

Abstract
Background
White blood cell count, which is inexpensive and widely available in clinical practice, has been proposed to provide prognostic information in coronary artery disease (CAD). Elevated levels of white blood cell subtypes may play different roles in atherothrombosis and predict cardiovascular outcomes.
Methods and Results
The association between white blood cell counts and mortality was evaluated in 823 subjects with angiographically demonstrated and clinically stable CAD in an observational–longitudinal study. The correlation among white blood cell counts and factor II plasma coagulant activity was analyzed in 750 subjects (554 CAD and 196 CAD‐free) not taking anticoagulant drugs. Subjects with overt leukocytosis or leukopenia were excluded. In the longitudinal study after a median follow‐up of 61 months, 160 (19.4%) subjects died, 107 (13.0%) of whom from cardiovascular causes. High levels of neutrophils, monocytes, eosinophils, and basophils were associated with an increased mortality rate. In multiadjusted Cox regression models, only neutrophils and basophils remained predictors of total and cardiovascular mortality. The associations remained significant after adjustment for traditional cardiovascular risk factors and by including D‐dimer and the chemokine CXCL12 in the regression models. Neutrophils and basophils were also significant predictors of factor II plasma coagulant activity variability after adjustment for blood cell counts, age, sex, inflammatory markers, CAD diagnosis, and prothrombin G20210A polymorphism. Factor II plasma coagulant activity was similarly increased in subjects with high neutrophil and basophil counts and in carriers of the prothrombin 20210A allele.
Conclusions
Both high neutrophil and basophil blood counts may predict mortality in patients with clinically stable CAD and are associated with enhanced factor II plasma coagulant activity, thereby suggesting underlying prothrombotic mechanisms.
Keywords: basophils, factor II plasma coagulant activity, neutrophils, secondary prevention of coronary artery disease, white blood cell count
Subject Categories: Chronic Ischemic Heart Disease, Secondary Prevention, Cardiovascular Disease, Prognosis, Biomarkers
Nonstandard Abbreviations and Acronyms
- CXCL12
C‐X‐C motif chemokine 12
- FII:c
factor II plasma coagulant activity
- NETs
neutrophil extracellular traps
- VHS
Verona Heart Study
Clinical Perspective
What Is New?
White blood cell subtypes have a different role in modulating procoagulant diathesis, with neutrophil and basophil count showing a direct correlation with factor II coagulant activity.
Basophil count is an independent predictor of total and cardiovascular mortality in patients with clinically stable coronary artery disease.
What Are the Clinical Implications?
Basophils, which are usually the less‐considered white blood cell subtype in clinical practice, may provide information about the individual procoagulant propensity and cardiovascular risk, potentially being a useful prognostic biomarker for patient‐tailored therapeutic choices.
In spite of major diagnostic and therapeutic improvements in the past decades, cardiovascular disease (CVD) is still responsible for 30% of deaths worldwide. Even when all of the traditional cardiovascular risk factors are reduced using the best available pharmacological therapy and lifestyle counseling, a substantial residual risk still remains unaddressed. 1 , 2
Atherosclerosis is the main pathological condition underlying CVD and is widely recognized as a chronic inflammatory disease driven by lipids and leukocytes. Leukocytes play a pivotal role in atherosclerosis, and consistently, white blood cell (WBC) count has been linked with CVD by several studies. 3 , 4 , 5 , 6
Counts of almost all subtypes of leukocytes have been associated with the risk of CVD, thereby suggesting WBC count as a potential predictor of cardiovascular risk. 7 , 8 Leukocytes have been implicated in all the phases of atherosclerosis development and progression, and their prognostic significance has been investigated in both primary and secondary cardiovascular prevention, including recurrent events and focusing on thrombotic complications. 9 , 10 , 11 Various biologic mechanisms could mediate the influence of leukocytes on CVD, from proteolytic and oxidative endothelial damage 7 to effects on vascular perfusion, 12 , 13 and from platelet aggregation to hypercoagulability with prothrombotic diathesis. 14 , 15 , 16
Atherosclerosis is well known to be associated with a detectable activation of the hemostatic system, 17 , 18 , 19 and the development of thrombosis after atherosclerotic plaque disruption is one of the main mechanisms of acute vessel occlusion and ischemic complications. Therefore, the crucial role of coagulation pathway in CVD is also widely recognized. On the potential link between leukocytes and coagulation, much of the evidence has emphasized the role of neutrophil extracellular traps (NETs). NETs are extracellular DNA fibers, including histones and neutrophil enzymes, which are well known for their antimicrobial role against infections. 20 , 21 NETs may represent a link between inflammation and thrombosis, 22 , 23 with seminal works showing that NETs provide a scaffold for thrombus formation and can increase platelet activation, tissue factor expression, and thrombin generation. 24 , 25 Differently, the role of other WBC subtypes in predicting CAD risk, particularly in supporting potential links between leukocytes and coagulation, still remain unclear, and basophils are among the less‐considered leukocyte subtypes in clinical practice.
The aims of the present study were to investigate both (1) the prognostic significance of WBC count in the setting of secondary prevention of coronary artery disease (CAD) and (2) the correlation between WBC counts and plasma coagulant activity of factor II (FII:c), which for its high concentration and position may be considered as an overall marker of the coagulation cascade.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Population
This observational study was performed within the framework of the VHS (Verona Heart Study), a local survey that aimed to search for new risk factors for CAD in subjects with angiographic documentation of the state of their coronary arteries. Details about enrollment criteria have been described elsewhere. 26 , 27 In brief, adult patients of both sexes undergoing a coronary angiography examination were recruited from those referred to the Institute of Internal Medicine and to the Institute of Cardiovascular Surgery of the University of Verona in Verona, Italy between June 1998 and June 2005. All of the subjects in the VHS are required to have no history of any acute illness in the month preceding the enrollment. Consistently, patients with CAD and acute coronary syndrome were excluded from this study. Subjects with severe renal failure (estimated glomerular filtration rate <30 mL/min) and those with severe hepatic impairment (clinically defined diagnosis of liver cirrhosis) were also excluded from this study.
WBC count was assessed as a possible predictor of total and cardiovascular mortality in 823 patients with angiographically proven and clinically stable CAD (80.2% men, mean age 61.8±9.7 years) for whom data of mortality during a 5‐year follow‐up period and blood cell count at enrollment were available. Subjects with overt leukocytosis (>10 000/μL) or leukopenia (<4000/μL) were excluded.
In the second part of this study, we analyzed the correlations between blood cell counts and FII:c. Therefore, we selected a total of 750 subjects who were not taking anticoagulant drugs at enrollment and for whom −80°C frozen, platelet‐poor, citrated plasma samples for the FII:c assay were available. Five hundred fifty‐four subjects had angiographically proven CAD, with at least one of the major epicardial coronary arteries (left anterior descending, circumflex, and right) affected with ≥1 significant stenosis (≥50% lumen reduction). All of the patients with CAD were taking antiplatelet drugs, mostly low‐dose aspirin. One hundred ninety‐six subjects had completely normal coronary arteries, having been submitted to coronary angiography for reasons other than CAD, mainly valvular heart disease (CAD free). These subjects were also required to have neither history nor clinical or instrumental evidence of atherosclerosis in vascular districts beyond the coronary bed. The angiograms were assessed by cardiologists who were unaware that the patients were to be included in the study.
All participants came from the same geographical area (northern Italy). The study was approved by the ethics committee of our institution (Azienda Ospedaliera Universitaria Integrata, Verona, Italy) and has been carried out according to the Declaration of Helsinki. Written informed consent was obtained from all the participants after a full explanation of the study.
Assessment of Outcome in Follow‐Up Analysis
Subjects were followed until death or until June 30, 2012. 28 Study subjects’ status was determined by searching in the National Population Register and by an ambulatory or telephone survey. Certification and date of death were obtained from the National Population Register. The causes of death were obtained from death certificates kept at the Italian Institute of Statistics. Death from cardiovascular causes was defined as death caused by ischemic heart disease, heart failure, peripheral vascular disease, or cerebrovascular disease.
