

Abstract
Although considerable efforts have been conducted to diagnose, improve, and treat cancer in the past few decades, existing therapeutic options are insufficient, as mortality and morbidity rates remain high. Perhaps the best hope for substantial improvement lies in early detection. Recent advances in nanotechnology are expected to increase the current understanding of tumor biology, and will allow nanomaterials to be used for targeting and imaging both in vitro and in vivo experimental models. Owing to their intrinsic physicochemical characteristics, nanostructures (NSs) are valuable tools that have received much attention in nanoimaging. Consequently, rationally designed NSs have been successfully employed in cancer imaging for targeting cancer-specific or cancer-associated molecules and pathways. This review categorizes imaging and targeting approaches according to cancer type, and also highlights some new safe approaches involving membrane-coated nanoparticles, tumor cell-derived extracellular vesicles, circulating tumor cells, cell-free DNAs, and cancer stem cells in the hope of developing more precise targeting and multifunctional nanotechnology-based imaging probes in the future.
Keywords: active targeting, nanotechnology-based imaging, passive targeting, targeted nanoplatforms, tumor biology
1. Introduction
Cancer is a complex disorder resulting from several alterations in biological processes and signaling pathways. Malignant tumors continue to be the source of a high level of morbidity and mortality throughout the world.[1] The cellular and molecular heterogeneity within a single tumor plays a key role in tumor progression and the failure of conventional therapies. However, researchers are still at the very beginning of understanding the full panorama of cancer biology, and whether cancer heterogeneity plays a role in specific cancer types.[2] Before targeted delivery approaches can be rationally designed, a better understanding of the biological processes involved in the biodistribution, release, and retention of targeted delivery systems inside the tumor is imperative. Along with the advances in cancer biology, new challenges and possible treatment options are at the forefront of cancer research. The complexity, heterogeneity, low concentrations of biomarkers, and unpredictable behavior of cancer calls remain major barriers to the development of novel treatment strategies.[3]
An optimized tumor-selective delivery system for imaging or therapeutic cargos would deliver the cargo to the correct target to achieve precise and efficient detection of the tumor resulting in the least possible systemic toxicity. However, targeted delivery faces many challenges due to the heterogeneity and diversity of the tumor targets. Keeping this in mind, categorization of tumor biomarkers (as an indicator of pathogenic processes, normal biological processes, or response to an exposure or therapeutic intervention) is necessary. An often used tumor selective delivery approach relies on the incorporation of the appropriate molecular species to target cancer cells.[4]
Quantitative measurement of contrast agents is desirable to establish automated algorithms and guidelines for the detection, real time monitoring, and evaluation of outcomes of cancer therapy. Imaging modalities based on contrast agents can be divided into four groups including: i) optical methods such as NIFI (near infrared fluorescent imaging), resonance energy transfer Raman, OCT (optical coherence tomography), and PAI (photoacoustic imaging); ii) magnetic methods such as MRI (magnetic resonance imaging), MAI (magneto-motive-approach imaging), and MPI (magnetic particle imaging); iii) acoustic methods such as USI (ultrasound imaging); and iv) nuclear methods such as CT (computed tomography), PET (positron emission tomography), SPECT (single photon emission computed tomography), and ϒ-imaging.[5] Many of the modalities used in clinics today, fall under the category of tomographic imaging, which relies on deeply penetrating radiation to probe structural and functional information of the imaged area in three dimensions. The clinical applications of many of these different imaging modalities have been reviewed (see refs. [5,6]). Despite all the benefits of clinical imaging techniques, these approaches are still faced with limitations to their clinical utility. Optical imaging approaches present several advantages, such as being less-expensive, higher spatial resolution, and avoiding harm associated with ionizing radiation. Nevertheless, the limited penetration depth of light restricts the use of optical contrast agents, especially in clinical application such as image-guided surgery, colonoscopy, and endoscopy.[7] The advantages and disadvantages of these imaging strategies are summarized in Table 1.
Table 1.
Conventional imaging modalities that are presently used in the clinic.
| Modality | Advantage | Limitation | Input Signal type | Resolution [mm] | Penetration depth |
|---|---|---|---|---|---|
| CT | Rapid, accurate, moderate cost, reproducible, widely available | Limited resolution, imaging interpretation difficult, ionizing radiation | X-ray | 25–200 μm (preclinical), 0.5–1 mm (clinical) | Unlimited |
| MRI | Soft tissue contrast, high resolution, customizable molecular targeting, cell tracking | High cost, large equipment required, limited sensitivity, requires contrast agent | Radio frequency | 25–100 μm (preclinical), ≈1 mm (clinical) | Unlimited |
| USI | Rapid, accurate, low cost, reproducibility, widely available | Limited resolution, image interpretation difficult, artifacts common | Sound waves | 10–00 μm (at ≈mm depth); 1–2 cm (at ≈cm depth) | 10 ms |
| PET | Quantification of metabolism and blood flow, high sensitivity, many radionuclide tracers available | High cost, limited availability, large equipment required, short tracer half-life, single process evaluation | Radionuclide (positron emitter) | <1 mm (preclinical), ≈5 mm (clinical) | Unlimited |
| PAT | Reduced tissue scattering, high resolution, non-ionizing/non-radioactive, no acoustic noise, high penetration depth, high resolution | Limited path length, dependence to temperature, weak absorption at short wavelengths. | Light | 5 μm-1 mm (depth-dependent) | <6 cm |
| SPECT | 3D imaging, widely available, highly sensitive, simultaneous imaging of multiple processes | Limited temporal resolution, few radionuclide tracers | Radionuclide (γ-ray emitter) | 0.5–2 mm (preclinical), 8–10 mm (clinical) | Unlimited |
| NIFI | Low cost, widely available | Photobleaching, low quantum yield, shallow tissue penetration | Ultraviolet to near infrared light | 2–3 mm | <2 cm |
Regarding these limitations, nanotechnology is a promising field at the forefront of cancer detection research. Considerable efforts have been made to create a variety of targeted molecular imaging nanoplatforms with unique features and capabilities. As opposed to conventional imaging techniques, tumor-selective imaging probes would deliver an optimized imaging agent to a specific target with high affinity, specificity, and sensitivity. Second, a lower but effective dosage of tumor-selective therapeutic nanoplatforms could efficiently localize in the tumor with minimized systemic toxicity. Moreover, it is possible to monitor and confirm whether the nanoplatform-based tumor-selective imaging probes have been properly delivered to the targeted site after injection. Compared to traditional imaging agents, the amount of injected tumor-selective imaging probes that actually reaches the tumor site can be quantitatively analyzed. The biodistribution of these probes within the body can also be monitored over a long period of time. Therefore, targeted nanoplatforms have the potential to significantly increase imaging contrast, enabling cancer detection at earlier stages, and allow monitoring of tumor response to conventional therapy or molecular targeted therapy.[8]
Some other benefits of using targeted nanoprobes are: 1) imaging nanoplatforms can be used to monitor the changes in the molecular microenvironment associated with tumors; 2) the integration of imaging and therapeutic capabilities provides a combined diagnostic-therapy that has been termed a “theranostic approach.”[1] Theranostic systems are able to reduce toxicity, enhance selectivity and targeting, generate data for diagnostics as well as enhance the therapeutic efficacy.[6]
One major aspect of cancer biology is concerned with the interactions between tumor cells, stroma, and the tumor microenvironment (TME). Passive and active strategies have been employed for the targeting of malignant tumors. The passive targeting mechanism, called “enhanced permeability and retention” (EPR) relies on intrinsic properties of the TME produced by angiogenesis processes, enzymes, etc. Active targeting of tumors involves molecular recognition taking place between the nanoplatform and tumor-specific biomarkers expressed on the TME, endothelial cell surface, and cancer cells. The interaction of molecular targets with nanoplatforms can provide: i) more precise selection of targeted contrast agents; ii) simultaneous detection and therapy; iii) more accurate evaluation of therapeutic outcomes; and iv) patient stratification to select those likely to benefit from targeted contrast agent imaging (personalized medicine).[9] The present review discusses the six most prevalent tumor types according to the National Cancer Institute (NCI). Additionally, brain and ovarian cancer (with high mortality rates) are discussed in detail.
Clinical trials exploiting cutting-edge, nanoparticle-based imaging platforms are increasing. The emerging challenges of current targeted imaging approaches are summarized. Novel strategies including dual-targeting of nanoplatforms, utilizing natural and more biocompatible imaging delivery systems, such as tumor-derived extracellular vehicles (TEV), membrane coated carriers, circulating tumor cells, tumor-associated DNA, and CSCs are covered.
2. Passive Targeting
Passive targeting exploits the accumulation of nanocarriers at the site of interest, such as tumors. The convection (or passive-diffusion process) is mediated by the transportation of nanocarriers via pores in leaky capillaries that are present in tumor masses, and in tissues that trigger angiogenesis. This process occurs in conjunction with the EPR effect. However, passive targeting cannot be classified as a type of selective targeting. The EPR effect does not only apply to tumors, but also to off-target organs such as the spleen, liver, and lungs.
2.1. Enhanced Permeability and Retention Effect
As the size of a tumor approaches 2 mm3, the delivery of nutrients and oxygen by simple diffusion is insufficient. Therefore, the formation of new blood vessels in the TME is essential to support the rapidly growth of malignant tumors.[10] The occurrence of hypoxia in the developing tumors due to lack of blood supply triggers the release of angiogenic growth factors from the neoplastic tissue, thus enabling further tumor growth. The imbalance between angiogenic growth factors and matrix metalloproteinase in neoplastic tissues results in pronounced vessel disorganization, which is associated with the formation of highly porous, large-gap junctions between the endothelial cells. Overall, the defective basement membranes and incomplete coverage with perivascular cells promotes leakage and accumulation of administered agents in tumor tissues.[11]
The smooth muscle layer that surrounds the endothelial cells (EC) in normal blood vessels is non-existent in tumor blood vessels.[12] Typical normal vasculature possesses tight, impermeable EC junctions. Molecules that are >2–4 nm in diameter are unable to pass through the junctions of normal vessels. Hence, larger NSs are generally excluded from normal tissues and organs. Neoplastic tissues with leaky vasculature permit entry of macromolecules up to 600 nm diameter into the tissue [11b,13]. Moreover, the defective lymphatic drainage system in tumor tissues enables NSs to remain in the neoplastic tissue for a prolonged period of time.[14] The EPR effect plays a key role in the delivery of targeted agents to the TME[15] (Figure 1).
Figure 1.
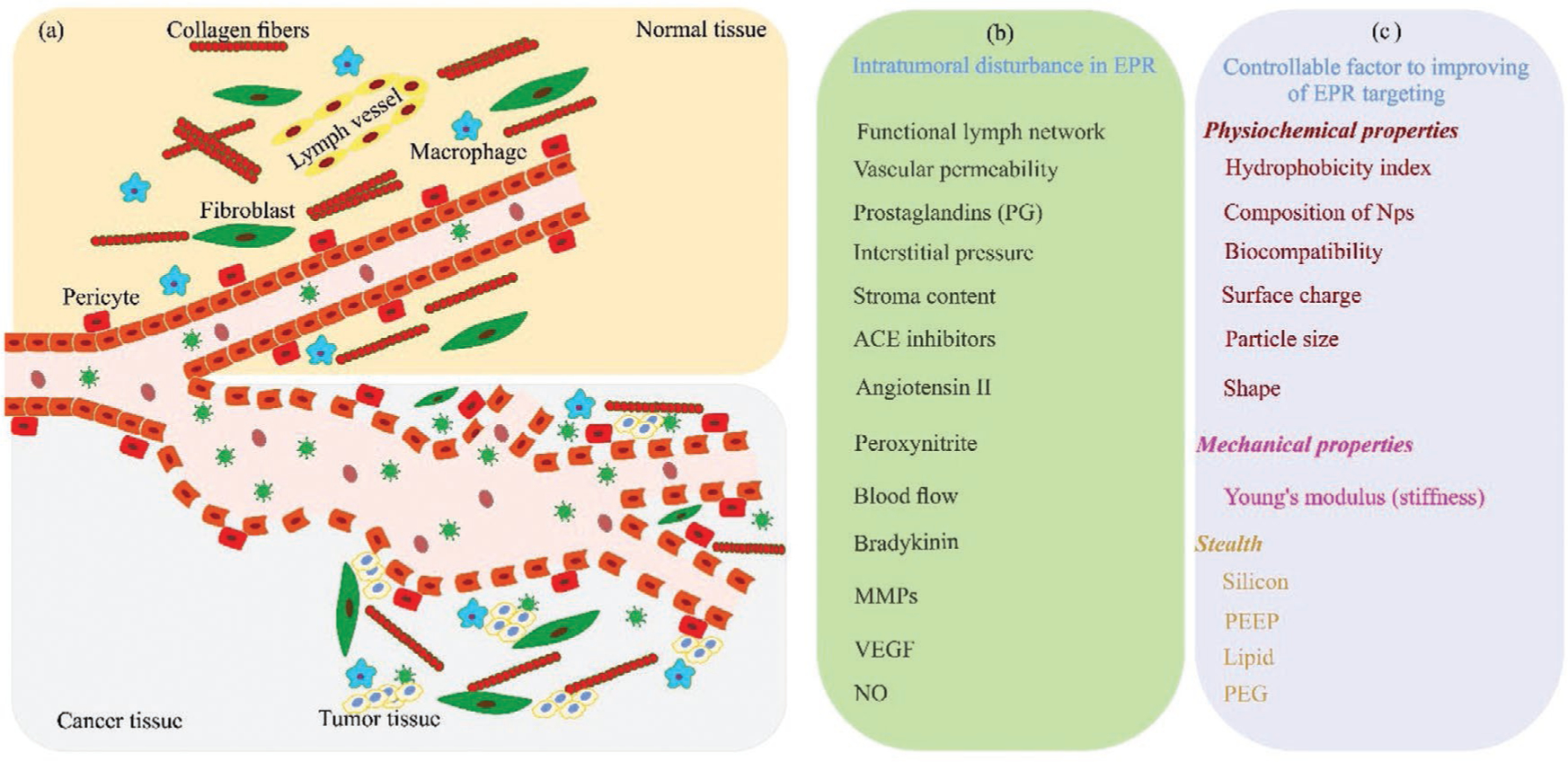
The EPR effect. a) Schematic illustration of the EPR effect in cancer and normal tissue. b) Altered tissue properties in cancerous tissue. c) Controllable factors to improve the EPR effect targeting. The stealth design of NPs aims to have maximum circulation half-life to ensure continuous delivery into the tumor site via the leaky vasculature. ACE, angiotensin-converting-enzyme; MMPs, matrix metalloproteinases; VEGF, vascular endothelial growth factors; NO, nitric oxide; PEG, poly ethylene glycol; PEEP, poly (ethyl ethylene phosphate).
The EPR effect was first observed in living animals using a fluorescent dye “Evans blue.” After injection of the dye, the tumor mass was selectively stained blue.[16] The EPR effect is highly variable, and differs according to the tumor type, mass, size, and the location of penetration and accumulation within the tumor mass. Furthermore, the mononuclear phagocyte system (MPS) and tumor-associated immune cell activity modulates the circulation profile of the targeted agent, tumor transport, accumulation, and release.
The pore size of the leaky vasculature varies significantly across different tumor types. Pore size plays a vital role in tumor accumulation of targeted agents. Because the pore sizes vary so greatly, designing a particle with an optimal shape and size is an extremely difficult task. For example, one study compared rhodamine B with MW 479 Da, with tetramethyl-rhodamine isothiocyanate conjugated-bovine serum albumin (TRITC-BSA) with MW 67 000 Da (Figure 2). The observation was that rhodamine B did not provide an appreciable amount of fluorescence in the tumor mass. However, TRITC-BSA emitted a considerable amount of fluorescence, which remained in the tumor, even 72 h after injection.[17] Another study used indocyanine green (ICG) for evaluating hepatic function in healthy mice.[18] The ICG that was initially bound to albumin and globulin was released as a free dye until it was cleared from the plasma (half-life <20 min; Figure 3). In contrast, albumin bound to ICG specifically accumulated in the tumor mass in mice. Over time, the accumulation of steadily ICG increased. This effect was not seen in normal tissue because the lymphatic system clears the dye from the bloodstream. Therefore, as the elapsed time increased, the image resolution increased.[17]
Figure 2.
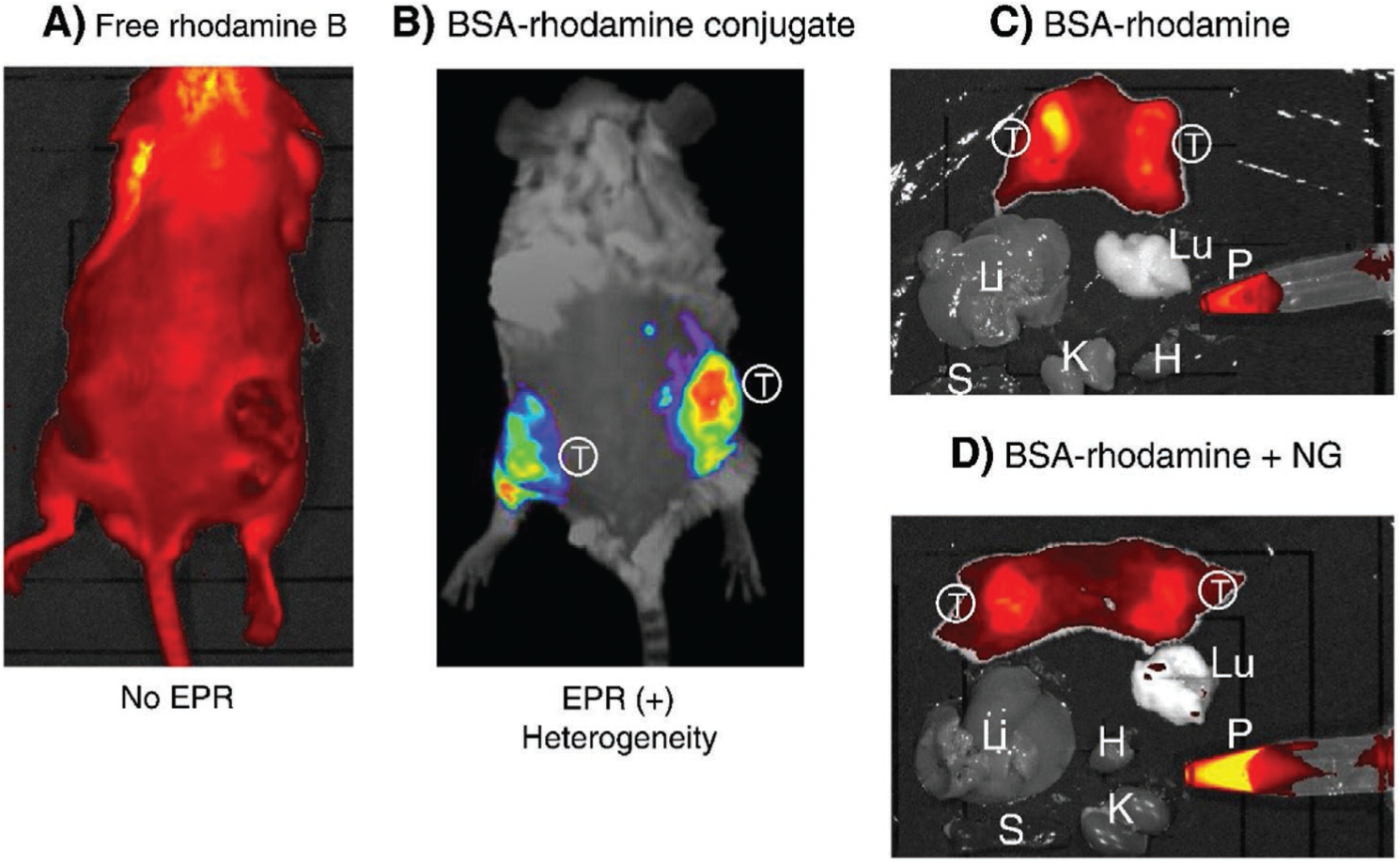
Fluorescence imaging based on EPR effect. The EPR effect-based uptake of a fluorescent imaging nanoprobe in the tumor was compared with uptake of the parental low molecular weight (LMW) fluorescent probe in vivo. A) 24 h after intravenous injection of the LMW fluorescent probe, rhodamine B into S-180 tumor-bearing mice, no distinct tumor is visible. B) Injection of TRITC-BSA (67 kDa) resulting in highly tumor selective fluorescence under the same experimental conditions. C) At 24 h, S-180 tumor-bearing mice were dissected, and each organ was imaged with an IVIS imaging system. Results showed that only tumor tissues showed significant fluorescence. D) Same as (C) except that nitroglycerin (NG) ointment was applied to the skin, and then the EPR effect and tumor targeting were evaluated. In (D), the cut surface of tumor tissues shows a more homogeneous tumor uptake of TRIT-BAS probe, and also more TRIT-BAS remained in the blood, which indicates that the EPR effect depends on time and would increase progressively. In (C) and (D), fluorescence is only seen in the tumor tissue. (S, spleen; T, tumor; Lu, lung; Li, liver; H, heart; P, plasma). Reproduced with permission.[17] Copyright 2013, Elsevier.
Figure 3.
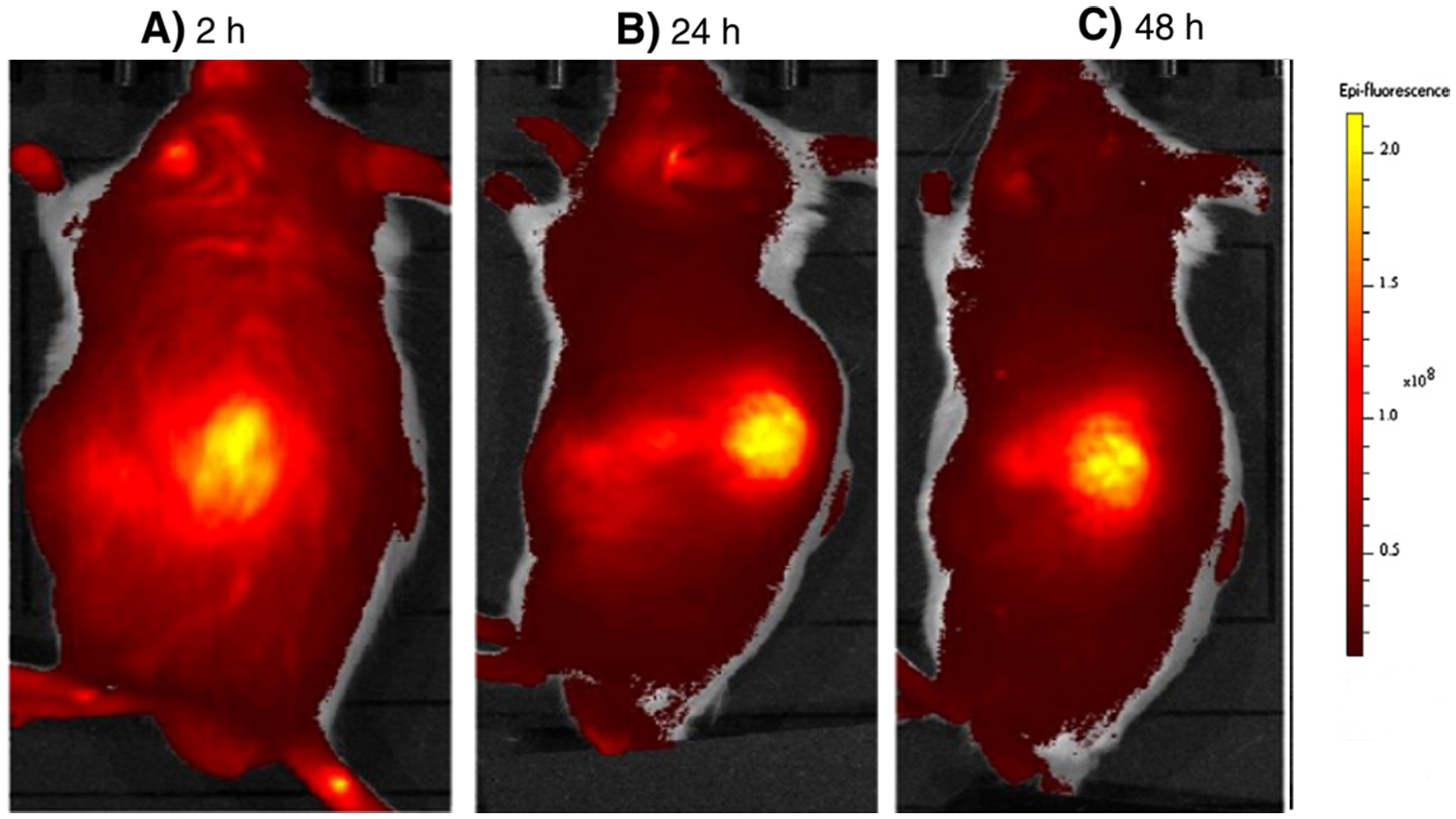
Tumor imaging with indocyanine green (ICG). ICG was injected intravenously into S-180 tumor-bearing mice and fluorescent imaging was carried out at 2, 24, and 48 h by IVIS imaging system. ICG bound to albumin and behaved as a macromolecule. As shown in the figures, the contrast of the tumor images increased as time passed. That is, nonspecific delivery of the agent to normal tissues was cleared via the lymphatic system thus improving the contrast of the tumor image (2 h vs 48 h). Reproduced with permission.[17] Copyright 2013, Elsevier.
The EPR effect is also dependent on the type, size, location, and total blood volume of the tumor. Blood volume is an especially important factor because it influences the biodistribution. In addition, an increase in blood volume is accompanied by an increase in tumor size, and a consequent decrease in tumor uptake of the nanoplatform. The EPR effect in humans has mostly been observed in squamous cell carcinoma of the head and neck, and was only rarely observed in cancers of the lung and breast.[19] The evaluation of the accumulation of various sizes of NPs in four different subcutaneous flank tumor models found that each tumor possessed unique accumulation properties. Another factor, which has a direct impact on tumor accumulation, is the density of microvessels within the tumor. The density of microvessels varies based on tumor type.[20]
In order to develop more efficient targeting methods taking advantage of the EPR effect, recent studies have focused on three topics: i) modification of the EPR effect by using anti-angiogenic agents; ii) reduction of interstitial fluid pressure; and iii) the application of external stimuli (i.e., ultrasound and temperature) for increasing the tumor permeability.[21]
In recent decades, although the number of preclinical studies on the EPR effect in tumor targeted therapy in laboratory animal models has steadily increased, nevertheless, any expected clinical translation has been rather limited. This is due to differences becoming appreciated between mouse (or rat) tumor models and real human tumors. These differences include: i) the metabolic rate and status, host lifespan, and the tumor size relative to the host; ii) the rate of tumor growth (doubling time), which is much faster in rodents; iii) the larger tumor-to-body weight ratio in mice compared to humans that alters the pharmacokinetic effects of nanocarriers; and iv) the heterogeneity of the TME-associated endothelial structure, blood flow rate in tumor regions, pericyte coverage, and ECM (extracellular matrix) density.[22]
3. Active Targeting
Active targeting agents can selectively transport NPs into the tumor mass and they bind to molecules expressed on the cancer cell surface with high affinity leading to endocytosis mediated cell uptake.[23] The surface-functionalized nanocarrier using specific ligands complements the passive targeting approach to improve the efficiency of nanocarrier delivery and tumor localization.[24] Conventional methods of active targeting consist of targeting molecules or receptors that are overexpressed in selected types of cancers.[25] They can be categorized into three subsets, including: i) targetable factors in the TME such as hypoxia, pH, MMP enzymes, and fibronectin; ii) targetable molecules on the surface of tumor EC such as vascular endothelial growth factor (VEGF) receptors, integrins, and vascular cell adhesion molecule-1 (VCAM-1); and iii) targetable molecules on/in the cancer cells themselves such as, transferrin receptor, folate receptor, epidermal growth factor receptor (EGFR), glucose transporter, and cathepsins.[22b]
4. Tumor Microenvironment Targeting
4.1. Hypoxia Targeting
The imbalance between blood vessel formation and the rate of tumor cell proliferation causes hypoxia due to oxygen-deprived conditions. Hypoxia leads to an upregulation in hypoxia inducible factor 1a (HIF-1a) and triggers the unfolded protein response (UPR). Both of these transcription elements influence the expression of genes associated with tumor initiation, progression, malignancy, metastasis, and tumor drug resistance[26] (Figure 4). The decreased O2 concentration causes the accumulation of reduced nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) in the TME resulting in further reduction of the remaining oxygen, and production of more reactive oxygen species (ROS).[27]
Figure 4.
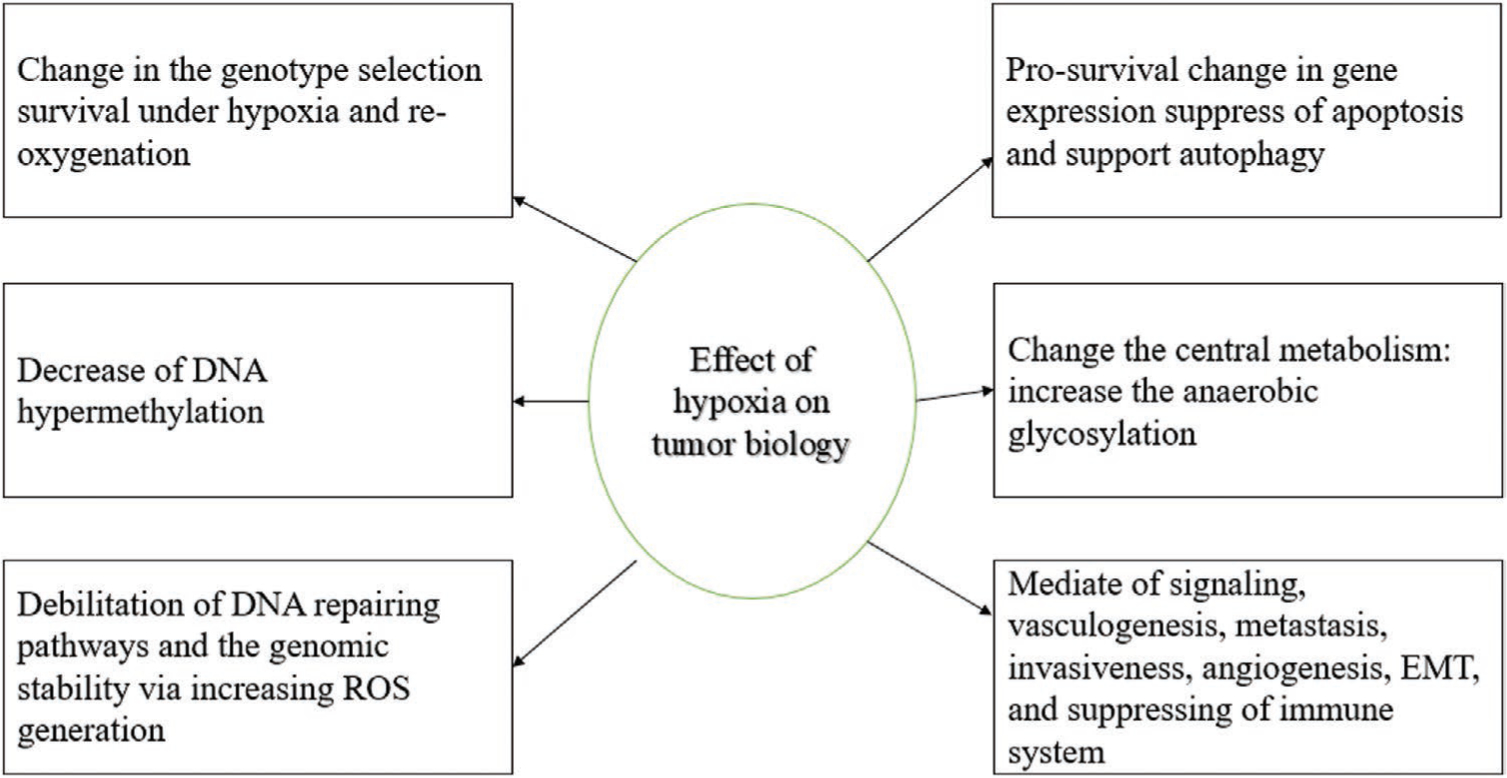
Effects of hypoxia on the tumor biology. TKR, tyrosine kinase receptor, ROS, EMT.[32]
The presence of hypoxia indicates that the cancer cells possess an insufficient blood supply to satisfy their present rate of growth. This fact results in only a small portion of drugs and contrast agents being actually delivered to the cancer site.[28] The redox balance and oxygen concentration are two intrinsic properties that are available for hypoxia targeting.[29] Different techniques are required to monitor the degree of hypoxia and to directly measure pO2 in the cells. The redox balance is mostly influenced by redox agents, such as cysteine and glutathione. The degree of hypoxia in cancerous tissue can be deduced by measuring the local concentration of reducting enzymes such as azoreductase and nitroreductase.[30] Chemical groups such as nitro, azo, and quinone groups can be used to target hypoxia and for reversible sensing between normoxia/hypoxia.
Hypoxia occurs in solid tumors, where O2 levels are <5 mm Hg.[31] Targeting systems have been designed to measure O2 concentrations within the clinically relevant range (0–15 mm Hg). The measurement or imaging of O2 concentration can be performed using three methods: i) a ratiometric sensing probe constructed from an O2-sensitive indicator and an O2-insensitive dye; ii) formation of FRET pairs between donor emission and acceptor absorption bands; and iii) the use of phosphorescence lifetime imaging. Since hypoxia plays a vital role in cancer progression, detecting and measuring it can be effective in cancer detection.[32] One study focused on hypoxia imaging, used a nanoprobe prepared from a poly (N-vinylpyr-rolidone) (PVP)-conjugated iridium (III) complex.[26] The PVP improved the retention time via the EPR effect, and allowed for continuous monitoring of tumor hypoxia. The use of an iridium (III) complex extended the phosphorescence emission (PPE) into the NIR region, improving the depth of penetration of the light. The concentration of O2 in normal tissue is high, causing the PPE to be quenched, but in cancerous tissue the hypoxia caused the PPE to be activated.[26]
While the low concentration of O2 does decrease cancer cell proliferation in the center of a solid tumor, the reduced O2 levels also provide a suitable habitat for anaerobic bacteria to proliferate in hypoxic tumors. The Luo group engineered anaerobic bacteria, including Bifidobacterium breve and Clostridium difficile to serve as cargo-carrying (upconversion nanorods) and antibody-directed (Au nanorod delivery) vehicles for imaging and photothermal ablation of tumors (Figure 5).[33] The in vivo results showed that the antibody-directed strategy had a longer retention time and was more effective for imaging and therapy compared to the cargo-carrying strategy.
Figure 5.
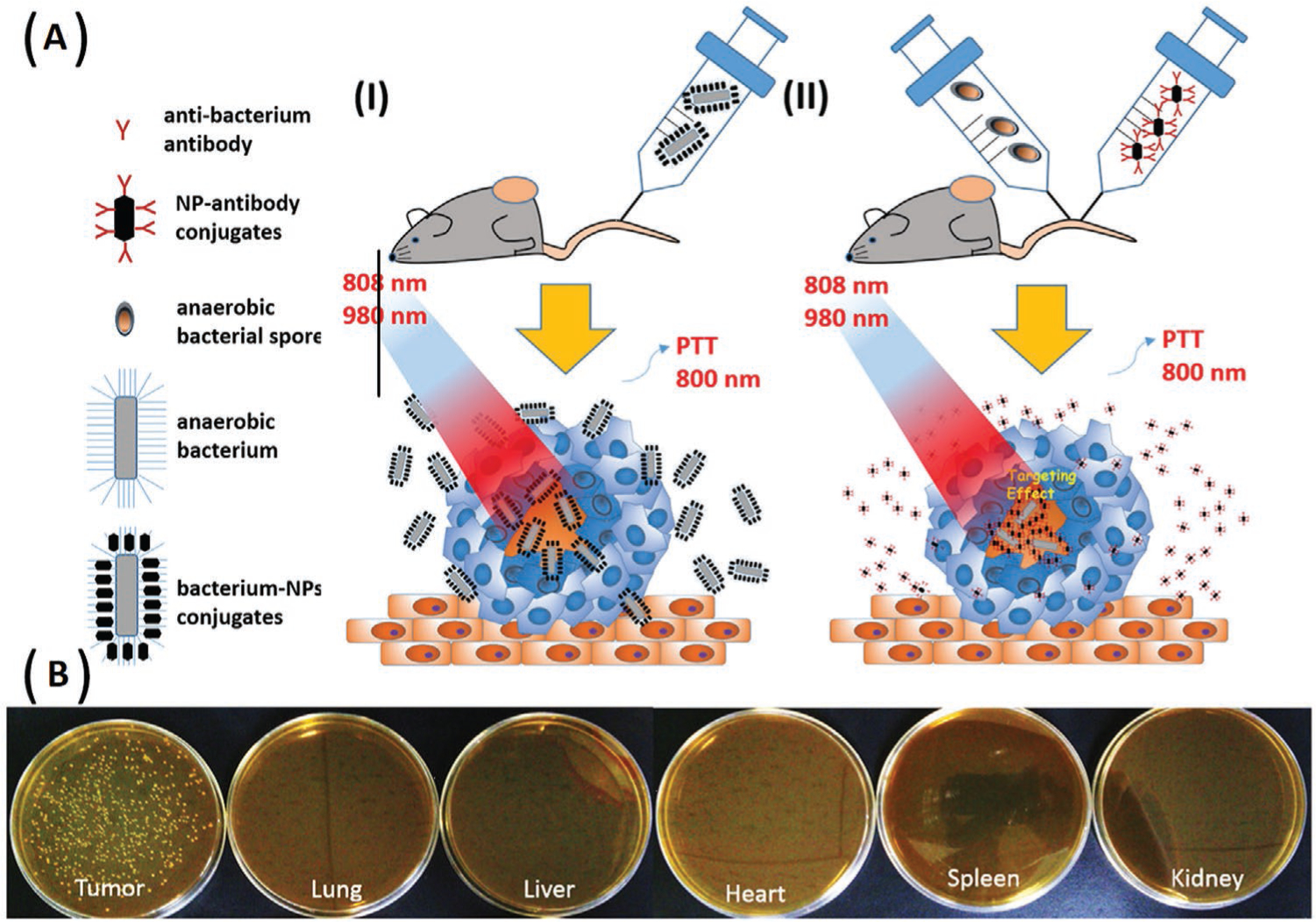
Use of anaerobic bacteria to target tumors. A) The scheme shows two approaches involving anaerobic bacteria to deliver functional NPs; I) a cargo-carrying method, a direct conjugation of NPs to B. breve bacteria, and II) an antibody-directed method involving conjugation of anti-Clostridium polyclonal antibodies onto NPs to trigger the germination of Clostridium spores. B) Selective growth of B. breve in tumor tissues. The tumor bearing mice were intravenously injected with B. breve and sacrificed after two days. The tumor tissues and five major organs (lung, spleen, heart, liver, and kidney) were cultured under an anaerobic environment at 37 °C Adapted with permission.[33] Copyright 2016, American Chemical Society.
