

Abstract

Here, a large-scale feasible, chromatography-free process to purge triphenylphosphine oxide (TPPO) from the crude product of Mitsunobu and Wittig reactions has been developed. Divergence in physicochemical properties like polarity and solubility of TPPO against the product was utilized to precipitate TPPO directly from the reaction mixture and eliminate by simple filtration on a kilogram scale at a pilot plant with high purity of the product.
Introduction
Triphenylphosphine oxide (Ph3P = O), commonly known as TPPO, is a byproduct of very popular and widely used name reactions such as Mitsunobu,1 Wittig,2 Staudinger,3 Appel,4 and Corey–Fuchs5 reactions, as shown in Figure 1. The alkene derivatives can be prepared from the corresponding aldehyde or ketone using triphenylphosphonium ylide (Wittig reagent) in the Wittig reaction. The Mitsunobu reaction is the most powerful tool for a chemist to construct C–O, C–N, C–S, C–X, C–C, and S–S with high stereospecificity. It involves triphenylphosphine (TPP), diethyl azodicarboxylate (DEAD), or diisopropyl azodicarboxylate (DIAD) and an appropriate reactive nucleophile with a broad range compatibility of functional groups.6,7
Figure 1.

Triphenylphosphine oxide (TPPO) as a byproduct in the renowned popular named reactions.
However, the Mitsunobu reaction constituting the significant and persistent problem of product purification precludes its use at a commercial scale. Product isolation is usually achieved by tedious, expensive, and time-consuming column chromatography, which is a powerful technique for purification of new chemical entities in almost all medicinal chemistry laboratories; however, this purification technique is not feasible for large-scale operations. Chromatography-free procedures have been summarized in few reviews8−10 such as modified phosphanes and fluorous phosphane; modified reagents can be removed by acid or base extraction by decomposition to volatile products or by phase-switching. These methods suffer lack of commercial availability and large-scale compatibility.
Results and Discussion
Given our continued interest in the process development of NCE of zidebactam analogues as β-lactamase inhibitors,11,12 O-alkylated hydroxylamine is a crucial intermediate, fundamentally obtained from the Mitsunobu reaction followed by Ing–Manske hydrazinolysis (Figure 2). We were interested in elaborating the “large-scale process-friendly” conditions for the Mitsunobu reaction, which are used routinely by medicinal chemists to synthesize novel molecules for functional group transformations.
Figure 2.
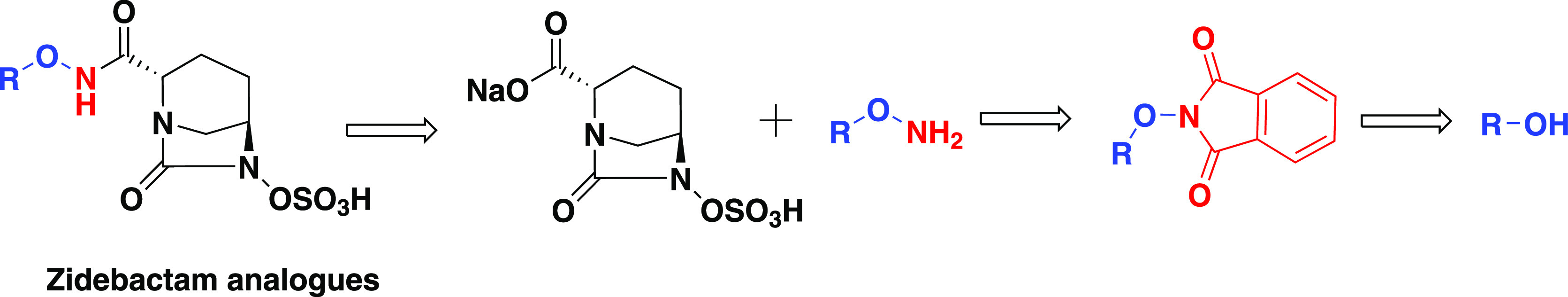
Retrosynthetic analysis for zidebactam analogues.
