

Abstract
Background
We determined if the sodium glucose co‐transporter 2 inhibitor empagliflozin attenuates pressure overload‐induced heart failure in non‐diabetic mellitus mice by direct cardiac effects and the mechanisms involved.
Methods and Results
Male C57BL/6J mice (4–6 months of age) were subjected to sham surgeries or transverse aortic constriction to produce cardiac pressure overload. Two weeks after transverse aortic constriction, empagliflozin (10 mg/kg per day) or vehicle was administered daily for 4 weeks. Empagliflozin increased survival rate and significantly attenuated adverse left ventricle remodeling and cardiac fibrosis after transverse aortic constriction. Empagliflozin also attenuated left ventricular systolic and diastolic dysfunction, evaluated by echocardiography, and increased exercise endurance by 36% in mice with transverse aortic constriction‐induced heart failure. Empagliflozin significantly increased glucose and fatty acid oxidation in failing hearts, while reducing glycolysis. These beneficial cardiac effects of empagliflozin occurred despite no significant changes in fasting blood glucose, body weight, or daily urine volume. In vitro experiments in isolated cardiomyocytes indicated that empagliflozin had direct effects to improve cardiomyocyte contractility and calcium transients. Importantly, molecular docking analysis and isolated perfused heart experiments indicated that empagliflozin can bind cardiac glucose transporters to reduce glycolysis, restore activation of adenosine monophosphate‐activated protein kinase and inhibit activation of the mammalian target of rapamycin complex 1 pathway.
Conclusions
Our study demonstrates that empagliflozin may directly bind glucose transporters to reduce glycolysis, rebalance coupling between glycolysis and oxidative phosphorylation, and regulate the adenosine monophosphate‐activated protein kinase mammalian target of rapamycin complex 1 pathway to attenuate adverse cardiac remodeling and progression of heart failure induced by pressure‐overload in non‐diabetic mellitus mice.
Keywords: cardiac hypertrophy, cardiac metabolism, cardiomyocytes, sodium glucose cotransporter 2, transverse aortic constriction
Subject Categories: Translational Studies, Heart Failure, Hypertrophy, Metabolism
Nonstandard Abbreviations and Acronyms
- ACC
acetyl‐coA carboxylase
- AMPK
adenosine monophosphate‐activated protein kinase
- GLUT
glucose transporter
- mTORC1
mammalian target of rapamycin complex 1
- NHE
sodium hydrogen exchanger
- SGLT2
sodium glucose co‐transporter 2
- TAC
transverse aortic constriction
Clinical Perspective
What Is New?
Our study indicates that empagliflozin attenuates adverse cardiac remodeling and fibrosis and progression of heart failure independent of reductions in blood glucose or diuresis in a non‐diabetic model of pressure overload.
Empagliflozin increases glucose and fatty acid oxidation in failing hearts and may directly bind glucose transporters to reduce cardiac glycolysis, rebalance coupling between glycolysis and oxidative phosphorylation, and regulate the adenosine monophosphate‐activated protein kinase mammalian target of rapamycin complex 1 pathway.
These studies provide new insights into the molecular, cellular, and metabolic mechanisms by which empagliflozin exerts cardioprotective effects in heart failure induced by pressure overload.
What Are the Clinical Implications?
Our findings suggest potential new pharmacological targets and therapeutic strategies for heart failure caused by pressure overload.
Treatment of heart failure (HF) remains a challenge for clinicians, as patients with HF have poor prognosis with a 5‐year survival rate of only about 50%. 1 Recently, sodium glucose co‐transporter 2 (SGLT2) inhibitors, a new class of antihyperglycemic drugs, have attracted significant attention because of their ability to reduce mortality and major adverse cardiovascular events in patients with HF. 2
The EMPA‐REG (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) clinical trial showed in patients with type 2 diabetes mellitus that empagliflozin, a highly‐selective SGLT2 inhibitor, reduced cardiovascular mortality, non‐fatal myocardial infarction or non‐fatal stroke by 13.2%, and hospitalization from congestive HF by 34.1%. 3 Dapagliflozin, another SGLT2 inhibitor, decreased the risk of worsening HF or death from cardiovascular causes in patients with HF and reduced ejection fraction, regardless of the presence or absence of diabetes mellitus. 4 These clinical trials demonstrate that SGLT2 inhibitors may improve outcomes in patients with HF, but their mechanisms of action are still poorly understood. Although SGLT2 inhibitors were developed mainly to treat type 2 diabetes mellitus, their beneficial cardiovascular effects do not appear to be explained entirely by reductions in blood glucose or diuresis since other antihyperglycemic drugs or diuretics do not evoke similar favorable outcomes in patients with HF. 5 , 6
We hypothesized that empagliflozin, a specific SGLT2 inhibitor, would improve cardiac function and protect against HF progression via direct effects on the heart independent of its antihyperglycemic or diuretic effects. We used non‐diabetic mellitus mice with HF induced by pressure‐overload to avoid potential effects of altered blood glucose concentration and insulin resistance which characterize patients with type 2 diabetes mellitus. Our studies demonstrate that empagliflozin attenuates adverse cardiac remodeling and progression of HF independent of reductions in blood glucose or diuresis. Importantly, our molecular docking studies and experiments in isolated perfused hearts suggest that empagliflozin directly binds glucose transporters to reduce glycolysis, rebalance coupling between glycolysis and oxidative phosphorylation, regulate the adenosine monophosphate‐activated protein kinase (AMPK) mammalian target of rapamycin complex 1 (mTORC1) pathway, and improve cardiac function in failing hearts.
Methods
All data and supporting materials have been provided within the article and supplementary files.
Animal Studies
Male C57Bl/6J mice (3–4 months of age) were obtained from Jackson Laboratory (Bar Harbor, ME). All animal procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The animal protocols in this study were approved by the Institutional Animal Care and Use Committee of University of Mississippi Medical Center (Approval number: 1406). Mice were housed for at least 1 week under standard housing conditions before experiments, with a 12‐hour day/night cycle, and food and drinking water ad libitum. Mice were euthanized by an overdose of anesthesia with 5% vol/vol isoflurane (inhalation) at the indicated time point.
Transverse Aortic Constriction Procedure
Mice were anesthetized with 2% to 3% vol/vol isoflurane by mechanical ventilation, and subcutaneously injected with bupivacaine (2 mg/kg) for one time before surgical incision. Then a midline incision was performed in the anterior portion of the neck. The sternum was cut from suprasternal notch down to the second intercostal space. The thymus was separated, the aortic arch was exposed, and a 7.0‐silk suture was wrapped around the arch between the innominate artery and left carotid artery over a 27‐gauge needle. The needle was then removed, and the wound was closed. 7 For the sham group, the procedure was similar, but without aortic constriction. Analgesic administration was given with one single injection of buprenorphine‐sustained release (0.5 mg/kg, subcutaneously) to relieve postoperative pain. After 2 weeks of transverse aortic constriction (TAC) or sham surgery, the mice were given 1× PBS as vehicle or empagliflozin (10 mg/kg per day by oral gavage) for the duration of the study which lasted an additional 4 weeks.
Transthoracic Echocardiography
Animals from each group were anesthetized with 1% to 2% isoflurane (inhalation), and transthoracic echocardiography (Vevo 3100, Visualsonics, Toronto, Canada) was performed to measure cardiac function, including systolic and diastolic parameters. Data from at least 3 cardiac cycles were collected. Left ventricular (LV) trace was performed to obtain an averaged ejection fraction, fractional shortening, cardiac output and thickness of the LV posterior wall during diastole for each group. Diastolic function was measured by the ratio of mitral valve E peak to A peak (E/A). Other parameters, including isovolumetric relaxation time (IVRT), isovolumetric contraction time (IVCT), and ejection time were also obtained by PW Doppler imaging. Myocardial performance index was calculated as: (IVCT+IVRT)/ ejection time. Global circumferential strain was obtained from parasternal short‐axis views, and the sampling points were placed manually along with the epicardial and endocardial layers during end‐systolic period. The software (Vevo LAB v3.2.0) subsequently identified tissue speckles and tracked their movement frame‐by‐frame throughout the cardiac cycle and calculated the global circumferential strain.
Exercise Endurance Test
At the end of experiments, the exercise capacity of mice was tested. Briefly, mice were acclimated to the treadmill (Columbus, Ohio) 2 days before performing a graded maximal exercise endurance test (GXT). On the first 2 days, mice were acclimated to the shock grid and the treadmill was engaged to a walking speed of 9 m/minutes for 5 minutes. On the third day, the GXT protocol was performed by placing the mice on the treadmill at 5° inclination with shock grid activated. The treadmill speed was then gradually increased until exhaustion of the mice as follows: (speed, duration, inclination) 9 m/minutes, 2 minutes, 5°; 12 m/minutes, 2 minutes, 10°; 15 m/minutes, 2 minutes, 15°; (18; 21; 23 m/minutes, 1 minutes each, 15°); and (+1 m/minute, each 1 minute thereafter, 15°). Exhaustion (end point of the GXT protocol) was defined as the point at which the mice maintained continuous contact with the shock grid for 5 seconds. Blood lactate concentration was measured from tail vein blood samples collected at resting (at least 1 hour before placement in the treadmill) and immediately after the GXT protocol.