Bifunctional therapeutic agents such as Pt (II) porphyrins can be effective agents for the imaging and therapy of cancer under hypoxic conditions. However, Pt (II) porphyrins showed aggregation in aqueous solutions. Addressing this problem, the hydrophilic starburst Pt(II) porphyrins (Pt-1, Pt-2, and Pt-3) with four cationic fluorene oligomeric arms could provide increased water solubility and prevent the aggregation of Pt(II) porphyrins.[34] Among the tested compounds, Pt-3 showed the best results for oxygen-sensing and the highest singlet oxygen quantum yield, and was chosen to serve as both a photosensitizer and an oxygen probe for simultaneous photodynamic therapy (PDT) and real time monitoring of cancer hypoxia (Figure 6).
Figure 6.
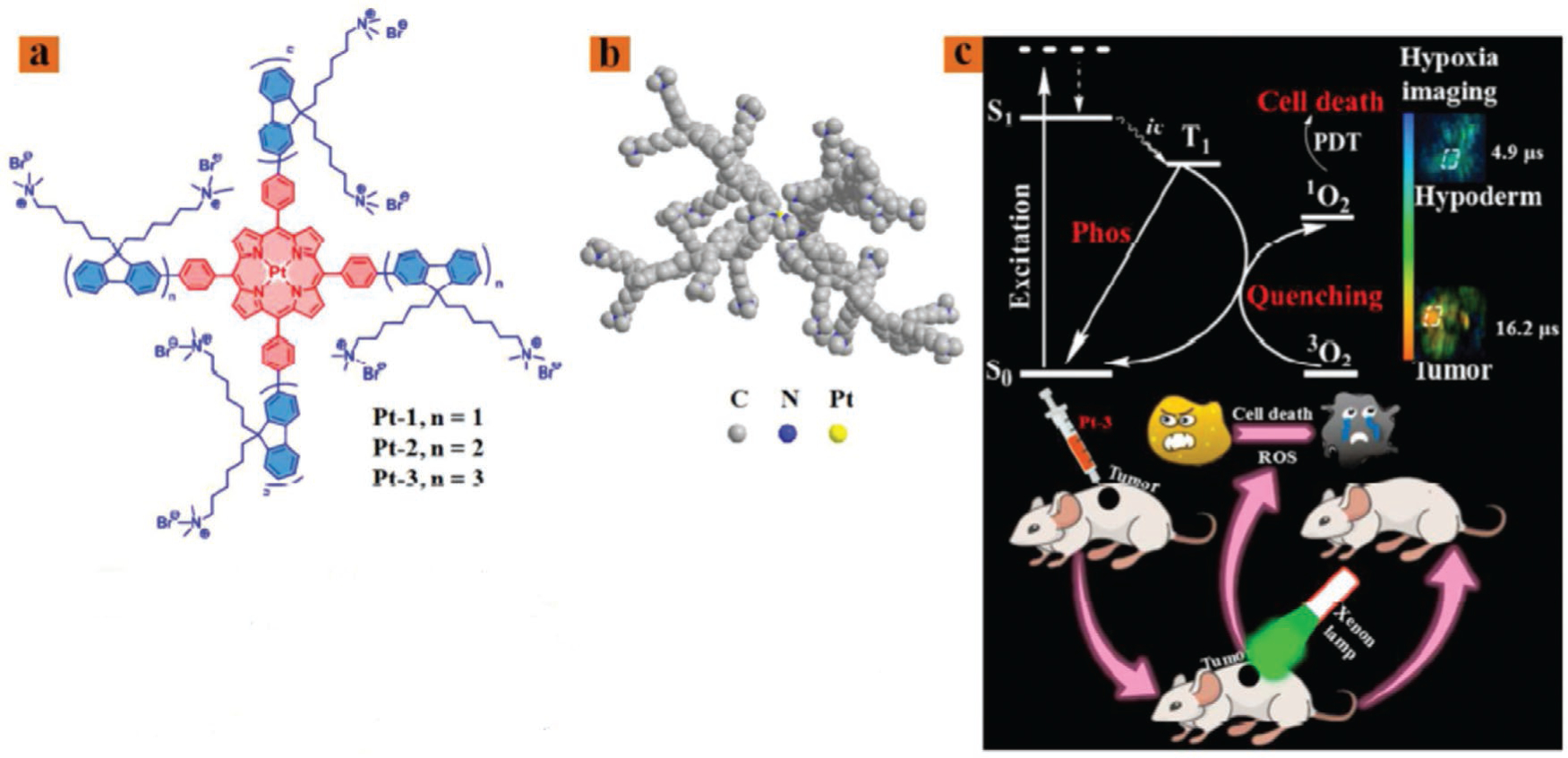
Bifunctional Pt (II) porphyrins. a) Structures of Pt-1, Pt-2, and Pt-3. b) The optimized 3D model of Pt-3 simulated by ChemBio3D. c) Schematic illustration of Pt-3 used as a bifunctional agent for tumor hypoxia imaging and PDT. Adapted with permission.[34] Copyright 2018, American Chemical Society.
Conventional cancer imaging methods can be invasive, and have low specificity and image resolution.[35] The designed hypoxia probe 1 (HyP-1) employing an N-oxide-based trigger could allow facile bio-reduction mediated by heme proteins (such as CYP450 enzyme) in the absence of oxygen, and amplify the photoacoustic (PA) signal (Figure 7). The HyP-1 allowed the production of a spectrally distinct signal for PA imaging. In vitro and in vivo results showed that in hypoxia conditions, HyP-1 had good selectivity for cancerous tissue and could be used as a multimodality imaging agent.[35] In order to achieve deeper tumor penetration, a study described a tunable nanocluster, a “bomb-like nanoprobe” equipped with both active hypoxia-targeting and passive tumor accumulation capability, with an initial size of 33 nm with a long half-life during blood circulation to release small molecule based hypoxic microenvironment-targeting. The CT imaging was assessed in animal models of pancreatic cancer and breast cancer, supporting the feasibility of deep hypoxic tumor targeting.[36]
Figure 7.
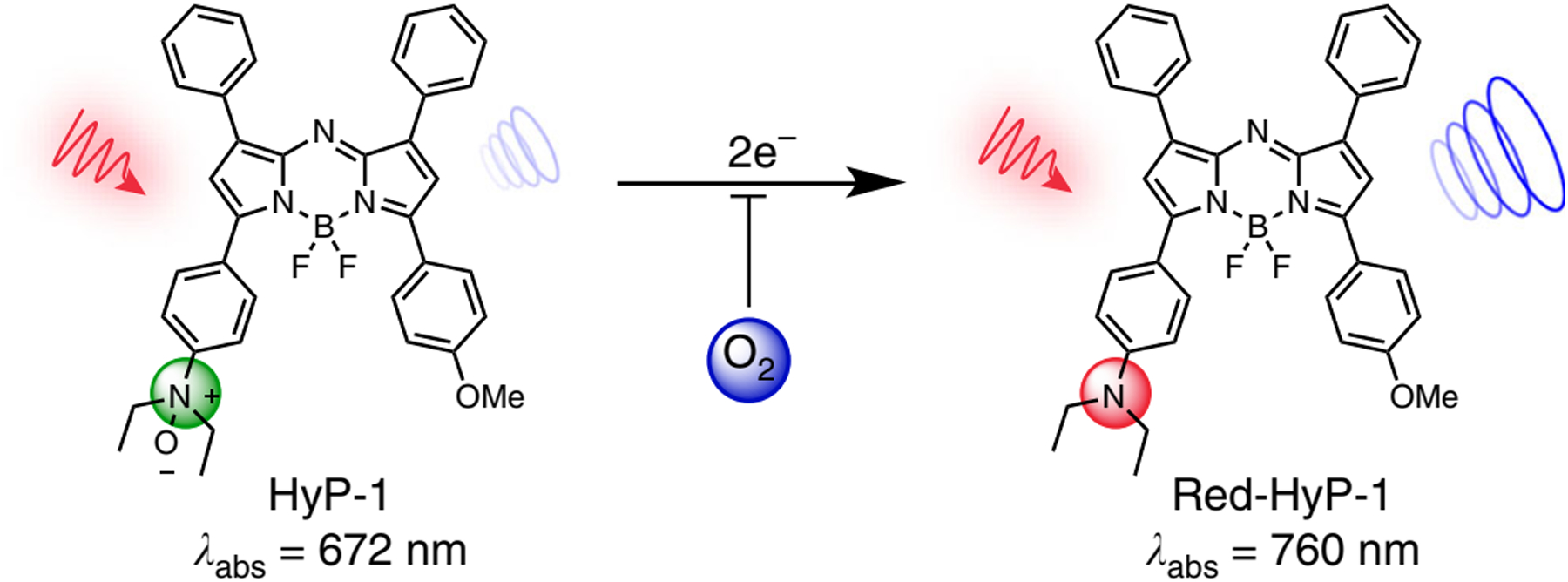
N-oxide-based hypoxia probe. Green HyP-1 N-oxide undergoes irreversible two-electron reduction by heme proteins (such as CYP450 enzyme) in the absence of O2, which binds competitively to the heme iron. Red-HyP-1 amine generates an enhanced photoacoustic signal (blue circles) upon irradiation at 770 nm (red arrow). Adapted with permission.[35] Copyright 2017, Nature.
4.2. pH Targeting
Mammalian cells import glucose as the primary source of energy metabolism. There are two possible metabolic pathways, the Pasteur effect and the Warburg effect. In the Pasteur effect, glycolysis is inhibited by oxygen, which allows for the glucose metabolite pyruvate to be converted into H2O, ATP, and CO2 by oxidative phosphorylation. The Warburg effect occurs under low oxygen conditions and involves aerobic glycolysis that converts glucose into lactic acid. Both these metabolic pathways are essential for the maintenance of energy and control of the pH in the extracellular space within the normal range (7.3–7.4). In the case of hypoxic cancerous tissue, the elevated glucose uptake and increased glycolysis lead to more production of lactic acid. Most of the glucose is converted into lactate, H+, and ATP. The produced lactate and H+ are exported into the extracellular space via the mono-carboxylate transporter and the sodium–hydrogen exchanger, respectively, resulting in the reduction of the pH range (6.2–6.9). The reduced pH in the TME induces tumor-progression, enhancement of angiogenesis, metastasis, migration, invasion, mutagenesis, and inhibits tumor cell apoptosis and antitumor immune response (reviewed in ref. [37]). Within cancerous tissue, the extracellular pH (pHe) varies depending on the type of tumor, tumor mass, and location of tumor (i.e., site) in the body (Table 2).
Table 2.
pHe values in different human tumor xenografts.[38]
| Xenograft cell line | pH range |
|---|---|
| Breast cancer | |
| SE (T60) | 6.76–6.84 |
| REI | 6.78–6.84 |
| JE | 6.8–6.84 |
| GA | 6.78–6.84 |
| BR | 6.7–6.84 |
| CH | 6.84–6.89 |
| MX-1 | 6.78–6.9 |
| Miscellaneous | |
| F8 (neurofibrosarcoma) | 6.84–6.96 |
| STO (pancreas) | 6.72–6.84 |
| LA (endometrium) | 6.79–6.84 |
| GE (thyroid) | 6.82–6.84 |
| MRI-H-212/B (melanoma) | 6.84–6.9 |
| H-MESO (mesothelioma) | 6.84–6.94 |
| Arterial blood | 7.36–7.44 |
| Lung cancer | |
| SE | 6.84–6.9 |
| KO | 6.84–6.97 |
| SCHRO | 6.68–6.84 |
| A 549 | 6.76–6.84 |
| LX-1 | 6.84–6.9 |
| LXFA 289 | 6.74–6.84 |
| LXFE 229 | 6.79–6.84 |
| SCLC | 6.84–6.89 |
| Sarcoma | |
| BO (osteogenic) | 6.75–6.84 |
| N4 (malignant fibrous histiocytoma) | 6.84–6.91 |
| Gastrointestinal cancer | |
| CXF 1103 (colon) | 6.84–6.97 |
| WiDr (colon, adenoma) | 6.74–6.84 |
| SP (stomach) | 6.84–7.01 |
The different pH ranges found in cancerous and normal tissues (Table 2) can be used for cancer targeting. The high stability of pH-sensitive NSs in the normal physiological pH range makes them effective targeting strategies for cancerous tissue. When the pH trigger point is reached, the cargo is rapidly released. The following approaches have been developed in order to achieve this goal: i) the use of ionizable chemical groups, such as amines, carboxylic acids, and phosphoric acids that can be incorporated in organic (polymers, lipids and peptides), inorganic (zinc oxide and calcium phosphate), and hybrid nanomaterials; ii) the use of acid-labile chemical linkers such as imine, cis-aconyl, orthoester, and hydrazone, which are covalently attached to the contrast agent and are stable when pH is neutral, but are hydrolyzed or degraded in acidic conditions; iii) carbon dioxide-generating precursors can react at low pH (HCO−3 + H+ → H2CO3 → H2O + CO2 ↑) producing carbon dioxide gas leading to the disintegration of the nanocarriers and the release of the contrast agents;[39] and iv) the use of pH-activatable contrast agents, such as LS662 an asymmetric cyanine, which are mostly synthetic organic chemical compounds and as fluorescence dyes, can take on active (on) and inactive (off) conformations. Under normal physiological conditions, these materials have an inactive conformation, but as soon as they enter the cancerous tissue, the conformation is exchanged to the active form. In fact, these materials can act as a contrast and targeting agent simultaneously.[40]
Yasuteru et al.[41] developed a tunable and pH-activatable fluorescent probe. They used 2, 6-dicarboxyethyl-1, 3, 5, 7-tetramethyl boron-dipyrromethene (BODIPY) as a fluorophore in order to tune the pH profile of the fluorescent probe, and to alter the functional group to the aminophenyl BODIPY. The designed probe could be used within the pH range 2–9 for different purposes in in vitro and in vivo studies. Wang et al.[42] employed a protonatable strategy, and prepared micelle NSs as ultra pH sensitive (UPS) nanoprobes for extracellular tumor imaging. The designed UPS, composed of an ultra pH-sensitive core (poly [ethylene glycol]-b-poly [2-(hexamethylenediamine) ethyl methacrylate] copolymer), had a sharp tunable pH (<0.25) response, with a near-infrared fluorescence (NIRF) dye (Cy5.5) as the fluorophore, and a targeting agent Arg-Gly-Asp (RGD) that bound to the αvβ3 integrin. At physiological pH (7.4), this fluorescent nanoprobe was self-quenched. At an acidic pH (6.9), the UPS nanoprobe showed a sharp and rapid response. The copolymer became protonated, the micelle was disrupted, and the fluorescent dye was activated (Figure 8).
Figure 8.
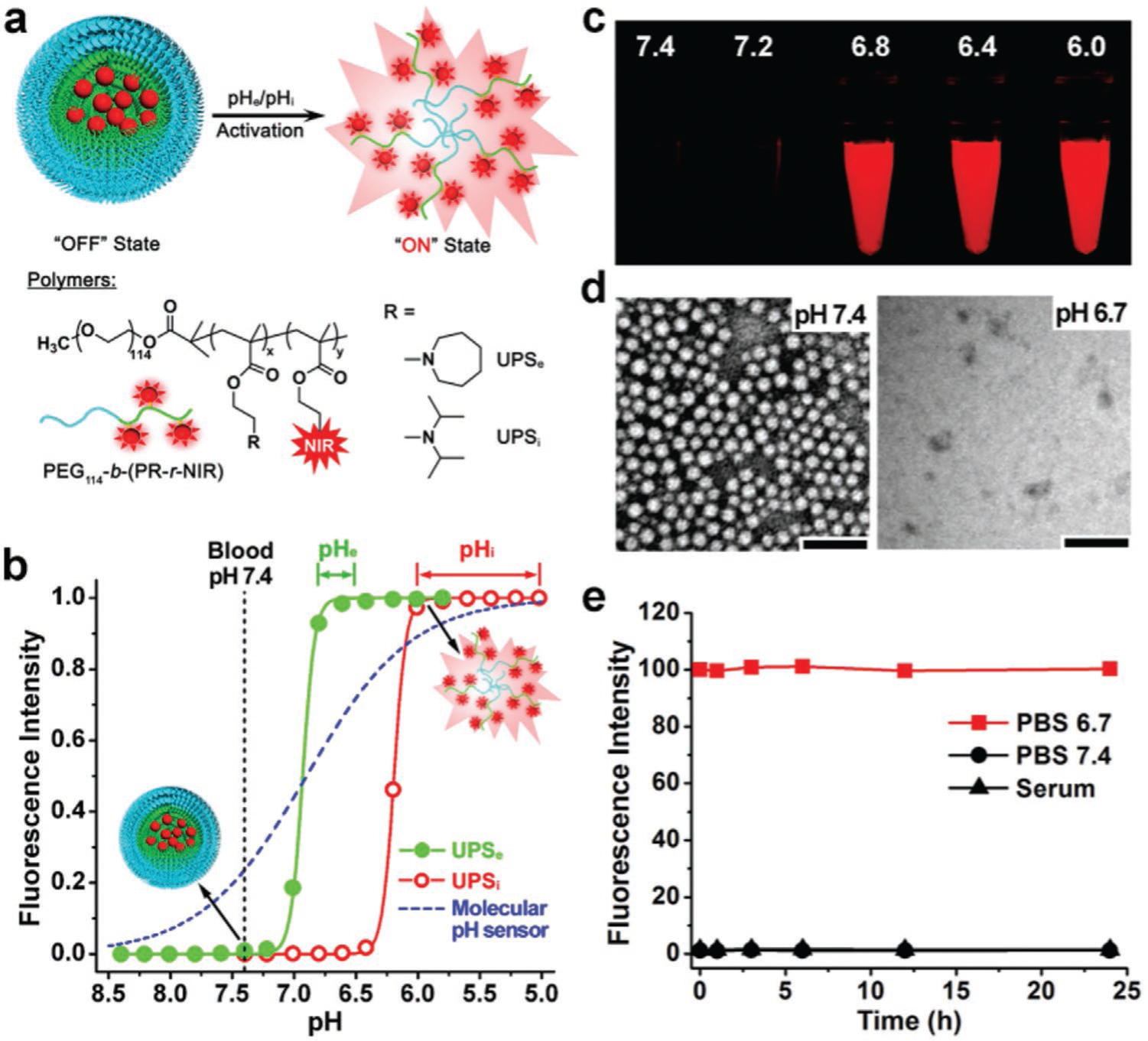
Preparation and characterization of an ultra pH sensitive nanoprobe. a) Structural composition of two types of nanoprobes, ultra pH sensitive extracellular (UPSe) and ultra pH sensitive intracellular (UPSi), with pH transitions at 6.9 and 6.2, respectively. The UPSe was specifically designed to be activated in tumor extracellular fluid (pH 6.5–6.8). The UPSi was activated inside acidic endocytic organelles (pH 5.0–6.0). Cy5.5 was used as a NIR fluorescence agent in the animal studies. b) Normalized fluorescence intensity as a function of pH for UPSe and UPSi nanoprobes. At high pH (7.4), both probes remain quenched. At pH below their transitions (6.9 and 6.2), the probes can be activated as a result of micellar dissociation. The blue dashed-line simulates the pH response of a small molecular pH sensor with a pKa of 6.9 based on the Henderson–Hassel Bach equation. For UPS, the pH response was extremely sharp. In contrast, small molecular pH sensors require 3 pH units for a comparable signal change. c) Fluorescent images of UPSe -Cy5.5 nanoprobe solution in different pH buffers. d) Transmission electron micrographs of UPSe nanoprobes at the normal pH and at pH 6.7. e) UPSe nanoprobes remain stable in fresh mouse serum over 24 h at 37 °C. Reproduced with permission.[42] Copyright 2014, Nature.
The development of a pH-activatable nanoprobe using PEGylated Mn2+-doped calcium phosphate NP, and poly (ethylene glycol)-b-poly (glutamic acid) (PEG-b-P (Glu)) block copolymers with improved mechanical properties was previously reported. Designing pH-sensitive MRI nanoprobes rapidly amplifies the magnetic resonance signals under pathological pH conditions. In acidic solid tumors, the designed NPs disintegrated and released Mn2+ ions. The relaxivity of Mn2+ after binding to the proteins was enhanced, which produced enhanced MRI contrast.[43]
The detection of cancer can be improved using dual-activatable imaging probes. Benedict et al.[44] prepared a pH-activatable fluorescence/MRI dual-modality imaging nanoprobe. They co-encapsulated MnO NPs as an MRI contrast agent and fluorescence quencher, plus coumarin-545T as a fluorophore in the hybrid silica nanoshells that were conjugated with folic acid (FA) for targeting of cancer cells (Figure 9). At normal pH, the MnO NPs remained within the nanosystem and the fluorophore was quenched. In cancerous tissue with low pH, the Mn2+ was released not only providing a strong T1 contrast enhancement, but also the coumarin fluorescence was recovered.[44]
Figure 9.
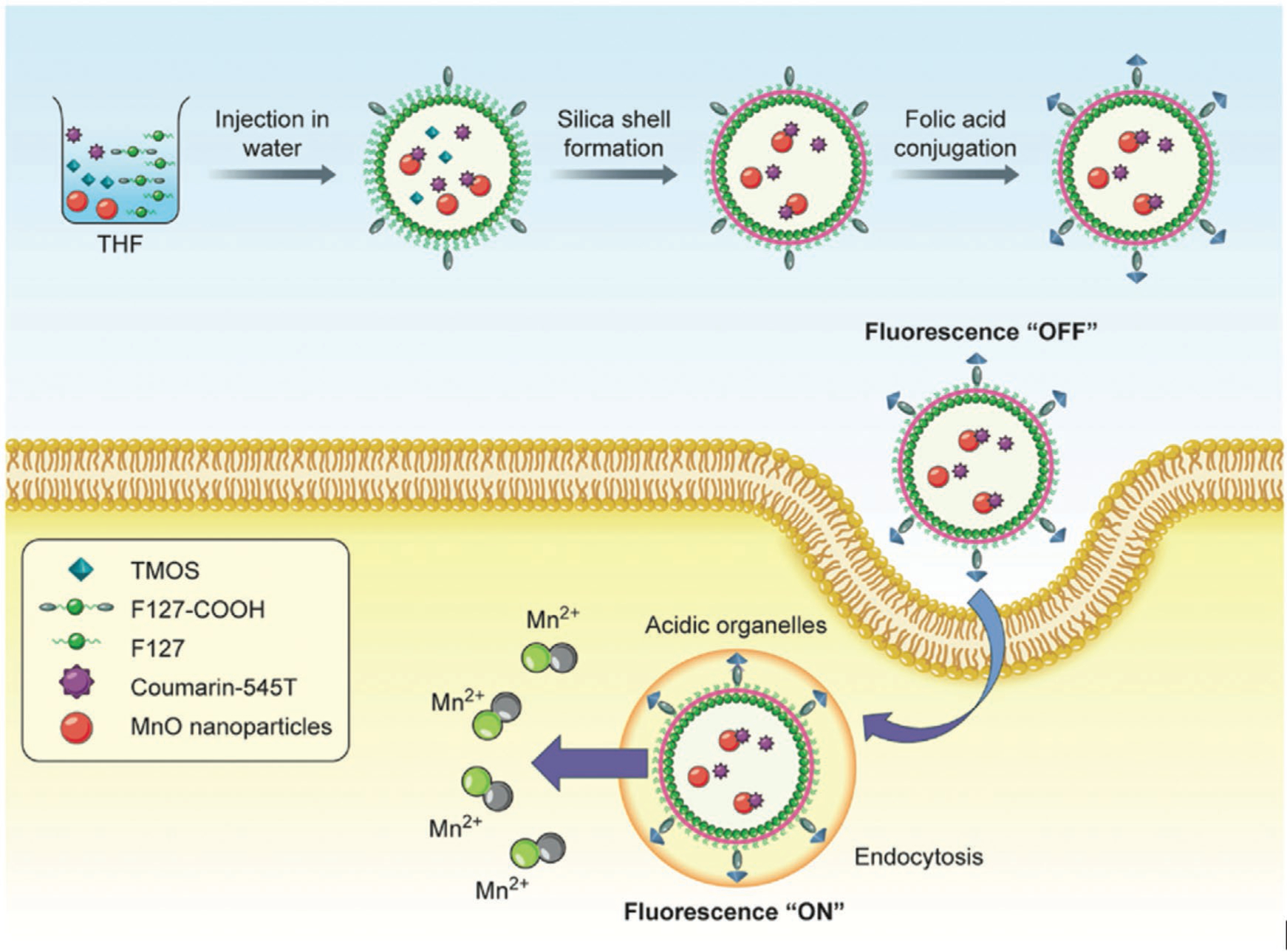
Schematic illustration of the preparation and working principle of nanosystems. First, monodisperse MnO NPs (MONP) were presynthesized. Next, a mixed payload of C545T as a fluorophore and MONP was encapsulated into a carboxylic acid functionalized silica nanoshell by an interfacial templating scheme. Finally, aminated FA was conjugated to the carboxylic acid groups for active targeting of cancer cells. Adapted with permission.[44] Copyright 2016, Wiley-VCH.
4.3. Matrix Metalloproteinase Targeting
MMPs are a family of zinc-containing endopeptidases that play an important role in the degradation of ECM proteins. In normal tissue, MMP expression is regulated by hormones, cytokines, cell-matrix (or cell–cell interactions), and growth factors. The MMPs are present in low quantities, and their activity is regulated by “tissue inhibitors of metalloproteinases” (TIMPs). However in tumors, the TIMP system becomes dysfunctional, and MMPs (including MMP 2, 3, 7, and 9) are overexpressed and activated depending on the stage and the type of cancer.[45] For example, the median concentration of MMP 2 in early-stage ovarian cancer is 0.47 μg mg−1, whereas the concentration of MMP 2 in end-stage ovarian cancer is 1.2 μg mg−1.[46]
While ECM components, such as collagen, fibrinogen, and gelatin are the natural substrates of MMPs, the large size of these proteins limits their use for targeting applications. MMP-sensitive peptides (MSPs) have been used as synthetic MMP substrates, composed of the correct amino acid sequence in short linear peptides that are easily incorporated into NSs. The selectivity and specificity of these MSPs depends on the sequence, which is recognized by the specific MMPs.[47]). Membrane-type (MT)-MMPs are a subfamily that are expressed on the cell membrane, and mediate pericellular proteolysis and cleavage of cell surface receptors. One study by Kondo et al.[48] used radiolabeled 18F-BODIPY650/665, a MT1-MMP peptide substrate coated with PEG to prevent cell uptake. The MT1-MMP peptide substrate was cleaved by MT1-MMPs and the PEG moiety was eliminated, allowing accumulation of the probe inside the tumor cells. This 18F-BODIPY650/665 could be used for dual optical imaging and PET. The results showed that MT1-MMPs were active in cancers and could be used as a targeting modality (Figure 10).
Figure 10.
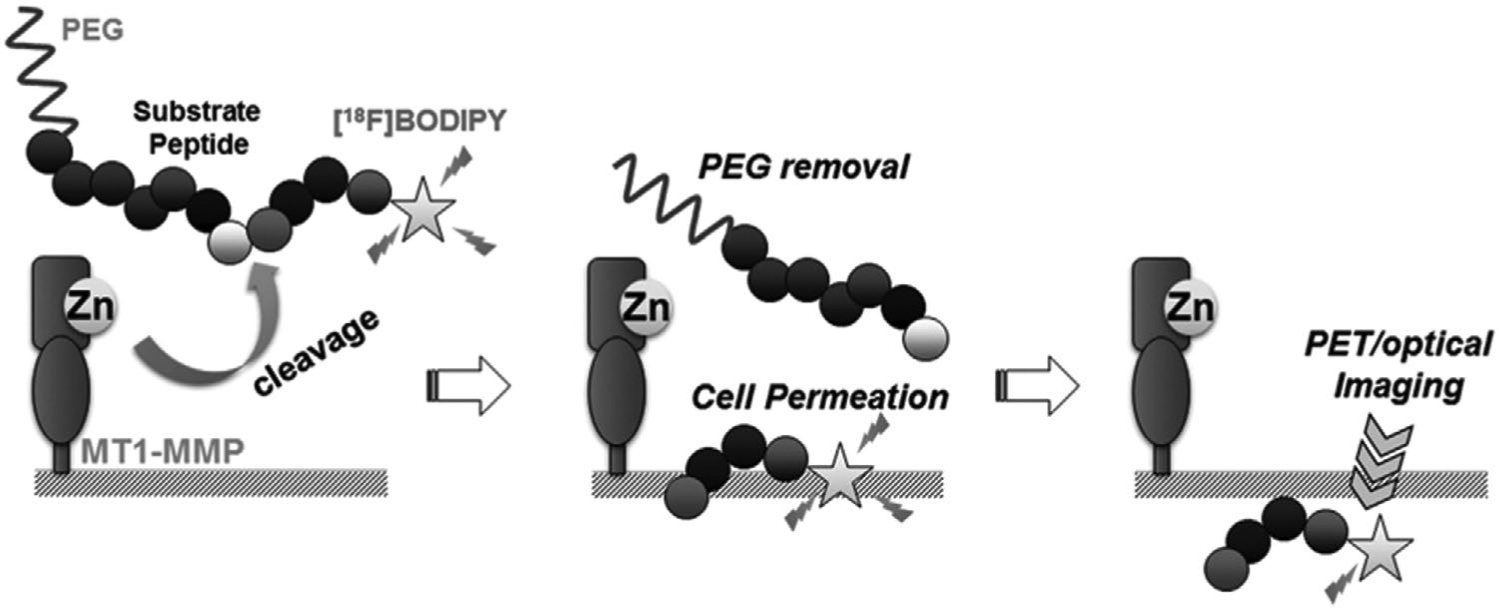
Mechanism of MT1-MMPs used as a targeting agent for cancer imaging. Since MT1-MMP substrate peptide is cleaved by MT1-MMPs in tumors, the PEG moiety is eliminated, which allows the probes to accumulate in tumor cells due to the high cellular membrane permeability of 18F-BODIPY that can be used for imaging of tumors. Adapted with permission.[48] Copyright 2015, Elsevier.
Kuo et al.[49] designed a NIRF sensitive probe for evaluating MMP-3 activity in an ovarian cancer cell line that may be used to detect ovarian cancer in its early stages. They used the cyanine dye as a fluorochrome and the amino terminus as a peptide substrate specific for MMP-3. Exposing the MMP-3 sensitive probe to MMP-3 enzyme significantly increased the NIRF emission intensity. More precise targeting of cancer can be achieved when MMPs are integrated with external/internal-responsive agents. A designed dual-stimulus responsive fluorescent nanoprobe, was fabricated from an asymmetric cyanine used as a pH-sensitive fluorescent dye, glycosyl-functionalized gold nanorods, and a specific peptide sequence as a linker and MMP substrate.[40b] The inactive form of the nanoprobe existed at pH 7.4, and in the presence of a low concentration of MMPs, while the fluorescence was activated in response to acidic pH and higher levels of MMPs as found in the TME (Figure 11).
Figure 11.
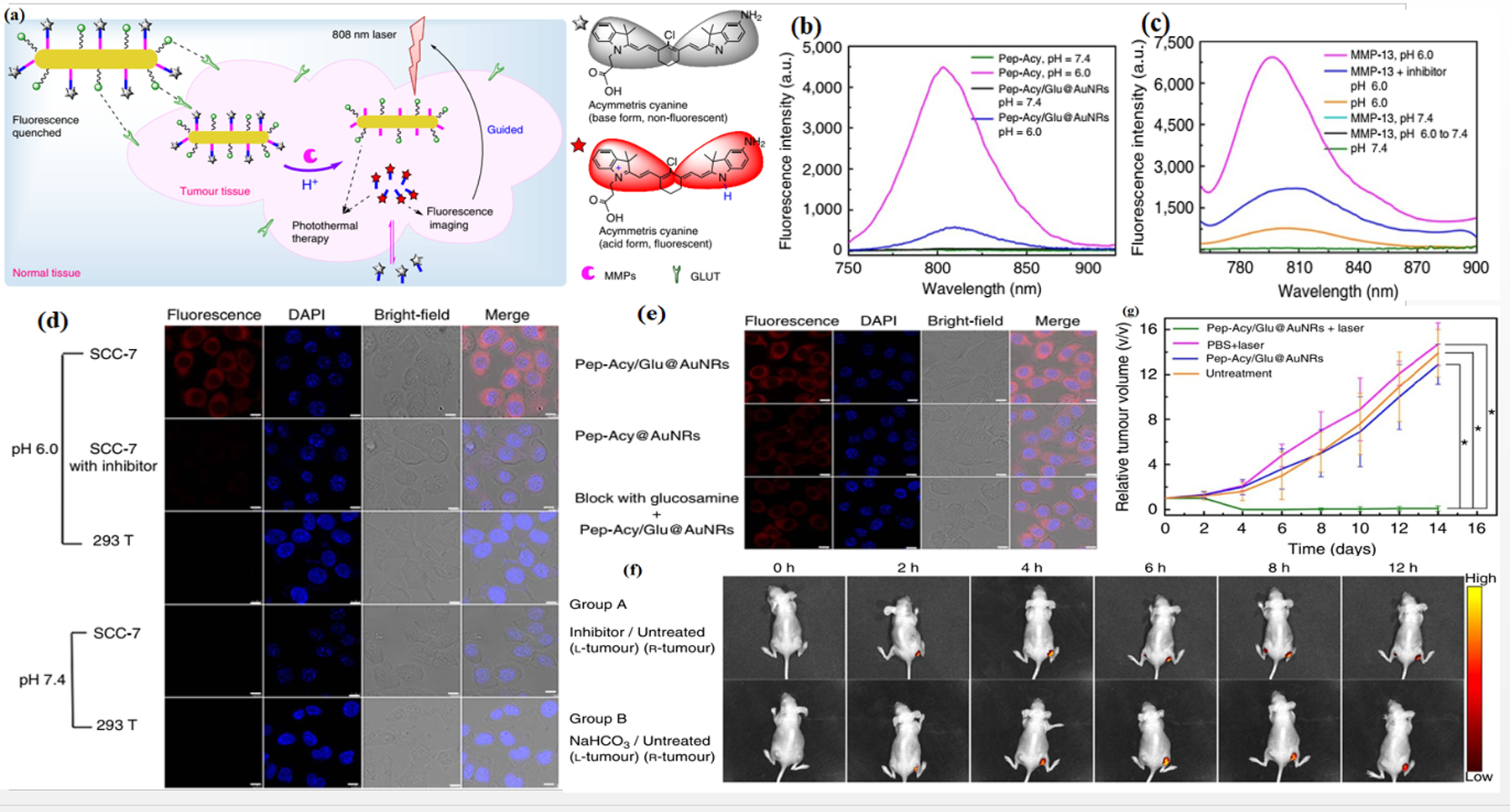
Imaging and photothermal therapy of tumors with a dual responsive nanoprobe. a) Scheme of the nanoprobe as a pH/MMP dual-stimulus responsive pH reversibly activated theranostic platform (Pep-Acy/Glu@AuNRs) for tumor-targeted precision imaging-guided photothermal therapy. b) Fluorescence spectra of theranostic platform and Pep-Acy. c) Fluorescence spectra of theranostic platform to pH and MMP-13. d) Cell internalization of theranostic platform in SCC-7 cells. e) Cell imaging of theranostic platform in SCC-7, 293 T and inhibitor pretreated SCC-7 cells. f) Theranostic platform-mediated in vivo fluorescence images in SCC-7 tumor-bearing mice. g) Comparative tumor volume change of groups of mice. Reproduced with permission.[40b] Copyright 2017, Nature.
4.4. Fibronectin Targeting
Fibronectin (FN) is a cell-adhesion glycoprotein found in the ECM and in various bodily fluids. FN regulates a wide spectrum of cellular and developmental functions, including growth, migration, proliferation, cell adhesion, and wound healing. FN is assembled from monomers consisting of three types of homologous repeat subunits (FNI, FNII, and FNIII domains) with different binding affinities to various ECM proteins.[50] FN contains 12 FNI, 2 FNII, and 15–17 FNIII domains (Figure 12). The two FN subunits are covalently linked together via disulfide bonds near their C-terminus. FN can be divided into two principal forms, cellular FN (cFN) that polymerizes into insoluble fibers in the ECM, and soluble plasma FN (pFN). The splicing sites are located in EDA (or EIIIA), EDB (or EIIIB), and IIICS (connecting segment) domains, and in regions between domains 15FNIII and 14FNIII. The expression of EDA and EDB domains is extremely restricted in normal human tissue, but is highly expressed in the ECM of many cancer types.
Figure 12.
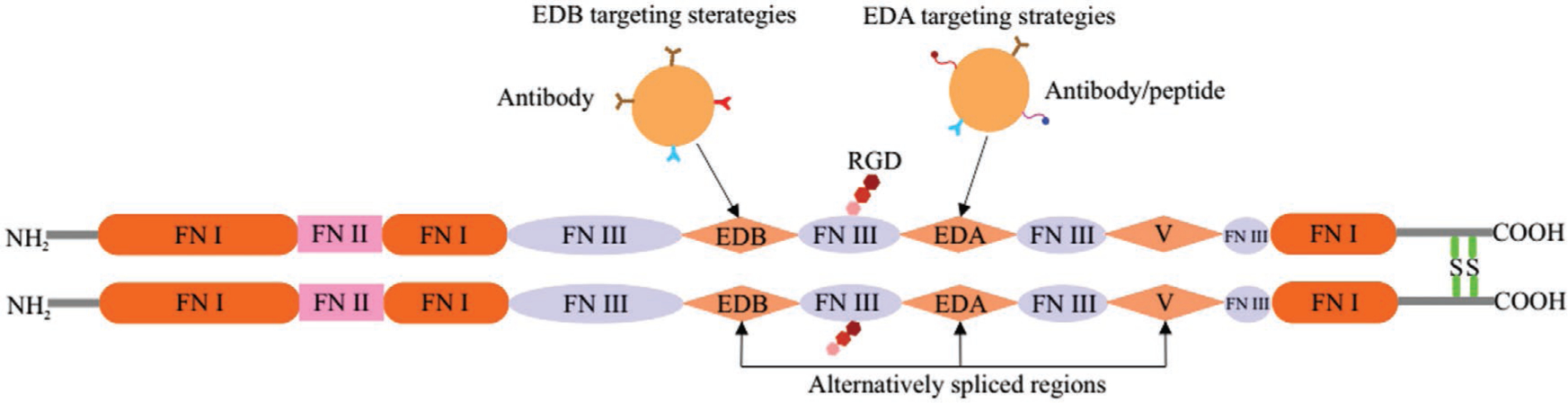
Schematic diagram of fibronectin. FN is composed of three types of repeats termed FNI (orange), FNII (pink) and FNIII (blue). Three FNIII domains, EDA, EDB, and the V region (light orange), can be alternatively spliced. EDA and EDB domains are markers of angiogenesis, a critical step in tumor progression. EBD targeting strategies consisting of antibody-based delivery (such as L19, BC-1) and EBA (F9), and peptide-based delivery can be used for therapy, imaging, and vaccination.