As shown in Scheme 1, an attempt to obtain the nucleophilic substitution effect of N-hydroxy phthalimide on the corresponding tosyl 4(13) or mesyl 5(14) derivatives failed because of a substantial low yield and an additional step. As these preliminary results were not competitive, we did not pursue it further and decided to focus on the Mitsunobu reaction.
Scheme 1. Initial Approach for the Synthesis of 6.

Reagents: (a) NsF, DBU, toluene; (b) CDI, NaBH4; (c) CH3C6H4SO2Cl, TEA; (d) CH3SO2Cl, TEA; (e) PhthNOH, base, DMF.
We reviewed the literature for the postreaction precipitation of TPPO to purify the product. Though the reported methods possess some advantages, there are several disadvantages such as follows: (a) the reaction solvent [tetrahydrofuran (THF), dichloromethane (DCM), acetonitrile, alcohol] must be removed completely before work-up, (b) the product must be soluble in highly nonpolar solvents such as cold hexanes or diethyl ether or a mixture of both,15 (c) treatment with metal chlorides (ZnCl216 or MgCl217,18) to obtain a TPPO–metal complex, (d) treatment with acidic reagents such as acetic acid19 or oxalyl chloride9 to afford immiscible TPPO-AcOH or triphenylphosphonium chloride, respectively, (e) an acid–base extraction of the product, and (f) using alkylative trapping on Merrifield resin.20,21
To overcome the above-mentioned disadvantages, it was decided to utilize the physicochemical properties of the product and byproducts in purification. It is known that TPPO is almost insoluble in deionized water, cyclohexane, petroleum ether, and hexane.22 TPPO can be removed with EtOH,23 MeOH,24 and isopropyl alcohol (IPA).25 It was also evident to take the advantage of hydrogen bond-induced effective crystallization of TPPO26 through the formation of cocrystals with several impurities/byproducts specifically 1,2-isopropylhydrazine dicarboxylate.27 Solubility screening of selected solvents for 6, TPPO, and TPPO-H2DIAD is tabulated in Table 1.
Table 1. Solubility Screening.
| solubility (mg/mL) | ||||||
|---|---|---|---|---|---|---|
| compound-6 |
TPPO-H2DIAD |
TPPO |
||||
| solvent | RT | 0 °C | RT | 0 °C | RT | 0 °C |
| 2-propanol | insoluble | insoluble | 60.4 | 13.3 | 46.5 | 25.4 |
| tetrahydrofuran | 124.5 | 101.0 | 157.5 | 61.0 | 169.7 | 105.0 |
| cyclohexane | insoluble | insoluble | insoluble | insoluble | insoluble | insoluble |
| toluene | 46.7 | 42.1 | 23.2 | 15.95 | 44.3 | 42.9 |
Fortunately, after preliminary screening of solvents, it was observed that cold toluene triggers effective precipitation of triphenylphosphine oxide-1,2-isopropylhydrazine dicarboxylate (TPPO-H2DIAD) and was successfully removed from the crude product at a lab scale. It was noted that the solubility of TPPO-H2DIAD increased with an increase in temperature. Therefore, at first, TPPO-H2DIAD was precipitated followed by crystallization of the product in a suitable solvent. Toluene18,23,28 was a solvent by choice: relatively inexpensive, promotes crystallization of weakly acidic derivatives with TPPO,27 and avoids operational time/cost of distillation. In addition, this change afforded almost a similar result like THF along with merits of feasibility at large-scale (easily recoverable and reusable) usage. The rate of the reaction was accelerated by increasing the reaction temperature from room temperature (RT).
Our goal was to find an optimal reaction condition, which would offer significant benefit in terms of purification through direct precipitation of TPPO-H2DIAD from the reaction mixture. Therefore, various conditions (combinations of solvents and temperature) were screened, as described in Table 2.