Biochemical Analysis
Samples of venous blood were drawn from each subject after an overnight fast at the time of enrollment before coronary angiography. Serum lipids, as well as other CAD risk factors, including hs‐CRP (high‐sensitivity C‐reactive protein), were determined at time of enrollment as previously described. 27
The 4‐variable version of the Modification of Diet in Renal Disease (MDRD) equation was used to estimate the estimated glomerular filtration rate from serum creatinine levels. 29
For coagulation analysis, blood was drawn into vacuum tubes containing 0.1 part 0.129 mol/L buffered sodium citrate per 10 parts blood. Venous blood samples collected at the time of enrollment were centrifuged for 15 minutes at 2500g, stored in 0.5‐mL aliquots, and frozen at −80°C within 1 hour of sample collection. Subsequently, when thawed out, plasma samples were centrifuged for 15 minutes at 1500g. FII:c was measured on a Behring coagulation timer (Dade Behring) by modification of the 1‐stage clotting method with the use of relative deficient plasma (Dade Behring).
D‐dimer plasma concentration was assessed by means of a particle‐enhanced, immuno‐turbidimetric assay, using the monoclonal antibody 8D3 (INNOVANCE D‐Dimer Assay; Siemens Healthcare Diagnostics Products, Marburg, Germany). 30 D‐dimer data were available for 435 out of 823 subjects (52.9%) within the group of patients with CAD followed in survival analysis.
Finally, plasma levels of C‐X‐C motif chemokine 12 (CXCL12) were also measured in a subgroup of subjects with CAD (530/823, 64.4%) for whom plasma samples were available. CXCL12 was assessed by a commercially available ELISA kit (R&D Systems Europe, Abingdon, UK). This kit measures the CXCL12‐1α variant, because this chemokine exists in 2 alternatively spliced variants. CXCL12‐1α is an 89 amino acid polypeptide, whereas CXCL12‐1β has the identical sequence with a 4‐residue C‐terminal extension. Plasma EDTA samples were processed according to the manufacturer’s instruction (product number SSA00) and measured in duplicates. The intra‐assay and interassay coefficients of variations were <5%.
Genotype Analysis for the Prothrombin G20210A Polymorphism
DNA was extracted using standard protocols, and genotyping was performed as previously indicated (for details see Olivieri et al 26 ). Genotype data were available for 717 out of the 823 patients with CAD (87.1%) followed in the survival analysis and for 714 out of the 750 subjects (95.2%) not taking anticoagulant drugs and with results of the FII:c assay. Because of the limited number of homozygous subjects (only 3 subjects had the genotype 20210AA, 2 in the survival analysis and 1 in FII:c analysis), the statistical calculations were performed categorizing the individuals as carrier or noncarrier of the variant.
Statistical Analysis
All calculations were performed using the IBM SPSS 23.0 (IBM, Armonk, NY) statistical package. Distributions of continuous variables in groups were expressed as mean±standard deviation. Skewed variables (ie, triglyceride, hs‐CRP) have been logarithmically transformed, and geometric mean with 95% CI was reported. Quantitative data were assessed using the Student t test or by ANOVA, with polynomial contrasts for linear trend and/or with Tukey post hoc comparison when appropriate. Correlations between continuous variables were assessed by Pearson test. Qualitative data were analyzed with the χ2 test or with χ2 for linear trend analysis when indicated.
Survival was assessed by using the Kaplan‐Meier method (log‐rank statistic) and Cox regression. Kaplan‐Meier curves were used for survival plots stratifying the CAD population on the basis of quartile distribution of blood cell counts for each leukocyte subtype. The proportional hazard assumption was tested by including time‐dependent covariates in the Cox model. A time‐dependent covariate was generated by creating interaction between blood cell counts and survival time. Multivariate Cox proportional hazards for both total and cardiovascular mortality were performed considering the lowest quartile as the reference group. First, all of the blood cell counts predicting mortality at univariate analysis were included in a Cox regression model with backward stepwise selection of variables (with P>0.10 as the critical value for excluding variables in the model) to disclose the most robust associations. Then, significant results were further assessed by Cox proportional hazard models adjusted for sex, age, and a priori identified potential confounders such as myocardial infarction history, smoke, diabetes mellitus, hypertension, plasma cholesterol and triglyceride, renal function (estimated glomerular filtration rate), and inflammatory status (hs‐CRP). Subjects with missing data were excluded from regression models. Hazard ratios (HRs) and 95% CIs are reported with 2‐tailed probability values.
Significant associations between FII:c and blood cell counts were evaluated by linear regression models estimating standardized β coefficients. To assess the predictors of FII:c variability, all of the leukocyte subtypes showing an association at univariate analysis were included in a sex, age, inflammatory marker (hs‐CRP), and CAD diagnosis‐adjusted regression model with backward stepwise selection of variables, with removal if P>0.10.
A value of P<0.05 was considered statistically significant.
Results
Leukocyte Subtypes and Survival in Subjects With CAD
In the longitudinal study, after a median follow‐up of 61 months, 160 of the 823 subjects with CAD (19.4%) died, 107 (13.0%) of whom for cardiovascular causes.
Stratifying the study population according to blood cell counts, both total and cardiovascular mortality increased progressively from the lowest to the highest quartile of neutrophil, eosinophil, basophil, and monocyte counts (Figure 1A and 1B). Hemoglobin levels also showed a significant association, with mortality rate decreasing from the lowest to the highest quartile (Figure S1), whereas no association was found for lymphocyte and platelet counts (data not shown). Time‐dependent covariates of blood cell counts were included in the Cox models and were not significant (P>0.05 in all of the analyses), thereby indicating that the proportional hazard assumption was not violated.
Figure 1. Total (A) and cardiovascular (CV) mortality (B) in the study population (n=823) stratified on the basis of quartile distribution of neutrophil, monocyte, eosinophil, and basophil cell counts.
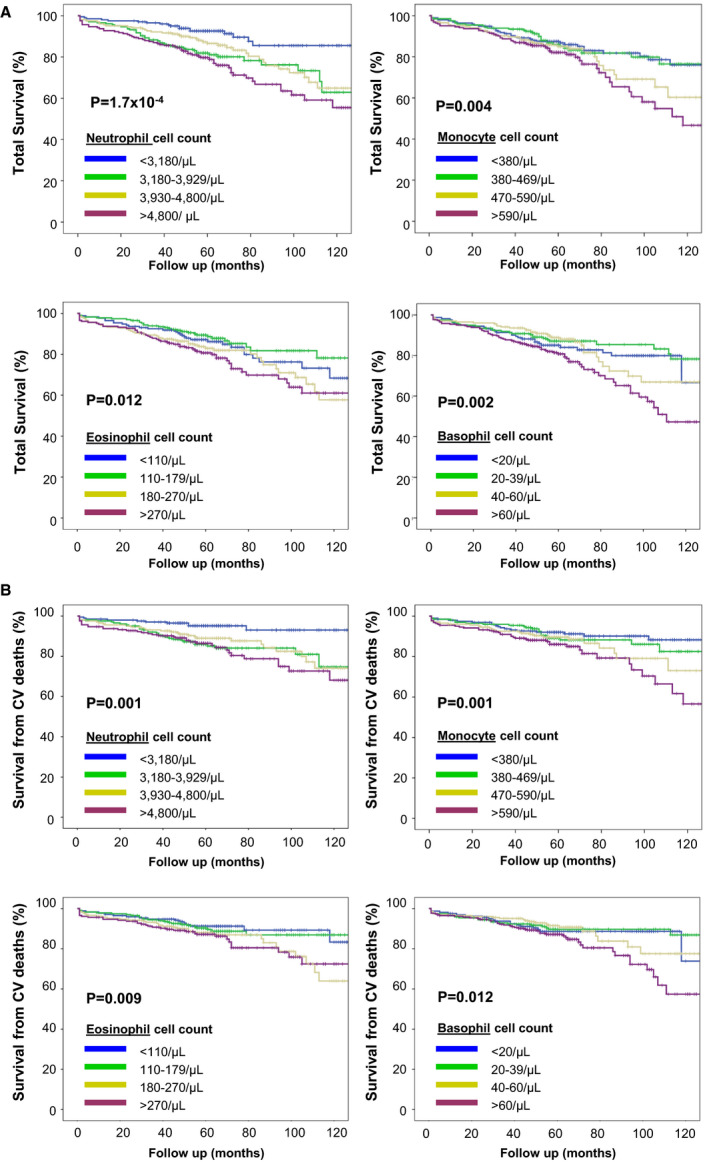
Lymphocyte cell count was not associated with mortality rate and is not reported in these graphs. P values were calculated by log rank for trend.