EDB-FN is absent in adult blood vessels, but is overexpressed during angiogenesis in normal and neoplastic tissues, making it an attractive marker for angiogenesis.[51] EDA-FN can also act as a marker of normal and tumor vasculature. Oncofetal forms of EDA-FN, EDB-FN or IIICS-FN, have been shown to be overexpressed in various cancer types. The changes in the expression and organization of FN in the ECM contribute to the “pre-metastatic niche,” and may dictate the pattern of metastatic spreading. The deposition of FN in the tumor ECM stimulates formation of a fibrin–fibronectin complex, which in turn facilitates the proliferation, angiogenesis, and metastasis of cancer.[52] During the epithelial-to-mesenchymal transition (EMT), transforming growth factor-beta (TGF-β) increases the expression of FN. The FN abundance can serve as a prognostic biomarker in human cancer. For example, in the case of invasive breast cancer, a significant correlation was found between the FN levels and the pathologic tumor stage, histologic grade, and patient survival rate.[53] Additionally, detection of EDA-FN in urine was shown to be a predictor of survival in bladder cancer patients.[54] Thus, FN is an attractive biomarker for molecular imaging for the early detection of high-risk cancer and for micro-metastasis.[55] FN has been used as a target to develop antibody-targeted platforms for accurate and specific delivery of imaging and therapeutic agents to metastatic sites.[56]
Zhou et al. developed a pentapeptide CREKA-targeted MRI contrast agent (CREKA-Tris (Gd-DOTA)3 (Gd-DOTA, 4,7,10-tris (carboxymethyl)-1,4,7,10-tetraazacyclododecane gadolinium) for breast cancer molecular imaging (Figure 13). The CREKA peptide sequence selectively bound to FN and the fibrin–FN complex. Compared with nontargeted controls, the targeted contrast agents were selective for the ECM of cancerous cells showing good and long-lasting enhancement of tumor contrast. Results showed that the CREKA-targeted imaging construct could act as a non-invasive, high-resolution molecular MRI probe to detect tumor micrometastases (≤0.5 mm).[57]
Figure 13.
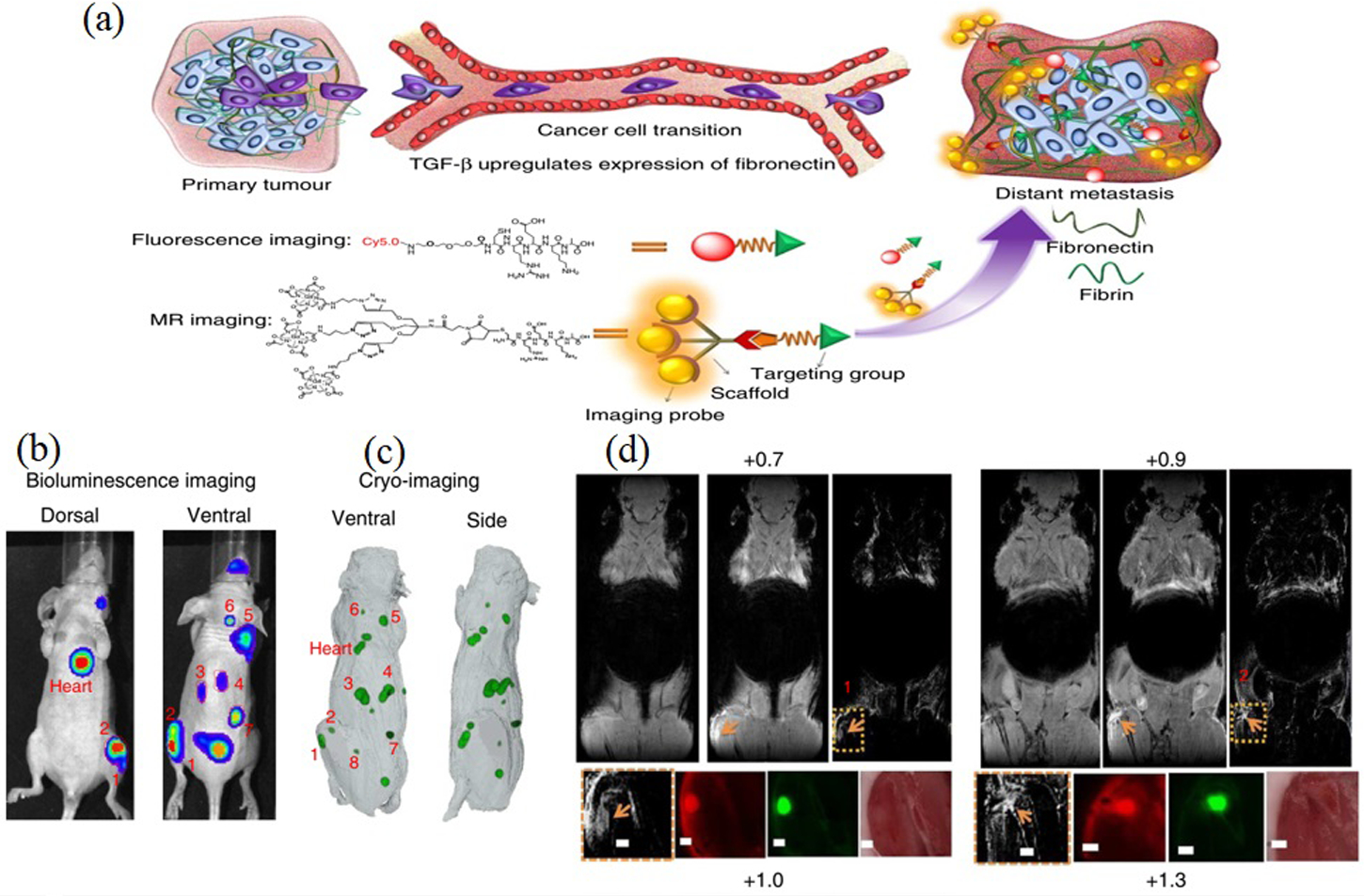
MRI detection of breast cancer micrometastases (BCMs) using fibrin-fibronectin targeted contrast agent, CREKA-Tris (Gd-DOTA) 3. A) BC metastasis is accompanied by upregulated fibronectin expression. By targeting overexpressed fibronectin, which forms complexes with fibrin, CREKA-Tris (Gd-DOTA) a targeted imaging probe, accumulates at sites of metastasis, producing tumor contrast enhancement in MRI, which was validated by high-resolution fluorescence imaging of CREKA-Cy5 also accumulated in metastases. B) MRI images of BCMs contrast enhanced with targeted imaging probe showing the coronal slices before and after CREKA-Tris (Gd DOTA) 3 injection, the subtraction images of the pre-injection from the post-injection images, and the amplified subtraction MRI images of metastatic sites. C and D) Corresponding GFP cryo-fluorescence images of the micrometastases and CREKA-Cy5.0 images validate the MRI detection of micrometastases. (Tumors are indicated by orange arrow; all scale bars, 1mm). Reproduced with permission.[57] Copyright 2017, Nature.
As mentioned above, EDB-FN is an EMT biomarker that can be identified by specific targeting ligands such as the ZD2 peptide sequence (Cys-Thr-Val-Arg-Thr-Ser-Ala-Asp). Han et al.[58] prepared a hydroxylated tri-gadolinium nitride metal-lofullerene (Gd3N@C80) that acted as a contrast agent, and the ZD2 peptide was used as a targeting ligand in the ZD2-Gd3N@ C80 probe, with the ability to detect aggressive tumors using MRI. The MRI data showed the designed probe allowed significantly decreased doses, and produced strong signal enhancement in aggressive triple-negative breast cancer (TNBC) in a mouse model.[58] One novel type of potentially clinically translatable molecular-targeted microbubble preparation (MBs) included an engineered 10th type III domain of the FN (MB-FN3 VEGFR2) scaffold-ligand to image VEGFR2 (vascular endothelial growth receptor 2)-associated neovasculature. The MB-FN3 VEGFR2 was developed for in vivo ultrasound molecular imaging (USMI) of breast cancer neovasculature with specific binding to VEGFR2, which was significantly higher in breast cancer compared to normal breast tissue. The FN3-scaffold could be produced via recombinant technology, with small size, solubility, lack of glycosylation, good stability, and disulfide bonds, leading to generation of small, high affinity ligands for USMI.[59]
4.5. Apoptosis Targeting
Many new therapeutic approaches for different diseases function by inhibiting or inducing apoptosis. Therefore, imaging systems capable of tracking of cell death (apoptosis) will become increasingly important. Different types of strategies for monitoring apoptosis have been developed based on a range of surrogate biomarkers. These include apoptosis signaling molecules such as caspases, as well as markers that are further downstream in the apoptosis cascade.[60] One strategy that can be used for apoptosis monitoring is therefore caspase targeting. Caspases are a family of cysteine protease peptidases (Figure 14) that take advantage of a cysteine residue as the catalytic nucleophile with exquisite specificity for cleaving target proteins at sites next to aspartic acid residues.[61] The concerted action of caspases is responsible for triggering apoptosis, a specific form of programmed cell death that is essential for embryonic development and is involved in the pathology of many diseases.
Figure 14.

Classification of caspases based on structure and function. Apoptotic caspase-2, −8, −9, and −10 are initiators, while caspase-3, −6, and −7 are key executioner caspases. Caspase-1, −4, −5, −11, and −12 are inflammatory caspases. Reproduced with permission.[61] Copyright 2015. Nature. Abbreviation: CARD, caspase recruitment domain; DED, death effector domain; L, large subunit; S, small subunit; S*, short form; L*, long form.
Annexins are a family of proteins that are able to bind to negatively charged phospholipids in the presence of calcium ions. Among the annexin family members, only annexin V shows the possibility of extracellular expression in addition to its intracellular localization.[62] During the early phase of programmed cell death, phosphatidylserine (PS) in the lipid bilayer of the cell membrane is flipped from the inner layer to the outer layer and exposed on the surface. Annexin V binds with high affinity to membranes with exposed PS, and has been used as a molecular imaging agent to visualize PS-expressing apoptotic cells.[63] Therefore, caspases and annexin V can be employed to track cell death within the TME, as a target and imaging agent, respectively.
Deju et al. designed a caspase-sensitive nano-aggregation fluorescent probe (C-SNAF). The designed probe consisted of: i) D-cysteine and 2-cyano-6-hydroxyquinoline (CHQ) moieties linked to an amino-luciferin scaffold; and ii) an L-DEVD capping sequence and a disulfide bond required for a two-step activation process involving caspase-3/7-mediated cleavage and intracellular thiol-mediated reduction. The results of in vitro and in vivo studies showed that in tumors that were unresponsive to therapy, inactive procaspase-3/7 dominated and could not cleave the L-DEVD (capping peptide) from C-SNAF, resulting in rapid clearance of the probe. However, in tumors that were therapy-responsive, increased membrane permeability and extensive activation of caspase-3/7 led to apoptotic cell death and increased fluorescence.[64] In another study, Zhang and co-workers used real time apoptosis imaging mediated by an AuHNRs-DTPP nanoplatform. They attached the chimeric peptide (DTPP) bearing the photosensitizer (protoporphyrin IX) to the surface of Au hollow nanorods for NIR-II photothermal therapy, real-time apoptosis imaging, and photodynamic therapy. Under 1064 nm laser irradiation, AuHNRs-DTPP exhibited high photothermal conversion efficiency. Results showed that the photosensitizer in DTPP was quenched after loading onto the surface, but upon exposure to caspase-3, the photosensitizer was released in an activated form allowing enhanced fluorescence for apoptosis imaging in vivo and photodynamic therapy.[65]
Lu et al. designed a novel nanoparticle labeled with annexin V and containing NOTA-maleimide aluminum [18F] fluoride, and evaluated it as a novel apoptosis targeting agent in vitro and in vivo. Results showed that the amount of the tracer binding to erythrocytes with exposed PS was 89.4%. The probe (18F-AlF-NOTA-MAL-Cys-Annexin V) had good specificity for apoptotic cells was suitable for further investigation in clinical apoptosis imaging.[66] Moreover, annexin V labeled with different types of radionuclides can be useful as radiotracers for in vivo tracing of apoptosis as SPECT and PET imaging agents. Annexin V and Annexin V derivatives have been radiolabeled with the radio-sotopes 111In, 123I, and 125I for SPECT imaging of apoptosis.[67]
5. Vasculature Targeting
Tumors cannot grow without a sufficient blood supply. To secure this blood supply, tumors take over already-existing blood vessels, and stimulate angiogenesis and sprouting of new vessels to reach the tumor.[68] The angiogenic switch is an important early event in tumor progression, leading to the beginning of neovascularization in premalignant lesions. In normal conditions, angiogenesis occurs in inflammatory conditions, tissue regeneration, as well as in cancer. Tumor angiogenesis is initiated by local hypoxia and then continues with the expression of other targetable factors, such a: VEGF, VEGFRs, platelet-derived growth factor (PDGF), angiopoietin, ephrins (EPH receptors), integrins (specially, αvβ3, and αvβ5), and endoglin (CD105), that work together to attract endothelial progenitor and supporting cells. Besides the maturation of new vessels, the endothelial tubes acquire supporting cells such as pericytes and smooth muscle cells, as well as ECM. Tumor vessels are leaky and tortuous, their diameter is irregular and their walls are thin. Deficient pericytes, or abnormal pericyte function, could be responsible for these morphological features in the tumor neovasculature. Therefore, angiogenesis and the involved factors are suitable candidate for targeted cancer imaging and evaluation of response to therapy.[69]
Hao et al. designed a radiolabeled NOTA-GO-TRC105 nanoprobe to target the neovasculature within the tumor mass. They used TRC105 as an antibody recognizing CD105 for targeting. The pharmacokinetics and tumor targeting efficacy of the graphene oxide (GO) conjugate was investigated with serial noninvasive PET imaging and biodistribution studies in vitro, in vivo, and ex vivo. The results showed that CD105 could be a promising vascular target for cancer imaging.[70] Wen and co-workers designed, synthesized, and engineered a “nanobomb” for targeting the tumor neovasculature. This nanobomb was rationally prepared via the encapsulation of vinyl azide (VA) into c(RGDfE) peptide-functionalized, hollow copper sulfide (HCuS) nanoparticles. The resulting RGD@HCuS(VA) nanoparticles were selectively internalized into integrin αvβ3-overexpressing tumor vascular endothelial cells, and dramatically increased the photoacoustic signal from the tumor neovasculature, with an increased signal-to-noise ratio. The probe allowed high-resolution photoacoustic angiography, combined with biodegradability, and led to precise destruction of tumor neovasculature without damaging normal tissue. This nanobomb had the potential for clinical translation to treat cancer patients with tumors accessible to NIR laser therapy while allowing simultaneous photoacoustic monitoring.[71] Grzegorz and co-workers designed targeted microbubbles, which could be used to effectively monitor response to different therapeutic regimens in animal models of pancreatic cancer. The microbubbles were targeted to endoglin (CD105), VEGFR2, or the VEGF-VEGFR complex via antibodies, and the signals were correlated with immunohistochemical expression of these markers, and also with the tumor microvessel density (MVD). They proposed that ultrasonic imaging using targeted microbubbles could be used to image tumor angiogenesis and the expression of neovascular markers in response to therapy.[72]
5.1. VEGF Targeting
VEGF is a marker of neovascularization that controls endothelial cell proliferation, survival, migration, invasion, vascular permeability, chemotaxis of bone marrow-derived progenitor cells, and vasodilation.[73] The VEGF family consists of VEGFA (commonly referred to as VEGF), VEGFB, VEGFC, VEGFD, and PGF (placenta growth factor), glycoproteins.[74] The VEGF ligand has three types of VEGF receptors: VEGFR1 (for VEGF), VEGFR2 (a positive regulator of angiogenesis), and VEGFR3 (for VEGFC and VEGFD).[75] VEGFRs are expressed in normal tissues in a controlled manner. However, in tumor tissue, the expression is strongly upregulated on the surface of ECs.[76] This localization can be used in the targeting and bioimaging of cancers. Woutor et al.[77]) used 89Zr and 111In radiolabeled bevacizumab (a monoclonal antibody (mAb), which binds to all the isoforms of VEGFA ligands). The results of micro-CT and micro-PET imaging showed the imaging probe had a significant tumor uptake in the ovarian xenograft tumor model, compared to non-specific 89Zr-IgG and 111In-IgG as control groups. 89Zr-bevacizumab not only allowed imaging for up to 168 h, but also enabled quantitative measurement of the tumor uptake.[78] In another study, Anton et al.[79] used IR Dye 800CW as a fluorescent dye and 89Zr as a radiolabel both bound to the bevacizumab antibody. They evaluated the tumor uptake and the optimal time for imaging to achieve the best contrast. In vivo fluorescence and PET imaging both showed that the fluorescent-labeled VEGF antibody could mediate highly specific and sensitive detection of tumors.
Another example was mAb VEGF-targeted bovine serum albumin-coated magnetic NPs (MNP@BSA), which have been used for targeting of VEGFR in brain cancer using MRI imaging.[80] The results indicated that MNP@BSA was effective in MRI visualization of intracranial gliomas, and could be used as a targeted contrast agent. Additionally, the level of VEFGR expression depended on the type of cancer. For example, the 124I-HuMV833 imaging probe was tested in ovarian and colon cancers. PET imaging results showed that uptake of the targeted imaging probes in ovarian tumors was greater than the uptake in colon cancer.[81]
5.2. Integrin Targeting
Integrins are a family of transmembrane glycoprotein cell surface receptors that facilitate bonding of the cell to the ECM and to immunoglobulins. These receptors contain 24 heterodimers on the cell surface, and are formed from 18 α-sub units and eight β-subunits. In the TME, integrins encourage tumor progression in several different ways, including tumor cell proliferation, survival, and invasion. Integrins are expressed on fibroblasts, marrow-derived cells, platelets, vascular endothelium, and perivascular cells, and facilitate cancer progression.[82] The classification of integrins is dependent on the type of receptors present. Experimental therapeutic compounds involving integrins have been reviewed in this article[83] (Figure 15). Under normal conditions, integrins mediate epithelial cell adhesion to the basement membrane, and are usually expressed at only low levels in adult epithelia. However, in epithelial cells originating from solid tumors, integrin expression is altered.[84] The profiles of overexpressed integrins and phenotypes in some human tumors are summarized in Table 3.
Figure 15.
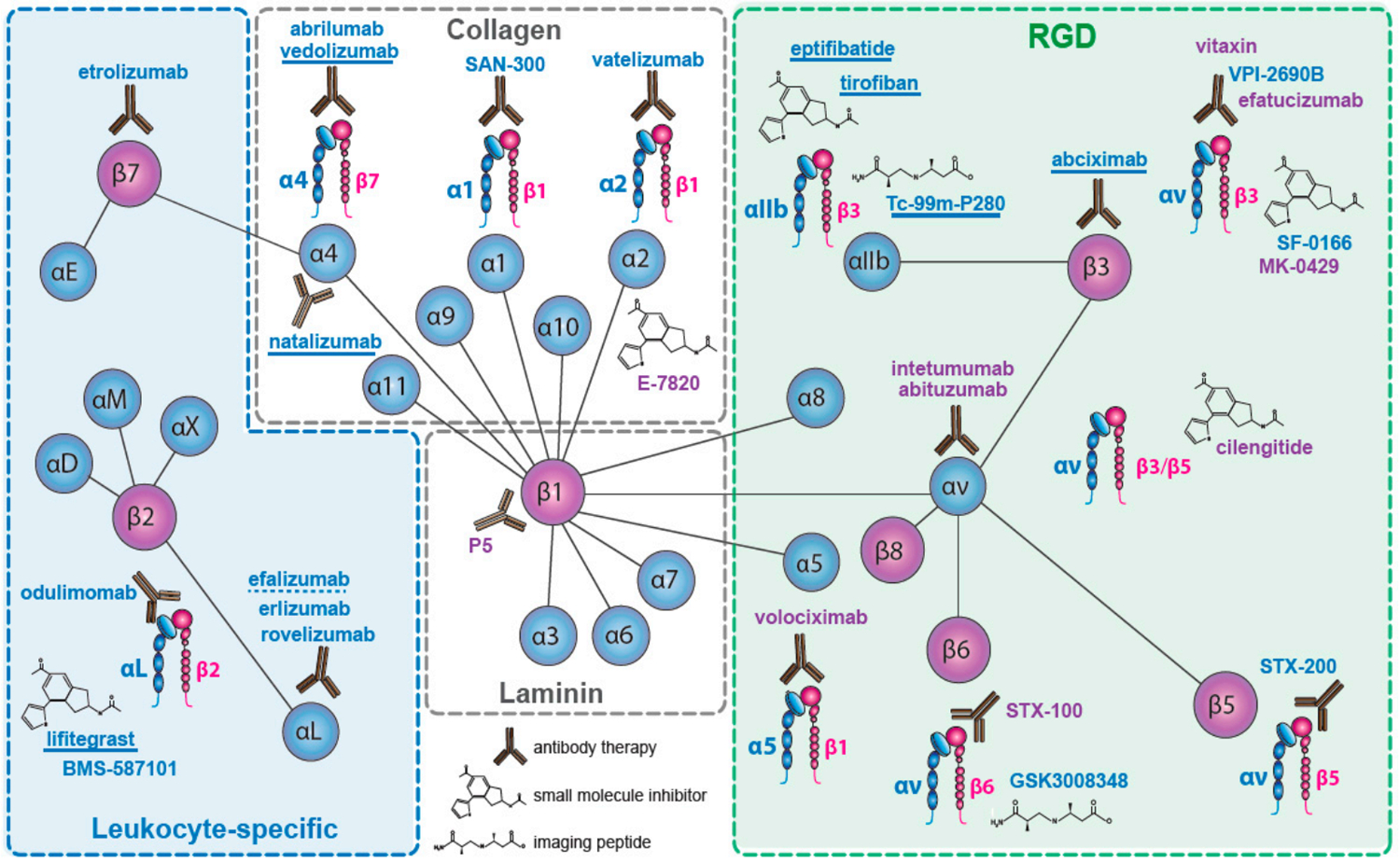
Classification of integrin-based targeting by type of receptors and compounds that target integrins. Adapted under the terms and conditions of the Creative Commons CC BY 4.0 International License.[83] Copyright 2017, The Authors, published by MDPI.
Table 3.
Overexpressed integrins and associated phenotypes in some human tumors.
| Tumor type | Integrins expressed | Major associated consequence | Reference |
|---|---|---|---|
| LCBMa) | αvβ6 and αvβ3 | High expression in endothelial cells, low expression in tumor cells | [85] |
| Prostate | αvβ3 and αvβ5 | High expression in peri-tumoral tissue depending on differentiation | [86] |
| Breast | α6β4 and αvβ3 | Correlated with increased tumor size and grade and decreased survival (α6β4). Increased bone metastasis (αvβ3) | [87] |
| Pancreatic | αvβ3 | Lymph node metastasis | [88] |
| Glioblastoma | αvβ3 and αvβ5 | Both expressed at the tumor-normal tissue margin with a possible role in invasion | [89] |
| Ovarian | α4β1 and αvβ3 | Increased peritoneal metastasis (α4β1) and tumor proliferation (αvβ3). | [90] |
| Cervical | αvβ3 and αvβ6 | Decreased patient survival | [91] |
| NSCLCb) | α5β1 | Decreased survival in patients with lymph node-negative tumors. | [92] |
| Melanoma | αvβ3 and α5β1 | Vertical growth phase and lymph node metastasis | [93] |
| Liver | αvβ6 | Differentiates cholangiocarcinoma from hepatocellular carcinoma | [94] |
| Colon | αvβ6 | Reduced patient survival | [95] |
Lung cancer brain metastases;
Non-small-cell lung carcinoma.
Different molecular ligands, such as the RGD, and Leu-Asp-Val (LDV) motifs can be used for the targeting of integrin receptors or subunits. The eight families of integrins, which play an important role in cancer progression, can all be targeted with the RGD tripeptide motif.[96] The selective accumulation of 125I-RGD-CR780-PEG5K NPs detected by SPECT, CT, photoacoustic, and fluorescence imaging showed that NPs were effective imaging probes and accumulated on αvβ3 integrins expressed in glioblastoma. Furthermore, the data collected from PAI showed that the probe selectively targeted angiogenic tumor vessels.[97] In another study, conjugated quantum dots (QDs)-cyclic RGD peptide (D-phenylalanine-lysine (cRGDfk) were used for targeting the αvβ3 integrin. Fluorescence imaging showed that cRGDfk-QDs had a highly selective uptake in tumor cells and tissues.[98]
More precise targeting of integrins could be achieved by designing 18F-FB-PEG3-GLU-RGD-BBN for dual targeting of the gastrin-releasing peptide receptor (GRPR) and integrin αvβ3. Results showed that this imaging probe had a high tumor accumulation with a favorable pharmacokinetic profile.[99] Binding of FN (a natural ligand) to α5β1 integrin requires the involvement of two small peptide sequences: PHSRN (Pro-His-Ser-Arg-Asn, synergistic binding site) and RGD (primary binding site). Zhao et al.[100] functionalized an α5β1-specific small peptide sequence that acted as a fibronectin mimetic, and PR-b (KSSPHSRN (SG)5 RGDSP), which was modified with β-alanine residues, conjugated to p-SCN-Bn-NOTA, and radiolabeled with 18F as a PET imaging probe. Both the imaging and biodistribution results suggested there was higher uptake of the designed probe in α5β1-positive tumors, compared to α5β1-negative tumors; and a higher α5β1-positive tumor uptake of the designed probe compared to the control probe. There was no significant difference between the designed and control probes in the uptake into the contralateral muscle.
5.3. Vascular Cell Adhesion Molecule-1 Targeting
Vascular cell adhesion molecule-1 (VCAM-1; CD106) was expressed on human CD34 hematological precursor cells and mediated their homing in the bone marrow stroma.[101] VCAM-1 was also expressed on the lateral and luminal side of endothelial cells, and mediated extravasation of leuko-cytes in inflammatory conditions.[102] Integrins have binding patterns for VCAM-1. Between them, α4β1 is most investigated.[103] VCAM-1 has two splice variations in humans, consisting of seven and six Ig-like domains (7d and 6d).[104] In comparison with VCAM-1 (7d), VCAM-1 (6d) binds to VLA-4 with higher affinity in soluble conditions. In mediating cell separation and adhesion, VCAM-1 (7d) is better and more effective.[105]
Under an inflammatory response, VCAM-1 is over-expressed and this may be mediated by ROS, Toll-like receptor (TLR) agonists, shear stress, cytokines, high concentrations of glucose, and oxidized low-density lipoprotein (oxLDL). Tumor tissue expression of VCAM-1 is variable. For example, in ECs and angiogenic vessels, VCAM-1 expression is upregulated and decreased, respectively. Additionally, on the tumor cell surface, VCAM-1 expression is aberrant, while its expression in the lymphatic ECs is constitutive.[106] Although the expression of VCAM-1 in some types of cancer is not completely predictable, circulating cancer cells can have sufficient VCAM-1 expression levels to make them promising candidates for targeted cancer imaging and therapy.[107] Micro-PET/CT results from one study showed that 68Ga-NOTA-VCAM-1ScFV had a higher uptake in the B16F10 cell line than in A375m cells, when used as an imaging nanoprobe.[108] They used LY2409881 as an IKKβ inhibitor (that can induce apoptosis of VCAM-1 positive cells) and DMSO as control groups. In the control group, uptake of the probe as a tracer consistently remained at the same level. However in the treated group, uptake of the tracer in the first week decreased and then slowly recovered until it reached the initial level. This study showed that VCAM-1 could be used as a targeted receptor for specific and selective cancer imaging.
VCAM-1 induces an inflammation-like effect in endothelial cells. In one study, Patel et al.[109] used radiolabeled iron oxide NPs conjugated with anti-VCAM-1 antibodies for evaluating the inflammatory tumor necrosis factor-a marker in a rat model of status epilepticus. Imaging results showed that the contrast agent rapidly and effectively bound to the vasculature of the inflamed brain tissue. The pattern of hypo-intensity in the MRI images was in agreement with the distribution of the contrast agent measured by phosphor-imaging and SPECT. Uddin et al.[110] used the VCAM-1 targeted antisense hairpin and DNA-functionalized gold NPs (AS-VCAM-1 hAuNP) for real time detection and imaging of VCAM-1 expression in retinal endothelial cells. An increase in VCAM-1 mRNA levels caused a fluorescence enhancement that was clearly visible and increased the signal/noise ratio. VCAM-1 was also over-expressed in the early stages of development of cancer micrometastases. An imaging probe based on iron oxide-(VCAM-1-MPIO) microparticles was developed and administered to mouse xenograft models of brain micrometastases of lung adenocarcinoma, melanoma, and human breast cancer as tumor models. The expression of VCAM-was higher in the metastases and was independent of the primary tumor type. The MRI imaging results showed that VCAM-1R targeting could be an approach to detect brain micrometastases arising from three different primary cancer types.[111] VCAM-1 is preferentially expressed in ovarian cancer metastases seeded on the peritoneal mesothelium with the potential to act as a marker in metastasis imaging, monitoring, and staging. Scalici et al.[112] designed a SPECT/CT imaging probe using a VCAM-1 targeted peptide (tVCAM-4 ([(VHPKQHRGGSPEG5K) 4K] 2-KK (DOTA)-βA-NH2)) and with 111In as a radiolabel. In vivo imaging results showed that there was a correlation between VCAM-1 expression levels and tumor stage. Clinically relevant imaging probes could quantify VCAM-1 expression levels as an indicator of ovarian cancer peritoneal metastasis and to monitor therapeutic response to platinum-based chemotherapy agents.
6. Cancer Cell Surface Marker Targeting
6.1. Transferrin Receptor Targeting
The transferrin receptor (TfR) is a homodimer (180 kDa) type II transmembrane glycoprotein that is integrated into the cell membrane, and plays an important role in iron uptake and homeostasis, and regulates cell growth via interaction with the iron-transporting protein transferrin.[113] Transferrin (Tf) is produced by the liver and transports iron ions throughout the body. Depending on the tumor cell iron requirements, higher expression of TfR has been shown in many malignancies compared to normally dividing cells (by up to 100-fold).[114] Thus, using Tf itself or anti-TfR antibodies could be employed to design different targeted theranostic agents for cancer cells. Biocompatible gadolinium biomineralized transferrin NPs (Gd@Tf NPs) were used to enhance T1 signal amplification for MRI by increasing the tumor targeting ability.[115] Interestingly, the T1 relaxivity of Gd@Tf NPs was much higher than that of Magnevist (a commercial MRI contrast agent), which were measured to be 17.42 mM−1 s−1 and 3–5 mM−1 s−1, respectively. This result could be due to the augmentation effect of protein on the relaxivity of Gd ions. Furthermore, compared to nontargeted NPs, Gd@Tf NPs enhanced the amplification of the T1 MR signal and showed better tumor localization in vivo. Gd@Tf NPs were excreted out of the body via the hepatobiliary system.
In another study, Wang et al.[116] developed self-assembled transferrin-IR780 NPs (Tf-IR780 NPs) for targeted imaging and phototherapy in colon cancer cells (CT26) and normal fibroblasts (L929). As expected, CT26 showed a significantly stronger red fluorescence in the cytoplasm compared to L929, indicating the targeting ability of Tf toward the overexpressed TfR on the surface of CT26 cells. The in vivo biodistribution profile of Tf-IR780 NPs in CT26 bearing mice demonstrated a strong signal in the tumor area at 12 h post-injection, and reached its maximum value after 48 h (Figure 16). The ex vivo imaging results revealed the accumulation of Tf-IR780 in tumor sites was much higher than other organs at 24 h post-injection.
Figure 16.
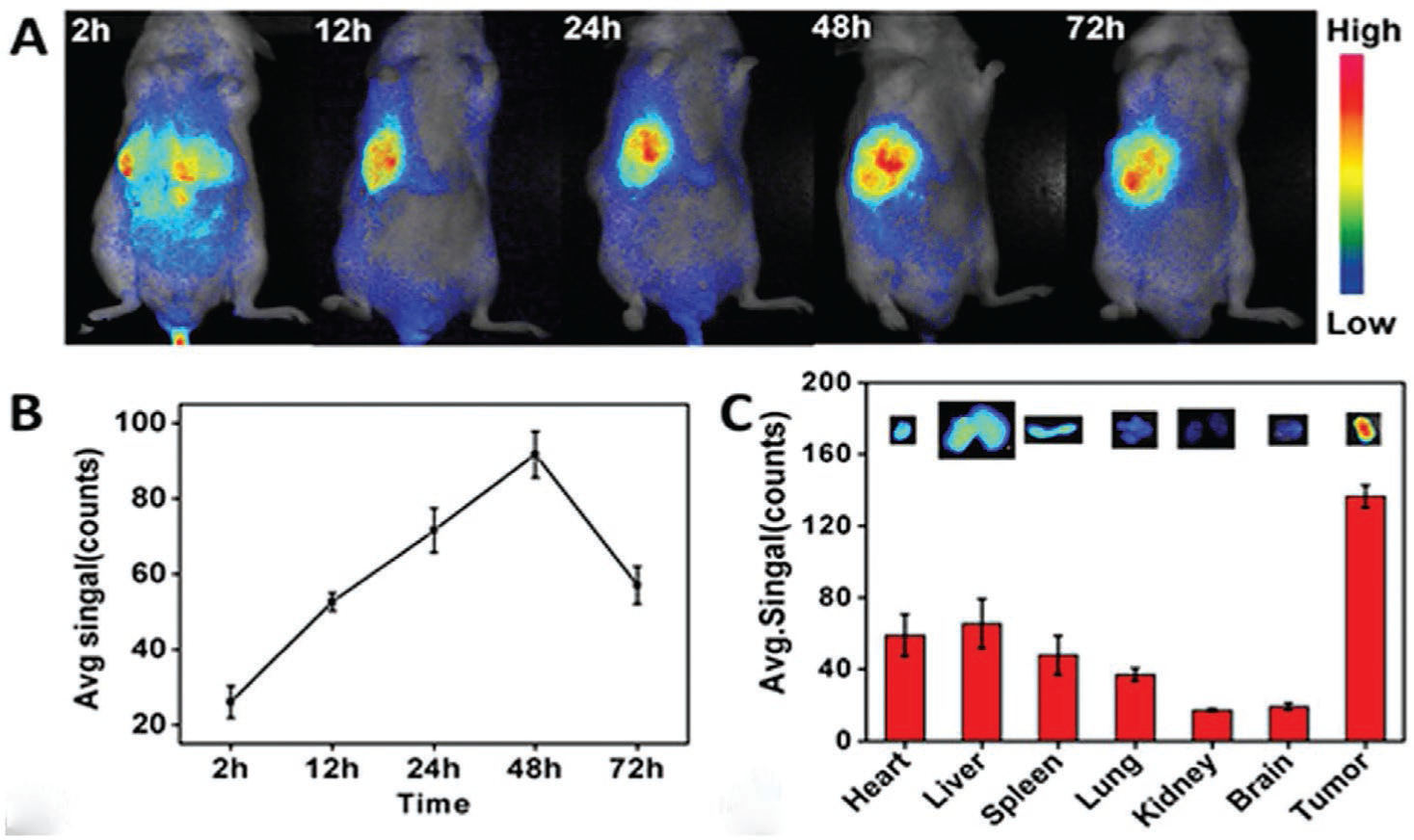
In vivo fluorescence imaging of Tf-IR780 NPs in tumor-bearing mice. A) In vivo NIR imaging and B) NIR intensity values of the mice bearing CT26 tumor injected with Tf-IR780 NPs (0.3 mg kg−1, IR780) at 2, 12, 24, 48, and 72 h post-injection, respectively; C) ex vivo imaging and NIR intensities of Tf-IR780 NPs in heart, liver, spleen, lung, kidney, brain, and tumor of the mice bearing CT26 tumor at 24 h post-injection. Adapted with permission.[116] Copyright 2016, Nature.
In order to achieve liver tumor imaging with bi-functional nanoprobes, Qi et al.[117] encapsulated SPIONs into PEG-poly(ε-caprolactone) (PEG-b-PCL) polymeric micelles, that were then decorated with Tf and the NIR fluorescent dye Cy5.5 to produce nanosized SPIO@PEG-b-PCL-Tf/Cy5.5 (SPPTC). No expression of TfR mRNA in the HL7702 normal cell line was observed, while there was good expression in the HePG2 carcinoma cells. The in vivo fluorescence imaging showed strong fluorescence at the tumor site 8 h post-injection of SPPTC, while there was no detectable signal in mice that were injected with nontargeted NPs (SPPC). To evaluate SPPTC as an MRI contrast agent, MRI images of tumor-bearing mice were taken before and after injection. SPPTC enhanced the contrast of the MR signal intensity by up to 54% at the tumor site, while it was measured to be only 16% in SPPC-treated group. This was explained by the accumulation of nontargeted NPs at the tumor via the EPR effect.
Ferritin is the natural iron storage protein possessing a cage-like structure and nanometer size (around 10 nm), with an affinity to the TfR type 1 (TfR1). Apoferritin (APF) is the version of ferritin that contains no iron, but has the same targeting ability.[118] Embedding melanin NPs (MNPs) and ferric ions into the cavity of APF were used to construct an efficient nanoplatform, AMF, for in vivo multimodality imaging (PET/MRI/PAI) of colon cancer.[119] The MNPs possessed excellent chelating ability for metal ions (Fe3+, 64Cu2+) that can be used for MRI and PET, and also had suitable optical characteristic to be used for PAI. The targeted AMF NPs exhibited higher cellular uptake in HT-29 cells, which had a high TfR1 expression compared to HepG2 cells, with a lower TfR1 expression. It was concluded that AMF increased the PET signal intensity 4 h post-injection in HT-29 tumor-bearing mice compared to controls. Similar results were achieved using MRI, and the relaxivity value of AMF was two times higher compared to the controls. Using PAI imaging with 500 μg mL−1 (based on MNP concentration), the PAI signal of AMF was twofold higher than MNPs, Fe-PEG-MNPs, and AMF without Fe.[119] This data suggested the TfR could be a target for future targeted cancer imaging.
