

Abstract
Cardiac fibroblasts are the primary cell type responsible for deposition of extracellular matrix in the heart, providing support to the contracting myocardium and contributing to a myriad of physiological signaling processes. Despite the importance of fibrosis in processes of wound healing, excessive fibroblast proliferation and activation can lead to pathological remodeling, driving heart failure and the onset of arrhythmias. Our understanding of the mechanisms driving the cardiac fibroblast activation and proliferation is expanding, and evidence for their direct and indirect effects on cardiac myocyte function is accumulating. In this review, we focus on the importance of the fibroblast‐to‐myofibroblast transition and the cross talk of cardiac fibroblasts with cardiac myocytes. We also consider the current use of models used to explore these questions.
Keywords: arrhythmias, cardiac fibroblasts, cardiomyocytes, fibrosis, heart failure, myofibroblast
Subject Categories: Arrhythmias, Electrophysiology, Animal Models of Human Disease, Fibrosis
Nonstandard Abbreviations and Acronyms
- Ang II
angiotensin II
- cFb
cardiac fibroblast
- Cx43
connexin 43
- ECM
extracellular matrix
- FSP1
fibroblast‐specific protein 1
- hiPSC
human induced pluripotent stem cell
- SMAD
small mothers against decapentaplegic
- TGF‐β
transforming growth factor‐β
- α‐SMA
α‐smooth muscle actin
Because of aging populations and lifestyle changes, the total number of deaths caused by cardiovascular diseases (CVDs) is increasing, by ≈21% from 2007 to 2017. 1 High mortality rates as well as a high level of morbidity contribute to the high economic burden of CVDs. 2 Many of these deadly and debilitating CVDs are associated with cardiac fibrosis, which can be classified as either diffuse interstitial or replacement fibrosis. 3 Replacement fibrosis occurs immediately following cardiac injury, such as myocardial infarction (MI), to form a fibrotic scar preventing rupture of the myocardium. 3 , 4 Interstitial fibrosis is associated with inflammation and more chronic conditions, such as cardiomyopathies or hypertension, and if the underlying condition is left untreated, results in irreversible replacement fibrosis. 3 , 4 , 5 Cardiac fibrosis contributes to both electrical and structural remodeling of the heart, which ultimately leads to decreased cardiac function, heart failure, and arrhythmias. 4 , 6 , 7 , 8 The relationship between fibrosis and CVDs has been the subject of multiple studies. However, with no efficient treatments directly targeting cardiac fibrosis, it presents an ever‐growing clinical challenge. 9 , 10 The fibrotic process is mediated by activation of cardiac fibroblasts (cFbs), an important nonexcitatory cell population in the heart, responsible for deposition of extracellular matrix (ECM) in health and disease. cFbs can influence cardiac function through direct and indirect effects on cardiomyocytes. 11 , 12 , 13 In this review, we summarize the current understanding of the interactions between cFbs and cardiomyocytes and discuss the key questions remaining.
Cardiac Fibroblasts
The human heart is composed of 5 major cell types: cardiomyocytes, fibroblasts, endothelial cells, macrophages, and smooth muscle cells. 14 , 15 , 16 Cardiomyocytes contribute roughly 30% to 40% by number and roughly 65% to 80% by volume in the adult mammalian heart. 16 , 17 , 18 , 19 , 20 , 21 Recent single‐cell sequencing studies highlight considerable heterogeneity between the different regions of the human heart, with ventricles having distinct cellular signatures compared with atria (47.1% cardiomyocytes compared with 31.1%, respectively). 16 , 20 Reports on proportion of cFbs in heart differ, from circa 20% to 60% in rats and humans. 6 , 15 , 16 , 19 cFb numbers increase with development, aging, and disease. Banerjee et al showed that, in both rats and mice, the number of fibroblasts increases postnatally until adulthood is reached (≈30% to ≈64% in rat and ≈10% to ≈25% in mouse). 15 An increase in cFbs has also been demonstrated in heart failure and following MI. 14 , 22 , 23
Comparison of transcriptomes has revealed more similarities between cFbs and cardiomyocytes than between other fibroblast cell types. 24 , 25 In addition, use of single‐cell RNA sequencing has identified several subpopulations of cFbs within the heart, displaying distinct genetic signatures. 16 As mesenchymal cells present in every tissue in our body, these subpopulations may be indicative of the various possible cFb lineages. 6 , 16 In the heart, cFbs are primarily derived from the proepicardium, but can also arise by epithelial‐to‐mesenchymal and endothelial‐to‐mesenchymal transition. 6 , 26 Because of this heterogeneity and their ubiquitous nature, markers specific to cFbs are still lacking. Vimentin is often used alongside specific morphological properties of cFbs, such as the lack of a basement membrane. Some reports have used FSP1 (fibroblast‐specific protein 1) and discoidin domain receptor to identify cFbs, but these are also found in leukocytes and fibrocytes, respectively. 6 , 27
Regardless of origin, resident cFbs are dispersed throughout the heart as strands and sheets that sit between cardiac muscle fibers acting as a scaffold. 28 , 29 Their main function is to maintain the ECM through regulation of matrix metalloproteinases and tissue inhibitors of metalloproteinases, and therefore mediate secretion and degradation of collagen. 30 During disease or injury (eg, following MI), cFbs undergo a transition from fibroblast to myofibroblast. 6 , 31 This activated phenotype is described as having a spindle shape, increased proliferation, expression of α‐smooth muscle actin (α‐SMA), and increased production of collagen. 32 , 33 Single‐cell sequencing in mouse hearts further defined these differences between the activated myofibroblast and nonactivated cFb. They found ≈30 genes differentially expressed between the 2 cell types, including periostin and collagens 1 and 3, all known to be associated with the myofibroblast. 34
The main role of this cell type is wound healing and repair. Following MI, formation of a fibrotic scar, in place of necrotic tissue, prevents rupture of the myocardium. 4 , 22 The expression of α‐SMA stress fibers provides these cells with the ability to contract. This causes shrinkage of the fibrotic scar and produces tension, which, in turn, increases the stiffness of the myocardium. 32 , 35 , 36 , 37 Although initially beneficial for maintenance of cardiac function and prevention of heart rupture, long‐term effects of excessive fibrosis are detrimental (see section on Cross Talk Between the Myofibroblast and the Cardiomyocyte below).