Including all the blood cell counts predicting mortality at univariate analysis in a Cox regression model with backward stepwise selection of variables, only neutrophils, basophils, and hemoglobin remained significant predictors of total and cardiovascular mortality (Table 1).
Table 1.
Total and Cardiovascular Mortality According to Quartile Distribution of Neutrophils, Basophils, and Hemoglobin*
| Quartile Distribution | Total Mortality, HR (95% CI) | P Value | Cardiovascular Mortality, HR (95% CI) | P Value | |
|---|---|---|---|---|---|
| Neutrophils, cell count/µL | <3180 | 1 | 1 | ||
| 3180–3929 | 2.26 (1.33–3.85) | 0.003 | 2.99 (1.46–6.12) | 0.003 | |
| 3930–4799 | 1.74 (1.01–3.01) | 0.047 | 2.13 (1.02–4.45) | 0.045 | |
| ≥4800 | 2.84 (1.70–4.76) | <0.001 | 2.84 (1.38–5.85) | 0.005 | |
| Basophils, cell count/µL | <20 | 1 | 1 | ||
| 20–39 | 0.99 (0.59–1.66) | 0.965 | 0.99 (0.53–1.87) | 0.983 | |
| 40–60 | 1.35 (0.81–2.26)) | 0.254 | 1.31 (0.71–2.45) | 0.388 | |
| ≥60 | 2.05 (1.31–3.20 | 0.002 | 1.79 (1.03–3.11) | 0.018 | |
| Hemoglobin, g/dL | <13.2 | 1 | 1 | ||
| 13.2–14.19 | 0.69 (0.47–1.03) | 0.068 | 0.50 (0.30–0.83) | 0.007 | |
| 14.2–15.09 | 0.43 (0.27–0.69) | <0.001 | 0.48 (0.28–0.81) | 0.006 | |
| ≥15.1 | 0.42 (0.27–0.45) | <0.001 | 0.35 (0.20–0.61) | <0.001 |
Data are presented as HR with 95% CIs. HR indicates hazard ratio.
Mortality HRs were estimated by Cox regression analysis according to quartiles of neutrophil, basophil, and hemoglobin levels. Subjects within the lowest quartile were considered as the reference group. The analyses were performed by including all blood cell counts predicting mortality at univariate analysis in a Cox regression model with backward stepwise selection of variables (with removal if P>0.10). Only significant results are reported.
Therefore, we focused our interest on neutrophil and basophil cell counts. Stratifying the study population according to neutrophil cell count, significant differences among the quartiles were found for sex, smoking habit, total and high‐density lipoprotein cholesterol, hs‐CRP, and D‐dimer (Table 2). Of note, subjects within the lowest quartile had a particularly reduced mortality rate (9.8% in the lowest quartile versus 27.5% in the highest quartile for total mortality and 5.2% in the lowest quartile versus 19.7% in the highest quartile for cardiovascular mortality). When the stratification was performed according to basophil count, differences among the quartiles were found for sex, smoking habit, renal function, CXCL12, and total and low‐density lipoprotein cholesterol (Table 3). Subjects within the highest quartile of basophil count had a particularly increased mortality rate (25.7% in the highest quartile versus 17.9% in the lowest quartile for total mortality and 18.8% in the highest quartile versus 12.5% in the lowest quartile for cardiovascular mortality). Considering that among granulocytes, eosinophils also have been linked with cardiovascular risk by previous studies 8 , 31 , 32 , 33 , data showing the population’s characteristics according to eosinophil cell count are presented in Table S1.
Table 2.
Clinical and Laboratory Characteristics of the Study Population in the Longitudinal Analysis According to Quartile Distribution of Neutrophil Cell Count
| Neutrophil Cell Count | P Value* | ||||
|---|---|---|---|---|---|
| <3180/µL | 3180–3929/µL | 3930–4799/µL | ≥4800/µL | ||
| Total mortality, % | 9.8 | 21.8 | 18.5 | 27.5 | 6.1×10−5 |
| Cardiovascular mortality, % | 5.2 | 16.6 | 14.4 | 19.7 | 2.9×10−4 |
| Age, y | 61.3±9.2 | 61.0±9.8 | 62.7±9.5 | 62.0±10.1 | NS |
| Male sex, % | 75.5 | 80.1 | 81.5 | 83.6 | 0.040 |
| BMI, kg/m2 | 26.7±3.2 | 26.8±3.9 | 26.7±3.3 | 26.5±3.7 | NS |
| Smoker, % | 59.4 | 69.7 | 71.0 | 70.7 | 0.017 |
| Hypertension, % | 67.5 | 66.0 | 65.2 | 66.3 | NS |
| Diabetes mellitus, % | 11.6 | 15.9 | 21.6 | 17.3 | NS |
| Myocardial infarction, % | 57.9 | 57.8 | 56.2 | 63.7 | NS |
| eGFR, mL/min | 75.6±16.1 | 73.5±17.3 | 74.5±18.3 | 73.2±22.5 | NS |
| Total cholesterol, mmol/L | 5.56±1.17 | 5.60±1.18 | 5.58±1.22 | 5.30±1.16 | 0.027 |
| LDL cholesterol, mmol/L | 3.74±1.00 | 3.72±1.06 | 3.76±1.09 | 3.66±0.92 | NS |
| HDL cholesterol, mmol/L | 1.24±0.31 | 1.17±0.32 | 1.22±0.30 | 1.12±0.31 | 0.004 |
| Triglycerides, mmol/L | 1.60 (1.49–2.62) | 1.76 (1.66–1.86) | 1.70 (1.60–1.80) | 1.69 (1.59–1.80) | NS |
| hs‐CRP, mg/L | 2.23 (1.90–2.62) | 2.98 (2.52–3.51) | 3.94 (3.29–4.72) | 7.04 (5.89–8.41) | 6.2×10−12 |
| D‐dimer, mcg/mL † | 0.53 (0.46–0.61) | 0.58 (0.51–0.67) | 0.58 (0.49–0.68) | 0.73 (0.62–0.85) | 0.004 |
| CXCL12, pg/mL ‡ | 2198 (2116–2282) | 2234 (2165–2306) | 2284 (2206–2364) | 2234 (2143–2330) | NS |
| Carriership of prothrombin 20210 A allele, % § | 4.4 | 3.4 | 5.5 | 8.0 | NS |
BMI indicates body mass index; CXCL12, C‐X‐C motif chemokine 12; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; hs‐CRP, high‐sensitivity C‐reactive protein; and LDL, low‐density lipoprotein.
By ANOVA with polynomial contrasts for linear trend or by χ2 for linear trend, when appropriate.
D‐dimer data were available for 435 out of 823 subjects (52.9%).
CXCL12 data were available for 530 out of 823 subjects (64.4%).
Prothrombin G20210A genotype data were available for 717 out of 823 subjects (87.1%).
Table 3.