6.2. Folate Receptor Targeting
The vitamin folic acid (FA) is transported into cells through receptor-mediated endocytosis mediated by the folate receptor (FR), which is overexpressed in cancer cell membranes compared to normal cells.[120] Different fluorescent nanomaterials such as semiconductor QDs,[121] carbon dots (CDs),[122] and small molecule organic dyes[123] have been decorated with FA to bind to cancer cells in vitro and in vivo. Liu et al.[124] reported the fabrication of a turn-on green fluorescent probe based on FA-modified CDS (FA-CDS) prepared by hydrogen bonding, to detect FR-positive cancer cells. The fluorescence intensity of CDs at 520 nm was gradually reduced by increasing the FA concentration, indicating that FA could quench the fluorescence of the CDs. Due to the weak interaction between FA and CDS, when FA binds to the FR, it detaches from the surface of the CDs, resulting in the recovery of the CD fluorescence. Thus, higher concentrations of FR, as found in tumor cells, resulted in a stronger fluorescence intensity. There was no significant fluorescence when normal cells were treated with FA-CDs. FA was also conjugated to rhodamine B-labeled poly(propylene fumarate)-co-poly(lactic-co-glycolic acid)-co-poly(ethylene glycol) NPs (PPF-PLGA-PEG-RhB-FA NPs) to track the NPs in both normal osteoblast MC3T3 cells and HeLa cancer cells.[125] In the normal cells, there was no significant difference between the fluorescence intensity of PPF-PLGA-PEG-RhB-FA and PPF-PLGA-PEG-RhB NPs. On the other hand, FA-conjugated NPs showed significantly higher fluorescence in cancer cells. Another study incorporated FA onto the surface of dye-loaded silica NPs as optical nanoprobes for in vitro and in vivo imaging. Depending on the variation of FR expression among the cell lines, their uptake for FA-conjugated silica NPs was different. In vivo imaging indicated that the targeted NPs preferentially accumulated at the site of pancreatic tumor-bearing mice, and there were either weak signals or no signals detected at 24 and 96 h post-injection, respectively. With the exception of the liver, there was no observable fluorescence in the brain, kidney, heart, and spleen, demonstrating good tumor specificity and targeted bio-distribution of FA-conjugated silica NPs.[126]
FA has also been used as a targeting moiety for many years using CT and MRI imaging modalities. In one such study, FA-linked polyethylenimine-entrapped gold NPs (FA-Au PENPs) were prepared for tumor CT imaging.[127] Unlike nontargeted Au PENPs, the tumor targeting ability of FA-Au PENPs via the FR was confirmed by confocal and ICP-OES. For targeted tumor CT imaging, the tumor bearing mice treated with FA-Au PENPs showed an obvious enhancement in CT contrast 5 h post-injection, with much higher CT values than nontargeted probes. In addition, 1 month later, H&E staining demonstrated that there were no histological changes in the liver, lungs, spleen, kidney, or heart of the mice, which indicated good in vivo biocompatibility of the FA-Au PENPs.
In another study, Zhang et al. synthesized FA-modified iron oxide (Fe3O4) NPs. The in vitro T2-weighted MR effect of the FA-modified Fe3O4 NPs on H460 lung carcinoma cells was evaluated using a 1.5 T MRI machine, in which the MR signal intensity of the cells showed a significant decrease as a function of Fe concentration, and the obtained images were much darker than those of the same cells treated with FA and nontargeted NPs. Moreover, at equal Fe concentrations, FR-positive cells absorbed more of the FA-modified Fe3O4 NPs compared to the FR-negative cells. MRI of H460 tumor-bearing mice injected with FA-modified Fe3O4 NPs at different time points was performed. There was a significant reduction in T2 signal intensity of H460 tumors at 0.85 h post-injection.[128] Overall, FR may hold great promise as a target for directed tumor imaging in the future.
6.3. EGFR Targeting
The tyrosine kinase EGFR is a 170 kDa transmembrane glycoprotein, which is activated by binding to endogenous ligands of the EGF family. EGFR plays a critical role in cell proliferation, division, inhibition of apoptosis, and angiogenesis, upon activation after internalization via clathrin-mediated endocytosis.[129] The over-expression of EGFR in diverse kinds malignant tumor cells has been demonstrated.[130] With a high affinity for EGFR (Kd = 2 nM), EGF proteins trigger cell proliferation in tumor cells.[131] Therefore, EGFR can be used for targeting cancer cells, using nanoplatforms that have been decorated with EGF proteins or EGFR antibodies for therapeutic and diagnostic applications. Moreover, anti-EGFR antibodies inhibit cell proliferation and trigger cell apoptosis by blocking the activation of EGFR. One study used cetuximab-800CW (anti-EGFR probe) as a fluorescent tracer for ex vivo colonoscopy using a NIR endoscopy platform.[132] The EGFR expression was about 51–69% higher in 78 low-grade dysplastic (LGD) adenomas than in normal colon crypts, and could be a promising tool for molecular-guided endoscopy. Gao et al.[133] encapsulated the t-BuPITBT-TPE fluorophore within DSPE-PEG NPs that were decorated with humanized mAb C225 (t-BuPITBT-TPE-C225 NPs), and used this complex for targeted imaging of EGFR overexpressing non-small cell lung cancer cells. The t-BuPITBT-TPE-C225 NPs were effectively internalized into EGFR overexpressing HCC827 cells showing a strong red fluorescence compared to only a very weak fluorescence in H23 cells, which express a significantly lower amount of EGFR on their surface.
Recently, multispectral optoacoustic tomography (MSOT) has been used to detect EGFR over-expression in orthotopic pancreatic xenografts, using a NIR EGF-conjugated CF-750 fluorescent probe.[134] Because MSOT is based on the photoacoustic features of the targeted tissue, it is not limited by photon scattering, resulting in high-resolution tomographic images. The specificity and bioactivity of the probe were investigated in different cell lines, including S2VP10L and MiaPaCa-2 cells, with high and low EGFR expression, respectively. After MSOT imaging of S2VP10L-tumor bearing mice, the EGF-conjugated CF-750 fluorescent probe showed the highest accumulation within the tumor 6 h post injection, with an average of 318 MSOT signal units. However, in mice implanted with MiaPaCa-2 tumors, the MSOT signal was only <10 MSOT signal units. These results indicate good binding and bioactivity of the EGF-conjugated CF-750 probe to EGFR in S2VP10 pancreatic tumor cells, and the ability of MSOT to detect the biodistribution of fluorescent dyes in living tissue.
Wang et al.[135] prepared a novel theranostic agent based on PEGylated SPIONs modified with anti-EGFR (Cetuximab) (anti-EGFR-PEG-SPIONs) for MRI and magnetic resonance-guided focused ultrasound surgery (MRgFUS) of lung cancer. They used this platform to address some limitations, such as low sensitivity of MRI for visualization of small tumors, and the poor efficiency of in vivo ultrasonic energy deposition. In vivo MRI employed two groups of H460 lung tumor bearing nude rats that were injected with anti-EGFR-PEG-SPIONs and PEGylated SPIONs (Figure 17). At 4 h post-injection of the targeted NPs, the T2 signal to noise ratio (SNR) showed a significant decrease at the tumor site compared to only a slight decrease with nontargeted NPs. This was explained by the targeting ability of anti-EGFR-PEG-SPIONs to the over-expressed EGFR on the H460 lung cancer cells. They also employed Prussian blue staining to confirm the targeted contrast agent had a higher deposition in tumor tissue than the non-targeted NPs (Figure 17c).
Figure 17.
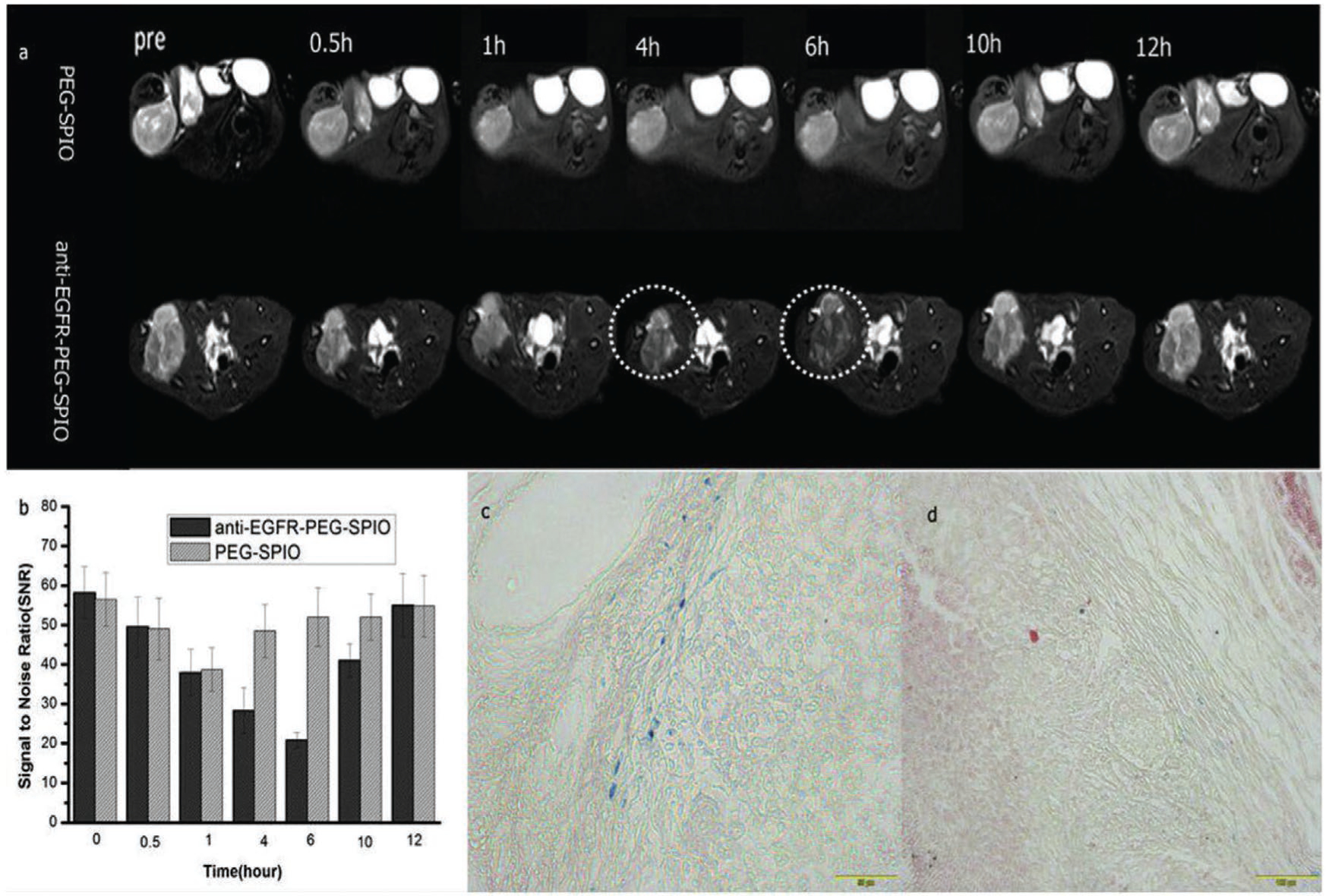
Tumor imaging with anti-EGFR-PEG-SPIONs. T2WI MRI images a) and SNR b) of tumor after injection of 0.1 mL targeted and nontargeted contrast agents at different time points (0.5, 1, 4, 6, 10, 12 h). The mean T2-weighted signal intensities were measured for each tumor. The relative SNR was calculated. Prussian blue staining of tumor tissues after 6 h injection of c) anti-EGFR-PEG-SPIONs and d) PEGylated SPIONs. Reproduced with permission.[135] Copyright 2017, Elsevier.
6.4. Glucose Transporter Targeting
Both primary and metastatic cancer cells consume higher amounts of glucose in order to provide themselves with energy, which is required for their rapid proliferation.[136] This increased glucose requirement results in upregulation of the glucose transporter (Glut) (e.g., Glut-1 and Glut-3) on the surface of cancer cells. This observation led to clinical imaging being revolutionized by the introduction of a new imaging approach termed FDG-PET. PET imaging is able to produce anatomical images with high resolution based on the preferential uptake of glucose into cancer cells compared to normal cells, employing the glucose analogue fluorodeoxyglucose labeled with the PET isotope 18-fluorine (half-life 10 min).[137] However, this technique is not considered highly specific for cancer, since other biological mechanisms (such as inflammation) result in higher metabolic uptake of glucose. Therefore, glucose imaging probes must distinguish between cancer and inflammation.
Inspired by PET, many researchers have taken advantage of the abnormal expression of Glut (as well as increased glucose metabolism) as a hallmark of cancer, in order to detect and image tumors. To this end, glucose (or its many derivatives) has been attached to different nanoplatforms, and their ability to target Gluts on cancer cells has been evaluated. Dreifuss et al.[138] employed glucose-functionalized gold nanoparticles (GF-GNPs) as a metabolically targeted CT contrast agent. They hypothesized that the cellular uptake of larger-sized GF-GNPs via GLUT-1 was unlikely, compared to the easier uptake of smaller glucose molecules. This led them to propose that GLU-1 induced a biological cascade that eventually resulted in the increased uptake of GF-GNPs, probably via endocytosis. Based on the CT images, the GF-GNPs could differentiate between cancer and inflammation, in a mouse model that combined both tumor and inflammation at different sites, possibly because of differences in the vasculature of the different pathologic conditions. Singh et al.[139] compared the mechanism of internalization between BSA-coated gold nanoclusters (BSA-AuNCs) and glucose-coated gold nanoclusters (Glu-AuNCs) in human epithelial carcinoma (A431) cells and the human keratinocyte cell line (HaCaT) as examples of cancerous and noncancerous cells, respectively. Based on fluorescence imaging, Glu-AuNCs were internalized by A431 cell via Glut-1 receptors, while there was significantly lower internalization by HaCaT cells. Likewise, BSA-AuNCs showed significantly higher cellular uptake in A431 cells than HaCaT cells, however, this internalization was dependent on the cell membrane potential, which affected the electrostatic interaction between cells and NPs. They further investigated the level of Glut-1 protein expression in both cell lines. As expected, the expression level of Glut-1 was 40% higher in A431 than HaCaT cells.
Recently, Zhao et al.[40b] reported the preparation of a novel dual-stimulus responsive nanoprobe for in vivo tumor-specific image-guided photothermal therapy. The nanoprobe called Pep-Acy/Glu@AuNRs, consisted of four constituents: i) gold nanorods (AuNRs) as the basic structure, photothermal therapy agent, and ultra-efficient fluorescent quencher; ii) an asymmetric fluorescent cyanine dye (Acy), which served as a tumor-specific imaging probe with pH-responsive near-infrared (NIR) absorption and fluorescence; iii) MMP-specific peptide (Pep) acting as a linker between the AuNRs and Acy; and iv) glycosyl residues (Glu) on the surface of AuNRs providing active tumor-targeting ability. In the presence of MMP type 13 at a pH of 6.0, there was intense fluorescence due to the detachment of Acy from the AuNRs because Pep was cleaved by MMP enzyme, and pH-sensitive activation of Acy into its acidic fluorescent form. Pep-Acy/Glu@AuNRs were not fluorescent either at pH 7.4 or in the absence of MMP-13. The in vitro cell internalization revealed that at pH 6.0, SCC-7 cells incubated with Pep-Acy/Glu@AuNRs showed 2.7-fold higher fluorescence intensity than the Glut-blocked SCC-7 cells, indicating the critical role of Glut in the cellular uptake of NPs via Glu interaction with Glut. They then explored the ability of Pep-Acy/Glu@AuNRs to allow in vivo precision tumor-targeting imaging in SCC-7 tumor-bearing nude mice (Figure 11). The fluorescence signal was intense and lasted for up to 12 h in the active tumors (R-tumor). In contrast, by manipulating the TME using either a MMP inhibitor (for Group A), or NaHCO3 (for Group B), there was almost no fluorescence signal detected in either group. These results demonstrated the various features of the designed nanoplatform, including dual-stimuli responsivity, accuracy, tumor targeting via the microenvironment, and overexpression of Glut within the tumor cells.
6.5. Cathepsin Targeting
Cathepsins (Cats) are a group of lysosomal peptidases with a cysteine residue at the active enzymic site. Cats are proteins that degrade the ECM and the basal membrane, and are essential for tumorigenesis. In humans, the Cat family comprises 11 members (Cat B, Cat C, Cat F, Cat H, Cat K, Cat L (Cat L1), Cat L2 (Cat V), Cat O, Cat S, Cat W, and CatZ [Cat X]). The majority of Cats are endopeptidases that cleave peptide bonds within their protein substrates (Cat C and Cat Z do not have any endopeptidase activity). Cat B possesses carboxypeptidase activity and Cat H possesses aminopeptidase activity. The functional contributions of the cysteine cathepsins to tumor invasion and metastasis are diverse. For example, Cat B, C, K, L, S, V, and X are all expressed in tumor-associated macrophages (TAM). Cats B, F, H, K, L, S, V, and X are all expressed in different tumor cells. Increased Cat expression is correlated with poor prognosis in breast (Cat B, L, C, and S), lung (Cat B, H, and S), ovarian cancer (Cat B), pancreatic (Cat B, L, and Z), osteosarcoma (Cat K), and colorectal cancer (Cat B, L, and S).[140]
Because Cat B an attractive target for the detection of tumor metastases, Ryu et al.[141] developed a Cat B-sensitive nanoprobe (Cat B-CNP) consisting of a self-quenched Cat B-sensitive fluorogenic peptide (Gly-Arg-Arg-Gly-Lys-Gly-Gly) probe conjugated onto the surface of tumor-targeting glycol-chitosan NPs. This platform facilitated sensitive and specific visualization of Cat B activity in cells and in vivo tumor models. Cat B-CNP demonstrated the potential to distinguish metastases in three metastatic mouse models including lung, liver, and peritoneal metastases. Metabolic glycol-engineering with a biorthogonal click reaction has been used to improve the tumor targeting efficiency of NPs as delivery vehicles for imaging agents or for anticancer drugs. This technique can develop metabolic agents that can create abnormal glycans expressed on the tumor-cell surface that can then be labeled with click chemistry approaches. Shim et al.[142] developed a Cat B-specific metabolic precursor consisting of a Cat B-specific cleavable substrate (Lys-Gly-Arg-Arg (KGRR)) conjugated to triacetylated N-azidoacetyl-D-mannose amine (RR-S-Ac3ManNAz) for creation of azide-containing glycans on tumor cells. Subsequently these azido-glycans could be labeled by NIRF dyes containing triple bonds using the click reaction. In vivo imaging results showed this system could be a promising tool for tumor specific active targeting.
An amino-functionalized metal–organic framework (MOF) could be an efficient delivery vehicle for cell imaging and chemo-photodynamic therapy. Liu et al.[143] developed a multifunctional MOF nanoprobe loaded with camptothecin (chemotherapy drug), FA (targeting moiety), and a chlorin (e6) (Ce6)-conjugated Cat B-substrate peptide as the activatable moiety. The MOF probe recognized FR-positive tumor cells, where Cat B activated the release of Ce6 as an imaging agent and a photosensitizer, and camptothecin as an anticancer drug.
Targeting cancerous tissue with iodinated CT contrast agents could be improved by taking advantage of enzyme overexpression using activity-based probe (ABP) methodology. A typical ABP includes a recognition element that is a substrate for a tumor-associated protease, a contrast agent, and a “war-head” (usually an electrophile that can form a covalent linkage between the target and the contrast agent). When CT is used for cancer imaging applications, its relatively low contrast requires the use of high concentrations of contrast agents. In order to overcome this limitation, Gaikwad et al., prepared a new class of iodinated nanoscale ABPs (IN-ABPs) that could enrich the concentration of iodine at the targeted tumor site by covalent attachment in the presence of Cats that are significantly overexpressed in cancer. The IN-ABPs were composed of a short targeting peptide sequence selective for specific Cats, an electrophilic moiety that allowed activity-dependent covalent binding, and tagged with dendrimers loaded with iodine atoms. IN-ABPs selectively bound to tumors in the presence of recombinant and intracellular Cats B, L, and S. They compared the in vivo biodistribution and tumor accumulation of IN-ABPs bearing 18 or 48 iodine atoms. The result of this study showed the synthetic feasibility and potential utility of ABPs as potent contrast agents for CT of tumors.[144] Employing a similar strategy, Tsvirkun et al.[145] developed nanosized Cat-targeted ABPs for functional CT imaging. Their probe consisted of various sizes of gold NPs with varying ratios of Cat-targeted substrate and PEG. The results showed that GNP-ABPs were a promising tool for enzymatic-based CT imaging.
7. Tumor Type-Specific Targeting
The TME is characterized by altered functions of ECM molecules, vascularized stroma, lymphatic networks, and abnormal cell phenotypes. The molecular imaging of specific cancer cell types plays a critical role in tumor detection, as described below. Depending on the type and stage of cancer, different antigens or receptors are overexpressed on the surface of the cancer cells and can be used in ligand-mediated targeted tumor imaging. Ligand targeting increases interactions between the contrast agent and the targeted cells, and also enhances the cell internalization of the agent without altering the overall biodistribution.[146] There are two types of cancer-specific ligands: i) serum markers that are mostly used for monitoring of patients with already diagnosed disease, predicting response to therapy, and determination of prognosis; ii) markers that exist within the tumor tissue (cell surface) and are used for molecular imaging and detection of cancer. Because serum markers have low sensitivity and are not useful for early detection, we have concentrated on the tumor tissue markers in most common cancer types with an annual incidence of 40 000 cases, and high mortality rates according to NCI data (https://www.cancer.gov/types/common-cancers).
7.1. Targeted Breast Cancer Imaging
Breast cancer is a multifaceted and heterogeneous disease with a high worldwide mortality rate.[147] Based on NCI data, breast cancer was the most common type of cancer diagnosed in 2018, with 266 120 new cases and 409 20 deaths.[148] Specific breast tumor tissue markers used in diagnosis and therapy, such as estrogen receptor (ER), progesterone receptor (PR), hormone receptor (HR), HER2 gene (also known as c-erbB-2 or neu), urokinase plasminogen activator (uPA), and plasminogen activator inhibitor 1 (PAI-1) are covered in Table 4. These are the most promising biomarkers in lymph node-negative breast cancer. However, only a select few of these markers have been clinically used for imaging. Depending on changes in the levels of ER, PR, HR, and HER2, breast cancer is often characterized into subtypes: luminal A (ER+/PR+/HER2−), luminal B (ER+/PR+/HER2+), HER2 overexpressing (ER−/PR−/HER2+), and TNBC (ER−/PR−/HER−).[149] Luminal tumors (≈70% of invasive breast cancers) respond to hormonal therapy, and the HER2 overexpressing subtype responds to targeted antibody therapy. TNBCs are more aggressive and difficult to treat, but may respond to chemotherapy. Breast cancer is classified into five stages (0, I, II, III, IV) with different marker expression, biology, and therapeutic responses. Hence, a full understanding of breast cancer subtypes is important for the success of treatment outcomes.
Table 4.
Summary of imaging platforms used for the detection of a range of human cancers.
| Biomarker (Target) | Ligand | Imaging platform | Status | References |
|---|---|---|---|---|
| Breast cancer | ||||
| gC1q receptor p32 protein | Peptide, CGNKRTRGC (LyP1) | Bi2S3-LyP-1 | In vitro/in vivo | [223] |
| Phosphatidylserine (PS) | monoclonal antibody (mAb), PGN635 | PGN-L-IO/DiR | In vitro/in vivo | [224] |
| urokinase plasminogen activator receptor (uPAR) | Amino-terminal fragments (ATF) | NIR830-ATF-IONP | In vivo | [225] |
| Epithelial cell adhesion molecule (EpCAM) | Aptamer | Apt-QD-Nut-NPs | In vitro/in vivo | [226] |
| Macrophage mannose receptor (MMR; CD206) | Anti-CD206 Ab | Dye-anti-CD206 | In vitro/in vivo | [227] |
| CD44 | Hyaluronic acid | HA-dOG-PTX-PM | In vivo | [228] |
| HER2 | Trastuzumab | 89Zr-Trastuzumab | Clinical trial | [229] |
| Gastrin releasing peptide (GRP) receptors | Bombesin (BBN) | DSPION-BBN | In vitro/in vivo | [230] |
| Chemokine Receptor CXCR4 | Pentixafor | 68Ga-Pentixafor | In vitro/in vivo | [231] |
| Sodium iodide symporter (NIS)-mediated nuclear reporter | I-124 | I-124 | In vitro/in vivo | [232] |
| CD146 | YY146 Ab | 64Cu-NOTA-YY146 | In vitro/in vivo | [163] |
| CD38 | IgG Ab | 89Zr-Df-IgG | In vitro/In vivo | [233] |
| Subtype somatostatin receptor 2 (SSTR2) | PA1 peptide | 68Ga-DOTA-PA1 | In vitro/In vivo | [234] |
| CD30 | Brentuximab vedotin (BV) | 89Zr-Df-BV | In vitro/In vivo | [235] |
| Monoclonal antibody, h173 | mAb, h173 | 64Cu-DOTA-h173 | In vitro/In vivo | [236] |
| Colorectal cancer | ||||
| Translocator protein (TSPO) | [18F] FEPPA (N-acetyl-N-(2-[18F]-fluoroethoxyben-zyl)-2-phenoxy-5-pyridinamine) | TSPO-PET tracer [18F] FEPPA | In Vitro | [237] |
| VEGFR-1 and NRP-1 (neuropilin-1) | CPQPRPLC Peptide | [99mTc] Tc-HYNIC-D(LPR) peptide for SPECT imaging | In vitro/In vivo | [238] |
| Metastatic SW620 membrane protein | SW620-specific DNA aptamer | Aptamer XL-33 | In vitro/Ex vivo | [239] |
| EpCAM | Anti EpCAM mAb | UCNP@SiO2 Rose Bengal (RB)- Linker-Protein G (LPG)-Anti EpCAM | In vitro | [240] |
| Colon cancer secretedprotein-2 (CCSP-2) | Anti-CCSP-2 antibody | Anti-CCSP-2 antibody-FPR-675 | In vitro/In vivo | [241] |
| Hexosaminidase | Probe for β-galactosidase | Hexosaminidase (HMRef-βGlcNAc) | In vitro/Ex vivo | [242] |
| Carcinoembryonic antigen (CEA) | Anti-CEA Ig G | IgG-conjugated fluorescent nanoparticles | In vivo | [243] |
| Claudin-1 | RTSPSSR Peptide | Cy5.5- a GGGSK Linker-Peptide | In vivo | [244] |
| Delta-like ligand 4 (Dll4) | Dll4 mAb (61B) | 61B-DOTA-64Cu PET probe | In vitro/In vivo | [245] |
| Prostate cancer | ||||
| PSMA | HBED (PSMA-targeted probe) | 68Ga-HBED-CC PSMA PET | In vivo (PCa patients) | [246] |
| PSMA | Anti-PASMA Ab | QD- PEG -PSMA Ab | In vivo | [247] |
| Gastric-releasing peptide receptors (GRPR) | Bombesin (BBN) | BBN-conjugated Cy5.5 - N-acetyl histidine - Glycol chitosan NPs | In vitro/In vivo | [248] |
| PSMA | xPSM-A9 and xPSM-A10 (RNA aptamers)/RGD/Ab | G4.5 PAMAM dendrimer- iron oxide | In vitro/In vivo | [249] |
| SPARC glycoprotein | M13 filamentous bacteriophage | M13-SBP-MNP | In vitro/In vivo | [250] |
| Androgen receptor (AR) | Peptide SP204 | Superparamagnetic iron oxide nanoparticles (SPIONs- SP204 | In vitro/In vivo | [183] |
| Prostate stem cell antigen (PSCA) | Anti PSCA mAb | GO-DEN(Gd-DTPA)-mAb | In vivo | [251] |
| Gastric-releasing peptide receptors (GRPR) | Bombesin (BBN) analogue (named SCH1) based on JMV594 peptide | 68Ga-NODAGA-SCH1 | In vitro/In vivo | [252] |
| Insulin-like growth factor 1 receptor (IGF1R) | Ab clone (1A2G11) | 64Cu-NOTA-1A2G11 | In vitro/In vivo | [253] |
| urokinase-type plasminogen activator receptor (uPAR) | AE105 Ab | 64Cu-DOTA-AE105 | In vitro/In vivo | [254] |
| GRPR | Peptides NOTA-BBN2 | 68Ga-NOTA-BBN2 | in vivo | [255] |
| Pancreatic cancer | ||||
| CD47 | iExosomes | iExosomes | In vitro/In vivo | [256] |
| Bombesin (BN) receptors | BN peptide | BN-CLIO(Cy5.5) | In vivo | [257] |
| Toll-like receptor 2 (TLR2) | TLR2 agonists | TLR2 agonists-IR800CW | In vitro | [258] |
| Insulin-like growth factor-1 (IGF1) receptor | IGF1 | IGF1-IONP-Dox | In vitro/In vivo | [189] |
| Claudin-4 | Anti-Claudin-4 | QDs- anti-Claudin-4 | In vitro | [259] |
| uPAR | Amino-terminal fragment (ATF) peptide | ATF-IONP-(GFLG)-Gem | In vitro/In vivo | [260] |
| KRAS2 mRNA | KRAS2 PNA-D (Cys-Ser-Lys-Cys) | [111In]DOTAn-Poly (diamidopropanoyl) m-KRAS2 PNA-D(Cys-Ser-Lys-Cys) | In vivo | [261] |
| Neurotensin receptors (NTRs) | Neurotensin (NT) | Aluminum-18F-NOTA-NT | In vivo | [262] |
| CA19.9 | Anti-CA19.9 Ab 5B1 | 89Zr-5B1 | In vivo | [263] |
| Bladder cancer | ||||
| CD44 | CD44v6 Ab | Liposomal nanoprobe | In vivo | [264] |
| CD47 | CD47 Ab | Antibody-functionalized SERS nanoparticles | In vitro/In vivo | [265] |
| Prostate stem cell antigen (PSCA) | Anti PSCA mAb | QD-PSCA | In vitro | [266] |
| Carbonic anhydrase IX (CAIX) | Anti-CAIX Ab | Anti-CAIX-Qdot625 | In vitro/ ex vivo | [267] |
| Argininosuccinate synthetase 1 (ASS1) | Fluoro-L-thymidine (FLT) | [18F]-fluoro-L-thymidine (FLT) | In vitro/In vivo | [268] |
| CD47 | CD47 Ab | Anti-CD47-FITC | In vivo | [200] |
| IL-5Rα | mAb A14 | 64Cu-A14 | In vitro/In vivo | [269] |
| Brain cancer | ||||
| Transferrin receptor | Transferrin peptide (Tfpep) | Tfpep-Au NPs | In vitro | [270] |
| TfR & HER2 | Poly (β-l-malic acid) polymeric nano-imaging agents (NIAs) | Gadolinium-DOTA- Poly (β-l-malic acid) polymeric nano-imaging agents (NIAs) | In vitro/In vivo | [271] |
| EGFR & CD105 | Denoted as Bs-F (ab) 2] | 64Cu-NOTA-Bs-F (ab)2 | In vitro/In vivo | [272] |
| Lipoprotein receptor-related protein (LRP) receptors & αvβ3 | Angiopep-2 peptides & cyclic [RGDyK] peptides | PAMAM-G5 dendrimer | In vitro/In vivo | [273] |
| LRP-1 | ANG | ANG -Tetrahedral DNA nanostructures (TDNs) | In vivo | [274] |
| LRP-1 | Angiopep-2 (ANG, TFFYGGSRGKRNNFKTEEY) | ANG/PLGA/DTX/ICG | In vitro/In vivo | [275] |
| CD146 | YY146, a high affinity anti-CD146 mAb | 89Zr-Df-YY146 | In vitro/ex vivo | [276] |
| Ovarian cancer | ||||
| IL-16 | anti-IL-16 Ab | Microbubbles- anti-IL-16 | In vivo | [277] |
| Death receptor 6 (DR6) | Anti-chicken DR6 Ab | Microbubbles-anti-chicken DR6 Ab | In vivo | [278] |
| CD276 | Anti-CD276 Ab | Microbubbles- anti-CD276 Ab | In vivo | [279] |
| HER2 | Trastuzumab | 89Zr-trastuzumab | In vivo | [280] |
| HER3 | HER3-antibody RG7116 | 89Zr-RG7116 | Phase I | [281] |
| Mesothelin | Anti-Mesothelin nanobody (NbG3a) | NbG3a-IONP | In vivo | [282] |
| HER-2 | anti-HER-2 mAb | Optical viral ghosts (OVGs)-ICG-anti-HER-2 | In vitro/In vivo | [283] |
| Cyclooxygenase-1 (COX-1) | P6 | [18F]-P6 | In vivo | [284] |
| Poly (ADP-ribose) polymerase (PARP) | FluorThanatrace (FTT) | [18F] FTT | In vitro/in vivo | [285] |
A recent report described a CD44-targeted nanomicellar payload delivery platform for selective tumor-specific imaging and therapy of triple negative breast cancer.[150] Several mAbs, e.g., trastuzumab (Herceptin) and pertuzumab, or small-molecule tyrosine kinase inhibitors (TKIs), such as neratinib and lapatinib, have been utilized for targeting HER2+-overexpressing breast tumors.[151] A range of specific molecules such as, “protein-phosphatase 2A-regulatory molecule (B) 55 β-subunit” (PP2A-B55β), P7170 (synthetic inhibitor), IL-15 receptor and it’s α subunit (IL15RA), and progesterone receptor (PgR) have been used for clinical targeting and inhibition of TNBC subtypes.
Radiolabeled fluoroestradiol (18F-FES) PET/CT imaging has been utilized for primary ER+ breast cancer detection, evaluation of metastases and monitoring response to endocrine therapy. FES is an estrogen hormone (EH) with the affinity to bind to ERα, and has been used as a targeted contrast agent for 18F-FES PET/CT imaging.[152] Heat shock proteins (Hsps) can function as a marker of breast cancer, and have been targeted using multifunctional NPs based on perfluoropolyether (PFPE)-conjugated peptide aptamers that specifically bind to Hsp70, and act as fluorescent and MRI contrast agents. The in vivo results demonstrated the platform possessed high tumor accumulation with a specific affinity to Hsp70. These peptide aptamers could effectively target the TME (surface of the tumor cells) and the interior of the tumor cells.[153]
Neu or HER2 is a proto-oncogene that is overexpressed in up to 30% of breast cancers. Kievit et al.[154] developed an imaging probe constructed from superparamagnetic iron oxide nanoparticles (SPIONs) coated with copolymer chitosan grafted PEG, and then conjugated with an anti-neu antibody. MR imaging demonstrated the probe was able to target neu receptors in vitro and also in vivo in a transgenic mouse model. Furthermore, this probe was able to recognize and tag spontaneous micrometastases in the liver, bone marrow, and lungs of tumor-bearing mice.
The overexpression of the chemokine receptor (CXCR4) plays an important role in breast cancer cell proliferation, invasion, and metastasis. One study developed 64Cu-doped gold nanoclusters conjugated to AMD3100, a ligand that specifically binds to CXCR4. The 64Cu-AuNC-AMD3100 was used for detection of lung metastasis in a mouse model bearing 4T1 metastatic breast cancer. The PET imaging results showed that the contrast agent had excellent affinity and sensitivity for targeting CXCR4, both in early stages of tumor, and in micro-metastases in the lungs.[155] Y1 receptors (Y1Rs) are also highly overexpressed in human breast cancer and its metastases. Fluorescent nanobubbles (NBs) were fabricated from tetradecafluorohexane and biodegradable photoluminescent polymers, and then conjugated to a PNBL-NPY ligand developed for specific targeting of Y1Rs both in vitro and in vivo. The results showed PNBL-NPY-modified NBs had good dispersity, biocompatibility, stability, and also possessed high affinity and specificity for Y1Rs.[156] In order to increase the specificity, probe circulation time, and precise targeting, a dual targeting strategy was described using hybrid GNRs conjugated to Herceptin (HER) and PEG. The imaging results showed good accumulation of the Her-PEG-GNRs in tumors compared to Her-GNR and PEG-GNR tested alone.[157]
7.2. Targeted Lung Cancer Imaging
Lung cancer is highly invasive and metastatic, with one of the cancer highest mortality rates worldwide.[158] According to the NCI, an estimated 234 030 new cases and 154 050 deaths from lung cancer were reported in 2018. Lung cancer is a heterogeneous disease and is difficult to diagnose early in many cases. The disease is often diagnosed only in advanced stages (stage III or VI). The lung cancer subtypes include i) squamous cell lung cancers (SQCLC), which account for ≈25–30% of all cases and arise from the main bronchi and spread to the carina; ii) adenocarcinomas (adenoCA), which represent about 40% of all lung cancers and arises from peripheral bronchi; iii) lung cell anaplastic carcinomas (LCAC), which represent about 10% of all lung cancers and lack classic glandular or squamous morphology in the tumor; and iv) small cell lung cancer (SCLC), which accounts for ≈10–15% of all lung cancers and arises from the lung neuroendocrine cells, and disseminates into the sub-mucosal lymphatic vessels and regional lymph nodes without any bronchial invasion. Based on histological data, lung cancer is divided into two classes, with different growth and spread profiles: non-small-cell lung carcinomas (NSCLC), which consist of adenoCA, LCAC, and SQCLC subtypes, and accounts for ≈85–90% of all lung cancers; and SCLC accounting for ≈10–15% of lung cancers. All lung cancer subtypes can become multifocal within the part of the lung they first occur (T3), spread throughout the lung of origin (T4), or spread to the contralateral lung (M1).[159]
Metastatic lung cancer can be diagnosed in inaccessible sites such as the bone, liver, or brain before any symptoms occur due to the primary lung lesion. Depending on genetic alterations, lung cancer can be classified in several ways, including: i) activation of mutations in proto-oncogenes such as BRAF, MEK, KRAS, PI3K, HER2, FAT2, GPR87, LYPD3, SLC7ALL, and especially EGFR; ii) amplification of proto-oncogenes, such as fibroblast growth factor receptor 1 (FGFR1) and discoidin domain receptor (DDR2) in SQCLC, and MET in adenoCA; iii) gene activation in anaplastic lymphoma kinase (ALK), rearranged during transfection (RET), or c-ros oncogene 1 (ROS1); iv) overexpression of miRNAs; v) inactivation of tumor suppressor genes (TSG), including RB1, CDKN2A, TP53, PTEN, FHIT, RASSF1A; and vi) increased telomerase activity.[160]
EGFR is mutated and overexpressed in almost 80% of NSCLC cases. Anti-EGFR Abs have been used as contrast agents in lung cancers. However, Ab production is difficult and costly, and Abs have rather low tumor penetration due to their large size. Therefore, a short peptide sequence (P75) was introduced as an EGFR-targeting peptide and used for CT/photoacoustic dual-modality image-guided photothermal therapy, Zhao et al.[161] designed P75-modified triangular gold NSs (P75-PEG-TGN). The in vitro and in vivo results showed high affinity to EGFR+ cancer cells, and increased accumulation on the tumor cell surface. The cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is a marker of immune T cells and also some lung cancer cells. The Ehlerding group[162] used 64Cu-radiolabeled ipilimumab (anti-CTLA-4 mAb) for PET imaging of human NSCLC cells. In vivo results showed the radiolabeled Ab effectively accumulated in CTLA-4+ NSCLC.
