

Abstract
Human β defensin type 3 (hBD-3) is a small cationic cysteine-rich peptide. It has a broad spectrum of antimicrobial activities. However, at high concentrations, it also shows hemolytic activity by interrupting red blood cells. To understand the selectivity of hBD-3 disrupting cell membranes, investigating the capability of hBD-3 translocating through different membranes is important. Since hBD-3 in the analogue form in which all three pairs of disulfide bonds are broken has similar antibacterial activities to the wild-type, this project investigates the structure and dynamics of an hBD-3 analogue in monomer, dimer, and tetramer forms through both zwitterionic and negatively charged lipid bilayers using molecular dynamics (MD) simulations. One tetramer structure of hBD-3 was predicted by running all-atom MD simulations on hBD-3 in water at a high concentration, which was found to be stable in water during 400 ns all-atom simulations based on root-mean-squared deviation, root-mean-squared fluctuation, buried surface area, and binding interaction energy calculations. After that, hBD-3 in different forms was placed inside different membranes, and then steered MD simulation was conducted to pull the hBD-3 out of the membrane along the z-direction to generate different configurational windows to set up umbrella-sampling (US) simulations. Because extensive sampling is important to obtain accurate free energy barriers, coarse-grained US MD simulations were performed in each window. Based on the long-term simulation result, membrane thinning was found near hBD-3 in different lipid bilayers and in different hBD-3 oligomer systems. By calculating the root-mean-squared deviation of the z-coordinate of hBD-3 molecules, rotation of the oligomer inside the bilayer and stretching of the oligomer structure along the z-direction were observed. Although reorientation of lipid heads toward the hBD-3 tetramer was observed based on the density profile calculation, the order parameter calculation shows that hBD-3 disrupts 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS) lipids more significantly and makes it less ordered than on 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipids. Calculating the free energy of hBD-3 through different lipid bilayers, it was found that generally hBD-3 encounters a lower energy barrier through negatively charged lipid membranes than the zwitterionic membrane. hBD-3 in different forms needs to overcome a lower energy barrier crossing the combined POPC+POPS bilayer through the POPS leaflet than through the POPC leaflet. Besides that, the potential of mean force result suggests that hBD-3 forms an oligomer translocating negatively charged lipid membranes at a low concentration. This study supplied new insight into the antibacterial mechanism of hBD-3 through different membranes.
1. Introduction
Defensins are cationic cysteine-rich small antimicrobial peptides (AMPs).1 Based on the disulfide bonding pattern, defensins can be classified into α, β, and θ categories. Only α and β defensins are human-related. While the α defensin has six cysteine residues forming three pairs of intramolecular disulfide bonds in the pattern of Cys1-Cys6, Cys2-Cys4, and Cys3-Cys5, the β defensin is in the form of Cys1-Cys5, Cys2-Cys4, and Cys3-Cys6. Human β defensin type 3 (hBD-3) has a charge density of +11, and it is mainly secreted by human epithelial cells, gut, and lungs.2,3 It has multimicrobicidal activities against yeast, fungi, and both Gram-positive and Gram-negative bacteria even at high salt concentrations.4 It has been suggested that hBD-3 kills bacteria by disrupting the bacterial cell membrane, and its antimicrobial activity is not affected by the absence of disulfide bonds present in the native hBD-3.5−8 In addition to killing bacteria and fungi, hBD-3 also exhibits chemotactic activities.9 Thus, in recent years, hBD-3 has attracted a lot of attention from researchers.8,10−12 Up to now, it has been demonstrated that AMPs such as hBD-3 can permeabilize bacterial membranes and cell walls; this ability correlates with their antibacterial effects, although the actual mode of the bacterial killing by AMPs remains an area where there is still a poor level of understanding.13,14 Along with its antimicrobial activities, however, hBD-3 shows hemolytic activity (breakdown of red blood cells) at high concentrations.4 Selective toxicity by killing bacterial pathogens without damaging host tissue is a feature that is crucial for AMPs. Thus, to understand the selectivity of hBD-3 on different cell membranes, it is very important to study hBD-3’s disruption capability through different kinds of lipid membranes, including both model mammalian cell membranes and model bacterial cell membranes. The result may supply useful insight into designing novel therapeutic agents.13 A biological membrane is complex in terms of chemical composition and structure.15 For example, phosphatidylcholines are the main structural lipids in animals and fungi but less common in plants and bacteria. Phosphatidylserine (PS) is the main negatively charged lipid in animals, accounting for 8–15 mol % of all phospholipids in cells. However, phosphatidylglycerol is a minor component of animal membranes, and its concentration is high in plants and bacteria. Thus, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS) bilayers are considered as good models for mammalian cell membranes, but POPG should be a good model for bacterial membranes. However, based on our survey, only limited experimental work has been performed on hBD-3 interacting with different lipid membranes. Lioi et al.10 did tests on hBD-3 disrupting human cell membranes, and they used PS to modulate the charge of the cell membrane. Because electrostatic interaction is the important driving force of hBD-3 interacting with different lipid membranes,9,16 in this project, we used POPC to build a zwitterionic model lipid membrane and POPS and POPC+POPS to build the negatively charged and combined negatively charged membranes, respectively, to represent the model bacterial membranes. Results in this work will be compared with experimental results obtained by Lioi et al.
Because defensins are highly positively charged, forming a dimer or higher-order oligomer can further increase their charge density. Defensins have been found to form a dimer or higher-order oligomers.17−19 Schibli et al.20 predicted the dimer structure of hBD-3 in solvent using the solution nuclear magnetic resonance (NMR) method and found that hBD-3 forms a dimer at a lower concentration than human β defensin type 1 (hBD-1) and type 2 (hBD-2). Besides forming a dimer, hBD-2 has been found to form a tetramer at high concentrations.18 However, the higher-order oligomer structure of hBD-3 is unknown yet.
The molecular dynamics (MD) simulation method has been widely applied to investigate the structure and dynamics of peptide disruption and insertion/translocation through lipid membranes.21−25 The force field applied in equilibrium AMP-lipid simulations is found to influence the free energy result and affect whether the pore formation can be observed during microsecond-long simulations.24 Yeasmin et al. found that hBD-3 forming an associated structure in a dimer compared to a monomer form is energetically favorable crossing the zwitterionic lipid bilayer center.26 Zhao et al. found that the translocation energy barrier of Bac2A-based AMPs depends on the peptide insertion orientation.27 General et al. found that there were different penetration paths for the peptide, which correspond to different translocation free energies.28
To understand the selectivity of hBD-3 on different cell membranes, running MD simulations to predict the structure, dynamics, and translocation free energy of hBD-3 crossing different lipid membranes is necessary. Because extensive sampling is extremely important to obtain accurate free energy barriers, which is only within reach for coarse-grained (CG) models,29 in this project, CG umbrella-sampling (US) simulations using the NAMD program30 were applied on hBD-3 crossing different model membranes. hBD-3 analogues in monomer, dimer, and tetramer forms interacting with three kinds of lipid bilayer systems were set up, including (1) a pure negatively charged lipid bilayer, represented by a POPS bilayer; (2) a pure zwitterionic bilayer, represented by a POPC lipid bilayer; and (3) a combined negatively charged POPS+POPC lipid bilayer, with the POPS on the top leaflet and the POPC lipid on the bottom leaflet, which also represents a bacterial membrane model when crossing the bilayer from the POPS leaflet to the POPC leaflet; while representing a mammalian cell membrane model when crossing the bilayer from the POPC leaflet to the POPS layer. The research group predicted the hBD-3 tetramer structure based on hBD-3 self-assembly in solvent as the input for US simulations. The hBD-3-lipid energy landscapes predicted in combination with the structure analysis result cast new insights into the interaction mechanism of hBD-3 with model bacterial and human red blood cell membranes.
