

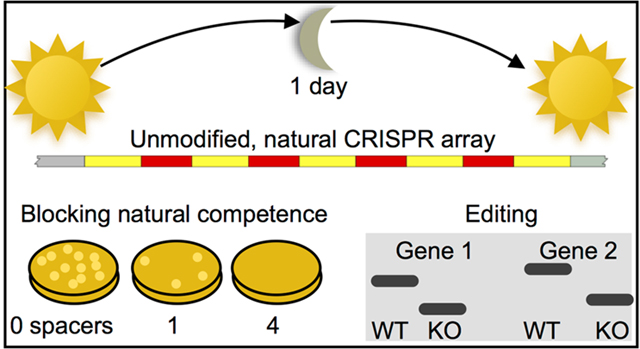
Abstract
CRISPR-Cas systems are prokaryotic immune systems that have proliferated widely not only in bacteria and archaea, but also much more recently, in human biological research and applications. Much work to date has utilized synthetic sgRNAs along with the CRISPR nuclease Cas9, but the discovery of array-processing nucleases now allows the use of more compact, natural CRISPR arrays in heterologous hosts, in addition to organisms with endogenous systems. Unfortunately, the construction of multiplex natural CRISPR arrays remains technically challenging, expensive, and/or time-consuming. This limitation hampers research involving natural CRISPR arrays in both native and heterologous hosts. To address this problem, we present a method to assemble CRISPR arrays that is simple, rapid, affordable, and highly scalable–we assembled 9-spacer arrays with 1 day’s worth of work. We used this method to harness the endogenous CRISPR-Cas system of the highly competent bacterium Acinetobacter baylyi, showing that while single spacers are not always completely effective at blocking DNA acquisition through natural competence, multiplex natural CRISPR arrays enable both nearly complete DNA exclusion and genome editing, including with multiple targets for both. In addition to demonstrating a CRISPR array assembly method that will benefit a variety of applications, we also find a potential bet-hedging strategy for balancing CRISPR defense versus DNA acquisition in naturally competent A. baylyi.
Graphical Abstract
CRISPR (clustered regularly interspaced short palindromic repeats)-Cas systems are adaptive immunity mechanisms that protect bacteria and archaea against invading nucleic acids, generally by detecting and cutting or degrading defined target sequences.1 CRISPR-Cas systems include Cas (CRISPR-associated) proteins, as well as their eponymous arrays of short direct repeats that alternate with similarly short DNA spacers. The spacer array is transcribed into a long precrRNA, which is then processed into individual crRNAs (CRISPR RNAs), each composed of a single spacer that is complementary to a particular nucleic acid target, and often a hairpin handle derived from a repeat. These crRNAs bind Cas effector proteins, such as Cas9, or multiprotein complexes, such as CASCADE. Once bound, they guide the effector to complementary DNA or RNA, depending on the system, which the effectors often cleave and/or degrade.
In short order, many laboratories have adapted CRISPR-mediated DNA cleavage for applications ranging from precise genome engineering to genetic circuits2 to targeted bacterial strain removal.3−6 Self-spreading CRISPR constructs have also been used to quickly generate homozygous diploid knockouts (the mutagenic chain reaction),7 and preliminary work suggests they could re-engineer entire populations through biased inheritance; that is, gene drives or active genetics.8−13
Spacer multiplexing is beneficial for nearly all of these applications.14 Targeting multiple sites on the same gene improves both mutagenesis and gene regulation,2 cleaving multiple target sites prevents emergence of resistant alleles,15 and multiple genes can be edited simultaneously. While natural CRISPR arrays are inherently multiplex, some including hundreds of spacers, multiplexing in synthetic biology applications has been comparatively limited. One reason is that constructing synthetic multiplex CRISPR arrays is technically challenging due to their extensive repetition. Addressing this difficulty, several strategies have been developed to assemble tandem arrays of synthetic sgRNA (single guide RNA) transcriptional units, but these were limited in array size or required time-consuming, sequential cloning for each additional spacer.16−19 Recently, others have shown that single-promoter sgRNA arrays can be assembled using tRNAs to direct processing and release of individual sgRNAs.20,21
Natural CRISPR arrays were largely abandoned in favor of such synthetic sgRNAs because this system only requires a single effector protein for use in non-native hosts, namely Cas9. However, the more recent discovery that other single-protein CRISPR effectors, including Cas12a (Cpf1) and Cas13a (C2c2), can process natural arrays without tracRNA means that natural, multiplex CRISPR arrays can be used in non-native hosts as easily as sgRNAs.22−24 In comparison to artificial sgRNA arrays, natural CRISPR arrays have several advantages for multiplexing. Natural arrays are much more compact, making them easier to package and deliver. Natural arrays also have a particular advantage for applications in prokaryotes, many of which already have their own endogenous CRISPR-Cas systems that can be retargeted using synthetic spacers.1 One attractive application could be to limit horizontal gene transfer, a major contributor to multidrug resistance and pathogenicity.25
Unfortunately, the signature palindromic repeats significantly complicate assembly of natural CRISPR arrays. This problem is particularly important because spacer design rules are not completely accurate even for the best studied Cas nucleases, so developing good arrays can require building and testing multiple designs.26 Recent approaches for assembling multiplex natural arrays have been limited to just a few spacers,4,27 imposed sequence constraints,28 or required sequential, time-consuming cloning steps for each additional spacer.5,28,29 Multiplex arrays can be assembled using very long single-stranded oligos (180 nt23), but these become significantly more expensive and unreliable as their length surpasses 60 nt. Another option is double-stranded DNA synthesis,5,24 but this can also be unreliable or require slower, more expensive cloned gene services. Such double-stranded DNA synthesis often takes longer or fails for sequences containing repetition and/or secondary structure,24,30 both of which are defining features of CRISPR arrays. Primed adaptation can generate multiplex arrays using the endogenous adaptation mechanism, but the results are stochastic, not designed.31,32 A recent one-pot method enables rapid assembly of nearly natural CRISPR arrays, but this still requires trimming the 3′ ends of spacers.33 This makes the method incompatible with systems that do not trim their spacers and thus require sequence complementarity throughout, including the most prevalent Type I systems.1 Array assembly therefore remains a key challenge in the field.14
As a testbed for multiplex harnessing of an endogenous CRISPR-Cas system, we used the highly naturally competent bacterium Acinetobacter baylyi,34 which is a largely non-pathogenic, but phenotypically similar, relative of the highly drug-resistant and clinically urgent pathogen A. baumannii.35−37 A. baylyi is an ideal platform for developing endogenous CRISPR applications, because it combines extensive natural competence, which promotes horizontal gene transfer, with a CRISPR-Cas system that prevents it. We first demonstrated that A. baylyi has a functional Type I−F CRISPR-Cas system, but that it is not fully effective against gene acquisition by natural competence when using single spacer arrays. To better harness the endogenous system, we developed a method to construct natural CRISPR arrays that is rapid (1 day), scalable (we had no difficulty reaching lengths of 9 spacers), affordable, and results in completely natural arrays with no sequence modifications, constraints, or considerations.