B‐type natriuretic peptide Assay
Serum B‐type natriuretic peptide levels in the different groups were measured using Enzyme Immunoassay Kit (Sigma‐Aldrich) following the manufacturer's instructions.
Immunoblotting
Immunoblotting was performed as previously described. 8 , 9 , 10 Briefly, protein concentration was measured using the Bradford dye‐binding method (Dye Reagent Concentrate, Bio‐Rad Protein Assay). The target proteins were separated by SDS‐PAGE, transferred to Millipore nitrocellulose membranes (BioRad, Hercules, CA). The membranes were incubated with primary antibodies at 4℃ overnight. Then, membranes were incubated with secondary antibodies at room temperature for 1 hour and signal intensity was determined using the Odyssey Infrared Imaging System (LI‐COR, Lincoln, NE).
Antibodies against phospho‐ACC (Cell signal, 3661), ACC (Cell signal, 3662), phospho‐AMPK (Cell signal, 2535), AMPK (Cell signal, 5831), phospho‐mammalian target of rapamycin (mTOR) (Cell signal, 2971), mTOR (Cell signal, 2983), GAPDH (Cell signal, 2118), phospho‐S6 ribosomal protein (Cell signal, 2217), S6 ribosomal protein (Cell signal, 2217), CD36 (cluster of differentiation 36) (Novus, NB400‐104), PPARα (peroxisome proliferator‐activated receptor‐alpha) (Novus, NBP1‐04676), SERCA2 (Cell signal, 9580), phospho‐CaMKII (Cell signal, 12716), phospholamban (Cell signal, 14562), and ryanodine receptor (Proteintech, 19765‐1‐AP) were used as primary antibodies. IRDye 800CW Donkey anti‐Rabbit immunoglobulin G (LI‐COR, 925‐32213) and IRDye 680RD Donkey anti‐Rabbit immunoglobulin G (LI‐COR, 925‐68073) were used as secondary antibodies.
Evaluation of Cardiac Hypertrophy and Fibrosis
Heart paraffin embedded tissue sections (5 μm) were prepared as described previously. 7 Briefly, fluorescein conjugated wheat germ agglutinin stain (Alexa Fluor‐488, Invitrogen, Carlsbad, CA) was used to evaluate cardiomyocyte size and myocyte nucleus was stained using 4′, 6‐diamidino‐2‐phenylindole. Masson trichrome stain (Sigma‐Aldrich) was used for detection of fibrosis.
Glucose and Fatty Acid Oxidation Measurements
Glucose and fatty acid oxidation in whole hearts was measured using a working heart system at the end of the experiments. The system was set to an afterload of 80 cm H2O. 8 , 9 , 11 , 12 , 13 Hearts were perfused at a preload of 15 mm Hg with Krebs–Henseleit buffer containing glucose (7 mmol/L), oleate (0.4 mmol/L), BSA (1%), and a low insulin concentration (10 lU/mL). [9, 10]‐3H‐oleate (50 µCi/L) and 14C‐glucose (20 µCi/L) was added to the buffer entering through the right pulmonary vein and pumped out through the aorta. The perfusion buffer was bubbled with 95% O2/5% CO2. The perfusate pumped out of the coronary venous was collected every 10 minutes to measure the radioactivity. Fatty acid oxidation was determined through the production of 3H2O from [9, 10]‐3H‐oleate. Metabolized 3H2O was separated from [9, 10]‐3H‐oleate by filtering through an anion exchange resin (BioRad; 1401241). Glucose oxidation was measured by metabolized 14CO2 dissolved in the buffer. To separate 14CO2 from 14C‐glucose, sulfuric acid was added to perfusate samples and 14CO2 was captured on hyamine hydroxide‐soaked filter paper. The 3H and 14C signals were measured by a scintillation counter (Beckman LS 6000).
Glucose Uptake and Glycolysis Measurements and Left Ventricular Developed Pressure
Glucose uptake and glycolysis were analyzed in the Langendorff heart perfusion system by measuring the production of 3H2O from D‐[2‐3H] glucose or D‐[5‐3H] glucose, respectively. Mice were injected with 1000 µ/kg heparin (Lake Zurich, IL) via i.p. 10 minutes before being euthanized. Isolated hearts were then retroperfused with radiolabeled Krebs‐Henseleit buffer. The buffer temperature was maintained at 37°C. Perfusate was collected every 10 minutes to test the radioactivity. During the ex vivo experiments, a fluid‐inflated balloon was inserted in the LV and connected to a LabChart 8 system (AdInstruments) to measure heart rate (HR) and LV developed pressure. The rate pressure product was calculated as LV developed pressure multiplied by HR (rate pressure product= LV developed pressure × HR). Metabolized 3H2O from 500 µL collected perfusate was separated from D‐[2‐3H] glucose or D‐[5‐3H] glucose by filtering through anion‐exchange 1‐X8 resin (BioRad; 1401441). The rate of glucose uptake and glycolysis was calculated by the amount of 3H2O production. Ten milliliters of scintillation fluid was added to each vial, and then mixed well. The radioactive signal was measured on a liquid scintillation counter (Beckman LS 6000, Brea, CA).
Myocardial ATP generation was calculated as that the oxidation of 1 molecule of oleic acid generating 118.5 ATP, 1 molecule of glucose 36 ATP and the glycolysis of 1 molecule of glucose generating 2 ATP.
Cardiomyocyte isolation and measurement of contractility and calcium transient
At the end of experiments, mice were injected with 1000 µ/kg of heparin for anticoagulation before being euthanized. 14 The heart was excised, cannulated, and connected to a heart perfusion apparatus (Radnoti, CA) and perfusion was initiated in the Langendorff mode. The heart was perfused with a Ca2+‐ free based buffer (pH 7.2, 37°C) containing: 135 mmol/L NaCl, 4 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L HEPES, 0.33 mmol/L NaH2PO4, 10 mmol/L glucose, 10 mmol/L 2,3‐butanedione monoxime (Sigma; B0753), and 5 mmol/L taurine (Sigma; T0625) and bubbled with 95% O2/5% CO2. After 3 to 5 minutes of perfusion, the buffer was replaced with similar buffer containing 0.3 mg/g body weight collagenase D (Roche; 11088858001), 0.4 mg/g body weight collagenase B (Roche; 11088807001), and 0.05 mg/g body weight protease type XIV (Sigma; P5147) dissolved in 25‐mL perfusion buffer. 15 After complete digestion of the heart (6–8 minutes), the heart was removed and minced. Extracellular Ca2+ was gradually added back to the cells gradually, starting at 0.06 mmol/L to a final concentration of 1.2 mmol/L. The interval of each Ca2+ concentration was 15 minutes. 13
Cardiomyocytes contractility was assessed using a SoftEdge Myocam system (IonOptix, Westwood, MA). Cardiomyocytes were placed in a chamber and stimulated with a supra‐threshold voltage at a frequency of 1 Hz. 16 , 17 IonOptix SoftEdge software was used to record the changes in sarcomere length and duration of shortening and re‐lengthening. The following parameters were used to evaluate cardiomyocyte contractility: resting sarcomere length; maximum velocity of shortening (−dL/dt); maximum velocity of re‐lengthening (+dL/dt); peak height, maximum change of sarcomere length during contraction; peak shortening, peak height normalized to resting sarcomere length. Intracellular Ca2+ transient was measured using a dual‐excitation, single emission photomultiplier system (IonOptix). Cardiomyocytes were treated with Fura 2‐AM (2 μmol/L) at 37°C for 20 minutes and then exposed to light emitted by a 75 W halogen lamp through either a 340‐ or 380‐nm filter while being stimulated to contract at a frequency of 1 Hz, and fluorescence emissions were detected. The following parameters were recorded: calcium baseline signal (F340/380); calcium peak signal, maximum change of calcium signal during contraction (ΔF340/380); percentage peak calcium change (%ΔF340/380), peak calcium signal normalized to baseline; maximum velocity of calcium change during contraction (+dF/dt); maximum velocity of calcium change during relaxation (‐dF/dt).
Molecular Docking
Binding ability of empagliflozin to potential targets including GLUT1 (glucose transporter 1), GLUT4, SGLT1 (sodium glucose co‐transporter 1), and NHE (sodium hydrogen exchanger) were examined by molecular docking analysis. Empagliflozin was prepared by using maestro Ligprep module and was docked into these protein models by using a standard docking module standard precision in Glide (Schrödinger software, 2015). Because of structural conservation between reported GLUT1 structures, the inhibitor co‐complex structures of GLUT1 (protein data bank [PDB] code: 5EQG) were chosen for detailed examination of potential docking with empagliflozin. Also, this structure was adopted to build a GLUT4 protein model (identity: 65.92%). Structural models of SGLT1 were created based on crystal structure of SiaT (PDB code: 5NV9) according to Bisignano et al. 18 As reported by Uthman et al., 18 NHE was also modeled with bacterial sodium‐proton antiporter (PDB code: 4CZB) as template. Homology models were performed by using SWISS‐MODEL server. 19 The ligand centroid in crystal structures or reported potential binding sites were chosen as the docking center. Glide GScore was used to rank the result and the lowest score was selected as the binding energy for the best binding mode.