Structural and electrical remodeling of the myocardium, attributed to chronic and proinflammatory conditions, such as hypertension, is commonly associated with cardiac fibrosis. 28 , 38 , 39 Inflammation has been demonstrated to lead to cardiac fibrosis via activation of profibrotic signaling cascades via interleukin 6 (IL‐6) (see section on Interleukin 6 below). 40 Stiffness itself can activate cFbs, leading to transition to myofibroblasts (Figure 1). Myofibroblasts themselves can secrete further profibrotic factors, driving progression to the kind of fibrosis seen following MI. 28 , 32 , 41 , 42 Alongside increased stiffness, fibrosis has also been observed to slow conduction and disrupt electrical wave propagation. Ultimately, these factors contribute to decreased cardiac function and increased susceptibility to arrhythmias. 43
Figure 1. Resident cardiac fibroblasts (cFbs) derive from endothelial‐to‐mesenchymal or epithelial‐to‐mesenchymal transition.
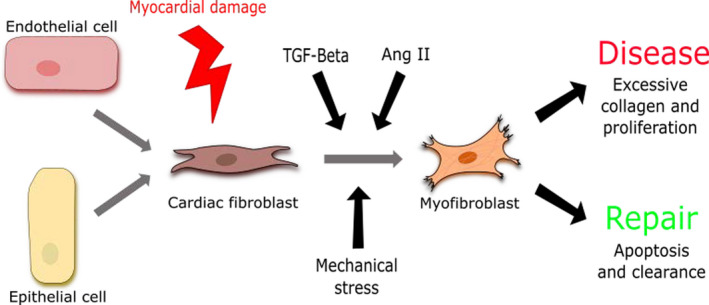
Injury to the myocardium initiates signaling pathways that trigger the activation of cFbs to myofibroblasts (MyoFbs). Loss of structural integrity via cardiomyocyte (CM) death also creates mechanical stress that mediates cFb to MyoFb activation. Consequences of MyoFb activation vary between repair and disease processes. There are multiple mechanisms for activation, including mechanical stimuli and paracrine factors, such as transforming growth factor‐β (TGF‐β) and angiotensin II (Ang II), from a variety of different sources, such as CMs and the cFbs themselves. 28 , 43
The transition from cFb to myofibroblast is a multifactorial process mediated by several factors (Figure 1). 28 , 37 Transforming growth factor‐β (TGF‐β) has been shown in several independent studies to be directly involved in this transition process. 44 , 45 , 46 Myofibroblasts, once activated, can themselves secrete TGF‐β, thus creating a detrimental positive feedback loop leading to sustained fibrosis. 47 , 48 , 49 The importance of tensile strength and mechanical stress in the activation of myofibroblasts has also been demonstrated (see section on Cross Talk Between the Myofibroblast and the Cardiomyocyte for more detail). 32 , 48 , 50 Perhaps not surprisingly, cFb activation can modulate cardiomyocyte function, and several modes of cFb‐cardiomyocyte communication have been uncovered.
Mechanisms of cFb‐Cardiomyocyte Communication
In the 1980s, multiple studies demonstrated cell‐cell interactions between excitatory and nonexcitatory cells in the heart. These were shown to be mediated via paracrine signaling and/or direct contacts between the cells. 51 , 52 From the mid‐2000s, there has been more focus on the effects of the nonexcitatory stromal cells on the electrophysiological features of the heart, with changes to conduction velocity and membrane depolarization observed during coculture. 53 This is perhaps not surprising considering that they make up a significant proportion of the total cell count. 54 More recently, multiple studies have identified that electrotonic communication occurs between cFbs and cardiomyocytes. This was demonstrated using cultured as well as freshly isolated cFbs in vitro, and in animal disease models in vivo. 13 , 53 , 55 , 56 The interaction and communication between cardiomyocytes and cFbs was shown to occur through several different modes (Figure 2). These include heterologous coupling through the formation of gap junctions or membrane nanotubes, and via mechanical forces and paracrine signaling. 13 , 53 , 57 , 58 However, the question still remains as to whether these coupling events occur in vivo in humans.
Figure 2. Figure depicting cardiomyocyte (CM)–myofibroblast (MyoFb) communcations.
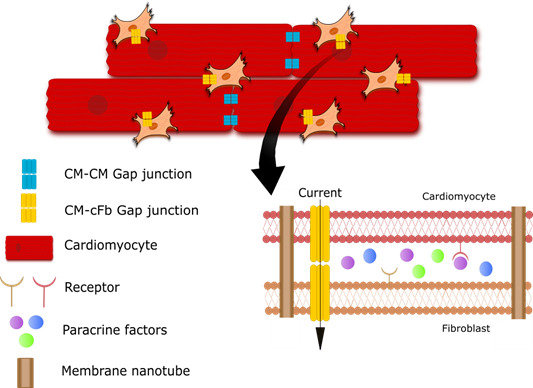
Gap junctions are present at distal junctions between CMs as well as elsewhere between CMs and MyoFbs. Gap junctions and membrane nanotubes allow exchange of molecules between the cytoplasms of cells. Paracrine factors secreted by cells bind receptors on neighbouring membranes, initiating signaling pathways. cFb indicates cardiac fibroblast.
