

Abstract
The scalable synthesis of the oxaquinolizidine marine natural product desmethylxestospongin B is based on the early application of Ireland-Claisen rearrangement, macrolactamization, and a late-stage installation of the oxaquinolizidine units by lactam reduction. The synthesis serves as the source of material to investigate calcium signaling and its effect on mitochondrial metabolism in various cell types, including cancer cells.
Keywords: xestospongins, cancer, metabolism, IP3R receptor, total synthesis
Graphical Abstract
Desmethylxestospongin B (dmXe B) is a complex marine natural product that exhibits unique properties as a modulator of calcium signaling with downstream effects on cancer cell mitochondrial metabolism. We report a scalable total synthesis of dmXe B by a flexible synthetic strategy that allows for control of oxidation levels and stereochemistry at C9/9’. We demonstrate that synthetic dmXe B is an effective lead compound for controlling mitochondrial metabolism and survival in cancer, inducing selective death of various cancer cells.
Xestospongins, along with araguspongins, are a group of natural products isolated from marine sponges of Xestospongia sp.1 Structurally, these compounds are comprised of two oxaquinolizidine units tethered by saturated alkylidene chains to form a macrocyclic core, and have variable oxidation and stereochemistry at C9/C9’ (Figure 1).1 While a range of biological activity was reported for xestospongins, we became intrigued by the mounting evidence for the unique ability of xestospongin B to influence mitochondrial metabolism by modulating calcium signaling between the endoplasmic reticulum (ER) and mitochondria through the inhibition of inositol-1,4,5-triphosphate receptors (IP3Rs).2 The constitutive activity of IP3Rs is essential for cellular bioenergetics in a variety of cell types. Its inhibition disrupts the constitutive calcium transfer from ER to mitochondria causing a drop in mitochondrial respiration that generates a bioenergetic crisis characterized by AMPK and autophagy activation.3 It has been shown that interruption of this communication leads to a selective, substantial cancer cell death leaving normal cells unaffected.4 One of the goals of this study was to evaluate the effect of desmethylxestospongin B on mitochondrial respiration in breast cancer cell lines, and the resultant selectivity in inducing cancer cell death leaving normal cells nearly unaffected.
Figure 1.

Structures of representative 1-oxaquinolizidine alkaloids.
While xestospongin B (Xe B) is no longer available from the natural sources, total synthesis provides a feasible source of material. We targeted desmethylxestospongin B (dmXe B),1b a natural product within this family of metabolites, based on the premise that the 3’-methyl group has no effect on the IP3R inhibitory activity, and obviating the need to install it would simplify the development of a scalable synthesis. While Xe C and Ar B are more accessible, they show lower specificity with notable side-effects on calcium homeostasis, and no data is available for Ar C.2b
Previous syntheses in this area by Baldwin5 and Hoye6 are characterized by remarkable brevity and strategic elegance, targeting C2-symmetrical, non-hydroxylated congeners such as araguspongine B (Ar B), and Xe C and A, and resolving controversy about the absolute configuration of these compounds. However, the simplifying C2-symmetry is broken in the biologically more intriguing Xe B and dmXe B by the presence of 9-OH and, in the former case, the 3´-CH3 groups.
The synthesis design is depicted in Scheme 1. The final assembly of the 1-oxaquinolizidine units was envisioned to take place by intramolecular N-alkylation of the amide groups in macrocyclic bis(lactam) 1 followed by lactam semireduction and hemiaminal formation. The bis(lactam) was planned to arise from combining two ω-amino acid precursors 2 and 3 by sequential amide formation. These precursors will be prepared from a common starting material, azidoalcohol 4. After its esterification with either 2-(benzyloxy)-5-chloropentanoic acid (5) or 5-chloropentanoic acid, intermediates 2 and 3 will be accessed via Ireland-Claisen rearrangement to establish stereogenic centers at C9’ and C9, respectively.
Scheme 1.

Synthesis design for dmXe B.
This synthesis design has the advantage of flexibility in accessing 1-oxaquinolizidine alkaloids with variable substitution at C9/9’ (with or without OH groups), as well as variable stereochemistry at the same positions.