Body Weight and Body Composition Analysis, and Measurements of Blood Pressure and Serum Ketone Body
These detailed methods can be found in Data S1.
Statistical Analysis
Data were expressed as mean±SEM. Two‐tailed Student t‐test, Mann–Whitney test, Dunn multiple comparison test, log‐rank test, non‐repeated and repeated measures 1‐way or 2‐way ANOVA followed by Tukey post‐hoc tests were used for comparisons (the statistical method for each comparison is indicated in the figure legends.) using Prism 7.0 (GraphPad Software, La Jolla, CA). A P value of <0.05 was considered as statistically significant.
Results
Empagliflozin Increased Survival Rate and Attenuated TAC‐Induced Cardiac Remodeling Without Altering Blood Glucose Concentration
Empagliflozin treatment increased survival rate from 50% to 63.2% after TAC surgery (vehicle and empagliflozin, respectively, Figure 1A). No deaths occurred in the sham and sham+empagliflozin groups. Empagliflozin did not significantly alter fasting blood glucose concentration in any of the groups (Figure 1B, Table S1). Furthermore, empagliflozin did not significantly alter food intake, 24‐hour urine volume and serum ketone bodies (Figure S1), body weight, or adiposity in these non‐diabetic mice compared with vehicle groups (Figure S2).
Figure 1. Empagliflozin treatment attenuated cardiac remodeling caused by transverse aortic constriction (TAC).
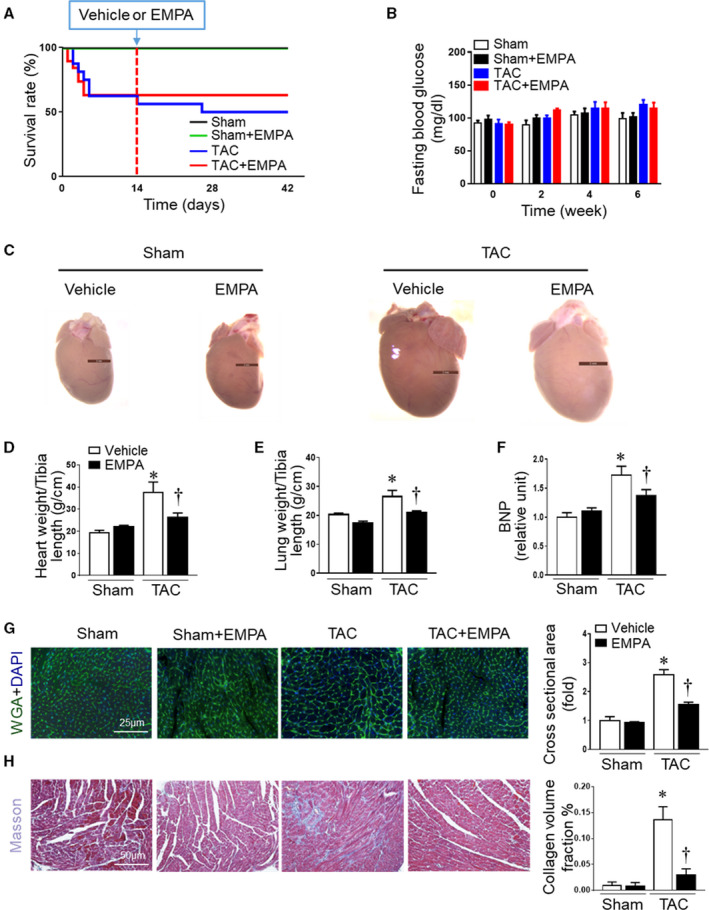
(A) Survival curve in different groups of mice after sham or TAC surgery subjected to vehicle or empagliflozin treatment (n=10–19). (B) Fasting blood glucose concentration among groups during baseline (time 0), 2, 4, and 6 weeks after surgery. Mice were fasted overnight, and blood glucose concentration was measured biweekly (n=10–19). There was no difference among any of the groups. (C) Representative hearts from sham+vehicle, sham+empagliflozin, TAC+vehicle, and TAC+empagliflozin mice at the end of experiments. (D) Dry heart weight and (E) lung weight in different groups (n=5–6). (F) Quantitative analysis of serum B‐type natriuretic peptide (n=6). (G) Left: Cardiomyocytes sizes were outlined by wheat germ agglutinin staining. Right: Quantitative analysis of wheat germ agglutinin staining (n=6). (H) Cardiac fibrosis was measured by Masson staining. Left: representative images of Masson staining. Right: quantitative analysis (n=6), Results are expressed as mean±SEM. TAC indicates transverse aortic constriction; BNP, B‐type natriuretic peptide; DAPI, 4′,6‐diamidino‐2‐phenylindole; and WGA, wheat germ agglutinin. *P<0.05 vs sham, † P<0.05 vs TAC. Log‐rank test (A). One‐way ANOVA (non‐repeated measures) (B and D–H).
Empagliflozin treatment attenuated TAC‐induced LV remodeling (Figure 1C) and reduced cardiac hypertrophy and lung dry weight compared with vehicle treatment (Figure 1D and 1E). The large increase in plasma concentration of B‐type natriuretic peptide, an HF biomarker, associated with TAC was also markedly attenuated by empagliflozin (Figure 1F). In addition, empagliflozin treatment in mice with TAC attenuated the size of individual cardiomyocytes and also reduced cardiac fibrosis compared with vehicle groups after TAC (Figure 1G and 1H, Table S1).
Empagliflozin Improved Cardiac Function and Increased Exercise Endurance in Mice With TAC‐Induced HF
We assessed cardiac function at baseline and at 2‐weeks post‐surgery before empagliflozin treatment was started. Compared with baseline values, TAC increased LV posterior wall thickness in diastole and reduced ejection fraction, fractional shortening, and cardiac output (Figure S3A–S3D) at 2 weeks. After 6 weeks of TAC, cardiac systolic function was reduced even further in vehicle‐treated mice (Figure 2A and 2C). Empagliflozin treatment for 4 consecutive weeks attenuated most of the impairment in systolic function, including reductions in ejection fraction, fractional shortening, and cardiac output (Figure 2A and 2C). In addition, TAC also caused diastolic dysfunction with a restrictive pattern, as evidenced by increased E/A ratio (Figure S3E, Figure 2B and 2C). We also found that myocardial performance index was increased after 6‐week TAC, but not significantly different after 2‐week TAC (Figure S3F). Four weeks of empagliflozin treatment not only attenuated development of HF with reduced ejection fraction, but also abolished the alterations in myocardial performance index and E/A ratio induced by TAC (Figure 2B and 2C). Empagliflozin also markedly attenuated TAC‐induced impairment of global circumferential strain, an index of LV function, compared with vehicle (Figure 2D, Table S2).
Figure 2. Empagliflozin treatment improved cardiac function and exercise endurance in heart failure.
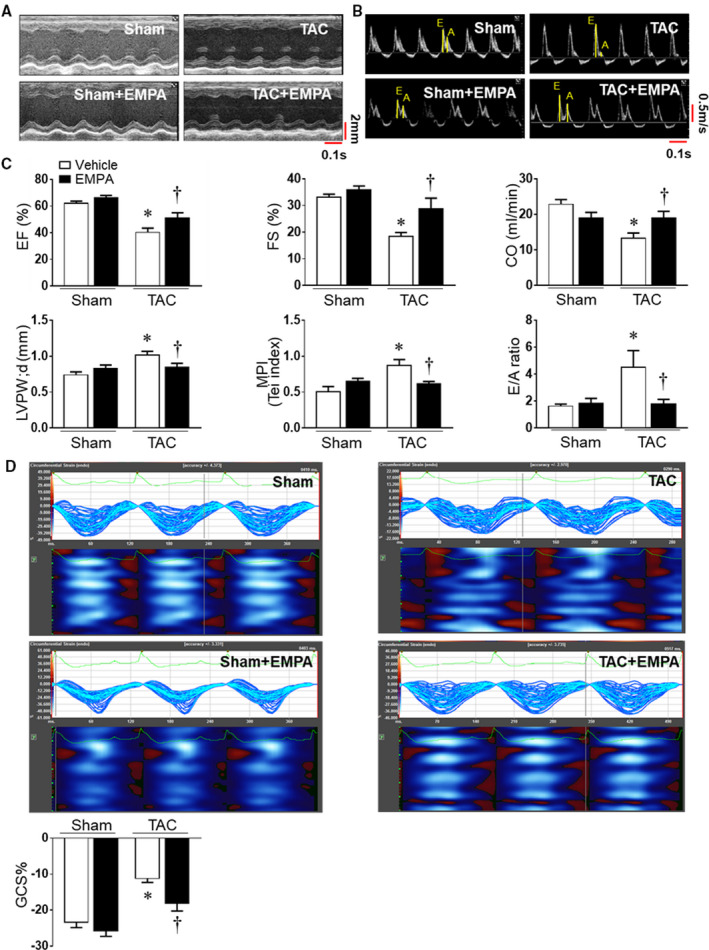
(A) and (B) Representative images of M‐mode of left ventricles and mitral valve flow Doppler in Sham and transverse aortic constriction groups. (C) Cardiac systolic and diastolic function were quantified, including ejection fraction, fraction shortening, cardiac output, myocardial performance index, and E/A ratio. The end‐diastolic left ventricular posterior wall thickness was also evaluated. (D) Cardiac speckle‐tracking analysis was used to evaluate cardiac contractility. Upper: the representative images of global circumferential strain among groups. Lower: Quantitative results. One‐way ANOVA (non‐repeated measures) (C–D). Results are expressed as mean±SEM, n=5–6 in each group. CO indicates cardiac output; EF, ejection fraction; FS, fraction shortening; GCS, global circumferential strain; LVPWd, LV posterior wall during diastole; and TAC, transverse aortic constriction. *P<0.05 vs sham, † P<0.05 vs transverse aortic constriction.
