

Abstract
Background
Cardiac surgery using cardiopulmonary bypass (CPB) frequently provokes a systemic inflammatory response syndrome, which is triggered by TLR4 (Toll‐like receptor 4) and TNF‐α (tumor necrosis factor α) signaling. Here, we investigated whether the adiponectin receptor 1 and 2 agonist AdipoRon modulates CPB‐induced inflammation and cardiac dysfunction.
Methods and Results
Rats underwent CPB with deep hypothermic circulatory arrest and were finally weaned from the heart‐lung machine. Compared with vehicle, AdipoRon application attenuated the CPB‐induced impairment of mean arterial pressure following deep hypothermic circulatory arrest. During the weaning and postweaning phases, heart rate and mean arterial pressure in all AdipoRon animals (7 of 7) remained stable, while cardiac rhythm was irretrievably lost in 2 of 7 of the vehicle‐treated animals. The AdipoRon‐mediated improvements of cardiocirculatory parameters were accompanied by increased plasma levels of IL (interleukin) 10 and diminished concentrations of lactate and K+. In myocardial tissue, AdipoRon activated AMP‐activated protein kinase (AMPK) while attenuating CPB‐induced degradation of nuclear factor κB inhibitor α (IκBα), upregulation of TNF‐α, IL‐1β, CCL2 (C‐C chemokine ligand 2), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and inducible nitric oxide synthase. Correspondingly, in cultured cardiac myocytes, cardiac fibroblasts, and vascular endothelial cells, AdipoRon activated AMPK, upregulated IL‐10, and attenuated activation of nuclear factor κB, as well as upregulation of TNF‐α, IL‐1β, CCL2, NADPH oxidase, and inducible nitric oxide synthase induced by lipopolysaccharide or TNF‐α. In addition, the treatment of cardiac myocytes with the AMPK activator 5‐aminoimidazole‐4‐carboxamide 1‐β‐D‐ribofuranoside resulted in a similar inhibition of lipopolysaccharide‐ and TNF‐α–induced inflammatory cell phenotypes as for AdipoRon.
Conclusions
Our observations indicate that AdipoRon attenuates CPB‐induced inflammation and impairment of cardiac function through AMPK‐mediated inhibition of proinflammatory TLR4 and TNF‐α signaling in cardiac cells and upregulation of immunosuppressive IL‐10.
Keywords: AdipoRon, cardiopulmonary bypass, ischemia/reperfusion, reactive oxygen species, systemic inflammatory response syndrome
Subject Categories: Basic Science Research, Animal Models of Human Disease, Inflammation, Ischemia, Oxidant Stress
Nonstandard Abbreviations and Acronyms
- AdipoR1
adiponectin receptor 1
- AdipoR2
adiponectin receptor 2
- AICAR
5‐aminoimidazole‐4‐carboxamide 1‐β‐D‐ribofuranoside
- AMPK
AMP‐activated protein kinase
- CCL2
C‐C chemokine ligand 2
- CPB
cardiopulmonary bypass
- DHCA
deep hypothermic circulatory arrest
- HLM
heart‐lung machine
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- HR
heart rate
- I/R
ischemia/reperfusion
- ICAM‐1
intercellular adhesion molecule 1
- IκB
nuclear factor κB inhibitor α
- iNOS
inducible nitric oxide synthase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NF‐κB
nuclear factor κB
- qPCR
quantitative real‐time polymerase chain reaction
- ROS
reactive oxygen species
- rRNA
ribosomal RNA
- TLR4
Toll‐like receptor 4
- VCAM‐1
vascular cell adhesion molecule 1
Clinical Perspective
What Is New?
Using a small animal model of cardiopulmonary bypass–induced systemic inflammatory response syndrome and in vitro experiments with cardiac myocytes, cardiac fibroblasts, and vascular endothelial cells, the present study demonstrates that the adiponectin receptor 1 and 2 agonist AdipoRon attenuates cardiopulmonary bypass–induced inflammation and cardiac dysfunction.
The beneficial effects of AdipoRon act through AMP‐activated protein kinase–mediated inhibition of Toll‐like receptor 4 and tumor necrosis factor α signaling in cardiac cells, as well as upregulation of immunosuppressive interleukin 10.
What Are the Clinical Implications?
These results indicate that AdipoRon is a promising therapeutic candidate to address cardiopulmonary bypass–induced systemic inflammatory response syndrome with the capacity to inhibit the underlying complex and multifactorial pathogenesis.
Cardiopulmonary bypass (CPB) using a heart‐lung machine (HLM) represents a standard technique in modern cardiac surgery. Systemic inflammatory response syndrome (SIRS), a serious disease condition characterized by heavy systemic inflammation, is a frequent complication affecting up to 10% of patients undergoing cardiac surgery employing CPB. 1 CPB‐induced SIRS results in various postoperative complications ranging from short‐term organ dysfunction to multiple organ failure and death. 2 Besides the surgical trauma, blood contact with nonphysiological surfaces, ischemia/reperfusion (I/R), as well as endotoxemia are major triggers of CPB‐induced SIRS. Several proinflammatory signaling pathways (eg, TLR4 [Toll‐like receptor 4] and TNF‐α [tumor necrosis factor α] signaling) and mediators (complement, cytokines, reactive oxygen species [ROS]) act synergistically, finally resulting in deleterious systemic activation of immune and nonimmune cells. 3 Numerous pharmacological strategies (eg, use of glucocorticoids, antioxidants, and antibodies against cytokines) have been developed to attenuate the emergence of SIRS. Until today, none of these approaches have proven to be clinically effective. 1 , 2 , 3
A tremendous number of preclinical and clinical studies under various (patho)physiological conditions have shown that the adipocytokine adiponectin mediates protective metabolic, anti‐inflammatory, and antiapoptotic effects with therapeutic relevance for CPB‐induced SIRS. 4 AdipoRon is a cost‐effective small molecule agonist for the ubiquitously expressed adiponectin receptor 1 (AdipoR1) and adiponectin receptor 2 (AdipoR2) enabling large‐scale clinical applications of AdipoR1‐ and AdipoR2‐mediated effects. 5 Like adiponectin, AdipoRon improves glucose and lipid metabolism as well as insulin sensitivity in vitro and in vivo by mechanisms involving the metabolic master switch AMP‐activated protein kinase (AMPK). 6 Moreover, in an animal study examining myocardial I/R AdipoRon—like adiponectin—reduced postischemic oxidative stress, attenuated cardiac tissue injury, and preserved left ventricular function. 7 These results make a convincing case that targeting adiponectin receptors with AdipoRon is an effective strategy to attenuate the multifactorial cross‐linked pathophysiology of CPB‐induced SIRS.
Thus, in the present study, using a rat model of CPB‐induced SIRS and cell culture experiments with cardiac cells, we investigated whether AdipoRon modulates CPB‐induced inflammation and cardiac dysfunction.
Methods
Data Availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Experimental Animals and Study Design
This study was approved by the supervisory body (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein‐Westfalen, Germany) and conducted in accordance with German and European guidelines of laboratory animal care (approval number 84‐02.04.2016.A085). All animals were kept under standard laboratory conditions and offered food and water ad libidum. Thirty male Wistar rats from Janvier Labs (Le Genest‐Saint‐Isle, France), weighing between 450 and 500 g, were randomly assigned to 3 experimental groups (sham, CPB‐Vehicle, and CPB‐AdipoRon) and subjected to CPB or sham treatment to achieve a size of n=7 per group. To guarantee comparable conditions between the experimental groups, exclusion criteria were defined before the start of the study. Animals were excluded from the study when ≥1 of the following conditions were fulfilled during the experimental procedure: low hemoglobin concentration <5.0 mg/dL during the entire procedure, persistent low mean arterial pressure (MAP) <40 mm Hg following deep hypothermic circulatory arrest (DHCA) not responding to norepinephrine application, prolonged ventricular fibrillation, and failure to convert to sinus rhythm during rewarming (no conversion until 35°C). Finally, 1 animal was excluded because of low hemoglobin concentration, 3 animals were excluded because of technical problems regarding the HLM or other components of the CPB circuit, and another 5 animals were excluded because of catecholamine‐refractory hypotension. All other animals were included in the study (sham: n=7; CPB‐Vehicle: n=7; and CPB‐AdipoRon: n=7).
CPB and Deep Hypothermic Circulatory Arrest
As previously described, 8 animals were intubated and underwent inhalative anesthesia with 2.5 Vol% isoflurane (AbbVie). For CPB, the median sacral artery and the right internal jugular vein were cannulated and connected to a customized small‐animal HLM (Humbs Engineering). The CPB circuit was primed with 8 mL of 6% hydroxyethyl starch (Fresenius Kabi). Moreover, the femoral artery was cannulated to monitor the MAP. After 30 minutes of systemic cooling, DHCA was induced for 45 minutes at a body core temperature of 16°C. Afterwards, CPB was restarted and the animals were subjected to 40 minutes of rewarming and subsequent reperfusion for 60 minutes at a body temperature of 37°C. Then, animals were weaned from CPB over a period of 20 minutes by stepwise 10% reductions of the CPB flow rate, which were performed every 2 minutes. Finally, animals were observed for 10 minutes without cardiocirculatory support, then euthanized and their organs harvested. Sham animals underwent vascular cannulation without initiation of CPB.
AdipoRon (12.5 mg/kg body weight, Sigma‐Aldrich) or vehicle (dimethyl sulfoxide, Sigma‐Aldrich) was applied twice via the shortcut access of the HLM at time points 10 minutes before the start of the CPB and with the beginning of the rewarming phase, respectively.