RESULTS
A. baylyi Contains a Functional Type I−F CRISPR-Cas System.
The A. baylyi genome contains a computationally identified Type I−F CRISPR-Cas system (Figure 1A),37 but its function has not been tested experimentally. Therefore, we first determined whether the endogenous CRISPR-Cas system can block horizontal gene transfer via natural competence. To test the system, we inserted single-spacer arrays targeting a kanamycin resistance gene into a previously used neutral locus in the genome.38 We tested four different spacers from both the top (T) and bottom (B) strands, each using the 5′-CC-protospacer-3′ protospacer-adjacent motif (PAM, 5′-antiprotospacer-GG-3′ on the complementary, targeted strand) previously shown to work in the Type I−F systems of E. coli,39 Pectobacterium astrosepticum,40 and Pseudomonas aeruginosa.41 When naturally competent cells carrying these single arrays were incubated with a self-replicating plasmid (pBAV-K1),42 there were still many kanamycin-resistant transformants, and only the T2 spacer reduced the transformation efficiency relative to a random spacer (Figure 1B, note the log scale). When they were challenged using a genomically integrating linear DNA construct (Vgr4-K1), again the T2 spacer worked well, now decreasing acquisition of kanamycin resistance by 1000-fold relative to a random spacer, but the others were less effective (Figure 1C). Escape clones did have somewhat smaller colony sizes, suggesting partial tolerance for ongoing self-targeting. All strains remained competent for Vgr4-K2, which contains a second kanamycin resistance gene with minimal homology to the first (Figure 1D).
Figure 1.

Synthetic A. baylyi CRISPR arrays blocking gene acquisition via natural competence. (A) The endogenous, Type I−F CRISPR locus in A. baylyi. (B−D) Cells containing individual spacer arrays (T1, T2, B1, or B2), a 4-spacer multiplex array including all individual spacers, or a random spacer were naturally transformed with the self-replicating plasmid pBAV-K1 (B), the integrating linear DNA Vgr4-K1 (C), or the nontargeted, integrating linear DNA Vgr4-K2 (D). Note the fraction of cells acquiring kanamycin resistance is shown on a log scale. Data includes two experimental replicates, each with three measurement replicates, error bars indicate propagated standard deviations (see Methods), and limits of detection were roughly 10−6. Statistical comparison to the random spacer was performed using multiple comparison analysis (Methods): *p <.01, **p < 10−6.
Construction of Multiplex CRISPR Arrays.
To increase the efficacy of the endogenous A. baylyi CRISPR-Cas system against incoming DNA, we next turned to multiplex arrays, which have been reported to increase CRISPR efficacy in a variety of contexts. However, constructing natural, multiplex Type I CRISPR arrays remains challenging for the reasons described above. Therefore, we developed a new method to assemble multiplex, completely natural arrays.
Our method is based on annealing and ligating single-stranded DNA oligos (Figure 2). The key insight is that despite extensive repetition, the correct order can be ensured by avoiding annealing or ligation within repeats. To achieve this, we design 60 nt top oligos that each include a single 28 nt repeat in their center and extend halfway (16 nt) into the spacer or flanking sequence on either side. These top oligos are joined together by annealing to 40 nt bottom bridge oligos, consisting of the reverse complement of each 32 nt spacer plus 4 nt of repeat on either side. The intentional gaps on the bottom strand avoid oligo annealing within repeats, and they are filled in later by PCR. We tested multiple conditions in optimizing our assembly protocol (Supporting Information, Figure S2), and our final optimized protocol is as follows (see Methods for more detail):
Figure 2.

Strategy for assembling multiplex, natural CRISPR arrays. Assembly strategy for a sample 3-spacer CRISPR array to be inserted into a vector using Gibson assembly or fusion PCR (A), or Golden Gate assembly (B). Each strategy shows the desired end product, the top and bottom oligos used for array annealing and ligation, and the PCR amplicons for insertion into a vector. Single-stranded primers (oligos) are shown as arrows pointing 5′ to 3′. Note that primers used for Golden Gate assembly (denoted “GG”) have an additional Golden Gate tail appended to their 5′ ends (see Methods). See Figure S1 for more detailed versions that include DNA sequence. (C) PCR amplified 9-spacer arrays using the Gibson (left) and Golden Gate (right) strategies. Colony PCR screening of E. coli clones for 9-spacer arrays inserted using Golden Gate (D) and Gibson (E) strategies, where the correct length is 914 bp. The ladder on all gels has 100 bp increments, with the 1 kb band marked by an asterisk.