Communication via Gap Junctions
Gap junctions are considered the main mechanism for electrotonic coupling in the heart. 59 They are ion channels that allow propagation of electrical signals between neighboring cells. Gap junctions are formed through the combination of 2 hemichannels, which are either homomeric or heteromeric. Depending on the composition of the 2 hemichannels, the junction will be either homotypic or heterotypic. 60 The combination of different connexins has demonstrated varying levels of channel permeability. 61 , 62 Connexin 40, connexin 43 (Cx43), and connexin 45 are the 3 main connexin subtypes found in the heart. Cx43 is the most widely expressed across the atrial and ventricular myocardium in both cardiomyocytes and cFbs. 59 , 60
Changes in the expression of Cx43, and in some cases connexin 45, have been observed to affect cardiac function. Studies have shown that the aging heart is accompanied by abnormalities in expression and distribution of Cx43. 63 , 64 When 10‐week‐old rat hearts were compared with those from 2‐year‐old rats, a decreased intensity of Cx43 expression was seen in the left ventricles. 64 Similar results were also seen in the atria of guinea pig hearts, accompanied by deterioration of adhesive junctions as well as gap junctions. 63 Furthermore, this age‐related decrease in Cx43 has been linked to slowed conduction velocity. 65 Together, these results show that through mediating expression of Cx43, aging can lead to conduction slowing and increased risk of arrhythmias.
These changes have also been observed following cardiac injury. Vasquez et al13 revealed that Cx43 and connexin 45 are present at cardiomyocyte‐fibroblast contact points in neonatal rat cells. In a model of cardiac injury, an increase in expression of Cx43 in cFbs, along with an observed increase in functional coupling between cardiomyocytes and cFbs, was detected. The same study also identified that myofibroblasts isolated from infarcted hearts, when cocultured with cardiomyocytes, decreased conduction velocities of the action potential and reduced action potential duration at 70% repolarization. More important, the authors showed that the effects on conduction and action potential duration were cFb density dependent, thus pointing toward a potential role in driving electrophysiological heterogeneity across the heart and thus contributing to increase in arrhythmias. 13 This cFb density dependency on cardiomyocyte electrophysiological features was also shown in modeling studies by Sanchez et al, with effects being most prominent in the left atrial pulmonary vein myocardium, a common site of atrial ectopy. 66
Zhang et al demonstrated in a mouse model that cFbs are activated following MI. Interestingly, these observations were apparent even in remote regions, away from the site of injury. 67 This implicates myofibroblasts in the postinfarct remodeling process, as established by other groups. 67 , 68 More important, Zhang et al demonstrated that cFbs from infarcted hearts displayed increased expression of Cx43 and intercellular cardiomyocyte coupling. However, in contrast, cardiomyocytes from these models exhibited downregulation and redistribution of Cx43 following MI. Decreased expression of Cx43 in cardiomyocytes has been shown to lead to increased propensity for abnormal conduction and susceptibility to arrhythmias. 67 , 69 These data together suggest that cFbs may be maintaining electrical coupling, to some extent, to preserve cardiac function. 67 Whether this adaptive process is ultimately detrimental to the heart requires further confirmation. With the availability of human induced pluripotent stem cell–derived cFbs and cardiomyocytes, there is an opportunity, and a clear need, for further studies robustly examining the interaction and effects of nonactivated and activated cFbs on cardiomyocyte electrophysiological features in human cell lines. 70
Communication via Membrane Nanotubes
Another mechanism that facilitates cFb‐cardiomyocyte communication is that of membrane nanotubes. This was established as a novel interaction by He et al, who demonstrated that membrane nanotubes, through the exchange of Ca2+, could facilitate both long‐ and short‐range connectivity between cardiomyocytes and cFbs and therefore regulate cardiac contractility. 57 Membrane nanotubes are long, thin, membrane‐bound connections that consist of F‐actin and, in some cells, microtubules. 71 , 72 They are suggested to help to control structural connectivity between cardiomyocytes and cFbs but also allow direct communication via the exchange of organelles, vesicles, and ions, such as Ca2+, across distances of up to several micrometers. In theory, membrane nanotubes have the advantage over gap junctions, which are limited to transfer of molecules of <1.2 kDa and only where cells are in close proximity. 57 Although membrane nanotubes have been identified in both cardiomyocytes and cFbs in vivo, 57 , 72 the proposed effects on contractility have not, and more research is required to identify their role and contributions to mediating electrical signals in the heart.
Communication via Paracrine Signaling
Cardiomyocytes and cFbs can also interact indirectly via paracrine signaling. 73 Considering that fibrosis is also demonstrated in areas remote to the injured myocardium, it is perhaps not surprising that paracrine factors may be involved in this process. 74 To further investigate this concept, several groups have used separated coculture methods, using physical inserts to separate the 2 cell types within the same well or cFb conditioned medium harvested from cultured cFbs. 13 , 75 , 76 These have demonstrated the involvement of paracrine mediators independently and alongside that of mechanical stress or physical coupling on electrophysiological activity. 13 , 75 LaFramboise et al showed that, following the addition of cFb conditioned medium, cardiomyocyte hypertrophy as well as reduction of spontaneous contractions were observed in neonatal cardiomyocytes. 75 Vasquez et al13 built on this work by comparing effects of conditioned media obtained from normal hearts and hearts with MI. They showed that conditioned medium from cFbs isolated from rat hearts with MI leads to slowed conduction velocities and a shortened action potential duration when compared with normal cFbs. 13
There is growing evidence in support of cFb‐produced paracrine factors playing a role in modulation of cardiac function and arrhythmogenesis. Many of these paracrine factors involved are known, however, mechanisms driving the observed changes in cardiac function and are under investigation. These will be discussed in the next section.
