

Abstract
Background
The cardiac ryanodine receptor type 2 (RyR2) is a large homotetramer, located in the sarcoplasmic reticulum (SR), which releases Ca2+ from the SR during systole. The molecular mechanism underlying Ca2+ sensing and gating of the RyR2 channel in health and disease is only partially elucidated. Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT1) is the most prevalent syndrome caused by RyR2 mutations.
Methods and Results
This study involves investigation of a family with 4 cases of ventricular fibrillation and sudden death and physiological tests in HEK 293 cells and normal mode analysis (NMA) computation. We found 4 clinically affected members who were homozygous for a novel RyR2 mutation, G3118R, whereas their heterozygous relatives are asymptomatic. G3118R is located in the periphery of the protein, far from the mutation hotspot regions. HEK293 cells harboring G3118R mutation inhibited Ca2+ release in response to increasing doses of caffeine, but decreased the termination threshold for store‐overload‐induced Ca2+ release, thus increasing the fractional Ca2+ release in response to increasing extracellular Ca2+. NMA showed that G3118 affects RyR2 tetramer in a dose‐dependent manner, whereas in the model of homozygous mutant RyR2, the highest entropic values are assigned to the pore and the central regions of the protein.
Conclusions
RyR2 G3118R is related to ventricular fibrillation and sudden death in recessive mode of inheritance and has an effect of gain of function on the protein. Despite a peripheral location, it has an allosteric effect on the stability of central and pore regions in a dose‐effect manner.
Keywords: normal mode analysis, ryanodine receptor type 2, sudden death, ventricular fibrillation
Subject Categories: Basic Science Research, Computational Biology, Calcium Cycling/Excitation-Contraction Coupling, Ion Channels/Membrane Transport, Physiology
Nonstandard Abbreviations and Acronyms
- NMA
normal mode analysis
- RyR2
ryanodine receptor type 2
- SOICR
store‐overload‐induced Ca2+ release
Clinical Perspective
What Is New?
The RyR2 novel mutation G3118R is related to stress‐induced autosomal recessive ventricular fibrillation and sudden death.
Despite a remote location, this mutation has a dose‐dependent allosteric effect on the pore and central region of the protein complex.
What Are the Clinical Implications?
The phenotype is life threatening in homozygotes; thus, carriers should be considered at low risk.
Prenatal evaluation in heterozygous couples is recommended.
Normal mode analysis may serve as a novel platform for the evaluation of mutations pathogenicity, in particular in large protein complexes such as RyR2, whereas other computational methods could be challenging to apply.
The cardiac ryanodine receptor type 2 (RyR2) is a large homotetramer, highly conserved ion channel that releases Ca2+ from the sarcoplasmic reticulum into the cytoplasm, which is needed for muscle contraction. 1 , 2 In 2001, dominant mutations in this protein were found to be related to catecholaminergic polymorphic ventricular tachycardia 1, a syndrome characterized by polymorphic ventricular tachycardia triggered by exertion or emotional stress. 2 In recent years, the spectrum of the disease associated with RyR2 mutations was markedly enlarged. 3 Disease‐causing RyR2 variants are clustered into 4 regions in the protein's linear sequence. 1 , 3 These are the N‐terminal region, which forms the gating ring at the cytosolic side, the pore region, and other transmembrane‐forming regions, including the C‐terminal domain. Most of the disease causing RyR2 mutations cause gain of function of the channel, by which Ca2+ release is enhanced during diastole, causing delayed afterdepolarizations and arrhythmias. Up until now, only autosomal dominant inheritance of the phenotypes caused by these mutations was described.
In this study, we describe a novel missense mutation, G3118R, in the RyR2 protein, which was found in members of a large family from a village near Jerusalem, Israel presenting with sudden death and documented ventricular fibrillation. Interestingly, the location of this mutation is within the far cytosolic region of the protein. The clinically affected individuals are homozygous for the mutation, whereas the heterozygous family members are asymptomatic. This unusual clinical scenario had prompted us to perform in vitro functional studies to determine the effect of the mutation on the protein function as well as computational analysis to simulate the dose effect of the mutation on RyR2 protein stability.
Methods
The data that support the findings of this study will be made available by the corresponding author to any researcher upon reasonable request.
Clinical Evaluation
Clinical assessments of the affected individuals and first‐degree relatives included resting ECGs, stress tests, 24‐hour Holters, and echocardiography. Historical data were collected during clinic visits, and a 5‐generation family tree was created to relate the families from 2 nearby villages.
Genetic Testing
Informed consent was obtained from the family in accordance with the Declaration of Helsinki as approved by our local institutional review board. Exonic sequences from DNA of proband IV2 (Figure 1A) were enriched with the SureSelect Human All Exon 50 Mb V5 Kit (Agilent Technologies, Santa Clara, CA). Sequences were generated on a HiSeq2500 (Illumina, San Diego, CA) as 125‐bp paired‐end runs. Read alignment and variant calling were performed with DNAnexus (Palo Alto, CA) by using default parameters with the human genome assembly hg19 (GRCh37) as reference. Exome analysis of the proband yielded a mean coverage of 96×. For the family members, amplicons containing the RyR2 variant were amplified by conventional polymerase chain reaction of genomic DNA, and analyzed by Sanger dideoxy nucleotide sequencing.