Phosphorylation: Mix 2 to 4 μL of each top oligo from 100 μM stock solutions (Figure S1A), and phosphorylate them using T4 polynucleotide kinase (PNK) and 1x T4 DNA ligase buffer at 37 °C for 15−60 min. This step can be skipped if ordering 5′ phosphorylated oligos. Phosphorylating the top oligos separately increases PNK activity, which is optimal on single-stranded DNA.
Annealing: Mix 1 part top oligos with 2−3 parts bottom oligos by molarity (Figure S1B), and perform a slow annealing starting from 90 °C. We used a thermocycler programmed to decrease to 37 °C by 0.1 °C/sec, but allowing a hot water bath to gradually cool should work as well.
Ligation: Add T4 DNA ligase and additional ligase buffer, and incubate at 37 °C for 30 min.
Clean up: Column purify the ligated array using a standard DNA purification column to remove unincorporated oligos.
Amplification: PCR amplify the array using primers appropriate for your cloning strategy of choice, for example, Gibson or Golden Gate assembly, using as high an annealing temperature as the primers will allow (Figure S2C−E).
Optional: Gel Purification: Run the raw ligation or amplified PCR product on an agarose gel, excise the correct band, and purify the DNA using a gel extraction kit. This step is optional for shorter arrays, but it can substantially increase accuracy for longer arrays.
Insert into vector: Insert the array into a vector using a method of your choice; for example, Golden Gate, Gibson assembly, or fusion PCR.
Transform: Transform the final construct into E. coli (for circular plasmids), or directly into A. baylyi (for linear constructs with genomic homology for recombination), spread on selective agar plates, and incubate overnight.
(Next day) Screen: On the following day, pick several colonies and PCR across the array to screen for assemblies of the correct length (Figure S2D,E).
The assembly steps can be completed in 1 day, and the resulting colonies can be screened the following day by PCR across the CRISPR array. This basic array assembly technique is compatible with multiple cloning strategies for insertion into a final vector. In developing our protocol, we successfully inserted the arrays into circular plasmids using both Gibson (Figure 2A and S1A) and Golden Gate (Figure 2B and S1B) cloning strategies, as well as into linear DNA fragments that we amplified via PCR.
Using our optimized protocol, we were able to quickly and accurately assemble a 9-spacer array (Figure 2C), using either Gibson or Golden Gate strategies to insert the array into the plasmid. For Golden Gate insertion, 11 of 16 picked colonies had the correct length array (Figure 2D), and for Gibson insertion, 8 of 16 picked colonies had the correct length (Figure 2E). Sanger sequencing confirmed that all arrays with the correct length were assembled in the correct order. Seven of the Golden Gate and two of the Gibson clones were completely correct, and the remainder had various indels or substitutions. Only one of the errors was at a junction between oligos, suggesting most may have occurred during oligo synthesis.
Multiplex Natural Arrays Enhance CRISPR Efficacy In Natural Competence.
To see if multiplex CRIPSR arrays more effectively interfere with natural competence in A. baylyi, we combined the four spacers targeting the kanamycin resistance gene into a single, 4-spacer natural array and inserted it into the A. baylyi genome. This 4 × Kan1 array was highly effective against both the self-replicating plasmid pBAV-K1 and the genomically integrating construct Vgr4-K1 (Figure 1B,C, Figure 3A). As for single-spacer arrays, the 4-spacer array was ineffective against a second, control kanamycin resistance gene with no homology to the targeted gene (Figure 1D, Figure 3B). The 4-spacer array allowed no escape transformants with the replicating plasmid, but we did obtain 2 escapes with the integrating construct. In one of these escapes, the inserted 4-spacer CRISPR array had been disrupted by the active insertion sequence IS1236.43 The other escape appeared to have a larger genomic deletion encompassing the array, as it had lost the spectinomycin resistance marker used to select for insertion of the array, and the entire region failed to amplify by PCR.
Figure 3.

Synthetic, multiplex A. baylyi CRISPR arrays block acquisition of multiple genes. Cells containing no exogenous CRISPR arrays (WT), 4-spacer arrays targeting kan1 and kan2, and an 8-spacer array targeting both kan genes (x-axis tick labels) were incubated with linear, genomically integrating DNA. Donor DNA constructs included Vgr4-Kan1 (A), Vgr4-Kan2 (B), both kan constructs (C), or a nontargeted beta-lactamase gene (D). Data include two experimental replicates, each with three measurement replicates, error bars indicate propagated standard deviations (see Methods), and limits of detection were roughly 10−6. **p < 10−7.
Next, we expanded our array to defend against both kanamycin resistance genes simultaneously, using an 8-spacer array. As a preliminary step, we constructed a 4-spacer array targeting the second kanamycin gene, added genomic homology arms via fusion PCR, and cloned the linear product into A. baylyi. Then we assembled an 8-spacer array targeting both kanamycin resistance genes. We easily assembled this 8-spacer array in a one-pot reaction, but we also assembled it from the individual 4-spacer arrays to demonstrate modular array construction. For the modular approach, we PCR amplified the cloned 4xKan2 array using a leftmost top primer that began with the first 16 bp of the final spacer in the 4xKan1 array rather than with the 5′ region of the vector, and then performed a fusion PCR of the three pieces Vector 5′−4xKan1, 4xKan2, and Vector 3′.
In contrast to single spacers (Figure 1), each 4-spacer array effectively blocked acquisition of its respective kanamycin resistance gene (Figure 3A,B), and only the 8-spacer array prevented acquisition of kanamycin resistance when both genes were present (Figure 3C). All arrays allowed acquisition of a nonhomologous beta-lactamase gene (Figure 3D). The modular construction shows that even if there is a size limit to this method, very large arrays can still be assembled in very few steps.
Markerless Genome Editing Using an Endogenous CRISPR-Cas System.