2. Results and Discussion
2.1. Tetramer Structure Prediction
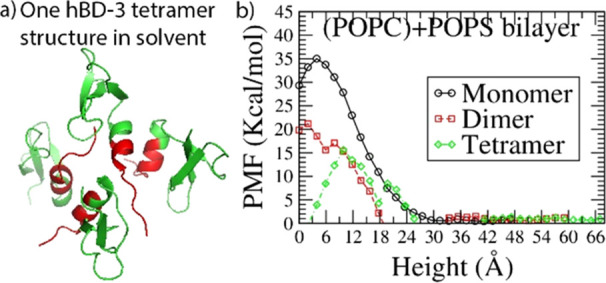
The hBD-3 monomer structure is available online with PDB ID of 1KJ6.20 Its sequence and charge density information is shown in Figure S1 in the Supporting Information. The dimer form of hBD-3 has been predicted previously,31 which is consistent with what Schibli et al.20 found in solution NMR experiments. To predict the higher-order oligomer structure of hBD-3, self-assembly simulation on eight hBD-3 molecules in solvent (with the details shown in the Methods section) was conducted. It was found that four of the hBD-3 molecules approached each other, bound on the head region, and formed a symmetric tetramer inside the box with the structure predicted after 200 ns simulation shown in Figure 1a. The root-mean-squared deviation (RMSD) was calculated based on the backbone Ca atom coordinates of the tetramer and its individual unit (after aligning the trajectory on the original ordered structure) after aligning on the initial structure. The tetramer structure was stable with an increase of RMSD by 1 Å during 200–400 ns simulation (as shown in Figure S2a). The root-mean-squared fluctuations (RMSFs) calculated based on the backbone Ca atoms of each unit are quite similar to each other (Figure 1b). The head and loop regions of each hBD-3 unit have more structural fluctuation than other parts of the peptide. As a comparison, the tetramer structures at 200 and 400 ns are shown in Figure S2b in the Supporting Information. Again, the head and loop regions show more deviation than other parts. These help to explain the small increase in RMSD, which should come from the structural change in the loop and head regions. In addition to that, the buried surface area (BSA) in the tetramer was calculated, as shown in Figure 1c. In the last 200 ns, the tetramer becomes more stable by increasing BSA slightly and with small fluctuations. Calculating the binding interaction energy of the tetramer, the result is shown in Figure 1d. The binding interaction energy decreases in the first 200 ns, and then in the 200–400 ns simulation, the binding interaction energy becomes more negative with less fluctuation; thus, the binding structure becomes more stable. Because the interaction between protein and solvent is important, the number of hydrogen bonds formed between the tetramer and water was calculated with the result shown in Figure 1e. Averagely 130 water molecules form hydrogen bonds with the tetramer in the 200–400 ns of the simulation. The distribution of water molecules and counter-ions in the tetramer system is also stable and even as shown in Figure S3a,b. These prove that the settings and simulations conducted on the tetramer system are reliable. Because we predicted the tetramer structure to supply input for the US simulation, the tetramer structure after 200 ns simulation (shown in Figure 1a) was used in the following US simulation.
Figure 1.

hBD-3 tetramer structure (a) in topview predicted from all-atom MD simulations using the program NAMD, with hBD-3 molecules shown in green and the head 10 residues highlighted in red, and (b) RMSF of residues on four units of the hBD-3 tetramer, (c) BSA of the hBD-3 tetramer, (d) binding interaction energy of the hBD-3 tetramer, and (e) number of hydrogen bonds formed between water and the tetramer during 400 ns all-atom simulations.
2.2. Initial and Last Structures, Rg, and RMSD Result of the hBD-3 Oligomer in Lipids
To avoid any influence of the initial insertion orientation on the free energy barrier result, the initial insertion orientations of the hBD-3 monomer, dimer, and tetramer in the POPS lipid bilayer in the all-atom model and CG model are shown in Figure 2 (left column and middle column), which always kept one hBD-3 unit in the same orientation, and the proteins were inserted into other lipid bilayers in the same orientation. After conducting microsecond-long CG simulations on the hBD-3 monomer/dimer/tetramer in different lipid bilayers in each window, the last structures in the POPS bilayer are shown in Figure 2 (right column). Compared with the initial structures from all-atom simulations and in CG simulations, structures of hBD-3 in different oligomerization states became less compact at 0 Å windows after 1000 ns CG simulations.
Figure 2.

Comparison of the first and last structures and orientation for the hBD-3 analogue in monomer (a), dimer (b), and tetramer (c) forms inside the POPS lipid bilayer at 0 Å windows in all-atom simulation (first column) and CG simulations (second column and third column). The same orientations are kept for the hBD-3 oligomer in different lipid bilayers. Water and ions are not shown for better visualization of the structure.
Calculating the Rg of protein based on the backbone Ca atom-containing bead (BAS bead) positions over the whole simulation time, the results at 0 Å windows for different systems and in different bilayers are shown in Figure S4 as examples. After 300 ns simulations, all the systems reached the equilibrated state. Calculating the average and the standard deviation of Rg over the sampling run (last 700 ns, while last 1000 ns for the tetramer in the POPC+POPS bilayer), the results in each window are shown in Figure S5. Overall, the Rg of the tetramer is higher than that of the monomer and dimer in different kinds of lipid bilayer systems, and the Rg of hBD-3 in the POPC bilayer is always smaller than that in other POPC+POPS or POPS bilayers. Similarly, the average RMSD and standard deviation of protein in different lipid bilayers were calculated, and the result is shown in Figure S6. Only the tetramer has large structure deviations in all three kinds of lipid bilayers while the structures of the monomer and dimer are relatively stable in different windows.
2.3. Redistribution of Lipid Head Beads toward Protein
Based on the simulation trajectories during the sampling run, the membrane thickness was calculated (with details in the Methods section) from evenly distributed 70,000 (or 100,000 for the tetramer in the POPC+POPS bilayer) frames generated. The membrane thickness result for all nine systems at different distances (d) from the center of mass (COM) of the hBD-3s is shown in Figure 3. The membranes have thinned out near the hBD-3 monomer in the height range of −18 to 18 Å, near the hBD-3 dimer in the height range of −24 to 24 Å, and near the hBD-3 tetramer in the height range of −30 to 30 Å in all three kinds of lipid bilayers. The extent of membrane thinning in the central windows is very close in different membranes, but the hBD-3 dimer caused relatively more significant membrane thinning than the monomer, which has slightly more significant membrane thinning than the tetramer. The reason could be that the hBD-3 dimer structure was restrained but not for the monomer or tetramer. Thus, the hBD-3 dimer structure is relatively more rigid than the other two. The closer the distance d from the lipids to the hBD-3, generally, the more significant the membrane thinning. In the lipid membrane region with a d larger than 30 Å, the membrane thinning disappeared in all the systems. Such a kind of membrane thinning around hBD-3 is consistent with our findings from all-atom simulations.26
Figure 3.

Membrane thickness within different distances d from the hBD-3 monomer in a pure POPC bilayer (a), pure POPS bilayer (b), and combined POPC and POPS bilayer (c); the hBD-3 dimer in the pure POPC bilayer (d), pure POPS bilayer (e), and combined POPC and POPS bilayer (f); and the hBD-3 tetramer in the pure POPC bilayer (g), pure POPS bilayer (h), and combined POPC and POPS bilayer (i) systems. d is the distance from the lipids to the COM of hBD-3s.
Although membrane thinning was observed in all nine kinds of proteins in lipid bilayer systems, the redistribution of lipid heads toward protein was only observed obviously in some of the systems by analyzing the POPC/POPS lipid head bead distribution around the lipid bilayer. The CNO bead is on the head of the POPS lipid, which is negatively charged, while the CHO bead is the head bead of the POPC lipid, which is zwitterionic. The average number density and the number density of CNO at 1000 ns, as well as the CNO distribution for the tetramer in the POPS bilayer at 0 and 1000 ns, are shown in Figure 4a–c. The slight CNO beads redistributed between the top and bottom layers are observed as shown in Figure 4b,c, which is consistent with the small peaks in the number density profile pointed out by the red arrows in the height range of −12 to 12 Å in Figure 4a.
Figure 4.

Number density profile of CNO beads (on POPS lipids) in the hBD-3 tetramer in the POPS system at the 0 Å window at (a) 1000 ns, with the CNO bead number density showing relocation across the bilayers pointed out using red arrows and the CNO bead distribution in the tetramer system at (b) 0 ns and at (c) 1000 ns, with all other molecules not shown.
However, the POPS lipid head only slightly reoriented toward the hBD-3 dimer (as shown by the 1000 ns result in Figure S7), and a negligible amount of POPS lipid reorientation toward the hBD-3 monomer was observed (1000 ns result shown in Figure S8). Similarly, only the POPC lipid head reorientation to the hBD-3 tetramer was observed at 1000 ns. There were only a slight amount of reorientation in the hBD-3 dimer system and a negligible amount in the hBD-3 monomer system (shown in Figures S9–11). The reorientation of the POPC head to the hBD-3 is consistent with findings from the all-atom MD simulation results in ref (26).