ECG, peripheral oxygen saturation, and rectal body temperature were monitored throughout the experimental procedure. Arterial electrolyte and blood gas analysis (ABL 700, Radiometer Copenhagen) as well as blood cell count (Vet abc, Scil animal care company) were analyzed at 5 predefined time points (T1: at the beginning of cannulation; T2: at a body temperature of 25°C in the cooling phase; T3: at a body temperature of 20°C in the rewarming phase; T4: at a body temperature of 35°C during the rewarming phase; and T5: at the end of the reperfusion phase). To maintain physiological vital parameters, sodium bicarbonate (8.4%, B. Braun Medical Inc), trometamol (36.34%, B. Braun Medical Inc), or carbon dioxide were applied or an altered ventilation scheme was used. In the rewarming and reperfusion phases at body temperatures of at least 30°C norepinephrine (Arterenol, Sanofi) was applied in case of MAP values <40 mm Hg.
A timeline of the experimental procedures including AdipoRon applications and sample takings is shown in Figure S1.
Troponin T Measurements
Serum levels of troponin T were measured using automated analyzers (Roche Diagnostics) at the Central Institute of Clinical Chemistry and Laboratory Diagnostics of Düsseldorf University Hospital.
Cell Culture
Neonatal cardiac myocytes and fibroblasts were prepared from hearts of 1‐ to 3‐day‐old Wistar rats and subsequently cultured as described. 9 Human aortic endothelial cells were obtained from PromoCell and cultured as recommended by the supplier. TLR4‐grade lipopolysaccharide was purchased from Enzo Life Sciences. AdipoRon, 5‐Aminoimidazole‐4‐carboxamide 1‐β‐D‐ribofuranoside (AICAR), and recombinant human TNF‐α were sourced from Sigma‐Aldrich.
Cytokine ELISA
Concentrations of the cytokines TNF‐α, IL (interleukin) 1β, IL‐6, and IL‐10 in blood plasma or cell culture supernatants were measured using ELISA kits from Biolegend, R&D Systems, and ThermoFisher Scientific, respectively.
Quantitative Real‐Time Polymerase Chain Reaction
RNA was extracted from left ventricular tissue samples or cultured cells by using TRIzol (Invitrogen) and the RNeasy Mini Kit (Qiagen) including a DNAse (Qiagen) treatment to remove residual genomic DNA. RNA concentration and purity was determined with an Infinite M1000 PRO plate reader (TECAN) and RNA integrity was checked using a 2100 Bioanalyzer (Agilent Technologies). cDNA was synthesized using the Quantitect Reverse Transcription Kit (Qiagen) in a T3000 thermocycler (Biometra). Quantitative real‐time polymerase chain reaction (qPCR) was performed in a StepOnePlus Real‐Time PCR System (Applied Biosystems) using the GoTaq PCR Master Mix (Promega) and self‐designed primers against mRNA transcripts encoding rat TNF‐α, IL‐1β, IL‐10, CCL2 (C‐C chemokine ligand 2), inducible nitric oxide synthase (iNOS), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox2 subunit), or hypoxanthine phosphoribosyltransferase 1 (HPRT1) as well as human TNF‐α, IL‐1β, IL‐10, CCL2, iNOS, intercellular adhesion molecule 1 (ICAM‐1), vascular cell adhesion molecule 1 (VCAM‐1), or 18S ribosomal RNA (rRNA). qPCR results were evaluated using the ΔCt method with HPRT1 or 18S rRNA as reference gene for normalization.
For detailed primer sequences please refer to Data S1.
Immunoblot
Proteins were extracted from left ventricular tissue or cells using Cell Lysis Buffer (Cell Signaling Technology) supplemented with proteinase and phosphatase inhibitors (cOmplete Mini and PhosSTOP, Roche). An amount of 50 µg of protein was separated by SDS‐PAGE (7–10% polyacrylamide gels) and transferred to nitrocellulose membranes (Bio‐Rad) via tank blot. Individual protein expression and activation was quantified using primary antibodies against phosphorylated AMPK, AMPK, cleaved caspase‐3, caspase‐3, phosphorylated nuclear factor κB inhibitor α, nuclear factor κB inhibitor (IκBα), β‐Actin and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) in combination with horseradish peroxidase–conjugated secondary antibodies (all Cell Signaling Technology). Protein bands were visualized using a chemiluminescence system (Thermo Fisher). Band intensities were quantified using ImageJ software (National Institutes of Health).
NF‐κB p65 Activation Assay
Activation of nuclear factor κB (NF‐κB) p65 in whole cell lysates was quantified using the TransAm NF‐κB p65 DNA‐binding ELISA (Active Motif).
Statistical Analysis
GraphPad PRISM version 6.07 (GraphPad Software) was used for statistical data analysis. Experimental data were presented as mean±SEM. Only nonparametric tests were applied to assess statistical differences, ie, Kruskal‐Wallis test and subsequent post hoc testing via 2‐sided Mann‐Whitney U test for pairwise comparisons between experimental groups. For Figures 1 and 2 as well as for the Table, statistical differences of analyzed markers were assessed by comparing the experimental groups CPB‐Vehicle and CPB‐AdipoRon at the indicated time points. Moreover, for reasons of clarity, values of P≥0.10 are not shown in Figures 1 and 2 or in Table. For Figures 3, 4, 5, 6, 7, 8, 9, 10 and Figures S2 through S4, statistical differences of marker expression levels between experimental groups were assessed by performing the indicated pairwise comparisons, which were of analytical interest for this study. Moreover, a Holm‐Bonferroni correction was conducted for the pairwise comparisons, which were performed in this study. As an exception, for Figure 1C, the statistical differences were assessed using Fisher exact test. Differences were considered statistically significant at a value of P<0.05.
Figure 1. AdipoRon improves cardiocirculatory recovery after deep hypothermic circulatory arrest and resistance to weaning from the heart‐lung machine.
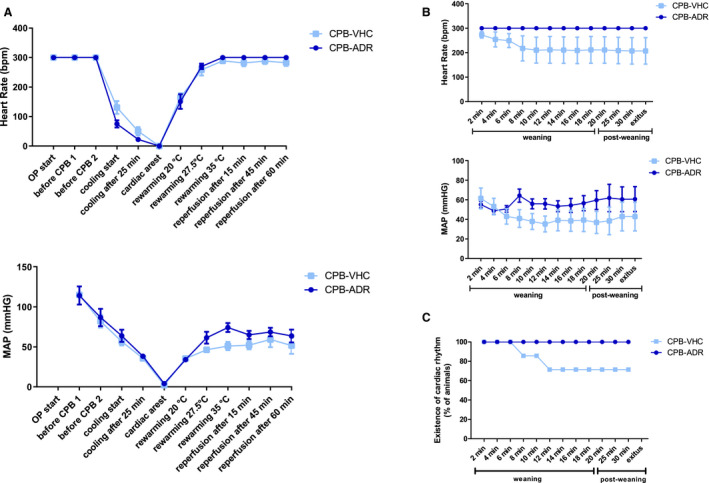
A, Heart rate (HR) and mean arterial pressure (MAP) values were recorded after the start of the operation (OP) throughout the cardiopulmonary bypass (CPB) procedure at the indicated predefined time points. B, HR and MAP values, as well as (C) existence of cardiac rhythm were recorded throughout the weaning and postweaning phases at the indicated predefined time points. HR measurements were restricted by an upper detection limit of 300 beats per minute (bpm). Results are presented as mean±SEM (n=7 animals per group). Differences of HR and MAP values were analyzed statistically by comparing the experimental groups CPB‐Vehicle and CPB‐AdipoRon at the indicated time points. No statistically significant intergroup differences were observed. Values of P≥0.10 are not shown.
Figure 2. AdipoRon modulates cardiopulmonary bypass (CPB)–induced changes in blood concentrations of K+, lactate, and cytokines.
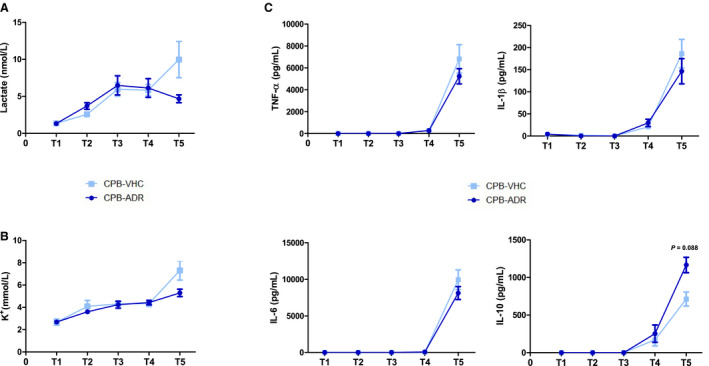
Blood levels of (A) lactate and (B) K+ as well as (C) the cytokines TNF‐α (tumor necrosis factor α), IL (interleukin) 1β, IL‐6, and IL‐10 were measured in AdipoRon‐ and vehicle‐treated rats before CPB (T1), at 25°C in the cooling phase (T2), at 20°C (T3) and 35°C (T4) in the rewarming phase, and after 60 minutes of reperfusion (T5). Results for T1 to T5 are presented as mean±SEM (n=7 animals per group). Differences of blood parameter values were analyzed statistically by comparing the experimental groups CPB‐Vehicle and CPB‐AdipoRon at the indicated time points. No statistically significant intergroup differences were observed. Values of P≥0.10 are not shown.
Table .