Figure 1. Family pedigree and clinical expression of G3118R mutation.
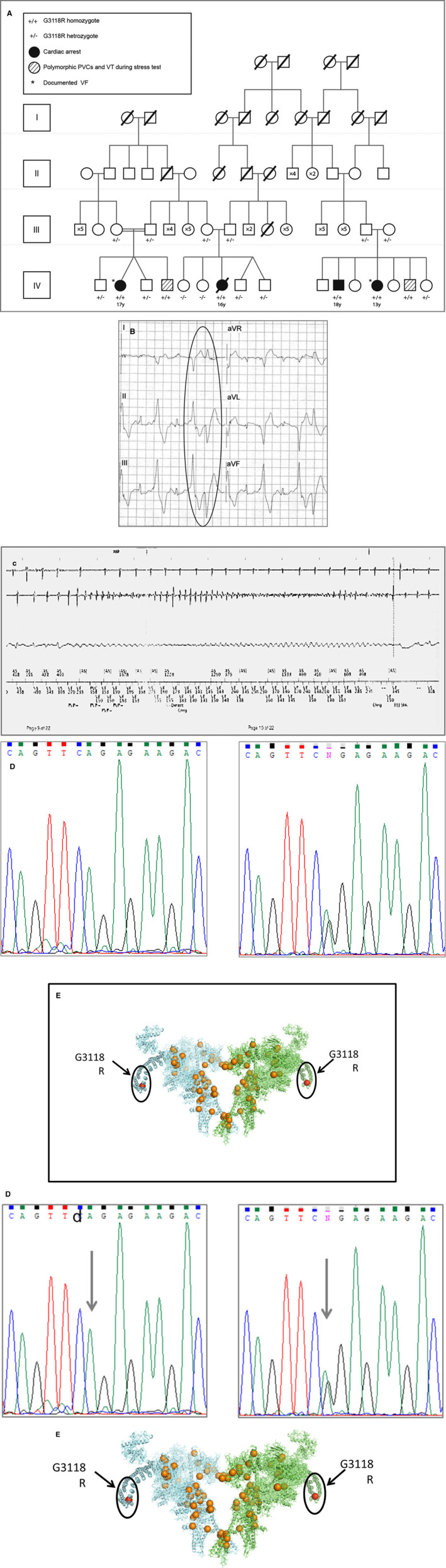
A, Family pedigree. B, Exercise‐induced ventricular bigeminy and bidirectional couplet of patient IV1. aVR, augmented Vector Right; aVL, augmented Vector Left; and aVF, augmented Vector Foot. C, Initiation of VF of patient IV13. D, Chromatograms of homozygous patient IV13 (left) showing adenine replacing guanine in position 237863752 of chromosome 1 in both alleles (arrow) and her heterozygous father (right). E, RyR2 three‐dimensional reconstruction, with G3118R amino acid region circled. PVCs, premature ventricular contractions; VF, ventricular fibrillation; and VT, ventricular tachycardia.
Two‐point logarithm of the odds–score analysis was performed using the Technion developed Superlink web‐based linkage analysis software (http://cbl‐link02.cs.technion.ac.il/superlinkattechnion/) under the following assumption: recessive mode of inheritance with a minor allele frequency of 0.00001 and 100% penetrance.
Construction of the RyR2‐G3118R Mutation
The G3118R point mutation in the mouse RyR2 was generated by using the overlap extension method with polymerase chain reaction. 4 , 5 Briefly, a BsiwI/AfeI fragment containing the G3118R mutation was generated by overlapping polymerase chain reaction. This fragment was then used to replace the corresponding wild‐type (WT) fragment in the BsiwI/NotI construct in pBluescript. The BsiwI/NotI construct containing G3118R was then subcloned to the full‐length RyR2 in pcDNA5/FRT/TO using BsiWI and NotI. The mutation G3118R was confirmed by DNA sequencing.
Generation of Stable Inducible HEK293 Cell Lines and Cell Culture
Stable inducible HEK293 cell lines expressing RyR2 WT or the G3118R mutant were generated using the Flp‐In T‐REx Core Kit from Invitrogen (Carlsbad, CA) as previously described. 6 , 7
[3H]Ryanodine Binding
[3H]Ryanodine binding to cell lysates prepared from HEK293 cells expressing the RyR2 WT or the RyR2‐G3118R mutant was carried out as previously described. 8
Caffeine‐Induced Ca2+ Release in HEK293 Cells
The free cytosolic Ca2+ concentration in transfected HEK293 cells was measured using the fluorescence Ca2+ indicator dye Fluo‐3 (Molecular Probes, Eugene, OR) as previously described. 7 , 9
Single‐Cell Luminal Ca2+ Imaging of HEK293 Cells
Luminal Ca2+ levels in HEK293 cells expressing RyR2‐WT or G3118R were measured using single‐cell Ca2+ imaging and the Förster resonance energy transfer–based endoplasmic reticulum (ER) luminal Ca2+‐sensitive cameleon protein D1ER as described previously. 10
Statistical Analysis
All values shown are mean±SEM unless indicated otherwise. To test for differences between groups, we used the Student t test (2‐tailed) or 1‐way ANOVA with a Dunnett's post hoc test. A P<0.05 was considered to be statistically significant.