Interestingly, in the combined POPC + POPS lipid bilayer, we observed the slight diffusion of CNO beads (shown in blue balls) from the top leaflet to the bottom leaflet (with CHO beads shown in chocolate balls) during 1000 ns CG simulations in the hBD-3 monomer, dimer, and tetramer systems (as shown in Figures S12–S14). This can agree with other researchers’ observations that lipid transport occurs at a microsecond-long trajectory using both experimental and simulation methods.32−34
2.4. hBD-3 Oligomer Stretching and Possible Rotation inside the Lipid Membranes
Outputting the structures of the hBD-3 monomer, dimer, and tetramer in the POPS lipid bilayer at 1000 ns and 0 Å windows and comparing them with those at 0 ns and 0 Å windows, the result is shown in Figure 2 (right column). The initial structures of the hBD-3 monomer/dimer/tetramer look more compact than their last structures, correspondingly. The output from the last hBD-3 dimer structures in seven different windows at 60 Å, 48 Å, 24 Å, 0 Å, −24 Å, −48 Å, and −60 Å in the POPS lipid bilayer is shown in Figure S15a, and the hBD-3 tetramer structures in seven different windows at 68 Å, 48 Å, 24 Å, 0 Å, −24 Å, −48 Å, and −68 Å in the POPS lipid bilayer in Figure S15b in the Supporting Information. Figure S15a,b shows that the shape of the hBD-3 dimer and tetramer changed in the POPS bilayer at different heights.
Calculating the average COM distances between hBD-3 units in the dimer and tetramer systems in different lipid bilayers, the result is shown in Figure 5a for the dimer, while in Figure 5b for the tetramer.
Figure 5.

COM distances between the hBD-3 analogue units in the dimer (a) and tetrameric forms (b) and root-mean-squared deviation of the z-coordinate (RMSZ) of the hBD-3 dimer (c) and tetramer (d) in pure POPC, pure POPS, and combined POPC (negative height) + POPS (positive height) systems.
The average distance between the hBD-3 units in the dimer does not change significantly along the height in three different types of lipid bilayers as shown in Figure 5. However, the distance increases when the dimer is in the height range of 36 Å ∼54 Å and −36 Å ∼ −54 Å and decreases until totally outside the pure POPS and mixed POPS and POPC lipid bilayer, which is in the height range of 54 Å ∼60 Å and −54 Å ∼ −60 Å. Such kind of unit distance decrease occurred in the range of 40 ∼48 Å and −40∼ −48 Å in the pure POPC lipid bilayer. This may be due to the strong electrostatic interaction of POPS and hBD-3, which pulls the hBD-3 toward the bilayer surface. However, the average distance between six pairs of hBD-3s in the tetramer is slightly higher at the center of the lipid bilayer compared to that outside in water except dissociation, with the longest distance being at the lipid–solvent interface, which is at around ±24 Å. This suggests the shifting of hBD-3 COM from each other for the tetramer inside the POPS bilayer. Dissociation of the tetramer outside of the POPS lipid bilayer was observed occasionally in some windows as shown in Figure 5b and Figure S15b. During 1000 ns simulations, the hBD-3 units shifted from each other to interact with the negatively charged POPS heads.
To quantitatively describe the rotation orientation along the z-direction (normal to lipid bilayer) and the relative shifting of the COM of each hBD-3 unit from the COM of the dimer/tetramer, a new property RMSZ is defined in this project. RMSZ represents root-mean-squared deviation of the z-coordinate of each hBD-3 unit COM from that of the whole hBD-3 dimer or tetramer. The RMSZ is calculated using eq 1
| 1 |
where N is the number of hBD-3 units in the oligomer. N is 2 for the dimer and 4 for the tetramer. Zi is the z-coordinate of the COM of each hBD-3 unit, and Zo is the z-coordinate of the COM of the hBD-3 oligomer. As shown in Figure S16, RMSZ is zero if all the units of the oligomer stay in the same height, and the larger the value of RMSZ, the more the scattering of units in the z-direction, or the more the rotation of the oligomer along the z-direction.
The RMSZ of the dimer and tetramer in different bilayers in sampling runs is shown in Figure S17 in the Supporting Information. The RMSZ of the hBD-3 dimer and tetramer in the POPS bilayer calculated is shown in Figure 5c,d. In all the lipid bilayer systems, hBD-3 units in the dimer and tetramer stay almost parallel to the x-y plane in the water–lipid interface regions (from −36 to −24 Å and from 24 to 36 Å) and in the water phase (54 Å ∼66 Å and −54 Å ∼ −66 Å) but are scattered in the water phase at positions not so far away from the lipid bilayer (−36 Å ∼ −54 Å and 36 Å ∼54 Å) and are moderately scattered or rotated inside the lipid bilayer. At the membrane center, the RMSZ result for different hBD-3 oligomers shows no dependence on the lipid membrane types in the range of 0 to 8–10 Å for the dimer and 0 to 10–15 Å for the tetramer. At the water–lipid interface, the units stay attached to the lipid surface because of the strong electrostatic interactions between positively charged hBD-3s and negatively charged POPS lipid head groups; thus, the RMSZ is small. However inside the lipid bilayer, the electrostatic interaction between the hBD-3 analogue and lipid head groups triggered the rotation of the hBD-3 dimer/tetramer and stretching of the hBD-3 units; thus, the RMSZ increases. In the solvent, water has a strong electrostatic interaction with the dimer/tetramer, which also can trigger the stretching and rotation of the hBD-3 oligomer. These can agree with the last structure result at the 0 Å window shown in Figure 2 (right column), and the result at the 0 Å window in Figure S18 in the POPC lipid bilayer system and in Figure S19 in the POPS + POPC bilayer. Also, because of the stronger interaction between the POPS lipid heads and the hBD-3 analogue, the stretching of the hBD-3 oligomer is slightly stronger when the dimer or the tetramer is out of the POPS or POPC+POPS lipid bilayer as shown in Figure 2 and Figures S18 and S19.
hBD-3 can form a symmetric dimer in solvent. Human α-defensin HNP-1 was found to form a dimer pore in the membrane.35 Also, reparameterizing the prediction from the SymmDock program36 has been applied to predict the symmetric oligomer structure of an insect defensin Sapecin and hBD-2 in the lipid membrane, which are close to the experimental result. Thus, SymmDock was applied to predict the possible symmetric oligomer structure of hBD-3 in the lipid membrane based on the symmetric hBD-3 dimer structure available.31 The smallest oligomer, an octamer of hBD-3, was predicted as the topview and sideview structures shown in Figure S20a,c. The octamer is formed by 4 hBD-3 dimers, with each dimer forming contacts in the β2 sheet region and aligned in a top-down direction inside the membrane. The orientation and structure of this octamer in the lipid bilayer in the topview and sideview are shown in Figure S20b,d respectively. Interestingly, the dimer alignment inside lipids agrees with the hBD-3 dimer inside the POPS lipid bilayer, which also forms a top-bottom alignment after microsecond-long simulation as shown in Figure 2(b, right), which is different from the initial insertion orientation.
2.5. Free Energy Result
Based on the sampling simulation trajectories in each window, the translocation free energy of hBD-3 analogues in different oligomerization statuses was calculated, with the average potential of mean force (PMF) result through both leaflets of the POPC lipid bilayers shown in Figure 6a, through the POPS lipid bilayer shown in Figure 6b, and PMF result through the POPC side of the mixed lipid bilayer shown in Figure 6c and through the POPS side of the mixed lipid bilayer in Figure 6d.
Figure 6.
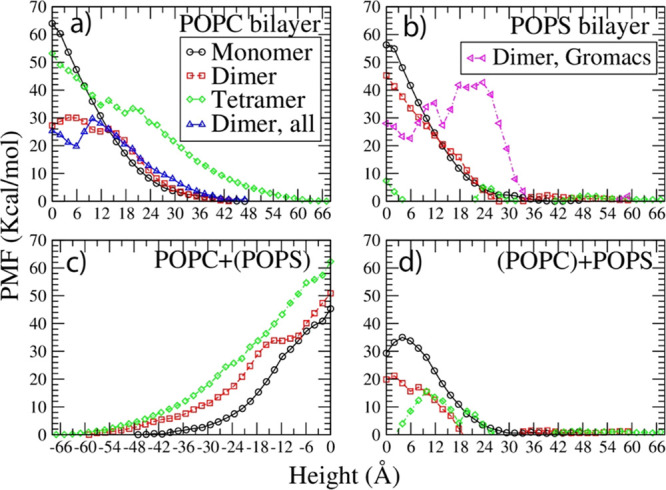
Translocation free energy result of hBD-3 analogues in monomer, dimer, and tetramer forms through POPC (a), POPS (b), and combined POPC (negative height) + POPS (positive height) lipid bilayers in (c) and (d).