CRISPR has been used for genome editing in many contexts, and we wanted to confirm that our natural arrays would enable editing of the A. baylyi genome as well. To do this, we constructed a 3-spacer array targeting the bap gene (ACIAD2866), which has been implicated in biofilm formation in Acinetobacter,44,45 and thus may be at least partially responsible for intractable clogging when using A. baylyi in microfluidics.46 We inserted the 3xBAP array into both pBAV1spec for cloning into E. coli, as well as into a linear construct with roughly 1 kb genomic homologies on either side for direct insertion into the A. baylyi genome. The pBAV1spec assembly transformed into E. coli was the correct length in eight of eight tested clones (Figure 4A, left half). We sequenced four of them, of which all had the correct spacer order, although one was missing two base-pairs. When we cotransformed this pBAV1spec-CRISPR3xBAP into A. baylyi along with a markerless bap deletion donor DNA (linear dsDNA with ∼1 kb homology arms on either side), both of two tested clones had the correct deletion (Figure 4B). Interestingly, we found that bap in our strain of A. baylyi ADP1 (ATCC 33305) was approximately 3 kb larger than in the published genome. This may have been due to a sequence assembly error or genomic instability, either of which could result from the many tandem repeats found in bap genes,44,45 but we did not pursue the discrepancy.
Figure 4.

Multiplex genome editing using an endogenous CRISPR-Cas system. Arrows indicate the expected bands for correct genomic edits, and asterisks indicate the 1 kb band of the ladder (not counted in lane numbering). (A) PCR screening of of 3×BAP (lanes 1−8) and 6×CRA-BAP (lanes 9−16) arrays in pBAV, cloned into E. coli. (B) PCR screening of two markerless bap deletions in A. baylyi using pBAV-CRISPR3×BAP. (C−F) PCR screening of markerless bap and double CRAΦ, bap deletions in A. baylyi using nonclonal, linear PCR products from array assembly. (C) Multiplex 3×BAP (lanes 1−8) and 6×CRA-BAP (lanes 9−16) arrays. (D) bap deletion screening for the same clones as in C. The deletion and wild type amplicons are roughly 4.5 and 12 kb, respectively. (E) CRAΦ deletion screening for the clones in lanes 9−16 of panels C and D. Product was only expected for CRAΦ deletion. (F) As in panel E, but circular CRAΦ phage screening. The 3 kb product was only expected if CRAΦ was present in its excised, circular episome form.
When using a linear construct to deliver the 3xBAP array into A. baylyi, we obtained far more clones than when using pBAV1spec (on the order of 1000 vs 36), which is expected because homologous recombination is more efficient than plasmid recircularization in A. baylyi natural competence.47 Of eight tested clones, seven had the correct size array (Figure 4C, left half) and seven had the correct BAP deletion (Figure 4D, left half), even despite the CRISPR array not having first been clonally verified.
Next, we attempted to delete two regions at once, creating a 6× array targeting both bap and the CRAΦ prophage, which binds the competence machinery when activated, complicating horizontal gene transfer experiments.48 The pBAV1spec-CRISPR6×CRA-BAP construct had the correct array length in 6 of 8 E. coli clones (Figure 4A, right half), but we were unable to use it to obtain a double genomic deletion in A. baylyi, likely due to the relative inefficiency of circular plasmids in natural transformation.
To increase our triple transformation efficiency, we used the genomically integrating, linear 6×CRA-BAP construct along with CRAΦ and bap deletion donor DNAs. Of eight tested clones, three had the correct array length (Figure 4C, right half). All three of those had both the desired genomic CRAΦ deletion (Figure 4E) and eliminated the excised, circular CRAΦ episome (Figure 4F). All three clones also had mutations in bap, although two of them had larger deletions (Figure 4D, right half), leaving one clone with both precise deletions. One of the larger bap deletions extended to the end of a nearby copy of the insertion sequence IS1236,43 and the other had a more complex rearrangement that appeared to involve an inversion of part of the genome. IS1236 is not present next to bap in the official genome sequence, but it was already there in our parental strain before the double deletion attempt. This is not completely unexpected, since IS1236 is known to be highly active in A. baylyi.49 If the correct editing rate were more important than speed, one could likely increase the percentage of clones with the correct edits by first clonally verifying the linear CRISPR array construct.
Construction of Cas12a Arrays.
Finally, we demonstrated that the method described here is generalizable to other natural CRISPR arrays, which use different repeat sequences and spacer lengths. For this demonstration, we chose Cas12a/Cpf1 arrays, which are processed by their respective single effector nuclease.50 The Cas12a CRISPR array unit for Franciscella novicida U112 is slightly longer than the A. baylyi array unit, with 36 bp repeats and 26−32 bp spacers.51 Nevertheless, we easily assembled a 4-spacer array with a full 68 bp unit length, targeting a beta lactamase gene (Figure S3A). All screened clones (eight of eight) had the full-length array in the correct order (Figure S3B) of which two were correct with no gaps.
DISCUSSION
The method presented here solves the challenge of rapid, affordable, and scalable construction of completely natural multiplex CRISPR arrays, with no sequence modifications and only minimal constraints. This should be highly beneficial for multiple applications in a variety of organisms, from basic research to applied tools. For applications using heterologous, array-processing Cas nucleases such as Cas12a, facile construction of multiplex natural arrays will help with gene regulation,22 genome engineering,23,24 and even population engineering.15
We designed this assembly method to include three key features that improve its accuracy and efficiency: unique ligation junctions, long annealing regions, and limited oligo length. In the first feature, the only ligation junctions are within the unique spacers on the top strand, which helps to ensure assembly in the correct order. We purposely left gaps in the repeat regions on the bottom strand to avoid ligation junctions within repeats. We tested the inclusion of an oligo covering the remaining 20 nt of the repeats to fill in the gaps on the bottom strand (repeat_RC), but this resulted in a smear of larger than expected ligation products, indicating increased ligation at incorrect junctions (Figure S2A). Furthermore, while developing this protocol we sequenced several correct-sized clones that had incorrect spacer order, but only when including the repeat_RC oligo.