Simulation of the Effect of Increasing Number of Affected RyR2 Monomers on the Protein Stability
To determine the protein mutation location within the three‐dimensional structure of the RyR2 protein, we used the tetrameric structure of the porcine RyR2 in the open and closed state, solved by cryo‐electron microscopy at 0.42 nm resolution recently published by Peng et al. 11 In the porcine protein, glycine residue in position 3119 is equivalent to the human 3118 residue. To assess the effect of G3118R mutation on the stability and the flexibility of the RyR2 protein, we performed a coarse‐grained normal mode (NMA) analysis on the complete tetramer RyR2 structure. To model the recessive mode of inheritance of our novel mutation, we generated 5 different RyR2 structures, starting from WT and proceeding to the tetramer bearing 1 to 4 mutated monomers. Each structure was based on the open state of the RyR2 receptor (Protein Data Bank accession number 5GOA), 11 and it generated a series of normal modes on which entropic differences calculations on C‐alpha atoms were performed according to the ENCoM coarse‐grained NMA method. 12 Course graining included calculations on each eighth residue; comparison with each fourth and each second residue for several experiments yielded similar mutant differences. The pore region of the tetramer is defined as 4486 to 4968, central region as 3613 to 4207, and N‐terminal domain region as 1 to 642. To cover the large domain between the N‐terminal domain and the central region, the calculations were also applied on the 643 to 2110 residues.
Results
Family Investigation
Between 2007 and 2017, 4 cases of cardiac arrest in young individuals were identified in a large family from a village near Jerusalem, Israel (Figure 1A), out of which 3 survived the event. One out of 4 resulted in the death of a 16‐year‐old girl while she was on a giant wheel in an amusement park (Figure 1A, patient IV7). The other three were aborted cardiac arrest cases. The clinical scenarios were of an 18‐year‐old man while helping friends to push a car (patient IV11), his 13‐year‐old sister immediately upon hearing about her brother's cardiac arrest (patient IV13), and a 17‐year‐old girl who had cardiac arrest while descending from a school bus (patient IV2) and was shocked by an automatic external defibrillator, which documented ventricular fibrillation. All survivors received an implantable cardiac defibrillator, and therapy with a beta blocker, metoprolol, was initiated. Resting ECG and echocardiography were normal in all clinically affected members; however, their exertional stress test demonstrated ventricular arrhythmia in the form of ventricular bigeminy and bidirectional couplets, which appeared at a rate of ≈100 beats per minute, increasing in frequency with increasing heart rate (Figure 1B).
In a cascade family clinical investigation, we identified 2 other siblings (patients IV4 and IV15) who had multifocal premature ventricular contractions and bidirectional couplets during stress tests. The parents of the affected individuals were asymptomatic, had normal baseline ECGs, echocardiography, and 24‐hour Holters. The father of patient IV3 had ventricular bigeminy and a single bidirectional couplet during his exertional stress test . Other parents' exertional stress tests were unremarkable.
During 9 years of follow up, patient IV13 had a breakthrough episode of ventricular fibrillation that terminated with shocks from her implantable cardiac defibrillator (Figure 1C). At this point, the dose of metoprolol was increased, after which no further ventricular fibrillation episodes occurred. This patient had atrial fibrillation episodes as well.
Genetic Analysis
Genetic analysis was performed for the clinically affected individuals and their family members. The clinically affected members were found homozygous for a missense mutation in exon 65 of the RyR2 gene, in which guanine was substituted for adenine at the base position 237863752 of the coding sequence (Chr1: 237863752G>A ‐Hg19‐) (Figure 1D). As a result of the mutation, glycine to arginine substitution occurred in residue 3118 of the RyR2 protein (p.G3118R_RyR2, NP_001026.2). The G3118 residue is conserved among species. The variant allele frequency is 0.00004293 according to the Genome Aggregation Database, and it is classified as a variant of unknown significance according to the American College of Medical Genetics and Genomics, and the Association for Molecular Pathology 2015 guidelines. 13 The heterozygous parents and siblings were asymptomatic, although one parent exhibited rare but typical arrhythmia. The 2‐point logarithm of the odds score for this family was 2.2476.
Residue G3118 is located within the cytosolic component of the RyR2 protein, far from areas where frequent cluster mutations were described (Figure 1E). Thus, we undertook studies in vitro to investigate the effect of this mutation on the protein function.
Functional Analysis of G3118R
[3H]ryanodine Binding
The effect of G3118R on cytosolic Ca2+ activation of RyR2 was investigated by the Ca2+‐dependent activation of [3H]Ryanodine binding in WT and the mutant. The results showed no difference between WT and the G3118 (Figure 2).
Figure 2. Effect of the RyR2‐G3118R mutation on the Ca2+ dependence of [3H]ryanodine binding.