Figure 6 shows that hBD-3 analogues in the monomer and dimer forms have to cross a positive energy barrier through the POPC bilayer, the POPS bilayer, and the POPC+POPS lipid bilayer. hBD-3 analogues in the tetramer form need to overcome a high-energy barrier in the POPC bilayer and the POPC+(POPS) side, but a very low energy barrier in the POPS bilayer or the (POPC) + POPS side. Moreover, crossing the POPS+POPC mixed lipid bilayer through the POPS side is slightly more energy favorable compared to through the POPC side for all three forms.
Figure 6a shows that the hBD-3 monomer, dimer, and tetramer need to overcome a high-energy barrier to cross the pure POPC bilayer. Comparing the PMF of the hBD-3 dimer in analogue form through the pure POPC bilayer from the CG US simulation prediction with the result from the all-atom MD simulation results in our previous work26 (shown in blue triangles in Figure 6a), very consistent agreement was reached. This proves that the simulation setup and methodology in the current project are reasonable.
Figure 6b shows the PMF profiles for the hBD-3 monomer, dimer, and tetramer crossing the POPS bilayers. Because of the electrostatic interaction between positively charged hBD-3 and negatively charged POPS lipids, the translocation energy barrier decreases as the oligomerization order of the hBD-3 analogue increases in the order of the hBD-3 monomer, hBD-3 dimer, and hBD-3 tetramer. The hBD-3 monomer needs to overcome the highest energy barrier to cross the POPS bilayer, while the hBD-3 tetramer overcomes the lowest energy barrier. Comparing the PMF result on the hBD-3 dimer through the POPS lipid bilayer using the CG method and standard MARTINI forcefield (shown in red squares in Figure 6b) with the prediction using the CG method and polarizable water model in combination with MARTINI forcefield (shown in magenta triangles in Figure 6b), an energy barrier with a similar height was predicted, despite the difference in the PMF shape. This proves that the settings and prediction from the current project are reasonable.
Similarly, the PMF of the hBD-3 in different forms in the combined lipid bilayers (POPS lipid on the top and POPC on the bottom layer) was analyzed with results shown in Figure 6c,d. hBD-3 needs to overcome a similar energy barrier crossing the combined lipid bilayer through the POPC leaflet in the monomer, dimer, and tetramer forms. However, it is easier to pass the combined lipid bilayer through the POPS leaflet in the tetramer form, than in the dimer form, than the monomer form because the higher the oligomer state, the stronger the electrostatic interaction between the hBD-3 analogue and the POPS bilayer.
When comparing the PMF results for the hBD-3 in different lipid bilayers, the result for the hBD-3 in the monomer form is shown in Figure S21a,b, in the dimer form in Figure S21c,d, and in the tetramer form in Figure S21e,f. The free energy barrier through different membranes, thus the cell selectivity, shows dependence on the oligomerization state of hBD-3; the higher the oligomerization order, the stronger the selectivity on the cell membrane. The cell selectivity is the most significant on the hBD-3 tetramer.
3. Discussion
3.1. Translocation Structure Information
In this project, an hBD-3 tetramer structure in solvent was predicted, and the structure was found to be stable in 400 ns all-atom simulations. Without imposing any restraint on the tetramer structure, the hBD-3 tetramer in the analogue form had dissociation occasionally inside different lipid bilayers during the long-term CG US simulations (as shown in Figure 5b). It was observed that the stretching and possible rotation of the COM of the hBD-3 analogue units in the dimer and tetramer forms inside the POPS lipid membrane were similar to those in the POPC membrane (as shown in Figures S15a,b and S18), which is slightly more significant than in the solvent phase except occasional dissociation. This demonstrates the effect of the media on the protein structure stability. To further prove that, we have put the tetramer (without any restraint on the tetramer structure) on the surface of the POPC bilayer and the POPS bilayer separately and ran all-atom MD simulations on both systems. After 100 ns all-atom MD simulations on each system, it was found that the tetramer did not bind with the POPC bilayer stably but dissociated after coming in contact with the lipid membrane surface. Thus, a sharp increase of the tetramer RMSD was observed (as the result shown in Figure S22a), which is also confirmed by the dissociated last structure of the tetramer in the POPC membrane system and a significant increase of the COM distance from the tetramer to the membrane upper surface (shown in Figure S22c,e). On the other hand, the hBD-3 tetramer can bind with the POPS bilayer, and the tetramer had structure deviation; gradually thus, the RMSD of the tetramer increased with time as shown in Figure S22b, and the tetramer dissociates into two dimers and sticks on the POPS membrane as the last structure shown in Figure S22d, and the COM distance result is shown in Figure S22f. In addition to that, the distance between unit pair COM in the tetramer was also calculated as shown in Figure S22g,h, which proves that the tetramer dissociated although units PROA and PROB and units PROB and PROD associated in the POPC bilayer system, while units PROA and PROC and units PROB and PROD associated in the POPS bilayer system. These prove that the interaction between the protein and media contributed to the structural change of the tetramer and dimer during simulations.
Because water and counter-ions are important molecules interacting with the protein and lipid membrane in the US simulation systems and in the tetramer in solvent all-atom simulation, their distributions around the protein and lipid membrane were analyzed and compared with those from the tetramer in the solvent system. The results for the CG systems at the 0 Å window are shown in Figure S23 for water, and in Figure S24 for ions, while in Figure S3a,b for the tetramer in all-atom systems. As can be seen, in the all-atom simulation systems, water and ions distributed evenly along the z-axis. The mass density of water is around 1.0 g/mL. In CG systems, water molecules distribute evenly outside of the lipid bilayer in different systems at the 0 Å windows. The counter-ions distribution is influenced by the charge of the lipid membrane. In the POPS+POPC combined bilayer, it has the POPS on the top leaflet and the POPC lipid on the bottom leaflet. Because POPS is negatively charged while POPC is zwitterionic, because of the attractive interaction between positively charged ions and negatively charged POPS, there are more ions around the POPS leaflet than around the POPC leaflet in monomer, dimer, and tetramer systems. In the pure POPS or pure POPC bilayers, almost symmetric distribution of ions around both leaflets was observed. These prove that the lipid membrane supplies a totally different environment from water, and the water and ions distribution in both tetramer-solvent and tetramer-membrane systems is reasonable.
The last structures of the hBD-3 dimer and tetramer in the POPS bilayer in the 0 Å window after microsecond-long CG simulation are shown in Figure 7a,b. The rotation of the hBD-3 dimer to align parallel to the membrane surface normal direction can agree with the SymmDock prediction.36 The tetramer structure is also quite different from the original symmetric tetramer structure predicted in solvent.
Figure 7.
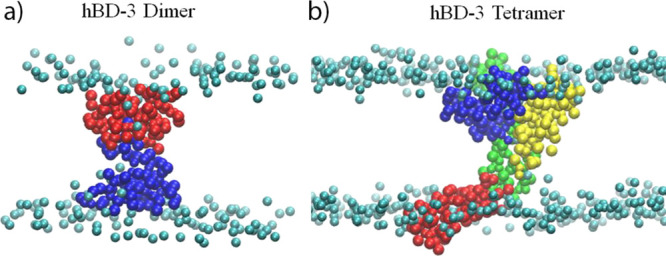
Last structures of the hBD-3 dimer (a) and tetramer (b) in the POPS bilayer system in the 0 Å window after microsecond-long simulations. Only protein and the head beads of POPS lipids are shown.
Analyzing the POPC and POPS lipid head distribution around the hBD-3, reorientation of both kinds of lipid heads toward the hBD-3 tetramer was observed, although only slightly in the dimer or monomer system. The reason should be that the hBD-3 tetramer has a much higher charge density than the dimer and monomer, thus causing more lipid head reorientation. Although membrane thinning can be observed in different systems for the hBD-3 in different forms (as shown in Figure 3), only when the hBD-3 forms a higher-order oligomer (the oligomerization order is 4), can the lipid reorientation be observable in CG simulations. The result proves that lipid reorientation to the hBD-3 oligomer is the common phenomenon during the membrane translocation process because of the interaction of hBD-3 and lipid heads. Despite the slight reorientation of POPC heads to hBD-3 in US simulations, interestingly the order parameter of POPC lipids in the hBD-3 dimer wild-type in the POPC bilayer all-atom simulation system in the 0 Å window did not decrease (even increased) compared to the pure POPC lipids (with the result shown in Figure S25a,b). Thus, hBD-3 embedded at the lipid bilayer center only caused limited disruption on POPC lipids. When calculating the order parameter of POPS lipids in the hBD-3 dimer system in the 0 Å window based on Gromacs CG simulation prediction, significant disruption of the hBD-3 dimer on POPS lipids was observed after microsecond-long simulation, compared with the order parameters of POPS in the pure lipid bilayer system as shown in Figure S25c,d. This proves that positively charged hBD-3 can disrupt negatively charged POPS lipids significantly during the translocation although not POPC lipids. Because the tetramer has an even higher charge density, it is expected that such kind of disruption on the POPS membrane should be even stronger.