AdipoRon Does Not Affect Physiological Blood Parameters During CPB
| CPB—Vehicle | CPB—AdipoRon | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T1 | T2 | T3 | T4 | T5 | T1 | T2 | T3 | T4 | T5 | ||
| Na+, mmol/L | Mean | 136.6 | 138.7 | 141.7 | 138.6 | 142.7 | 137.7 | 137.4 | 140.1 | 137.1 | 138.9 |
| SEM | 1.4 | 0.5 | 0.8 | 1.4 | 2.9 | 1.3 | 1.0 | 1.3 | 2.3 | 1.9 | |
| Ca2+, mmol/L | Mean | 0.31 | 0.82 | 0.87 | 0.70 | 0.84 | 0.36 | 0.80 | 0.85 | 0.81 | 0.80 |
| SEM | 0.05 | 0.02 | 0.06 | 0.04 | 0.05 | 0.07 | 0.04 | 0.03 | 0.07 | 0.02 | |
| Cl−, mmol/L | Mean | 93.2 | 101.6 | 105.0 | 102.2 | 106.8 | 93.0 | 101.0 | 105.8 | 104.3 | 107.5 |
| SEM | 2.1 | 0.7 | 1.2 | 0.9 | 1.6 | 2.9 | 0.4 | 1.1 | 0.9 | 4.5 | |
| Glucose, mg/dL | Mean | 189.6 | 221.0 | 242.7 | 344.0 | 198.9 | 219.9 | 252.3 | 254.6 | 371.4 | 230.9 |
| SEM | 16.0 | 17.8 | 24.2 | 19.1 | 45.9 | 18.5 | 9.6 | 11.1 | 17.6 | 29.5 | |
| Hemoglobin, g/dL | Mean | 11.5 | 8.5 | 7.7 | 8.6 | 8.2 | 11.9 | 8.6 | 7.8 | 8.8 | 8.4 |
| SEM | 0.3 | 0.2 | 0.2 | 0.2 | 0.3 | 0.6 | 0.2 | 0.1 | 0.1 | 0.3 | |
| Hematocrit, % | Mean | 26.2 | 24.5 | 23.1 | 24.9 | 23.3 | 27.0 | 25.0 | 21.6 | 24.0 | 23.5 |
| SEM | 2.1 | 0.7 | 1.0 | 1.0 | 1.3 | 1.4 | 0.9 | 0.4 | 1.0 | 1.4 | |
| Erythrocytes, 106/µL | Mean | 4.8 | 4.5 | 4.1 | 4.6 | 4.2 | 4.9 | 4.7 | 4.1 | 4.6 | 4.5 |
| SEM | 0.4 | 0.1 | 0.1 | 0.1 | 0.2 | 0.3 | 0.2 | 0.1 | 0.2 | 0.2 | |
| White blood cells, 103/mm3 | Mean | 6.5 | 4.6 | 2.8 | 6.6 | 9.4 | 6.3 | 5.0 | 2.6 | 7.4 | 7.2 |
| SEM | 2.7 | 1.9 | 1.1 | 2.7 | 3.8 | 2.8 | 2.2 | 1.2 | 3.0 | 2.9 | |
Physiological blood parameters were measured in AdipoRon‐ and vehicle‐treated rats before CPB (T1), at 25°C in the cooling phase (T2), at 20°C (T3) and 35°C (T4) in the rewarming phase, and after 60 minutes of reperfusion (T5). Results for T1 to T5 are presented as mean±SEM (n=7 animals per group). Differences of blood parameter values were analyzed statistically by comparing the experimental groups cardiopulmonary bypass (CPB)‐Vehicle and CPB‐AdipoRon at the indicated time points. No statistically significant intergroup differences were observed. P values ≥0.10 are not shown.
Figure 3. AdipoRon attenuates cardiopulmonary bypass (CPB)–induced myocardial inflammation.
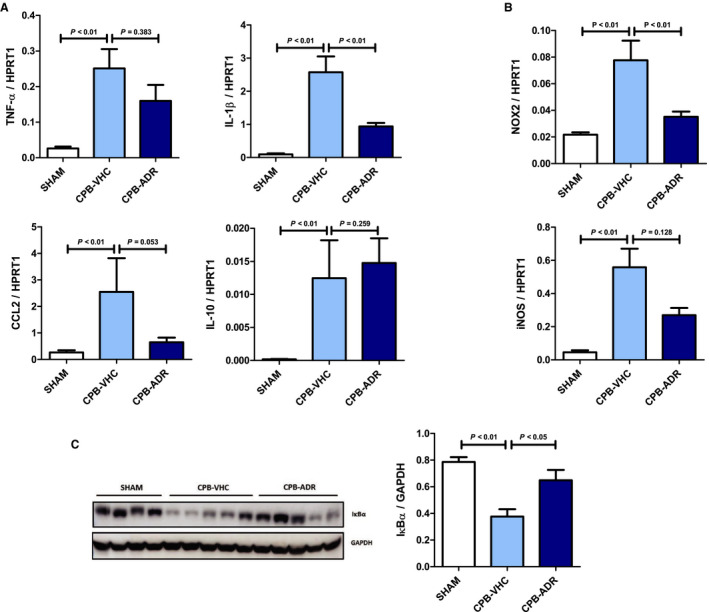
Myocardial mRNA expression of (A) the cytokines/chemokines TNF‐α (tumor necrosis factor α), IL (interleukin) 1β, CCL2 (C‐C chemokine ligand 2), and IL‐10, as well as (B) the enzymes nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox2 subunit) and inducible nitric oxide synthase (iNOS) was measured relative to HPRT1 (hypoxanthine phosphoribosyltransferase 1) by quantitative real‐time polymerase chain reaction in AdipoRon‐ and vehicle‐treated animals after completion of the experimental CPB procedure. C, Expression of nuclear factor κB inhibitor α (IκBα) in myocardial tissue of AdipoRon‐ and vehicle‐treated rats was analyzed by immunoblot. Left panel: representative pictures of the IκBα (molecular weight: 39 kDa) and GAPDH, molecular weight: 37 kDa) band patterns. Right panel: column bars indicate quantified levels of IκBα/GAPDH expression ratios. Results are presented as mean+SEM (n=7 animals per group). Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 4. AdipoRon does not affect cardiopulmonary bypass (CPB)–induced myocardial injury.
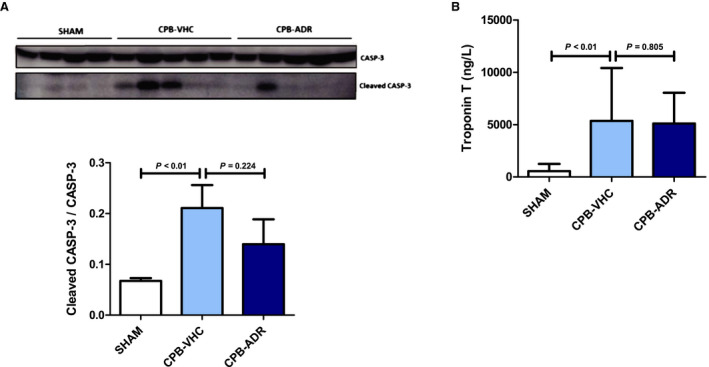
A, Myocardial activation of apoptosis marker caspase‐3 (CASP‐3) in AdipoRon‐ and vehicle‐treated animals was analyzed by immunoblot after completion of the experimental CPB procedure. Upper panel: representative pictures of the uncleaved (molecular weight: 35 kDa) and cleaved CASP‐3 (molecular weight: 17 kDa) band patterns. Lower panel: column bars indicate quantified levels of cleaved CASP‐3/CASP‐3 expression ratios. B, Serum levels of troponin T were measured at the end of the reperfusion phase (T5) in AdipoRon‐ and vehicle‐treated animals. Results are presented as mean+SEM (n=7 animals per group). Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 5. AdipoRon upregulates cardiopulmonary bypass (CPB)–induced myocardial activation of AMP‐activated protein kinase (AMPK).
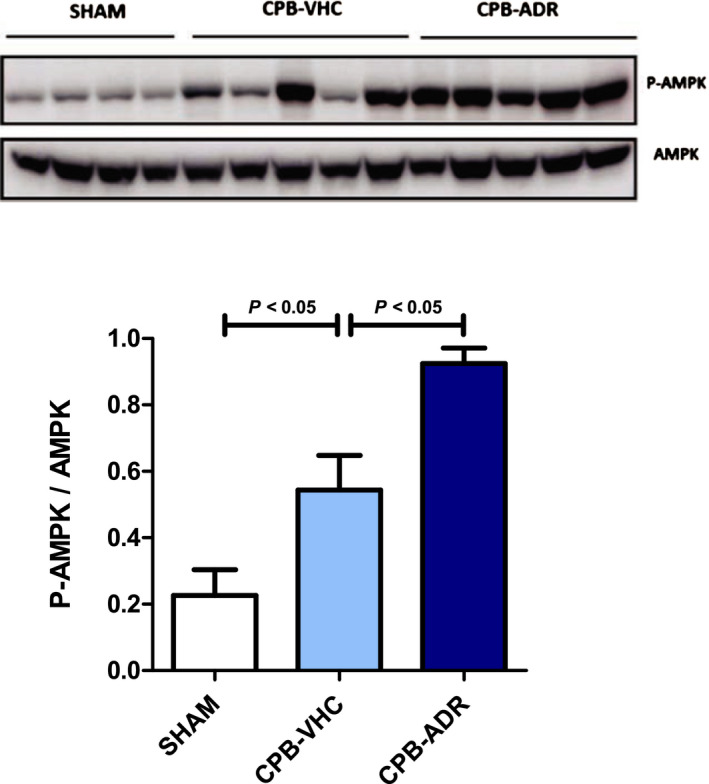
Phosphorylation of AMPK in myocardial tissue of AdipoRon‐ and vehicle‐treated rats was analyzed by immunoblot after completion of the experimental CPB procedure. Upper panel: representative pictures of the resulting phosphorylated AMPK (P‐AMPK; molecular weight: 62 kDa) and AMPK (molecular weight: 62 kDa) band patterns. Lower panel: column bars indicate quantified P‐AMPK/AMPK expression ratios. Results are presented as mean+SEM (n=7 animals per group). Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 6. AdipoRon inhibits the emergence of a systemic inflammatory response syndrome–associated inflammatory phenotype in cardiac myocytes.