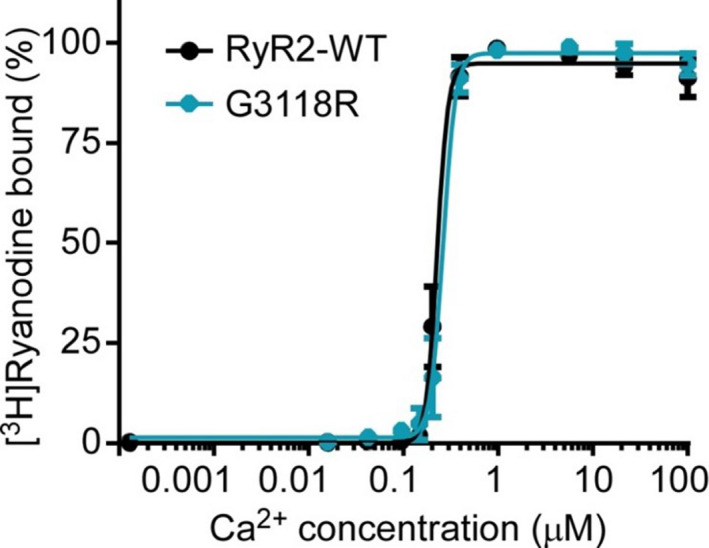
[3H]ryanodine binding to whole‐cell lysates prepared from HEK293 cells transfected with the RyR2‐WT or the RyR2‐G3118R mutant cDNA was performed using various Ca2+ concentrations, 500 mmol/L KCl, and 5 nmol/L [3H]ryanodine. The amounts of [3H]ryanodine bound at different Ca2+ concentrations were normalized to the maximal binding. Data were analyzed using the Student t test (2‐tailed) and expressed as mean±SEM (n=3). WT, wild‐type.
Caffeine Assay
Changes in intracellular Ca2+ levels in response to incremental application of caffeine were compared between the 3 cell lines by measuring peak fluorescence amplitudes after each increment normalized to the peak amplitude for maximal Ca2+ release induced in each experiment (wt n=7, G3118R n=9 and RyR2‐WT/G3118R [n=7]). As shown in Figure 3, the level of Ca2+ release in HEK293 cells transfected with both RyR2‐WT and RyR2‐G3118R increased progressively with each consecutive addition of caffeine and then decreased with further additions of caffeine. HEK293 cells harboring G3118R mutation showed a slightly suppressed Ca2+ release response to caffeine compared to WT/G3118R and to RyR2‐WT (Figure 3D).
Figure 3. Effect of RyR2‐G3118R on caffeine‐induced Ca2+ release.
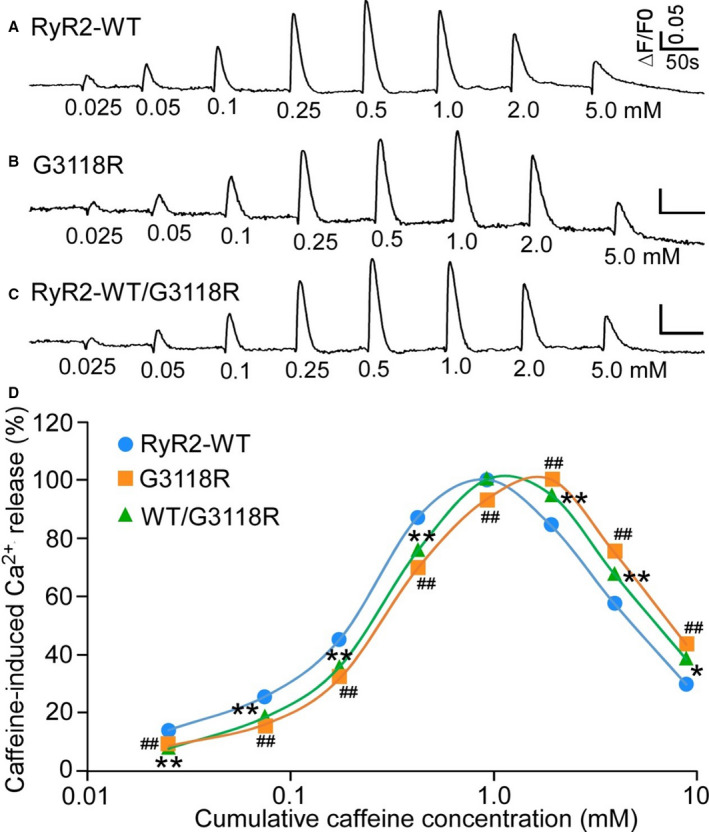
HEK293 cells were transfected with RyR2 WT (A) or G3118R (B), or cotransfected with RyR2‐WT and G3118R (C). The fluorescence intensity of the fluo‐3‐loaded transfected cells was monitored continuously before and after each caffeine addition. The amplitude of each caffeine peak was normalized to that of the maximum peak for each experiment. D, The relationships between caffeine‐induced Ca2+ release and cumulative caffeine concentrations in HEK293 cells transfected with RyR2‐WT or G3118R, or cotransfected with RyR2‐WT/G3118R. Data were calculated using 1‐way ANOVA with a Dunnett's post hoc test, expressed as mean±SEM from RyR2 WT (n=7), G3118R (n=9), and RyR2‐WT/G3118R (n=7). (*P<0.05, **P<0.01, ## P<0.01, vs WT). WT, wild‐type.