1-Chloro-3-butene served as the starting point for the synthesis of 4 (Scheme 2a). Jacobsen hydrolytic kinetic resolution of 1-chlorobutene oxide, obtained by epoxidation with MCPBA, delivered (S)-6 in 58% yield and 92% ee.7 The epoxide was distributed divergently, with part of it being advanced to allylic alcohol 7 with dimethylmethylenesulfur ylide.8 After reductive para-methoxybenzylation (PMB) under electrophilic conditions and displacement of the chloro group, iodide 8 was obtained in a 71% overall yield.9 The development of electrophilic benzylation was required due to the propensity of 7 to undergo intramolecular etherification during alkoxide formation under a number of reaction conditions. Cuprate coupling with the remainder of chloro epoxide (S)-6 with the reagent formed from 8 was accomplished in 78% yield.10 Subsequent substitution of chloride with sodium azide, O-silylation, and removal of the PMB group delivered 58.1 grams of 4 in 87% yield.
Scheme 2.

Preparation of early stage intermediates 4 and 14.
In order to access non-symmetrical congeners such as dmXe B possessing a C9 hydroxy group, the synthesis of an α-benzyloxy acyl chloride 14 was required. Due to the thermal instability of ester enolates generated from α-alkoxy esters, alkylation of t-butyl α-benzyloxyacetate with iodide 10 was accomplished at –95 °C in moderate yield of 45% under optimized reaction conditions. After desilylation, mesylation, and substitution with LiCl, ester 13 was obtained in 86% yield. Cleavage of the t-butyl ester and chlorodehydroxylation with oxalyl chloride completed the synthesis of acyl chloride 14 (93% yield, 10.2 grams).
In preparation for the bis(macrolacram) assembly, the individual amino acid synthons were obtained by Ireland-Claisen rearrangement (ICR) of two esters prepared from allylic alcohol 4 (Scheme 3a and b).11 Intermediate 2 was obtained from ester 15 (82% yield, dr 10:1) under reaction conditions developed specifically for α-alkoxy esters, where we found that the best chelation-controlled outcome is achieved with KN(SiMe3)2/PhMe combination, while, surprisingly, LDA/THF affords non-chelation controlled diastereomer with good selectivity, despite the expected higher propensity of lithium to coordinate with oxygen Lewis bases.12 ICR of ester 16 gave rise to carboxylic acid 17 (87% yield, dr 6:1), which, after forming the methyl ester with Me3SiCHN2, was advanced to amino ester 3 by azide reduction with SnCl2/PhSH (82% yield).13
Scheme 3.

Fragment union and completion of the synthesis.
Fragment union was achieved by amide formation from acid 2 and amine 3, followed by another azide reduction under the same conditions, ester hydrolysis, and macrolactamization.14 At this stage, the minor diastereomers formed during ICR were separable, and macrocyclic bis(lactam) 1 was isolated in 35% yield as a single diastereomer.
Completion of the synthesis follows a bidirectional strategy, starting with the formation of two valerolactam moieties by intramolecular N-alkylation of 1 (LiN(SiMe3)2, THF, 85% yield). Removal of the silyl groups preceded the crucial reductive 1-oxaquinolizidine formation with Li-NH3 reagent, which simultaneously accomplished O-debenzylation.15 Notably, numerous alternative reagents attempted to achieve semireduction of the lactams (iBu2AlH,16 NaAlH2(OR)2,17 IrCl(CO)(PPh3)2/(HMe2Si)2O),18 consistently resulted in complete reduction to piperidine. Partial α-dehydroxylation leading to a minor amount of 22 was also observed.
The final hydrogenation of the double bonds required optimization, as the typical conditions using Pd/C in various solvents proved inconsistent and occasionally resulted in an intractable mixture of products. However, hydrogenation of 21 and 22 with Rh/Al2O3 in ethyl acetate19 reliably and cleanly delivered dmXe B (94% yield) and araguspongine B, (Ar B, 97% yield), respectively.
To further highlight the generality of the strategy, we completed the synthesis of bis-hydroxylated Ar C, which has not been accessed previously by synthesis (Scheme 4, See Supporting Information for the synthesis of 23 and 24). In this case, we opted for p-methoxybenzyl (PMB) ether at C9/9’ to test an alternative deprotection/reductive oxaquinolizine construction method. We also utilized allyl ester in 24 to simplify access to the free acid, as the methyl ester proved resistant to hydrolysis. Thus, after acid 23 and amine 24 were combined by amide formation, azide reduction, acid deallylation, and macrolactamization afforded bis-lactam 25. Six-membered lactam closure was accomplished with LiN(SiMe3)2 as previously, and removal of TBS and PMB groups delivered 26 in high yield. Finally, reduction of the lactam with Li-NH3 and hydrogenation completed the synthesis of Ar C.