Clinical and Laboratory Characteristics of the Study Population in the Longitudinal Analysis According to Quartile Distribution of Basophil Cell Count
| Basophil Cell Count | P Value* | ||||
|---|---|---|---|---|---|
| <20/µL | 20–39/µL | 40–60/µL | ≥60/µL | ||
| Total mortality, % | 17.9 | 15.7 | 16.3 | 25.7 | 0.024 |
| Cardiovascular mortality, % | 12.5 | 10.8 | 11.9 | 18.8 | 0.040 |
| Age, y | 62.1±10.4 | 61.4±9.6 | 61.4±8.9 | 62.1±9.9 | NS |
| Male sex, % | 73.5 | 81.6 | 80.8 | 82.8 | 0.045 |
| BMI, kg/m2 | 26.8±4.0 | 26.4±3.4 | 26.9±3.4 | 26.6±3.5 | NS |
| Smoker, % | 59.4 | 69.7 | 71.0 | 70.7 | 0.017 |
| Hypertension, % | 66.5 | 66.7 | 67.0 | 65.3 | NS |
| Diabetes mellitus, % | 18.3 | 16.8 | 17.0 | 15.1 | NS |
| Myocardial infarction, % | 61.1 | 58.5 | 59.2 | 57.6 | NS |
| eGFR, mL/min | 77.3±20.9 | 76.4±17.6 | 72.5±17.7 | 71.9±18.6 | 0.007 |
| Total cholesterol, mmol/L | 5.11±1.10 | 5.42±1.16 | 5.63±1.20 | 5.73±1.18 | 9.9×10−7 |
| LDL cholesterol mmol/L | 3.40±0.83 | 3.55±1.03 | 3.83±1.03 | 3.91±1.04 | 2.2×10−5 |
| HDL cholesterol, mmol/L | 1.20±0.37 | 1.18±0.28 | 1.18±0.31 | 1.19±0.29 | NS |
| Triglycerides, mmol/L | 1.61 (1.49–1.73) | 1.67 (1.57–1.77) | 1.73 (1.62–1.84) | 1.71 (1.61–1.81) | NS |
| hs‐CRP, mg/L | 4.78 (3.88–5.88) | 3.38 (2.72–4.20) | 3.13 (2.64–3.71) | 3.80 (3.26–4.42) | NS |
| D‐dimer, µg/mL† | 0.63 (0.55–0.73) | 0.62 (0.54–0.73) | 0.54 (0.46–0.64) | 0.61 (0.52–0.71) | NS |
| CXCL12, pg/mL‡ | 2147 (2040–2260) | 2214 (2129–2304) | 2247 (2179–2318) | 2287 (2217–2359) | 0.020 |
| Carriership of prothrombin 20210 A allele, % § | 5.3 | 6.6 | 4.9 | 4.7 | NS |
BMI indicates body mass index; CXCL12, C‐X‐C motif chemokine 12; eGFR, estimated glomerular filtration rate; HDL, high‐density lipoprotein; hs‐CRP, high‐sensitivity C‐reactive protein; and LDL, low‐density lipoprotein.
By ANOVA with polynomial contrasts for linear trend or by χ2 for linear trend, when appropriate.
D‐dimer data were available for 435 out of 823 subjects (52.9%).
CXCL12 data were available for 530 out of 823 subjects (64.4%).
Prothrombin G20210A genotype data were available for 717 out of 823 subjects (87.1%).
According to these previous analyses, the best cutoff values discriminating between different mortality rates were identified at the 25th percentile for neutrophils, the 75th percentile for basophils, and the 25th percentile for hemoglobin. Subjects with a neutrophil count higher than the lowest quartile, a basophil count within the highest quartile, or hemoglobin levels within the lowest quartile had an increased risk of both total and cardiovascular mortality (Table 4). The association of neutrophils and basophils with both total and cardiovascular mortality remained significant after adjustment for sex, age, and even all of the main cardiovascular risk factors including hs‐CRP levels (neutrophils ≥3.180/µL: HR, 1.91 (1.07–3.43) for total mortality, and HR, 2.28 (1.02–5.10) for cardiovascular mortality; basophils ≥60/µL: HR, 1.78 (1.21–2.62) for total mortality, and HR, 1.71 (1.05–2.77) for cardiovascular mortality (Table 4). Considering the widely recognized relationship between inflammatory markers and WBC counts, further analyses were performed. As expected, the risk of total and cardiovascular mortality was raised progressively by increasing hs‐CRP levels (Figure S2), and hs‐CRP remained a significant predictor of mortality in the fully adjusted Cox regression model (data not shown). Stratifying the study population on the basis of hs‐CRP levels (median level as threshold value) and either neutrophil (25th percentile as threshold value) or basophil cell count (75th percentile as threshold value), the highest risk of mortality was evident in subjects having both high hs‐CRP and high neutrophil or basophil cell count (Figures S3 and S4).
Table 4.
Total and Cardiovascular Mortality According to Neutrophils, Basophils, and Hemoglobin Levels in Different Cox Regression Models*
| Cox Regression Models | Total Mortality HR, (95% CI) | P Value | Cardiovascular Mortality, HR (95% CI) | P Value | |
|---|---|---|---|---|---|
| Neutrophils ≥3180/µL | Unadjusted | 2.36 (1.48–3.78) | <0.001 | 3.27 (1.71–6.28) | <0.001 |
| Age and sex adjusted | 2.29 (1.43–3.66) | 0.001 | 3.13 (1.63–6.00) | 0.001 | |
| Full adjusted † | 1.91 (1.07–3.43) | 0.030 | 2.28 (1.02–5.10) | 0.045 | |
| Basophils ≥60/µL | Unadjusted | 1.84 (1.34–2.53) | <0.001 | 1.79 (1.22–2.64) | 0.003 |
| Age and sex adjusted | 1.77 (1.29–2.44) | <0.001 | 1.70 (1.16–2.51) | 0.007 | |
| Full adjusted † | 1.78 (1.21–2.62) | 0.004 | 1.71 (1.05–2.77) | 0.031 | |
| Hemoglobin ≥13.2 g/dL | Unadjusted | 0.59 (0.42–0.81) | 0.001 | 0.61 (0.41–0.91) | 0.016 |
| Age and sex adjusted | 0.62 (0.44–0.87) | 0.006 | 0.70 (0.40–1.08) | 0.100 | |
| Full adjusted † | 0.72 (0.48–1.09) | 0.119 | 0.87 (0.52–1.45) | 0.592 |
Data are presented as HR with 95% CIs. HR indicates hazard ratio.
Estimated by Cox regression models comparing high versus low levels of neutrophil, basophil, and hemoglobin.
Estimated by Cox regression models comparing high versus low levels of neutrophil, basophil, and hemoglobin. adjusted for age, sex, myocardial infarction history, smoke, diabetes mellitus, hypertension, estimated glomerular filtration rate, plasma lipids (cholesterol and triglyceride), and high‐sensitivity C‐reactive protein levels.
In further subgroup analyses, the association of neutrophils and basophils with mortality remained significant after adjustment for the prothrombin G20210A polymorphism (n=717), D‐dimer (n=435), and CXCL12 plasma levels (n=530).
The carriership of the prothrombin 20210A allele was not associated with an increased risk of mortality (Figure S5). The association of both neutrophils and basophils with total mortality remained significant after adjustment for the carriership of the prothrombin 20210A allele (neutrophils ≥3.180/µL: HR, 2.09 [1.27–3.46]; basophils ≥60/µL: HR, 1.86 [1.31–2.65]).
D‐dimer levels correlated directly with neutrophil count (R=0.146, P=0.002), whereas no correlation was found with basophil count (Table S2). High levels of D‐dimer were associated with an increased risk of mortality by Kaplan‐Meier survival curves (Figure S6). Nonetheless, the association of both neutrophils and basophils with total mortality remained significant after adjustment for D‐dimer levels (neutrophils ≥3.180/µL: HR, 2.43 [1.25–4.72]; basophils ≥60/µL: HR, 2.29 [1.49–3.53]).
CXCL12 levels correlated directly with basophil count (R=0.110, P=0.011), whereas no correlation was found with neutrophil count (Table S3). CXCL12 levels were not significantly associated with mortality, although subjects within the highest quartile tended to have an increased mortality rate (Figure S7). The association of both neutrophils and basophils with total mortality remained significant after adjustment for CXCL12 levels (neutrophils ≥3.180/µL: HR, 3.06 [1.52–6.13]; basophils ≥60/µL: HR, 1.88 [1.22–2.88]).
Finally, the study population was stratified in 4 groups on the basis of both neutrophil and basophil cell count, by differently combining patients in the lowest neutrophil quartile and patients in the highest basophil quartile (Figure 2). Subjects with either high neutrophil (≥3.180/µL) or basophil cell count (≥60/µL) had an increased mortality rate (P<0.001 by log rank for trend), but subjects with both high neutrophil and basophil levels had the worst survival rate, with a 3‐fold increased risk of total mortality (full‐adjusted HR, 2.95 with 95% CI, 1.44–6.03) and even a 4‐fold increased risk of cardiovascular mortality (full‐adjusted HR, 4.01 with 95% CI, 1.38–11.71).
Figure 2. Total (A) and cardiovascular (CV) (B) mortality by combining high or low cell counts of neutrophils and basophils (C).