We next tested if TAC‐induced HF would be associated with impaired exercise capacity during GXT, and if empagliflozin would improve exercise capacity. TAC decreased exercise capacity by ≈50% in vehicle treated mice compared with the sham group (time until exhaustion: 10.8±1.3 versus 21.3±0.9 minutes, P<0.01). Empagliflozin increased exercise endurance by ≈36% (14.7±1.2 minutes) (Figure 3A). We also observed significantly reduced VO2 max, an index of maximum oxygen uptake during GXT, in the vehicle‐treated TAC group (Figure 3B); empagliflozin treatment partially restored VO2 max toward normal in TAC mice (Figure 3B). TAC also shortened the time needed for the respiratory exchange rate to increase >1.0 when compared with the sham group (8.9 versus 14.6 minutes, P=0.01) (Figure 3C) and resulted in the highest blood lactate concentration immediately after the GXT (Figure 3D). Empagliflozin treatment caused only a modest, non‐significant delay in the time until respiratory exchange rate exceeded 1.0 in the TAC+empagliflozin group (P=0.22) (Figure 3C), but it markedly reduced blood lactate concentration at the end of GXT (Figure 3D, Table S3).
Figure 3. Empagliflozin treatment improved mice exercise capacity after 6‐week TAC.
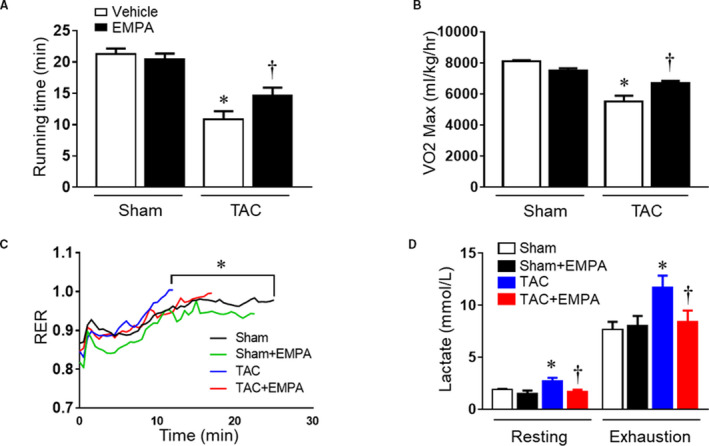
(A) Running time and (B) maximum oxygen uptake were measured in different groups. (C) Respiratory exchange ratio curves from 4 groups. (D) Blood lactate level at resting status and exhaustion in different groups; 2‐way ANOVA (repeated measures). Results are expressed as mean±SEM, n=5–6 in each group. RER indicates respiratory exchange ratio; and TAC, transverse aortic constriction. *P<0.05 vs sham, † P<0.05 vs TAC. One‐way ANOVA (non‐repeated measures) (A–C).
We also measured the systemic arterial and left ventricle pressures in these mice at the end of the experiments. Since there is a gradient between the pressures before and after the aortic constriction, we measured the systemic arterial pressure through the left carotid artery, while measuring the intra‐LV pressure through the apex of the heart. Although blood pressure beyond the aortic constriction initially is reduced after aortic constriction, within a few days the systemic arterial pressure beyond the constriction returns to nearly normal because of compensatory increases in pressure behind the constriction that raise intraventricular pressures. 20 , 21 Empagliflozin treatment, however, did not significantly alter systemic arterial pressure or intraventricular pressure (Figure S4).
Empagliflozin Improved Cardiomyocyte Contractility and Calcium Transients in Mice With TAC‐Induced HF
At the end of the experimental protocol, we isolated cardiomyocytes from each group to measure their contractility and calcium transients (Table S4). We found no differences in resting sarcomere length among the groups. However, maximum cardiomyocyte contraction (Peak ∆Length) and contraction (‐dl/dt) and relaxation (+dl/dt) velocities were significantly reduced in TAC mice treated with vehicle (Figure 4A and 4F). Empagliflozin treatment in TAC mice improved cardiomyocyte contractility parameters including maximum contraction, velocity of contraction and relaxation (Figure 4A and 4F).
Figure 4. Contractile function and calcium transients from isolated cardiomyocytes in sham, sham+empagliflozin, transverse aortic constriction and transverse aortic constriction +empagliflozin groups.
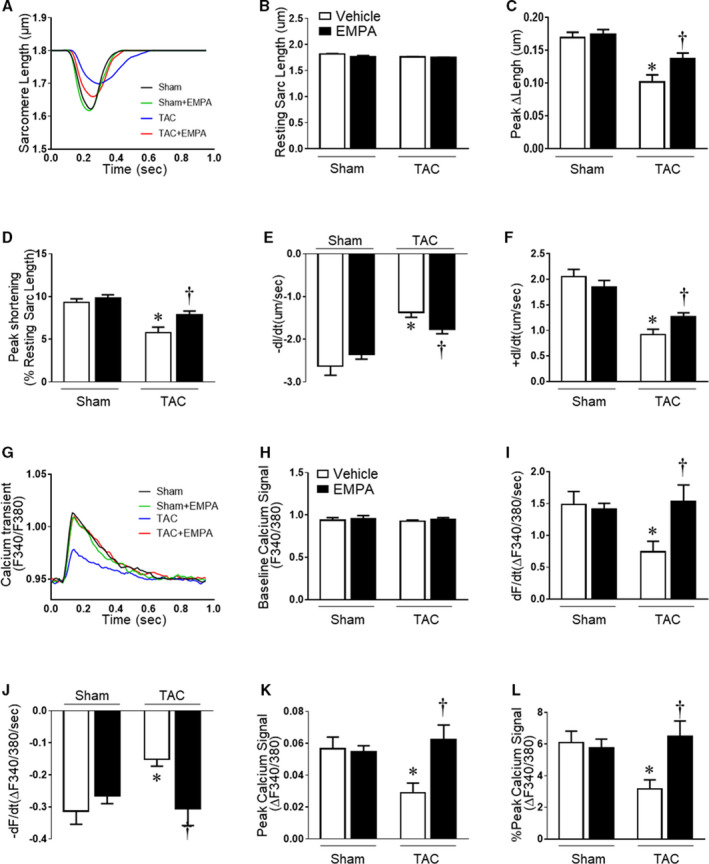
(A) Representative traces of cell contraction by time using 1Hz. (B) Resting sarcomere length; (C) Maximum cardiomyocyte contraction (Peak ∆length). (D) Shortening length normalized by resting sarcomere length (Peak shortening); (E) Maximum contraction velocity (−dL/dt); (F) Maximum relaxation velocity (+dL/dt); (G) Representative fluorescent traces of F340/380 ratios of cardiomyocytes loaded with Fura 2‐AM calcium transient using 1 Hz stimulation; (H) Baseline calcium signal (F340/380); (I) Maximum calcium change velocity during contraction (+dF/dt); (J) Maximum calcium change rate during relaxation(−dF/dt). (K) Maximum calcium amplitude transient (Peak calcium signal); (L) Maximum calcium transient amplitude normalized by baseline calcium signal (% Peak calcium signal). Results are expressed as mean±SEM, n=5–6 mice per group (40–92 cells were measured for each animal). One‐way ANOVA (non‐repeated measures) (B–F and H–L). TAC indicates transverse aortic constriction. *P<0.05 vs sham, † P<0.05 vs transverse aortic constriction.
Baseline calcium transients in cardiomyocytes were similar in all groups during resting conditions (Figure 4G and 4H). During contraction, however, the velocity of calcium increasing and decreasing transients during contraction and relaxation, respectively, were attenuated in TAC mice treated with vehicle compared with the sham group (Figure 4I and 4J). Empagliflozin completely reversed the reduced calcium transients in TAC+empagliflozin mice to values comparable with sham groups (Figure 4I and 4J). In addition, empagliflozin increased calcium peak signals to values observed in sham groups, while cardiomyocytes from TAC vehicle mice showed reduced calcium peak signal (Figure 4K and 4I). We also investigated calcium‐related pathways. The expression of SERCA2a, phospholamban, phospho‐CAMKII, and ryanodine receptor 2 were reduced after TAC, while only ryanodine receptor 2 was upregulated by empagliflozin treatment after TAC (Figure S5).