Scheme 4.

The synthesis of araguspongine C (Ar C).
Having established access to synthetic dmXe B, we proceeded to evaluate its effect on IP3R-mediated calcium release, mitochondrial respiration and selective anticancer activity in breast cancer cell lines, and how it compares to Xe B isolated previously from a marine sponge. As shown in Figure 2a, 30 min incubation with either dmXe B or Xe B completely abolished IP3R calcium release induced by ATP in MDA-MB-231 cells. Subsequently, the normal cell line MCF10A and a cancer cell line MDA-MB-231 were treated with increasing concentrations of dmXe B and Xe B for 24 h, and oxygen consumption rates (OCR) were determined using Seahorse system. As shown in Figure 2b, both compounds decrease OCR similarly in a dose-dependent fashion. A similar effect was observed in a triple-negative cell line BT-549 and the luminal A MCF7 cell line (See Supporting Information).
Figure 2.

Average cytosolic calcium increases in response to ATP (100 μM) in MDA-MB-231 cells treated with ethanol (vehicle, control) or 5 μM Xe B (upper panel, red trace) or dmXe B (bottom panel, blue trace) for 1 h. Mean ± SEM of 3 independent experiments. In each experiment, 100 cells were analyzed. b. Basal oxygen consumption rate (OCR) of MDA-MB-231 (upper panel) and MCF10A (bottom panel) cells incubated for 24h with increasing concentration of either Xe B (red) or dmXe B (blue). The black bar represents cells basal OCR without treatment. Mean ± SEM of 3 independent experiments with 10 replicates each. *p < 0.05, **p < 0.01, ***p < 0.001 compared to respective control. c. MDA-MB-231 (upper panel) and MCF10A (lower panel) were treated with increasing concentrations of either Xe B (upper panel, red) or dmXe B (lower panel, blue) for 24 h and cell death was determined by propidium iodide incorporation by flow cytometry. Mean ± SEM of 3 independent experiments, each in triplicate. *p < 0.05, ***p < 0.001 compared to respective control. d. Representative plates of three different experiments with MDA-MB-231 cells treated with 7.5 μM of either Xe B (upper panel) or dmXe B (bottom panel).
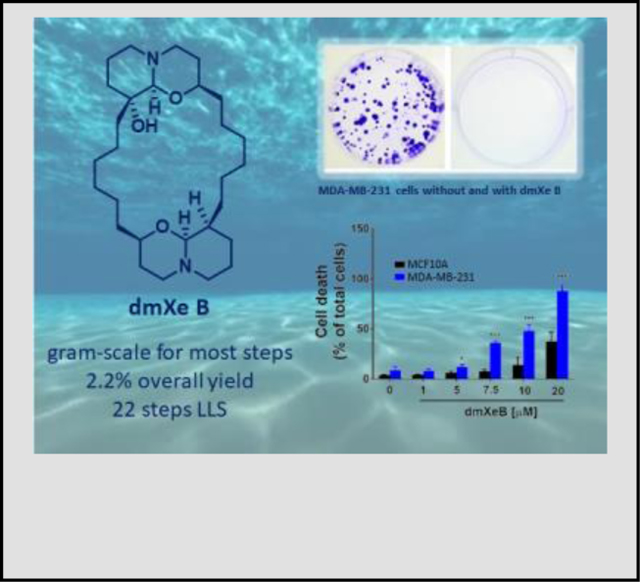
We have also demonstrated that dmXe B induces selective cell death in breast cancer cell lines. Several breast cancer cell lines that represent heterogeneity of breast cancer and the normal cell line MCF10A were treated with different concentrations of either Xe B or dmXe B for 24 h and cell death was measured by propidium iodide incorporation through flow cytometry. As shown in Figure 2c, 7.5 μM Xe B induced a significant increase in cell death in MDA-MB-231 cells, without affecting the normal cell line MCF10A. At 10 μM, Xe B is still more effective in MDA-MB-231 cells, but some effects in MCF10A cells become measurable, indicating reduced selectivity. On the other hand, dmXe B in fact shows somewhat increase selectivity, inducing cell death in MDA-MB-231 at 5 μM, which is sustained at 10 μM with no effect on normal MCF10 cells (Figure 2c). At 20 μM, MDA-MB-231 cells are still more sensitive, with almost 100% of the population dead, whereas MCF10A shows an initial increase in cell death. In cell lines representative of luminal A (MCF7) and luminal B (ZR75) breast cancer cell lines Xe B and dmXe B display selective cell death at concentrations between 5 and 10 μM. Similarly to the observations with MDA-MB-231 cell line, dmXe B at 20 μM concentration caused an almost complete cell death in ZR75 and MCF7 cell lines, while also affecting the normal cell line MCF10A to a much lesser extent. The BT-549 cells are more resistant to both compounds, showing only selectivity at 7.5 μM. Finally, we determined whether dmXe B affected the ability of cancer cells to proliferate indefinitely (one of the hallmarks of cancer), as demonstrated previously for Xe B.4 MDA-MB-231 cells were treated with either compound for 24 h and then one thousand cells were collected and reseeded. After one week, we evaluated colony formation and found that at 7.5 μM the synthetic dmXe B, similarly to natural Xe B, completely abolished this phenomenon (Figure 2d).