Cross Talk Between the Myofibroblast and the Cardiomyocyte
Following identification of such communications between cFbs‐cardiomyocytes, the importance of defining the consequences of these mechanisms became apparent. The primary focus to date has been on TGF‐β. An important mediator in the differentiation of cFb to myofibroblast, it has also been shown to have direct effects on cardiomyocyte function. 47 , 72 , 77 However, there is also evidence for the involvement of angiotensin II (Ang II), IL‐6, and mechanical stimuli in the communication processes. 72 , 73
Role of TGF‐β
TGF‐β is an important cytokine involved in many different cellular processes, including proliferation, differentiation, and migration. 77 Its expression is altered by several stimuli, such as mechanical stretch, hormones, and cytokines. In the heart, it is the driving force for myofibroblast activation and is therefore crucial during injury and wound healing. 44 , 78 TGF‐β receptor stimulation activates multiple downstream pathways, via both small mothers against decapentaplegic (Smad)– (Figure 3) and non–Smad‐mediated transcription (Figure 4), to induce cFb proliferation, collagen synthesis, and myofibroblast activation. 4 , 47 , 79 Multiple studies have shown that TGF‐β activates cFbs, at least to some extent, demonstrated by the expression of α‐SMA fibers and a contractile phenotype. 4 , 25 , 47
Figure 3. Schematic of transforming growth factor‐β (TGF‐β) activation via small mothers against decapentaplegic (Smad) signaling.
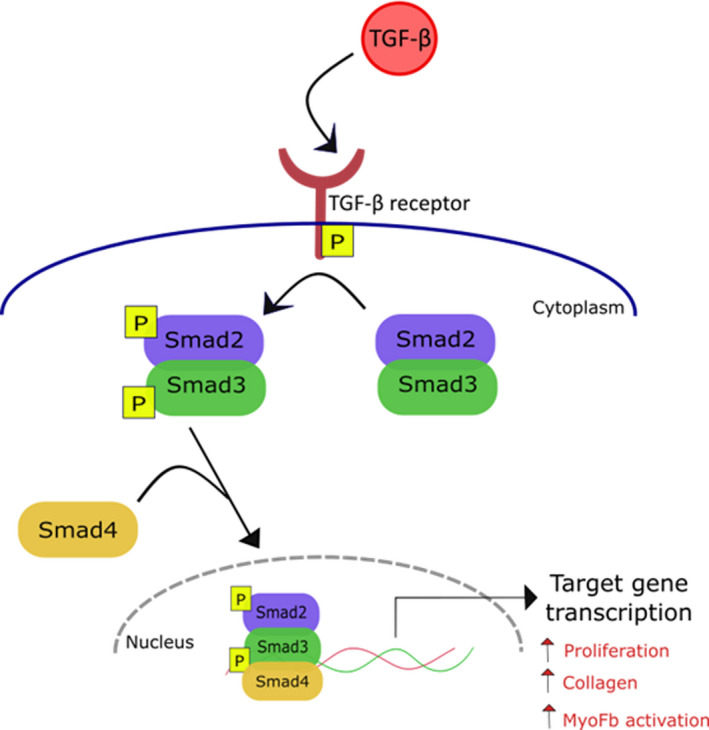
TGF‐β binds its receptor, initiating phosphorylation (p) of Smad2/3. This complex then binds Smad4 and translocates to the nucleus to induce transcription of target genes involved in proliferation, collagen production, and activation of cardiac fibroblasts to myofibroblasts (MyoFbs). 42 , 75
Figure 4. Schematic of transforming growth factor‐β (TGF‐β) activation via noncanonical (small mothers against decapentaplegic–independent) signaling.
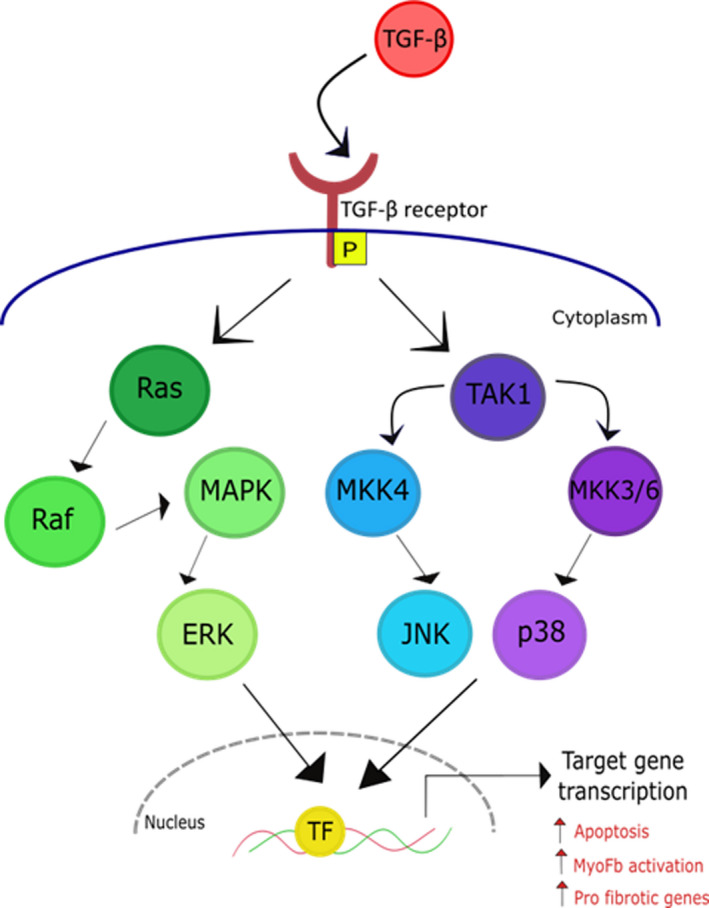
TGF‐β binds its receptor, initiating activation of Ras or TAK1 and further downsteam activation of extracellular signal‐regulated kinase (ERK), JNK, or p38. This leads to promotion of transcription factors (TFs) and the transcription of target genes involved in myofibroblast (MyoFb) activation, fibrosis, and cardiomyocyte apoptosis. 79 MAPK indicates mitogen‐activated protein kinase.