Other CD markers (i.e., CD30, CD48, CD146, CD44, and CD133) are overexpressed on the surface of lung cancer cells. Overexpression of CD146 (or MUC18) is associated with metastatic potential and is detectable in 50–75% of lung cancers. The YY146 mAb was radiolabeled using 64Cu (64Cu–NOTA-YY146) as a targeted contrast agent for in vitro and in vivo PET imaging of CD146+ intrapulmonary metastases of NSCLC cells.[163] Additionally, the delta-opioid receptor (бOR; a member of the G protein receptor family) is overexpressed in human lung cancer cells, but not expressed in normal lung cells. Cohen et al.[164] described an imaging system using synthetic Dmt-Tic peptide (a бOR antagonist) and IR800 NIR dye that had excellent affinity for бOR for in vitro lung cancer cell imaging. More examples are provided in Table 4.
7.3. Targeted Colorectal Cancer Imaging
Colorectal cancer (CRC) is the second-most and third-most common cancer in the United States for women and men, respectively. More-developed regions of the world have a higher incidence than less-developed regions. Based on the standardized incidence rate (ASRi), the majority of patients with sporadic CRC are >50 years of age, and both genetic factors (e.g., mutations in the DNA mismatch-repair genes, proto-oncogenes, and tumor suppressor genes) and environmental factors (e.g., smoking, alcohol intake, and increased body weight) contribute to the etiology of CRC. Furthermore, epigenetic alterations seem to affect gene expression to trigger changes in benign polyps into malignant tumors.[165] Nowadays, CRC diagnosis relies on assessment of patient symptoms and is followed by an instrumental approach if needed (i.e., colonoscopy, capsule endoscopy, CT colonography, and measurement of prognostic/predictive biomarkers of CRC). CRC biomarkers can be categorized into diagnostic, pharmacological, predictive, prognostic risk/predisposition, screening, and surrogate response biomarkers.[166] The most important DNA biomarkers are microsatellite instability (MSI), aberrant methylation of septin 9 (SEPT9) (a GTPase), mutation of adenomatous polyposis coli (APC), and Kirsten rat sarcoma (KRAS).[167] CRC (colon and rectal cancers) and their stages are important for the treatment of CRC. Surgery and targeted therapy using mAbs against over-expressed factors (e.g., EGFR, VEGF-A, HER) is the mainstay potentially curative treatment for patients with non-metastatic tumors (stages I–III), while fusion proteins that target multiple proangiogenic growth factors have been utilized for metastatic CRC (Stage IV).[168] Beyond conventional imaging modalities, PET-CT is a novel molecular imaging approach employing radiolabeled Abs or Ab fragments to detect CRC overexpressing EGFR[169] (Table 4).
If metastatic disease becomes clinically established, the long-term patient outcomes are not favorable, and current imaging rarely detects the early stages of cancer development at either the primary or metastatic sites. NPs have been found to accumulate in tumors in high amounts.[170] CRC diagnosis and treatment could also by improved by employing NPs or nanoprobes. Detection of small polyp was enabled using a nano-beacon composed of polystyrene NPs with coumarin 6 dyes encapsulated within the core, and a surface decorated with poly(N-vinylacetamide) (PNVA), and coated with peanut agglutinin (PNA). These NPs showed a high binding affinity to the CRC-associated Thomsen–Friedenreich (TF) antigen. The designed nano-beacon could be used for the clinical detection of hidden polyps, early quantitative detection of CRC, and for distinguishing adenomas and adenocarcinomas from normal colonic tissue.[171]
The multiple vibrational modes of NIR emission can be improved by using QDs. Unlike organic dyes, QDs can allow for multiplexed imaging due to the narrow-band emissions. Development of a protease-activatable QD (PbS/CdS/ZnS core/shell/shell) probe emitting in the NIR-II spectral region (PA-NIRQD) showed selective fluorescence activation and a high signal peak in the presence of MMP enzyme activity at tumor sites in a colon cancer mouse model.[172] In order to overcome poor tissue penetration of light and the background auto-fluorescence of traditional fluorescence-based imaging probes, one study used multifunctional silica-based nanocapsules, which contained two distinct triplet-triplet annihilation upconversion (TTA-UC) chromophore pairs, and were then conjugated with TCP (a vasculature-targeting peptide for CRC). The experimental results demonstrated that this platform bound only to CRC cells with differential-color imaging and greater accumulation at targeted tumor sites, and was a promising tool for CRC diagnosis within the heterogeneous TME.[173]
Images generated using fluorescent microscopy/endomicroscopy (such as two-photon microscopy [TPM]) have high resolution, which enables visualization of biological processes (such as cell trafficking and cell–cell interaction). The morphology of biopsies taken from diseased colon could be visualized without fixation and staining. Beack et al.[174] developed PNA-conjugated hyaluronate (HA) with high affinity to CD44/CD44v6 receptors, for colon cancer detection and to enable image-guided endoscopic resection of a large colorectal polyp. TPM of rhodamine B (RhoB) fluorescence has been used for bioimaging of CRC. Another strategy to improve detection of smaller or non-polypoid lesions that have miss rates of up to 24% during colonoscopy, is to combine advanced imaging technology and targeted molecular probes, preferably using biomarkers that apply to the whole surface area of the colon. c-Met is a human cell membrane tyrosine kinase that is overexpressed in the early stages of the colorectal adenoma-carcinoma progression. GE-137 is a fluorescently labeled peptide agent with a high affinity for c-Met. After being conjugated to a fluorescent cyanine dye and administered to mice and human patients, fluorescence colonoscopy enabled visualization of neoplastic polyps.[175] The altered pHe of cancer tissue could lead to drug resistance and has been considered as an imaging target. One study used fluorescent probes, two-photon probes (XBH1–3), and a two-photon microscope for the in-situ measurement of pHe. Ex vivo and in vivo results suggested that the XBH1 platform selectively stained cells in the acidified cancer tissue. This probe could directly monitor pH values both inside and outside the cells in colon cancer tissue, as well provide information on morphological aspects.[176]
7.4. Targeted Prostate Cancer Imaging
Prostate cancer (PCa) is the second most frequently diagnosed solid-organ malignancy in men in the United States and the second most common in males worldwide. Age range (50–74 years), race (African–American race), and family history (e.g., BRCA mutations) are the most established risk factors for prostate cancer.[177] FDA-approved prostate specific antigen (PSA or human kallikrein-3), ProPSA, and prostate cancer antigen 3 (PCA3) are noninvasive biomarkers that are currently used for prostate cancer detection.[178] However, most modalities have poor sensitivity and specificity at low PSA levels. Advancements in the field of molecular imaging are important for developing multimodality imaging for biopsy guidance aimed at early detection of PCa, or recurrence posttreatment (Table 4).
Prostate-specific membrane antigen (PSMA) is a membrane glycoprotein that is strongly upregulated at all stages of PCa. Numerous studies have employed Abs targeting PSMA for improving the imaging sensitivity. At present, only the radiolabeled anti-PSMA Ab targeting the intracellular epitope (7E11) (ProstaScint, Jazz Pharmaceuticals, USA) has been approved by the FDA. The rapid clearance of PET tracer labeled, anti-PSMA Ab from off-target tissues made it an ideal tracer for PCa detection, staging, and clinical decisions.[179] The heterogeneity of PCa motivated an increased focus on the tumor vasculature for imaging. Agemy et al.[180] designed a PCa vasculature homing-based (synoptic) targeting agent using iron oxide NPs coated with CREKA, a blood clotting peptide that recognizes the fibrin-fibronectin complexes. The CREKA-PEG-NPs self-amplified their tumor accumulation, enhanced tumor imaging, and allowed for optimized treatment.
Hepsin (HPN) is a type II transmembrane serine protease that is expressed in the precursor lesions of prostate cancer, high-grade prostatic intraepithelial neoplasia (HG-PIN), and hormone-refractory metastatic tumors. HPN binding peptides conjugated to imaging nanoprobes bound to PCa with high affinity in vivo. In situ histochemical analysis of patient tissues demonstrated the potential of this nanoprobe as an imaging agent for PCa.[181] Likewise, GRPRs are overexpressed in prostate tumor cells. One study conjugated a GRPR bombesin (Bom) peptide to the PET isotope 64Cu. In vitro micro-PET/CT imaging results confirmed the binding specificity of this platform to GRPR on the prostate cancer cell surface. Furthermore, in vivo results demonstrated that these NPs exhibited no acute toxicity in treated mice, suggesting that Bom-PEG-[64Cu] CuS NPs were ideally suited for PET imaging of orthotopic prostate tumors.[182] Another study synthesized SP204 and PC204 peptide-conjugated SPIONs that accumulated in a PCa xenograft model, with potential for PCa-targeted imaging and diagnosis.[183]
The robust molecular structure of tobacco mosaic virus (TMV) offers a versatile platform for theranostic applications. The Hu group[184] synthesized a bimodal imaging agent by loading the internal cavity of TMV self-assembled NPs with a NIRF dye Cy7.5 dysprosium ions (Dy3+) to produce a complex. The imaging probe was then conjugated with Asp-Gly-Glu-Ala (DGEA) peptide that targets integrin α2β1. NIRF imaging and T2-mapping (using ultra-high-field MRI [UHFMRI]) confirmed that this biocompatible probe effectively targeted PC-3 PC cells and tumors. The Dy-Cy7.5-TMV-DGEA was suitable for multi-scale MRI scanning of the entire body, particularly in the context of UHFMRI.
7.5. Targeted Pancreatic Cancer Imaging
Pancreatic cancer (PC) is a gastrointestinal tumor and is the fourth leading cause of cancer mortality in the United States due to its late diagnosis, early metastasis, and resistance to chemotherapy. The 5 year patient survival rate for all patients is less than 5%.[185] Based on NIH statistics, 55 440 newly diagnosed cases and 44 330 deaths were reported in the year 2018. The pancreas acts as both an endocrine and exocrine gland. Tumors originating from endocrine tissue are termed islet cell tumors (or neuroendocrine). More than 90% of PC tumors originate from the ductal epithelium of the pancreas, called pancreatic ductal adenocarcinoma (PDAC).[186]
Several tyrosine kinases, including VEGFR-2, c-KIT, FGFR-1, colony stimulating factor 1 receptor (CSF1R), and SRC are overexpressed on the surface of PC cells. Other receptors (such as SRC, CSF1R, VEGFR-2, c-KIT, PDGFR, TβRI, TβRII, and FGFR-1) are also overexpressed on the surface of PC cells.[187] PC is diagnosed in three stages (I, II, and III). The relative expression of targetable molecules differs in each stage[188] (Table 4). PC has a dense tumor stromal barrier, which limits diffusion and the accessibility of contrast agents (as well as drugs) to the cancer cells. Thus, targeting of both stromal and PC cells is required. In PC, insulin-like growth factor 1 receptor (IGF1R) is overexpressed in both stromal and tumor cells. Zhou et al.[189] used iron oxide NPs, IGF1 as a ligand, and doxorubicin to form IGF1-IONPx-Dox. Non-invasive MRI results demonstrated that it could act as an effective theranostic system to improve PC targeted imaging and therapy.
P32 (gC1qR) is a multifunctional cellular receptor protein that is overexpressed on the surface of PC. The Jiang group designed multifunctional core-shell magnetic nanospheres prepared from iron oxide NPs and silica labeled with FITC and LyP-1 peptide that targets the P32 receptor called (Fe3O4@SiO2-FITC@mSiO2-LyP-1; Figure 18). In vivo MRI and fluorescence imaging confirmed specific accumulation of the designed nanospheres in the tumor tissue, allowing MRI of orthotopic PC xenografts.[190] Despite its name, prostate stem cell antigen (PSCA) has been reported to be overexpressed in primary pancreatic ductal adenocarcinoma. PSCA has been employed to distinguish PC from chronic pancreatitis, and higher PSCA levels have been correlated with poor prognosis and metastasis of PCa. Zettlitz et al.[191] developed a dual-labeled probe based on anti-PSCA A2 cys-diabody (A2cDb) with a specific conjugation site for IRDye800CW and random 124I-labeling (124I-A2cDb-800). In mice bearing PC xenograft tumors, immunoPET allowed non-invasive, whole-body imaging to localize PCs, and NIRF image guidance could allow identification of tumor margins during resection.
Figure 18.
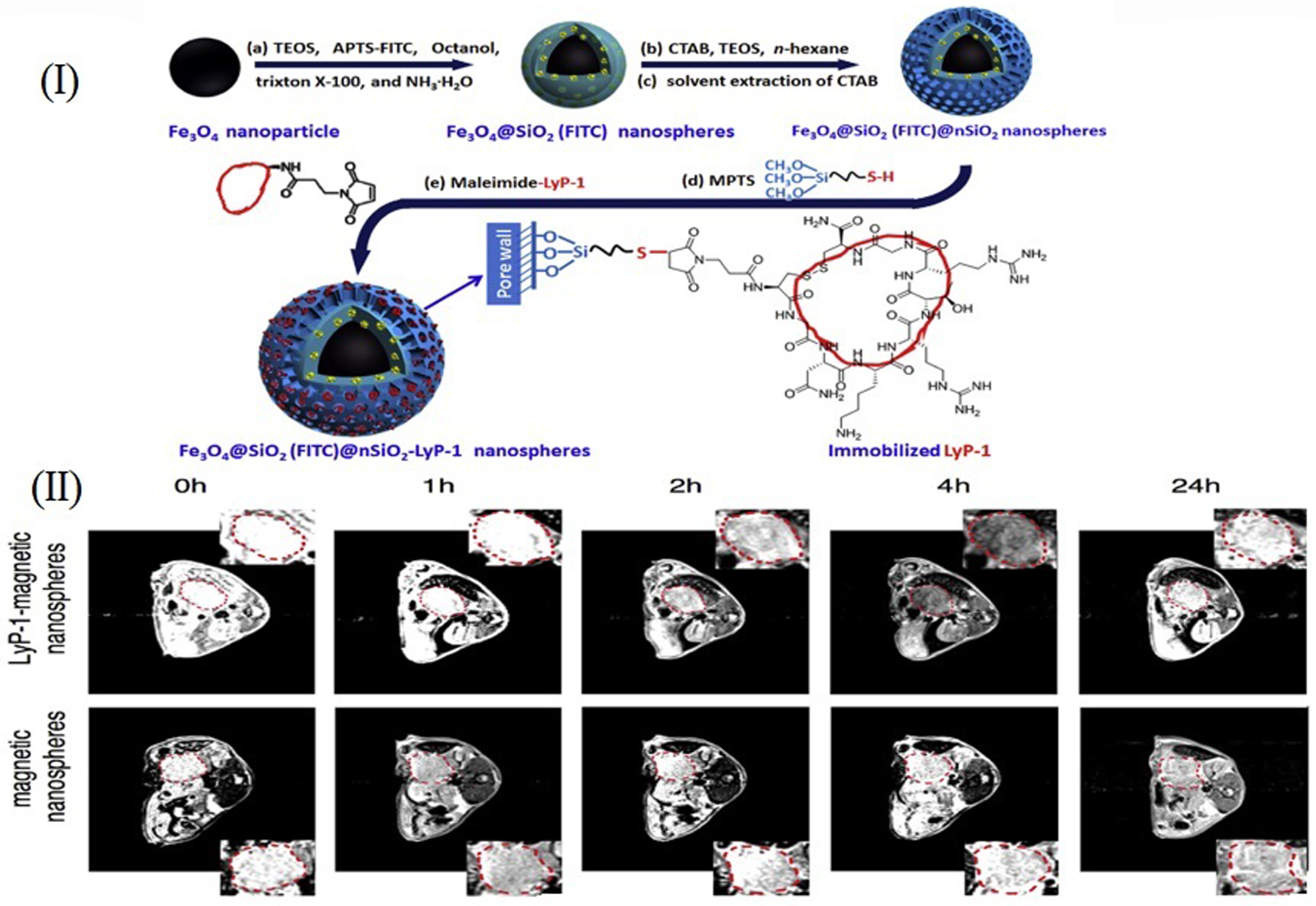
Tumor targeting with multifunctional silica nanospheres. (I) Synthetic route and structure of multifunctional nanospheres. a) Coating a layer of FITC-incorporated silica via the co-condensation of TEOS and APTS-FITC. b) Further growth of a CTAB/SiO2 composite layer using CTAB as a structure directing agent. c) Removal of CTAB producing mesopores in the outer shell. d) Insert thiol groups via the surface modification of NS with MTPS. e) Immobilization of LyP-1 via the “Click” reaction between thiol groups anchored on the NS and the terminal maleimide group in the cyclic LyP-1 derivative. (II) T2 weighted MRI of orthotopic pancreatic cancer before and after admin istration of the Fe3O4@SiO2-FITC@mSiO2 or Fe3O4@SiO2-FITC@mSiO2-LyP-1 systemically at different time points (The inset is enlarged picture of corresponding tumor region). Reproduced with permission.[190] Copyright 2017, Elsevier.
Another overexpressed marker in PCa is the receptor for advanced glycation end products (RAGE), which plays a critical role in the transition of premalignant epithelial precursor cells to pancreatic ductal adenocarcinoma. The Kim group has synthesized a fluorescent dye (Cy5), labeled anti-RAGE scFv antibody, with high binding affinity to murine RAGE and no internalization in PC cell lines. The anti-RAGE scFv successfully visualized RAGE expression in a KRASG12D mouse bearing PC tumors. In vivo biodistribution studies used the 64Cu-labeled scFv Ab fragment in a syngeneic mouse model, demonstrating receptor specific uptake in RAGE-overexpressing tumors. PET imaging data showed anti-RAGE scFv had a high affinity to RAGE in vivo.[192]
7.6. Targeted Bladder Cancer Imaging
Nearly 380 000 new cases and 150 000 deaths caused by bladder cancer are reported annually. Bladder cancer is the fifth most common type of cancer, and is responsible for nearly 3% of all cancer related deaths in the United States.[193] Bladder cancer develops as two distinct forms, papillary and non-papillary, which are pathologically and clinically distinct. The majority of bladder cancers are superficial papillary lesions (NMIBC: non-muscle-invasive bladder cancer) that originate from hyperplastic changes in the mucosa (referred to as low-grade intraepithelial neoplasia), and account for ≈70–80% of cases. In the early stages of NMIBC, tumors penetrate the epithelial basement membrane, but have not invaded into the bladder wall. The opposite is true for most high grade, muscle-invasive bladder cancers. These tumors can be multifocal and tend to recur after local excision. However, they usually do not metasta-size to other organs.[194]
Aggressive bladder cancers are usually the solid non-papillary type, which originate from in situ precursor lesions (i.e., dysplasia or severe carcinoma in situ). These tumors frequently give rise to distant organ metastasis and are more likely to invade the bladder wall. Clinically, the non-papillary and papillary forms are separately classified, however, there is some overlap between them. Patients with external papillary tumors generally experience multiple recurrences, but only a small fraction progress to high-grade invasive bladder tumors. Conversely, the majority of high-grade invasive bladder cancers develop in patients with no history of superficial papillary lesions. This dual-track concept of bladder carcinogenesis was developed on the basis of correlation between pathological and clinical observations.[194,195]
Classification of bladder cancer into different subtypes is based on several factors. Damrauer et al. divided bladder cancer into luminal and basal subtypes,[196] while Sjödahl et al.[197] classified bladder cancer, according to four mRNA expression profiles, into five major subtypes: urobasal A (UroA); urobasal (UroB); genetically unstable (GU); squamous cell carcinoma-like (SCCL); and infiltrating. According to the Cancer Genome Atlas (TCGA), bladder cancer can contain four defined expression clusters, (I–IV). Choi et al.[198] classified bladder cancer into three luminal, basal, and p53like subtypes. Figure 19 schematically depicts the subtype classification, overlap between them, subtype markers, and possible targets.
Figure 19.
Different subtypes classification of bladder cancer. UNC, University of North Carolina; MDA, MD Anderson Cancer Center; TCGA, the Cancer Genome Atlas; Genom. Unst., genomically unstable; Inf. Epi., infiltrated epithelial; Inf. Mes., infiltrated mesenchymal; UroA, urobasal A; UroB, urobasal B; SCC, squamous cell carcinoma; Sc/Ne, small cell/neuroendocrine; TFs, transcription factors. Adapted with permission.[199] Copyright 2015, Elsevier.
Studies have shown that CD47 is overexpressed in >80% of bladder cancer cells. CD47 binds to the signal regulatory protein α, which is expressed on dendritic cells and macrophages to provide signals to prevent phagocytosis. Targeting CD47 in the cancer tissue can be accomplished using different ligands (such as anti-CD47 Abs). In order to evaluate the expression and function of CD47 in bladder cancer, Pan et al. evaluated fluorescently labeled anti-CD47 Ab as an intravesical imaging contrast agent. The results of fluorescence imaging, confocal microscopy, and cystoscopy showed the imaging agent possessed high sensitivity and specificity for CD47-targeted imaging.[200]
Chemokine receptors (CKRs) are a superfamily of small transmembrane G-protein coupled receptors involved in inflammatory and immune reactions. Different chemokine receptors, including CCR1-CCR10, CXCR1-CXCR6, XCR1, and CX3CR1, have been identified. CXCR4 is the only type of CKR that is upregulated in MIBC tissue samples. Currently, CXCR4 could be a new molecular probe target with high affinity for imaging of high-grade superficial bladder cancer. Nishizawa et al.[201] used T140 (14-mer peptide) as an antagonistic ligand for developing a TY14003 molecular probe for targeting of CXCR4. The in vivo results of fluorescent imaging indicated that the probe was promising for detection of flat high-grade superficial bladder cancer lesions.
NMIBC lesions are generally localized to the bladder lumen, while a targeted-imaging probe can only detect luminal surface biomarkers. CA9 and CD47 are biomarkers that are expressed on the luminal surface. Davis et al. synthesized gold-silica NPs as surface-enhanced Raman scattering (SERS) capable NPs, that were targeted with Abs s420-anti-CA9, s440-anti-CD47, and s421-anti-IgG4 for active and passive targeting (Figure 20). The main results of this study were i) evidence of passive targeting of intra-vesical NPs; ii) the EPR effect operated for topically applied NPs; and iii) the bladder tissue could be classified as normal or cancerous using multiplexed molecular SERS imaging.[202] More examples of targeted imaging systems for detection of bladder cancer are summarized in Table 4.
Figure 20.
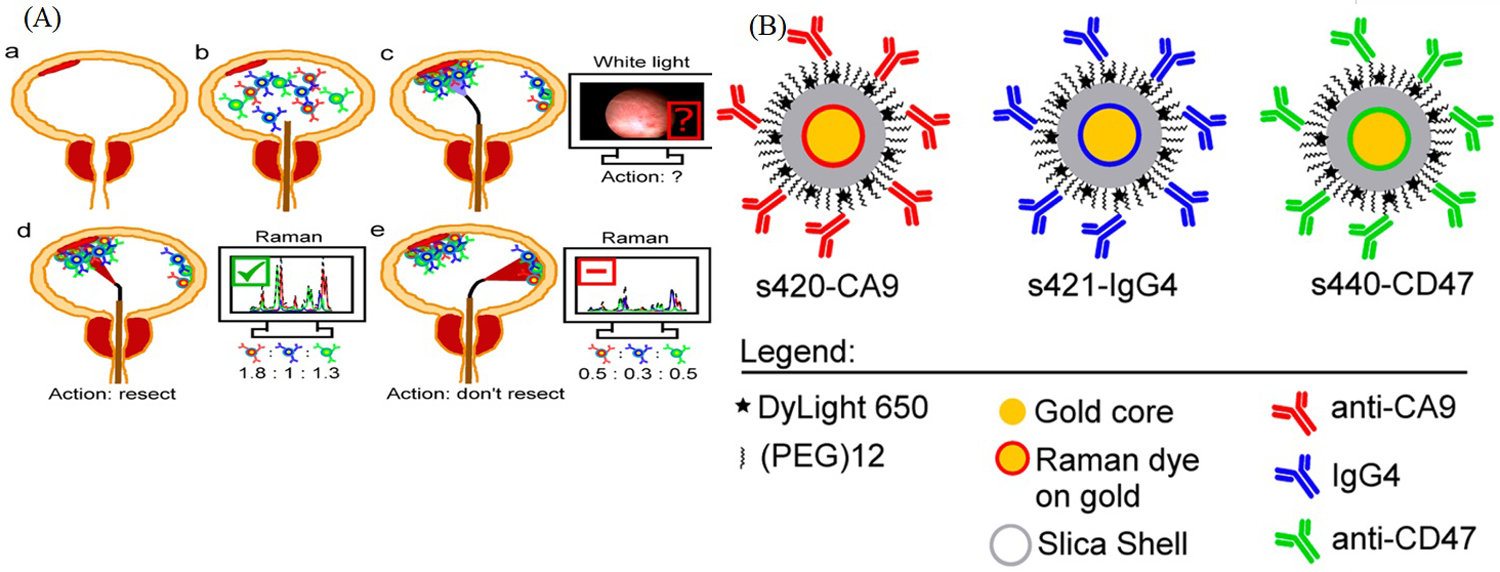
SERS NPs for bladder cancer imaging. A) Proposed application of intraluminal SERS NPs. a) Patient presents with potential NMIBC (red color tissue). b) Before cystoscopy, intraluminal SERS nanoparticles are administered. Each NP color represents a different targeting mechanism (passive, blue; CA9, red; and CD47, green). c) Patient receives standard of care, which is guided transurethral resection. Regions ambiguous on white light cystoscopy (WLC) are subsequently interrogated with Raman endoscopy. d, e) Based on absolute and relative binding levels of each channel, flat lesions can be identified, and cancer tissue is resected. B) Schematic representation of the SERS NPs. The blue IgG4 NPs are used as negative experimental control for active binding of CA9- and CD47-targeted SERS NPs. Adapted with permission.[202] Copyright 2018, American Chemical Society.
7.7. Targeted Brain Cancer Imaging
Brain cancer describes a heterogeneous group of primary and metastatic tumors occurring in the central nervous system (CNS). The annual incidence of primary malignant brain tumors is ≈24 000 cases worldwide. Brain cancer is the leading cause of death in children under the age of 15.[203] The failure of early diagnosis is mostly due to the absence of targeted imaging systems with high selectivity, and poor treatment outcome is due to the failure of current chemotherapy regimens or incomplete surgical resection (because of the inherent infiltrative character of brain tumors).[204] Primary brain tumors (composed of cells derived from astrocytes, oligodendrocytes, or ependymal cells) are known as astrocytoma, oligodendrogliomas, and ependymomas, respectively.[205]
Unlike the normal capillaries in the brain, the tight junctions between the ECs of brain tumors are seriously compromised, producing a leaky tumor vasculature, while the high intratumoral interstitial pressure, limits drug penetration from the bloodstream into the brain tumor. Moreover, the remaining blood–brain barrier (BBB) limits transportation of targeted agents. In order to overcome these problems, multitargeting imaging probes need to display high permeability across the BBB and overcome other penetration impediments. Targeting brain tumors can be improved by targeting receptors, such as integrin αvβ3, or aminopeptidase N. These receptors are distributed on proliferating ECs within the brain tumor (sites that are in direct contact with circulating NPs in the blood-stream) and are overexpressed on brain capillary ECs.[89,206]
Glioblastoma multiforme (GBM), is the most malignant grade (IV) of astrocytoma, and requires complete surgical resection for long-term cures. Although traditional chemotherapy does not work well against GBM, innovative dual-targeted imaging and therapy nanoconstruct have been investigated with high permeability across the BBB. Ni et al.[207] developed a bimodal imaging agent for MR/fluorescence imaging of intracranial GBM, benefiting from the MRI and upconversion luminescence (UCL) capabilities of upconversion nanoparticles (UCNPs). CD13 is overexpressed in glioma, and can be recognized by a tumor-homing NGR peptide motif. Huang et al.[208] synthesized an ANG-conjugated PEG-CdSe/ZnS quantum dot-based imaging probe. Fluorescence imaging results showed that the probe could cross the BBB and target CD13-overexpressing glioma tumors. The PEGylated UCNPs were modified with angiopep-2 (ANG/PEG-UCNPs) as a targeting ligand with high affinity to the low-density lipoprotein receptor related protein (LRP), overexpressed on both BBB and glioblastoma cells. The ANG/PEG-UCNP platform displayed higher transcytosis across the BBB and endocytosis into glioblastoma cells compared to nontargeted PEG-UCNPs, with no significant cytotoxic effect. The MR images of glioblastoma-bearing mice showed the T1-weighted contrast was enhanced at the tumor site 1 h post-injection of ANG/PEG-UCNPs. The tumor was barely visible in mice injected with PEG-UCNPs or the clinically employed Gd-DTPA contrast agent. The results were better than those with the commonly used fluorescent dye 5-ALA. In a similar study taking advantage of the EPR effect in brain tumors and its angiogenic blood vessels, Li et al.[209] fabricated a dual-modality Gd-Ag2S nanoprobe to take advantage of the deep tissue penetration of MR and high spatiotemporal resolution of fluorescence imaging, in order to help surgeons conduct more precise surgery for GBM.
However, these studies did not address how the NPs could cross the tight junctions of the BBB. Diaz et al.[210] employed MRI-guided transcranial focused ultrasound (TcMRgFUS) as a non-invasive technique to increase the permeability of the BBB to allow SERS imaging. They used silica shell-coated GNPs, where after BBB disruption using TcMRgFUS, the delivery of 50 and 120 nm GNPs to the tumor periphery was achieved without any vascular damage. This approach could pave the way for specific delivery of a wide range of therapeutic and diagnostic agents. Applying a resonating magnetic field allowed magneto-responsive nanoplatforms (such as magnetic-fluid-loaded liposomes [MFLs]) to target and monitor malignant brain tumors.[211] The in vivo results showed that after 4 h exposure to a focused 190 Tm−1 magnetic field gradient, MFLs could pass through the BBB, and accumulated only in U87 human glioblastoma xenografts, and were retained therein for almost 24 h, as shown by MRI. There was no sign of MFLs in other areas of the brain (Figure 21).
Figure 21.
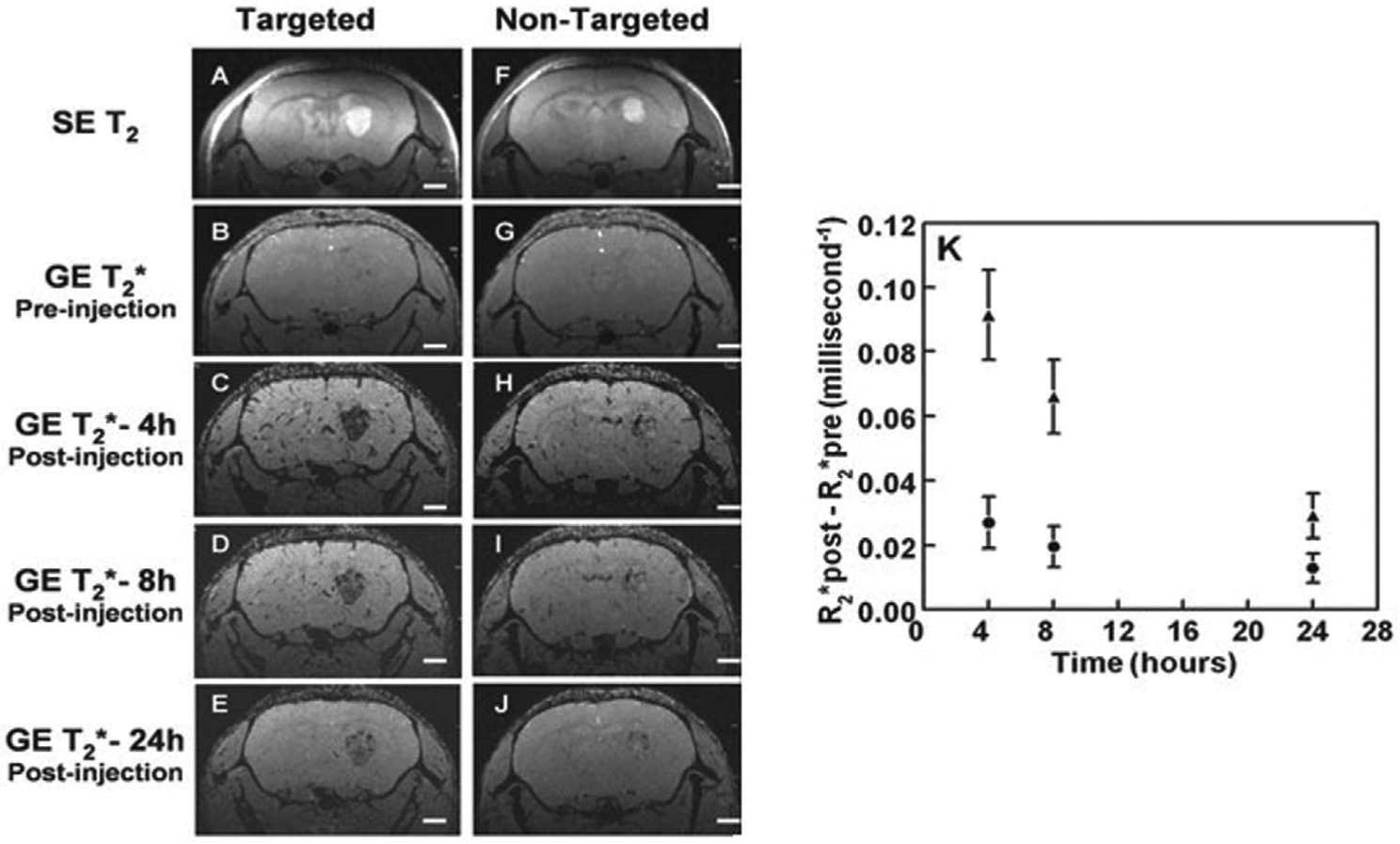
Glioblastoma imaging with magnetic field responsive MFLs and MRI. Series of brain MR images versus time from glioblastoma bearing mice injected with MFLs and at 4 h post injection receiving magnetic targeting by external application of 0.4 T magnet (A–E) or not (F–J); the spin echo T2–weighted (SE T2) baseline acquisitions performed before MFLs injection show the tumor locations as hyper-intense lesions (A,F); the T2*-weighted gradient echo (GE T2*) sequences reveal the presence of the contrast agent as hypointense areas (B–E and G–J); the persistence of the hyposignal at the targeted tumor level remains clearly visible 24 h post injection (E). Relaxation rate difference (R2*post − R2*pre) for the targeted (▴) and nontargeted (•) tumors as a function of the time period following MFLs administration (K); the magnet was removed at 4 h for the targeted tumor. White bars represent 1 mm. Reproduced with permission.[211] Copyright 2015, Wiley-VCH.
GLUT-1 and ASCT2 (an important L-isomer-selective amino-acid transporter) are found in high density in the BBB and in brain tumors. Zhang et al.[212] prepared CDs that were tagged with L-Asp, glucose, and/or L-Glu. These groups allowed the CDs to cross the BBB via ACT2 and GLUT-1 transporters. Since the RGD tripeptide is known to act as an αvβ3 integrin targeting agent, they asked whether CD-Asp, could also act as a RGD-like functional group and bind to the αvβ3 integrin on the immature ECs in the glioma. In vitro and in vivo results showed that the CD-Asp NPs could act as an excellent fluorescence imaging and targeting agent for safe and noninvasive glioma imaging.
SPIONs possess negative contrast capability in MRI, and have emerged as a versatile agent in magnetic targeting. Xu et al.[213] described a theranostic liposome (QSC-Lip) preparation based on QDs, SPIONs, and cilengitide (CGT, a cyclic RGD pentapeptide) for in vivo dual-MRI/NIR imaging. The data revealed that the QSC-Lip imaging probe not only produced negative-contrast enhancement in gliomas using MRI, but also created tumor-localized fluorescence under magnetic targeting, and could be used to guide the surgical resection of the glioma. More examples of targeted brain cancer imaging are summarized in Table 4.
7.8. Targeted Ovarian Cancer Imaging
Ovarian cancer is known as the “silent lady killer,” and is the fifth leading cause of cancer-related death in women. Because this cancer is frequently diagnosed at the later stages of the disease (stage 3 III or stage IV), it has the highest morbidity and mortality of all gynecological cancers. Epithelial ovarian cancer (accounting for ≈90% of cases) is classified into four histological subtypes: serous, endometrioid, clear-cell, and mucinous carcinomas. Of these types, high-grade serous carcinoma (HGSC) is the most commonly diagnosed, and unlike the other subtypes, it probably originates in the fallopian tubes.[214] The risk of developing ovarian cancer is determined by genetic factors, age, postmenopausal hormonal therapy, infertility, and nulliparity. In terms of screening, germline mutations in BRCA1 or BRCA2 present a high risk for developing ovarian cancer. In women with an average risk of developing ovarian cancer, the biomarker CA125 has been the primary focus for screening. The combination of CA125 blood test and radiographic imaging (transvaginal ultrasonography) has been evaluated as a screening strategy.[214]
Based on grade, size, symptoms, etc., several markers including VEGFR, EGFR, and PDGF, and their receptors (PDGFR, KIT pathways, ERBB2, and α-folate receptor [αFR]) have been selected to be implemented in targeted therapy.[215] These therapeutic strategies could be improved by using targeted imaging techniques. SPECT and PET are molecular imaging techniques that have been used in the imaging of ovarian cancer. Ovarian cancer-specific molecules including cell surface receptors, hormone receptors, receptor tyrosine kinases, angiogenic and immune-related factors can be labeled using radioactive nuclides.[215] Although 18F-FDG PET has been studied in the diagnosis of ovarian cancer, it is not thought to be a good option for the primary diagnosis of ovarian cancer. Hence, discovering other options is necessary. Some ovarian tumors show overexpression of ERα (≈70% of patients). The use of PET imaging mediated by 18F-FES demonstrated that this platform could provide reliable information about tumor ERα status and whether endocrine therapy could be employed.[216] HER3 overexpression has been found to be a mediator of tumor resistance to HER1 and HER2-targeted therapies in both breast and ovarian cancer. However, imaging of this receptor using a radiolabeled anti-HER3 mAb showed a long biological half-life and relatively poor tumor penetration. The Chiara Da Pieve group chose to use an affibody with rapid clearance by the kidneys, biocompatibility, and good specificity and affinity. They developed an [18F] aluminum fluoride radiolabeling procedure for the HER3-targeted affibody (ZHER3:8698). This platform showed successful tumor targeting with clear visualization of HER3-over-expressing xenografts in tumor-bearing mice, 1 h post injection.[217] The overexpression of folate receptor-α (FR-α) found in 90–95% of epithelial ovarian cancers prompted the investigation of an FR-α–targeted fluorescent agent for intraoperative tumor-specific fluorescence imaging in ovarian cancer surgery. Nanoemulsions (NEs) were loaded with imaging contrast and decorated with folatet-PEG3400-DSPE in platinum (Pt) resistant ovarian cancer cells.[218] Another study used folate-FITC as an FR-α targeted fluorescent imaging agent in patients with ovarian cancer.[219] They proved that FR-α–FITC had a good pharmacodynamic profile after systemic administration in patients, and could improve tumor staging and allow real-time visualization of the tumor tissue during surgery.