Empagliflozin Increased Cardiac Oxidative Phosphorylation in Mice With TAC‐Induced HF
At the end of the experimental protocol, we measured cardiac energy metabolism including glucose and fatty acid oxidation, glucose uptake, and glycolysis (Table S5). TAC mice treated with vehicle exhibited reduced glucose and fatty acid oxidation compared with sham groups (Figure 5A and 5B). Empagliflozin treatment in TAC mice increased glucose and fatty acid oxidation (Figure 5A and 5B). Surprisingly, empagliflozin treatment inhibited glycolysis and glucose uptake in both sham+empagliflozin and TAC+empagliflozin groups. TAC alone was associated with slightly reduced glucose uptake and glycolysis, but this difference was not significant (P=0.11) (Figure 5C and 5D). ATP production was reduced in hearts from TAC mice treated with vehicle when compared with sham group whereas empagliflozin partially restored total ATP production in hearts from TAC mice. (Figure 5E). Cardiac hemodynamic parameters of these isolated perfused hearts are also shown in Figure 5F and Table S5.
Figure 5. Chronic empagliflozin treatment improved oxidative phosphorylation in failing hearts.
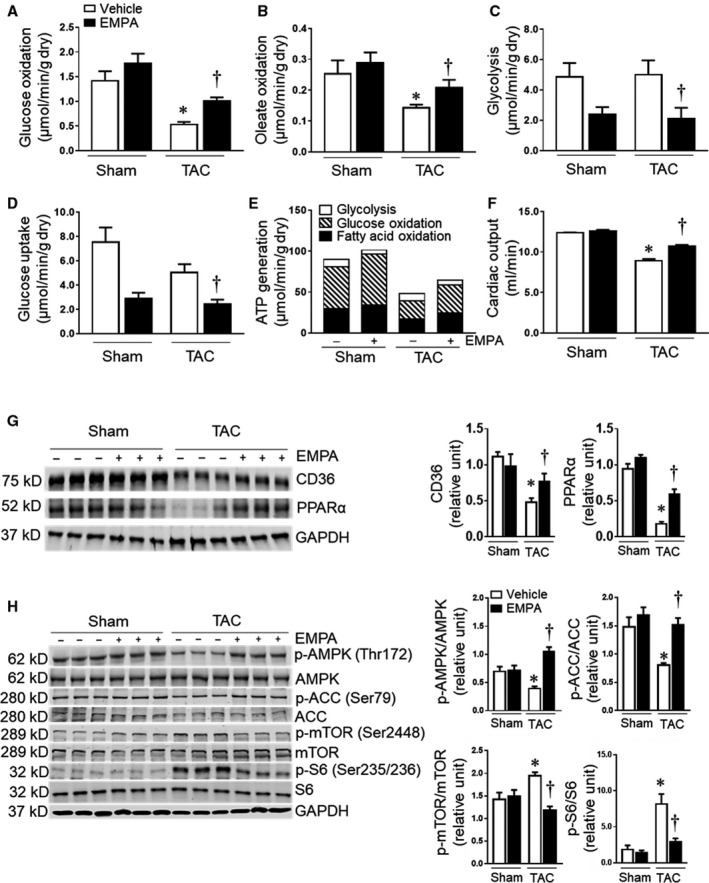
(A) to (D) Mean glucose and oleate acid oxidation, glycolysis, and glucose uptake in isolated perfused hearts from mice in different groups at the end of the protocol (n=5–6). (E) Calculated ATP generation in Sham, Sham+empagliflozin, TAC and TAC+empagliflozin groups (n=5–6). (F) The cardiac output of the isolated perfused hearts (n=5–6) in each group. (G) Left: representative blots of CD36, PPARα, and GAPDH. Right: Quantitative analysis (n=5–6). (H) Left: the representative blots of AMP‐activated protein kinase, acetyl‐coA carboxylase, mammalian target of rapamycin complex 1, S6 ribosomal protein and GAPDH were shown. Right: quantitative analysis (n=5–6). Results are expressed as mean±SEM; One‐way ANOVA (non‐repeated measures) (A–H). ACC indicates acetyl‐coA carboxylase; AMPK, adenosine monophosphate‐activated protein kinase; CD36, cluster of differentiation 36; mTOR, mammalian targe of rapamycin; S6, ribosomal protein S6; and TAC, transverse aortic constriction. *P<0.05 vs sham, † P<0.05 vs transverse aortic constriction.
We also investigated the expression of CD36 and PPARα, which are associated with fatty acid intake and oxidation, in these hearts. We found reduced CD36 and PPARα expression in the TAC group treated with vehicle, and empagliflozin partially restored their expression in the TAC+empagliflozin group without altering the expression in sham+empagliflozin mice (Figure 5G, Table S6).
Empagliflozin Restored AMPK Activity and Decreased mTORC1in Mice With TAC‐Induced HF
To further explore the beneficial cardiac effects of empagliflozin in HF, we investigated molecular pathways activated in the hearts of empagliflozin‐treated mice. We found that TAC decreased phosphorylated adenosine monophosphate‐activated protein kinase (AMPK)/AMPK ratio while increasing phosphorylated mammalian target of rapamycin complex 1 (mTORC1)/mTORC1 and p‐S6/S6 ratio in vehicle treated mice compared with the sham control group (Figure 5H). These changes were completely reversed by empagliflozin treatment. Acetyl‐coA carboxylase (ACC) was reduced in TAC mice treated with vehicle, and empagliflozin treatment increased p‐ACC/ACC ratio back to normal levels in TAC+empagliflozin group (Figure 5H, Table S6).
Molecular Docking Analysis of Empagliflozin Binding in the Heart and Effects on Glucose Uptake and Glycolysis in Isolated Perfused Hearts
Since there are no known SGLT2 receptors in the heart, we used molecular docking analysis to predict potential binding targets for empagliflozin in the heart. We found that empagliflozin had higher affinity for GLUT1 and GLUT4 (GScore was −7.920 and −7.273, respectively), and lower affinities for SGLT1 and sodium hydrogen exchanger 1 (NHE1) (−6.756 and −4.443) (Figure 6A and 6E, Figure S6A and S6B). When the binding features of empagliflozin were compared with the compound in the GLUT1 crystal structure (PDB:4PYP), their glucoside groups were well aligned. Also, empagliflozin shared high similarity with GLUT1 inhibitor in the crystal structure (PDB:5EQG) in the binding mode. (Figure S6C and S6D). This analysis further supports our model which predicts that empagliflozin binds to glucose transporters.
Figure 6. Empagliflozin has direct affinity to glucose transporters in the heart and reduces cardiac glucose uptake.
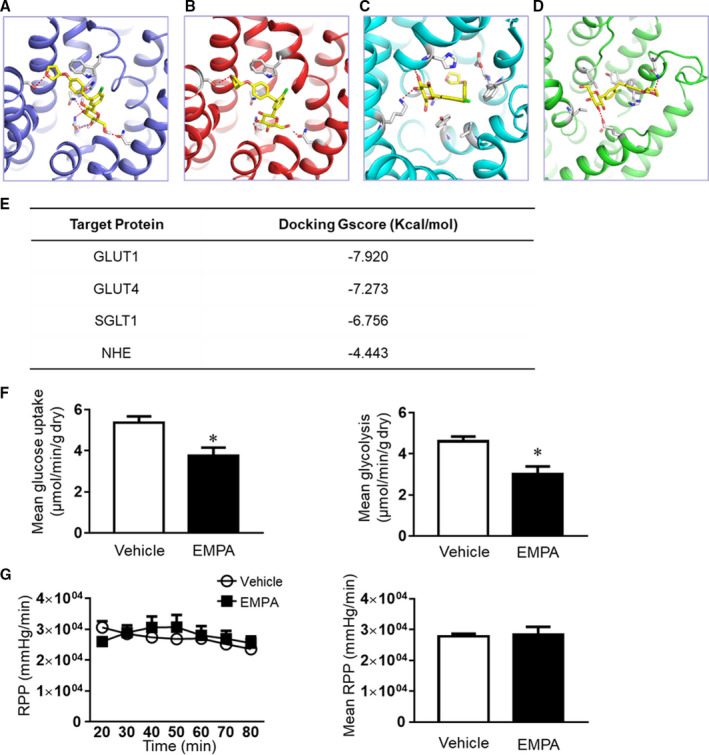
Molecular docking results showed that empagliflozin can bind (A) GLUT1 (glucose transporter 1); (B) GLUT4; (C) sodium glucose co‐transporter 1 (SGLT1); (D) NHE (sodium hydrogen exchanger). (E) Docking energy of each binding is shown. (F) Glucose uptake and glycolysis were reduced by acute treatment of empagliflozin in isolated perfused hearts. (G) The rate pressure product was slightly increased by direct treatment of empagliflozin in isolated perfused hearts. Results were expressed as mean±SEM, n=6 in each group. GLUT1, glucose transporter 1; NHE, sodium hydrogen exchanger; RPP, rate pressure product; and SGLT1, sodium glucose co‐trasporter 1. *P<0.05 vs vehicle. Mann–Whitney test (F–G).