In 1988, Thompson et al reported the importance of TGF‐β in cardiac disease in rats. They showed a marked increase in TGF‐β in cardiomyocytes following MI with increased localization to the border zone. 80 Several studies since then have reported TGF‐β contributing to both fibrotic and hypertrophic effects in the myocardium, both in vitro and in vivo. 4 , 73 , 77 Furthermore, higher levels of TGF‐β are detected in hypertrophic hearts and in heart failure models. 22 , 81 These effects have been identified in direct coculture with cFbs and myofibroblasts and through the use of cFb/myofibroblast conditioned medium. In the presence of a TGF‐β receptor inhibitor, these effects were reduced or even reversed. These studies clearly highlight the role of TGF‐β in profibrotic and prohypertrophic processes. 73 , 82
Nakajima et al also observed the effects of TGF‐β to be more prominent in the atria compared with the ventricles. Through use of models with constitutive TGF‐β activity, they concluded that TGF‐β alone was not sufficient in promoting ventricular fibrosis in their mouse model. 77 Rahmutula et al later built on this work using a similar model. They observed an increased expression in TGF‐β signaling proteins in both atria and ventricles, although expression of fibrotic genes was only increased in the atria. This expression was associated with an increased atrial susceptibility to fibrosis and arrhythmia. Similar observations were made in human atrial tissue. 83 Interestingly, it has also been demonstrated that TGF‐β maintains a positive feedback loop by acting on cardiomyocytes to sustain its own expression and maintain myofibroblast proliferation. 73 , 84 Competitive inhibition of TGF‐β receptors can prevent the cFb activation process. 22 Several independent studies have explored targeting TGF‐β signaling as a potential antifibrotic therapeutic strategy. Kuwahara et al observed, in pressure overloaded mouse hearts, that fibrosis could be decreased using an anti–TGF‐β neutralizing antibody. 49 Similarly, Khalil et al demonstrated that deletion of Smad2/3 attenuated the fibrotic response in pressure overloaded mouse hearts. However, increased mortality in TGF‐β receptor knockdown models and following long‐term pharmacological inhibition after MI limits its therapeutic use. 22 , 44
Despite this, a study by Davies et al further implicated non‐Smad signaling in atrial remodeling and arrhythmogenesis, specifically with age. Through use of a conditional mouse knockdown of Mkk4, they demonstrated MKK4 as a negative regulator of TGF‐β signaling through activation of the JNK pathway. They showed that, with age and with Mkk4 deletion, there were significantly greater levels of fibrotic tissue, TGF‐β1 expression, TGF‐β receptor expression, and effects on matrix metalloproteinase and tissue inhibitor of metalloproteinase expression levels, providing a substrate for arrhythmogenesis. Furthermore, they discovered that, in patients with atrial fibrillation, there were decreased levels of MKK4. 85
More recently, TGF‐β has also been linked to the WNT signaling pathway in the context of fibrosis and cardiac tissue remodeling. 86 Several groups have observed a role for WNT proteins in the activation of myofibroblasts. Blyszczuk et al demonstrated this through the suppression of TGF‐β–mediated myofibroblast activation by blocking WNT signaling. 87 Others have observed stimulation of proliferation and upregulation of profibrotic genes through activation of the WNT pathway. 86 , 88 , 89 These data, together with those of Davies et al, 85 suggest that although TGF‐β itself may not be an appropriate target for pharmacological intervention, deciphering downstream targets could in fact provide new avenues for treatment.
The influence of TGF‐β on ionic handling in the heart is less well defined. In coculture with cardiomyocytes, both cFbs (defined as α‐SMA–negative Fbs) and myofibroblasts have been reported to alter Ca2+ transients. Myofibroblasts decreased Ca2+ transient amplitudes, whereas surprisingly, cFbs increased them. This effect was blocked by a TGF‐β receptor inhibitor. TGF‐β treatment of cardiomyocytes alone was not able to recapitulate the effects seen in coculture systems, indicating that TGF‐β itself does not drive these Ca2+ transient alterations. 73 Kaur et al observed a peak Na+ current increase of ≈40% following exposure to TGF‐β. TGF‐β effects on the current magnitude were voltage independent, and these effects were reversed in presence of a TGF‐β neutralizing antibody. 76 Whether these effects alter action potential morphological features and conduction or whether TGF‐β leads to changes in intracellular Na+ concentrations is unclear.
The effects of TGF‐β on cardiac pathogenesis have been recognized for many years through promotion of fibrosis and cardiomyocyte hypertrophy. 49 , 81 , 85 The precise role of TGF‐β in cardiac disease is only now becoming more evident. Numerous studies and the modern ‐omics approaches have highlighted TGF‐β involvement in the modulation of multiple signaling pathways, important in activating the myofibroblast. 44 , 90 , 91 Novel use of computer modeling has enabled generation of in silico fibrosis models, 92 allowing dissection of the TGF‐β signalosome and potentially identification of novel downstream targets for antifibrotic therapy.