The second feature is long (20 nt) annealing regions that allow more rapid and specific annealing and ligation than the usual 4 bp Golden Gate overlaps, particularly at the 37 °C where T4 DNA ligase has optimal activity. The long annealing regions also allow the user to choose spacers without constraints imposed by the requirement for junction orthogonality, since such long sequences should be highly specific. This allows for very easy, plug-and-play oligo design. Third, the longest oligos must only be the unit length of the CRISPR array, which for A. baylyi is 60 nt. Oligos of this length are relatively reliable, affordable, and rapidly delivered from most DNA synthesis vendors.
A final advantage lies in cost-saving oligo reusability. Unlike ad-hoc construction strategies, this method places the ligation junctions in the same location for every spacer-repeat unit, meaning that many oligos can be reused for alternate array designs without checking for compatibility. For example, our 4xKan1 and 4xKan2 arrays were easily joined with just one additional oligo. This modular assembly demonstrates that verified subarrays can easily be joined with just one additional day of work.
It should be noted that this method does have a few limitations. First, while it can generate arrays of defined spacer order, it cannot generate randomized array libraries. Second, arrays with multiple copies of the same or highly homologous spacers could be challenging to assemble. Third, spacers that have strong secondary structures or that are complementary to either other spacers or the repeat could pose a challenge.
The PCR amplification step following ligation both enriches the correct size product and produces a double-stranded construct with no gaps. A fully double-stranded insert is particularly important for Gibson Assembly-based insertion into the vector because of the required exonuclease, but we also found it to be important for Golden Gate insertion. Without PCR amplification, Golden Gate insertion of a 6× array yielded clones containing a range of incorrectly sized inserts (compare Figure S2 panels D and E). Interestingly, these incorrect arrays almost always contained spacers that were in the correct order, but truncated at the 5′ end. We suspect the 5′-specific truncation may involve a gap repair process within the E. coli host that may be mediated by repeats and directionally biased by plasmid replication.
In prokaryotes with endogenous CRISPR-Cas systems, this method will improve the study and understanding of the ecological importance of CRISPR in its natural context, including the antagonistic interplay between CRISPR and horizontal gene transfer (HGT).25,52−54 This seemingly contradictory pair of abilities has raised evolutionary questions about trade-offs between the acquisition of new traits via HGT, versus CRISPR-mediated exclusion of foreign DNA.55,56 This interaction is important for microbial evolutionary theory, but when the transferring genes confer antibiotic resistance or pathogenicity, it also directly impacts human health.57 Here, we demonstrated that in the highly competent A. baylyi, the CRISPR-HGT interaction is not straightforward. While multiplex arrays effectively blocked exogenous DNA uptake, weaker single spacers reduced, but did not eliminate, HGT. This suggests that for A. baylyi, one solution to the CRISPR-HGT conundrum is to hedge their bets. Single spacers provide some protection against incoming targeted DNA, but particularly for weaker spacers or when multiple spacers compete for limited CASCADE complexes,58 some targeted DNA can still be acquired. When the tolerance is only partial, the targeted protospacer (or the CRISPR machinery) will eventually mutate to eliminate genomic self-targeting and alleviate growth costs, allowing ongoing exploration of the genetic diversity in the environment.
METHODS
Array Construction.
We designed spacers to match target sequences preceded by CC on the nontargeted strand using a computational tool to ensure they were maximally orthogonal to the rest of the A. baylyi genome.59 Briefly, the algorithm searches for all possible spacers in the target sequence that have the appropriate PAM, and then scans them against the host genome to find the most similar sequence, giving greater weight to bases in the PAM-proximal seed sequence. The best match (highest score) against the host genome is assigned as the score for that spacer. We chose spacers from among the lowest scoring (most genome-orthogonal) sequences to cover the entire target and include both DNA strands. For a random spacer, we found the lowest scoring sequence among a computer-generated, random pool. Oligos were designed according to the diagrams in Figure 2 and Figure S1, and their sequences are given in Table S1. Spacer sequences are shown in Table S2. We ordered standard quality, desalted oligos normalized to 100 uM in TE buffer from ValueGene, Eton Bio, and Integrated DNA Technologies. All enzymes and buffers were from New England Biolabs. To construct arrays, we used the following optimized procedure:
Phosphorylate oligos by mixing 1−2 μL of each top-strand oligo along with 1× T4 ligase buffer and 1 μL of T4 polynucleotide kinase (NEB). Polynucleotide kinase buffer will not work without supplementary ATP. Incubate at 37 degrees for 30−60 min.
Anneal oligos by mixing 1 part phosphorylated top oligos with 2 to 3 parts bottom oligos, heating to 85 °C, and slowly cooling back to 37 °C at 0.1 °C per second in a thermocycler.
Ligate by adding 1 μL of T4 DNA ligase and another 1× ligase buffer. Incubate at 37 °C for another 30−60 min.
Remove unligated oligos using a PCR purification column (Lamda Biotech).
PCR amplify the ligation product using primers as shown in Figure 2 and Figure S1. We used Q5 DNA polymerase and the manufacturer’s recommended protocol, annealing at 72 °C, extending for 20 s, and running for 20 cycles. A high annealing temperature is critical to recover the correct product; primers can be checked at http://tmcalculator.neb.com/.
Purify the PCR product either directly or after excising the correct band from a gel, using a column-based PCR or gel purification kit (Qiagen).