Table 2. Screening of the Mitsunobu Reaction on a 50 g Scale (Scheme 2).
| entry | solvent | volume (mL) | temperature (°C) | TPPO-H2DIAD (dry) by filtration (g) | product isolation | yield (%) |
|---|---|---|---|---|---|---|
| 1 | THF | 250 | 0–5 | 0 | column | 71 |
| 2 | toluene | 500 | 25–30 | 0 | column | 70 |
| 3 | toluene | 500 | –5–0 | 5.5 | column | 70 |
| 4 | toluene | 200 | 25–30 | 10 | column | 70 |
| 5 | toluene | 200 | –5–0 | 75 | IPA (125 mL) | 68 |
It was identified that the concentration of the reaction mass plays a very crucial role. Precipitation of TPPO-H2DIAD is inversely proportional to the concentration of the reaction mass; a significant drop in precipitation was observed with a diluted reaction mass. The tuning of lower temperature seemed to be promising for removing TPPO-H2DIAD directly from the reaction. The notable observation was that TPPO-H2DIAD could not be removed at RT or higher temperature. Also, multiple solvents and solvent mixtures were explored to precipitate the desired product selectively. Highly pure 6 was readily isolated from the crude by treatment of IPA followed by simple filtration.
N-Boc-4-hydroxy-l-proline methyl ester (1) was transformed to N-boc-4-fluoro-l-proline29 (2) and treated with 1,1′-carbonyldiimidazole (CDI)/sodium borohydride (NaBH4) instead of popular pyrophoric lithium aluminum hydride (LAH) to afford reduced alcohol 3, as shown in Scheme 1. The Mitsunobu reaction was carried out in toluene at 0–50 °C. Precipitation of TPPO-H2DIAD was induced by cooling the reaction mixture along with continuous stirring. The solid was filtered and the filtrate was concentrated to remove toluene. Large-scale feasible IPA was used to crystallize 6 as a white solid at a pilot scale (Scheme 2). Using a simple work-up process, most of the impurities from the Mitsunobu reaction were successfully removed (Figure 3). Quantitative % content (w/w) of compound-6 and TPPO during the work-up process was estimated by high-performance liquid chromatography (HPLC) and is tabulated in Table 3.
Scheme 2. Pilot-Plant Synthesis of 6.

Figure 3.

Flowchart and the HPLC chromatograph of the work-up process for 6.
Table 3. Quantitative % Content (w/w) of Compound-6 and TPPO by HPLC.
| % content (w/w) | compound-6 | TPPO |
|---|---|---|
| reaction mixture | 20.55 | 29.33 |
| TPPO- H2DIAD | 0.14 | |
| toluene residue | 24.86 | 6.25 |
| pure compound-6 | 0.05 | |
| IPA residue | 13.21 | 10.97 |
Notwithstanding the popularity of the Wittig reaction, a similar purification process was also investigated to prepare the alkene derivative. TPPO was efficiently removed without column chromatography in the Wittig reaction for the synthesis of vitamin A on a large scale. The pure vitamin A acetate product could then easily be obtained via crystallization after removing some of the heptane and TPPO separated from the MeOH/water phase.30 Relatively inexpensive cyclohexane was used instead of n-heptane, THF, or DCM to isolate TPPO directly from the reaction mass without utilizing alcoholic phase separation. The keto derivative 8(31) was obtained from hydroxyl derivative 7. However, the large-scale validated Corey–Kim oxidation condition32 developed in our lab at elevated temperature (−5 to −10 °C) was chosen instead of the routine cryogenic (≥−30 °C) condition with an appropriate ratio of toluene/DCM instead of TCCA/TEMPO. Potassium tert-butoxide was added to the suspension of the Wittig reagent in cyclohexane followed by the addition of 8 at RT. The reaction mass was cooled, filtered to eliminate TPPO, and the filtrate was washed with brine. A cyclohexane layer was concentrated and treated with hexane to remove the small amount of TPPO invariably present after filtration. A hexane layer was concentrated under reduced pressure to afford methylene derivative 9 at a pilot scale (Scheme 3).
Scheme 3. Synthesis of 9 and 10.

Reagents: (a) NCS/DMS/TEA; (b) PPh3CH3Br, KOtBu; (c) 1M TBAF, THF; (d) PhthNOH, PPh3, DIAD.