Angiotensin II
Ang II is a hormone that acts as part of the renin‐angiotensin system and is involved in regulating blood pressure and sodium homeostasis. 93 , 94 Inhibition of Ang II signaling through use of angiotensin‐converting enzyme inhibitors has long been used to treat hypertension. 95 However, evidence over the years has also suggested an important role for Ang II in ventricular remodeling following cardiac injury. 96 , 97 Schieffer et al showed that, by blocking angiotensin receptors and by angiotensin‐converting enzyme inhibition, ventricular remodeling was attenuated through reduction of cardiac hypertrophy and fibrosis. 96
Recent evidence implicates Ang II as a powerful profibrotic factor, as secreted by cFbs/myofibroblasts. 98 , 99 Indeed, several independent studies demonstrate that exposure to Ang II stimulates cFb proliferation and, ultimately, fibrotic processes. 97 , 99 , 100 Cao et al observed that Ang II significantly upregulated α‐SMA in neonatal rat cFbs and that this correlated with an initial increase in Cx43 expression, indicative of myofibroblast activation. 90 In the heart, myofibroblasts have been identified as a significant source of Ang II, implying the existence of a positive‐feedback mechanism. 100 Ang II also stimulates the release of TGF‐β and IL‐6 from cFbs, which has been shown to further escalate the fibrotic process and the hypertrophic response seen in cardiomyocytes. 99 , 101 In patients with hypertensive heart disease, the angiotensin‐converting enzyme inhibitor, lisinopril, reduced myocardial fibrosis, irrespective of its effects on left ventricular hypertrophy. 102 These clinical findings validate some of the preclinical evidence in favor of Ang II being a major driver of cardiac fibrosis.
Interleukin 6
IL‐6 is a cytokine involved in processes of differentiation, growth, and survival. 103 Studies show that the IL‐6 signaling pathway is crucial to cardiac function and cardioprotection following acute injury. However, continuous activation has been associated with attenuated cardiac function attributable to processes including cardiomyocyte hypertrophy and fibrosis. 104 , 105 IL‐6 is secreted by both cardiomyocytes and cFbs, with increased expression seen in myocardial hypertrophy. 106 , 107 Although cardiomyocytes and cFbs lack IL‐6 receptors, local IL‐6 secretion is likely to promote recruitment of the inflammatory cells to myocardium. Indeed, IL‐6 has clearly been demonstrated to play a prominent role in inflammation. 40 , 103
A study conducted by Ma et al used a macrophage/cFb coculture model to observe the effects of Ang II and IL‐6 on the activation of cFbs. They observed that cFbs were the main source of IL‐6 following stimulation of Ang II and that this was reliant on the presence of macrophages. It was also determined that the presence of macrophages stimulated α‐SMA expression and collagen synthesis, and therefore activation of myofibroblasts, in an IL‐6–dependent manner. 108 These results dictate in part a role for IL‐6 in cardiac fibrosis through inflammatory processes. However, it was subsequently demonstrated that following cardiac injury, cardiomyocytes are stimulated to produce IL‐6, in turn thus stimulating proliferation of cFbs and further IL‐6 secretion. This feedback loop fuels cardiac fibrosis and hypertrophy, leading to decreased cardiac function. 99 , 106 , 107 Furthermore, Meléndez et al demonstrated that elevations in IL‐6 resulted in cardiac fibrosis and a large degree of ventricular stiffness. 40
The role of IL‐6 has also been investigated using complete genetic knockouts. These mice displayed significant differences when compared with wild type. The hearts were larger while also having thinner chamber walls. Echocardiographic analysis showed cardiac dilation, implying decreased cardiac function. Increased collagen deposition was also observed in knockout mice, along with a higher percentage of cFbs. However, interestingly, abrogation of IL‐6 led to decreased cellular interactions between cardiomyocytes‐cFbs because of a loss of cellular adhesion. 104 Remarkably, a more recent study, performed by McArthur et al, observed an upregulation of Cx43 expression following stimulation with IL‐6. Although this was only significant when in combination with the soluble IL‐6 receptor, it does point to a greater involvement in these cellular interactions. 109 This evidence supports the involvement of inflammation, and specifically IL‐6, in myocardial disorder through cardiomyocyte hypertrophy, fibrosis, and a loss of cardiomyocyte‐cFb interactions. However, mechanisms driving the sustained IL‐6 response that leads to these pathogenic transitions are unclear and require further investigation.
Role of Mechanical Stimuli
The Young modulus is a mechanical property that describes the elasticity of a material. 110 The healthy myocardium has a Young modulus ranging from 10 to 30 kPa. 48 , 111 , 112 However, following disease or injury, the development of a collagenous fibrotic scar increases the Young modulus to ≥100 kPa. 35 , 113 The protein α‐SMA is associated with an increase in contractile force, which is needed for wound contraction following collagen deposition and is therefore used as an activation marker for myofibroblasts. 41 , 42
It is now commonly accepted that, under standard tissue culture conditions, because of the stiffer plastic surfaces (≈3 GPa), cFbs undergo activation to myofibroblasts. Several independent studies demonstrated increased expression of α‐SMA after only a few hours of culture in rat cFbs. 13 , 41 , 112 Further evidence for the role of mechanical signals in the cFb‐to‐myofibroblast activation process has been demonstrated through the use of multiple substrates of varying Young moduli to mimic healthy and pathological cardiac environments. Use of higher Young moduli substrates (≥100 kPa) leads to increased proliferation of cFbs, higher levels of α‐SMA, and increased deposition of collagen. 41 , 112 How these mechanical signals drive the conversion to myofibroblast phenotype is unclear. 41 Despite this, most studies to date use culture conditions with stiffer surfaces, thus presumably working with activated cFbs. Future studies should take into account substrate stiffness when designing experiments using cFbs to better match physiological conditions (see section on Cardiomyocyte and Cardiac Fibroblast In Vitro Culture Models). Some groups have begun to experiment with polyacrylamide‐ or poly(ethylene glycol)‐based hydrogels, and others are using silicone. 48 , 114 In addition, with several commercially sourced culture dishes with softer Young moduli available, these experiments are now more achievable.