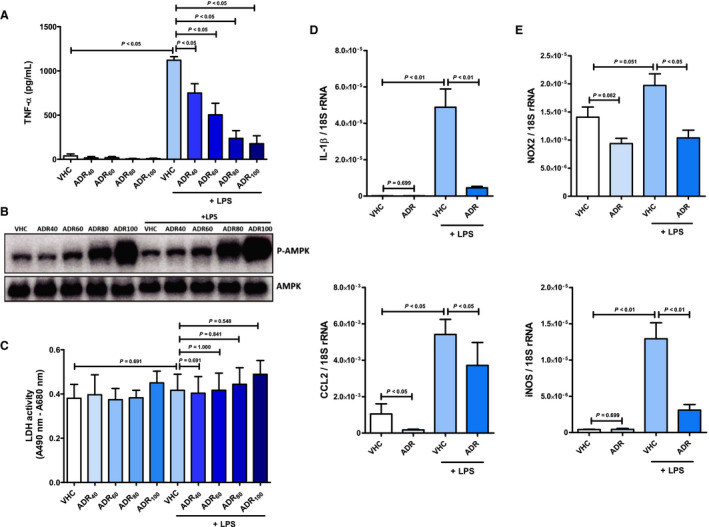
Cardiac myocytes were preincubated with AdipoRon (concentration range 40–100 µmol/L) or vehicle (dimethyl sulfoxide) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. A, Concentration of TNF‐α (tumor necrosis factor α) in culture supernatants, (B) phosphorylation status of AMP‐activated protein kinase (AMPK; molecular weight: 62 kDa) in cell lysates, and (C) content of lactate dehydrogenase (LDH) in culture supernatants was quantified by ELISA (n=5), immunoblot (n=2), and activity assay (n=5), respectively. Cardiac myocytes were preincubated with AdipoRon (80 µmol/L) or vehicle for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. mRNA expression of (D) the cytokine IL (interleukin) 1β and the chemokine CCL2 (C‐C chemokine ligand 2), as well as (E) the enzymes nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox2 subunit) and inducible nitric synthase (iNOS) were measured relative to 18S ribosomal RNA (rRNA) by quantitative real‐time polymerase chain reaction (n=6–7). Results are presented as mean+SEM. Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 7. AdipoRon inhibits the emergence of a systemic inflammatory response syndrome–associated inflammatory phenotype in cardiac fibroblasts.
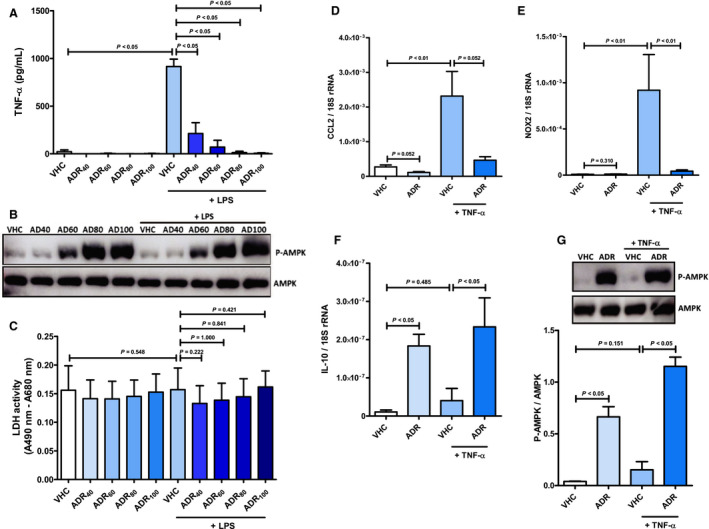
Cardiac fibroblasts were preincubated with AdipoRon (concentration range 40–100 µmol/L) or vehicle (dimethyl sulfoxide) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. A, Concentration of TNF‐α (tumor necrosis factor α) in culture supernatants, (B) phosphorylation status of AMP‐activated protein kinase (AMPK; molecular weight: 62 kDa) in cell lysates and (C) content of lactate dehydrogenase (LDH) in culture supernatants was quantified by ELISA (n=5), immunoblot (n=2), and activity assay (n=5), respectively. Cardiac fibroblasts were preincubated with AdipoRon (80 µmol/L) or vehicle for 2 hours before stimulation with TNF‐α (10 ng/mL) for 1 hour. mRNA expression of (D) the chemokine CCL2 (C‐C chemokine ligand 2), (E) the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox2 subunit), and (F) the cytokine IL (interleukin) 10 was measured relative to 18S ribosomal RNA (rRNA) by quantitative real‐time polymerase chain reaction (n=6). G, Phosphorylation of AMPK in cell lysates was analyzed by immunoblot (n=5). Upper panel: representative pictures of the resulting phosphorylated AMPK (P‐AMPK; molecular weight: 62 kDa) and AMPK (molecular weight: 62 kDa) band patterns. Lower panel: column bars indicate quantified P‐AMPK/AMPK expression ratios. Results are presented as mean+SEM. Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 8. AdipoRon inhibits the emergence of a systemic inflammatory response syndrome–associated inflammatory phenotype in vascular endothelial cells.
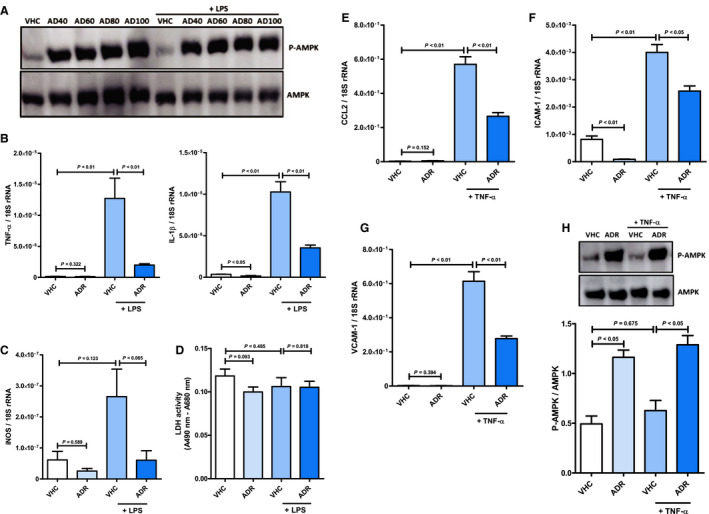
Vascular endothelial cells were preincubated with AdipoRon (concentration range 40–100 µmol/L) or vehicle (dimethyl sulfoxide) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. A, Phosphorylation status of AMP‐activated protein kinase (AMPK; molecular weight: 62 kDa) in cell lysates was quantified by immunoblot (n=2). Vascular endothelial cells were preincubated with AdipoRon (80 µmol/L) or vehicle for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. mRNA expression of (B) the cytokines TNF‐α (tumor necrosis factor α) and IL (interleukin) 1β and (C) the enzyme inducible nitric oxide synthase (iNOS) was measured by quantitative real‐time polymerase chain reaction (n=6). D, The content of lactate dehydrogenase (LDH) in culture supernatants was quantified by activity assay (n=6). Vascular endothelial cells were preincubated with AdipoRon (80 µmol/L) or vehicle for 2 hours before stimulation with TNF‐α (10 ng/mL) for 1 hour. mRNA expression of (E) the chemokine CCL2 (C‐C chemokine ligand 2) and the adhesion molecules (F) intercellular adhesion molecule 1 (ICAM‐1) and (G) vascular cell adhesion molecule 1 (VCAM‐1) was measured relative to 18S ribosomal RNA (rRNA) by quantitative real‐time polymerase chain reaction (n=6). H, Phosphorylation of AMPK in cell lysates was analyzed by immunoblot (n=5). Upper panel: representative pictures of the resulting phosphorylated AMPK (P‐AMPK; molecular weight: 62 kDa) and AMPK (molecular weight: 62 kDa) band patterns. Lower panel: column bars indicate quantified P‐AMPK/AMPK expression ratios. Results are presented as mean+SEM. Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 9. AdipoRon inhibits systemic inflammatory response syndrome–associated proinflammatory TLR4 (Toll‐like receptor 4) and TNF‐α (tumor necrosis factor α) signaling in cardiac myocytes by mechanisms involving upregulation of IL (interleukin) 10 and activation of AMP‐activated protein kinase (AMPK).