Effect of G3118R Mutation on Store‐Overload‐Induced Ca2+ Release
The effect of G3118 on the store‐overload‐induced Ca2+ oscillations or Ca2+ waves, a well described property of the RyR2 channel, 14 were studied. ER Ca2+ dynamics were monitored using a Förster resonance energy transfer–based ER luminal Ca2+‐sensing protein D1ER. Increasing extracellular Ca2+ from 0 to 2 mmol/L induced spontaneous ER Ca2+ oscillations in RyR2 WT‐expressing HEK‐293 cells (Figure 4, shown as downward deflections of the Förster resonance energy transfer signal). Store‐overload‐induced Ca2+ release (SOICR) occurred when the ER luminal Ca2+ reached the SOICR activation threshold, (F act) and terminated when the ER luminal Ca2+ decreased to the SOICR termination threshold, (F term). The activation and termination thresholds were calculated as described in Figure 4A. Fractional Ca2+ release (activation threshold–termination threshold) represents the fraction of Ca2+ release during SOICR. The termination threshold for Ca2+ release is significantly reduced in G3118R‐expressing HEK‐293 cells, which resulted in a higher fractional Ca2+ release than G3118R (Figure 4D and 4E). These results are consistent with gain of function of the mutated channel.
Figure 4. The RyR2‐G3118R mutation impairs the termination of Ca2+ release.
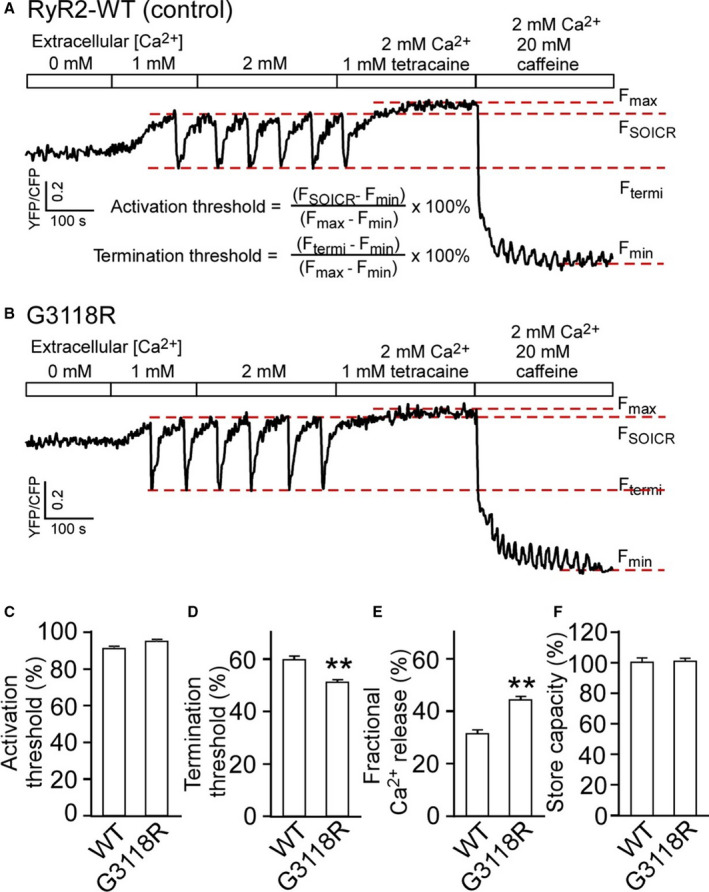
Stable, inducible HEK293 cell lines expressing RyR2 WT and G3118R were transfected with the FRET‐based endoplasmic reticulum luminal Ca2+‐sensing protein D1ER and induced using tetracycline. FRET recordings from representative RyR2‐WT (A) and G3118R (B) expressing cells are shown. FSOICR indicates the FRET level at which SOICR occurs, and Ftermi represents the FRET level at which SOICR terminates. The maximum FRET signal Fmax is defined as the FRET level after tetracaine treatment. The minimum FRET signal Fmin is defined as the FRET level after caffeine treatment. C and D, The activation and termination thresholds were determined as shown in (A). E, The fractional Ca2+ release is defined as activation threshold–termination threshold. F, The store capacity was calculated by subtracting Fmin from Fmax. Data were analyzed using the Student t test (2‐tailed) and expressed as mean±SEM from RyR2‐WT (n=5) and G3118R (n=4). (**P<0.01, vs WT). FRET, Förster resonance energy transfer; SOICR, store‐overload‐induced Ca2+ release; and WT, wild‐type.
Structural Analysis of the RyR2 Protein
To assess the effect of G3118R mutation on the stability and the flexibility of RyR2 protein we performed a coarse‐grained NMA on the complete tetramer RyR2 structure, based on a published structure of the porcine RyR2. 11 To model the recessive mode of inheritance of G3118R novel mutation, 5 different RyR2 structures were generated, starting from WT and proceeding to the tetramer bearing 1 to 4 mutated monomers. G3118R mutation affects the protein structure in a dose‐dependent manner (Figure 5). In the most critical pore region, mutation in only one monomer suffices to change the protein flexibility from the WT values, and this effect was found for each of the additional mutations up to 4 mutated monomers (P<0.01 for each addition, Figure 5A). Slightly less dramatic effect is seen in the broader central region, in which statistical significance has been shown to be between 1 to 3, 2 to 4, and 1 to 4 mutated monomers (P<0.01 for each, Figure 5B), suggesting that 2 additional mutations are needed for the significant difference in the protein entropy values. In more peripheral domains of the tetramer, RyR2 bearing 1 mutation is significantly different from RyR2 bearing 4 (P<0.01, Figure 5C and 5D). These results indicate that G3118R mutation affects the protein flexibility in an allosteric and dose‐dependent manner. Visualization of the mutant tetramer is shown in Figure 6. The pore and the central region present high entropic values due to the G3118 mutation in a model of homozygous state of all four monomers bearing the G3118R substitution. These data indicate that the novel G3118R mutation in the RyR2 channel has a significant effect on the protein structure, which accumulates in a dose‐dependent manner. Despite the peripheral location of the mutation, its structural impact is allosteric, affecting mostly the central and the crucial part of RyR2 tetramer, the Ca2+ pore.
