 22, 8916–8919. 10.1021/acs.orglett.0c03301. [DOI] [PubMed] [Google Scholar]
- Adero P. O.; Amarasekara H.; Wen P.; Bohé L.; Crich D. The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev. 2018, 118, 8242–8284. 10.1021/acs.chemrev.8b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo C.; Bennet A. J. The physical organic chemistry of glycopyranosyl transfer reactions in solution and enzyme-catalyzed. Curr. Opin. Chem. Biol. 2019, 53, 145–157. 10.1016/j.cbpa.2019.08.003. [DOI] [PubMed] [Google Scholar]
- Crich D. Mechanism of a chemical glycosylation reaction. Acc. Chem. Res. 2010, 43, 1144–1153. 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- Chatterjee S.; Moon S.; Hentschel F.; Gilmore K.; Seeberger P. H. An Empirical Understanding of the Glycosylation Reaction. J. Am. Chem. Soc. 2018, 140, 11942–11953. 10.1021/jacs.8b04525. [DOI] [PubMed] [Google Scholar]
- Mydock L. K.; Demchenko A. V. Mechanism of chemical O-glycosylation: from early studies to recent discoveries. Org. Biomol. Chem. 2010, 8, 497–510. 10.1039/B916088D. [DOI] [PubMed] [Google Scholar]
- Frihed T. G.; Bols M.; Pedersen C. M. Mechanisms of Glycosylation Reactions Studied by Low Temperature Nuclear Magnetic Resonance. Chem. Rev. 2015, 115, 4963–5013. 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- Crich D.; Sun S. Are Glycosyl Triflates Intermediates in the Sulfoxide Glycosylation Method? A Chemical and 1H, 13C, and 19F NMR Spectroscopic Investigation. J. Am. Chem. Soc. 1997, 119 (46), 11217–11223. 10.1021/ja971239r. [DOI] [Google Scholar]
- Crich D.; Chandrasekera N. S. Mechanism of 4,6-O-Benzylidene-Directed β-Mannosylation as Determined by α-Deuterium Kinetic Isotope Effects. Angew. Chem., Int. Ed. 2004, 43, 5386–5389. 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- Huang M.; Garrett G. E.; Birlirakis N.; Bohé L.; Pratt D. A.; Crich D. Dissecting the mechanisms of a class of chemical glycosylation using primary 13C kinetic isotope effects. Nat. Chem. 2012, 4, 663–667. 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Retailleau P.; Bohé L.; Crich D. Cation Clock Permits Distinction Between the Mechanisms of α- and β-O- and β-C-Glycosylation in the Mannopyranose Series: Evidence for the Existence of a Mannopyranosyl Oxo-carbenium Ion. J. Am. Chem. Soc. 2012, 134, 14746–14749. 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adero P. O.; Furukawa T.; Huang M.; Mukherjee D.; Re-tailleau P.; Bohé L.; Crich D. Cation Clock Reactions for the Determination of Relative Reaction Kinetics in Glycosylation Reactions: Applications to Gluco- and Mannopyranosyl Sulfoxide and Trichloroacetimidate Type Donors. J. Am. Chem. Soc. 2015, 137, 10336–10345. 10.1021/jacs.5b06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuckendorff M.; Bols P. S.; Barry C. B.; Frihed T. G.; Pedersen C. M.; Bols M. β-Mannosylation with 4,6-benzylidene protected mannosyl donors without preactivation. Chem. Commun. 2015, 51, 13283–13285. 10.1039/C5CC04716A. [DOI] [PubMed] [Google Scholar]
- van der Vorm S.; Hansen T.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. The influence of acceptor nucleophilicity on the glycosylation reaction mechanism. Chem. Sci. 2017, 8, 1867–1875. 10.1039/C6SC04638J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vorm S.; van Hengst J. M. A.; Bakker M.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. Mapping the Relationship between Glycosyl Acceptor Reactivity and Glycosylation Stereoselectivity. Angew. Chem., Int. Ed. 2018, 57, 8240–8244. 10.1002/anie.201802899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Lodder G.; Mazurek J.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. Equatorial Anomeric Triflates from Mannuronic Acid Esters. J. Am. Chem. Soc. 2009, 131, 12080–12081. 10.1021/ja905008p. [DOI] [PubMed] [Google Scholar]