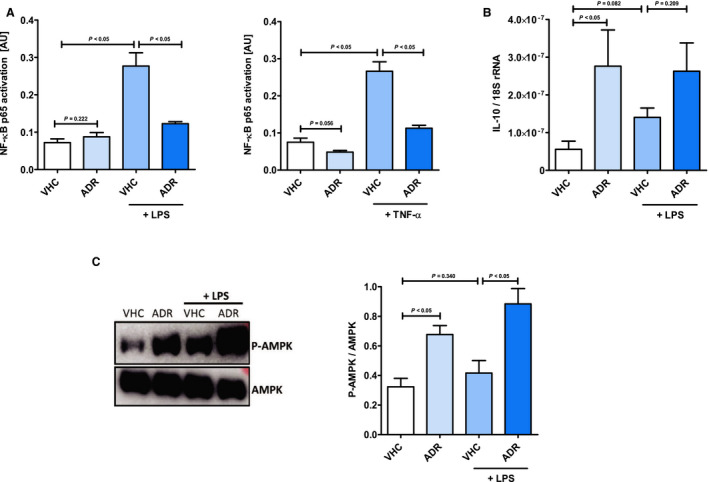
A, Cardiac myocytes were preincubated with AdipoRon (80 µmol/L) or vehicle (dimethyl sulfoxide) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour (left panel) or TNF‐α (10 ng/mL) for 15 minutes (right panel). Activation of nuclear factor κB (NF‐κB) p65 in cell lysates was measured by DNA‐binding ELISA (n=5). Cardiac myocytes were preincubated with AdipoRon (80 µmol/L) or vehicle ) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. B, mRNA expression of the cytokine IL (interleukin) 10 was quantified relative to 18S ribosomal RNA (rRNA) by quantitative real‐time polymerase chain reaction (n=6–7). C, Phosphorylation of AMPK in cell lysates was analyzed by immunoblot (n=5). Left panel: representative pictures of the resulting phosphorylated AMPK (P‐AMPK; molecular weight: 62 kDa) and AMPK (molecular weight: 62 kDa) band patterns. Right panel: column bars indicate quantified phospho‐AMPK/AMPK expression ratios. Results are presented as mean+SEM. Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Figure 10. 5‐Aminoimidazole‐4‐carboxamide 1‐β‐D‐ribofuranoside (AICAR) inhibits the emergence of a systemic inflammatory response syndrome–associated inflammatory phenotype in cardiac myocytes.
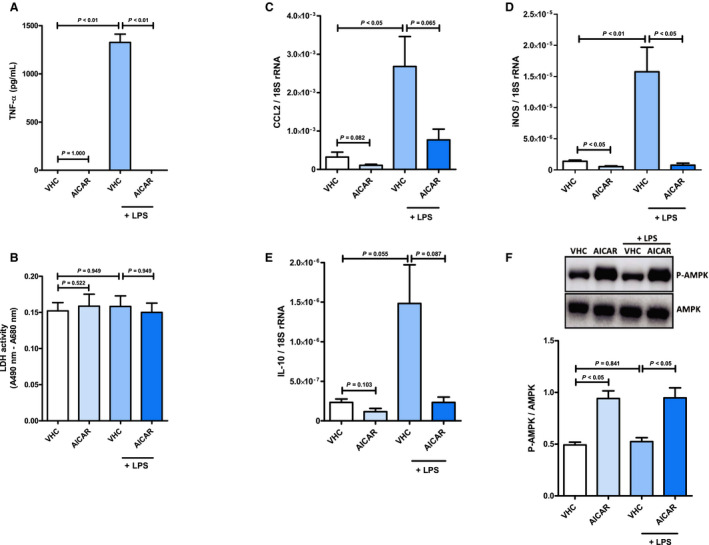
Cardiac myocytes were preincubated with AICAR (1 mmol/L) or vehicle (H2O) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. A, Concentration of TNF‐α (tumor necrosis factor α) in culture supernatants and (B) content of lactate dehydrogenase (LDH) in culture supernatants was quantified by ELISA (n=7) and activity assay (n=7), respectively. Cardiac myocytes were preincubated with AICAR (1 mmol/L) or vehicle (H2O) for 2 hours before stimulation with lipopolysaccharide (1 mg/mL) for 1 hour. mRNA expression of (C) the chemokine CCL2 (C‐C chemokine ligand 2), (D) the enzyme inducible nitric oxide synthase (iNOS), and (E) the cytokine IL (interleukin) 10 was measured relative to 18S ribosomal RNA (rRNA) by quantitative real‐time polymerase chain reaction (n=6). F, Phosphorylation of AMP‐activated protein kinase (AMPK) in cell lysates was analyzed by immunoblot (n=5). Upper panel: representative pictures of the resulting phosphorylated AMPK (P‐AMPK; molecular weight: 62 kDa) and AMPK (molecular weight: 62 kDa) band patterns. Lower panel: column bars indicate quantified P‐AMPK/AMPK expression ratios. Results are presented as mean+SEM. Differences of marker expression levels between experimental groups were analyzed statistically by performing the indicated pairwise comparisons.
Results
AdipoRon Improves Cardiocirculatory Recovery After DHCA
CPB initiated a similar decrease of heart rate (HR) and MAP during cooling in both AdipoRon‐ and vehicle‐treated animals, finally resulting in DHCA (Figure 1A). In the rewarming phase, cardiac rhythm was resumed and at 35°C, HR of both groups had returned to initial values, which were maintained until the end of the reperfusion phase, respectively. Throughout the reperfusion phase, the HR values of AdipoRon‐treated animals remained at the upper detection limit of 300 beats per minute, which was slightly higher than those of the vehicle group. MAP reverted only in part to its initial values following DHCA in both groups. However, AdipoRon application improved MAP recovery, resulting in numerically but not significantly increased values from time point “27.5°C rewarming” throughout the remaining rewarming and reperfusion phases.
AdipoRon Improves Cardiocirculatory Resistance to Weaning From the HLM
During the entire process of weaning from the HLM and the postweaning observational period, HR and MAP values of the AdipoRon group remained stable at the levels observed at the end of the reperfusion phase (Figure 1B). In contrast, shortly after the initiation of weaning in 2 of 7 animals in the vehicle group, the cardiac rhythm was irretrievably lost, ie, asystole, or persistent ventricular fibrillation occurred (Figure 1C). This subsequently resulted in average lower values of HR and MAP in animals in the vehicle group.
Effects of AdipoRon on Blood Levels of Electrolytes, Metabolites, and Hemoglobin
Blood levels of Na+ were stable throughout the CPB procedure for both experimental groups (Table). Moreover, the time courses of Ca2+, Cl−, and glucose concentration showed no differences. Hemoglobin and hematocrit experienced a similar hemodilution‐associated downfall pattern for both experimental groups. In vehicle‐treated animals, the application of CPB with DHCA resulted in a significant increase in plasma levels of lactate, a marker for ischemia‐induced hypoxic metabolism, from 1.3 nmol/L at T1 to 10.0 nmol/L at T5 (Figure 2A). From T1 to T4 lactate levels were similar in AdipoRon‐treated animals. However, at T5, lactate levels of AdipoRon‐treated animals (4.7 nmol/L) were numerically but not significantly lower compared with those in the vehicle group. Increased blood release of K+ into the circulation is an unspecific marker of CPB‐induced organ injury resulting from I/R and inflammation. In animals in the vehicle group, the CPB procedure induced a significant increase in plasma levels of K+ from 2.7 nmol/L at T1 to 7.3 nmol/L at T5 (Figure 2B). From T1 to T4, K+ levels were again similar in AdipoRon‐treated animals, while at T5, the K+ levels (5.3 nmol/L) were numerically but not significantly lower when compared with animals in the vehicle group.
AdipoRon Does Not Affect Compensatory Drug Application
For norepinephrine (Arterenol), sodium bicarbonate (NaBic) and trometamol (TRIS), amounts applied to animals of both experimental groups were not different, respectively (Figure S2).
Effects of AdipoRon on CPB‐Induced Changes of Circulating Cytokine Concentrations
As a result of the CPB procedure, we observed increases in circulating white blood cell counts (Table) and plasma concentrations of the proinflammatory cytokines TNF‐α, IL‐1β, and IL‐6, as well as the anti‐inflammatory cytokine IL‐10, following DHCA from T4 to T5 (Figure 2C) in animals in the vehicle group. While the changes in white blood cell counts and plasma levels of TNF‐α, IL‐1β, and IL‐6 were not affected by AdipoRon, plasma concentrations of IL‐10 were upregulated by trend at T5 (P=0.088) in AdipoRon‐treated animals.
AdipoRon Attenuates CPB‐Induced Myocardial Inflammation
Because of the AdipoRon‐induced improvement of cardiocirculatory parameters, we also looked for associated changes in myocardial inflammation. We observed a significant CPB‐induced upregulation in myocardial expression of cytokines such as TNF‐α, IL‐1β and IL‐10, the chemokine CCL2, as well as the enzymes NADPH oxidase and iNOS, which mediate production of ROS and reactive nitrogen species (Figure 3A and 3B). Whereas IL‐10 was not affected by AdipoRon, the CPB‐induced increase in myocardial expression of TNF‐α (numerically, P=0.383), IL‐1β (P<0.01), and CCL2 (by trend, P=0.053) was attenuated in AdipoRon‐treated animals, respectively. Moreover, myocardial expression of NADPH oxidase and iNOS was attenuated significantly (NADPH oxidase, P<0.01) or numerically (iNOS, P=0.128) in animals in the AdipoRon group.
NF‐κB is a central transcription factor of various proinflammatory signaling pathways controlling the expression of target genes such as TNF‐α, IL‐1β, and CCL2. 10 Proteosomal degradation of the inhibitor protein IκBα results in nuclear translocation and increased transcriptional activity of NF‐κB. Our analysis by immunoblot revealed that employment of CPB resulted in substantial degradation of IκBα in myocardial tissue (Figure 3C). Consistent with its inhibitory effect on the myocardial expression of several inflammatory markers, application of AdipoRon significantly attenuated IκBα degradation (P<0.05) as an essential prerequisite for NF‐κB activation.
AdipoRon Does Not Affect CPB‐Induced Myocardial Injury
Persistent upregulation of proinflammatory cytokines and chemokines as well as sustained release of ROS and reactive nitrogen species eventually triggers myocardial tissue injury. A key element in the process of inflammation‐associated apoptotic tissue injury is activation of caspase‐3 via proteolytic cleavage. Our analysis by immunoblot revealed that myocardial cleavage of caspase‐3 was significantly upregulated (P<0.01 versus sham) in the rat model of CPB‐induced SIRS (Figure 4A). In contrast to the AdipoRon‐mediated inhibitory effects on myocardial inflammation, the CPB‐induced increase in myocardial activation of caspase‐3 was only attenuated numerically (P=0.224) in animals in the AdipoRon group. Moreover, the CPB‐induced increase in circulating levels of troponin T (P<0.01) was not affected by AdipoRon, further indicating absence of significant inhibitory action with regard to myocardial injury (Figure 4B).