Hazard ratio (HR) with 95% CI were calculated by comparing subjects with high cell counts of both neutrophils and basophils (G4) versus those with low cell counts of both neutrophils and basophils (G1). P values were calculated by log rank for trend. HRs were estimated by sex‐ and age‐adjusted and full‐adjusted Cox regression models (by including sex, age, myocardial infarction history, smoke, diabetes mellitus, hypertension, plasma cholesterol and triglyceride, estimated glomerular filtration rate, and high‐sensitivity C‐reactive protein). B indicates basophils; and N, neutrophils.
Leukocyte Subtypes and Plasma Coagulant Activity of Factor II
On the associations between blood cell counts and FII:c, the general characteristics of the study sample are summarized in Table S4. Neutrophil, basophil, lymphocyte, and platelet levels correlated directly with FII:c at univariate analysis (Table 5). However, after adjustment for blood cell counts, age, sex, hs‐CRP, and CAD diagnosis, among leukocyte subtypes, only neutrophils (standardized β coefficient=0.085; P=0.021) and basophils (standardized β coefficient=0.073; P=0.042) remained significant predictors of FII:c variability (Table 5). These associations were confirmed even after adjustment for the carriership of the prothrombin 20210A allele for both neutrophils (standardized β coefficient=0.109; P=0.003) and basophils (standardized β coefficient=0.107; P=0.004), as well as in the subgroup of patients with CAD (neutrophil standardized β coefficient=0.165; P<0.001 and basophil standardized β coefficient=0.118; P=0.005).
Table 5.
Associations Between Blood Cell Counts and Factor II Coagulant Activity at Univariate Analysis and by Including All Blood Cell Counts in Sex, Age, High‐Sensitivity C‐Reactive Protein, and Coronary Artery Disease–Adjusted Regression Model*
| Standardized β Coefficient | P Value | |
|---|---|---|
| Univariate analysis | ||
| Hematocrit | 0.029 | NS |
| Hemoglobin | −0.021 | NS |
| Red blood cells | 0.038 | NS |
| Platelets | 0.254 | 1.8×10−12 |
| White blood cells, total | 0.148 | 4.5×10−5 |
| Lymphocytes | 0.081 | 0.027 |
| Monocytes | −0.001 | NS |
| Neutrophils | 0.134 | 2.3×10−4 |
| Eosinophils | 0.028 | NS |
| Basophils | 0.113 | 0.002 |
| Adjusted analysis † | ||
| Platelets | 0.198 | 7.9×10−8 |
| Neutrophils | 0.085 | 0.021 |
| Basophils | 0.073 | 0.042 |
CAD indicates coronary artery disease, NS, not significant.
The analyses were performed in 750 subjects (554 CAD and 196 CAD‐free) not taking anticoagulant drugs.
Only significant associations are reported.
Stratifying the second study population according the threshold values defined in the previous longitudinal study, FII:c increased progressively along neutrophil or basophil quartiles, from the lowest to the highest (Figure 3A and 3B). The effect of neutrophil and basophil was independent and additive, with the lowest FII:c in subjects with low levels of both neutrophils (<3180/µL) and basophils (<60/µL) and the highest FII:c in those with high levels of both neutrophils (≥3180/µL) and basophils (≥60/µL), as shown in Figure 3C. As expected, carriers of the prothrombin 20210A allele had higher FII:c levels, whereas no significant association was found with either neutrophil or basophil blood cell counts (Table S5). Stratifying the study sample on the basis of neutrophils/basophils and prothrombin G20210A polymorphism, subjects with high counts of both neutrophils and basophils had increased levels of FII:c, similar to the carriers of the prothrombin 20210A allele (Figure 4).
Figure 3. Factor II plasma coagulant activity (FII:c) in subjects not taking anticoagulant drugs (n=750) stratified on the basis of quartile distribution of neutrophil (A) and basophil cell counts (B), or by combining neutrophil and basophil levels (C).

P values were calculated by ANOVA for linear trend. B indicates basophils; and N, neutrophils.
Figure 4. Factor II plasma coagulant activity (FII:c) in subjects not taking anticoagulant drugs stratified within the study sample according high/low counts of neutrophils and basophils (only subjects with concordant high or low counts of both neutrophils and basophils were considered for this analysis) and the carriership of the prothrombin G20210A polymorphism.

P values were calculated by ANOVA with polynomial contrast for linear trend and by ANOVA with Tukey post hoc comparison*.
Discussion
In the present study, both neutrophil and basophil counts, within normal range values, were predictors of total and cardiovascular mortality in patients with stable CAD. In the second part of this study, the same 2 leukocytes subtypes were directly correlated with FII:c, thereby suggesting a link with procoagulant diathesis potentially playing a role in acute thrombotic complications of CAD.
The result of an increased mortality rate according to higher WBC count in patients with CAD is consistent with previous findings showing an association between leucocytes and cardiovascular risk. 3 , 4 , 5 , 6 There is a long‐existing and large body of medical literature supporting a biologically plausible relationship between CAD and WBC count, especially on neutrophils, 34 , 35 indicated as predictor of incident cardiovascular events stronger than other leukocyte components. 36 However, the role of different WBC subtypes in predicting CAD risk remains unclear, with mixed and sometimes controversial results. 37 In our study, many WBC counts were associated with mortality rate at univariate analysis, but only neutrophils and basophils, the latter scarcely investigated before, remained significant predictors by including all blood cell components in the regression model and after adjustment for the traditional cardiovascular risk factors. The results were also significant by including in the regression model hs‐CRP plasma levels, which are the best‐known biomarker of inflammatory status. The risk of mortality was particularly increased in subjects having both high hs‐CRP levels and either neutrophil or basophil counts. All of these results suggest that both neutrophil and basophil cell counts may have a prognostic value in CAD beyond and independent of the usual inflammatory markers.
It is worth noting that in subgroup analyses, these results remained significant after adjustment for the prothrombin G20210A polymorphism (ie, a widely known genetic determinant of procoagulant diathesis 26 ), D‐dimer (ie, one of the most extensively investigated biomarkers of coagulation/thrombosis 38 ), and CXCL12 plasma levels (ie, a chemokine that modulates leukocytes trafficking and homing and has recently been proposed as a potential causal mediator of CAD 39 , 40 ), thereby further supporting the potential prognostic role of neutrophils and basophils in CAD.
Considering the relationship found between neutrophil/basophil counts and FII:c, we also evaluated a major genetic determinant of FII:c, namely, the prothrombin G20210A polymorphism. The associations of neutrophil and basophil counts with both FII:c and mortality were independent of the prothrombin G20210A polymorphism. In our study sample, the carriership of the A allele, which is characterized by high FII:c levels, was not associated with an increased risk of mortality. However, we should take into account that only 38 out of 717 genotyped subjects were carriers of the A allele (36 heterozygous and 2 homozygous carriers), with 5 death events observed in this subgroup. Therefore, our analysis was clearly underpowered to explore the potential prognostic significance of the prothrombin G20210A polymorphism in subjects with CAD. Studies on larger population samples are needed to adequately address that issue.
In the longitudinal study, neutrophil cell count, but not that of basophils, correlated with D‐dimer plasma levels. D‐dimer is a reliable and sensitive index of fibrin deposition and stabilization, thus being indicative of in vivo thrombus formation at one point in time. However, its plasma concentration may be high in several other conditions unrelated to thrombosis including inflammatory disorders. 38 In our subanalysis, high D‐dimer levels were associated with an increased risk of mortality at univariate analysis. However, the associations of neutrophil and basophil counts with mortality were substantially unchanged after adjustment for D‐dimer levels.