Insert the array into a vector. For Gibson assembly, we mixed 2 μL of total DNA (with equimolar parts) with 2 μL of 2× master mix and incubated at 50 °C for 1 h. For Golden Gate assembly, we mixed 4 μL total DNA (with equimolar parts), 0.5 μL of T4 DNA ligase buffer, 0.25 μL of T4 DNA ligase, and 0.25 μL of BsaI, and incubated for 30−50 cycles of 1 min each at 37 and 24 °C, followed by 10 min at 50 °C. Vectors were prepared by PCR using primers as shown in Figure 1 and Figure S2, and gel extracted. Whenever the vector PCR was derived from a plasmid, we used the primers Vector 3′F and Vector 5′R and treated the product with DpnI. For linear constructs used in direct transformation into A. baylyi, the vector consisted of approximately 1 kb homology arms on either side of the array. In these cases, we either directly mixed the three pieces (5′ arm, array, and 3′ arm) in a full-length PCR reaction, or first prejoined the three pieces via either Gibson or Golden Gate assembly, and then PCR amplified and gel extracted the full construct.
For modular assembly of the 8xKan array, we first assembled both 4xKan1 and 4xKan2 arrays and inserted them into the genomic integration vector as above. Next, we PCR amplified the 5′ part of the 4xKan1 construct through the array using the primers pp_5′F and Kan1_B2_RC, as well as the 4xKan2 construct using using the primers Kan1_B2-R-Kan2_T1 and Array_R. Then we performed a 3-piece PCR with primers Vector_5′F and Vector_3′R to fuse (i) Vector 5′−4xKan1, (ii) 4xKan2, and (iii) the vector 3′ piece (amplified using primers Vector_3′F and pp_3′R).
To assemble FnCas12a arrays, we followed the same procedure described above, using the Golden Gate insertion strategy.
Cell Culture, Transformations, and Screening.
We grew all cells in LB media at 30 or 37 °C. A. baylyi strain ADP1 was obtained from ATCC (stock #33305) and for E. coli we used a lab strain of MG1655. The kan1 gene was aminoglycoside O-phosphotransferase APH(3′)-IIIa, and the kan2 gene was aminoglycoside O-phosphotransferase APH(3′)-IIa. These two genes have no significant similarity as determined by BLAST alignment. For transformation of A. baylyi via natural competence, we washed overnight cultures, resuspended in fresh LB, and incubated 50 μL of cells plus DNA at 37 °C for 2 to 4 h. All data plotted in the same figure used the same concentration of donor DNA, generally 0.2−1 ng/μL. To quantify the fraction of transformed cells, we performed five 10-fold serial dilutions and spotted three measurement replicates of 2 μL each at each dilution level onto 2% agar LB plates containing the appropriate (or no) antibiotic selection (20 ug/mL of kanamycin and/or spectinomycin). Each experiment was repeated on two separate days. Lower agar concentrations did not work well for colony counting, because the motile cells began to spread and colonies became less well-defined. Only colonies visible after 20 h at 30 °C were counted.
We inserted CRISPR arrays into a neutral genomic region that had been used previously, replacing genomic coordinates 2,159,575−2,161,720, covering ACIAD2187, ACIAD2186, and part of ACIAD2185.38 The integration site for CRISPR-targeted kanamycin resistance genes was another region that we have found to be neutral in our lab conditions, ACIAD3427. The upstream homology arm covered coordinates 3,341,420−3,342,480, and the downstream homology arm covered 3,342,641−3,343,720. Our replicating plasmid was the broad host pBAV1k,42 which we modified to spectinomycin resistance when using it to carry CRISPR arrays. In our arrays, we included the 80 bp upstream of the endogenous CRISPR array to include any leader sequences or regulatory elements. For markerless genomic deletions, we constructed linear donor DNA by PCR fusing approximately 1 kb regions upstream and downstream of the targeted gene.
For PCR screening of clonal CRISPR arrays in E. coli, we picked individual colonies into 50 μL of water, and used 1 μL directly in a PCR reaction. For A. baylyi, we found we did not obtain clean results unless we first used a genomic miniprep kit to purify DNA (Promega Wizard). We inverted the colors for all agarose gels to assist visualization.
Statistical Analysis.
To calculate error bars for ratios on logarithmic plots, we used error propagation as described previously.46 For each experimental replicate (each with three measurement replicates; i.e., 2 μL spots), we took the log base 10 of each data point, found the standard deviations for both transformed and total cell count measurement replicates (σ1 and σ2), and calculated the standard deviation of the ratio as . To find the total variance across experimental replicates from different days, we used the error propagation formula
where the subscript c denotes experimental replicates, f is the fraction transformed, and nc is the number of measurement replicates for each experiment (here, 3 spotting replicates). Performing calculations on a logarithmic scale creates a problem when some, but not all, measurement replicates are below the limit of detection, because zeros create infinities. In these cases, we set the zeros to half the limit of detection as a conservative estimate for the purposes of plotting, since excluding them would artificially increase the average for that experiment.
We performed significance tests as described previously.46 In Figures 1 and 3, we performed multiple comparison tests using the Matlab function multcompare, using the error propagated means and variances (on log10 scales) and Tukey’s HSD criterion. Where data were below the limit of detection, we tested for differences from that limit of detection.
Supplementary Material
Funding
R.C. and J.H. were supported by the National Institutes of Health Grant No. R01-GM069811.
Footnotes
The authors declare the following competing financial interest(s): R. Cooper and J. Hasty have filed a provisional patent 62/983,273 based on the technique described in this manuscript.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.9b00489.
Figure S1, a more detailed version of Figure 2a,b; Figure S2, optimization of array assembly; Figure S3, construction of a 4-spacer FnCas12a array; Table S1, oligos/primers used; Table S2, CRISPR spacers used; annotated DNA sequences for both the linear, genomically integrating vector pp2.1-CRISPR8xkan-Spec-pp2.2 and the replicating plasmid pBAV1spec-CRISPR3×CRA-Spec (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acssynbio.9b00489
Contributor Information
Robert M. Cooper, BioCircuits Institute, University of California, San Diego, La Jolla, California 92093, United States; San Diego Center for Systems Biology, La Jolla, California 92093, United States.