- Rönnols J.; Walvoort M. T. C.; van der Marel G. A.; Codée J. D. C.; Widmalm G. Chair interconversion and reactivity of mannuronic acid esters. Org. Biomol. Chem. 2013, 11, 8127–8134. 10.1039/c3ob41747f. [DOI] [PubMed] [Google Scholar]
- Santana A. G.; Montalvillo-Jiménez L.; Diaz-Casado L.; Mann E.; Jiménez-Barbero J.; Gómez A. M.; Asensio J. L. Single-Step Glycosylations with 13C-Labelled Sulfoxide Donors: A Low-Temperature NMR Cartography of the Distinguishing Mechanistic Intermediates. Chem. - Eur. J. 2021, 27, 2030–2042. 10.1002/chem.202003850. [DOI] [PubMed] [Google Scholar]
- Moremen K. W.; Haltiwanger R. S. Emerging structural insights into glycosyltransferase-mediated synthesis of glycans. Nat. Chem. Biol. 2019, 15, 853–864. 10.1038/s41589-019-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albesa-Jové D.; Romero-García J.; Sancho-Vaello E.; Contreras F.-X.; Rodrigo-Unzueta A.; Comino M.; Carreras-González A.; Arrasate P.; Urresti S.; Biarnés X.; Planas A.; Guerin M. E. Structural Snapshots and Loop Dynamics along the Catalytic Cycle of Glycosyltransferase GpgS. Structure 2017, 25, 1034–1044. 10.1016/j.str.2017.05.009. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Rovira C. Reaction Mechanisms in Carbohydrate-Active Enzymes: Glycoside Hydrolases and Glycosyltransferases. Insights from ab Initio Quantum Mechanics/Molecular Mechanics Dynamic Simulations. J. Am. Chem. Soc. 2015, 137, 7528–7547. 10.1021/jacs.5b01156. [DOI] [PubMed] [Google Scholar]
- Ishida T.; Parks J. M.; Smith J. C. Insight into the Catalytic Mechanism of GH11 Xylanase: Computational Analysis of Substrate Distortion Based on a Neutron Structure. J. Am. Chem. Soc. 2020, 142, 17966–17980. 10.1021/jacs.0c02148. [DOI] [PubMed] [Google Scholar]
- Rovira C.; Males A.; Davies G. J.; Williams S. J. Mannosidase mechanism: at the intersection of conformation and catalysis. Curr. Opin. Struct. Biol. 2020, 62, 79–92. 10.1016/j.sbi.2019.11.008. [DOI] [PubMed] [Google Scholar]
- Alonso-Gil S.; Coines J.; André I.; Rovira C. Conformational itinerary of sucrose during hydrolysis by retaining amylosucrase. Front. Chem. 2019, 7, 269. 10.3389/fchem.2019.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirke J. C. K.; Crich D. Glycoside Hydrolases Restrict the Side Chain Conformation of Their Substrates to Gain Additional Transition State Stabilization. J. Am. Chem. Soc. 2020, 142, 16965–16973. 10.1021/jacs.0c05592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Gil S.; Males A.; Fernandes P. Z.; Williams S. J.; Davies G. J.; Rovira C. Computational Design of Experiment Unveils the Conformational Reaction Coordinate of GH125 α-Mannosidases. J. Am. Chem. Soc. 2017, 139, 1085–1088. 10.1021/jacs.6b11247. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Biarnés X.; Planas A.; Rovira C. The Conformational Free-Energy Landscape of β-d-Mannopyranose: Evidence for a 1S5 → B2,5 → oS2 Catalytic Itinerary in β-Mannosidases. J. Am. Chem. Soc. 2010, 132, 16058–16065. 10.1021/ja105520h. [DOI] [PubMed] [Google Scholar]
- Biarnés X.; Ardèvol A.; Iglesias-Fernández J.; Planas A.; Rovira C. Catalytic Itinerary in 1,3–1,4-β-Glucanase Unraveled by QM/MM Metadynamics. Charge Is Not Yet Fully Developed at the Oxocarbenium Ion-like Transition State. J. Am. Chem. Soc. 2011, 133, 20301–20309. 10.1021/ja207113e. [DOI] [PubMed] [Google Scholar]