The described methodology to remove TPPO directly from the reaction mass may not be suitable for each and every product of these types of reactions; indeed the concept is effective and will help chemists to remove maximum TPPO directly from the reaction mass by additional manipulations. It may be possible to envisage the current process to purify products of Appel, Staudinger, and Corey–Fuchs reactions. The utilization of this concept with other TPPO generating reactions has not yet been tested, but it is anticipated that the described methodology has potential for achieving a practical solution to remove TPPO without column chromatography. In this paper, we have described, along with full experimental details, the preparation of 6, 10 (Mitsunobu reaction), and 9 (Wittig reaction).
Conclusions
The present method illustrates one of the ideal approaches to isolate products with the highest purity by implementing an appropriate solvent, reaction concentration, and temperature depending on the polarity and solubility of products to improve the process development of NCE. However, precise conditions truly depend on the specific substrate at hand. The process showcased product isolation at a pilot scale without chromatography of two renowned named reactions wherein TPPO removal is a bottleneck. We anticipate that the present process will provide an initial isolation tool for other reactions that generate TPPO as a byproduct.
Experimental Procedures
(2S,4S)-tert-Butyl 4-Fluoro-2-(hydroxymethyl)pyrrolidine-1-carboxylate (3)
CDI Adduct Preparation
A suspension of 2 (5.0 kg, 21.44 mol) in THF (25L) and CDI (5.3 kg 32.68 mol) was added in 7 equal portions and agitated for 3 h at room temperature. After completion of the reaction, CDI adduct was cooled at −5 to 0 °C. In another flask, NaBH4 (0.82 kg, 21.68 mol) was added in 3 equal portions to water (15 L) at 0 to 5 °C under agitation; the above chilled CDI adduct solution was added dropwise at 5–10 °C and stirred for 3 h at room temperature. After completion of the reaction, it was acidified with 50% HCl in water (19 L) till pH ∼ 3–4. DCM (25 L) was charged and stirred for 15 min. The DCM layer was then separated and washed with 5% aq sodium bicarbonate (10 L) followed by water (15 L). The organic layer was concentrated under reduced pressure to afford 3 (4.25 kg, 90%) as a pale yellow low melting solid. 1H NMR (a mixture of rotamers, 400 MHz, DMSO-d6): δ 5.31–5.18 (m, 1H), 4.77 (s, 1H), 3.77 (bs, 1H), 3.66–3.58 (m, 1H), 3.56–3.39 (m, 3H), 3.17–3.16 (s,1H), 2.27–2.15 (m, 2H), 1.40–1.36 (s, 9H); 13C NMR (125 MHz, DMSO-d6): 153.79–153.55, 94.25, 93.44, 92.88, 92.06, 78.74, 62.12–61.93, 57.90, 53.33–53.15, 33.78–33.08, 28.06; ESMS: 219.9 (M + H); HPLC: ∼94.77%; SOR [α]D20: −34.56 (c 0.5, MeOH).
tert-Butyl (2S,4S)-2-{[(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)oxy]methyl}-4-fluoro-pyrrolidine-1-carboxylate (6)
To a stirred solution of 3 (4.0 kg, 18.26 mol) in toluene (16 L) in a jacketed flask, N-hydroxy phthalimide (2.98 kg, 18.26 mol) and TPP (7.17 kg, 27.39 mol) were charged at room temperature and cooled to 0–5 °C. DIAD (5.53 kg, 27.39 mol) [Caution: DIAD is a health hazard and causes acute toxicity; must be handled with appropriate precautions!] was added dropwise at below 25 °C and stirred for 30 min. The reaction mass was heated for 3 h at 50 to 55 °C. After completion of the reaction, the reaction mass was cooled at −5 to 0 °C. TPPO-H2DIAD (9.13 kg, wet) was filtered over PNF (Pressure Nutsche Filter) and washed with chilled toluene (4 L). The organic layer was distilled out under reduced pressure to get an oily mass and treated with IPA (10 L) at 50–55 °C for 30 min. It was then cooled, filtered, and washed with chilled IPA (3 L) followed by drying under vacuum at 50–55 °C in a rotocone vacuum dryer (RCVD) for 6 h to afford 6 (4.52 kg, 68%) as a white powder. 1H NMR (a mixture of rotamers, 400 MHz, CDCl3): δ 7.83–7.75 (bs, 4H), 5.33–5.23 (m, 1H), 4.54–4.46 (m, 1H), 4.32–4.18 (m, 1H), 4.11–4.09 (m, 1H), 3.71–3.54 (m, 2H), 2.82–2.75 (s, 1H), 2.29–2.21 (s, 1H), 1.43–1.39 (s, 9H); 13C NMR (125 MHz, CDC13): δ 163.59, 163.38, 154.11–153.88, 134.54–134.42, 128.96–128.77, 123.49, 94.01, 93.23, 92.61, 91.84, 80.37–80.12, 54.42,53.74–53.11, 35.34–35.18, 34.33–34.17, 28.31–28.18; ESMS: 264.9 (M + H); HPLC: ∼99.50%; SOR [α]D20: −57.18 (c 0.5, DCM).