In summary, we developed a scalable synthesis of dmXe B, taking advantage of a convergent strategy allowing the preparation of xestospongins with variable stereochemistry and oxidation at C9/9’, as attested to by the synthesis of Ar C, which has not been accessed by previously described strategies. The hallmarks of the strategy include a strategic application of ICR to achieve the desired makeup of the C9/9’ stereocenters, assembly of the macrocyclic core by macrolactamization, and late-stage 1-oxaquinolizidine construction by amide reduction. With dmXe B in hand, we established that it is an effective inhibitor of the constitutive ER-to-mitochondria calcium transfer, and subsequently confirmed its ability to induce selective cancer cell death in a variety of cell lines, including metastatic cancer, while leaving normal cells nearly unaffected. According to a recent study, these effects are associated with a drop in the mitochondrial activity of the calcium-sensitive α-ketoglutarate dehydrogenase, a key enzyme in oxidative phosphorylation and reductive carboxylation required for cancer cell survival.20
Supplementary Material
Acknowledgements
This work was supported by ANID/FONDECYT grants 1160332 and 1200255, ANID/FONDAP grant 15150012 (CC), UC CRCC grant CRR-19-577249, and the NIGMS R01-077379 (AZ). MP is a recipient of the Tantiwiwat Fellowship, Mabe Memorial Fellowship, and UCSB Travel grant.We thank Dr. Hongjun Zhou for the assistance with the NMR spectrometry, Dr. Dmitri Uchenik for the assistance with mass spectroscopy, and Dr. Guang Wu for the assistance with X-ray crystallography.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Nakagawa M, Endo M, Tanaka N, Gen-Pei L, Tetrahedron Lett. 1984, 25, 3227–3230. [Google Scholar]; b) Moon S-S, MacMillan JB, Olmstead MM, Ta TA, Pessah IN, Molinski TF, J. Nat. Prod 2002, 65, 249–254. [DOI] [PubMed] [Google Scholar]; c) Kobayashi M, Kawazoe K, Kitagawa I, Chem. Pharm. Bull 1989, 37, 1676- . [DOI] [PubMed] [Google Scholar]; d) Reddy MVR, Faulkner DJ, Nat. Prod. Lett 1997, 11, 53–59. [Google Scholar]; e) Venkateswarlu Y, Reddy MVR, Rao JV, J. Nat. Prod 1994, 57, 1283–1285. [Google Scholar]; f) Kobayashi M, Yasuhisa M, Aoki S, Murakami N, Kitagawa I, In Y, Ishida T, Heterocycles 1998, 47, 195–203. [Google Scholar]
- [2].a) Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN, Neuron 1997, 19, 723–733. [DOI] [PubMed] [Google Scholar]; b) Jaimovich E, Mattei C, Liberona JL, Cardenas C, Estrada M, Barbier J, Debitus C, Laurent D, Molgo J, FEBS Lett. 2005, 579, 2051–2057. [DOI] [PubMed] [Google Scholar]