As well as acting as a stimulus for activation, mechanical forces have also been implicated in communicative processes in a phenomenon known as mechanoelectric feedback. The effect of mechanical forces on cardiomyocyte‐cardiomyocyte communications is relatively well investigated 115 , 116 ; however, the role of mechanical signal transmission between cardiomyocytes and cFbs is yet to be elucidated. Intercalated discs are structures found at the distal ends of cardiomyocytes, mediating the mechanoelectric feedback between cardiomyocytes. 117 , 118 , 119 They consist of the fascia adherens, desmosomes, and gap junctions. 117 Fascia adherens junctions are constructed from adhesion proteins, known as cadherins, that link the intercalated discs to the actin cytoskeleton within the cardiomyocytes. 118 , 119 Mutations in cadherins, as well as plakoglobin and desmoplakin, have significantly affected electrical coupling.
Normal electrical coupling of cardiomyocytes has some dependence on normal mechanical coupling, and thus it is reasonable to assume that the mechanoelectric phenomenon also mediates heterocellular coupling of cFbs and cardiomyocytes. 119 , 120 This has been evidenced in part through the application of pulsatile stretch, which induced higher expression of proteins involved in both mechanical and electrical coupling in neonatal rat cardiomyocytes. 116 Zhuang et al used a custom‐made stretch apparatus to apply 10% pulsatile stretch to cells for either 6 or 72 hours. They observed an increase in Cx43 expression, which corresponded to a relatively rapid increase in conduction velocity. This increase in electrical coupling could contribute to the formation of arrhythmia through dysregulated conduction in heterogeneously contracting cardiac tissue. 116 Conversely, Thompson et al showed that inhibiting myofibroblast contraction and blocking mechanosensitive channels in monolayers of cocultured myofibroblasts and neonatal rat ventricular cardiomyocytes lead to a minimal increase in myofibroblast membraneous expression of Cx43, a significant increase in cadherin expression, and an increase in conduction velocity. 84 It is difficult to reconcile these divergent findings. Pulsatile stretch and reduced cellular tension are both likely to play a role in disease progression; however, the pathophysiological relevance of these studies remains to be confirmed.
Multiple studies have also shown that growing cFbs or cardiomyocytes on stiffer substrates causes an increase in expression of proteins mediating cell‐cell contact. 84 , 119 N‐cadherin is most commonly found between myofibroblasts and cardiomyocytes. By applying pulsatile stretch to cultures of cardiomyocytes and/or myofibroblasts, there was an upregulation in expression of adhesion proteins, such as N‐cadherin, in both cardiomyocytes and myofibroblasts. 116 , 119 Thompson et al have also demonstrated that reduced expression of N‐cadherin, via short‐interfering RNA, reversed conduction slowing by myofibroblasts. 121
Multiple lines of evidence confirm that environmental stiffness affects structural and functional properties of the myocardium. Increased mechanical forces can lead to altered cardiomyocyte electrophysiological features and pathological fibrosis. 32 , 112 Better understanding of the mechanisms and key players driving cardiac fibrosis is necessary if we are to develop targeted therapeutic agents.
Cardiomyocyte and cFb in Vitro Culture Models
Current in vitro cell culture models do not recapitulate the stiffness characteristics of healthy or diseased myocardium, nor do they regularly consider multiple cells types that exist in the heart and their interplay. Considering that standard plastic dishes, because of high tensile strength, can lead to fibroblast activation, some previous work may require validation. 13 , 72 Indeed, activation state of the cFb can significantly modulate structural and functional myocardial properties. 13 , 73 cFbs and myofibroblasts (TGF‐β–treated or α‐SMA–positive cells) have differential effects on electrophysiological features in cardiomyocytes. More important, in coculture with cardiomyocytes, myofibroblasts often cause a slowing of conduction. 73 , 84 , 121 However, defining what is the tensile strength of healthy human myocardium is not straightforward. Tensile strengths in the range of 5 to 50 kPa have been reported. 36 , 41 , 111 , 112 Determination of the Young modulus in healthy and diseased human myocardial tissue can be compromised by handling after surgery but equally by different stages of disease. Furthermore, whether the Young modulus varies between the different chambers of the heart is yet to be investigated and should be considered moving forward.