tert-Butyl (2S,4S)-2-{[(1,3-Dioxo-1,3-dihydro-2H-isoindol-2-yl)oxy]methyl}-4-methylene-pyrrolidine-1-carboxylate (10)
To a stirred solution of 9 (50 g, 0.23 mol) in toluene (200 mL), N-hydroxy phthalimide (38.24 g, 0.23 mol) and TPP (90.39 g, 0.345 mol) were charged at room temperature and cooled to 0–5 °C. DIAD (69.75 g, 0.345 mol) [Caution: DIAD is a health hazard and causes acute toxicity; must be handled with appropriate precautions!] was added dropwise at below 25 °C and stirred for 30 min. The reaction mass was heated for 3 h at 50 to 55 °C. After completion of the reaction, the reaction mass cooled at −5 to 0 °C. TPPO-H2DIAD (90.0 g, wet) was filtered and washed with chilled toluene (50 mL). The organic layer was distilled under reduced pressure to obtain an oily mass (HPLC: 83.68%). Crude was purified by silica gel column purification to afford 10 (58.17 g, 70%) as colorless oil, which turned to a white solid upon extended exposure to high vacuum. 1H NMR (400 MHz, CDC13): δ 7.83–7.75 (bs, 4H), 5.13–5.07 (m, 2H), 4.33–4.30 (m, 2H), 4.05–3.87 (m, 3H), 2.95–2.89 (m, 2H), 3.71–3.54 (m, 2H), 1.44–1.40 (s, 9H); 13C NMR (125 MHz, CDC13): δ 163.31, 154.06–153.67, 144.27, 143.03, 134.51, 128.90–128.77, 123.48, 108.74, 107.93, 80.02–79.8, 54.95, 50.97, 50.29, 35.52, 34.51, 28.27, 21.91; ESMS: 359.1 (M + H); HPLC: 95.08%.
(S)-tert-Butyl 2-(tert-Butyl(ethoxy)dimethyl silane)-4-oxopyrrolidine-1-carboxylate (8)
A suspension of N-chlorosuccinimide (3.63 kg, 27.19 mol) in DCM (9 L) and toluene (9 L) was agitated and cooled to −15 °C. A solution of dimethyl sulfide (1.96 kg, 31.71 mol) in toluene (3 L) was added dropwise at below −5 °C. After 30 min, a solution of 7 (3 kg, 9.06 moles) in toluene (24 L) was added dropwise at −5 to −10 °C and stirred for 2 h. Triethylamine (3.21 kg, 31.71 mol) was added at −5 to −10 °C by controlling the addition rate and stirred for 30 min. After completion of the reaction, saturated aq NaHCO3 (30 L) was added at −5 °C and stirred for 30 min. The reaction was allowed to warm to room temperature. The organic layer was separated and washed with 15% aq NaCl (20 L). The organic phase was concentrated under reduced pressure to afford 8 (2 kg, 67%) as a pale yellow liquid. 1H NMR (a mixture of rotamers, 400 MHz, CDCl3): δ 4.47–4.46 (m, 1H), 3.95–3.65 (m, 2H), 3.58–3.50 (m, 1H), 3.41–3.32 (m, 1H), 3.21–3.18 (m, 1H), 2.07–2.05 (m, 1H), 1.89–1.86 (m, 1H), 1.47–1.44 (s, 10H), 0.86 (s, 19H), 0.04 (s, 12 H); 13C NMR (125 MHz, CDC13): δ 154.58, 78.95, 70.46, 69.75, 64.43, 63.58, 57.67, 55.04–54.69, 38.16, 37.23, 28.50, 27.55, 25.84–25.74, 18.16–17.97; SOR [α]D20 + 3.2 (c 1.0, CHCl3).