- [3].Cárdenas C, Miller RA, Smith I, Bui T, Molgo J, Müller M, Vais H, Cheung KH, Yang J, Parker I, Thompson C, Birnbaum M, Hallows KR, Foskett JK Cell 2010, 142, 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cardenas C, Muller M, McNeal A, Lovy A, Jaňa F, Bustos G, Smith N, Molgo J, Diehl A, Ridky TW, Foskett JK. Cell Rep. 2016, 14, 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baldwin JE, Melman A, Lee V, Firkin CR, Whitehead RC, J. Am. Chem. Soc 1998, 120, 8559–8560. [Google Scholar]
- [6].a) Hoye TR, North JT, Yao LJ, J. Am. Chem. Soc 1994, 116, 2617–2618. [Google Scholar]; b) Hoye TR, Ye Z, Yao LJ, North JT, J. Am. Chem. Soc 1996, 118, 12074–12081. [Google Scholar]
- [7].Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN, J. Am. Chem. Soc 2002, 124, 1307–1315. [DOI] [PubMed] [Google Scholar]
- [8].Alcaraz L, Hartnett JJ, Mioskowski C, Martel JP, Le Gall T, Shin DS, Falck JR, Tetrahedron Lett. 1994, 35, 5449–5452. [Google Scholar]
- [9].Morimoto Y, Iwahashi M, Nishida K, Hayashi Y, Shirahama H, Angew. Chem. Int. Ed. Engl 1996, 35, 904–906. [Google Scholar]
- [10].Lipshutz BH, Kozlowski JA, Parker DA, Nguyen SL, McCarthy KE, J. Organomet. Chem 1985, 285, 437–447. [Google Scholar]
- [11].a) Ireland RE, Mueller RH, Willard AK, J. Am. Chem. Soc 1976, 98, 2868–2877. [Google Scholar]; b) Ilardi EA, Stivala CE, Zakarian A, Chem. Soc. Rev 2009, 38, 2133–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Podunavac M, Lacharity JJ, Jones KE, Zakarian A, Org. Lett 2018, 20, 4867–4870. [DOI] [PubMed] [Google Scholar]
- [13].Bartra M, Romea P, Urpi F, Vilarrasa J, Tetrahedron, 1990, 46, 587–594. [Google Scholar]
- [14].El-Faham A, Albericio F, Chem. Rev 2011, 111, 6557–6602. [DOI] [PubMed] [Google Scholar]
- [15].a) Allan KM, Stoltz BM, J. Am. Chem. Soc 2008, 130, 17270–17271. [DOI] [PubMed] [Google Scholar]; b) Evans DA, Illig CR, Saddler JC, J. Am. Chem. Soc 1986, 108, 2478–2479. [DOI] [PubMed] [Google Scholar]
- [16].a) Kim S, Ahn KH, J. Org. Chem 1984, 49, 17–17-1724. [Google Scholar]; b) Ahn KH, Lee SJ Tetrahedron Lett. 1992, 33, 507–510. [Google Scholar]
- [17].a) Roussi F, Quirion J-C, Tomas A, Husson H-P, Tetrahedron 1998, 54, 10363–10378. [Google Scholar]; b) Amat M, Llor N, Hidalgo J, Escolano C, Bosch J, J. Org. Chem 2003, 68, 1919–1928. [DOI] [PubMed] [Google Scholar]
- [18].a) Gergory AW, Chambers A, Hawkins A, Jakubec P, Dixon DJ, Chem.-Eur. J 2015, 21, 111–114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tan PW, Seayad J, Dixon DJ, Angew. Chem. Int. Ed 2016, 55, 13436–13440. [DOI] [PubMed] [Google Scholar]
- [19].a) Berkowitz WF, Choudhry SC, Hrabie JA, J. Org. Chem 1982, 47, 824–829. [Google Scholar]; b) Golan O, Goren Z, Biali SE J. Am. Chem. Soc 1990, 112, 9300–9307. [Google Scholar]
- [20].Cardenas C, Lovy A, Silva-Pavez E, Urra F, Mizzoni C, Ahumada-Castro U, Bustos G, Jaña F, Cruz P, Farias P, Mendoza E, Huerta H, Murgas P, Hunter M, Rios M, Cerda O, Georgakoudi I, Zakarian A, Molgó J, Fosket JK, Sci. Signaling 2020, 13, doi: 10.1126/scisignal.aay1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.