Combining the overexpressed CA-125 membrane marker with ultrasound (US) contrast agents could allow the detection of early stage ovarian cancer. The Yong Gao group formulated CA-125 targeted nanobubbles (NBs) to detect CA-125+ ovarian cancer.[220] Their results demonstrated that the targeted NBs were stable, specific, and selectively bound to CA-125+ ovarian cancer cells in vitro, with strong accumulation in ovarian cancer tissue in vivo and long-lasting contrast enhancement. Human epididymis protein 4 (HE4) is one of two U.S. FDA-approved serum biomarkers in ovarian cancer. Tissue concentrations of HE4 are greater than serum concentrations, hence HE4 may be a target for ovarian cancer imaging. Recently, Williams et al.[221] developed a carbon nanotube (CNT)-based probe using an immobilized Ab that recognized HE4. NIR bandgap photoluminescence from CNTs between 800 and 1600 nm successfully allowed detection of HE4 in patient serum and ascites samples, and in orthotopic murine models of ovarian cancer. HER-2 is expressed in a high percentage of ovarian cancers, and systemic delivery of an HER-2 affibody attached to magnetic iron oxide nanoparticles (IONPs) into mice bearing HER-2 positive SKOV3 tumors demonstrated potential for image-guided surgery.[222] However, it would be desirable to simultaneously target multiple cell surface biomarkers to increase the specificity and sensitivity for ovarian cancer detection (Table 4).
8. Tumor-Specific Imaging Probes in Clinical Trials
There are many nano-delivery vehicles in ongoing clinical trials for the delivery of therapeutic agents to tumors. However most of these are not typically surface-modified with targeting moieties, or equipped for tumor detection and imaging. Tumor-selective imaging probes need to satisfy critical safety and toxicity standards, and overcome limitations, such as sub-optimum pharmacokinetics, resource-intensive scale-up, reimbursement issues, and an evolving regulatory framework for good manufacturing practice. Addressing these criteria is essential for evaluating the probes that are undergoing preclinical testing, or are transitioning into early-phase clinical trials. Some of these probes are being investigated in phase 1 clinical trials in patients with solid tumors, while other specific cancer indications are being explored in advanced clinical trials (phases 2 and 3) [7b] (summarized in Table 5).
Table 5.
Summary of nanotechnology-assisted cancer specific molecular imaging probes in clinical trials.
| Imaging modality | Nanoplatform | Cancer type | Receptor | Status | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| PET/CT | [18F] Fluoroestradiol (FES) | Breast cancer | ER+ | Phase 1 | NCT02559544 |
| PET/CT | 68Ga-NOTA-BBN-RGD | Breast cancer | GRPR | Phase 1 | NCT02749019 |
| PET | [18F]-ML-10 | Metastatic brain cancer | EPR | Phase 2 | NCT00805636 |
| PET/CT | (99m Tc) ECDG | Lung cancer | EPR | Phase 3 | NCT01394679 |
| PET/CT | 68Ga-NOTA-3P-TATE-RGD | Lung cancer | Integrin αvβ3 | Early Phase 1 | NCT02817945 |
| PET/CT | 124I | Breast cancer | NIS | Early Phase 1 | NCT01360177 |
| PET/CT | 89Zr-trastuzumab | Breast cancer | HER2 | Phase 1 | NCT02286843 |
| PET | 64Cu- TP3805 | Bladder carcinoma | EPR | Early Phase 1 | NCT03039413 |
| PET | 68Ga-labeled HBED-CC PSMA | Prostate cancer | PSMA | Phase 1 Phase 2 | NCT02611882 |
| dPET-CT | 18F-FMISO | NSCLC | hypoxia | – | NCT01617980 |
| PET | 89Zr-trastuzumab | Breast cancer | HER2 | – | NCT01081600 |
| PET/CT | (18F-3c) ([18F]ISO-1) | Breast cancer | Sigma-2 receptor | Phase 1 | NCT02762110 |
| PET | 64Cu-DOTA-AE105 | Breast, Prostate and Bladder cancer | uPAR | Early Phase 1 | NCT02139371 |
| PET/CT | [18F]ISO-1 | Breast cancer | Sigma-2 receptor | – | NCT03057743 |
| IOI | OTL38 | Ovarian cancer | Folate Receptor-α | Phase 2 | NCT02317705 |
| PET | 68Ga-NOTA-AE105 | Breast, Prostate and Bladder cancer | uPAR | Phase 1 | NCT02437539 |
| PET | 89Zr-GSK2849330 | cancers | HER3 | Phase 1 | NCT02345174 |
| PET | 89Zr-AMG211 | Gastrointestinal cancer | CEA, CD66e and CD3 | Phase 1 | NCT02760199 |
| PET/CT | 68Ga-PSMA | Prostate cancer | PSMA | Phase 2 | NCT03689582 |
| SPECT/CT | 99mTc-ABH2 | Breast cancer | HER2 | Early Phase 1 | NCT03546478 |
| MRI/PET | 64Cu-MM-302 | Brain solid tumors | HER2 | Early Phase 1 | NCT02735798 |
| PET/CT | 18F-EF5 | Ovarian cancer | Hypoxia | – | NCT01881451 |
| PET/CT | 68Ga-NODAGA-Ac-Cys-ZEGFR:1907 | Cancers | EGFR | – | NCT02916329 |
| NIRF | OTL38 | Lung cancer | Folate Receptor | Phase 2 | NCT02872701 |
| PET | 89Zr-labeled KN035 | Solid tumors | PD-L1 | – | NCT03638804 |
| PET | 68Ga-Labeled F(ab’) 2- Trastuzumab | Solid tumors | HER2 | Phase 1 | NCT00613847 |
| PET/CT | 68Ga-PSMA | Recurrent Prostate Carcinoma | PSMA | Phase 3 | NCT03582774 |
| PET/CT | 18F-FMISO | NSCLC | Hypoxia | – | NCT02016872 |
| PET/CT | 89Zr-daratumumab | Multiple Myeloma | CD38 | Phase 1 Phase 2 | NCT03665155 |
| PET | 18-F-MISO | Prostate Adenocarcinoma | Hypoxia | Phase 2 | NCT01898065 |
| PET/CT | 18F-DCFPyL | RCC | PSMA | – | NCT02687139 |
| NIRF | bevacizumab-IRDye800CW | Breast cancer | VEGF | Phase 1 | NCT01508572 |
| PET/CT | 89Zr-trastuzumab | Breast cancer | HER2 | – | NCT02286843 |
| NIRF | Bevacizumab-IRDye800CW | Rectal cancer | VEGF | Phase 1 | NCT01972373 |
| NIRF | Indocyanine green | Lung cancer | EPR | Phase 1 | NCT00264602 |
| PET/CT | 18F-FDG | NSCLC | Metabolism targeting | Phase 3 | NCT02938546 |
| PET | 18F-Fluoroazomycin arabinoside | Tongue cancer | Hypoxia | Phase 1 Phase 2 | NCT03181035 |
| Theranostic | 177Lu-PP-F11N | Thyroid cancer | cholecystokinin-2 receptors | Phase 1 | NCT02088645 |
| PET/CT | 18F-DCFPyL | Prostate cancer | PSMA | Early Phase 1 | NCT02691169 |
| Ultrasound Imaging | Perflutren Lipid Microsphere | Prostate cancer | EPR | Phase 2 | NCT02967458 |
| PET/CT | 18Fluciclatide | Solid tumors | αvβ3 | Phase 1 Phase 2 | NCT01176500 |
| PET/CT | 64Cu-plerixafor | Cancers | CXCR4 | Early Phase 1 | NCT02069080 |
| Ultrasound Imaging | BR55 | Prostate cancer | VEGFR2 | Phase 1 Phase 2 | NCT02142608 |
| PET/MRI | [89Zr]-Df-Trastuzumab | Breast cancer | HER2 | Early Phase 1 | NCT03321045 |
| PET/CT | 111In-folic acid | Prostate cancer | Folate receptor | – | NCT00003763 |
| NIRF | OTL38 | Ovarian cancer | Folate receptor | Phase 3 | NCT03180307 |
Abbreviations: CEA, human carcinoembryonic antigen; PSMA, prostate specific membrane antigen; GRPR, gastrin-releasing peptide receptor; uPAR, urokinase plasminogen activator receptor; NSCLC, non-small-cell-lung cancer; RCC, renal cell carcinoma; (18F-3c) ([18F]ISO-1), N-(4-(6,7-dimethoxy-3,−4-dihydroisoquinolin-2(1H)-yl) butyl)-2-(2-[18F]-fluoroethoxy)-5-methylbenzamide; ECDG, ethylenedicysteine-deoxyglucose; NIRF, near infrared fluorescent image NIS, [Na+I-symporter, sodium iodide symporter]; [18F]-ML-10, 2-(5-fluoro-pentyl)-2-methyl-malonic-acid. All information obtained from (https://clinicaltrials.gov).
Bevacizumab (Avastin) is an anti-VEGF-A MAb that is used in the clinic for several purposes. PET imaging using (89Zr)-bevacizumab has indicated that VEGF-A is a suitable target for imaging purposes in various tumor types. For the first time, Weele et al.[286] developed and tested the safety of clinical grade fluorescent-labeled Bevacizumab-800CW for non-invasive NIFR imaging of VEGF-A in patients with high-grade dysplasia in Barrett’s esophagus. The aim of this project was to validate the formulation, production, quality control, stability, extended characterization, and preclinical safety of a fluorescent imaging agent suitable for first-in human application (Clinical Trial identifier: NCT02129933).
C dots (Cornell dots) are 6–7 nm diameter, core–shell, hybrid silica particles that could allow simultaneous PET/optical imaging for the detection of metastatic melanoma. One study reported an ultra-small, cancer-selective, silica-based imaging probe, which was recently approved for first-in-human clinical trials, and could overcome a number of limitations of conventional imaging probes. This multimodal platform consisted of Cy5 fluorescent dye and 124I in a nanoconstruct functionalized with the cRGDyK peptide that targets αvβ3 integrin receptors. As part of a larger pilot study consisting of 30 patients from Memorial Sloan Kettering Cancer Center, these probes were evaluated for intraoperative mapping of sentinel lymph nodes in patients with melanoma, breast, cervical, and uterine cancer. Its applications included real-time lymphatic drainage patterns and intraoperative detection and imaging of nodal metastases of melanoma (Clinical Trial identifier: NCT02106598).[7b,287]
The kinase insert domain receptor (KDR) is an important regulator of neoangiogenesis in human tumors. Willmann et al.[288] carried out the first human clinical trial using USMI in 24 women with ovarian cancer and 21 women with breast cancer using KDR targeted contrast microbubbles (MBKDR). The imaging probe was injected intravenously, and USMI was conducted from 5 to 29 min post injection. USMI mediated by MBKDR was well tolerated by all the patients without any safety concerns. Among the 40 patients undergoing analysis, KDR expression determined by immunohistochemical (IHC) staining matched well with the imaging data for both cancer types (EudraCT Number: 2012-000699-40).
9. Challenges and Future Directions
Currently, cancer patients are categorized based on the site and tissue of origin of the disease. However, it is becoming increasingly clear that the wide heterogeneity seen in tumors and in patients calls for the use of more targeted therapy as opposed to conventional cancer treatment methods. Cancer is a worldwide health issue, and there is an urgent need to identify more effective and non-invasive biomarkers for early diagnosis, prognosis, and therapeutic targeting based on individual patient characteristics. Several relatively new approaches may be used to facilitate imaging for cancer detection and treatment, as described below.
9.1. Dual-Targeted Imaging Platforms
Conventional single-targeted delivery systems have several limitations, including a lack of specificity for cancer cells, inability to cope with the emergence of drug resistance, and the lack of commonality between targetable receptors in different cancer types. Dual targeting strategies are promising alternatives to single-targeted delivery systems, taking advantage of two different types of cell surface receptors or TME-associated properties. Bi-specific Abs that incorporate amino acid sequences that recognize two different antigen epitopes for dual targeting have been shown to enhance targeting and optimize tumor specificity. CD105 and TF are two biomarkers which are both over-expressed in pancreatic cancer. Luo et al.[289] designed a dual receptor-targeted construct consisting of a bi-specific heterodimer of Fab’ antibody fragments recognizing CD105 and tissue factor, using a click chemistry approach. It was dual-labeled with NIRF and PET imaging reporters (64Cu-NOTA-heterodimer-ZW800) and used for the imaging of pancreatic tumors (Figure 22). The PET imaging results showed higher tumor uptake in comparison with either Fab fragment homodimer used alone. PET and NIRF imaging allowed for clear delineation of the cancer. However, the NIRF signal was significantly weaker than the PET signal.[289]
Figure 22.
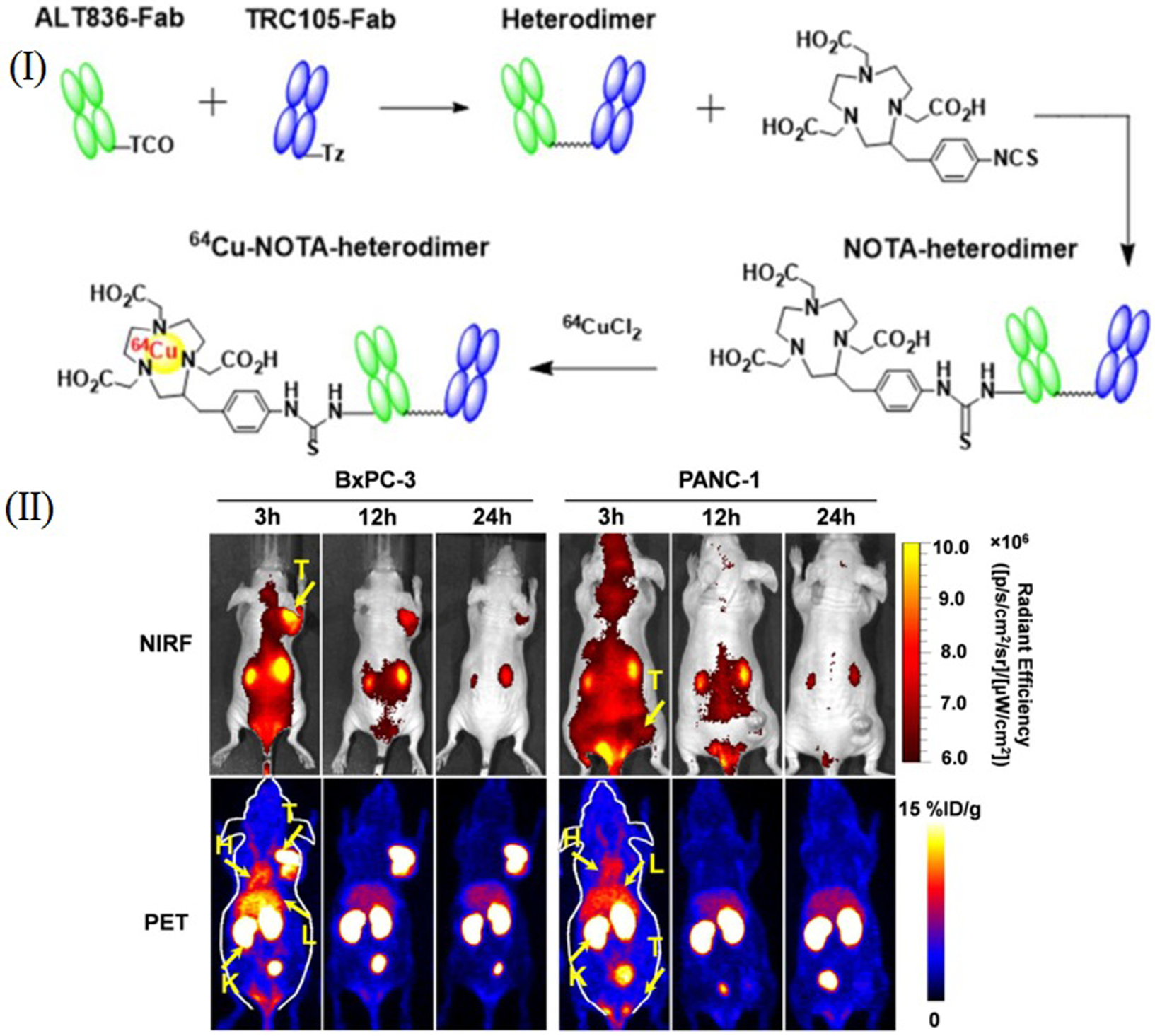
Dual NIRF and PET imaging of pancreatic tumors. I) Manufacturing of 64Cu-labeled heterodimer and homodimers of ALT836-F (ab′) 2 and TRC105-F (ab′) 2. II) NIRF/PET imaging in mice bearing PANC-1 or BxPC-3 tumors with 64Cu-NOTA heterodimer-ZW800 as a targeted imaging probe. Serial maximum intensity projections PET/NIRF images of mice bearing PANC-1 or BxPC-3 tumors at 3, 12, and 24 h following injection of targeted imaging probe. Reproduced with permission.[289] Copyright 2017, American Chemical Society.
Many chemotherapeutic agents enter into cells and are active only in the nucleus. Therefore, it is crucial to develop an improved delivery system to be able to target the nucleus. Surface ligand density is also an important factor that must be considered in order to achieve optimum real-time imaging of tumors. In this context, bi-specific targeted imaging constructs may be a promising approach to overcome physical barriers, enhance biocompatibility, lengthen circulation time, and improve cellular uptake, for clinical diagnosis and treatment.[290] The Xiaoting Liu group[291] constructed a dual-targeted DNA tetrahedron nanocarrier loaded with Dox, with two aptamers, one to bind to MUC-1 on the cell surface, and another AS1411 to bind to nucleolin. The Dox@MUC1-Td-AS1411 construct was used for breast cancer cell imaging and drug delivery. Fluorescence imaging results showed that MUC1-Td-AS1411 could differentiate MUC1+ from MUC1− cells. The Dox-loaded drug platform was effectively delivered into the nucleus, thereby killing the breast cancer cells.
Peptide targeting can also provide a modular strategy for targeting tumor tissue and molecular imaging of extracellular protease activity in vivo. For instance, activatable CPP (ACPP) is a MMP-cleavable linker that can be used in combination with cyclic-RGD binding to integrin αvβ3 for the targeting of the ECM in murine breast tumors. The cyclic-RGD-PLGC(Me)AG-ACPP loaded with chemotherapy agents allowed imaging and potent chemotherapeutic activity in mouse tumor models.[292] To achieve higher sensitivity and specificity of contrast imaging, and to overcome the poor tumor penetration of a VEGFR2 single-targeted agent, the Jing Du group[293] developed a novel dual-targeted US imaging agent using C3F8-flled PLGA NBs that were attached to dual anti-VEGFR2 and anti-HER2 mAbs. This construct could effectively penetrate the leaky tumor vasculature to target the cancer cells, and led to higher US imaging contrast compared to either of the single-targeted NBs in tumor-bearing mice.
9.2. Cell Membrane-Coated Imaging Agents
The complexity of biological interactions and the synthetic nature of most NPs have led to relatively poor performance of some imaging platforms within body. Overcoming these barriers using a more biomimetic design, including components of the cell membrane and its derivatives, can produce nanovesicles that can be more effectively transported within the body and interact with complex biological systems. Membrane-derived molecules and components including lipids, simple sugars (e.g., mannose, galactose, sialic acid), and peptides (e.g., CD47, MUC1, fibronectin-binding protein B, HER2/neu, etc.) have been widely explored for functionalizing NPs[294] (Figure 23). In order to replicate natural membrane structures, new approaches have focused on membrane-bound biomacromolecules, carbohydrate chains, and proteins.[294]
Figure 23.
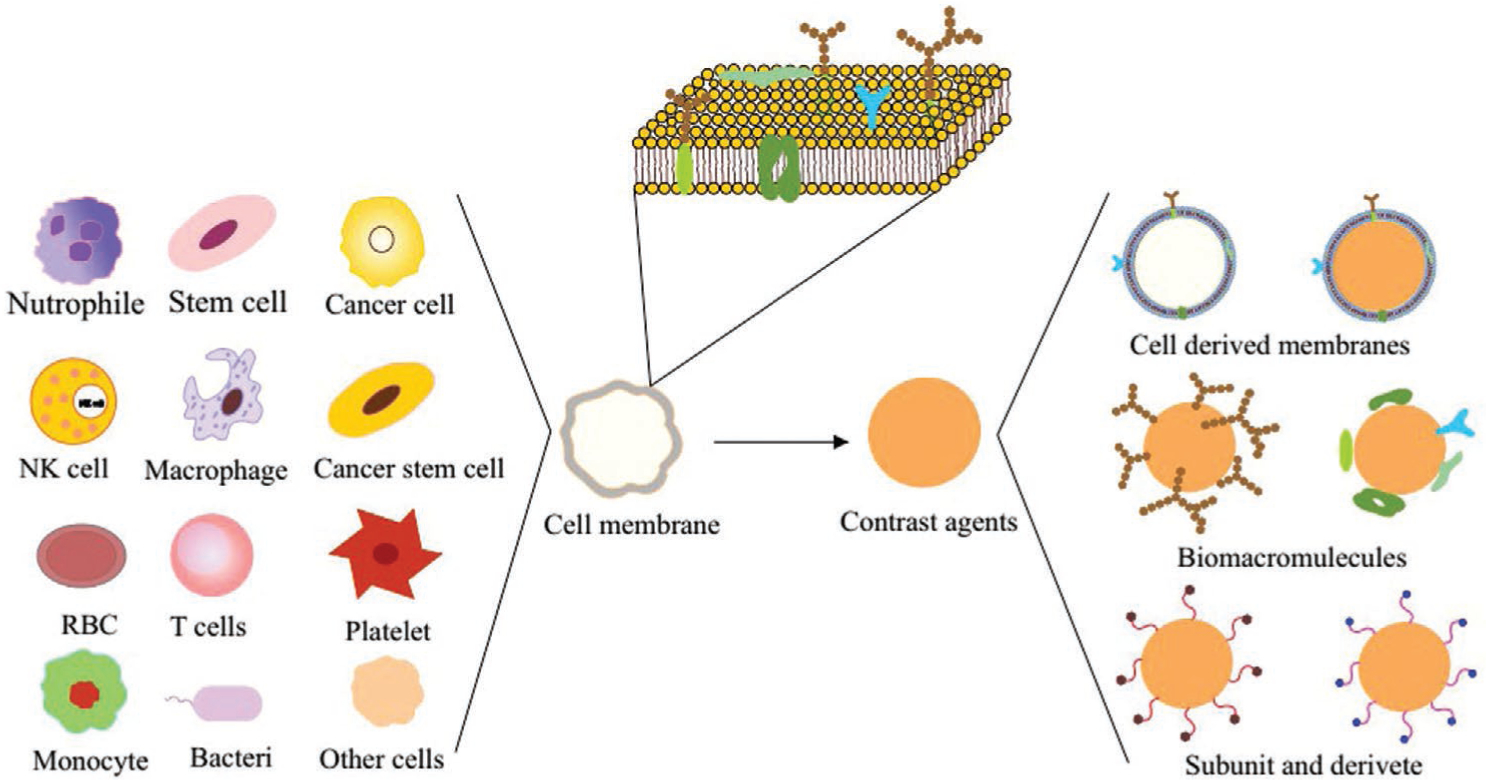
Scheme illustrating the synthesis of different types of cell membrane-coated nanoparticles.
One strategy to produce biocompatible and non-immunogenic NPs, involves using a layer of cell membrane coated around a preformed NP core. The NPs could be further functionalized with tumor-homing ligands, enhancing their circulation, active targeting, and therapeutic efficacy. Red blood cells (RBC), cancer cells, stem cells, white blood cells (WBC), and platelet cells have all been used as a source for the membrane material used as the NP coating. Each type of particle has the potential to create the next-generation of nanotherapeutics and nanodiagnostics platforms.[295] In order to overcome the BBB, the Zhilan Chai group[296] incorporated the biotinylated form of DCDX (candoxin-derived peptide) into streptavidin/RBC membrane-coated NPs loaded with DOX. Targeting efficiency was studied in an in vitro BBB model, and in vivo studies demonstrated that the platform was capable of traversing the BBB to target brain tumors, resulting in a significant increase in the median survival of glioma-bearing mice. Because cancer cell membranes carry the full array of cancer cell membrane antigens, these antigens could be coupled to NPs and loaded with an immunological adjuvant. The resulting formulation can be used to promote a tumor-specific immune response (i.e., taken up by antigen presenting cells, APCs) for use in cancer vaccine applications.[297] Cell membrane-coated NP platforms can bridge the gap between synthetic and natural biological materials. The surface modification of imaging contrast agents with polymers could activate the immune system to different degrees. Therefore, cell membrane-coated imaging platforms could be a new approach to diagnosis and therapy. An imaging system prepared by coating UCNPs with cancer cell membranes (CCM) was shown to enhance the binding to the identical source cancer cells by flow cytometry and UCL imaging.[298] Macrophages are capable of tumor homing and can avoid reticuloendothelial system (RES) uptake, therefore cell membrane vesicles derived from macrophages (MM-vesicles) were coated onto Fe3O4NPs for photothermal therapy (PTT). Fe3O4@MM NPs showed good biocompatibility, immune system evasion, and breast cancer targeting arising from the source macrophages.[299]
One dual-modality image-guided cancer theranostic system, was described by the Yanyu Huang group,[300] who developed a multifunctional smart nanosystem based on CCM vesicles (derived from leukemic cells) mixed with IDINPs and loaded with DOX-GFP-SPIO/ICG. The in vitro results showed that the CCM-camouflaged IDINPs produced ROS, induced cell death, and were “disguised” as leukemic cells, thus avoiding phagocytosis by macrophages in vivo. Furthermore, NIR laser and X-ray irradiation triggered the release of DOX from the CCM/ IDINPs in GSH-enriched tumor cells with an efficient tumor-homing targeting capability in vivo. The loading of SPIO and ICG into the CCM/IDINPs enabled precise MRI and NIR imaging of the CCM/IDINPs in the tumor.
Cancer-associated fibroblasts (CAFs) make up the majority of tumor stromal cells in the TME, and are induced by several pathways operating in cancer biology. Semiconducting polymer NPs (SPNs), have been used as theranostic/imaging agents, and upon laser irradiation can generate not only NIR fluorescence and PA signals for imaging, but also singlet oxygen (1O2) and heat for combined photodynamic and photothermal therapy. In one recent study, the Jingchao Li group[301] camouflaged SPNs with fibroblast cell membranes for enhanced multimodal cancer photo theranostic. In vivo fluorescence and PA imaging of tumors in living mice revealed that the platform preferentially targeted CAFs, providing amplified NIR fluorescence and PA signals for tumor imaging, and enhanced the phototherapeutic efficiency of treatment. Likewise, mesenchymal stem cell (MSC) membrane-derived vesicles, with long circulation times and good tumor targeting properties, were studied to camouflage polydopamine (PDA)-coated hydrophobic Fe3O4 NPs, as an image-guided photothermal and siRNA delivery platform. The experimental results showed that the Fe3O4@PDA-siRNA@MSCs NPs displayed good MSC-mimicking ability for tumor targeting, photothermal conversion efficiency, allowed MR imaging, and also silenced the target gene in a DU145 xenograft mouse model.[302]
9.3. Circulating Marker-Based Imaging
9.3.1. Tumor-Derived Extracellular Vesicles
Tumor-derived extracellular vesicles (TEVs) include microvesicles, exosomes, ectosomes, oncosomes, and so on. Exosomes are nano-sized (30–150 nm) vesicles that are secreted by many different cell types from both the host and the tumor, and have been detected in blood, urine, saliva, and ascites fluid. They are proposed to play an important role in intercellular communication. They also reflect the phenotypic state of the parental cell, such as genetic or signaling alterations that can occur in cancer cells. Exosomes are surrounded by a bilayer lipid membrane, and contain many bioactive molecules such as proteins, enzymes, lipids, mRNAs, circular RNA, and microRNAs. Exosomes can pass through tissue barriers within the body and can carry out horizontal transfer of biological information between cancer cells. Exosomes are involved in cancer development and progression by various mechanisms, including angiogenesis, EMT, migration, metastasis, immune escape, and expansion of therapy-resistant cancer cells.[303] Circulating tumor-derived EVs can act as noninvasive biomarkers by measurement of their cargos, for instance caveolin-1/S100B in melanoma, EpCAM in ovarian cancer, glypican-1 in pancreatic cancer, integrin α6β4 and integrin α6β1 in lung metastases, miR-17–92a in colon cancer recurrence, etc.[304] In comparison with traditional tissue biopsies, the higher sensitivity and specificity of exosomes as tumor-specific diagnostic markers could be used for early stage cancer diagnosis, monitoring, and prognostic evaluation.
Exosomes possess inherent biocompatibility, the ability to evade the immune system, resistance to degradation, stability in the blood circulation (due to the possession of a negative zeta potential), and the ability to target particular cell types via recognition of transmembrane proteins expressed on the exosome surface. Drugs including curcumin, paclitaxel, doxorubicin, exogenous siRNAs, and antitumor miRNAs are some examples of cargos that have been delivered when encapsulated in EVs employed as delivery vehicles.[305] The exosomal membrane can be further modified by attaching targeting moieties to enhance tissue-specific homing and facilitate targeted drug delivery. For example, mesenchymal cell-derived exosomes were engineered to carry siRNA specific to oncogenic KRASG12D (a common mutation in pancreatic cancer) and could enhance micropinocytosis by a CD47 dependent pathway, increasing the overall survival rate of mice with pancreatic cancer.[256] Bose et al.[306] constructed Cy5-antimiR-21-loaded TEVs derived from 4T1 cells that were used to camouflage gold-iron oxide NPs (GIONs). The multifunctional TEV-GION-NP theranostic platforms acted as a multimodal contrast agent for T2-weighted MRI in vitro. The in vivo biodistribution, tumor accumulation, and antitumor activity suggested it was promising for cancer imaging and therapy (Figure 24).
Figure 24.
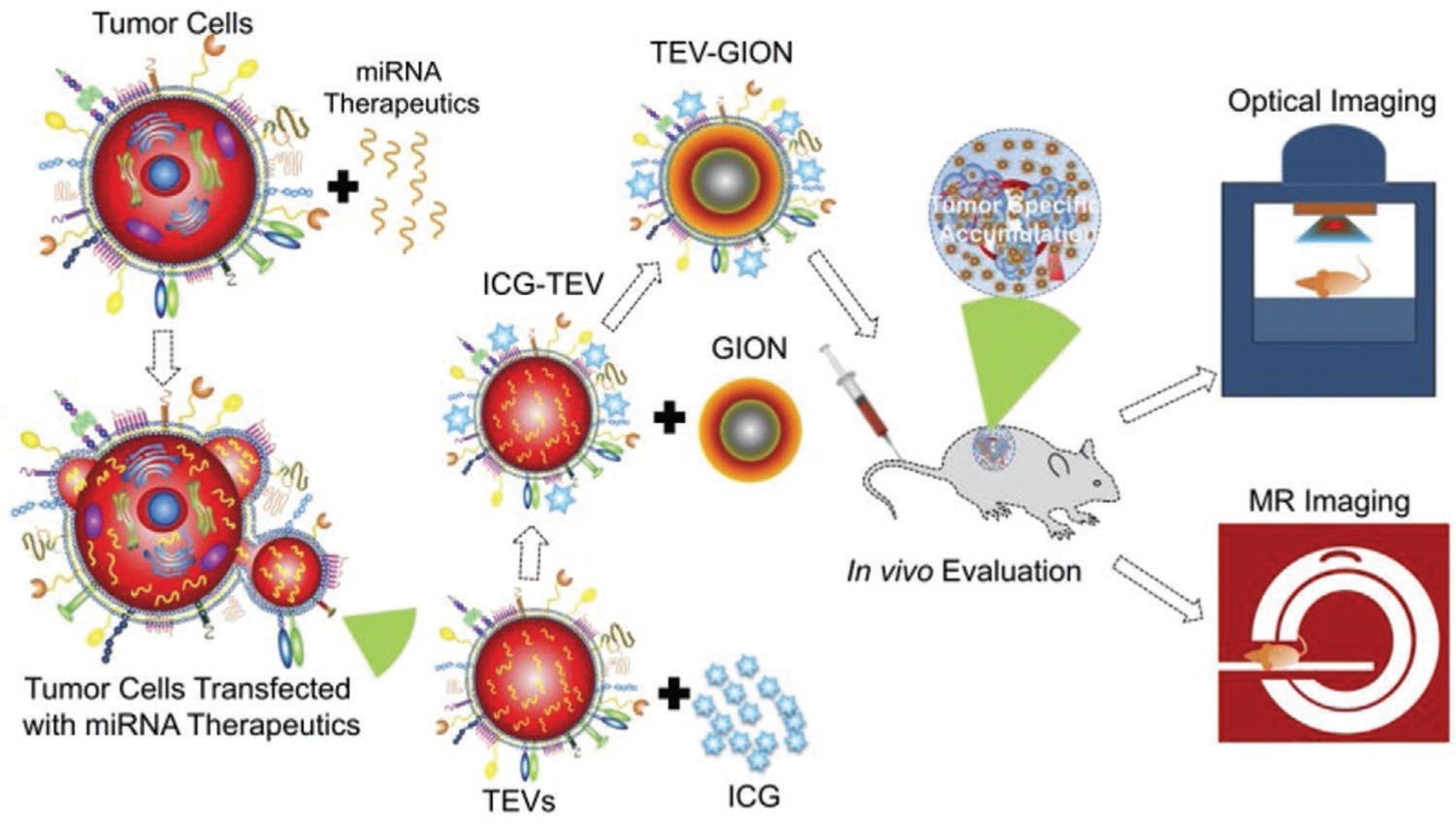
Scheme of preparation of TEV-GION-NPs nanotheranostic platform and applications for therapy and imaging. Reproduced with permission.[307] Copyright 2018, American Chemical Society.
The biodistribution and targeting of EVs administered via various delivery routes, has been studied by in vivo tracking of the EVs to target organs. Monitoring over time has been performed both directly (e.g., lipophilic tracer dyes, radionuclides, and magnetic particles) and indirectly (e.g., transduction of a reporter gene).[308] Specific labeling of EVs has been carried out by expression of fluorescent proteins fused with the EV membrane proteins CD63 proteins, C1C2 peptide, and luciferase mRNA.[309] The bioengineering of the parental cells and the use of extracellular vesicle mimetics (EVMs or artificial nanovesicles) might be helpful to improve the performance of EVs, and overcome the problem of the small quantities of exosomes naturally produced by cells.[305] The development of exosomal proteomics related to cancer, as well as improved microfluidic techniques for detecting and isolating exosomes, will likely improve their utility for cancer diagnosis.
9.3.2. Circulating Tumor Cells and Cell-Free Nucleic Acids
Another novel and non-invasive approach to the early diagnosis of cancer is the detection of liquid biopsy-based biomarkers, such as cell-free DNA (cfDNA) or circulating tumor DNA (ctDNA). cfDNA can be isolated from the plasma and urine of cancer patients. However, the dynamic changes (both qualitative and quantitative) in cfDNAs occurring throughout the different stages of cancer progression, require to be fully understood before they can be used as biomarkers for cancer and for identifying cancer relapse. The average length of cfDNA fragments found in the blood of healthy individuals and in patients diagnosed with malignant tumors is 70–200 bp and 1–200 kb, respectively.[310] These high- and low molecular-weight DNA strands are likely be derived from the necrotic and apoptotic cells that enter the circulation.[311] Circulating biomarkers are of great interest, especially when biopsies of the primary or metastatic tumor are not available. They could provide a longitudinal analysis method for molecular profiling of cancer cells, assessing minimal residual disease in the non-metastatic setting, and monitoring response to systemic therapy.[312]
CtDNAs are generally detected using microarray-based comparative genome hybridization (CGH), single nucleotide polymorphism (SNP) analysis, massively parallel sequencing (MPS), or next-generation sequencing (NGS). The low sensitivity and the expense of these methods are limitations for the widespread and accurate detection of cfDNAs.[313] One strategy to improve the methylation-specific PCR (MSP) technique, is to use fluorescence-based (i.e., TaqMan) probes to facilitate the quantitative detection of DNA methylations without requiring further manipulation in the PCR step.[314] Similar to TaqMan probes, QDs possessing high photostability and a large dynamic range, have also been used as FRET donors to detect methylated DNA. 5-Amino-propargyl-2′-deoxycytidine 5′-triphosphate coupled to a Cy5 fluorescent dye served as a FRET acceptor in an assay for methylated DNA targets. The sensitivity of the Cy5-dCTP QD-FRET system was best when using multilabeled products.[315]
Whole circulating tumor cells (CTCs) are a rare and heterogeneous population of cancer cells found in peripheral blood, which are a marker of tumor dissemination and progression. They are an attractive surrogate biomarker that could be useful in cancer diagnosis and as a prognostic indicator. The HER2 status in patients with breast cancer has been established by the analysis of CTCs.[312] The assessment of ctDNAs and CTC biomarkers is currently being incorporated into clinical trials. The in vivo monitoring of CTCs via targeted imaging modalities might provide more information about the role of these markers in tumor metastasis, mechanisms of drug resistance, and improved patient assessment.
9.4. Targeted Cancer Stem Cell Imaging
CSCs represent a minor sub-population (≈1% of all cancer cells) within human tumors. They possess the highest tumorigenic potential, can generate heterogeneous progeny within the bulk cancer fraction, and are thought to be responsible for much (if not most) treatment resistance. CSCs are characterized by features including the ability for self-renewal, developing into multiple lineages, and the potential to proliferate extensively.[316] CSCs are also involved in invasion and distant metastasis through the EMT/MET phenomenon. For CSCs targeting, different types of strategies such as: i) targeting of tumor microenvironment markers/factors (Bone microenvironment/Immune-niche Angiogenesis/Spleen microenvironment/Lym-phoid tissue/Hypoxia/Vasculogenesis), ii) targeting key signaling pathways (Wnt/NF-kB/Notch/PI3k/AKT/ MAPK/TGF-β/mTOR/Hedgehog/Ras/Raf/Mek/Erk), and iii) over-expressed specific biomarkers (Table 6); can be used and might provide information about tumor prognosis and response in the future.[316–317] Some CSC targeting strategies are shown in Figure 25.
Table 6.