3.2. Translocation Free Energy Barrier Information
In this project, we predicted the PMF of the hBD-3 through both zwitterionic lipid membranes (i.e., mammalian cell membrane) represented by the POPC lipid bilayer and negatively charged lipid membranes (i.e., bacterial cell membrane) represented by the POPS lipid bilayer. Moreover, we also predicted the PMF of the hBD-3 through a combined lipid bilayer, which has POPS lipids on one leaflet and POPC lipids on the other leaflet. Because a mammalian cell membrane is overall neutrally charged or has a negatively charged group on the inner layer, crossing the combined bilayer from the POPC side can represent the hBD-3 crossing a mammalian cell membrane. In that instance, we found a positive energy barrier for the hBD-3 in dimer/tetramer/monomer forms crossing the combined membrane, which is similar to that through the pure POPC lipid bilayer case as shown in Figure 6. The tetramer is supposed to overcome a lower energy barrier than the monomer because it can form an association during the translocation and also because the tetramer system has a higher protein-to-lipid ratio than the monomer system. However interestingly, Figure 6c shows that the tetramer even needs to overcome a higher energy barrier than the monomer to cross the combined lipid bilayer from the POPC side. On the other hand, translocating through the combined lipid bilayer from the POPS side can represent disruption on a bacterial lipid membrane, and we found that a lower energy barrier than the pure POPS lipid bilayer is required for the hBD-3s, in the order of tetramer, dimer, and monomer. Because of that, the energy barrier difference for the combined lipid bilayer from the POPC leaflet side and the POPS leaflet side became more energy favorable for the tetramer to cross the bacterial membrane from the POPS side, than for the dimer and the least for the monomer. This supports a stronger cell selectivity on the combined bilayer. Because the real cell membrane is a mixture of different lipids, the result from the combined bilayer membrane system suggests that hBD-3 can have a stronger cell selectivity on the real mammalian and bacterial cell membranes than represented by the pure POPC or pure POPS bilayer.
The AMP-to-lipid number ratio has been found to affect the disruption capability of the AMP on the membrane. During the melittin disrupting dipalmitoylphosphatidylcholine bilayer, the transmembrane water pores form spontaneously when above a critical peptide-to-lipid ratio in MD simulations running all-atom Gromacs simulations using GROMOS forcefield.37 Melittin has been found to induce the formation of 25–30 Å diameter pores in POPC vesicles at a peptide/lipid molar ratio of 1:50 using the leakage of coencapsulated marker experimental method.38 At a peptide-to-lipid molar ratio of 1:117, nine pores with a lifetime of 40 ps open per second per lipid vesicle (composed of egg yolk phosphatidylglycerol) in the initial phase were predicted for magainin2 (an AMP) using an experimental method by the efflux of a fluorescent dye (calcein).39 With the peptide/lipid ratio increase, the membrane disruption capability of magainin2 increases sharply. Calculating the peptide/lipid ratio in hBD-3 systems as data shown in Table S2, the dimer system has the highest peptide-to-lipid number ratio (1:100 to 1:107 in three different bilayer systems), then the next is the tetramer system (1:150 to 1:160), and the lowest is the monomer system (1:200 to 1:214). Thus, it is not surprising to see that the hBD-3 dimer needs to cross a lower energy barrier than the tetramer and the monomer through the POPC bilayer as shown in Figure 6a. Because the peptide-to-lipid ratio is very low in the current systems worked on, we do not expect to see any water pore formation in any of our systems. This agrees with our findings from the water number density calculation with results shown in Figures S26–27. Calculating the water density profile in all-atom and CG simulations, only a negligible amount of water stays in the 0 Å window in the hBD-3 dimer through the POPC bilayer using all-atom MD simulations (the trajectory is from our previous work published in ref (26)), but no water in the CG simulations was detected, no matter using a polarizable water model or not. Hence, it is not surprising to see a positive hBD-3 translocation energy barrier through different lipid bilayers in the dimer, tetramer, and monomer forms.
In addition to that, forming oligomerization also plays a role as suggested in our previous work.26 In negatively charged bilayers such as the POPS bilayer or POPS side+(POPC) bilayer as shown in Figure 6b,d, the free energy barrier decreases in the same order that the oligomerization order increases. This suggests that crowding on the lipid membrane and forming oligomerization are important for small AMPs translocating the bacterial lipid membrane.40 Forming an oligomer is a prerequisite for the hBD-3 to cross the negatively charged lipid membranes, either prior to or after binding to the membrane surface, considering the tetramer structure change outside and inside the membrane as shown in Figure S15b and the low concentration of hBD-3 on negatively charged lipid membrane systems in this work.
Experimentally, hBD-3 in a very low concentration can disrupt the bacterial membrane, but only at high concentrations it can disrupt the zwitterionic lipid membrane.11 Because hBD-3 can form a dimer even at a low concentration, and possibly also form a higher-order cluster around the bacterial lipid membrane, it can disrupt the bacterial membrane severely.
hBD-3 shows hemolytic activity (breakdown of red blood cells) at high concentrations.4 In this project, the hBD-3 dimer system has the highest hBD-3/lipid ratio, thus the highest concentration. However, the hBD-3 dimer needs to encounter a high positive energy barrier through the POPC bilayer and through the POPC + (POPS) bilayer in the height range studied. Thus, at an hBD-3-to-lipid number ratio of 1:100 or 1:107, hBD-3 possesses no hemolytic activity (crossing the mammalian cell membrane) at all.
3.3. Comparison with Experimental Work and Related
Lioi et al. carried out experiments on the disruption of hBD-3 on human monocyte membranes and PS-modified cell membranes.10 They found that a small amount of PS expressed on the outer side of the human cell membrane can increase its susceptibility to the hBD-3 attack. In this work, the combined POPS + POPC bilayer can best represent the cell membrane with PS expressed on the outer side. Based on our prediction, hBD-3 needs to cross a much lower energy barrier through such kind of membrane from the POPS leaflet than through the zwitterionic membrane (POPC bilayer) or from the POPC leaflet. This agrees with findings of Lioi et al. from experiments, and our MD simulation result shed light on the hBD-3 membrane selectivity.
In this project, to set up the US simulations on a tetramer translocation through different membranes, a tetramer structure was predicted using the MD simulation method, because there is no program available to predict the tetramer structure without knowing possible binding sites between units in the tetramer. In 400 ns all-atom MD simulations, the tetramer formed on the head of the hBD-3 wild-type and became stable especially in the second 200 ns simulation based on RMSD, RMSF, BSA, and binding interaction energy results. This structure may not be the most stable tetramer structure for hBD-3, which can be comparable to that using the experimental method. However, it is good enough to function as the initial input for the US simulation in this project. More work will be performed in the future to predict the higher-order oligomer structures of human defensins.
4. Conclusions
In this project, the structure, dynamics, and free energy of the hBD-3 analogue in monomer, dimer, and tetramer forms through anionic POPS, zwitterionic POPC, and combined POPC + POPS lipid bilayers are investigated using CG MD simulations. Lipids around the hBD-3s tend to reorient toward the hBD-3, which causes membrane thinning near the peptide. hBD-3 disrupts POPS lipids significantly, but not on POPC lipids. The stretching of hBD-3 units over the bilayer and slight rotation of the hBD-3 dimer and tetramer inside the lipid bilayer were observed from the RMSZ result. The free energy results suggest that it is more energetically favorable for the hBD-3 to cross the bacterial-membrane-representing lipid bilayers than through the mammalian-membrane-representing lipid bilayers. It is suggested that the protein-to-lipid ratio plays a more important role in the hBD-3 free energy barrier crossing the zwitterionic bilayer; the higher the protein-to-lipid ratio, the lower the energy barrier, while the oligomerization state of hBD-3 plays a role in determining the translocation free energy barrier through the negatively charged bilayer. The higher the oligomerization order, the lower the free energy barrier. Because, experimentally, hBD-3 can transpass the bacterial cell membrane, the structural analysis result in this project suggests that forming a cluster structure will be energetically favorable for the hBD-3 to cross the bacterial membrane at a low concentration.