Figure 5. Mean entropy difference of G3118R mutants.
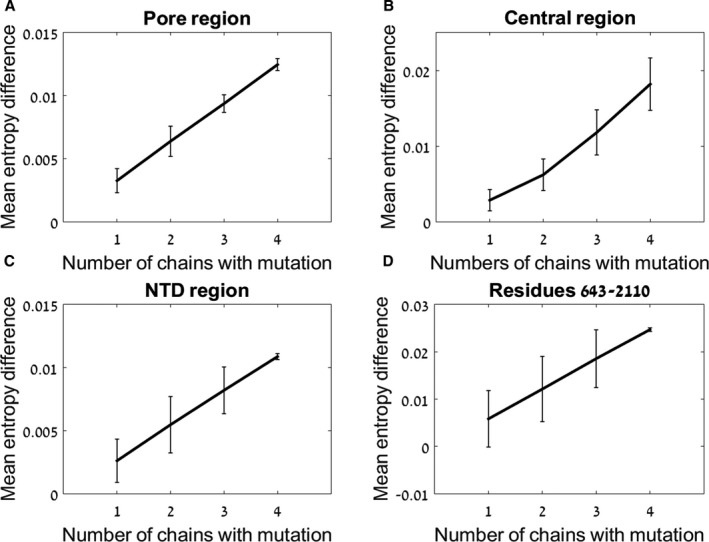
The effect of 1 to 4 G3118R mutated monomers on protein flexibility is shown. The values are presented as mutant‐WT on the ordinate. The number of mutated monomers is shown on the abscissa. The differences in the pore region (A), central region (B), NTD region (C), and region of 643 to 2110 (D) are shown. NTD, N‐terminal domain; and WT, wild‐type.
Figure 6. Structural flexibility of G3118R mutant showing the effect of 4 mutated monomers on protein flexibility based on entropic calculations.
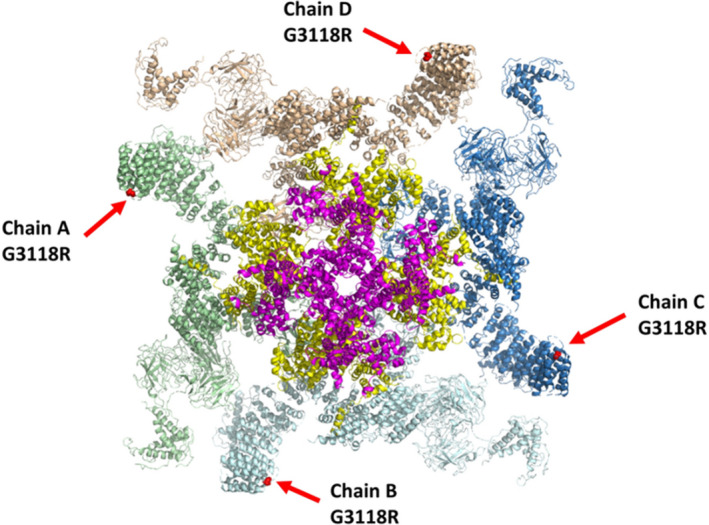
The four monomers are shown in different colors (pale green, light blue, dark blue, white). G3118R mutation location is shown as a red dot pointed at with a red arrow. Residues of the pore region with highest entropic values are colored pink, and residues of the central region with highest entropic values are colored yellow. The numbering is according to the human protein.
Discussion
In the present study we report for the first time the clinical phenotypes and cellular physiology associated with the novel RyR2‐G3118R mutation in a large family, with 4 members who presented with sudden death, ventricular and atrial fibrillation, and 2 more with exercise‐induced polymorphic premature ventricular contractions and bidirectional ventricular couplets. Interestingly, and unlike other RyR2 disease‐causing mutations, all of the clinically affected members are homozygous for the mutation, whereas the heterozygous members are asymptomatic. This may indicate that the effect of this mutation on the channel function is milder than other mutations' and requires that all 4 monomers of RyR2 tetramer are mutated. The structural analysis of the mutant RyR2 model supports this assumption.