In the present work, neutrophil and basophil counts predicted mortality independent of the chemokine CXCL12 levels. CXCL12, also known as stromal derived cell factor‐1, is present in many tissues and constitutively expressed by bone marrow stromal cells. CXCL12 plays a role in recruitment and differentiation of bone marrow progenitor cells and in trafficking and homing of leukocytes, 41 , 42 including neutrophils and basophils. 43 , 44 Consistent with earlier studies indicating that CXCL12 may be important for the recruitment and activation of the basophils, 45 in our study sample, CXCL12 levels correlated with basophil count. Certainly, CXCL12 is not a main cytokine regulating the production of granulocytes in the bone marrow and their release into the bloodstream. However, this chemokine has been identified as a potential causal mediator of CAD in humans with proatherogenic properties, 39 , 40 and the genomic locus 10q11, hosting the CXCL12 gene, has been linked with CAD by genome‐wide association studies. 46 Among WBC subtypes, neutrophils showed the strongest association with mortality. This result was expected, because neutrophils play a crucial role in inflammatory and innate immunity response, and several studies have shown a harmful proinflammatory role of neutrophils in the development and progression of CAD. 8 , 47 As proof of concept, therapies reducing neutrophils’ inflammatory response, such as colchicine (an agent inhibiting neutrophil chemotaxis by interfering with the formation of microtubules) 48 and canakinumab (a human monoclonal antibody against interleukin‐1‐β), 49 have been shown to reduce the cardiovascular risk. Consistent with such biological backgrounds, neutrophil counts, even within what is considered the normal range, have been associated with cardiovascular events in various study cohorts. 50
The link between neutrophils and cardiovascular events goes beyond inflammation, and recent interest has been addressed to hemostatic pathways. Neutrophils are not only able to activate platelets via ROS production, but also can stimulate coagulation by several mechanisms. Neutrophils increase the expression of tissue factor by endothelial cells, degrade the tissue factor pathway inhibitor by means of neutrophil‐derived proteinases (eg, cathepsin G), 47 and release NETs, 20 , 22 , 24 , 51 , 52 , 53 , 54 , 55 , 56 which may provide a trigger and support for thrombus formation. Consistent with these concepts, we found a strong direct correlation between neutrophil count and FII:c, which may reflect the global activation of a coagulation cascade. 26 The association between neutrophil cell expansion and prothrombotic diathesis, although with some still controversial results, is now widely recognized. 57 Nonetheless, to the best of our knowledge, the present work is the first highlighting a direct correlation between neutrophil count and FII:c. Although our data on neutrophils are substantially confirmatory of previous findings of scientific literature, the major novelty in the present study is represented by the results on basophils, which is the less‐considered and less‐understood leukocyte subtype in clinical practice. Basophilia is an unusual condition, potentially related to various and different clinical conditions, like hypersensitivity reactions, myeloproliferative disorders, and hypothyroidism. Data about basophils and CAD are scanty so far. In a Japanese study cohort investigating associations between leukocyte counts and CAD risk factors, high basophil count was linked with lipid abnormalities, in particular high triglyceride plasma levels. 58 In the UK Biobank study cohort, the group of subjects who died from either cardiovascular or noncardiovascular causes also had slightly higher basophil counts at univariate analysis. 59 In our longitudinal analysis, basophil cell count was shown for the first time to be an independent predictor of total and cardiovascular mortality in CAD. In the second part of our study, basophil cell count was associated with FII:c variability. The effects of basophil and neutrophils appeared to be additive, with the highest risk of mortality, as well as the largest increase of FII:c levels, in subjects with both high basophil and high neutrophil counts as compared with those with both low basophil and low neutrophil counts (Figures 2 and 3, respectively). Notably, subjects with both neutrophil and basophil cell counts had increased FII:c, similar to the carriers of the prothrombin 20210A allele, a well‐known prothrombotic gene variant. This is the first report linking basophil with prothrombotic diathesis and cardiovascular risk. It is worth noting that there are some molecular grounds that may support the biological plausibility of such a relationship. As neutrophils, basophils also can release extracellular DNA traps facilitating coagulation activation and thrombus formation. 60 , 61 Basophil granules contain polyphosphate, 62 which is now recognized as a procoagulant player in hemostasis and can accelerate blood clotting by activating the contact pathway and promoting the activation of factor V. 63 Basophils can synthetize platelet‐activating factor, 64 which regulates platelet aggregation and has been implicated in different pathophysiologic mechanisms, including thrombosis and tissue ischemia. 65 Finally, it is intriguing to observe that mast cells, the tissue counterparts of basophils, have been proposed to play a pathophysiological role in thrombosis and CVD, also beyond allergic acute coronary syndrome (the so‐called Kounis syndrome 66 ), and novel therapeutic strategies to prevent cardiovascular events via targeting of mast cells have been speculated. 67 , 68
In summary, in the present study, we confirm the potential prognostic value of WBC counts in CVD. In our study cohort of subjects with clinically stable CAD, subjects with high neutrophil and basophil counts had an increased risk of total and cardiovascular mortality. High levels of neutrophil and basophil counts were also associated with increased FII:c, thereby suggesting a prothrombotic diathesis, potentially representing a link between these leukocyte subtypes and cardiovascular risk. Such results may be particularly intriguing in terms of the data on basophils, which are usually the most neglected leukocyte subtype of blood cell count in clinical practice. However, the interpretation of these results should be taken with caution.
The present study had important limitations, including the relatively limited sample size and the low prevalence of women, as well as the lack of FII:c data in patients with CAD of the longitudinal study. The limitations because of the small sample size appeared particularly evident in subgroup analyses. For instance, in the case of the prothrombin G20210A polymorphism, the limited number of the carriers of the A allele did not allow us to draw any conclusions about the survival analysis. Moreover, there was the lack of some laboratory analyses, like plasma levels of cell‐free DNA or myeloperoxidase‐DNA complexes, which are considered as potential markers of NETs and could be useful in investigating the complex relationships between leukocytes and coagulation. 69 Finally, we relied on a single measurement of blood cell count and, although we excluded overt leukocytosis or leukopenia, such value could be affected by short‐term physiological changes. On the other hand, our study possessed some strengths such as the clear‐cut, angiographically demonstrated phenotypes of the study population and the complete adjustment for the traditional cardiovascular risk factors, including markers of inflammation. On the associations between blood cell counts and FII:c, we should take into account that all patients with CAD were taking antiplatelet drugs, mostly low‐dose aspirin, and aspirin has been shown to impair generation of thrombin in clotting blood. 49 However, it is worth noting that FII:c was assessed in platelet‐poor plasma, thereby limiting the potential influence of platelet‐related mechanisms on coagulation. Moreover, the associations between FII:c and neutrophil/basophil counts were confirmed after adjustment for CAD diagnosis, even in the subgroup of patients with CAD, who were all exposed to antiplatelet drugs. We should also take into account that thrombin has been proposed as a growth factor for several cell types, including leukocytes. 70 , 71 , 72 , 73 However, the results showing the independent effects on FII:c in respect of the prothrombin G20210A polymorphism, which in turn was not associated with either neutrophil and basophil counts, support more the hypothesis that neutrophils and basophils may influence FII:c, rather than FII:c could stimulate cellular populations of neutrophils and basophils. As with any observational study, the results cannot imply causation, and further studies are needed to replicate and clarify these findings. Nonetheless, our results may have significant clinical implications by supporting the potential prognostic importance of WBC counts in the setting of secondary prevention of CAD. These inexpensive and widely available tests may help to identify subjects at increased cardiovascular risk and, hypothetically, also those who could have the largest benefit from supplementary therapeutic approaches (eg, anti‐inflammatory drugs or low‐dose anticoagulants).
Sources of Funding
This work was supported by the Cariverona Foundation (project B36J16002570003).
Disclosures
None.
Supporting information
Tables S1–S5
Figures S1–S7
Acknowledgments
This study was performed (in part) in the LURM (Laboratorio Universitario di Ricerca Medica) Research Center, University of Verona. The authors thank Maria Zoppi for her invaluable secretarial help and Patrizia Pattini for her excellent technical help.
(J Am Heart Assoc. 2021;10:e018243. DOI: 10.1161/JAHA.120.018243.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018243
For Sources of Funding and Disclosures, see page 12.