5. Methods
5.1. hBD-3 Tetramer Structure Preparation
To find out the higher-order oligomer structure of hBD-3, eight hBD-3 molecules were placed inside a rectangular water box with the closest distance between atoms on hBD-3 to the water box edge being no less than 12 Å and the closest distance between atoms on different hBD-3 molecules being at least 8 Å. After that, counter-ions (used to neutralize the system) and 0.15 M of NaCl were added. In total, 20,821 TIP3P water molecules, 59 SOD, and 147 CLA ions were implemented in the box. The details of the simulation are shown in Table S1 in the Supporting Information. The initial simulation system snapshot is shown in Figure S29 in the Supporting Information. After a brief energy minimization using the conjugate gradient method, all-atom MD simulations using the program NAMD version 2.12 and CHARMM36 forcefield41,42 have been performed at a desired temperature of 300 K and pressure of 1 atm (NPT ensemble) to watch the movements of hBD-3 inside the simulation box. A modified Nosé–Hoover method was applied to control the pressure while the temperature was controlled by Langevin dynamics.43,44 A time step of 2 fs and periodic boundary conditions were applied. The nonbond cutoff is 12 Å, the switch distance is 10 Å, and the pair list distance is 14 Å. The long-range electrostatic interaction was calculated using the particle mesh Ewald (PME) method.
The RMSD of the tetramer and its units was calculated using the VMD program45 after aligning the trajectory on the initial tetramer/unit structure. The RMSF of each unit of the tetramer was calculated using the VMD program and a tcl code. The BSA for the tetramer was calculated in two steps using the vmd program and tcl code using Richards and Lee’s method with a water probe size of 1.4 Å.46 First, the total solvent accessible surface area of the tetramer (ASAtetramer) was calculated based on the tetramer’s trajectory. Second, the accessible surface area of each unit in the tetramer (ASAunit1, ASAunit2, ASAunit3, and ASAunit4) was calculated individually. Then, the BSA was calculated using eq 2:
| 2 |
Furthermore, based on the 400 ns MD simulation trajectories of the tetramer in solvent, the total pairwise interaction energy was calculated using the MM-GBSA method47 by applying NAMD and the NAMD energy plugin of the VMD program.45 This interaction energy (Ebinding) is calculated using eq 3
| 3 |
Etetramer is the potential energy of the tetramer, and Eunit1,Eunit2,Eunit3, and Eunit4 are the potential energy of the 1st, 2nd, 3rd, and 4th unit in the tetramer. < > is the ensemble average over simulation time. In the MM-GBSA method, the solvent effect was counted using the generalized Born implicit solvent model (GBIS).48
5.2. Initial All-Atom and CG System Setup
Because hBD-3 wild-type and analogue forms have similar antibacterial activities, in this project, hBD-3 analogues in three kinds of oligomerization forms were studied: monomer, dimer, and tetramer. In total, nine all-atom MD systems were set up using the CHARMM-GUI program49−51 by inserting the hBD-3 analogues at the center of the POPS lipid bilayer, POPC bilayer, and POPS (top leaflet) + POPC(bottom leaflet) bilayer. The initial insertion orientation of the hBD-3 monomer, dimer, and tetramer is shown in Figure 2 (left column), which always kept one hBD-3 unit in the same orientation, and the proteins were inserted into other lipid bilayers in the same orientation. No protonation on glutamate residues was applied in the CHARMM-GUI setup steps. The details of the system setup are shown in Table S1 in the Supporting Information.
After setting up nine all-atom MD systems using the CHARMM-GUI program, the systems were directly converted into the CG systems using a residue-based coarse-grained (RBCG) method51−53 implemented via the CGTools plugin of VMD.45 CG water molecules were added into monomer/dimer/tetramer systems to make sure that there was enough water above and below the protein even when they were totally outside of the lipid bilayers, with water thickness above/below lipid bilayer information shown in Table S2 and box size information shown in Table S3. The initial insertion orientation of the hBD-3 monomer, dimer, and tetramer in the CG simulations is shown in Figure 2 (middle column).
To check the stability of the tetramer interacting with the lipid membranes, two more all-atom simulations were set up using the CHARMM-GUI program by placing one tetramer above the POPC and POPS bilayers separately by at least 15 Å and in the same orientation as above to set up the initial simulation systems. In addition to that, two pure lipid bilayer systems were set up using the CHARMM-GUI program as well: one is a pure POPC lipid bilayer and the other is a pure POPS lipid bilayer. The details of simulation systems are shown in Table S1 in the Supporting Information. The CHARMM36-modified forcefield was applied. The temperature was set at 310.15 K to make sure that the different lipid membranes stay in the lamellar fluid phase throughout the simulation. After that, NAMD all-atom MD simulations ver 2.12 were performed for at least 100 ns on each system in an NPZAT ensemble so that the membrane surface area in the x-y plane remained unaltered, and the pressure normal to the bilayer was held fixed, which can allow the z-axis to expand and contract to achieve a constant Pz. Electrostatic interactions were calculated using the PME method54 with a real space cutoff of 12 Å, and the same cutoff was applied for the Lennard–Jones interaction calculations. The SHAKE algorithm was applied to control the lengths of all bonds involving hydrogens. The integration time step was 2 fs with a trajectory output frequency of 100 ps.
5.3. Steered Molecular Dynamics (SMD) Simulation and US
To generate multiple configurations along the z-axis direction for the US simulations, the protein was pulled up from the center of the membrane to the outside of the simulation box along the bilayer normal and also pulled down to the outside of the bilayer using SMD. Constant forces of 0.05, 0.1, and 0.15 kcal/(mol·Å) were applied in the z-direction (the normal of the lipid bilayer) on the BAS beads, which contain backbone CA atoms of the hBD-3 monomer, dimer, and tetramer, respectively. hBD-3 units have been secured together in the dimer structure throughout the SMD and US simulations by restraining the distance between two hydrogen-bond-forming residues (Q29) using a force constant of 10 kcal/(mol·Å2) because it is known that hBD-3 forms a symmetric dimer structure with the hydrogen bonds formed by the Q29 residue pair.20 No restraints were set up for the hBD-3 tetramer because the tetramer structure is stable in solvent, and microsecond-long simulation can help to check the stability of the tetramer structure in the lipid membrane. Different windows were prepared based on the height-distance from the COM of the protein to the COM of the lipid bilayer at an interval of 2 Å. To include the configurations of the hBD-3 oligomer staying totally outside of the lipid bilayer, the height (distance) range set up for the tetramer system is from −68 to 68 Å, while from −60 to 60 Å for the dimer systems and – 48 to 48 Å for the monomer systems. The details of different systems are shown in Table S2.
MD simulations of CG systems were performed with NAMD 2.12 and the modified MARTINI force field52 at 310.15 K and 1 atm using an NPT ensemble. Each window was energy-minimized and equilibrated for around 20 ns with restraints on the protein height from the lipid bilayer mass center using a force constant of 3 kcal/ (mol·Å2). Nonbonded interactions were calculated using a cutoff of 12 Å, with shifting through the interaction range for electrostatic interaction and shifting beginning at 9 Å for Lennard–Jones interaction. The dielectric constant was set to 15.0. The pair list distance was updated at least once per 10 steps and with a 14 Å pair list cutoff. In all the systems, Langevin dynamics was used with a damping coefficient of 1 ps–1 to maintain constant temperature. A constant pressure of 1 atm is maintained with a Nosé–Hoover Langevin piston, using a piston period of 2000 fs and a decay time of 1000 fs. A time step of 10 fs was used. The deviation of distance between the COM of the protein and lipid bilayer in different windows was extracted from the simulation output, and results were combined using the 0.93 edition of the Weighted Histogram Method (WHAM) program55 with a tolerance of 0.00001. Equation 4 was used in the WHAM to calculate the free energy
| 4 |
where k is the force constant, x is the height of protein, and xo is the target height of the window, i.e., desired distance between the COM of the protein to the COM of the lipid bilayer in the window.
5.4. RMSD, Membrane Thickness, Rg, Order Parameter, Free Energy Calculation, and Structure Analysis
RMSD from CG simulation was calculated based on the BAS bead coordinates with time after aligning the trajectory on the initial structure. The radius of gyration (Rg) was calculated based on the positions of the BAS bead, which contains backbone CA atoms with time.
The membrane thickness was calculated based on the distance between CNO/CHO beads on the head of the POPS/POPC lipids on the top leaflet and on the bottom leaflet as the sketch shown in Figure S28 in the Supporting Information. The RMSD and Rg of protein were also calculated based on the BAS bead (which includes the CA atoms in each residue) positions. The density profile was calculated using the density plugin of the VMD program56 based on bead positions in CG simulations.