Jeff Hasty, BioCircuits Institute, Molecular Biology Section, Division of Biological Sciences, and Department of Bioengineering, University of California, San Diego, La Jolla, California 92093, United States; San Diego Center for Systems Biology, La Jolla, California 92093, United States.
REFERENCES
- (1).Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJM, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, and Koonin EV (2015) An updated evolutionary classification of CRISPR−Cas systems. Nat. Rev. Microbiol 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Didovyk A, Borek B, Tsimring L, and Hasty J. (2016) Transcriptional regulation with CRISPR-Cas9: principles, advances, and applications. Curr. Opin. Biotechnol 40, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yosef I, Manor M, Kiro R, and Qimron U. (2015) Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. U. S. A 112, 7267–7272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti VA, and Marraffini LA (2014) Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol 32, 1146–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gomaa AA, Klumpe HE, Luo ML, Selle K, Barrangou R, and Beisel CL (2014) Programmable Removal of Bacterial Strains by Use of Genome-Targeting CRISPR-Cas Systems. mBio 5, e00928–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Citorik RJ, Mimee M, and Lu TK (2014) Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol 32, 1141–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gantz VM, and Bier E. (2015) The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science 348, 442–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gantz VM, Jasinskiene N, Tatarenkova O, Fazekas A, Macias VM, Bier E, and James AA (2015) Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc. Natl. Acad. Sci. U. S. A 112, E6736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hammond A, Galizi R, Kyrou K, Simoni A, Siniscalchi C, Katsanos D, Gribble M, Baker D, Marois E, Russell S, Burt A, Windbichler N, Crisanti A, and Nolan T. (2016) A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat. Biotechnol 34, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).DiCarlo JE, Chavez A, Dietz SL, Esvelt KM, and Church GM (2015) Safeguarding CRISPR-Cas9 gene drives in yeast. Nat. Biotechnol 33, 1250–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li M, Bui M, Yang T, Bowman CS, White BJ, and Akbari OS (2017) Germline Cas9 expression yields highly efficient genome engineering in a major worldwide disease vector, Aedes aegypti. Proc. Natl. Acad. Sci. U. S. A 114, E10540–E10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kyrou K, Hammond AM, Galizi R, Kranjc N, Burt A, Beaghton AK, Nolan T, and Crisanti A. (2018) A CRISPR-Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol 36, 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Grunwald HA, Gantz VM, Poplawski G, Xu X-RS, Bier E, and Cooper KL (2019) Super-Mendelian inheritance mediated by CRISPR−Cas9 in the female mouse germline. Nature 566, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Adiego-Perez B, Randazzo P, Daran JM, Verwaal R,´ Roubos JA, Daran-Lapujade P, and van der Oost J. (2019) Multiplex genome editing of microorganisms using CRISPR-Cas. FEMS Microbiol. Lett 366, 2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Champer J, Liu J, Oh SY, Reeves R, Luthra A, Oakes N, Clark AG, and Messer PW (2018) Reducing resistance allele formation in CRISPR gene drive. Proc. Natl. Acad. Sci. U. S. A 115, 5522–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kabadi AM, Ousterout DG, Hilton IB, and Gersbach CA (2014) Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 42, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vidigal JA, and Ventura A. (2015) Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat. Commun 6, 8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cao J, Wu L, Zhang S-M, Lu M, Cheung WKC, Cai W, Gale M, Xu Q, and Yan Q. (2016) An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res., gkw660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dow LE, Fisher J, O’Rourke KP, Muley A, Kastenhuber ER, Livshits G, Tschaharganeh DF, Socci ND, and Lowe SW (2015) Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol 33, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xie K, Minkenberg B, and Yang Y. (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. U. S. A 112, 3570–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Port F, and Bullock SL (2016) Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat. Methods 13, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tak YE, Kleinstiver BP, Nuñez JK, Hsu JY, Horng JE, Gong J, Weissman JS, and Joung JK (2017) Inducible and multiplex gene regulation using CRISPR−Cpf1-based transcription factors. Nat. Methods 14, 1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, and Zhang F. (2017) Multiplex gene editing by CRISPR−Cpf1 using a single crRNA array. Nat. Biotechnol 35, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Campa CC, Weisbach NR, Santinha AJ, Incarnato D, and Platt RJ (2019) Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods 16, 887–893. [DOI] [PubMed] [Google Scholar]
- (25).Marraffini LA, and Sontheimer EJ (2008) CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, and Root DE (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Caliando BJ, and Voigt CA (2015) Targeted DNA degradation using a CRISPR device stably carried in the host genome. Nat. Commun 6, 6989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Luo ML, Mullis AS, Leenay RT, and Beisel CL (2015) Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression. Nucleic Acids Res. 43, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cress BF, Toparlak ÖD, Guleria S, Lebovich M, Stieglitz JT, Englaender JA, Jones JA, Linhardt RJ, and Koffas MAG (2015) CRISPathBrick: Modular Combinatorial Assembly of Type II-A CRISPR Arrays for dCas9-Mediated Multiplex Transcriptional Repression in E. coli. ACS Synth. Biol 4, 987–1000. [DOI] [PubMed] [Google Scholar]