References
- 1. Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. DOI: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- 2. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, et al. Heart disease and stroke statistics‐2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. DOI: 10.1161/CIR.0000000000000558. [DOI] [PubMed] [Google Scholar]
- 3. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. DOI: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 4. Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. DOI: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 5. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. DOI: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Niccoli G, Montone RA, Sabato V, Crea F. Role of allergic inflammatory cells in coronary artery disease. Circulation. 2018;138:1736–1748. DOI: 10.1161/CIRCULATIONAHA.118.035400. [DOI] [PubMed] [Google Scholar]
- 7. Madjid M, Awan I, Willerson JT, Casscells SW. Leukocyte count and coronary heart disease: implications for risk assessment. J Am Coll Cardiol. 2004;44:1945–1956. DOI: 10.1016/j.jacc.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 8. Madjid M, Fatemi O. Components of the complete blood count as risk predictors for coronary heart disease: in‐depth review and update. Tex Heart Inst J. 2013;40:17–29. [PMC free article] [PubMed] [Google Scholar]
- 9. Koren‐Morag N, Tanne D, Goldbourt U. White blood cell count and the incidence of ischemic stroke in coronary heart disease patients. Am J Med. 2005;118:1004–1009. DOI: 10.1016/j.amjmed.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 10. Haim M, Boyko V, Goldbourt U, Battler A, Behar S. Predictive value of elevated white blood cell count in patients with preexisting coronary heart disease: the Bezafibrate Infarction Prevention Study. Arch Intern Med. 2004;164:433–439. DOI: 10.1001/archinte.164.4.433. [DOI] [PubMed] [Google Scholar]
- 11. Dragu R, Huri S, Zukermann R, Suleiman M, Mutlak D, Agmon Y, Kapeliovich M, Beyar R, Markiewicz W, Hammerman H, et al. Predictive value of white blood cell subtypes for long‐term outcome following myocardial infarction. Atherosclerosis. 2008;196:405–412. DOI: 10.1016/j.atherosclerosis.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 12. Barron HV, Cannon CP, Murphy SA, Braunwald E, Gibson CM. Association between white blood cell count, epicardial blood flow, myocardial perfusion, and clinical outcomes in the setting of acute myocardial infarction: a thrombolysis in myocardial infarction 10 substudy. Circulation. 2000;102:2329–2334. DOI: 10.1161/01.CIR.102.19.2329. [DOI] [PubMed] [Google Scholar]
- 13. Shoenfeld Y, Pinkhas J. Leukopenia and low incidence of myocardial infarction. N Engl J Med. 1981;304:1606. [DOI] [PubMed] [Google Scholar]
- 14. Bovill EG, Bild DE, Heiss G, Kuller LH, Lee MH, Rock R, Wahl PW. White blood cell counts in persons aged 65 years or more from the Cardiovascular Health Study. Correlations with baseline clinical and demographic characteristics. Am J Epidemiol. 1996;143:1107–1115. DOI: 10.1093/oxfordjournals.aje.a008687. [DOI] [PubMed] [Google Scholar]
- 15. Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface‐adherent platelets via sequential action of P‐selectin and the beta 2‐integrin CD11b/CD18. Blood. 1996;88:146–157. DOI: 10.1182/blood.V88.1.146.146. [DOI] [PubMed] [Google Scholar]
- 16. de Gaetano G, Cerletti C, Evangelista V. Recent advances in platelet‐polymorphonuclear leukocyte interaction. Haemostasis. 1999;29:41–49. [DOI] [PubMed] [Google Scholar]
- 17. Meade TW, Mellows S, Brozovic M, Miller GJ, Chakrabarti RR, North WR, Haines AP, Stirling Y, Imeson JD, Thompson SG. Haemostatic function and ischaemic heart disease: principal results of the Northwick Park Heart Study. Lancet. 1986;2:533–537. DOI: 10.1016/S0140-6736(86)90111-X. [DOI] [PubMed] [Google Scholar]
- 18. Kannel WB. Overview of hemostatic factors involved in atherosclerotic cardiovascular disease. Lipids. 2005;40:1215–1220. DOI: 10.1007/s11745-005-1488-8. [DOI] [PubMed] [Google Scholar]
- 19. Borissoff JI, Spronk HM, ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364:1746–1760. DOI: 10.1056/NEJMra1011670. [DOI] [PubMed] [Google Scholar]
- 20. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. DOI: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 21. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. DOI: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 22. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–2776. DOI: 10.1182/blood-2013-10-463646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mozzini C, Garbin U, Fratta Pasini AM, Cominacini L. An exploratory look at NETosis in atherosclerosis. Intern Emerg Med. 2017;12:13–22. DOI: 10.1007/s11739-016-1543-2. [DOI] [PubMed] [Google Scholar]
- 24. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880–15885. DOI: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. 2015;36:1405–1414. DOI: 10.1093/eurheartj/ehv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olivieri O, Martinelli N, Baroni M, Branchini A, Girelli D, Friso S, Pizzolo F, Bernardi F. Factor II activity is similarly increased in patients with elevated apolipoprotein CIII and in carriers of the factor II 20210A allele. J Am Heart Assoc. 2013;2:e000440. DOI: 10.1161/JAHA.113.000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olivieri O, Martinelli N, Girelli D, Pizzolo F, Friso S, Beltrame F, Lotto V, Annarumma L, Corrocher R. Apolipoprotein C‐III predicts cardiovascular mortality in severe coronary artery disease and is associated with an enhanced plasma thrombin generation. J Thromb Haemost. 2010;8:463–471. DOI: 10.1111/j.1538-7836.2009.03720.x. [DOI] [PubMed] [Google Scholar]
- 28. Martinelli N, Girelli D, Baroni M, Guarini P, Sandri M, Lunghi B, Tosi F, Branchini A, Sartori F, Woodhams B, et al. Activated factor VII‐antithrombin complex predicts mortality in patients with stable coronary artery disease: a cohort study. J Thromb Haemost. 2016;14:655–666. DOI: 10.1111/jth.13274. [DOI] [PubMed] [Google Scholar]
- 29. Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of diet in renal disease study group. Ann Intern Med. 1999;130:461–470. DOI: 10.7326/0003-4819-130-6-199903160-00002. [DOI] [PubMed] [Google Scholar]
- 30. Tosi F, Micaglio R, Sandri M, Castagna A, Minguzzi D, Stefanoni F, Chiariello C, Franzese I, Luciani GB, Faggian G, et al. Increased plasma thrombin potential is associated with stable coronary artery disease: an angiographically‐controlled study. Thromb Res. 2017;155:16–22. DOI: 10.1016/j.thromres.2017.04.021. [DOI] [PubMed] [Google Scholar]
- 31. Uderhardt S, Ackermann JA, Fillep T, Hammond VJ, Willeit J, Santer P, Mayr M, Biburger M, Miller M, Zellner KR, et al. Enzymatic lipid oxidation by eosinophils propagates coagulation, hemostasis, and thrombotic disease. J Exp Med. 2017;214:2121–2138. DOI: 10.1084/jem.20161070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marx C, Novotny J, Salbeck D, Zellner KR, Nicolai L, Pekayvaz K, Kilani B, Stockhausen S, Bürgener N, Kupka D, et al. Eosinophil‐platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood. 2019;134:1859–1872. DOI: 10.1182/blood.2019000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mackman N. Eosinophils, atherosclerosis, and thrombosis. Blood. 2019;134:1781–1782. DOI: 10.1182/blood.2019003027. [DOI] [PubMed] [Google Scholar]
- 34. Rana JS, Boekholdt SM, Ridker PM, Jukema JW, Luben R, Bingham SA, Day NE, Wareham NJ, Kastelein JJ, Khaw KT. Differential leucocyte count and the risk of future coronary artery disease in healthy men and women: the EPIC‐Norfolk Prospective Population Study. J Intern Med. 2007;262:678–689. DOI: 10.1111/j.1365-2796.2007.01864.x. [DOI] [PubMed] [Google Scholar]
- 35. Horne BD, Anderson JL, John JM, Weaver A, Bair TL, Jensen KR, Renlund DG, Muhlestein JB. and Intermountain Heart Collaborative Study G . Which white blood cell subtypes predict increased cardiovascular risk? J Am Coll Cardiol. 2005;45:1638–1643. DOI: 10.1016/j.jacc.2005.02.054. [DOI] [PubMed] [Google Scholar]