More than 170 RyR2 disease‐causing mutations have been published. 15 These are mostly clustered into 4 regions in the protein sequence, 1 and typically cause gain of function of the protein by diastolic Ca2+ leakage from the sarcoplasmic reticulum, which leads to cytosolic Ca2+ overload, driving the membranous sodium–calcium exchanger, NCX, and creating delayed afterdepolarizations, triggered activity, and ventricular arrhythmias. 1 Several mechanisms have been proposed to underlie the gain of function effect of catecholaminergic polymorphic ventricular tachycardia 1–related mutations. These include a decrease of the channel affinity to the accessory protein FKBP1B, a protein that stabilizes the closed state of the channel, thus increasing RyR2 sensitivity to cytosolic Ca2+ after protein kinase A phosphorylation 16 , 17 modification of SOICR, so that the channel opens at a reduced level of luminal Ca2+ 14 , 18 and profound disruption in the three‐dimensional conformation of RyR2, 19 such as domain unzipping or reduced stickiness of RyR2 domains, which enhances Ca2+ sensitivity, thus facilitating spontaneous Ca2+ release. 20 , 21 The mechanism by which G3118R mutation affects the protein function might be different, because it is located outside cluster mutations regions. G3118R mutation mildly inhibited Ca2+ release in response to the administration of increasing doses of caffeine, a known agonist of RyR2, in HEK293 cells, but decreased Ca2+ SOICR termination threshold, thus increasing the fractional Ca2+ release in response to increasing extracellular Ca2+. Ca2+ release termination threshold was previously described to be modulated by calmodulin binding to RyR2, 22 and by one of NH2‐terminal domains, 23 but is probably not relevant to G3118R, which is located outside the calmodulin binding domains. The slightly suppressed caffeine response might be due to the delayed termination of Ca2+ release, so that the maximum peak of caffeine‐induced Ca2+ release is shifted to the right or requires higher caffeine concentration.
To better understand the effect of this mutation on protein structure and function, we studied the changes in the stability and flexibility of RyR2 by using the NMA computational model. Flexibility is important to protein function; it has been described to be involved in protein folding and allosteric interactions. 24 , 25 , 26 , 27 The NMA method can be used to sample the ensemble of accessible conformations of a given protein, which results from its flexibility. 12 , 28 Thus, it would be a method of choice, especially when applied to large protein complexes, such as RyR2. The use of coarse‐graining, based on the protein structure solved by cryo‐electron microscopy at a resolution of 0.42 nm is also acceptable, because vibrational protein modes usually have a highly collective characteristics. 29 Inserting G3118R into an increasing number of the protein monomers has demonstrated that this peripheral mutation, located in a region whose function is unknown, causes a major allosteric effect on the pore and the central regions, and that this effect accumulates in a dose‐dependent manner. The structural model and calculations support the recessive mode of inheritance of the novel G3118R mutation, suggesting a structural mechanism of cumulative effect. In this study, we limited ourselves to describing structural differences between protein mutants. In the future we aim to focus on protein dynamics and larger rearrangements, such as opening and closing the Ca2+ channel. For this purpose, additional methods, such as NMA combined with elastic networks 30 , 31 , 32 or molecular dynamics–based computational electrophysiology, 33 , 34 may be required.
conclusions
RyR2 G3118R is a novel mutation located far from the known cluster mutation regions. It is related to sudden cardiac death and atrial and ventricular fibrillation, and the clinical expression is evident only in homozygotes. The mutation causes gain of function of the protein by reducing the Ca2+‐dependent termination of Ca2+ release, thus increasing the fraction of Ca2+ released from the sarcoplasmic reticulum. Computational analysis of the stability and the flexibility of RyR2 protein demonstrates a major allosteric effect of G3118R mutation on the pore region, which accumulates in a dose‐dependent manner.
Sources of Funding
None.
Disclosures
None.
(J Am Heart Assoc.2021;10:e017128. DOI: 10.1161/JAHA.120.017128.)
For Sources of Funding and Disclosures, see page 11.