To determine if empagliflozin can directly bind to cardiac GLUT1 and GLUT4 proteins, we isolated hearts from an additional group of healthy mice and perfused them with Krebs‐Henseleit vehicle buffer with or without empagliflozin (2.5 μmol/L) using the isolated perfusion heart system. We found that 1‐hour of acute empagliflozin treatment significantly reduced glucose uptake and glycolysis in isolated perfused hearts (Figure 6F). Although glucose uptake and glycolysis were reduced by empagliflozin, the cardiac function indicated by rate pressure product was not impaired (Figure 6G, Table S7). The dose of empagliflozin was determined from previous pharmacokinetic studies in mice. 22 , 23 , 24 , 25
In addition, acutely treating the cardiomyocytes with empagliflozin (0.5 μmol/L) 23 did not alter contractility and calcium transient, however, cariporide, the specific NHE1 antagonist significantly increased calcium transient with slightly decreasing contractility in cardiomyocytes (Figure S7A and S7H).
Discussion
The mechanisms for the beneficial outcomes observed with SGLT2 inhibitors in HF have been the subject of much speculation and have remained unclear. 5 , 26 , 27 Our study indicates that despite no known SGLT2 receptors in the heart, the SGLT2 inhibitor empagliflozin has direct cardioprotective actions to attenuate cardiac hypertrophy while improving myocardial ATP generation, cardiac function, and exercise endurance in non‐diabetic mellitus mice with pressure overload‐induced HF. An important finding of our study, based on molecular docking analysis and isolated perfused heart experiments, is that empagliflozin directly binds glucose transporters to reduce glucose uptake and glycolysis, rebalance myocardial oxidative phosphorylation, restore AMPK activity, and enhance cardiomyocyte calcium transients and contractility in failing hearts. Our study therefore provides new insights on the molecular and cellular mechanisms by which empagliflozin improves cardiac function in heart failure caused by pressure overload.
Empagliflozin Treatment Improves Cardiac Function and Attenuates TAC‐Induced HF in the Absence of Changes in Blood Glucose or Body Weight
Several mechanisms have been previously proposed for the beneficial effects of SGLT2 inhibitors in HF, including attenuation of hyperglycemia, induction of natriuresis/diuresis, inhibition of cardiomyocyte NHE, and increased ketone body metabolism. 3 , 27 , 28 , 29 Although SGLT2 inhibitors reduce fasting blood glucose in diabetic mellitus animals and in people with type 2 diabetes mellitus, previous studies have also shown beneficial effects of these drugs in the absence of major changes in blood glucose and in non‐diabetic mellitus models of HF. 26 , 30 , 31 In the present study, we found that chronic empagliflozin treatment in non‐diabetic mellitus mice had no significant effect on fasting blood glucose, suggesting that the beneficial actions on cardiac function in HF were not be mediated by empagliflozin’s antihyperglycemic effect. Empagliflozin treatment also promotes mild weight loss in patients with diabetes mellitus, 32 which could contribute, at least in part, to its cardiac protective actions. However, empagliflozin did not significantly alter food intake, body weight, or adiposity in non‐diabetic mellitus mice in the present study.
We also measured the concentration of serum ketone body (beta hydroxybutyrate) in the different groups. While some studies have found that empagliflozin increases ketone body production, 33 , 34 we did not find significant changes in ketone body concentration between vehicle and empagliflozin groups. One potential explanation for these differences may be the duration of empagliflozin administration which was 6 weeks in our study compared with longer periods of treatment in some other animal experiments or clinical trials. 33 , 34
Since there appears to be no expression of SGLT2 receptors in the heart, some studies have credited the beneficial cardiac effects of empagliflozin mainly to its ability to evoke natriuresis via a glycosuria osmotic effect that reduces blood pressure in patients with diabetes mellitus. 27 Although empagliflozin‐induced diuresis could be beneficial in HF, other diuretics and antihypertensive agents that are even more effective in causing diuresis and lowering blood pressure have not been as successful in slowing disease progression of HF and improving outcomes. 35 In addition, empagliflozin treatment did not affect mean systemic arterial pressure or intra ventricular pressure in our study. Thus, it seems unlikely that the powerful cardioprotective effect of empagliflozin found in our study can be primarily attributed to empagliflozin’s modest diuretic or blood pressure lowering effects.
Although there are no known SGLT2 receptors in the heart, our study and previous studies suggest that SGLT2 inhibitors may have direct effects in the heart. Uthman and colleagues 36 , 37 reported that SGLT2 inhibitors can suppress cardiac NHE1 activity. SGLT2 inhibitors may bind to NHE1 to reduce sodium and calcium overload in failing hearts to improve cardiac function. 36 , 37 However, the few clinical studies that examined whether inhibition of NHE1 expression or activity may protect the heart against ischemic injury or HF are controversial. 38 , 39 Our study showed that acute treatment of cardiomyocytes with the NHE1 inhibitor cariporide slightly decreased contractility, and increased calcium transients, indicating an uncoupling of contraction and calcium transients in cardiomyocytes. However, we did not observe a similar acute effect of empagliflozin in cardiomyocytes indicating differences in their acute modes of action.
Molecular Docking Analysis and Studies in Isolated, Perfused Hearts Suggest That Empagliflozin Binds Glucose Transporters and Improves Oxidative Phosphorylation in TAC‐Induced HF
Our studies suggest that empagliflozin improved myocardial oxidative phosphorylation via a direct cardiac effect. This finding is reinforced by our previous study showing that empagliflozin treatment directly protected the heart from ischemia‐reperfusion injury in ex vivo and in vitro experiments. 23 Although, previous reports suggest that SGLT2 receptors are not expressed in the heart, our molecular docking analysis predicts other potential binding targets for empagliflozin in the heart. This analysis suggested that empagliflozin can dock with GLUT1, GLUT4, SGLT1, and NHE, all of which are involved in cellular glucose and sodium homeostasis. 8 However, the molecular docking analysis indicated that empagliflozin has much higher affinity for GLUT1 and GLUT4 compared with SGLT1 and NHE.
Direct binding of empagliflozin to cardiac GLUT1 and GLUT4 proteins may influence cardiac glucose uptake and glycolysis. To test this potential mechanism, we performed ex vivo experiments using isolated perfused hearts and acutely treated them with empagliflozin to quantify glucose uptake and glycolysis. Our results showed that cardiac glucose uptake and glycolysis were significantly reduced by empagliflozin compared with vehicle treatment, indicating that empagliflozin may bind to glucose transporters in the heart and suppress rather than enhance their activity.
Our study and others 40 suggest that appropriate suppression of glucose uptake and glycolysis in HF may, at some stages, be beneficial. During HF, fatty acid oxidation is reduced, and cardiac energy supply is increasingly dependent on glucose metabolism. 41 However, excessive glycolysis may worsen mitochondrial uncoupling and in later stages of HF oxidative metabolism is severely impaired. 42 Thus, attenuating excessive glycolysis can improve coupling between glycolysis and oxidative phosphorylation, 43 as observed in our study.
In the present study, empagliflozin treatment in failing hearts increased expression of CD36 and PPARα proteins, which are important for fatty acid uptake and oxidation. 44 , 45 These findings further support the concept that empagliflozin enhances fatty acid uptake and oxidation in failing hearts and helps to restore the balance between energy demand and supply, thus improving cardiac performance.
Empagliflozin Activated AMPK, Inhibited mTORC1, and Attenuated Cardiac Hypertrophy and Adverse Remodeling in TAC‐Induced HF
Our data showed that empagliflozin treatment markedly attenuated cardiac hypertrophy while restoring AMPK activity and inhibiting phosphorylation of mTORC1 in TAC‐induced HF. The mTORC1 pathway is essential for cell growth and protein synthesis. It can be activated as a cellular adaptive response to induce cardiac hypertrophy during pressure overload, but persistent activation of mTORC1 can cause excessive cardiac hypertrophy and HF. 46 AMPK can downregulate mTORC1 by phosphorylating Raptor and tuberous sclerosis complex 2. 47 , 48 Thus, the effect of empagliflozin to attenuate cardiac hypertrophy and adverse remodeling in HF may be attributable to decreased mTORC1 subsequent to increased AMPK. Empagliflozin inhibition of glucose uptake may induce activation of AMPK in these hearts since inhibiting glucose transporters can suppress the pentose phosphate pathway resulting in activation of AMPK. 49 Our previous study also showed that empagliflozin can activate AMPK through liver kinase B1 pathway in hearts. 23 However, the mechanisms by which empagliflozin activates AMPK are still unclear and will require additional studies.
AMPK and mTORC1 are important regulators of cellular metabolism. 29 Our results showed that ACC, another downstream kinase of AMPK, was activated by empagliflozin in TAC‐induced HF. ACC regulates fatty acid oxidation by acting with carnitine palmitoyltransferase I. 50 The increased phosphorylated ACC induced by empagliflozin in TAC groups may therefore facilitate fatty acid oxidation. Furthermore, inhibition of mTORC1, as observed during empagliflozin treatment, may block the metabolic shift from fatty acid oxidation to glycolysis in failing hearts. 47 Of interest, empagliflozin did not over‐activate AMPK or inhibit mTORC1 in sham groups but in HF empagliflozin treatment restored phosphorylation of AMPK and mTORC1 back to almost normal levels. Similarly, empagliflozin treatment did not persistently increase glucose or fatty acid oxidation, but rebalanced glycolysis and oxidative phosphorylation. By enhancing AMPK activity and inducing a cascade of downstream biochemical and molecular changes, empagliflozin may have multiple beneficial actions, such as reducing inflammation and oxidative stresses in HF and other cardiovascular diseases.