AdipoRon Modulates Myocardial Activation of AMPK
Activation of AMPK by phosphorylation of its α subunit ensures cellular homeostasis by mediating protective metabolic, anti‐inflammatory, and antiapoptotic effects. Moreover, AMPK is a central component of signaling cascades originating from AdipoR1/R2. 5 Our analysis by immunoblot (Figure 5) revealed that the CPB procedure itself induced a significant activation of myocardial AMPK (P<0.05). However, the application of AdipoRon triggered a further upregulation (P<0.05) of myocardial AMPK activation.
AdipoRon Inhibits Emergence of a SIRS‐Associated Inflammatory Phenotype in Cardiac Cells
Endotoxemia and massive release of proinflammatory cytokines such as TNF‐α constitute major triggers of CPB‐induced SIRS. 3 To evaluate mechanistically whether AdipoRon acts cardioprotective during CPB‐induced SIRS by inhibiting the emergence of an inflammatory phenotype in cardiac myocytes, cells were preincubated with AdipoRon doses ranging from 40 to 100 µmol/L before stimulation with lipopolysaccharide or TNF‐α. Treatment of cardiac myocytes with AdipoRon dose‐dependently suppressed the lipopolysaccharide‐induced release of TNF‐α (Figure 6A) while simultaneously upregulating phosphorylation, ie, activation of AMPK (Figure 6B). Correspondingly, AdipoRon‐mediated suppression of lipopolysaccharide‐induced TNF‐α release as well as induction of AMPK activation were also observed for cardiac fibroblasts (Figure 7A and 7B). In vascular endothelial cells, maximal AMPK activation was already achieved at an AdipoRon concentration of 40 µmol/L (Figure 8A). Based on the results of the associated cytotoxicity measurements by lactate dehydrogenase assay for cardiac myocytes (Figure 6C) and fibroblasts (Figure 7C), which indicated numerically but not significantly increased lactate dehydrogenase activity in supernatants of cardiac cells preincubated with 100 µmol/L AdipoRon, an AdipoRon dose of 80 µmol/L was finally chosen for all further in vitro experiments, including the experiments with vascular endothelial cells. As part of these experiments, expression analysis by qPCR revealed that preincubation of cardiac myocytes with AdipoRon attenuated the lipopolysaccharide‐induced upregulation of the cytokine IL‐1β (Figure 6D; P<0.01), the chemokine CCL2 (P<0.05), as well as the enzymes NADPH oxidase (Figure 6E; P<0.05) and iNOS (P<0.01). Moreover, AdipoRon inhibited the TNF‐α–induced upregulation of IL‐1β (P<0.001), CCL2 (P<0.001), NADPH oxidase (P<0.01), and iNOS expression (numerical, P=0.114) in cardiac myocytes (Figure S3). In cardiac fibroblasts, we observed corresponding AdipoRon‐mediated attenuation of TNF‐α–induced C CL2 (P=0.052) and NADPH oxidase (P<0.01) expression as shown in Figure 7D and 7E, respectively. Finally, also in vascular endothelial cells, AdipoRon mediated a comprehensive spectrum of anti‐inflammatory effects as evidenced by attenuation of lipopolysaccharide‐induced upregulations of TNF‐α (Figure 8B; P<0.01), IL‐1β (P<0.01), and iNOS (Figure 8C; P=0.065) expression without causing toxic effects, as evidenced by unchanged lactate dehydrogenase activity in culture supernatants (Figure 8D). In addition, AdipoRon also inhibited the TNF‐α–induced upregulations of CCL2 (Figure 8E; P<0.01), ICAM‐1 (Figure 8F; P<0.05), and VCAM‐1 (Figure 8G; P<0.01) in vascular endothelial cells.
AdipoRon Inhibits Proinflammatory TLR4 and TNF‐α Signaling in Cardiac Cells by Mechanisms Involving Upregulation of IL‐10 and Activation of AMPK
NF‐κB is a key regulatory transcription factor in both the proinflammatory TLR4 and TNF‐α signaling pathways. Using an NF‐κB p65 subunit DNA‐binding ELISA, we observed a significant induction of NF‐κB p65 activation (P<0.05) following stimulation of cardiac myocytes with lipopolysaccharide or TNF‐α (Figure 9A). Moreover, we witnessed lipopolysaccharide‐ and TNF‐α–induced upregulations (by trend, P=0.086) of IκBα phosphorylation as prerequisite for NF‐κB p65 activation (Figure S4). Corroborating our results of the aforementioned expression analysis, AdipoRon preincubation inhibited both activation of NF‐κB p65 (P<0.05) and phosphorylation of IκBα (by trend, P=0.086), respectively. Simultaneously, AdipoRon treatment of cardiac myocytes induced a significant upregulation of the anti‐inflammatory cytokine IL‐10 under baseline (P<0.05) and a numerical, not significant, upregulation (P=0.209) under proinflammatory culture conditions (Figure 9B). Moreover, in cardiac fibroblasts, the AdipoRon‐induced upregulation of IL‐10 was significant for both baseline (P<0.05) and proinflammatory (P<0.05) culture conditions (Figure 7F). However, in vascular endothelial cells, there was no IL‐10 expression detectable by qPCR.
Furthermore, as observed for myocardial AMPK in the animal model of CPB‐induced SIRS, the comprehensive anti‐inflammatory effects of AdipoRon were accompanied by significantly upregulated phosphorylation, ie, activation, of AMPK in cardiac myocytes (Figure 9C) and fibroblasts (Figure 7G) as well as vascular endothelial cells (Figure 8H).
Finally, to further clarify the key mechanistic role of AMPK for mediation of anti‐inflammatory and cardioprotective effects induced by AdipoRon in our study, we performed additional in vitro experiments examining effects of the AMPK activator AICAR on the development of a SIRS‐associated inflammatory phenotype in cardiac myocytes. As observed for AdipoRon, treatment of cardiac myocytes with 1‐mmol/L AICAR significantly blocked the lipopolysaccharide‐induced release of TNF‐α (Figure 10A) without mediating toxic side effects, as evidenced by unchanged lactate dehydrogenase activity in culture supernatants (Figure 10B). Moreover, expression analysis by qPCR revealed that—as observed for AdipoRon—preincubation of cardiac myocytes with AICAR attenuated the lipopolysaccharide‐induced upregulation of inflammatory markers such as the chemokine CCL2 (Figure 10C; P=0.065) and the enzyme iNOS (Figure 10D; P<0.05). However, in contrast to AdipoRon, AICAR did not upregulate expression of the anti‐inflammatory cytokine IL‐10 in cardiac myocytes (Figure 10E), ie, the IL‐10 expression remained unchanged under baseline conditions and was downregulated by trend (P=0.087) under proinflammatory culture conditions in response to AICAR stimulation. Finally, as expected, AICAR significantly upregulated (P<0.05, respectively) phosphorylation, ie, activation, of AMPK in cardiac myocytes (Figure 10F) under both baseline and proinflammatory culture conditions.
Discussion
In this study, we report that application of the synthetic adiponectin receptor agonist AdipoRon is beneficial in a rat model of CPB‐induced SIRS by modulating systemic and myocardial inflammation thus attenuating impairment of myocardial function.
AMPK is a key mediator of metabolic, anti‐inflammatory, and cytoprotective/organoprotective effects with relevance for CPB‐induced SIRS. It inhibits energy‐consuming and stimulates energy‐producing metabolic processes avoiding hypoxanthine accumulation during ischemia and thus attenuating ROS generation after reperfusion. 11 Correspondingly, synthetic activators of AMPK have been shown to ameliorate I/R‐induced inflammation and injury in cardiac myocytes in vitro 12 and in vivo. 13 Consistent with the fact that AMPK is a central component of signaling cascades originating from AdipoR1/R2, 5 our analysis by immunoblot shows that AdipoRon application in the model of CPB‐induced SIRS actually results in increased myocardial activation of AMPK. This finding indicates that administered AdipoRon is readily delivered to the heart where it effectively activates AMPK‐dependent cardioprotective processes.
An associated major finding of our study is that AdipoRon modulates CPB‐induced inflammation at both systemic and cardiac level. As part of the inflammatory cascade of CPB‐induced SIRS, large amounts of various immunomodulatory cytokines are released systemically. 3 Correspondingly, we observed substantial increases in plasma levels of TNF‐α, IL‐1β, IL‐6, and IL‐10 following DHCA in our rat model of CPB‐induced SIRS. Whereas TNF‐α, IL‐1β, and IL‐6 were not affected by AdipoRon, it upregulated plasma levels of the anti‐inflammatory IL‐10, which has been shown to attenuate development of organ failure in human SIRS. 14 In the heart, we observed CPB‐induced increases in myocardial expression of TNF‐α, IL‐1β, CCL2, and ROS‐ and reactive nitrogen species–producing NADPH oxidase and iNOS, which were mostly attenuated significantly or by trend following AdipoRon application. Moreover, AdipoRon inhibited the CPB‐induced myocardial degradation of IκBα as prerequisite for activation of transcription factor NF‐κB, which controls the expression of target genes such as TNF‐α, IL‐1β, CCL2, NADPH oxidase, and iNOS in immune and nonimmune cells. 10 These findings are consistent with a previous study by Zhang et al, 15 which demonstrated that application of AdipoRon attenuated the upregulation of left ventricular NADPH oxidase expression triggered by myocardial I/R. Moreover, systemic overexpression of adiponectin in murine autoimmune myocarditis resulted in comprehensive inhibition of myocardial inflammation as evidenced by diminished expression of proinflammatory cytokines and chemokines such as TNF‐α, IL‐1β, and CCL2. 16 Although AdipoRon did attenuate myocardial inflammation and impairment of cardiac function in rats subjected to CPB with DHCA, these effects were not accompanied by a limitation of myocardial injury in the present study. However, in 2 earlier studies examining the effects of selenium 17 and DNAse I 18 in a rat model of CPB‐induced SIRS, we observed that treatment‐induced improvements of ROS formation/persistence and inflammation do not necessarily go along with immediate alleviation of cardiac and pulmonary tissue injury in our disease model. Thus, whether an attenuation of cardiac injury associated with AdipoRon‐mediated improvements of CPB and DHCA‐related inflammation may occur at later time points has to be examined in future studies including an extended postweaning observational period.