(S)-tert-Butyldimethylsilyloxymethyl-N-tert-butyloxycarbonyl-4-methylenepyrrolidine (9)
To a stirred suspension of methyltriphenylphosphonium bromide (2.38 kg, 6.66 mol) in cyclohexane (10 L), potassium tert-butoxide (0.75 kg, 6.66 mol) was added in 3 equal portions (slight exotherm observed) to obtain a bright yellow color suspension and stirred for 1 h at RT. A solution of 8 (2 kg, 6.06 moles) in cyclohexane (6 L) was added dropwise at below 30 °C and stirred for 4 h. After completion of the reaction, the reaction mass was cooled to 0 °C, filtered through celite, and washed with cyclohexane (2 L). The filtrate was washed with 15% aq NaCl (2 × 10 L). The organic phase was concentrated under reduced pressure and agitated with hexane (3 L) for 30 min. It was then filtered and washed with hexane (1 L). The obtained filtrate was concentrated under reduced pressure to afford 9 (1.45 kg, 73%) as a pale yellow liquid. 1H NMR (a mixture of rotamers, 400 MHz, CDC13): δ 4.95–4.89 (m, 2H), 4.05–3.91 (m, 2H), 3.82–3.79 (m, 1H), 3.63–3.57 (m,1H), 3.37–3.36 (m,1H), 2.57–2.54 (m, 2H), 1.45–1.43 (s, 11H), 0.86 (s, 12 H), 0.01 (s, 8H); 13C NMR (125 MHz, CDC13): δ 154.59, 154.02, 145.80, 144.49, 107.07, 106.22, 79.41–79.18, 70.44, 69.73,64.43, 36.57, 63.25, 58.30–57.68, 55.04–54.73, 51.43, 50.75, 38.15, 37.27, 34.87, 34.19, 28.49, 25.84–25.72, 18.15–17.95; Mass: 328.0 (M + H); SOR [α]D20: −22.0 (c 1.0, CHCl3).
Acknowledgments
The authors thank the CRL-3 group for discussion/suggestions, Dr. Vipul Rane and Dr. Vinod Ahirrao for analytical support, and the pilot plant team for scale-up support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01996.
Copies of spectral data of intermediates and the final products (PDF)
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mitsunobu O.; Yamada M.; Mukaiyama T. Preparation of esters of phosphoric acid by the reaction of trivalent phosphorus compounds with diethyl azodicarboxylate in the presence of alcohols. Bull. Chem. Soc. Jpn. 1967, 40, 935–939. 10.1246/bcsj.40.935. [DOI] [Google Scholar]
- Wittig G.; Geissler G. Zur Reaktionsweise des Pentaphenyl-phosphors und einiger Derivate. Justus Liebigs Ann. Chem. 1953, 580, 44–57. 10.1002/jlac.19535800107. [DOI] [Google Scholar]
- Staudinger H.; Meyer J. New organic compounds of phosphorus. III. Phosphine-methylene derivatives and phosphinimines. Helv. Chim. Acta. 1919, 2, 635–646. 10.1002/hlca.19190020164. [DOI] [Google Scholar]
- Appel R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P-N Linkage. Angew. Chem., Int. Ed. 1975, 14, 801–811. 10.1002/anie.197508011. [DOI] [Google Scholar]
- Corey E. J. A synthetic method for formyl-> ethynyl conversion. Tetrahedron Lett. 1972, 13, 3769–3772. 10.1016/S0040-4039(01)94157-7. [DOI] [Google Scholar]
- Mitsunobu O.; Yamada Y. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. 10.1246/bcsj.40.2380. [DOI] [Google Scholar]