A list of known solid tumor CSC-related molecular markers for targeted therapy and diagnosis.
| Breast | Colorectal | Glioma | Lung | Ovarian | Pancreatic | Prostate | Bladder |
|---|---|---|---|---|---|---|---|
| a6-Integrin | ABCB5+ | a6-Integrin | ABCG2+ | CD24− | ABCG2+ | a6b1-integrin | Side |
| ALDH1+ | ALDH1+ | CD15+ | ALDH1+ | CD44+ | ALDH1+ | a6-integrin | Population |
| CD24− | b-Catenin | CD90+ | CD90+ | CD117+ | c-Met+ | ALDH1+ | |
| CD44+ | CD24− | CD13+ | CD117+ | CD133+ | CD24+ | CD44+ | |
| CD90+ | CD26+ | Nestin+ | CD133+ | CD44+ | CD133+ | ||
| CD133+ | CD29+ | CD133+ | CD166+ | ||||
| Hedgehog-Gli | CD44+ | CXCR4+ | Trop2 | ||||
| CD133+ | EpCAM+ | ||||||
| CD166+ | Nestin+ | ||||||
| EpCAM+ | |||||||
| LGR5+ |
Figure 25.
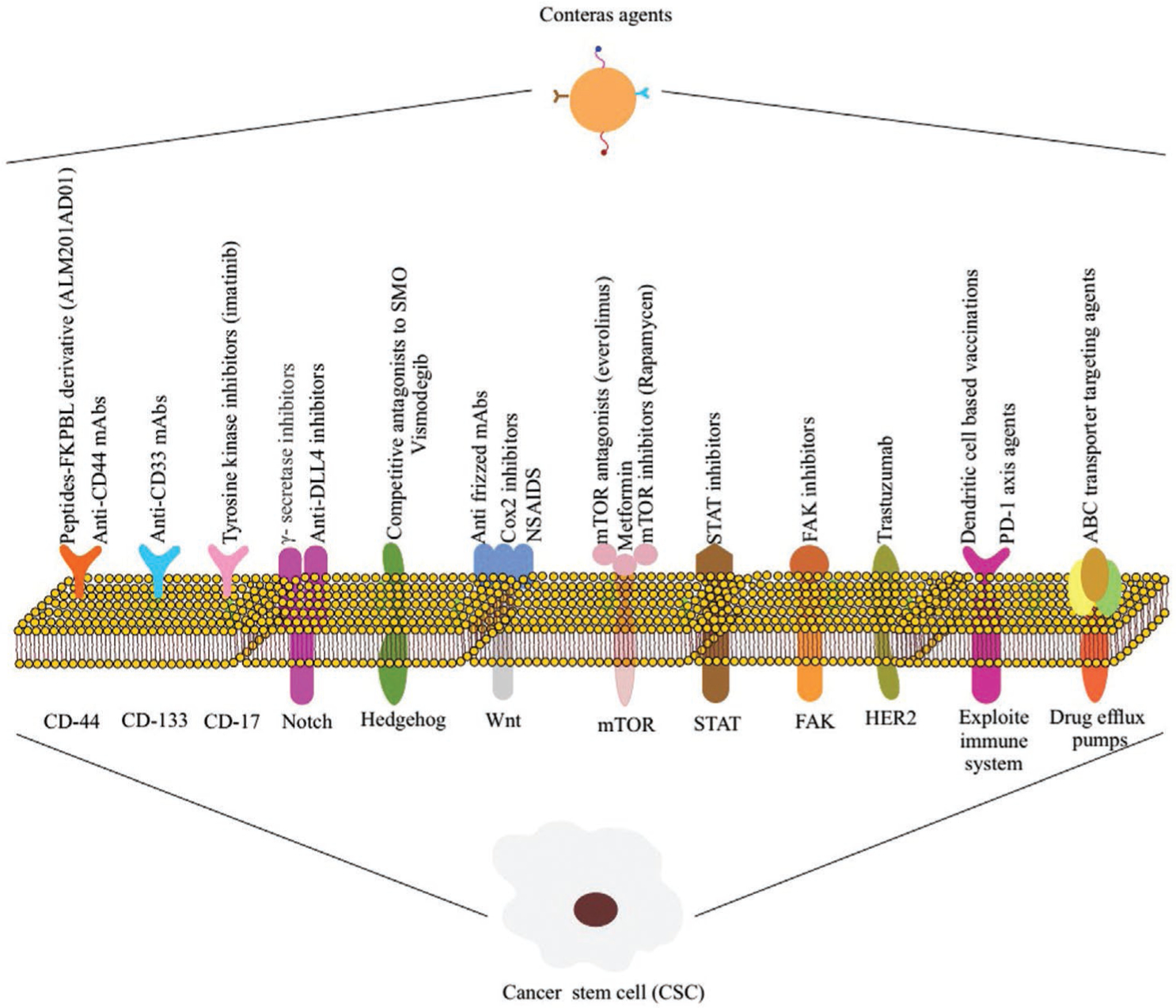
Strategies to target cancer stem cells. Many strategies aimed at eradicating CSCs have been developed. Targeting strategies consist of: cell surface markers, modulation of the immune system, cell signaling pathways, and inhibiting drug efflux pumps to sensitize cells to imaging.
Therapies that target CSCs in combination with conventional chemotherapy have already reached clinical trials. However, within the field of imaging and diagnosis, CSCs remain a topic of intense debate. Similar to cancer cells, in vivo imaging modalities using optical, nuclear, and magnetic resonance reporters are currently being employed to investigate the complexity underlying the behavior of CSCs. Furthermore, since CSCs are very rare in biological samples, the main concern in optical imaging is choosing a sufficiently sensitive reporter molecule and the best imaging modality. The leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) is considered to be a bona fide marker of CSCs. Researchers have used 89Zr immunoPET to evaluate and select efficient anti-LGR5 mAbs (8F2 and 9G5) for the development of Ab-drug conjugates (ADCs), imaging, and monitoring of LGR5-positive tumor response to therapy.[318] They demonstrated that the 8F2-based ADC was more effective for toxin delivery to LGR5-positive tumors, and suggested 89Zr-labeled anti-LGR5 mAbs could be used to stratify tumors, for best response to LGR5-targeted ADC therapy. Another study evaluated the potential of a PCa-specific PC204 peptide to target CD133, and the epithelial cell adhesion molecule (EpCAM), two known transmembrane glycoprotein markers that are overexpressed on PCa CSCs.[183] They found that PC204 had a strong affinity for EpCAM+, CD133+, and CD133− CSCs in the PCa cell line, and may be a promising molecular imaging platform for resistant solid tumors.
As an example of a surface marker for targeting CSC signaling pathways, the Tang group[319] targeted the ϒ-secretase enzyme, which plays an important role in the Notch signaling pathway. They used N-[N-(3, 5-fluorophenylacetyl-L-alanyl)]-S-phenylglycine-methyl ester (DAPT) as an inhibitor of ϒ-secretase, plus HA to increase the biocompatibility and biodegradability. More specifically, HA could bind to CD44+ CSCs. In vivo MR and PA imaging results showed that the nanoprobe accumulated in the CSC microenvironment. Another approach for targeting CSCs is the use of reporter genes. The Liu group used luc2 fused to the eGFP coding sequence for designing a dual-function bioluminescence-fluorescence imaging reporter probe for breast CSCs. In vitro and in vivo results demonstrated the reporter gene was suitable for CSC targeting.[320] Proteasome activity is another candidate as a target for CSCs. Considering the fact that 26S proteasome activity is reduced in CSCs, Vlashi et al.[321] engineered cancer cells to a express fluorescent fusion protein ZsGreen-ornithine decarboxylase that accumulated in cells with reduced 26S proteasome activity. The ZsGreen-positive cells could be tracked using in vivo fluorescence imaging. Results showed that the proteasome could be a suitable candidate for targeting of CSCs. The development of better techniques with higher imaging resolution, and better contrast to localize CSCs will be required for their clinical detection and eradication.
10. Perspectives and Conclusion
Selective tumor targeting and effective delivery systems utilizing NSs have resulted in the development of novel targeting methods. Targeted nanodelivery systems are able to reach, detect, and treat various types of tumors. Many types of targets and targeting agents overlap among different human tumors. Thus, the exploration of novel molecular targets enables us to improve delivery to tumors with decreased off-target activity and less toxicity. With the aim of categorizing cancer targeting based on tumor biology, we have divided targeting strategies into five subsets including, passive targeting, TME targeting, endothelial cell targeting, general cancer cell targeting, and specific cancer cell targeting. We have also discussed new techniques and methods used for more precise cancer targeting. Despite many recent advancements in targeted delivery, there is still a long way to go, and there are many problems to overcome. These include, targeted delivery structures may still possess toxicity, the targeting moieties may not be specific, resistance or relapse is often observed in patients treated with targeted nanosystems, and most tumors currently cannot be targeted with the presently established targeted delivery systems.
Although passive and active targeting improves the accumulation and cellular uptake of NCs in tumor sites and cancer cells, even small differences in the NC size have an impact on cellular uptake and localization. In order to establish links between the nanosized particles and the targeting mechanism, the NCs need to be the ideal size to be transported out of the vasculature, penetrate into the tumors, and localize to the intended cellular compartment. On the other hand, the amount of targeted agent that can be delivered is largely independent of the percentage of the administered dose and is dependent on the precision of the targeting and the balance between passive and active targeting. In addition, an insignificant amount of the administered NCs actually interact with the cancer cells (<14 out of 1 million NPs injected intravenously), indicating that the majority of intratumoral NCs are either trapped in the ECM or taken up by TAMs. These off-target delivery limitations demand the re-evaluation of current targeting strategies using more quantitative approaches.
Going forward, the consideration of the cancer type, subtype, and stage are critical steps in the diagnostic process by: i) helping the clinician develop a treatment plan; ii) giving an indication of prognosis; iii) aiding the evaluation of the results of treatment; iv) facilitating the exchange of information between treatment centers; and v) contributing to further investigation regarding human cancer. Researchers need to focus on carrier-dependent targeting, combination targeting, protocols for patient selection, and routes to enable rapid and efficient clinical translation.
Acknowledgements
This work was financially supported by Proteomics and Molecular Medicine Research Center, Tabriz University of Medical Sciences. MRH was supported by US NIH Grants R01AI050875 and R21AI121700. The authors are grateful to Dr. Ebrahim Azizi from Michigan University for fruitful advice and comments. Also, special thanks to Mehdi Shahgolzari who prepared the designed figures.
Biographies

Mehdi Azizi obtained his B.Sc. (chemistry) from Razi University, Iran in 2012, and M.Sc. (medical nanotechnology) from Tehran University of Medical Sciences, Iran in 2016. He is currently a Ph.D. candidate in the Department of Medical Nanotechnology at Tabriz University of Medical Sciences, and works with the Dr. Roya Salehi and Dr. Mohammad Mehrmohammadi group at the Department of Biomedical Imaging at Wayne State University. His current research involves the synthesis, development, and application of smart theranostic nanoplatforms.

Hassan Dianat-Moghadam is currently a Ph.D. candidate in the Medical Biotechnology Department at the Tabriz University of Medical Sciences, and works with Prof. Mohammad Nouri’s group at the Stem Cells Research Center. He obtained his B.Sc. (medical laboratory sciences) from Shiraz University of Medical Sciences in 2012, and M.Sc. (medical biotechnology) from Tehran University of Medical Sciences in 2016. His Ph.D. work is focused on cancer stem cell isolation and targeting them with immunoliposome loaded with novel therapeutic drugs.

Michael R. Hamblin was a principal investigator at the Wellman Center for Photomedicine at Massachusetts General Hospital, an associate professor of dermatology, Harvard Medical School, and is now a visiting professor at the University of Johannesburg, South Africa. His research concerns photodynamic therapy and photobiomodulation.
Footnotes
Conflict of Interest
MRH Scientific Advisory Boards: Transdermal Cap Inc, Cleveland, OH; BeWell Global Inc, Wan Chai, Hong Kong; Hologenix Inc. Santa Monica, CA; LumiThera Inc, Poulsbo, WA; Vielight, Toronto, Canada; Quantum Dynamics LLC, Cambridge, MA; Global Photon Inc, Bee Cave, TX; Medical Coherence, Boston MA; NeuroThera, Newark DE; JOOVV Inc, Minneapolis-St. Paul MN; AIRx Medical, Pleasanton CA; FIR Industries, Inc. Ramsey, NJ; UVLRx Therapeutics, Oldsmar, FL; Ultralux UV Inc, Lansing MI.
Contributor Information
Mehdi Azizi, Proteomics Research Centre, Tabriz University of Medical Sciences, Tabriz 5165665811, Iran; Department of Medical Nanotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Golgasht Street, Tabriz 516615731, Iran; Student Research Committee, Tabriz University of Medical Sciences, Tabriz 5165666532, Iran.
Hassan Dianat-Moghadam, Stem Cell Research Center, Tabriz University of Medical Sciences, Tabriz 5165665621, Iran.
Roya Salehi, Department of Medical Nanotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Golgasht Street, Tabriz 516615731, Iran.
Masoud Farshbaf, Drug Applied Research Center, Tabriz University of Medical Sciences, Tabriz 6581151656, Iran.
Disha Iyengar, U-BiND Systems Laboratory, Department of Pharmaceutical Sciences, Wayne State University, Detroit, MI 48201, USA; Northville High School, Northville, MI 48168, USA.
Samaresh Sau, U-BiND Systems Laboratory, Department of Pharmaceutical Sciences, Wayne State University, Detroit, MI 48201, USA.
Arun K. Iyer, U-BiND Systems Laboratory, Department of Pharmaceutical Sciences, Wayne State University, Detroit, MI 48201, USA
Hadi Valizadeh, Department of Medical Nanotechnology, Faculty of Advanced Medical Sciences, Tabriz University of Medical Sciences, Golgasht Street, Tabriz 516615731, Iran.
Mohammad Mehrmohammadi, Department of Biomedical Engineering, Wayne State University Detroit, MI 48202, USA.
Michael R. Hamblin, Wellman Center for Photomedicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA Laser Research Centre, Faculty of Health Science, University of Johannesburg, Doornfontein, Johannesburg 2028, South Africa.
References
- [1].Li C, Nat. Mater 2014, 13, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, Caldas C, Califano A, Doherty M, Elsner M, Nat. Med 2015, 21, 846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shi J, Kantoff PW, Wooster R, Farokhzad OC, Nat. Rev. Cancer 2017, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Group BDW, Atkinson AJ Jr., Colburn WA, DeGruttola VG, DeMets DL, Downing GJ, Hoth DF, Oates JA, Peck CC, Schooley RT, Clin. Pharmacol. Ther 2001, 69, 89.11240971 [Google Scholar]
- [5].Smith BR, Gambhir SS, Chem. Rev 2017, 117, 901. [DOI] [PubMed] [Google Scholar]
- [6].Huang H, Lovell JF, Adv. Funct. Mater 2017, 27, 1603524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Hong G, Antaris AL, Dai H, Nat. Biomed. Eng 2017, 1, 0010; [Google Scholar]; b) Park S.-m., Aalipour A, Vermesh O, Yu JH, Gambhir SS, Nat. Rev. Mater 2017, 2, 17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen H, Zhang W, Zhu G, Xie J, Chen X, Nat. Rev. Mater 2017, 2, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].van der Meel R, Sulheim E, Shi Y, Kiessling F, Mulder WJ, Lammers T, Nat. Nanotechnol 2019, 14, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bergers G, Benjamin LE, Nat. Rev. Cancer 2003, 3, 401. [DOI] [PubMed] [Google Scholar]
- [11].a) Greish K, J. Drug Targeting 2007, 15, 457; [DOI] [PubMed] [Google Scholar]; b) Cho K, Wang X, Nie S, Shin DM, Clin. Cancer Res 2008, 14, 1310; [DOI] [PubMed] [Google Scholar]; c) Iyer AK, Khaled G, Fang J, Maeda H, Drug Discovery Today 2006, 11, 812. [DOI] [PubMed] [Google Scholar]
- [12].Skinner SA, Tutton PJ, O’Brien PE, Cancer Res. 1990, 50, 2411. [PubMed] [Google Scholar]
- [13].Talekar M, Kendall J, Denny W, Garg S, Anti-Cancer Drugs 2011, 22, 949. [DOI] [PubMed] [Google Scholar]
- [14].Weissleder R, Nahrendorf M, Pittet MJ, Nat. Mater 2014, 13, 125. [DOI] [PubMed] [Google Scholar]
- [15].a) Matsumura Y, Maeda H, Cancer Res. 1986, 46, 6387; [PubMed] [Google Scholar]; b) Maeda H, Matsumura Y, Crit. Rev. Ther. Drug Carrier Syst 1989, 6, 193. [PubMed] [Google Scholar]
- [16].Xiao K, Luo J, Li Y, Xiao W, Lee JS, Gonik AM, Lam KS, Nanosci. Nanotechnol. Lett 2010, 2, 79. [Google Scholar]
- [17].Maeda H, Nakamura H, Fang J, Adv. Drug Delivery Rev 2013, 65, 71. [DOI] [PubMed] [Google Scholar]
- [18].Nakamura H, Liao L, Hitaka Y, Tsukigawa K, Subr V, Fang J, Ulbrich K, Maeda H, J. Controlled Release 2013, 165, 191. [DOI] [PubMed] [Google Scholar]
- [19].Hansen AE, Petersen AL, Henriksen JR, Boerresen B, Rasmussen P, Elema DR, Rosenschöld P. M. a., Kristensen AT, Kjær A, Andresen TL, ACS Nano 2015, 9, 6985. [DOI] [PubMed] [Google Scholar]
- [20].Perry JL, Reuter KG, Luft JC, Pecot CV, Zamboni W, DeSimone JM, Nano Lett. 2017, 17, 2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nichols JW, Bae YH, J. Controlled Release 2014, 190, 451. [DOI] [PubMed] [Google Scholar]
- [22].a) Gillies RJ, Schomack PA, Secomb TW, Raghunand N, Neoplasia 1999, 1, 197; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Danhier F, J. Controlled Release 2016, 244, 108. [DOI] [PubMed] [Google Scholar]
- [23].Nabil G, Bhise K, Sau S, Atef M, El-Banna HA, Iyer AK, Drug Discovery Today 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rosenblum D, Joshi N, Tao W, Karp JM, Peer D, Nat. Commun 2018, 9, 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yousef S, Alsaab HO, Sau S, Iyer AK, Heliyon 2018, 4, e01071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zheng X, Wang X, Mao H, Wu W, Liu B, Jiang X, Nat. Commun 2015, 6, 5834. [DOI] [PubMed] [Google Scholar]
- [27].Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT, Cell Metab. 2005, 1, 401. [DOI] [PubMed] [Google Scholar]
- [28].Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF, Clin. Cancer Res 2005, 11, 8782. [DOI] [PubMed] [Google Scholar]
- [29].Chitneni SK, Palmer GM, Zalutsky MR, Dewhirst MW, J. Nucl. Med 2011, 52, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cui L, Zhong Y, Zhu W, Xu Y, Du Q, Wang X, Qian X, Xiao Y, Org. Lett 2011, 13, 928. [DOI] [PubMed] [Google Scholar]
- [31].a) Liu J, Sun Y-Q, Huo Y, Zhang H, Wang L, Zhang P, Song D, Shi Y, Guo W, J. Am. Chem. Soc 2013, 136, 574; [DOI] [PubMed] [Google Scholar]; b) Lee MH, Yang Z, Lim CW, Lee YH, Dongbang S, Kang C, Kim JS, Chem. Rev 2013, 113, 5071. [DOI] [PubMed] [Google Scholar]
- [32].Harris AL, Nat. Rev. Cancer 2002, 2, 38. [DOI] [PubMed] [Google Scholar]
- [33].Luo C-H, Huang C-T, Su C-H, Yeh C-S, Nano Lett. 2016, 16, 3493. [DOI] [PubMed] [Google Scholar]
- [34].Lv Z, Zou L, Wei H, Liu S, Huang W, Zhao Q, ACS Appl. Mater. Interfaces 2018. [DOI] [PubMed] [Google Scholar]
- [35].Knox HJ, Hedhli J, Kim TW, Khalili K, Dobrucki LW, Chan J, Nat. Commun 2017, 8, 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Huo D, Liu S, Zhang C, He J, Zhou Z, Zhang H, Hu Y, ACS Nano 2017, 11, 10159. [DOI] [PubMed] [Google Scholar]
- [37].a) Gatenby RA, Gillies RJ, Nat. Rev. Cancer 2004, 4, 891; [DOI] [PubMed] [Google Scholar]; b) Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A, Bailey K, Balagurunathan Y, Rothberg JM, Sloane BF, Cancer Res. 2013, 73, 1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Manchun S, Dass CR, Sriamornsak P, Life Sci. 2012, 90, 381. [DOI] [PubMed] [Google Scholar]
- [39].Liu J, Ma H, Wei T, Liang X-J, Chem. Commun 2012, 48, 4869. [DOI] [PubMed] [Google Scholar]
- [40].a) Gilson RC, Tang R, Som A, Klajer C, Sarder P, Sudlow GP, Akers WJ, Achilefu S, Mol. Pharmaceutics 2015, 12, 4237; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao X, Yang C-X, Chen L-G, Yan X-P, Nat. Commun 2017, 8, 14998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Urano Y, Asanuma D, Hama Y, Koyama Y, Barrett T, Kamiya M, Nagano T, Watanabe T, Hasegawa A, Choyke PL, Nat. Med 2009, 15, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X, Zhao T, Sumer BD, DeBerardinis RJ, Gao J, Nat. Mater 2014, 13, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mi P, Kokuryo D, Cabral H, Wu H, Terada Y, Saga T, Aoki I, Nishiyama N, Kataoka K, Nat. Nanotechnol 2016, 11, 724. [DOI] [PubMed] [Google Scholar]
- [44].Hsu BYW, Ng M, Tan A, Connell J, Roberts T, Lythgoe M, Zhang Y, Wong SY, Bhakoo K, Seifalian AM, Adv. Healthcare Mater 2016, 5, 721. [DOI] [PubMed] [Google Scholar]
- [45].a) Nagase H, Visse R, Murphy G, Cardiovasc. Res 2006, 69, 562; [DOI] [PubMed] [Google Scholar]; b) Egeblad M, Werb Z, Nat. Rev. Cancer 2002, 2, 161; [DOI] [PubMed] [Google Scholar]; c) Choi JW, Ahn SE, Rengaraj D, Seo HW, Lim W, Song G, Han JY, Oncol. Lett 2011, 2, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schmalfeldt B, Prechtel D, Härting K, Späthe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W, Clin. Cancer Res 2001, 7, 2396. [PubMed] [Google Scholar]
- [47].a) Sternlicht MD, Werb Z, Annu. Rev. Cell Dev. Biol 2001, 17, 463; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tu Y, Zhu L, in Smart Pharmaceutical Nanocarriers (Ed.: Torchilin V), World Scientific, Singapore: 2016, p. 83; [Google Scholar]; c) Turk BE, Huang LL, Piro ET, Cantley LC, Nat. Biotechnol 2001, 19, 661. [DOI] [PubMed] [Google Scholar]
- [48].Kondo N, Temma T, Deguchi J, Sano K, Ono M, Saji H, J. Controlled Release 2015, 220, 476. [DOI] [PubMed] [Google Scholar]
- [49].Wang K-H, Wang Y-M, Chiu L-H, Chen T-C, Tsai Y-H, Zuo CS, Chen K-C, Changou CA, Lai W-FT, PLoS One 2018, 13, e0192047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Atukorale PU, Covarrubias G, Bauer L, Karathanasis E, Adv. Drug Delivery Rev 2017, 113, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Petrini I, Barachini S, Carnicelli V, Galimberti S, Modeo L, Boni R, Sollini M, Erba PA, Oncotarget 2017, 8, 4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].a) Sceneay J, Smyth MJ, Möller A, Cancer Metastasis Rev. 2013, 32, 449; [DOI] [PubMed] [Google Scholar]; b) Zhou F, Drabsch Y, Dekker TJ, De Vinuesa AG, Li Y, Hawinkels LJ, Sheppard K-A, Goumans M-J, Luwor RB, De Vries CJ, Nat. Commun 2014, 5, 3388. [DOI] [PubMed] [Google Scholar]
- [53].Bae YK, Kim A, Kim MK, Choi JE, Kang SH, Lee SJ, Hum. Pathol 2013, 44, 2028. [DOI] [PubMed] [Google Scholar]
- [54].Arnold SA, Loomans HA, Ketova T, Andl CD, Clark PE, Zijlstra A, Clin. Exp. Metastasis 2016, 33, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Malik G, Knowles LM, Dhir R, Xu S, Yang S, Ruoslahti E, Pilch J, Cancer Res. 2010, 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].a) Han Z, Lu Z-R, J. Mater. Chem. B 2017, 5, 639; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rybak J-N, Roesli C, Kaspar M, Villa A, Neri D, Cancer Res. 2007, 67, 10948. [DOI] [PubMed] [Google Scholar]
- [57].Zhou Z, Qutaish M, Han Z, Schur RM, Liu Y, Wilson DL, Lu Z-R, Nat. Commun 2015, 6, 7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Han Z, Wu X, Roelle S, Chen C, Schiemann WP, Lu Z-R, Nat. Commun 2017, 8, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Abou-Elkacem L, Wilson KE, Johnson SM, Chowdhury SM, Bachawal S, Hackel BJ, Tian L, Willmann JK, Theranostics 2016, 6, 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, Bogyo M, Nat. Med 2009, 15, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Shalini S, Dorstyn L, Dawar S, Kumar S, Cell Death Differ. 2015, 22, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].van Genderen HO, Kenis H, Hofstra L, Narula J, Reutelingsperger CP, Biochim. Biophys. Acta Mol. Cell Res 2008, 1783, 953. [DOI] [PubMed] [Google Scholar]
- [63].Boersma HH, Kietselaer BL, Stolk LM, Bennaghmouch A, Hofstra L, Narula J, Heidendal GA, Reutelingsperger CP, J. Nucl. Med 2005, 46, 2035. [PubMed] [Google Scholar]
- [64].Ye D, Shuhendler AJ, Cui L, Tong L, Tee SS, Tikhomirov G, Felsher DW, Rao J, Nat. Chem 2014, 6, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhang W, Cai K, Li X, Zhang J, Ma Z, Foda MF, Mu Y, Dai X, Han H, Theranostics 2019, 9, 4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Lu C, Jiang Q, Hu M, Tan C, Yu H, Hua Z, Oncotarget 2017, 8, 51086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].a) Watanabe H, Murata Y, Miura M, Hasegawa M, Kawamoto T, Shibuya H, Nucl. Med. Commun 2006, 27, 81; [DOI] [PubMed] [Google Scholar]; b) Lahorte C, Van De Wiele C, Bacher K, Van Den Bossche B, Thierens H, Van Belle S, Slegers G, Dierckx R, Nucl. Med. Commun 2003, 24, 871; [DOI] [PubMed] [Google Scholar]; c) Ke S, Wen X, Wu Q-P, Wallace S, Charnsangavej C, Stachowiak AM, Stephens CL, Abbruzzese JL, Podoloff DA, Li C, J. Nucl. Med 2004, 45, 108. [PubMed] [Google Scholar]
- [68].Hanahan D, Folkman J, Cell 1996, 86, 353. [DOI] [PubMed] [Google Scholar]
- [69].a) Hellström M, Gerhardt H, Kalén M, Li X, Eriksson U, Wolburg H, Betsholtz C, J. Cell Biol 2001, 153, 543; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ruoslahti E, Nat. Rev. Cancer 2002, 2, 83. [DOI] [PubMed] [Google Scholar]
- [70].Hong H, Yang K, Zhang Y, Engle JW, Feng L, Yang Y, Nayak TR, Goel S, Bean J, Theuer CP, ACS Nano 2012, 6, 2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gao W, Li S, Liu Z, Sun Y, Cao W, Tong L, Cui G, Tang B, Biomaterials 2017, 139, 1. [DOI] [PubMed] [Google Scholar]
- [72].Korpanty G, Carbon JG, Grayburn PA, Fleming JB, Brekken RA, Clin. Cancer Res 2007, 13, 323. [DOI] [PubMed] [Google Scholar]
- [73].Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Nature 2005, 438, 820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].a) Dvorak HF, J. Clin. Oncol 2002, 20, 4368; [DOI] [PubMed] [Google Scholar]; b) Hicklin DJ, Ellis LM, J. Clin. Oncol 2005, 23, 1011. [DOI] [PubMed] [Google Scholar]
- [75].a) Alitalo K, Carmeliet P, Cancer Cell 2002, 1, 219; [DOI] [PubMed] [Google Scholar]; b) Ellis LM, Hicklin DJ, Nat. Rev. Cancer 2008, 8, 579. [DOI] [PubMed] [Google Scholar]
- [76].Senger DR, Van De Water L, Brown LF, Nagy JA, Yeo K-T, Yeo T-K, Berse B, Jackman RW, Dvorak AM, Dvorak HF, Cancer Metastasis Rev. 1993, 12, 303. [DOI] [PubMed] [Google Scholar]
- [77].Gerber H-P, Ferrara N, Cancer Res. 2005, 65, 671. [PubMed] [Google Scholar]
- [78].Nagengast WB, de Vries EG, Hospers GA, Mulder NH, de Jong JR, Hollema H, Brouwers AH, van Dongen GA, Perk LR, Hooge M, J. Nucl. Med 2007, 48, 1313. [DOI] [PubMed] [Google Scholar]
- [79].van Scheltinga AGT, van Dam GM, Nagengast WB, Ntziachristos V, Hollema H, Herek JL, Schröder CP, Kosterink JG, Lub-de Hoog MN, de Vries EG, J. Nucl. Med 2011, 52, 1778. [DOI] [PubMed] [Google Scholar]
- [80].Abakumov MA, Nukolova NV, Sokolsky-Papkov M, Shein SA, Sandalova TO, Vishwasrao HM, Grinenko NF, Gubsky IL, Abakumov AM, Kabanov AV, Nanomed.: Nanotechnol., Biol. Med 2015, 11, 825. [DOI] [PubMed] [Google Scholar]
- [81].Jayson GC, Zweit J, Jackson A, Mulatero C, Julyan P, Ranson M, Broughton L, Wagstaff J, Hakannson L, Groenewegen G, JNCI, J. Natl. Cancer Inst 2002, 94, 1484. [DOI] [PubMed] [Google Scholar]
- [82].Desgrosellier JS, Cheresh DA, Nat. Rev. Cancer 2010, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Raab-Westphal S, Marshall JF, Goodman SL, Cancers 2017, 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Curr. Biol 2006, 16, 1796. [DOI] [PubMed] [Google Scholar]
- [85].Berghoff AS, Kovanda AK, Melchardt T, Bartsch R, Hainfellner JA, Sipos B, Schittenhelm J, Zielinski CC, Widhalm G, Dieckmann K, Clin. Exp. Metastasis 2014, 31, 841. [DOI] [PubMed] [Google Scholar]
- [86].a) McCabe N, De S, Vasanji A, Brainard J, Byzova T, Oncogene 2007, 26, 6238; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Heß K, Böger C, Behrens H-M, Röcken C, Ann. Diagn. Pathol 2014, 18, 343. [DOI] [PubMed] [Google Scholar]
- [87].a) Diaz LK, Cristofanilli M, Zhou X, Welch KL, Smith TL, Yang Y, Sneige N, Sahin AA, Gilcrease MZ, Mod. Pathol 2005, 18, 1165; [DOI] [PubMed] [Google Scholar]; b) Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, Anderson RL, Breast Cancer Res. 2006, 8, R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hosotani R, Kawaguchi M, Masui T, Koshiba T, Ida J, Fujimoto K, Wada M, Doi R, Imamura M, Pancreas 2002, 25, e30. [DOI] [PubMed] [Google Scholar]
- [89].Bello L, Francolini M, Marthyn P, Zhang J, Carroll RS, Nikas DC, Strasser JF, Villani R, Cheresh DA, Black PM, Neurosurgery 2001, 49, 380. [DOI] [PubMed] [Google Scholar]
- [90].a) Slack-Davis JK, Atkins KA, Harrer C, Hershey ED, Conaway M, Cancer Res. 2009, 69, 1469; [DOI] [PubMed] [Google Scholar]; b) Landen CN, Kim T-J, Lin YG, Merritt WM, Kamat AA, Han LY, Spannuth WA, Nick AM, Jennnings NB, Kinch MS, Neoplasia 2008, 10, 1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gruber G, Hess J, Stiefel C, Aebersold D, Zimmer Y, Greiner R, Studer U, Altermatt H, Hlushchuk R, Djonov V, Br. J. Cancer 2005, 92, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Adachi M, Taki T, Higashiyama M, Kohno N, Inufusa H, Miyake M, Clin. Cancer Res 2000, 6, 96. [PubMed] [Google Scholar]
- [93].a) Takayama S, Ishii S, Ikeda T, Masamura S, Doi M, Kitajima M, Anticancer Res. 2005, 25, 79; [PubMed] [Google Scholar]; b) Hieken TJ, Ronan SG, Farolan M, Shilkaitis AL, Das Gupta TK, Cancer 1999, 85, 375. [DOI] [PubMed] [Google Scholar]
- [94].Patsenker E, Wilkens L, Banz V, Österreicher C, Weimann R, Eisele S, Keogh A, Stroka D, Zimmermann A, Stickel F, J. Hepatol 2010, 52, 362. [DOI] [PubMed] [Google Scholar]
- [95].Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio AM, J. Clin. Invest 2005, 115, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].a) Nieberler M, Reuning U, Reichart F, Notni J, Wester H-J, Schwaiger M, Weinmüller M, Räder A, Steiger K, Kessler H, Cancers 2017, 9, 116; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Humphries JD, Byron A, Humphries MJ, J. Cell Sci 2006, 119, 3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Tang L, Sun X, Liu N, Zhou Z, Yu F, Zhang X, Sun X, Chen X, ACS Appl. Nano Mater 2018, 1, 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Tang R, Xue J, Xu B, Shen D, Sudlow GP, Achilefu S, ACS Nano 2015, 9, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Liu Z, Yan Y, Chin FT, Wang F, Chen X, J. Med. Chem 2008, 52, 425. [DOI] [PubMed] [Google Scholar]
- [100].Jin Z-H, Furukawa T, Kumata K, Xie L, Yui J, Wakizaka H, Fujibayashi Y, Zhang M-R, Saga T, Biol. Pharm. Bull 2015, 38, 1722. [DOI] [PubMed] [Google Scholar]
- [101].Alvarez P, Carrillo E, Vélez C, Hita-Contreras F, Martínez-Amat A, Rodríguez-Serrano F, Boulaiz H, Ortiz R, Melguizo C, Prados J, Biomed Res. Int 2013, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wittchen ES, Front. Biosci 2009, 14, 2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].a) Costa MF, Bornstein VU, Candéa AL, Henriques-Pons A, Henriques MG, Penido C, Eur. J. Immunol 2012, 42, 1250; [DOI] [PubMed] [Google Scholar]; b) Kon S, Atakilit A, Sheppard D, Exp. Cell Res 2011, 317, 1774; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Barthel SR, Annis DS, Mosher DF, Johansson MW, J. Biol. Chem 2006, 281, 32175. [DOI] [PubMed] [Google Scholar]
- [104].Hession C, Tizard R, Vassallo C, Schiffer SB, Goff D, Moy P, Chi-Rosso G, Luhowskyj S, Lobb R, Osborn L, J. Biol. Chem 1991, 266, 6682. [PubMed] [Google Scholar]
- [105].Barreiro O, Yáñez-Mó M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sánchez-Madrid F, J. Cell Biol 2002, 157, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Schlesinger M, Bendas G, Int. J. Cancer 2015, 136, 2504. [DOI] [PubMed] [Google Scholar]
- [107].Chen Q, Zhang XH-F, Massagué J, Cancer Cell 2011, 20, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zhang X, Liu C, Hu F, Zhang Y, Wang J, Gao Y, Jiang Y, Zhang Y, Lan X, Mol. Pharmaceutics 2018, 15, 609. [DOI] [PubMed] [Google Scholar]
- [109].Patel N, Duffy B, Badar A, Lythgoe M, Årstad E, Bioconjugate Chem. 2015, 26, 1542. [DOI] [PubMed] [Google Scholar]
- [110].Uddin MI, Jayagopal A, Wong A, McCollum GW, Wright DW, Penn JS, Nanomedicine 2018, 14, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Cheng VW, Soto MS, Khrapitchev AA, Perez-Balderas F, Zakaria R, Jenkinson MD, Middleton MR, Sibson N, Clin. Cancer Res 2018, 24, 1954. [DOI] [PubMed] [Google Scholar]
- [112].Scalici JM, Thomas S, Harrer C, Raines TA, Curran J, Atkins KA, Conaway MR, Duska L, Kelly KA, Slack-Davis JK, J. Nucl. Med 2013, 54, 1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Sugyo A, Tsuji AB, Sudo H, Nomura F, Satoh H, Koizumi M, Kurosawa G, Kurosawa Y, Saga T, Oncol. Rep 2017, 37, 1529. [DOI] [PubMed] [Google Scholar]
- [114].Zhao T, He X-W, Li W-Y, Zhang Y-K, J. Mater. Chem. B 2015, 3, 2388. [DOI] [PubMed] [Google Scholar]
- [115].Zhao Y, Peng J, Li J, Huang L, Yang J, Huang K, Li H, Jiang N, Zheng S, Zhang X, Nano Lett. 2017, 17, 4096. [DOI] [PubMed] [Google Scholar]
- [116].Wang K, Zhang Y, Wang J, Yuan A, Sun M, Wu J, Hu Y, Sci. Rep 2016, 6, 27421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Qi H, Li Z, Du K, Mu K, Zhou Q, Liang S, Zhu W, Yang X, Zhu Y, Nanoscale Res. Lett 2014, 9, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhen Z, Tang W, Guo C, Chen H, Lin X, Liu G, Fei B, Chen X, Xu B, Xie J, ACS Nano 2013, 7, 6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Yang M, Fan Q, Zhang R, Cheng K, Yan J, Pan D, Ma X, Lu A, Cheng Z, Biomaterials 2015, 69, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Weitman SD, Lark RH, Coney LR, Fort DW, Frasca V, Zurawski VR, Kamen BA, Cancer Res. 1992, 52, 3396. [PubMed] [Google Scholar]
- [121].Hai X, Wang Y, Hao X, Chen X, Wang J, Sens. Actuators, B 2018, 268, 61. [Google Scholar]
- [122].Hassan M, Gomes VG, Dehghani A, Ardekani SM, Nano Res. 2018, 11, 1. [Google Scholar]
- [123].Gao X, Zhai M, Guan W, Liu J, Liu Z, Damirin A, ACS Appl. Mater. Interfaces 2017, 9, 3455. [DOI] [PubMed] [Google Scholar]
- [124].Liu Q, Xu S, Niu C, Li M, He D, Lu Z, Ma L, Na N, Huang F, Jiang H, Ouyang J, Biosens. Bioelectron 2015, 64, 119. [DOI] [PubMed] [Google Scholar]