SIRS‐associated myocardial inflammation has been shown to decrease cardiac contractility and impair myocardial compliance, resulting in acute heart failure, because myocyte contractility is inhibited by proinflammatory cytokines, ROS, and reactive nitrogen species. 19 Consistent with diminished myocardial inflammation in animals in the AdipoRon group, we observed that AdipoRon treatment attenuates CPB‐induced impairment of HR and MAP following DHCA. Moreover, during the weaning and postweaning phases, HR and MAP values in all animals in the AdipoRon group (7 of 7) remained at levels observed immediately before the start of the weaning phase, while in 2 of 7 of the dimethyl sulfoxide–treated animals the cardiac rhythm was irretrievably lost. These AdipoRon‐mediated enhancements of HR and MAP seem to result in improved tissue perfusion and oxygen supply, as following DHCA we observed reduced blood levels of lactate—a marker of ischemia‐induced hypoxic metabolism 20 —in AdipoRon‐treated animals.
Endotoxemia is a major trigger of CPB‐induced SIRS as systemically released endotoxin/lipopolysaccharide potently elicits inflammatory responses via activation of TLR4 signaling. 21 Moreover, proinflammatory cytokines such as TNF‐α are central mediators reinforcing CPB‐induced SIRS, 3 resulting in deleterious sustained hyperactivation of immune and nonimmune cells. The results of our in vitro experiments using cardiac cells provide clear mechanistic indications on how AdipoRon acts anti‐inflammatory and cardioprotective in the animal model of CPB‐induced SIRS. As observed in vivo, in cultured cardiac myocytes, cardiac fibroblasts, and vascular endothelial cells—representing the major SIRS‐relevant types of cardiac cells—AdipoRon treatment dose‐dependently and comprehensively inhibited the upregulation of various SIRS‐related proinflammatory markers such as TNF‐α, IL‐1β, CCL2, NADPH oxidase, and iNOS, while simultaneously attenuating phosphorylation of IκBα and subsequent activation of the proinflammatory transcription factor NF‐κB triggered by lipopolysaccharide and TNF‐α, respectively. Corresponding inhibitory effects with regard to TLR4 and TNF‐α signaling in cardiac myocytes have been previously described for adiponectin. 16 , 22 Moreover, AdipoRon induced a significant upregulation of the anti‐inflammatory cytokine IL‐10 in cardiac myocytes and fibroblasts. Although we observed no comparable in vivo effect for myocardial tissue, AdipoRon application induced by trend increased plasma levels of IL‐10, which has been shown to contribute to the immunoinhibitory effects of adiponectin. 23 Furthermore, as observed for myocardial tissue in vivo, the anti‐inflammatory effects of AdipoRon in cardiac myocytes, cardiac fibroblasts and vascular endothelial cells were accompanied by significant activation of AMPK, the key mediator of anti‐inflammatory and cardioprotective effects triggered by AdipoR1/R2 signaling. 5 Similar to our study examining the effects of AdipoRon, Wang et al 24 observed that administration of the AMPK activator AICAR in a rat model of CPB was associated with increased activation of myocardial AMPK and AMPK‐associated downstream signaling pathways. Thus, AICAR attenuated CPB‐induced myocardial injury and dysfunction as evidenced by plasma levels of cardiac troponin I, echocardiographic measurement of left ventricular ejection fraction, and left ventricular end‐systolic dimension. Moreover, in various studies employing murine models of lipopolysaccharide‐induced septic myocardial injury in vivo, administration of AMPK activators such as AICAR or metformin resulted in comprehensive cardioprotection 25 , 26 , 27 associated with suppression of TLR4 signaling–induced myocardial inflammation and injury. In addition, in a murine model of myocardial I/R, Kim et al 28 demonstrated that activation of myocardial AMPK and associated downstream signaling triggered by the small molecule activator A769662 protects the heart against injury by inhibiting myocardial apoptosis and necrosis. Accordingly, Zhang et al 7 showed that AdipoRon‐induced attenuation of myocardial oxidative stress and tissue injury as well as preservation of left ventricular function following myocardial‐specific I/R is essentially mediated by AMPK. Finally, systemic adiponectin overexpression attenuates myocardial inflammation in murine autoimmune myocarditis through AMPK‐mediated inhibition of proinflammatory TLR4 and TNF‐α signaling. 16 , 22 The results of our in vitro experiments examining the effects of the AMPK activator AICAR on the emergence of a SIRS‐associated inflammatory phenotype in cardiac myocytes further support our hypothesis that AdipoRon attenuates CPB‐induced SIRS by activating AMPK, as AICAR—similar to AdipoRon—induced a comprehensive spectrum of anti‐inflammatory effects inhibiting lipopolysaccharide‐induced upregulation of various SIRS‐associated proinflammatory markers such as TNF‐α, CCL2, and iNOS. The fact that AICAR did not induce a simultaneous upregulation of the anti‐inflammatory cytokine IL‐10 in cardiac myocytes, neither under baseline nor under proinflammatory culture conditions, indicates that the AdipoRon‐induced upregulation of IL‐10 is independent of AMPK activation. This is conceivable, since AMPK represents the major mediator of signaling originating from AdipoR1/R2, but otherwise also an activation of various AMPK‐independent downstream signaling pathways has been observed. 29 However, the similarity of AICAR‐ and AdipoRon‐mediated inhibitory effects with regard to emergence of TNF‐α and TLR4 signaling–induced inflammatory phenotypes in cardiac myocytes illustrates that AMPK is the essential mediator of AdipoRon‐related immunomodulation and cardioprotection in our rat model of CPB‐induced SIRS.
Several limitations of our study have to be considered. First, it is a pilot study that examined only a single AdipoRon dose/application regimen that included only a relatively small number of animals. Moreover, regarding the evaluation of myocardial function, there were no hemodynamic measurements included and the significance of HR measurements was restricted by an upper detection limit. Furthermore, our study only analyzed acute effects of AdipoRon, ie, its impact on long‐term parameters of cardiac recovery—such as remodeling and hemodynamics 30 —was not assessed. In addition, because of our lack of experience with the postweaning stability of the animals we chose, a postweaning observational period, which was possibly too short to assess whether the observed protective effects of AdipoRon, are maintained long enough to attenuate the long‐term detrimental effects of CPB‐induced SIRS and to make the AdipoRon‐induced improvements in cardiocirculatory resistance fully visible. However, to allow a meaningful analysis of the protective long‐term effects of AdipoRon with regard to cardiac inflammation, injury, remodeling, and hemodynamics, we plan to expand the analytical setup of our rat model of CPB‐induced SIRS, ie, animals will be weaned from the CPB and then wake up from anesthesia to be observed for up to 14 days postoperatively. Moreover, this approach would allow to examine the effects of additional repeated AdipoRon administrations during the follow‐up observational phase on the long‐term course of the CPB‐induced SIRS. Besides, the clinical application of CPB with DHCA is currently restricted to extended aortic surgery and complex pediatric cardiac surgery. However, DHCA enables extended periods of global ischemia and facilitates the subsequent restoration of cardiac rhythm. Moreover, the establishment of CPB with nonhypothermic cardiac arrest is challenging in small animal models. 31 Finally, possibly because of the relatively small sizes of the treatment groups in our study, we repeatedly observed effects of AdipoRon treatment in our in vivo and in vitro experiments, which lacked statistical significance.
Conclusions
Our study indicates that AdipoRon is a promising therapeutic candidate to address CPB‐induced SIRS with the capacity to attenuate the underlying complex and multifactorial pathogenesis. Thus, it will be worth testing alternative dose/application regimens and novel synthetic agonists with further improved AdipoR1/AdioR2 binding affinity and specificity as well as optimized absorption, distribution, metabolism, and excretion properties.
Sources of Funding
This work was supported by grant number 34/2015 of the Research Committee Funding Program of the Medical Faculty, Heinrich Heine University Düsseldorf (Düsseldorf, Germany) to Jenke. Yazdanyar was supported by a research grant provided by the Christiane and Claudia Hempel‐Foundation for Clinical Stem Cell Research (Düsseldorf, Germany) to Akhyari, Jenke, and Lichtenberg.
Disclosures
None.
Supporting information
Data S1
Figures S1–S4
Acknowledgments
The authors thank the Susanne Bunnenberg Heart Foundation for the financial contribution supporting the acquisition of laboratory equipment at the Cardiovascular Center of Düsseldorf University Hospital. Open access funding was enabled and organized by ProjektDEAL.
(J Am Heart Assoc. 2021;10:e018097. DOI: 10.1161/JAHA.120.018097.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018097
For Sources of Funding and Disclosures, see page 16.