References
- 1. Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. [DOI] [PubMed] [Google Scholar]
- 3. Roston TM, Yuchi Z, Kannankeril PJ, Hathaway J, Vinocur JM, Etheridge SP, Potts JE, Maginot KR, Salerno JC, Cohen MI, et al. The clinical and genetic spectrum of catecholaminergic polymorphic ventricular tachycardia: findings from an international multicentre registry. Europace. 2018;20:541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site‐directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. [DOI] [PubMed] [Google Scholar]
- 5. Zhao M, Li P, Li X, Zhang L, Winkfein RJ, Chen SR. Molecular identification of the ryanodine receptor pore‐forming segment. J Biol Chem. 1999;274:25971–25974. [DOI] [PubMed] [Google Scholar]
- 6. Liu Y, Wei J, Wong King Yuen SM, Sun B, Tang Y, Wang R, Van Petegem F, Chen SRW. CPVT‐associated cardiac ryanodine receptor mutation G357S with reduced penetrance impairs Ca2+ release termination and diminishes protein expression. PLoS One. 2017;12:e0184177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y, Zhou Q, Xie C, Zhang J, Guo A, et al. The ryanodine receptor store‐sensing gate controls Ca2+ waves and Ca2+‐triggered arrhythmias. Nat Med. 2014;20:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li P, Chen SR. Molecular basis of Ca(2)+ activation of the mouse cardiac Ca(2)+ release channel (ryanodine receptor). J Gen Physiol. 2001;118:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lieve KVV, Verhagen JMA, Wei J, Bos JM, van der Werf C, Rosés I Noguer F, Mancini GMS, Guo W, Wang R, van den Heuvel F, et al. Linking the heart and the brain: neurodevelopmental disorders in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2019;16:220–228. [DOI] [PubMed] [Google Scholar]
- 10. Jones PP, Jiang D, Bolstad J, Hunt DJ, Zhang L, Demaurex N, Chen SR. Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store‐overload‐induced Ca2+ release. Biochem J. 2008;412:171–178. [DOI] [PubMed] [Google Scholar]
- 11. Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SR, Yan N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016;354:312–323. DOI: 10.1126/science.aah5324. [DOI] [PubMed] [Google Scholar]
- 12. Frappier V, Chartier M, Najmanovich RJ. ENCoM server: exploring protein conformational space and the effect of mutations on protein function and stability. Nucleic Acids Res. 2015;43:395–400. DOI: 10.1093/nar/gkv343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store‐overload‐induced Ca2+ release (SOICR). Proc Natl Acad Sci USA. 2004;101:13062–13067. DOI: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Medeiros‐Domingo A, Bhuiyan ZA, Tester DJ, Hofman N, Bikker H, van Tintelen JP, Mannens MM, Wilde AA, Ackerman MJ. The RYR2‐encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise‐induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54:2065–2074. DOI: 10.1016/j.jacc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wehrens XHT, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song L‐S, Rosemblit N, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise‐induced sudden cardiac death. Cell. 2003;113:829–840. DOI: 10.1016/S0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 17. Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–3214. DOI: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 18. Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR. Enhanced store overload‐induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. [DOI] [PubMed] [Google Scholar]
- 19. Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, Oda T, Kobayashi S, Ikemoto N, Matsuzaki M. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–772. DOI: 10.1161/CIRCULATIONAHA.107.718957. [DOI] [PubMed] [Google Scholar]
- 20. Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Kobayashi S, et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation‐linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–1424. DOI: 10.1161/CIRCRESAHA.109.209312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suetomi T, Yano M, Uchinoumi H, Fukuda M, Hino A, Ono M, Xu X, Tateishi H, Okuda S, Doi M, et al. Mutation‐linked defective interdomain interactions within ryanodine receptor cause aberrant Ca2⁺ release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation. 2011;124:682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tian X, Tang Y, Liu Y, Wang R, Chen SR. Calmodulin modulates the termination threshold for cardiac ryanodine receptor‐mediated Ca2+ release. Biochem J. 2013;455:367–375. DOI: 10.1042/BJ20130805. [DOI] [PubMed] [Google Scholar]
- 23. Liu Y, Sun B, Xiao Z, Wang R, Guo W, Zhang JZ, Mi T, Wang Y, Jones PP, Van Petegem F, et al. Roles of the NH2‐terminal domains of cardiac ryanodine receptor in Ca2+ release activation and termination. J Biol Chem. 2015;290:7736–7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daniel RM, Dunn RV, Finney JL, Smith JC. The role of dynamics in enzyme activity. Annu Rev Biophys Biomol Struct. 2003;32:69–92. DOI: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- 25. Teilum K, Olsen JG, Kragelund BB. Functional aspects of protein flexibility. Cell Mol Life Sci. 2009;66:2231–2247. DOI: 10.1007/s00018-009-0014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mittag T, Kay LE, Forman‐Kay JD. Protein dynamics and conformational disorder in molecular recognition. J Mol Recognit. 2010;23:105–116. [DOI] [PubMed] [Google Scholar]
- 27. Tzeng SR, Kalodimos CG. Protein dynamics and allostery: an NMR view. Curr Opin Struct Biol. 2011;21:62–67. DOI: 10.1016/j.sbi.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 28. Lange OF, Lakomek NA, Farès C, Schröder GF, Walter KF, Becker S, Meiler J, Grubmüller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC‐derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. DOI: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 29. Mahajan S, Sanejouand YH. On the relationship between low‐frequency normal modes and the large‐scale conformational changes of proteins. Arch Biochem Biophys. 2015;567:59–65. DOI: 10.1016/j.abb.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 30. Seo S, Jang Y, Qian P, Liu WK, Choi JB, Lim BS, Kim MK. Efficient prediction of protein conformational pathways based on the hybrid elastic network model. J Mol Graph Model. 2014;47:25–36. DOI: 10.1016/j.jmgm.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 31. Lee BH, Seo S, Kim MH, Kim Y, Jo S, Choi MK, Lee H, Choi JB, Kim MK. Normal mode‐guided transition pathway generation in proteins. PLoS One. 2017;12:e0185658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng W, Brooks BR, Hummer G. Protein conformational transitions explored by mixed elastic network models. Proteins. 2007;69:43–57. [DOI] [PubMed] [Google Scholar]
- 33. Kutzner C, Köpfer DA, Machtens JP, de Groot BL, Song C, Zachariae U. Insights into the function of ion channels by computational electrophysiology simulations. Biochim Biophys Acta. 2016;1858:1741–1752. [DOI] [PubMed] [Google Scholar]
- 34. Machtens JP, Briones R, Alleva C, de Groot BL, Fahlke C. Gating charge calculations by computational electrophysiology simulations. Biophys J. 2017;112:1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]