Additionally, we found that empagliflozin improves calcium amplitude in cardiomyocytes after TAC. The immunoblotting results showed that empagliflozin increased expression of ryanodine receptor 2, rather than SERCA2a, phospholamban, or phospho‐CAMKII, although these findings differ somewhat from those of Mustroph et al. 51 Possible reasons for these differences are different mouse ages, duration of TAC, and empagliflozin administration protocol.
Empagliflozin has also been suggested to protect against endoplasmic reticulum stress caused by pressure overload in TAC‐induced HF. 52 However, the exact mechanisms by which empagliflozin and other SGLT2 inhibitors improve outcomes in HF have not been fully elucidated and remain an important area for further investigation.
In summary, our study demonstrated that chronic empagliflozin treatment can attenuate progression of TAC‐induced HF in non‐diabetic mellitus mice via multiple direct actions on the heart that improve its energy supply and attenuate hypertrophy and fibrosis (Figure 7). Empagliflozin directly binds glucose transporters to restore coupling between glycolysis and oxidative phosphorylation, enhances calcium transients and contractility of isolated cardiomyocytes, and improves cardiac function in failing hearts. Furthermore, inhibition of glucose uptake by empagliflozin may increase AMPK activity and inhibit mTORC1 to attenuate cardiac hypertrophy and protect the heart from HF progression.
Figure 7. Empagliflozin binds glucose transporters and improves cardiac metabolism in heart failure.
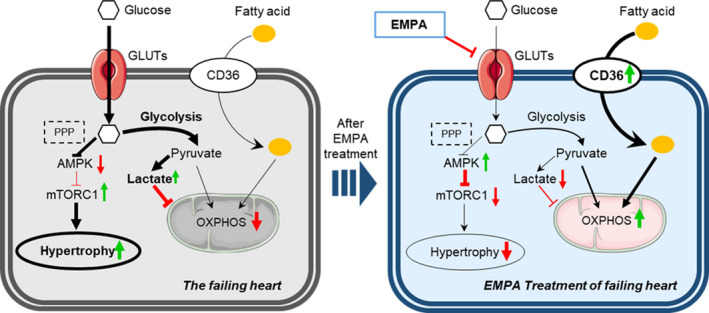
In failing hearts, empagliflozin binding to glucose transporters (eg, GLUT1, GLUT4) reduces the relative excessive glycolysis, increases CD36 (cluster of differentiation 36) expression to restore fatty acid uptake, and improves oxidative phosphorylation. Furthermore, decreased glucose uptake may lead to impaired pentose phosphate pathway, which can activate AMP‐activated protein kinase and inhibit mammalian target of rapamycin complex 1 to attenuate cardiac hypertrophy. AMPK indicates adenosine monophosphate‐activated protein kinase; CD36, cluster of differentiation 36; GLUTs, glucose transporters; mTORC1, mammalian target of rapamycin complex 1; OXPHOS, oxidative phosphorylation; and PPP, the pentose phosphate pathway.
Sources of Funding
This work was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health [grant number P01HL051971]; the National Institute of General Medical Sciences [grant numbers P20GM104357, U54GM115428, and R01GM124108]; the National Institute of Diabetes and Digestive and Kidney Diseases [grant numbers R01 DK121411 and R00 DK113280]; and the National Institute on Aging [grant number R01AG049835].
Disclosures
None.
Supporting information
Supplemental Methods
Tables S1–S7
Figures S1–S7
Acknowledgments
We thank Jingwen Zhang (University of South Florida) for technical support on immunofluorescence; Jia Liu (University of South Florida) for technical support on the Langendorff preparation; and Elizabeth R. Flynn and Sydney P. Moak (University of Mississippi Medical Center) for assistance with western blots, and statistical analysis.
(J Am Heart Assoc. 2021;10:e06021. DOI: 10.1161/JAHA.120.018298.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018298.
For Sources of Funding and Disclosures, see page 15.
References
- 1. Taylor CJ, Ordonez‐Mena JM, Roalfe AK, Lay‐Flurrie S, Jones NR, Marshall T, Hobbs FDR. Trends in survival after a diagnosis of heart failure in the United Kingdom 2000–2017: population based cohort study. BMJ. 2019;364:l223. DOI: 10.1136/bmj.l223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA‐REG OUTCOME(R) trial. Eur Heart J. 2016;37:1526–1534. DOI: 10.1093/eurheartj/ehv728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fitchett D, Inzucchi SE, Wanner C, Mattheus M, George JT, Vedin O, Zinman B, Johansen OE. Relationship between hypoglycaemia, cardiovascular outcomes, and empagliflozin treatment in the EMPA‐REG outcome(R) trial. Eur Heart J. 2020;41:209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. DOI: 10.1056/NEJMoa1911303. [DOI] [PubMed] [Google Scholar]
- 5. Zelniker TA, Braunwald E. Mechanisms of cardiorenal effects of sodium‐glucose cotransporter 2 inhibitors: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2020;75:422–434. DOI: 10.1016/j.jacc.2019.11.031. [DOI] [PubMed] [Google Scholar]
- 6. Cherney DZ, Odutayo A, Aronson R, Ezekowitz J, Parker JD. Sodium glucose cotransporter‐2 inhibition and cardiorenal protection: JACC review topic of the week. J Am Coll Cardiol. 2019;74:2511–2524. DOI: 10.1016/j.jacc.2019.09.022. [DOI] [PubMed] [Google Scholar]
- 7. Cates C, Rousselle T, Wang J, Quan N, Wang L, Chen X, Yang L, Rezaie AR, Li J. Activated protein c protects against pressure overload‐induced hypertrophy through AMPK signaling. Biochem Biophys Res Comm. 2018;495:2584–2594. DOI: 10.1016/j.bbrc.2017.12.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, Cates C, Liu Q, Zheng Y, Li J. Sestrin2 prevents age‐related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. 2017;31:4153–4167. DOI: 10.1096/fj.201700063R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang H, Sun W, Quan N, Wang L, Chu D, Cates C, Liu Q, Zheng Y, Li J. Cardioprotective actions of notch1 against myocardial infarction via lkb1‐dependent AMPK signaling pathway. Biochem Pharmacol. 2016;108:47–57. DOI: 10.1016/j.bcp.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morrison A, Chen L, Wang J, Zhang M, Yang H, Ma Y, Budanov A, Lee JH, Karin M, Li J. Sestrin2 promotes LKB1‐mediated AMPK activation in the ischemic heart. FASEB J. 2015;29:408–417. DOI: 10.1096/fj.14-258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma Y, Wang J, Gao J, Yang H, Wang Y, Manithody C, Li J, Rezaie AR. Antithrombin up‐regulates amp‐activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb Haemost. 2015;113:338–349. DOI: 10.1160/TH14-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu H, Li X, Ren D, Tan Y, Chen J, Yang L, Chen R, Li J, Zhu P. The cardioprotective effects of carvedilol on ischemia and reperfusion injury by AMPK signaling pathway. Biomed Pharmacother. 2019;117:109106. DOI: 10.1016/j.biopha.2019.109106. [DOI] [PubMed] [Google Scholar]
- 13. Li X, Liu J, Hu H, Lu S, Lu Q, Quan N, Rousselle T, Patel MS, Li J. Dichloroacetate ameliorates cardiac dysfunction caused by ischemic insults through AMPK signal pathway‐not only shifts metabolism. Toxicol Sci. 2019;167:604–617. DOI: 10.1093/toxsci/kfy272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ma Y, Li J. Metabolic shifts during aging and pathology. Compr Physiol. 2015;5:667–686. DOI: 10.1002/cphy.c140041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jain R, Liao M. Isolation, culture, and functional analysis of adult mouse cardiomyocytes. Methods Mol Med. 2007;139:251–262. DOI: 10.1007/978-1-59745-571-8_16. [DOI] [PubMed] [Google Scholar]
- 16. Moshal KS, Kumar M, Tyagi N, Mishra PK, Metreveli N, Rodriguez WE, Tyagi SC. Restoration of contractility in hyperhomocysteinemia by cardiac‐specific deletion of NMDA‐R1. Am J Physiol‐Heart Circ Physiol. 2009;296:H887–H892. DOI: 10.1152/ajpheart.00750.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moshal KS, Tipparaju SM, Vacek TP, Kumar M, Singh M, Frank IE, Patibandla PK, Tyagi N, Rai J, Metreveli N, et al. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am J Physiol Heart Circ Physiol. 2008;295:H890–H897. DOI: 10.1152/ajpheart.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bisignano P, Ghezzi C, Jo H, Polizzi NF, Althoff T, Kalyanaraman C, Friemann R, Jacobson MP, Wright EM, Grabe M. Inhibitor binding mode and allosteric regulation of Na(+)‐glucose symporters. Nat Commun. 2018;9:5245. DOI: 10.1038/s41467-018-07700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. Swiss‐model: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. DOI: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu J‐H, Hagaman J, Kim S, Reddick RL, Maeda N. Aortic constriction exacerbates atherosclerosis and induces cardiac dysfunction in mice lacking apolipoprotein E. Arterioscler Thromb Vasc Biol. 2002;22:469–475. DOI: 10.1161/hq0302.105287. [DOI] [PubMed] [Google Scholar]