- Fletcher S. The Mitsunobu reaction in the 21 st century. Org. Chem. Front. 2015, 2, 739–752. 10.1039/C5QO00016E. [DOI] [Google Scholar]
- Dembinski R. Recent Advances in the Mitsunobu Reaction: Modified Reagents and the Quest for Chromatography-Free Separation. Eur. J. Org. Chem. 2004, 13, 2763–2772. 10.1002/ejoc.200400003. [DOI] [Google Scholar]
- Byrne P. A.; Rajendran K. V.; Muldoon J.; Gilheany D. G. A convenient and mild chromatography-free method for the purification of the products of Wittig and Appel reactions. Org. Biomol. Chem. 2012, 10, 3531–3537. 10.1039/c2ob07074j. [DOI] [PubMed] [Google Scholar]
- Dandapani S.; Curran D. P. Separation-Friendly Mitsunobu Reactions: A Microcosm of Recent Developments in Separation Strategies. Chem. - Eur. J. 2004, 10, 3130–3138. 10.1002/chem.200400363. [DOI] [PubMed] [Google Scholar]
- Tadiparthi R.; Patil V. J.; Dixit P.; Patel M. V.. A Process for Preparation of (2s,5r)-n-(2-Amino ethoxy)-6-(sulfooxy)-7-oxo-1,6-diazabicyclo [3.2.1] octane-2-carboxamide. WO Patent WO2016120752A12016.
- Tadiparthi R.; Dond B.; Birajdar S.; Kale A.; Patil V. J.. A Process for Preparation of (2s,5r)-7-Oxo-n-[(2s)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide. WO Patent WO20150793292015.
- Zhang G.; Ren B.; Wang H.; Zhao H.; Guo Y.; Wang Z.; Zhou C.. 5-Amino-4-carbamoyl-pyrazole Compounds as Selective and Irreversible t790m over wt-egfr Kinase Inhibitors and Use Thereof. WO Patent WO2016008411A12016.
- Kaplaneris N.; Koutoulogenis G.; Raftopoulou M.; Kokotos C. G. 4-Fluoro and 4-hydroxy pyrrolidine-thioxotetrahydropyrimidinones: Organocatalysts for green asymmetric transformations in brine. J. Org. Chem. 2015, 80, 5464–5473. 10.1021/acs.joc.5b00283. [DOI] [PubMed] [Google Scholar]
- Dodge J. A.; Nissen J. S.; Presnell M. A. General Procedure for Mitsunobu Inversion of Sterically Hindered Alcohols: Inversion of Menthol. (1S, 2S, 5R)-5-Methyl-2-(1-Methylethyl) Cyclohexyl 4-Nitrobenzoate: Cyclohexanol, 5-methyl-2-(1-methylethyl)-, 4-nitrobenzoate,[1S-(1α, 2α, 5β)]. Org. Synth. 2003, 73, 110 10.1002/0471264180.os073.11. [DOI] [Google Scholar]
- Batesky D. C.; Goldfogel M. J.; Weix D. J. Removal of triphenylphosphine oxide by precipitation with zinc chloride in polar solvents. J. Org. Chem. 2017, 82, 9931–9936. 10.1021/acs.joc.7b00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukin K.; Kishore V.; Gordon T. Development of a Scalable Synthesis of Oxadiazole Based S1P1 Receptor Agonists. Org. Process Res. Dev. 2013, 17, 666–671. 10.1021/op300345v. [DOI] [Google Scholar]
- Isola A. M.; Holman N. J.; Tometzki G. B.; Watts J. P.; Koser S.; Klintz R.; Münster P.. Triphenylphosphine Oxide Complex Process. U.S. Patent US6011181A12000.
- Fukumoto T.; Yamamoto A.. (Shin-Etsu Chemical Co., Ltd., Tokyo, Japan). Phosphine Oxide Removal from Compounds Formed by a Wittig Reaction. WO Patent WO5292973A11994.