The order parameters of the POPC and POPS lipids in both pure membrane and hBD-3 systems based on all-atom simulations were calculated using the CHARMM program with eq 5:
| 5 |
Here, SCH is the lipid acyl chain order parameters, and θ is the angle between the C–H bond vector on the lipid acyl chain and the bilayer normal (typically, the z-axis in a membrane simulation). The angular brackets represent molecular and temporal ensemble averages.
To calculate the order parameter of the POPS lipids from Gromacs CG simulations, the last structure of the protein in the lipid membrane in the 0 Å window was generated from Gromacs simulation and then converted into all-atom simulation using the CHARMM-GUI program. Based on 11 ns all-atom MD simulation trajectories, the order parameter of POPS lipids was calculated using the CHARMM program. Although we wish to use the same method to convert the NAMD CG simulation-predicted last structures into an all-atom system and then do order parameter calculation, this did not work.
As shown by RMSD and Rg results, all the systems reached the equilibrated state after 300 ns simulations except the tetramer in the POPC+POPS bilayer system, which reached the equilibrated state after around 1000 ns; thus in total, 2000 ns simulations were conducted on this system as shown. The free energy was calculated using the WHAM program based on the last 700 ns trajectories from all the windows while the last 1000 ns from the tetramer in the POPC+POPS bilayer system. Similarly, the last 1000 ns trajectories were applied to perform RMSD, Rg, distance, and RMSZ calculations for this system. In the hBD-3 in the POPC+POPS mixed bilayer system, 0 Å to 48 Å/60 Å/68 Å is considered the height range for the PMF from the POPS leaflet, abbreviated as (POPC) + POPS side, while −48 Å/–60 Å/–68 Å to 0 Å is considered the height range for PMF from the POPC leaflet, abbreviated as POPC+ (POPS).
5.5. Extra CG Simulations Using Polarizable Water and Gromacs Simulations
Because a polarizable water forcefield has been shown to predict the free energy accurately,57 MARTINI 2.2 forcefield along with the polarizable water model were applied to repeat the US simulations on the hBD-3 dimer in the analogue form translocating through the POPS lipid bilayer using the Gromacs 2019.1.d package.
The initial system was set up in CHARMM-GUI by inserting the hBD-3 dimer analogue at the center of the POPS lipid bilayer. To have enough amount of water molecules covering the top and bottom of the protein during the translocation process, a water thickness of 110 Å above and below the lipid bilayer was applied. Initially, the system was energy-minimized using the steepest descent algorithm and then equilibrated for 0.25 ns by restraining the protein and lipid with a harmonic restraint of force constant of 1000 kJ·mol–1·nm–2 and 200 kJ.mol–1.nm–2, respectively, followed by a total of 5 ns equilibration by gradually reducing the restraint to zero and increasing the timestep from 2 to 20 fs. The temperature and pressure of the system were kept constant at 310.15 K and 1 atm using V-rescale and Berendsen pressure coupling methods.58
Then, the protein was pulled from the center of the lipid bilayer at a constant velocity of 0.00005 Å/fs to the top and bottom of the lipid bilayer, while the lipid head was restrained with a harmonic restraint of force constant of 50 kJ· mol–1·nm–2. In total, 61 configurations were generated with the protein at different heights from the lipid bilayer center at a uniform spacing of 2 Å. Each window was equilibrated for 20 ns restraining the height of the protein from the center of the lipid bilayer with the NPT ensemble.
After a short equilibration, each window was run for 1000 ns restraining the height of the protein from the center of the lipid bilayer with a force constant of 6 kcal/mol. Å2 (2510.4 kJ.mol–1.nm–2) in an NPT ensemble. The temperature was coupled at 310.15 K using the V-rescale thermostat algorithm with a time constant of 1 ps, while the pressure was coupled at 1 atm using the Parrinello–Rahman algorithm with a time constant of 12 ps and isothermal compressibility of 3 × 10–4 bar–1. Nonbonded van der Walls interaction was treated at a cutoff of 11 Å, and the neighbor list was updated once in every 20 steps. Electrostatic interactions were treated using the PME algorithm54 with a dielectric constant of 2.5 and Fourier spacing of 1.5 Å. During the simulations, the bond length involving hydrogen atoms was constrained using the LINCS algorithm. The WHAM was used to combine all the windows to find the PMF curve using a tolerance of 0.00001.
Acknowledgments
This work was supported by the Center for Energy Systems Research located at Tennessee Tech University and the TTU Faculty Research Grant to Dr. Zhang. The simulations were performed using supercomputer time partially from XSEDE via an award to Zhang L. (MCB160041) and partially using the high-performance computers at Tennessee Technological University. The discussion with Dr. Zhenlu Li at Case Western Reserve University helped with the initial CG work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01803.
Additional tables and figures; CG and all-atom simulation system setup and simulation details; hBD-3 sequence, charge and disulfide bonding pattern; RMSD, BSA, and RMSF of the hBD-3 tetramer; membrane thickness calculation sketch; Rg, average RMSD of the hBD-3 monomer, dimer, and tetramer; average number density of CNO and CHO beads in different lipid bilayers; hBD-3 oligomer last structures; RMSZ calculation sketch and result; hBD-3 octamer structure predicted using SymmDock; translocation free energy of hBD-3 in different oligomerization states; RMSD of tetramer binding on the membrane surface; order parameters of POPC lipids; water and ion number density profiles in different systems; and initial system snapshot of the tetramer in solvent simulation (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kaiser V.; Diamond G. Expression of mammalian defensin genes. J. Leukocyte Biol. 2000, 68, 779–784. [PubMed] [Google Scholar]
- Huttner K. M.; Bevins C. L. Antimicrobial peptides as mediators of epithelial host defense. Pediatr. Res. 1999, 45, 785–794. 10.1203/00006450-199906000-00001. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Diamond G. The role of cationic antimicrobial peptides in innate host defenses. Trends Microbiol. 2000, 8, 402–410. 10.1016/S0966-842X(00)01823-0. [DOI] [PubMed] [Google Scholar]
- Harder J.; Bartels J.; Christophers E.; Schroder J.-M. Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- Wu Z.; Hoover D. M.; Yang D.; Boulgue C.; Santamaria F.; Oppenheim J. J.; Lubkowski J.; Lu W. Engineering disulfide bridges to dissect antimicrobial and chemotactic activities of human-defensin 3. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 8880–8885. 10.1073/pnas.1533186100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Zhou L.; Li J.; Suresh A.; Verma C.; Foo Y.; Yap E.; Tan D.; Beuerman R. Linear analogues of human beta-defensin 3: Concepts for design of antimicrobial peptides with reduced cytotoxicity to mammalian cells. ChemBioChem 2008, 9, 964–973. 10.1002/cbic.200700560. [DOI] [PubMed] [Google Scholar]
- Klüver E.; Schulz-Maronde S.; Scheid S.; Meyer B.; Forssmann W.; Adermann K. Structure-activity relation of human beta-defensin 3: Influence of disulfide bonds and cysteine substitution on antimicrobial activity and cytotoxicity. Biochemistry 2005, 44, 9804–9816. 10.1021/bi050272k. [DOI] [PubMed] [Google Scholar]
- Hoover D.; Wu Z.; Tucker K.; Lu W.; Lubkowski J. Antimicrobial characterization of human -defensin 3 derivatives. Antimicrob. Agents Chemother. 2003, 47, 2804–2809. 10.1128/AAC.47.9.2804-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z.; Dubyak G. R.; Jia X.; Lubkowski J. T.; Weinberg A. Human β-defensin-3 structure motifs that are important in CXCR4 antagonism. FEBS J. 2013, 280, 3365–3375. 10.1111/febs.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioi A. B.; Rhodriguez A. L. R.; Funderburg N. T.; Feng Z.; Weinberg A.; Sieg S. F. Membrane damage and repair in primary monocytes exposed to human β-defensin-3. J. Leukocyte Biol. 2012, 92, 1083–1091. 10.1189/jlb.0112046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan T. K.; Lay F. T.; Poon I. K. H.; Hinds M. G.; Kvansakul M.; Hulett M. D. Human defensin 3 contains an oncolytic motif that binds PI(4,5)P2 to mediate tumour cell permeabilisation. Oncotarget 2016, 7, 2054–2069. 10.18632/oncotarget.6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudheendra U. S.; Dhople V.; Datta A.; Kar R. K.; Shelburne C. E.; Bhunia A.; Ramamoorthy A. Membrane disruptive antimicrobial activities of human beta defensin-3 analogs. Eur. J. Med. Chem. 2015, 91, 91–99. 10.1016/j.ejmech.2014.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerikova M.; El-Tibi I. P.; Maslarska V.; Bozhanov S.; Tachkov K. Antimicrobial activity, mechanism of action, and methods for stabilisation of defensins as new therapeutic agents. Biotechnol. Biotechnol. Equip. 2019, 33, 671–682. 10.1080/13102818.2019.1611385. [DOI] [Google Scholar]