- 36. Wheeler JG, Mussolino ME, Gillum RF, Danesh J. Associations between differential leucocyte count and incident coronary heart disease: 1764 incident cases from seven prospective studies of 30,374 individuals. Eur Heart J. 2004;25:1287–1292. DOI: 10.1016/j.ehj.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 37. Lassale C, Curtis A, Abete I, van der Schouw YT, Verschuren WMM, Lu Y, Bueno‐de‐Mesquita HBA. Elements of the complete blood count associated with cardiovascular disease incidence: findings from the EPIC‐NL cohort study. Sci Rep. 2018;8:3290. DOI: 10.1038/s41598-018-21661-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tripodi A. D‐dimer testing in laboratory practice. Clin Chem. 2011;57:1256–1262. DOI: 10.1373/clinchem.2011.166249. [DOI] [PubMed] [Google Scholar]
- 39. Sjaarda J, Gerstein H, Chong M, Yusuf S, Meyre D, Anand SS, Hess S, Pare G. Blood CSF1 and CXCL12 as causal mediators of coronary artery disease. J Am Coll Cardiol. 2018;72:300–310. DOI: 10.1016/j.jacc.2018.04.067. [DOI] [PubMed] [Google Scholar]
- 40. Döring Y, van der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, Atzler D, Santovito D, Rader DJ, Saleheen D, et al. CXCL12 derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation. 2019;139:1338–1340. DOI: 10.1161/CIRCULATIONAHA.118.037953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T. Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science. 1993;261:600–603. DOI: 10.1126/science.8342023. [DOI] [PubMed] [Google Scholar]
- 42. Bleul CC, Fuhlbrigge RC, Casasnovas JM, Aiuti A, Springer TA. A highly efficacious lymphocyte chemoattractant, stromal cell‐derived factor 1 (SDF‐1). J Exp Med. 1996;184:1101–1109. DOI: 10.1084/jem.184.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strydom N, Rankin SM. Regulation of circulating neutrophil numbers under homeostasis and in disease. J Innate Immun. 2013;5:304–314. DOI: 10.1159/000350282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yamaguchi M, Koketsu R, Suzukawa M, Kawakami A, Iikura M. Human basophils and cytokines/chemokines. Allergol Int. 2009;58:1–10. DOI: 10.2332/allergolint.08-RAI-0056. [DOI] [PubMed] [Google Scholar]
- 45. Jinquan T, Jacobi HH, Jing C, Reimert CM, Quan S, Dissing S, Poulsen LK, Skov PS. Chemokine stromal cell‐derived factor 1alpha activates basophils by means of CXCR4. J Allergy Clin Immunol. 2000;106:313–320. DOI: 10.1067/mai.2000.108108. [DOI] [PubMed] [Google Scholar]
- 46. Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AFR, Barbalic M, Gieger C, et al. Large‐scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. DOI: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gaul DS, Stein S, Matter CM. Neutrophils in cardiovascular disease. Eur Heart J. 2017;38:1702–1704. DOI: 10.1093/eurheartj/ehx244 [DOI] [PubMed] [Google Scholar]
- 48. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low‐dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61:404–410. DOI: 10.1016/j.jacc.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 49. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. DOI: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 50. Adamsson Eryd S, Smith JG, Melander O, Hedblad B, Engstrom G. Incidence of coronary events and case fatality rate in relation to blood lymphocyte and neutrophil counts. Arterioscler Thromb Vasc Biol. 2012;32:533–539. DOI: 10.1161/ATVBAHA.111.240416. [DOI] [PubMed] [Google Scholar]
- 51. Jimenez‐Alcazar M, Kim N, Fuchs TA. Circulating extracellular DNA: cause or consequence of thrombosis? Semin Thromb Hemost. 2017;43:553–561. DOI: 10.1055/s-0036-1597284. [DOI] [PubMed] [Google Scholar]
- 52. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–1862. DOI: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet‐dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–1961. DOI: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. von Brühl M‐L, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. DOI: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, Liu X, Tesmenitsky Y, Shvartz E, Sukhova GK, et al. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ Res. 2018;123:33–42. DOI: 10.1161/CIRCRESAHA.117.312494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Massberg S, Grahl L, von Bruehl M‐L, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–896. DOI: 10.1038/nm.2184. [DOI] [PubMed] [Google Scholar]
- 57. Noubouossie DF, Reeves BN, Strahl BD, Key NS. Neutrophils: back in the thrombosis spotlight. Blood. 2019;133:2186–2197. DOI: 10.1182/blood-2018-10-862243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mochizuki K, Miyauchi R, Misaki Y, Kasezawa N, Tohyama K, Goda T. Associations between leukocyte counts and cardiovascular disease risk factors in apparently healthy Japanese men. J Nutr Sci Vitaminol (Tokyo). 2012;58:181–186. DOI: 10.3177/jnsv.58.181. [DOI] [PubMed] [Google Scholar]
- 59. Welsh C, Welsh P, Mark PB, Celis‐Morales CA, Lewsey J, Gray SR, Lyall DM, Iliodromiti S, Gill JMR, Pell J, et al. Association of total and differential leukocyte counts with cardiovascular disease and mortality in the UK biobank. Arterioscler Thromb Vasc Biol. 2018;38:1415–1423. DOI: 10.1161/ATVBAHA.118.310945. [DOI] [PubMed] [Google Scholar]
- 60. Morshed M, Hlushchuk R, Simon D, Walls AF, Obata‐Ninomiya K, Karasuyama H, Djonov V, Eggel A, Kaufmann T, Simon H‐U, et al. NADPH oxidase‐independent formation of extracellular DNA traps by basophils. J Immunol. 2014;192:5314–5323. DOI: 10.4049/jimmunol.1303418. [DOI] [PubMed] [Google Scholar]
- 61. Yousefi S, Morshed M, Amini P, Stojkov D, Simon D, von Gunten S, Kaufmann T, Simon HU. Basophils exhibit antibacterial activity through extracellular trap formation. Allergy. 2015;70:1184–1188. DOI: 10.1111/all.12662. [DOI] [PubMed] [Google Scholar]
- 62. Moreno‐Sanchez D, Hernandez‐Ruiz L, Ruiz FA, Docampo R. Polyphosphate is a novel pro‐inflammatory regulator of mast cells and is located in acidocalcisomes. J Biol Chem. 2012;287:28435–28444. DOI: 10.1074/jbc.M112.385823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Smith SA, Morrissey JH. Polyphosphate: a new player in the field of hemostasis. Curr Opin Hematol. 2014;21:388–394. DOI: 10.1097/MOH.0000000000000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lie WJ, Homburg CH, Kuijpers TW, Knol EF, Mul FP, Roos D, Tool AT. Regulation and kinetics of platelet‐activating factor and leukotriene C4 synthesis by activated human basophils. Clin Exp Allergy. 2003;33:1125–1134. DOI: 10.1046/j.1365-2222.2003.01726.x. [DOI] [PubMed] [Google Scholar]
- 65. Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet‐activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med. 2002;30:S294–S301. DOI: 10.1097/00003246-200205001-00020. [DOI] [PubMed] [Google Scholar]
- 66. Hermans MAW, van Lennep JER, van Daele PLA, Bot I. Mast cells in cardiovascular disease: from bench to bedside. Int J Mol Sci. 2019;20:3395. DOI: 10.3390/ijms20143395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ponomaryov T, Payne H, Fabritz L, Wagner DD, Brill A. Mast cells granular contents are crucial for deep vein thrombosis in mice. Circ Res. 2017;121:941–950. DOI: 10.1161/CIRCRESAHA.117.311185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bot I, Shi GP, Kovanen PT. Mast cells as effectors in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:265–271. DOI: 10.1161/ATVBAHA.114.303570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, Neufeld EJ. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations. Pediatr Blood Cancer. 2010;54:273–278. DOI: 10.1002/pbc.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schwartz SM. Serum‐derived growth factor is thrombin? J Clin Invest. 1993;91:4. DOI: 10.1172/JCI116197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zimmerman GA, McIntyre TM, Prescott SM. Thrombin stimulates the adherence of neutrophils to human endothelial cells in vitro. J Clin Invest. 1985;76:2235–2246. DOI: 10.1172/JCI112232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Green D, Karpatkin S. Role of thrombin as a tumor growth factor. Cell Cycle. 2010;9:656–661. DOI: 10.4161/cc.9.4.10729. [DOI] [PubMed] [Google Scholar]
- 73. Shimaya Y, Shimada M, Shutto Y, Fujita T, Murakami R, Nakamura N, Yamabe H, Okumura K. Thrombin stimulates synthesis of macrophage colony‐stimulating factor, granulocyte‐macrophage colony‐stimulating factor and granulocyte colony‐stimulating factor by human proximal tubular epithelial cells in culture. Nephron Extra. 2012;2:1–8. DOI: 10.1159/000335751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figures S1–S7