- Connolly T. J.; Auguscinski W.; Fung P.; Galante R.; Liu W.; McGovern L.; Sebastian A.; Shen X.; Shi X.; Wilk B.; Varsalona R. Development of a pilot-plant-scale synthesis of an alkylated dihydrobenzothiadiazole S, S-dioxide: incorporation of a late-stage Mitsunobu reaction. Org. Process Res. Dev. 2010, 14, 868–877. 10.1021/op100113j. [DOI] [Google Scholar]
- Lipshutz B. H.; Blomgren P. A. Efficient Scavenging of Ph3P and Ph3PO with High-Loading Merrifield Resin. Org. Lett. 2001, 3, 1869–1871. 10.1021/ol0159219. [DOI] [PubMed] [Google Scholar]
- Hu F. H.; Wang L. S.; Cai S. F. Solubilities of triphenylphosphine oxide in selected solvents. J. Chem. Eng. Data 2009, 54, 1382–1384. 10.1021/je800842z. [DOI] [Google Scholar]
- de Koning P. D.; McAndrew D.; Moore R.; Moses I. B.; Boyles D. C.; Kissick K.; Stanchina C. L.; Cuthbertson T.; Kamatani A.; Rahman L.; Rodriguez R. Fit-for-purpose development of the enabling route to crizotinib (PF-02341066). Org. Process Res. Dev. 2011, 15, 1018–1026. 10.1021/op200131n. [DOI] [Google Scholar]
- Cardoso F. S.; Mickle G. E.; da Silva M. A.; Baraldi P. T.; Ferreira F. B. Application of In Situ FTIR for the Preparation of 17-α-Estradiol via Mitsunobu Reaction. Org. Process Res. Dev. 2016, 20, 306–311. 10.1021/acs.oprd.5b00394. [DOI] [Google Scholar]
- Banks A.; Breen G. F.; Caine D.; Carey J. S.; Drake C.; Forth M. A.; Gladwin A.; Guelfi S.; Hayes J. F.; Maragni P.; Morgan D. O.; Oxley P.; Perboni A.; Popkin M. E.; Rawlinson F.; Roux G. Process Development and Scale Up of a Glycine Antagonist. Org. Process Res. Dev. 2009, 13, 1130–1140. 10.1021/op9001824. [DOI] [Google Scholar]
- Etter M. C.; Baures P. W. Triphenylphosphine oxide as a crystallization aid. J. Am. Chem. Soc. 1988, 110, 639–640. 10.1021/ja00210a076. [DOI] [Google Scholar]
- Mukherjee S.; Bordawekar S.; Nere N. Investigation of variable impurity profile from a mitsunobu reaction using insights from kinetic modeling, multi-phase interactions, and computational fluid dynamics. Ind. Eng. Chem. Res. 2016, 55, 4867–4877. 10.1021/acs.iecr.6b00344. [DOI] [Google Scholar]
- Hopes P.; Langer T.; Millard K.; Steven A. Decoration of an α-Resorcylate Nucleus as Part of the Manufacture of a Glucokinase Activator. Org. Process Res. Dev. 2018, 22, 996–1006. 10.1021/acs.oprd.8b00175. [DOI] [Google Scholar]
- Tamboli Y.; Kashid B.; Yadav R. P.; Rafeeq M.; Merwade A. Y. Large-Scale Practical Synthesis of Boc-Protected 4-Fluoro-l-Proline. Org. Process Res. Dev. 2020, 24, 1609–1613. 10.1021/acs.oprd.0c00080. [DOI] [Google Scholar]
- Wüstenberg B.; Müller M. A.; Schütz J.; Wyss A.; Schiefer G.; Litta G.; John M.; Hähnlein W.. Vitamins, 2. Vitamin A (Retinoids). In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley, 2000; pp 1–26. [Google Scholar]
- Del Valle J. R.; Goodman M. Asymmetric hydrogenations for the synthesis of Boc-protected 4-alkylprolinols and prolines. J. Org. Chem. 2003, 68, 3923–3931. 10.1021/jo034214l. [DOI] [PubMed] [Google Scholar]
- Gangakhedkar K.; Diwan F.; Varangaonkar A.; Deo K.. Process for Preparation of Ketolide Compounds. WO Patent WO2012127351A12012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.