- Nin-Hill A.; Rovira C. The Catalytic Reaction Mechanism of the β-Galactocerebrosidase Enzyme Deficient in Krabbe Disease. ACS Catal. 2020, 10, 12091–12097. 10.1021/acscatal.0c02609. [DOI] [Google Scholar]
- Darby J. F.; Gilio A. K.; Piniello B.; Roth C.; Blagova E.; Hubbard R. E.; Rovira C.; Davies G. J.; Wu L. Substrate Engagement and Catalytic Mechanisms of N-Acetylglucosaminyltransferase V. ACS Catal. 2020, 10, 8590–8596. 10.1021/acscatal.0c02222. [DOI] [Google Scholar]
- Gómez H.; Polyak I.; Thiel W.; Lluch J. M.; Masgrau L. Retaining Glycosyltransferase Mechanism Studied by QM/MM Methods: Lipopolysaccharyl-α-1,4-galactosyltransferase C Transfers α-Galactose via an Oxocarbenium Ion-like Transition State. J. Am. Chem. Soc. 2012, 134, 4743–4752. 10.1021/ja210490f. [DOI] [PubMed] [Google Scholar]
- Yang M.; Fehl C.; Lees K. V.; Lim E.-K.; Offen W. A.; Davies G. J.; Bowles D. J.; Davidson M. G.; Roberts S. J.; Davis B. G. Functional and informatics analysis enables glycosyltransferase activity prediction. Nat. Chem. Biol. 2018, 14, 1109–1117. 10.1038/s41589-018-0154-9. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Rovira C. The Molecular Mechanism of Enzymatic Glycosyl Transfer with Retention of Configuration: Evidence for a Short-Lived Oxocarbenium-Like Species. Angew. Chem., Int. Ed. 2011, 50, 10897–10901. 10.1002/anie.201104623. [DOI] [PubMed] [Google Scholar]
- Yan L.; Liu Y. The Retaining Mechanism of Xylose Transfer Catalyzed by Xyloside α-1,3-Xylosyltransferase (XXYLT1): A Quantum Mechanics/Molecular Mechanics Study. J. Chem. Inf. Model. 2020, 60, 1585–1594. 10.1021/acs.jcim.9b00976. [DOI] [PubMed] [Google Scholar]
- Ren W.; Farren-Dai M.; Sannikova N.; Swiderek K.; Wang Y.; Akintola O.; Britton R.; Moliner V.; Bennet A. J. Glycoside hydrolase stabilization of transition state charge: new directions for inhibitor design. Chem. Sci. 2020, 11, 10488–10495. 10.1039/D0SC04401F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield D. M.; Douglas S. P.; Tang T.-H.; Csizmadia I. G.; Pang H. Y. S.; Moolten F. L.; Krepinsky J. J. Differential reactivity of carbohydrate hydroxyls in glycosylations. 11. The likely role of intramolecular hydrogen bonding on glycosylation reactions. Galactosylation of nucleoside 5′-hydroxyls for the syntheses of novel potential anticancer agents. Can. J. Chem. 1994, 72, 2225–2238. 10.1139/v94-284. [DOI] [Google Scholar]
- Nerinckx W.; Desmet T.; Claeyssens M. A hydrophobic platform as a mechanistically relevant transition state stabilising factor appears to be present in the active centre of all glycoside hydrolases. FEBS Lett. 2003, 538, 1–7. 10.1016/S0014-5793(03)00148-0. [DOI] [PubMed] [Google Scholar]
- Yamamoto-Katayama S.; Ariyoshi M.; Ishihara K.; Hirano T.; Jingami H.; Morikawa K. J. Mol. Biol. 2002, 316, 711–723. 10.1006/jmbi.2001.5386. [DOI] [PubMed] [Google Scholar]
- Iglesias-Fernández J.; Hancock S. M.; Lee S. S.; Khan M.; Kirkpatrick J.; Oldham N. J.; McAuley K.; Fordham-Skelton A.; Rovira C.; Davis B. G. A front-face ‘SNi synthase’ engineered from a retaining ’double-SN2′ hydrolase. Nat. Chem. Biol. 2017, 13, 874–881. 10.1038/nchembio.2394. [DOI] [PubMed] [Google Scholar]
- Montalvillo-Jimenez L.; Santana A. G.; Corzana F.; Jimenez-Oses G.; Jimenez-Barbero J.; Gomez A. M.; Asensio J. L. Impact of Aromatic Stacking on Glycoside Reactivity: Balancing CH/π and Cation/π Interactions for the Stabilization of Glycosyl Oxocarbenium Ions. J. Am. Chem. Soc. 2019, 141, 13372–13384. 10.1021/jacs.9b03285. [DOI] [PubMed] [Google Scholar]