- (30).Hughes RA, and Ellington AD (2017) Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology. Cold Spring Harbor Perspect. Biol 9, a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Strotksaya A, Semenova E, Savitskaya E, and Severinov K. (2015) Rapid Multiplex Creation of Escherichia coli Strains Capable of Interfering with Phage Infection Through CRISPR. Methods Mol. Biol 1311, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Strotskaya A, Savitskaya E, Metlitskaya A, Morozova N, Datsenko KA, Semenova E, and Severinov K. (2017) The action of Escherichia coli CRISPR−Cas system on lytic bacteriophages with different lifestyles and development strategies. Nucleic Acids Res. 28, gkx042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liao C, Ttofali F, Slotkowski RA, Denny SR, Cecil TD, Leenay RT, Keung AJ, and Beisel CL (2019) Nat. Commun 10, 2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Elliott KT, and Neidle EL (2011) Acinetobacter baylyi ADP1: Transforming the choice of model organism. IUBMB Life 63, 1075–1080. [DOI] [PubMed] [Google Scholar]
- (35).Chen T-L, Siu L-K, Lee Y-T, Chen C-P, Huang L-Y, Wu RC-C, Cho W-L, and Fung C-P (2008) Acinetobacter baylyi as a pathogen for opportunistic infection. J. Clin. Microbiol 46, 2938–2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chen TL, Siu LK, Wu RCC, Shaio MF, Huang LY, Fung CP, Lee CM, and Cho WL (2007) Comparison of one-tube multiplex PCR, automated ribotyping and intergenic spacer (ITS) sequencing for rapid identification of Acinetobacter baumannii. Clin. Microbiol. Infect 13, 801–806. [DOI] [PubMed] [Google Scholar]
- (37).Touchon M, Cury J, Yoon E-J, Krizova L, Cerqueira GC, Murphy C, Feldgarden M, Wortman J, Clermont D, Lambert T, Grillot-Courvalin C, Nemec A, Courvalin P, and Rocha EPC (2014) The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol. Evol 6, 2866–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Murin CD, Segal K, Bryksin A, and Matsumura I. (2012) Expression Vectors for Acinetobacter baylyi ADP1. Appl. Environ. Microbiol 78, 280–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Almendros C, Guzman NM, Diez-Villasenor C, Garcia-Martinez J, and Mojica FJM (2012) Target Motifs Affecting Natural Immunity by a Constitutive CRISPR-Cas System in Escherichia coli. PLoS One 7, e50797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS, Richter C, Przybilski R, Pitman AR, and Fineran PC (2013) Cytotoxic chromosomal targeting by CRISPR/Cas systems can reshape bacterial genomes and expel or remodel pathogenicity islands. PLoS Genet. 9, e1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rollins MF, Schuman JT, Paulus K, Bukhari HST, and Wiedenheft B. (2015) Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res. 43, 2216–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bryksin AV, and Matsumura I. (2010) Rational Design of a Plasmid Origin That Replicates Efficiently in Both Gram-Positive and Gram-Negative Bacteria. PLoS One 5, e13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gerischer U, D’Argenio DA, and Ornston LN (1996) IS 1236, a newly discovered member of the IS3 family, exhibits varied patterns of insertion into the Acinetobacter calcoaceticus chromosome. Microbiology 142, 1825–1831. [DOI] [PubMed] [Google Scholar]
- (44).Loehfelm TW, Luke NR, and Campagnari AA (2008) Identification and Characterization of an Acinetobacter baumannii Biofilm-Associated Protein. J. Bacteriol 190, 1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).De Gregorio E, Del Franco M, Martinucci M, Roscetto E, Zarrilli R, and Di Nocera PP (2015) Biofilm-associated proteins: news from Acinetobacter. BMC Genomics 16, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cooper RM, Tsimring L, and Hasty J. (2017) Inter-species population dynamics enhance microbial horizontal gene transfer and spread of antibiotic resistance. eLife 6, 8053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Palmen R, Vosman B, Buijsman P, Breek CK, and Hellingwerf KJ (1993) Physiological characterization of natural transformation in Acinetobacter calcoaceticus. J. Gen. Microbiol 139, 295–305. [DOI] [PubMed] [Google Scholar]
- (48).Renda BA, Chan C, Parent KN, and Barrick JE (2016) Emergence of a Competence-Reducing Filamentous Phage from the Genome of Acinetobacter baylyi ADP1. J. Bacteriol 198, 3209–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Renda BA, Dasgupta A, Leon D, and Barrick JE (2015) Genome instability mediates the loss of key traits by Acinetobacter baylyi ADP1 during laboratory evolution. J. Bacteriol 197, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Fonfara I, Richter H, Bratovic M, Le Rhun A, and Charpentier E. (2016) The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521. [DOI] [PubMed] [Google Scholar]
- (51).Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, and Zhang F. (2015) Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Bikard D, Hatoum-Aslan A, Mucida D, and Marraffini LA (2012) CRISPR Interference Can Prevent Natural Transformation and Virulence Acquisition during In Vivo Bacterial Infection. Cell Host Microbe 12, 177–186. [DOI] [PubMed] [Google Scholar]
- (53).Jiang W, Maniv I, Arain F, Wang Y, Levin BR, and Marraffini LA (2013) Dealing with the Evolutionary Downside of CRISPR Immunity: Bacteria and Beneficial Plasmids. PLoS Genet. 9, e1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, and Sontheimer EJ (2013) Processing-Independent CRISPR RNAs Limit Natural Transformation in Neisseria meningitidis. Mol. Cell 50, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).García-Martínez J, Maldonado RD, Guzman NM, and́ Mojica FJM (2018) The CRISPR conundrum: evolve and maybe die, or survive and risk stagnation. Microb Cell 5, 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Weinberger AD, and Gilmore MS (2012) CRISPR-Cas: To Take Up DNA or Not–That Is the Question. Cell Host Microbe 12, 125–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Garcia-Gutierrez E, Almendros C, Mojica FJM, Guzman NM, and Garcia-Martinez J. (2015) CRISPR Content Correlates with the Pathogenic Potential of Escherichia coli. PLoS One 10, e0131935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Stachler A-E, Turgeman-Grott I, Shtifman-Segal E, Allers T, Marchfelder A, and Gophna U. (2017) High tolerance to self-targeting of the genome by the endogenous CRISPR-Cas system in an archaeon. Nucleic Acids Res. 45, 5208–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Didovyk A, Borek B, Hasty J, and Tsimring L. (2016) Orthogonal Modular Gene Repression in Escherichia coli Using Engineered CRISPR/Cas9. ACS Synth. Biol 5, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.