References
- 1. Boeken U, Feindt P. Is a sirs/sepsis syndrome after cardiac surgery a consequence of extracorporeal circulation? Z Herz Thorax Gefasschir. 2008;22:110–118. [Google Scholar]
- 2. Born F, Pichlmaier M, Peterß S, Khaladj N, Hagl C. Systemic inflammatory response syndrome in heart surgery: new possibilities for treatment through the use of a cytokine adsorber during ECC? Kardiotechnik. 2014;23:41–46. [Google Scholar]
- 3. Paparella D, Yau TM, Young E. Cardiopulmonary bypass induced inflammation: pathophysiology and treatment. An update. Eur J Cardiothorac Surg. 2002;21:232–244. DOI: 10.1016/S1010-7940(01)01099-5. [DOI] [PubMed] [Google Scholar]
- 4. Holland WL, Scherer PE. Cell biology. Ronning after the adiponectin receptors. Science. 2013;342:1460–1461. DOI: 10.1126/science.1249077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buechler C, Wanninger J, Neumeier M. Adiponectin receptor binding proteins–recent advances in elucidating adiponectin signalling pathways. FEBS Lett. 2010;584:4280–4286. DOI: 10.1016/j.febslet.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 6. Okada‐Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami KI, Matsuda K, Yamaguchi M, Tanabe H, Kimura‐Someya T, Shirouzu M, et al. A small‐molecule adipor agonist for type 2 diabetes and short life in obesity. Nature. 2013;503:493–499. DOI: 10.1038/nature12656. [DOI] [PubMed] [Google Scholar]
- 7. Zhang Y, Zhao J, Li R, Lau WB, Yuan Y‐X, Liang B, Li R, Gao E‐H, Koch WJ, Ma X‐L, et al. AdipoRon, the first orally active adiponectin receptor activator, attenuates postischemic myocardial apoptosis through both AMPK‐mediated and AMPK‐independent signalings. American Journal of Physiology‐Endocrinology and Metabolism. 2015;309:E275–E282. DOI: 10.1152/ajpendo.00577.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pinto A, Jahn A, Immohr MB, Jenke A, Dohrn L, Kornfeld M, Lichtenberg A, Akhyari P, Boeken U. Modulation of immunologic response by preventive everolimus application in a rat CPB model. Inflammation. 2016;39:1771–1782. DOI: 10.1007/s10753-016-0412-5. [DOI] [PubMed] [Google Scholar]
- 9. Vetter R, Kott M, Schulze W, Rupp H. Influence of different culture conditions on sarcoplasmic reticular calcium transport in isolated neonatal rat cardiomyocytes. Mol Cell Biochem. 1998;188:177–185. [PubMed] [Google Scholar]
- 10. Oeckinghaus A, Ghosh S. The NF‐kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collard CD, Gelman S. Pathophysiology, clinical manifestations, and prevention of ischemia‐reperfusion injury. Anesthesiology. 2001;94:1133–1138. DOI: 10.1097/00000542-200106000-00030. [DOI] [PubMed] [Google Scholar]
- 12. Yeh CH, Chen TP, Wang YC, Lin YM, Fang SW. AMP‐activated protein kinase activation during cardioplegia‐induced hypoxia/reoxygenation injury attenuates cardiomyocytic apoptosis via reduction of endoplasmic reticulum stress. Mediators Inflamm. 2010;2010:130636. DOI: 10.1155/2010/130636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cieslik KA, Taffet GE, Crawford JR, Trial J, Mejia Osuna P, Entman ML. AICAR‐dependent AMPK activation improves scar formation in the aged heart in a murine model of reperfused myocardial infarction. J Mol Cell Cardiol. 2013;63:26–36. DOI: 10.1016/j.yjmcc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kawai S, Sakayori S, Watanabe H, Nakagawa T, Inoue G, Kobayashi H. The role of interleukin‐10 in systemic inflammatory response syndrome with sepsis. J Infect Chemother. 1998;4:121–127. DOI: 10.1007/BF02491513. [DOI] [Google Scholar]
- 15. Zhang Y, Zhao J, Li R, Lau WB, Yuan YX, Liang B, Li R, Gao EH, Koch WJ, Ma XL, et al. AdipoRon, the first orally active adiponectin receptor activator, attenuates postischemic myocardial apoptosis through both AMPK‐mediated and AMPK‐independent signalings. Am J Physiol Endocrinol Metab. 2015;309:E275–E282. DOI: 10.1152/ajpendo.00577.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jenke A, Wilk S, Poller W, Eriksson U, Valaperti A, Rauch BH, Stroux A, Liu P, Schultheiss H‐P, Scheibenbogen C, et al. Adiponectin protects against Toll‐like receptor 4‐mediated cardiac inflammation and injury. Cardiovasc Res. 2013;99:422–431. DOI: 10.1093/cvr/cvt118. [DOI] [PubMed] [Google Scholar]
- 17. Steinbrenner H, Bilgic E, Pinto A, Engels M, Wollschlager L, Dohrn L, Kellermann K, Boeken U, Akhyari P, Lichtenberg A. Selenium pretreatment for mitigation of ischemia/reperfusion injury in cardiovascular surgery: influence on acute organ damage and inflammatory response. Inflammation. 2016;39:1363–1376. DOI: 10.1007/s10753-016-0368-5. [DOI] [PubMed] [Google Scholar]
- 18. Weber C, Jenke A, Chobanova V, Yazdanyar M, Chekhoeva A, Eghbalzadeh K, Lichtenberg A, Wahlers T, Akhyari P, Paunel‐Gorgulu A. Targeting of cell‐free DNA by DNase I diminishes endothelial dysfunction and inflammation in a rat model of cardiopulmonary bypass. Sci Rep. 2019;9:19249. DOI: 10.1038/s41598-019-55863-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y, Ge S, Peng Y, Chen X. Inflammation and cardiac dysfunction during sepsis, muscular dystrophy, and myocarditis. Burns Trauma. 2013;1:109–121. DOI: 10.4103/2321-3868.123072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee D, Sohn H, Park ZY, Oh S, Kang Y, Lee KM, Kang M, Jang Y, Yang SJ, Hong Y, et al. A lactate‐induced response to hypoxia. Cell. 2015;161:595–609. DOI: 10.1016/j.cell.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 21. Riddington DW, Venkatesh B, Boivin CM, Bonser RS, Elliott TS, Marshall T, Mountford PJ, Bion JF. Intestinal permeability, gastric intramucosal pH, and systemic endotoxemia in patients undergoing cardiopulmonary bypass. JAMA. 1996;275:1007–1012. DOI: 10.1001/jama.1996.03530370045029. [DOI] [PubMed] [Google Scholar]
- 22. Bobbert P, Scheibenbogen C, Jenke A, Kania G, Wilk S, Krohn S, Stehr J, Kuehl U, Rauch U, Eriksson U, et al. Adiponectin expression in patients with inflammatory cardiomyopathy indicates favourable outcome and inflammation control. Eur Heart J. 2011;32:1134–1147. DOI: 10.1093/eurheartj/ehq498. [DOI] [PubMed] [Google Scholar]
- 23. Wolf AM, Wolf D, Rumpold H, Enrich B, Tilg H. Adiponectin induces the anti‐inflammatory cytokines IL‐10 and IL‐1RA in human leukocytes. Biochem Biophys Res Commun. 2004;323:630–635. DOI: 10.1016/j.bbrc.2004.08.145. [DOI] [PubMed] [Google Scholar]
- 24. Wang Z, Cao Y, Yin Q, Han Y, Wang Y, Sun G, Zhu H, Xu M, Gu C. Activation of AMPK alleviates cardiopulmonary bypass‐induced cardiac injury via ameliorating acute cardiac glucose metabolic disorder. Cardiovasc Ther. 2018;36:e12482. DOI: 10.1111/1755-5922.12482. [DOI] [PubMed] [Google Scholar]
- 25. Tzanavari T, Varela A, Theocharis S, Ninou E, Kapelouzou A, Cokkinos DV, Kontaridis MI, Karalis KP. Metformin protects against infection‐induced myocardial dysfunction. Metabolism. 2016;65:1447–1458. DOI: 10.1016/j.metabol.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vaez H, Rameshrad M, Najafi M, Barar J, Barzegari A, Garjani A. Cardioprotective effect of metformin in lipopolysaccharide‐induced sepsis via suppression of toll‐like receptor 4 (TLR4) in heart. Eur J Pharmacol. 2016;772:115–123. DOI: 10.1016/j.ejphar.2015.12.030. [DOI] [PubMed] [Google Scholar]
- 27. Liu G, Wu K, Zhang L, Dai J, Huang W, Lin L, Ge P, Luo F, Lei H. Metformin attenuated endotoxin‐induced acute myocarditis via activating AMPK. Int Immunopharmacol. 2017;47:166–172. DOI: 10.1016/j.intimp.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 28. Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, Zaha V, Sakamoto K, Young LH. A small molecule AMPK activator protects the heart against ischemia–reperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. DOI: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity‐related diseases. Cell Metab. 2013;17:185–196. DOI: 10.1016/j.cmet.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 30. Zhang N, Wei WY, Liao HH, Yang Z, Hu C, Wang SS, Deng W, Tang QZ. Adiporon, an adiponectin receptor agonist, attenuates cardiac remodeling induced by pressure overload. J Mol Med (Berl). 2018;96:1345–1357. DOI: 10.1007/s00109-018-1696-8. [DOI] [PubMed] [Google Scholar]
- 31. Jungwirth B, de Lange F. Animal models of cardiopulmonary bypass: development, applications, and impact. Semin Cardiothorac Vasc Anesth. 2010;14:136–140. DOI: 10.1177/1089253210370491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figures S1–S4
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.