- 21. Stanek KA, Coleman TG, Murphy WR. Overall hemodynamic pattern in coarctation of the abdominal aorta in conscious rats. Hypertension. 1987;9:611–618. DOI: 10.1161/01.HYP.9.6.611. [DOI] [PubMed] [Google Scholar]
- 22. Steven S, Oelze M, Hanf A, Kroller‐Schon S, Kashani F, Roohani S, Welschof P, Kopp M, Godtel‐Armbrust U, Xia N, et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017;13:370–385. DOI: 10.1016/j.redox.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu Q, Liu J, Li X, Sun X, Zhang J, Ren D, Tong N, Li J. Empagliflozin attenuates ischemia and reperfusion injury through LKB1/AMPK signaling pathway. Mol Cell Endocrinol. 2020;501:110642. DOI: 10.1016/j.mce.2019.110642. [DOI] [PubMed] [Google Scholar]
- 24. Tahara A, Takasu T, Yokono M, Imamura M, Kurosaki E. Characterization and comparison of sodium‐glucose cotransporter 2 inhibitors in pharmacokinetics, pharmacodynamics, and pharmacologic effects. J Pharmacol Sci. 2016;130:159–169. DOI: 10.1016/j.jphs.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 25. Oelze M, Kroller‐Schon S, Welschof P, Jansen T, Hausding M, Mikhed Y, Stamm P, Mader M, Zinssius E, Agdauletova S, et al. The sodium‐glucose co‐transporter 2 inhibitor empagliflozin improves diabetes‐induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS One. 2014;9:e112394. DOI: 10.1371/journal.pone.0112394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Byrne NJ, Parajuli N, Levasseur JL, Boisvenue J, Beker DL, Masson G, Fedak PWM, Verma S, Dyck JRB. Empagliflozin prevents worsening of cardiac function in an experimental model of pressure overload‐induced heart failure. JACC. 2017;2:347–354. DOI: 10.1016/j.jacbts.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bertero E, Prates Roma L, Ameri P, Maack C. Cardiac effects of sglt2 inhibitors: the sodium hypothesis. Cardiovasc Res. 2018;114:12–18. DOI: 10.1093/cvr/cvx149. [DOI] [PubMed] [Google Scholar]
- 28. Griffin M, Rao VS, Ivey‐Miranda J, Fleming J, Mahoney D, Maulion C, Suda N, Siwakoti K, Ahmad T, Jacoby D, et al. Empagliflozin in heart failure: diuretic and cardio‐renal effects. Circulation. 2020;142:1028–1039. DOI: 10.1161/CIRCULATIONAHA.120.045691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mazer CD, Hare GM, Connelly PW, Gilbert RE, Shehata N, Quan A, Teoh H, Leiter LA, Zinman B, Jüni P, et al. Effect of empagliflozin on erythropoietin levels, iron stores and red blood cell morphology in patients with type 2 diabetes and coronary artery disease. Circulation. 2019;141:704–707. DOI: 10.1161/CIRCULATIONAHA.119.044235. [DOI] [PubMed] [Google Scholar]
- 30. Lim VG, Bell RM, Arjun S, Kolatsi‐Joannou M, Long DA, Yellon DM. SGLT2 inhibitor, canagliflozin, attenuates myocardial infarction in the diabetic and nondiabetic heart. JACC Basic Transl Sci. 2019;4:15–26. DOI: 10.1016/j.jacbts.2018.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, Dyck JE, Uddin GM, Oudit GY, Mayoux E, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC. 2018;3:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenstock J, Jelaska A, Zeller C, Kim G, Broedl UC, Woerle HJ; investigators E‐RBt . Impact of empagliflozin added on to basal insulin in type 2 diabetes inadequately controlled on basal insulin: a 78‐week randomized, double‐blind, placebo‐controlled trial. Diabetes Obes Metab. 2015;17:936–948. DOI: 10.1111/dom.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yurista SR, Sillje HHW, Oberdorf‐Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, van Goor H, van Veldhuisen DJ, de Boer RA, Westenbrink BD. Sodium‐glucose co‐transporter 2 inhibition with empagliflozin improves cardiac function in non‐diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. 2019;21:862–873. DOI: 10.1002/ejhf.1473. [DOI] [PubMed] [Google Scholar]
- 34. Kappel BA, Lehrke M, Schutt K, Artati A, Adamski J, Lebherz C, Marx N. Effect of empagliflozin on the metabolic signature of patients with type 2 diabetes mellitus and cardiovascular disease. Circulation. 2017;136:969–972. DOI: 10.1161/CIRCULATIONAHA.117.029166. [DOI] [PubMed] [Google Scholar]
- 35. Pellicori P, Carubelli V. Diuretic treatment in patients with chronic heart failure: evidences, experiences, and current perspectives. G Ital Cardiol. 2017;18:129–138. DOI: 10.1714/2663.27298. [DOI] [PubMed] [Google Scholar]
- 36. Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, Jancev M, Hollmann MW, Weber NC, Coronel R, et al. Class effects of sglt2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia. 2018;61:722–726. DOI: 10.1007/s00125-017-4509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uthman L, Nederlof R, Eerbeek O, Baartscheer A, Schumacher C, Buchholtz N, Hollmann MW, Coronel R, Weber NC, Zuurbier CJ. Delayed ischaemic contracture onset by empagliflozin associates with nhe1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc Res. 2019;115:1533–1545. DOI: 10.1093/cvr/cvz004. [DOI] [PubMed] [Google Scholar]
- 38. Mentzer RM Jr, Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, Haverich A, Knight J, Menasche P, Myers ML, et al. Sodium‐hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the expedition study. Ann Thorac Surg. 2008;85:1261–1270. DOI: 10.1016/j.athoracsur.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 39. Chaitman BR. A review of the guardian trial results: clinical implications and the significance of elevated perioperative CK‐MB on 6‐month survival. J Card Surg. 2003;18(Suppl 1):13–20. DOI: 10.1046/j.1540-8191.18.s1.3.x. [DOI] [PubMed] [Google Scholar]
- 40. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Riehle C, Abel ED. Glut1 deficiency in cardiomyocytes does not accelerate the transition from compensated hypertrophy to heart failure. J Mol Cell Cardiol. 2014;72:95–103. DOI: 10.1016/j.yjmcc.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. DOI: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. 2017;360:12–18. DOI: 10.1016/j.yexcr.2017.03.052. [DOI] [PubMed] [Google Scholar]
- 43. Shiratori R, Furuichi K, Yamaguchi M, Miyazaki N, Aoki H, Chibana H, Ito K, Aoki S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism‐dependent manner. Sci Rep. 2019;9:18699. DOI: 10.1038/s41598-019-55296-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nahle Z, Hsieh M, Pietka T, Coburn CT, Grimaldi PA, Zhang MQ, Das D, Abumrad NA. CD36‐dependent regulation of muscle FOXO1 and PDK4 in the PPAR delta/beta‐mediated adaptation to metabolic stress. J Biol Chem. 2008;283:14317–14326. DOI: 10.1074/jbc.M706478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kaimoto S, Hoshino A, Ariyoshi M, Okawa Y, Tateishi S, Ono K, Uchihashi M, Fukai K, Iwai‐Kanai E, Matoba S. Activation of PPAR‐ALPHA in the early stage of heart failure maintained myocardial function and energetics in pressure‐overload heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H305–H313. DOI: 10.1152/ajpheart.00553.2016. [DOI] [PubMed] [Google Scholar]
- 46. Hua Y, Zhang Y, Ceylan‐Isik AF, Wold LE, Nunn JM, Ren J. Chronic AKT activation accentuates aging‐induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol. 2011;106:1173–1191. DOI: 10.1007/s00395-011-0222-8. [DOI] [PubMed] [Google Scholar]
- 47. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, et al. Mtorc1 regulates cardiac function and myocyte survival through 4E‐BP1 inhibition in mice. J Clin Investig. 2010;120:2805–2816. DOI: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. DOI: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 49. Ren Y, Shen HM. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019;25:101154. DOI: 10.1016/j.redox.2019.101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Beauloye C, Bertrand L, Horman S, Hue L. AMPK activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovasc Res. 2011;90:224–233. DOI: 10.1093/cvr/cvr034. [DOI] [PubMed] [Google Scholar]
- 51. Mustroph J, Wagemann O, Lucht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F, et al. Empagliflozin reduces Ca/calmodulin‐dependent kinase ii activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018;5:642–648. DOI: 10.1002/ehf2.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou Y, Wu W. The sodium‐glucose co‐transporter 2 inhibitor, empagliflozin, protects against diabetic cardiomyopathy by inhibition of the endoplasmic reticulum stress pathway. Cell Physiol Biochem. 2017;41:2503–2512. DOI: 10.1159/000475942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods
Tables S1–S7
Figures S1–S7