- Andersson D. I.; Hughes D.; Kubicek-Sutherland J. Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updat. 2016, 26, 43–57. 10.1016/j.drup.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Enkavi G.; Javanainen M.; Kulig W.; Róg T.; Vattulainen I. Multiscale Simulations of Biological Membranes: The Challenge To Understand Biological Phenomena in a Living Substance. Chem. Rev. 2019, 119, 5607–5774. 10.1021/acs.chemrev.8b00538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhople V.; Krukemeyer A.; Ramamoorthy A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1499–1512. 10.1016/j.bbamem.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Hoover D. M.; Chertov O.; Lubkowski J. The structure of humanβ-defensin-1: New insights into structural properties of β-defensins. J. Biol. Chem. 2001, 276, 39021–39026. 10.1074/jbc.M103830200. [DOI] [PubMed] [Google Scholar]
- Hoover D. M.; Rajashankar K. R.; Blumenthal R.; Puri A.; Oppenheim J. J.; Chertov O.; Lubkowski J. The structure of human β-defensin-2 shows evidence of higher order oligomerization. J. Biol. Chem. 2000, 275, 32911–32918. 10.1074/jbc.M006098200. [DOI] [PubMed] [Google Scholar]
- Hill C.; Yee J.; Selsted M.; Eisenberg D. Crystal structure of defensin HNP-3, an amphiphilic dimer: Mechanisms of membrane permeabilization. Science 1991, 251, 1481–1485. 10.1126/science.2006422. [DOI] [PubMed] [Google Scholar]
- Schibli D. J.; Hunter H. H.; Aseyev V.; Starner T.; Wiencek J. M.; McCray P. B. Jr.; Tack B.; Vogel H. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 2002, 277, 8279–8289. 10.1074/jbc.M108830200. [DOI] [PubMed] [Google Scholar]
- Woo S. Y.; Lee H. All-atom simulations and free-energy calculations of coiled-coil peptides with lipid bilayers: Binding strength, structural transition, and effect on lipid dynamics. Sci. Rep. 2016, 6, 22299 10.1038/srep22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbart J.; Roux B. Determination of membrane-insertion free energies by molecular dynamics simulations. Biophys. J. 2012, 102, 795–801. 10.1016/j.bpj.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Shorthouse D.; Koldso̷ H.; Sansom M. S. P. Free energy landscape of lipid interactions with regulatory binding sites on the transmembrane domain of the EGF receptor. J. Phys. Chem. B 2016, 120, 8154–8163. 10.1021/acs.jpcb.6b01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; Hong C. K.; Wang Y.; Tieleman D. P. Antimicrobial peptide simulations and the influence of force field on the free energy for pore formation in lipid bilayers. J. Chem. Theory Comput. 2016, 12, 4524–4533. 10.1021/acs.jctc.6b00265. [DOI] [PubMed] [Google Scholar]
- Rzepiela A. J.; Sengupta D.; Goga N.; Marrink S. J. Membrane poration by antimicrobial peptides combining atomistic and coarse-grained descriptions. Faraday Discuss. 2010, 144, 431–443. 10.1039/B901615E. [DOI] [PubMed] [Google Scholar]
- Yeasmin R.; Buck M.; Weinberg A.; Zhang L. Translocation of human β defensin type 3 through a neutrally charged lipid membrane: A free energy study. J. Phys. Chem. B. 2018, 122, 11883–11894. 10.1021/acs.jpcb.8b08285. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Zhao C.; Liang G.; Zhang M.; Zheng J. Engineering antimicrobial peptides with improved antimicrobial and hemolytic activities. J. Chem. Inf. Model. 2013, 53, 3280–3296. 10.1021/ci400477e. [DOI] [PubMed] [Google Scholar]
- General I. J.; Asciutto E. K. Structure and dynamics of Penetratin’s association and translocation to a lipid bilayer. AIP Adv. 2017, 7, 035008 10.1063/1.4978263. [DOI] [Google Scholar]
- May A.; Pool R.; van Dijk E.; Bijlard J.; Abeln S.; Heringa J.; Feenstra K. A. Coarse-grained versus atomistic simulations: realistic interaction free energies for real proteins. Struct. Bioinform. 2014, 30, 326–334. 10.1093/bioinformatics/btt675. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kalé L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Different dynamics and pathway of disulfide bonds reduction of two human defensins, a molecular dynamics simulation study. Proteins. 2017, 85, 665–681. 10.1002/prot.25247. [DOI] [PubMed] [Google Scholar]
- Kol M. A.; de Kruijff B.; de Kroon A. I. P. M. Phospholipid flip-flop in biogenic membranes: what is needed to connect opposite sides. Semin. Cell. Dev. Biol. 2002, 13, 163–170. 10.1016/S1084-9521(02)00044-7. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Vattulainen I. Molecular Mechanism for Lipid Flip-Flops. J. Phys. Chem. B 2007, 111, 13554–13559. 10.1021/jp077094k. [DOI] [PubMed] [Google Scholar]
- Fattal E.; Nir S.; Parente R. A.; Szoka F. C. Jr. Pore-Forming Peptides Induce Rapid Phospholipid Flip-Flop in Membranes. Biochemistry 1994, 33, 6721–6731. 10.1021/bi00187a044. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lu W.; Hong M. The Membrane-Bound Structure and Topology of a Human α-Defensin Indicate a Dimer Pore Mechanism for Membrane Disruption. Biochemistry 2010, 49, 9770–9782. 10.1021/bi101512j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W.; Schneidman D.; Zhang N.; Ma B.; Nussinov R. Probing Oligomerized Conformations of Defensin in the Membrane. Methods Mol. Biol. 2017, 1529, 353–362. 10.1007/978-1-4939-6637-0_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta D.; Leontiadou H.; Mark A. E.; Marrink S.-J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317. 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Ladokhin A. S.; Selsted M. E.; White S. H. Sizing membrane pores in lipid vesicles by leakage of coencapsulated markers: pore formation by melittin. Biophys. J. 1997, 72, 1762–1766. 10.1016/S0006-3495(97)78822-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K.; Murase O.; Miyajim K. Kinetics of pore formation by an antimicrobial peptide, Magainin 2, in phospholipid bilayers. Biochemistry 1995, 34, 12553–12559. 10.1021/bi00039a009. [DOI] [PubMed] [Google Scholar]
- Prince A.; Sandhu P.; Kumar P.; Dash E.; Sharma S.; Arakha M.; Jha S.; Akhter Y.; Saleem M. Lipid-II independent antimicrobial mechanism of nisin depends on its crowding and degree of oligomerization. Sci. Rep. 2016, 6, 37908 10.1038/srep37908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyna G. J.; Tobias D. J.; Klein M. L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. 10.1063/1.467468. [DOI] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Lee B.; Richards F. M. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971, 55, 379–400. 10.1016/0022-2836(71)90324-X. [DOI] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug. Discov. 2015, 10, 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner D. E.; Chan K. Y.; Phillips J. C.; Schulten K. Parallel generalized Born implicit solvent calculations with NAMD. J. Chem. Theory Comput. 2011, 7, 3635–3642. 10.1021/ct200563j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Jo S.; Lim J. B.; Klauda J. B.; Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Cheng X.; Swails J. M.; Yeom M.-S.; Eastman P. K.; Lemkul J. A.; Wei S.; Buckner J.; Jeong J. C.; Qi Y.; Jo S.; Pande V. S.; Case D. A.; Brooks C. L. III; MacKerell A. D. Jr.; Klauda J. B.; Im W. CHARMM-GUI input generator for NAMD, Gromacs, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S.-J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- Shih A. Y.; Freddolino P. L.; Arkhipov A.; Schulten K. Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations. J. Struct. Biol. 2007, 157, 579–592. 10.1016/j.jsb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Grossfield A.An implementation of WHAM: The weighted histogram analysis method, Version 2.0.9. 2014. Available at membrane.urmc.rochester.edu/content/wham. Accessed Dec. 1, 2020.
- Giorgino T. Computing 1-D atomic densities in macromolecular simulations: The density profile tool for VMD. Comput. Phys. Commun. 2014, 185, 317–322. 10.1016/j.cpc.2013.08.022. [DOI] [Google Scholar]
- Yesylevskyy S. O.; Schäfer L. V.; Sengupta D.; Marrink S. J. Polarizable Water Model for the Coarse-Grained MARTINI Force Field. PLoS Comput. Biol. 2010, 6, e1000810 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.