- [125].Liu X, MillerII AL, Yaszemski MJ, Lu L, RSC Adv. 2015, 5, 33275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Yin F, Zhang B, Zeng S, Lin G, Tian J, Yang C, Wang K, Xu G, Yong K-T, J. Mater. Chem. B 2015, 3, 6081. [DOI] [PubMed] [Google Scholar]
- [127].Zhou B, Yang J, Peng C, Zhu J, Tang Y, Zhu X, Shen M, Zhang G, Shi X, Colloids Surf., B 2016, 140, 489. [DOI] [PubMed] [Google Scholar]
- [128].Zhang Z, Hu Y, Yang J, Xu Y, Zhang C, Wang Z, Shi X, Zhang G, Mol. Imaging Biol 2016, 18, 569. [DOI] [PubMed] [Google Scholar]
- [129].McCawley LJ, O’Brien P, Hudson LG, Endocrinology 1997, 138, 121. [DOI] [PubMed] [Google Scholar]
- [130].Nicholson R, Gee J, Harper M, Eur. J. Cancer 2001, 37, 9. [DOI] [PubMed] [Google Scholar]
- [131].Kitai Y, Fukuda H, Enomoto T, Asakawa Y, Suzuki T, Inouye S, Handa H, J. Biotechnol 2011, 155, 251. [DOI] [PubMed] [Google Scholar]
- [132].Tjalma JJ, Garcia-Allende PB, Hartmans E, van Scheltinga AGT, Boersma-van Ek W, Glatz J, Koch M, Van Herwaarden YJ, Bisseling TM, Nagtegaal ID, J. Nucl. Med 2016, 57, 480. [DOI] [PubMed] [Google Scholar]
- [133].Gao M, Su H, Lin G, Li S, Yu X, Qin A, Zhao Z, Zhang Z, Tang BZ, Nanoscale 2016, 8, 15027. [DOI] [PubMed] [Google Scholar]
- [134].Hudson SV, Huang JS, Yin W, Albeituni S, Rush J, Khanal A, Yan J, Ceresa BP, Frieboes HB, McNally LR, Cancer Res. 2014, 74, 6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wang Z, Qiao R, Tang N, Lu Z, Wang H, Zhang Z, Xue X, Huang Z, Zhang S, Zhang G, Biomaterials 2017, 127, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hay N, Nat. Rev. Cancer 2016, 16, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Evans NR, Tarkin JM, Buscombe JR, Markus HS, Rudd JH, Warburton EA, Nat. Rev. Neurol 2017, 13, 676. [DOI] [PubMed] [Google Scholar]
- [138].Dreifuss T, Motiei M, Betzer O, Popovtzer A, Abourbeh G, Mishani E, Popovtzer R, Presented at Nanoscale Imaging, Sensing, Actuation Biomed. Appl. XIV, San Francisco, CA, February 2017. [Google Scholar]
- [139].Singh S, Colloids Surf., B 2017, 155, 25. [DOI] [PubMed] [Google Scholar]
- [140].a) Mohamed MM, Sloane BF, Nat. Rev. Cancer 2006, 6, 764; [DOI] [PubMed] [Google Scholar]; b) Olson OC, Joyce JA, Nat. Rev. Cancer 2015, 15, 712. [DOI] [PubMed] [Google Scholar]
- [141].Ryu JH, Na JH, Ko HK, You DG, Park S, Jun E, Yeom HJ, Seo DH, Park JH, Jeong SY, Biomaterials 2014, 35, 2302. [DOI] [PubMed] [Google Scholar]
- [142].Shim MK, Yoon HY, Ryu JH, Koo H, Lee S, Park JH, Kim JH, Lee S, Pomper MG, Kwon IC, Angew. Chem 2016, 128, 14918. [DOI] [PubMed] [Google Scholar]
- [143].Liu J, Zhang L, Lei J, Shen H, Ju H, ACS Appl. Mater. Interfaces 2017, 9, 2150. [DOI] [PubMed] [Google Scholar]
- [144].Gaikwad HK, Tsvirkun D, Ben-Nun Y, Merquiol E, Popovtzer R, Blum G, Nano Lett. 2018, 18, 1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tsvirkun D, Ben-Nun Y, Merquiol E, Zlotver I, Meir K, Weiss-Sadan T, Matok I, Popovtzer R, Blum G, J. Am. Chem. Soc 2018, 140, 12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC, Adv. Drug Delivery Rev 2014, 66, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Perou CM, Sørlie T, Eisen MB, Van De Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Nature 2000, 406, 747. [DOI] [PubMed] [Google Scholar]
- [148].Smith RA, Andrews KS, Brooks D, Fedewa SA, Manassaram-Baptiste D, Saslow D, Brawley OW, Wender RC, Ca-Cancer J. Clin 2018, 68, 297. [DOI] [PubMed] [Google Scholar]
- [149].a) C. G. A. Network, Nature 2012, 490, 61;23000897 [Google Scholar]; b) Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schütz F, Breast Cancer Res. 2008, 10, R65; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) de Macêdo Andrade AC, Júnior CAF, Guimarães BD, Barros AWP, de Almeida GS, Weller M, BMC Women Health 2014, 14, 110; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Molina R, Barak V, van Dalen A, Duffy MJ, Einarsson R, Gion M, Goike H, Lamerz R, Nap M, Sölétormos G, Tumor Biol. 2005, 26, 281. [DOI] [PubMed] [Google Scholar]
- [150].Wang Z, Sau S, Alsaab HO, Iyer AK, Nanomedicine 2018, 14, 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Watson SS, Dane M, Chin K, Tatarova Z, Liu M, Liby T, Thompson W, Smith R, Nederlof M, Bucher E, Cell Systems 2018, 6, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Fowler AM, Linden HM, J. Nucl. Med 2017, 58, 560. [DOI] [PubMed] [Google Scholar]
- [153].Zhang C, Moonshi SS, Wang W, Ta HT, Han Y, Han FY, Peng H, Král P, Rolfe BE, Gooding JJ, ACS Nano 2018, 12, 10201. [DOI] [PubMed] [Google Scholar]
- [154].Kievit FM, Stephen ZR, Veiseh O, Arami H, Wang T, Lai VP, Park JO, Ellenbogen RG, Disis ML, Zhang M, ACS Nano 2012, 6, 2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Zhao Y, Detering L, Sultan D, Cooper ML, You M, Cho S, Meier SL, Luehmann H, Sun G, Rettig M, ACS Nano 2016, 10, 5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Balasubramanian P, Kinders RJ, Kummar S, Gupta V, Hasegawa D, Menachery A, Lawrence SM, Wang L, Ferry-Galow K, Davis D, PLoS One 2017, 12, e0175414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Eghtedari M, Liopo AV, Copland JA, Oraevsky AA, Motamedi M, Nano Lett. 2008, 9, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Herbst RS, Morgensztern D, Boshoff C, Nature 2018, 553, 446. [DOI] [PubMed] [Google Scholar]
- [159].a) Travis WD, Brambilla E, Riely GJ, J. Clin. Oncol 2013, 31, 992; [DOI] [PubMed] [Google Scholar]; b) Lemjabbar-Alaoui H, Hassan OU, Yang Y-W, Buchanan P, Biochim. Biophy. Acta 2015, 1856, 189; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Robinson CG, Bradley JD, Semin. Radiat. Oncol 2010, 20, 178. [DOI] [PubMed] [Google Scholar]
- [160].a) Shtivelman E, Hensing T, Simon GR, Dennis PA, Otterson GA, Bueno R, Salgia R, Oncotarget 2014, 5, 1392; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Engl N. J. Med 2004, 350, 2129; [DOI] [PubMed] [Google Scholar]; c) Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Proc. Natl. Acad. Sci. U. S. A 2004, 101, 13306; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Science 2004, 304, 1497; [DOI] [PubMed] [Google Scholar]; e) Cohen AS, Khalil FK, Welsh EA, Schabath MB, Enkemann SA, Davis A, Zhou J-M, Boulware DC, Kim J, Haura EB, Oncotarget 2017, 8, 113373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Zhao Y, Liu W, Tian Y, Yang Z, Wang X, Zhang Y, Tang Y, Zhao S, Wang C, Liu Y, ACS Appl. Mater. Interfaces 2018, 10, 16992. [DOI] [PubMed] [Google Scholar]
- [162].Ehlerding EB, England CG, Majewski RL, Valdovinos HF, Jiang D, Liu G, McNeel DG, Nickles RJ, Cai W, Mol. Pharmaceutics 2017, 14, 1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].England CG, Jiang D, Hernandez R, Sun H, Valdovinos HF, Ehlerding EB, Engle JW, Yang Y, Huang P, Cai W, Mol. Pharmaceutics 2017, 14, 3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Cohen AS, Patek R, Enkemann SA, Johnson JO, Chen T, Toloza E, Vagner J, Morse DL, Bioconjugate Chem. 2015, 27, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, van de Velde CJH, Watanabe T, Nat. Rev. Dis. Primers 2015, 1, 15065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Chand M, Keller DS, Mirnezami R, Bullock M, Bhangu A, Moran B, Tekkis PP, Brown G, Mirnezami A, Berho M, World J Gastrointest. Oncol 2018, 10, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Ahlquist DA, Sargent DJ, Loprinzi CL, Levin TR, Rex DK, Ahnen DJ, Knigge K, Lance MP, Burgart LJ, Hamilton SR, Ann. Intern. Med 2008, 149, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Yiu AJ, Yiu CY, Anticancer Res. 2016, 36, 1093. [PubMed] [Google Scholar]
- [169].Makris NE, Boellaard R, van Lingen A, Lammertsma AA, van Dongen G, Verheul HM, Menke CW, Huisman MC, J. Nucl. Med 2015, 56, 249. [DOI] [PubMed] [Google Scholar]
- [170].Toy R, Bauer L, Hoimes C, Ghaghada KB, Karathanasis E, Adv. Drug Delivery Rev 2014, 76, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Nakase H, Sakuma S, Fukuchi T, Yoshino T, Mohri K, Miyata K, Kumagai H, Hiwatari K-I, Tsubaki K, Ikejima T, Int. J. Nanomed 2017, Volume 12, 1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Jeong S, Song J, Lee W, Ryu YM, Jung Y, Kim S-Y, Kim K, Hong SC, Myung SJ, Kim S, Nano Lett. 2017, 17, 1378. [DOI] [PubMed] [Google Scholar]
- [173].Kwon OS, Song HS, Conde J. o., Kim H.-i., Artzi N, Kim J-H, ACS Nano 2016, 10, 1512. [DOI] [PubMed] [Google Scholar]
- [174].Beack S, Cho M, Kim Y-E, Ahn G-O, Hahn SK, Bioconjugate Chem. 2017, 28, 1434. [DOI] [PubMed] [Google Scholar]
- [175].Burggraaf J, Kamerling IM, Gordon PB, Schrier L, De Kam ML, Kales AJ, Bendiksen R, Indrevoll B, Bjerke RM, Moestue SA, Nat. Med 2015, 21, 955. [DOI] [PubMed] [Google Scholar]
- [176].Si HY, Cho MK, Kang JS, Noh C-K, Shin SJ, Lim CS, Kim HM, Anal. Chem 2018, 90, 3898. [DOI] [PubMed] [Google Scholar]
- [177].a) Sathianathen NJ, Konety BR, Crook J, Saad F, Lawrentschuk N, Nat. Rev. Urol 2018, 1; [DOI] [PubMed] [Google Scholar]; b) Hajipour H, Hamishehkar H, Rahmati-Yamchi M, Shanehbandi D, Ahmad SNS, Hasani A, Int. J. Cancer Manag 2018, 11. [Google Scholar]
- [178].Chistiakov DA, Myasoedova VA, Grechko AV, Melnichenko AA, Orekhov AN, Semin. Cancer Biol 2018, 52, 9. [DOI] [PubMed] [Google Scholar]
- [179].Li R, Ravizzini GC, Gorin MA, Maurer T, Eiber M, Cooperberg MR, Alemozzaffar M, Tollefson MK, Delacroix SE, Chapin BF, Prostate Cancer Prostatic Dis. 2017, 1. [DOI] [PubMed] [Google Scholar]
- [180].Agemy L, Sugahara KN, Kotamraju VR, Gujraty K, Girard OM, Kono Y, Mattrey RF, Park J-H, Sailor MJ, Jimenez AI, Blood 2010, 116, 2847, blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Kelly KA, Setlur SR, Ross R, Anbazhagan R, Waterman P, Rubin MA, Weissleder R, Cancer Res. 2008, 68, 2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Cai H, Xie F, Mulgaonkar A, Chen L, Sun X, Hsieh J-T, Peng F, Tian R, Li L, Wu C, Nanomedicine 2018, 13, 2777. [DOI] [PubMed] [Google Scholar]
- [183].Yeh C-Y, Hsiao J-K, Wang Y-P, Lan C-H, Wu H-C, Biomaterials 2016, 99, 1. [DOI] [PubMed] [Google Scholar]
- [184].Hu H, Zhang Y, Shukla S, Gu Y, Yu X, Steinmetz NF, ACS Nano 2017, 11, 9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Siegel RL, Miller KD, Jemal A, Ca-Cancer J. Clin 2015, 65, 5. [DOI] [PubMed] [Google Scholar]
- [186].a) Singh D, Upadhyay G, Srivastava RK, Shankar S, Biochim. Biophys. Acta 2015, 1856, 13; [DOI] [PubMed] [Google Scholar]; b) Lee CJ, Heidt DG, Simeone DM, in Encyclopedia of Cancer, Springer, Cham, Switzerland: 2008, p. 2254. [Google Scholar]
- [187].a) Patra CR, Bhattacharya R, Mukhopadhyay D, Mukherjee P, Adv. Drug Delivery Rev 2010, 62, 346; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, Nat. Rev. Dis. Primers 2016, 2, 16022. [DOI] [PubMed] [Google Scholar]
- [188].Makohon-Moore A, Iacobuzio-Donahue CA, Nat. Rev. Cancer 2016, 16, 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Zhou H, Qian W, Uckun FM, Wang L, Wang YA, Chen H, Kooby D, Yu Q, Lipowska M, Staley CA, ACS Nano 2015, 9, 7976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Jiang Y, Liu S, Zhang Y, Li H, He H, Dai J, Jiang T, Ji W, Geng D, Elzatahry AA, Biomaterials 2017, 115, 9. [DOI] [PubMed] [Google Scholar]
- [191].Zettlitz KA, Tsai W-TK, Knowles SM, Kobayashi N, Donahue TR, Reiter RE, Wu AM, J. Nucl. Med 2018, 59, 1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Kim H-Y, Wang X, Kang R, Tang D, Boone BA, Zeh III HJ, Lotze MT, Edwards WB, PLoS One 2018, 13, e0192821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM, Int. J. Cancer 2010, 127, 2893. [DOI] [PubMed] [Google Scholar]
- [194].Czerniak B, Dinney C, McConkey D, Annu. Rev. Pathol.: Mech. Dis 2016, 11, 149. [DOI] [PubMed] [Google Scholar]
- [195].Knowles MA, Hurst CD, Nat. Rev. Cancer 2015, 15, 25. [DOI] [PubMed] [Google Scholar]
- [196].Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Sjödahl G, Lauss M, Lövgren K, Chebil G, Gudjonsson S, Veerla S, Patschan O, Aine M, Fernö M, Ringnér M, Clin. Cancer Res 2012. [DOI] [PubMed]
- [198].Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee I-L, Cancer Cell 2014, 25, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Aine M, Eriksson P, Liedberg F, Höglund M, Sjödahl G, Eur. Urol 2015, 68, 921. [DOI] [PubMed] [Google Scholar]
- [200].Pan Y, Volkmer J-P, Mach KE, Rouse RV, Liu J-J, Sahoo D, Chang TC, Metzner TJ, Kang L, Van De Rijn M, Sci. Transl. Med 2014, 6, 260ra148. [DOI] [PubMed] [Google Scholar]
- [201].Nishizawa K, Nishiyama H, Oishi S, Tanahara N, Kotani H, Mikami Y, Toda Y, Evans BJ, Peiper SC, Saito R, Int. J. Cancer 2010, 127, 1180. [DOI] [PubMed] [Google Scholar]
- [202].Davis RM, Kiss B, Trivedi DR, Metzner TJ, Liao JC, Gambhir SS, ACS Nano 2018, 12, 9669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, Ca-Cancer J. Clin 2018. [DOI] [PubMed]
- [204].Antuña AR, Vega MA, Sanchez CR, Fernandez VM, J. Neurol. Surg. Part A Central Eur. Neurosurg 2018, 79, 101. [DOI] [PubMed] [Google Scholar]
- [205].Mischel PS, Cloughesy TF, Nelson SF, Nat. Rev. Neurosci 2004, 5, 782. [DOI] [PubMed] [Google Scholar]
- [206].a) Roose T, Netti PA, Munn LL, Boucher Y, Jain RK, Microvasc. Res 2003, 66, 204; [DOI] [PubMed] [Google Scholar]; b) Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E, Cancer Res. 2000, 60, 722. [PMC free article] [PubMed] [Google Scholar]
- [207].Ni D, Zhang J, Bu W, Xing H, Han F, Xiao Q, Yao Z, Chen F, He Q, Liu J, ACS Nano 2014, 8, 1231. [DOI] [PubMed] [Google Scholar]
- [208].Huang N, Cheng S, Zhang X, Tian Q, Pi J, Tang J, Huang Q, Wang F, Chen J, Xie Z, Nanomedicine 2017, 13, 83. [DOI] [PubMed] [Google Scholar]
- [209].Li C, Cao L, Zhang Y, Yi P, Wang M, Tan B, Deng Z, Wu D, Wang Q, Small 2015, 11, 4517. [DOI] [PubMed] [Google Scholar]
- [210].Diaz RJ, McVeigh PZ, O’Reilly MA, Burrell K, Bebenek M, Smith C, Etame AB, Zadeh G, Hynynen K, Wilson BC, Nanomedicine 2014, 10, e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Marie H, Lemaire L, Franconi F, Lajnef S, Frapart YM, Nicolas V, Frébourg G, Trichet M, Ménager C, Lesieur S, Adv. Funct. Mater 2015, 25, 1258. [Google Scholar]
- [212].Zheng M, Ruan S, Liu S, Sun T, Qu D, Zhao H, Xie Z, Gao H, Jing X, Sun Z, ACS Nano 2015, 9, 11455. [DOI] [PubMed] [Google Scholar]
- [213].Xu HL, Yang JJ, ZhuGe DL, Lin MT, Zhu QY, Jin BH, Tong MQ, Shen BX, Xiao J, Zhao YZ, Adv. Healthcare Mater 2018, 7, 1701130. [DOI] [PubMed] [Google Scholar]
- [214].Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY, Nat. Rev. Dis. Primers 2016, 2, 16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Reyners A, Broekman K, Glaudemans A, Brouwers A, Arts H, van der Zee A, de Vries E, Jalving M, Ann. Oncol 2016, 27, i23. [DOI] [PubMed] [Google Scholar]
- [216].van Kruchten M, de Vries EF, Arts HJ, Jager NM, Bongaerts AH, Glaudemans AW, Hollema H, de Vries EG, Hospers GA, Reyners AK, J. Nuclear Med 2015, 56, 50. [DOI] [PubMed] [Google Scholar]
- [217].Da Pieve C, Allott L, Martins CD, Vardon A, Ciobota DM, Kramer-Marek G, Smith G, Bioconjugate Chem. 2016, 27, 1839. [DOI] [PubMed] [Google Scholar]
- [218].Patel NR, Piroyan A, Nack AH, Galati CA, McHugh M, Orosz S, Keeler AW, O’Neal S, Zamboni WC, Davis B, Mol. Pharmaceutics 2016, 13, 1996. [DOI] [PubMed] [Google Scholar]
- [219].Van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W, Sarantopoulos A, De Jong JS, Arts HJ, van der Zee AG, Nat. Med 2011, 17, 1315. [DOI] [PubMed] [Google Scholar]
- [220].Gao Y, Hernandez C, Yuan H-X, Lilly J, Kota P, Zhou H, Wu H, Exner AA, Nanomedicine 2017, 13, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Williams RM, Lee C, Galassi TV, Harvey JD, Leicher R, Sirenko M, Dorso MA, Shah J, Olvera N, Dao F, Sci. Adv 2018, 4, eaaq1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Satpathy M, Wang L, Zielinski R, Qian W, Lipowska M, Capala J, Lee GY, Xu H, Wang YA, Mao H, Small 2014, 10, 544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Kinsella JM, Jimenez RE, Karmali PP, Rush AM, Kotamraju VR, Gianneschi NC, Ruoslahti E, Stupack D, Sailor MJ, Angew. Chem., Int. Ed 2011, 50, 12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Zhang L, Zhou H, Belzile O, Thorpe P, Zhao D, J. Controlled Release 2014, 183, 114. [DOI] [PubMed] [Google Scholar]
- [225].Xi L, Grobmyer SR, Zhou G, Qian W, Yang L, Jiang H, J. Biophotonics 2014, 7, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Das M, Duan W, Sahoo SK, Nanomedicine 2015, 11, 379. [DOI] [PubMed] [Google Scholar]
- [227].Sun X, Gao D, Gao L, Zhang C, Yu X, Jia B, Wang F, Liu Z, Theranostics 2015, 5, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zhong Y, Goltsche K, Cheng L, Xie F, Meng F, Deng C, Zhong Z, Haag R, Biomaterials 2016, 84, 250. [DOI] [PubMed] [Google Scholar]
- [229].Ulaner GA, Hyman DM, Ross DS, Corben A, Chandarlapaty S, Goldfarb S, McArthur H, Erinjeri JP, Solomon SB, Kolb H, J. Nucl. Med 2016, 57, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Jafari A, Salouti M, Shayesteh SF, Heidari Z, Rajabi AB, Boustani K, Nahardani A, Nanotechnology 2015, 26, 075101. [DOI] [PubMed] [Google Scholar]
- [231].Derlin T, Jonigk D, Bauersachs J, Bengel FM, Clin. Nuclear Med 2016, 41, e204. [DOI] [PubMed] [Google Scholar]
- [232].Oh S-G, Lee SB, Ji HD, Lee J-E, Singh TD, Kim SK, Jeong SY, Ahn B-C, Lee J, Lee S-W, J. Nucl. Med 2017, 58, 62.27469362 [Google Scholar]
- [233].Ehlerding EB, England CG, Jiang D, Graves SA, Kang L, Lacognata S, Barnhart TE, Cai W, Mol. Pharmaceutics 2017, 14, 2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Liu F, Liu T, Xu X, Guo X, Li N, Xiong C, Li C, Zhu H, Yang Z, Mol. Pharmaceutics 2018, 15, 619. [DOI] [PubMed] [Google Scholar]
- [235].Kang L, Jiang D, Ehlerding EB, Barnhart TE, Ni D, Engle JW, Wang R, Huang P, Xu X, Cai W, Mol. Pharmaceutics 2018, 15, 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Liu S, Li D, Guo J, Canale N, Li X, Liu R, Krasnoperov V, Gill PS, Conti PS, Shan H, Mol. Pharmaceutics 2014, 11, 3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Berroterán-Infante N, Balber T, Fürlinger P, Bergmann M, Lanzenberger R, Hacker M, Mitterhauser M, Wadsak W, ACS Med. Chem. Lett 2018, 9, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Rezazadeh F, Sadeghzadeh N, Abedi SM, Abediankenari S, Nucl. Med. Biol 2018, 62–63, 54. [DOI] [PubMed] [Google Scholar]
- [239].Li X, An Y, Jin J, Zhu Z, Hao L, Liu L, Shi Y, Fan D, Ji T, Yang CJ, Anal. Chem 2015, 87, 4941. [DOI] [PubMed] [Google Scholar]
- [240].Liang L, Care A, Zhang R, Lu Y, Packer NH, Sunna A, Qian Y, Zvyagin AV, ACS Appl. Mater. Interfaces 2016, 8, 11945. [DOI] [PubMed] [Google Scholar]
- [241].Kim J, Do E.-j., Moinova H, Bae SM, Kang JY, Hong S-M, Fink SP, Joo J, Suh Y-A, Jang SJ, Neoplasia 2017, 19, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Matsuzaki H, Kamiya M, Iwatate RJ, Asanuma D, Watanabe T, Urano Y, Bioconjugate Chem. 2016, 27, 973. [DOI] [PubMed] [Google Scholar]
- [243].Tiernan JP, Ingram N, Marston G, Perry SL, Rushworth JV, Coletta PL, Millner PA, Jayne DG, Hughes TA, Nanomedicine 2015, 10, 1223. [DOI] [PubMed] [Google Scholar]
- [244].Rabinsky EF, Joshi BP, Pant A, Zhou J, Duan X, Smith A, Kuick R, Fan S, Nusrat A, Owens SR, Cell. Mol. Gastroenterol. Hepatol 2016, 2, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Zhou B, Wang H, Liu R, Wang M, Deng H, Giglio BC, Gill PS, Shan H, Li Z, Mol. Pharmaceutics 2015, 12, 3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Rauscher I, Maurer T, Souvatzoglou M, Beer AJ, Vag T, Wirtz M, Weirich G, Wester H-J, Gschwend JE, Schwaiger M, Clinical Nuclear Medicine 2016, 41, e397. [DOI] [PubMed] [Google Scholar]
- [247].Gao X, Cui Y, Levenson RM, Chung LW, Nie S, Nat. Biotechnol 2004, 22, 969. [DOI] [PubMed] [Google Scholar]
- [248].Lee C-M, Jeong H-J, Cheong S-J, Kim E-M, Kim DW, Lim ST, Sohn M-H, Pharm. Res 2010, 27, 712. [DOI] [PubMed] [Google Scholar]
- [249].Lo S-T, Kumar A, Hsieh J-T, Sun X, Mol. Pharmaceutics 2013, 10, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Ghosh D, Lee Y, Thomas S, Kohli AG, Yun DS, Belcher AM, Kelly KA, Nat. Nanotechnol 2012, 7, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Guo L, Shi H, Wu H, Zhang Y, Wang X, Wu D, An L, Yang S, Carbon 2016, 107, 87. [Google Scholar]
- [252].Sun Y, Ma X, Zhang Z, Sun Z, Loft M, Ding B, Liu C, Xu L, Yang M, Jiang Y, Bioconjugate Chem. 2016, 27, 1857. [DOI] [PubMed] [Google Scholar]
- [253].Hong H, Nayak TR, Shi S, Graves SA, Fliss BC, Barnhart TE, Cai W, Mol. Pharmaceutics 2014, 11, 3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Persson M, Juhl K, Rasmussen P, Brandt-Larsen M, Madsen J, Ploug M, Kjaer A, Mol. Pharmaceutics 2014, 11, 2796. [DOI] [PubMed] [Google Scholar]
- [255].Richter S, Wuest M, Bergman CN, Krieger S, Rogers BE, Wuest F, Mol. Pharmaceutics 2016, 13, 1347. [DOI] [PubMed] [Google Scholar]
- [256].Kamerkar S, LeBleu VS, Sugimoto H, Yang S, Ruivo CF, Melo SA, Lee JJ, Kalluri R, Nature 2017, 546, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Montet X, Weissleder R, Josephson L, Bioconjugate Chem. 2006, 17, 905. [DOI] [PubMed] [Google Scholar]
- [258].Huynh AS, Chung WJ, Cho H-I, Moberg VE, Celis E, Morse DL, Vagner J, J. Med. Chem 2012, 55, 9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Qian J, Yong K-T, Roy I, Ohulchanskyy TY, Bergey EJ, Lee HH, Tramposch KM, He S, Maitra A, Prasad PN, J. Phys. Chem. B 2007, 111, 6969. [DOI] [PubMed] [Google Scholar]
- [260].Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M, Nie S, Mao H, Yang L, ACS Nano 2013, 7, 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Amirkhanov NV, Zhang K, Aruva MR, Thakur ML, Wickstrom E, Bioconjugate Chem. 2010, 21, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Wang M, Zhang H, Wang H, Feng H, Deng H, Wu Z, Lu H, Li Z-B, Mol. Pharmaceutics 2018, 15, 975. [DOI] [PubMed] [Google Scholar]
- [263].Houghton JL, Abdel-Atti D, Scholz WW, Lewis JS, Mol. Pharmaceutics 2017, 14, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Zhang D, Wang Z, Wang L, Wang Z, Wang H, Li G, Qiao Z-Y, Xu W, Wang H, ACS Appl. Mater. Interfaces 2018, 10, 28331. [DOI] [PubMed] [Google Scholar]
- [265].Davis RM, Kiss B, Trivedi DR, Metzner TJ, Liao JC, Gambhir SS, ACS Nano 2018, 12, 9669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Yuan R, Rao T, Cheng F, Yu WM, Ruan Y, Zhang XB, Larré S, Exp. Ther. Med 2018, 16, 4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Wang J, Fang R, Wang L, Chen G, Wang H, Wang Z, Zhao D, Pavlov VN, Kabirov I, Wang Z, Cell. Physiol. Biochem 2018, 47, 1565. [DOI] [PubMed] [Google Scholar]
- [268].Allen MD, Luong P, Hudson C, Leyton J, Delage B, Ghazaly E, Cutts R, Yuan M, Syed N, Nigro CL, Cancer Res. 2014, 74, 896. [DOI] [PubMed] [Google Scholar]
- [269].Paquette M, Vilera-Perez L-G, Beaudoin S, Ekindi-Ndongo N, Boudreaut P-L, Bonin M-A, Battista M-C, Bentourkia M. h., Lopez AF, Lecomte R, OncoImmunology 2017, 6, e1331195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Dixit S, Novak T, Miller K, Zhu Y, Kenney ME, Broome A-M, Nanoscale 2015, 7, 1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Patil R, Ljubimov AV, Gangalum PR, Ding H, Portilla-Arias J, Wagner S, Inoue S, Konda B, Rekechenetskiy A, Chesnokova A, ACS Nano 2015, 9, 5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Luo H, Hernandez R, Hong H, Graves SA, Yang Y, England CG, Theuer CP, Nickles RJ, Cai W, Proc. Natl. Acad. Sci. USA 2015, 112, 12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Yan H, Wang L, Wang J, Weng X, Lei H, Wang X, Jiang L, Zhu J, Lu W, Wei X, ACS Nano 2011, 6, 410. [DOI] [PubMed] [Google Scholar]
- [274].Tian T, Li J, Xie C, Sun Y, Lei H, Liu X, Xia J, Shi J, Wang L, Lu W, ACS Appl. Mater. Interfaces 2018, 10, 3414. [DOI] [PubMed] [Google Scholar]
- [275].Hao Y, Wang L, Zhao Y, Meng D, Li D, Li H, Zhang B, Shi J, Zhang H, Zhang Z, Macromol. Biosci 2015, 15, 1571. [DOI] [PubMed] [Google Scholar]
- [276].Hernandez R, Sun H, England CG, Valdovinos HF, Barnhart TE, Yang Y, Cai W, Mol. Pharmaceutics 2016, 13, 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Barua A, Yellapa A, Bahr JM, Adur MK, Utterback CW, Bitterman P, Basu S, Sharma S, Abramowicz JS, Biomed Res. Int 2015, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Barua A, Edassery SL, Bahr JM, Bitterman P, Basu S, Sharma S, Abramowicz JS, Int. J. Gynecol. Cancer 2016, 26, 1375. [DOI] [PubMed] [Google Scholar]
- [279].Lutz AM, Bachaval SV, Drescher CW, Pysz MA, Willmann JK, Gambhir SS, Clin. Cancer Res 2014, 20, 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Munnink THO, de Korte MA, Nagengast WB, Timmer-Bosscha H, Schröder CP, de Jong JR, van Dongen GA, Jensen MR, Quadt C, Lub-de Hooge MN, Eur. J. Cancer 2010, 46, 678. [DOI] [PubMed] [Google Scholar]
- [281].Bensch F, Lamberts LE, Lub-de Hooge MN, Terwisscha van Scheltinga AG, de Jong JR, Gietema JA, Schroder CP, Smeenk MM, Jacob W, Abiraj K, J. Clin. Oncol 2014, 32, 11095. [Google Scholar]
- [282].Prantner AM, Yin C, Kamat K, Sharma K, Lowenthal AC, Madrid PB, Scholler N, Mol. Pharmaceutics 2018, 15, 1403. [DOI] [PubMed] [Google Scholar]
- [283].Guerrero Y, Bahmani B, Jung B, Vullev V, Kundra V, Anvari B, Proc. SPIE 2013, 8568, 856819. [Google Scholar]
- [284].Perrone MG, Malerba P, Uddin MJ, Vitale P, Panella A, Crews BC, Daniel CK, Ghebreselasie K, Nickels M, Tantawy MN, Eur. J. Med. Chem 2014, 80, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Makvandi M, Pantel A, Schwartz L, Schubert E, Xu K, Hsieh C-J, Hou C, Kim H, Weng C-C, Winters H, J. Clin. Invest 2018, 128, 2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].ter Weele EJ, van Scheltinga AGT, Linssen MD, Nagengast WB, Lindner I, Jorritsma-Smit A, de Vries EG, Kosterink JG, Lub-de Hooge MN, Eur. J. Pharm. Biopharm 2016, 104, 226. [DOI] [PubMed] [Google Scholar]
- [287].Benezra M, Penate-Medina O, Zanzonico PB, Schaer D, Ow H, Burns A, DeStanchina E, Longo V, Herz E, Iyer S, J. Clin. Invest 2011, 121, 2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Willmann JK, Bonomo L, Carla AT, Rinaldi P, Rindi G, Valluru KS, Petrone G, Martini M, Lutz AM, Gambhir SS, J. Clin. Oncol 2017, 35, 2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Luo H, England CG, Goel S, Graves SA, Ai F, Liu B, Theuer CP, Wong HC, Nickles RJ, Cai W, Mol. Pharmaceutics 2017, 14, 1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Dianat-Moghadam H, Heidarifard M, Jahanban-Esfahlan R, Panahi Y, Hamishehkar H, Pouremamali F, Rahbarghazi R, Nouri M, J. Controlled Release 2018, 288, 62. [DOI] [PubMed] [Google Scholar]
- [291].Liu X, Wu L, Wang L, Jiang W, Talanta 2018, 179, 356. [DOI] [PubMed] [Google Scholar]
- [292].Crisp JL, Savariar EN, Glasgow HL, Ellies LG, Whitney MA, Tsien RY, Mol. Cancer Ther 2014, 13, 1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Du J, Li X-Y, Hu H, Xu L, Yang S-P, Li F-H, Sci. Rep 2018, 8, 3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Fang RH, Jiang Y, Fang JC, Zhang L, Biomaterials 2017, 128, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].a) Hu C-MJ, Fang RH, Luk BT, Chen KN, Carpenter C, Gao W, Zhang K, Zhang L, Nanoscale 2013, 5, 2664; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li R, He Y, Zhang S, Qin J, Wang J, Acta Pharm. Sin. B 2017, 7, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Chai Z, Hu X, Wei X, Zhan C, Lu L, Jiang K, Su B, Ruan H, Ran D, Fang RH, J. Controlled Release 2017, 264, 102. [DOI] [PubMed] [Google Scholar]
- [297].Fang RH, Hu C-MJ, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L, Nano Lett. 2014, 14, 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Rao L, Bu LL, Cai B, Xu JH, Li A, Zhang WF, Sun ZJ, Guo SS, Liu W, Wang TH, Adv. Mater 2016, 28, 3460. [DOI] [PubMed] [Google Scholar]
- [299].Meng Q-F, Rao L, Zan M, Chen M, Yu G-T, Wei X, Wu Z, Sun Y, Guo S-S, Zhao X-Z, Nanotechnology 2018, 29, 134004. [DOI] [PubMed] [Google Scholar]
- [300].Huang Y, Mei C, Tian Y, Nie T, Liu Z, Chen T, NPG Asia Mater. 2018, 10, 1002. [Google Scholar]
- [301].Li J, Zhen X, Lyu Y, Jiang Y, Huang J, Pu K, ACS Nano 2018, 12, 8520. [DOI] [PubMed] [Google Scholar]
- [302].Mu X, Li J, Yan S, Zhang H, Zhang W, Zhang F, Jiang J, ACS Biomater. Sci. Eng 2018. [DOI] [PubMed] [Google Scholar]
- [303].a) Li W, Li C, Zhou T, Liu X, Liu X, Li X, Chen D, Mol. Cancer 2017, 16, 145; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D, Cancer Cell 2016, 30, 836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Sun Y-Z, Ruan J-S, Jiang Z-S, Wang L, Wang S-M, Biomed Res. Int 2018, 2018, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Gangadaran P, Hong CM, Ahn B-C, Front. Pharmacol 2018, 9, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].JC Bose R, Uday Kumar S, Zeng Y, Afjei R, Robinson E, Lau K, Bermudez A, Habte F, Pitteri SJ, Sinclair R, ACS Nano 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].JC Bose R, Uday Kumar S, Zeng Y, Afjei R, Robinson E, Lau K, Bermudez A, Habte F, Pitteri SJ, Sinclair R, ACS Nano 2018, 12, 10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Gangadaran P, Hong CM, Ahn B-C, Biomed Res. Int 2017, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Hyenne V, Lefebvre O, Goetz JG, Cell Adhes. Migr 2017, 11, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Han X, Wang J, Sun Y, Genomics, Proteomics Bioinf. 2017, 15, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Lu T, Li J, Am. J. Cancer Res 2017, 7, 2318. [PMC free article] [PubMed] [Google Scholar]
- [312].De Mattos-Arruda L, Cortes J, Santarpia L, Vivancos A, Tabernero J, Reis-Filho JS, Seoane J, Nat. Rev. Clin. Oncol 2013, 10, 377. [DOI] [PubMed] [Google Scholar]
- [313].Ilie M, Hofman V, Long E, Bordone O, Selva E, Washetine K, Marquette CH, Hofman P, Ann. Transl. Med 2014, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW, Nucleic Acids Res. 2000, 28, 32e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Bailey VJ, Keeley BP, Zhang Y, Ho YP, Easwaran H, Brock MV, Pelosky KL, Carraway HE, Baylin SB, Herman JG, ChemBioChem 2010, 11, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Batlle E, Clevers H, Nat. Med 2017, 23, 1124. [DOI] [PubMed] [Google Scholar]
- [317].Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP, Nat. Rev. Clin. Oncol 2015, 12, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Azhdarinia A, Voss J, Ghosh SC, Simien JA, Hernandez Vargas S, Cui J, Yu WA, Liu Q, Carmon KS, Mol. Pharmaceutics 2018, 15, 2448. [DOI] [PubMed] [Google Scholar]
- [319].Tang J, Zhou H, Liu J, Liu J, Li W, Wang Y, Hu F, Huo Q, Li J, Liu Y, ACS Appl. Mater. Interfaces 2017, 9, 23497. [DOI] [PubMed] [Google Scholar]
- [320].Liu H, Patel MR, Prescher JA, Patsialou A, Qian D, Lin J, Wen S, Chang Y-F, Bachmann MH, Shimono Y, Proc. Natl. Acad. Sci. USA 2010, 201006732. [Google Scholar]
- [321].Vlashi E, Kim K, Lagadec C, Donna LD, McDonald JT, Eghbali M, Sayre JW, Stefani E, McBride W, Pajonk F, JNCI, J. Natl. Cancer Inst 2009, 101, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]