Most experiments are conducted using murine cardiomyocytes and cFbs, despite significant differences in electrophysiological features when compared with humans, which have been reviewed extensively elsewhere. 122 , 123 These species differences are thought to contribute to high failure rates of cardiac drugs in clinical trials. 124 The use of human induced pluripotent stem cell–derived cardiomyocytes (hiPSC‐CMs) may circumvent these problems to some extent. However, hiPSC‐CMs show an immature phenotype, including spontaneous beating, reduced expression of the potassium channels, and a more depolarized resting membrane potential. 124 The cardiac differentiation process is also a relatively lengthy one. 125 Methods aimed at improving differentiation efficiency and hiPSC‐CM yield are becoming routine, whereas many groups have shown tangible improvements in maturity. 126 , 127 , 128 Yang et al treated hiPSC‐CMs for 2 weeks with fatty acids and observed a more mature phenotype. This included larger and less circular cell size and a significant increase in sarcomere length. Also observed were increases in calcium transient amplitude and twitch force. 129 A slightly more established method is the use of mechanical forces during the differentiation process. Through increased substrate stiffness or application of stretch, several independent studies have demonstrated more mature characteristics. 130 , 131 , 132
Cellular interactions are of course also to be considered when developing a culture model for experimentation. Novel methods, allowing differentiation of human induced pluripotent stem cell–derived cFbs, have been developed, opening up opportunities for much needed human coculture experiments. 133 , 134 Furthermore, the roles of other cell types are also beginning to be explored. 20 Hulsmans et al observed the presence of Cx43 at cell‐cell contacts between cardiomyocytes and macrophages. They also demonstrated that, in the presence of macrophages, cardiomyocytes had a more depolarized resting membrane potential and decreased action potential duration. 135 Use of endothelial and fibroblast coculture methods has also been reported to aid in induced pluripotent stem cell–derived cardiomyocyte maturation. 136 , 137 , 138 , 139
Consideration of the effects of ECM on cardiomyocyte and cFb structure and function is also necessary. The varying composition of the ECM can alter its properties, such as stiffness, and consequently exert differential effects on cardiomyocytes and cFbs. 28 , 35 It is therefore important to replicate the ECM cellular environment as closely as possible in vitro to create representative culture models. Research groups often use collagen coatings during cell culture as collagen is highly expressed in the myocardium. 112 However, following injury, it has been demonstrated that the collagen composition of the ECM changes to express more collagen 1 than collagen 3. 42 , 140 Equally, ECM protein fibronectin was shown to be required for TGF‐β–induced myofibroblast differentiation. 48 ECM also has positive effects on cardiomyocyte organization in culture, aiding in cellular alignment, similar to that seen in vivo. This has been demonstrated through use of ridged collagen plates, where cardiomyocytes are allowed to connect to the ECM, forming laminae. 141
More frequently, hiPSC‐CMs are being used in a 3‐dimensional context to mimic the more complex tissue environment that exists in vivo. This concept of cardiac tissue engineering allows interrogation of both cell‐ECM and cell‐cell interactions, not only between cardiomyocytes but also between other cardiac cell types. 72 , 142 A recent study by Lee et al established a 3‐dimensional microtissue, using both cardiomyocytes and cFbs, to develop a novel model of cardiac fibrosis. 143 Research such as this allows more accurate interrogation of cardiac disease and creates new platforms for discovery of more tailored medicine through use of patient‐derived models.
Is the Myofibroblast Phenotype Reversible?
Previously, it was thought that the myofibroblast was a terminally differentiated phenotype. The only method of reestablishing tissue homeostasis was that of apoptotic clearance after wound healing. However, more recent studies suggest that dedifferentiation or phenotype reversibility is possible and could hold the answer to identifying new therapeutic targets. 47
Several pathways have been identified in the cFb‐myofibroblast activation process, with TGF‐β being the main target of interest. Indeed, competitive inhibition of TGF‐β receptors leads to a decrease in expression of α‐SMA fibers, a marker for myofibroblast activation. 47 , 48 Other research has shown that softer substrates, around or just below what would be expected of the healthy myocardium, also lead to a decrease in expression of α‐SMA fibers. 37 , 48 α‐SMA expression levels in these studies did not return to those of quiescent cFbs, suggesting that there may be an intermediate stage, termed the protomyofibroblast, 42 , 144 as observed in a liver fibrosis study. 145 This cFb plasticity was only demonstrated in freshly isolated cells, so the myofibroblast reversibility in long‐term culture on harder substrates needs to be investigated. 22 , 32 , 48 Interestingly, a study by Nagaraju et al observed that myofibroblasts obtained from patients with heart failure had less α‐SMA expression following treatment with a TGF‐β receptor inhibitor. Whether this was dedifferentiation or simply a loss of α‐SMA fibers is unclear. 22
Prolonged activation of myofibroblasts leads to sustained fibrotic processes within the myocardium, leading to deterioration of heart function. Fibrosis, although ultimately detrimental, is necessary immediately following the myocardial injury, and myofibroblasts are a crucial part of this process. Inhibition of TGF‐β following myocardial injury has led to increased mortality in a mouse model. 22 Full genetic knockouts of TGF‐β or certain signaling components (ie, Smad2) have embryonic lethality. 44 , 146 Therefore, reversal of the cFb‐to‐myofibroblast transition may not be an appropriate therapeutic approach for myocardial injury, although it may be more useful for treatment of chronic CVD, thus preventing the progression toward heart failure. 22
Conclusions
Studies over the past 20 years have started to unveil the complexity of the cell‐cell interactions within the myocardium. In this article, we focus on the role of the cFb and the activated myofibroblast in altering the structure and function of the myocardium. Yet, there are still conflicting views as to the effects of these 2 cell types on cardiac electrophysiological features and whether these phenomena occur in humans. To advance the work further, we will require use of the full range of cellular and animal model systems available, while acknowledging the particular strengths and weaknesses each possesses.
There are difficulties involved with in situ experimentation; there are also limitations involved with culturing cFbs in vitro, in particular their propensity for differentiation/activation to myofibroblasts. In addition, considering the immaturity of hiPSC‐CM models, the importance of developing better, more representative fibrosis models is necessary.
Another issue that cannot be avoided is the immense complexity of the pathogenesis of fibrosis. With multiple different and often intersecting pathways, it will be difficult to identify a single valid target to reduce or revert the process. However, only by integrating existing biological and computational platforms with systems biology approaches will further progress be made. Many of these processes are conserved across organs and, therefore, identifying common mechanisms could have huge clinical benefit for many different diseases involving fibrosis.
Sources of Funding
This work was supported by the British Heart Foundation grants (PG/17/55/33087, RG/17/15/33106, FS/19/12/34204, and FS/19/16/34169 to Dr Pavlovic; SP/15/9/31605, PG/14/59/31000, RG/14/1/30588, RM/13/30157, and P47352/CRM to Dr Denning; and FS/12/40/29712 to Dr Gehmlich); the Wellcome Trust Grants (109604/Z/15/Z to Dr Pavlovic and 201543/B/16/Z to Dr Gehmlich); Animal Free Research UK (AFR19‐20293 to Drs Pavlovic and Denning); National Centre for the Replacement, Refinement, and Reduction of Animals in Research (CRACK‐IT:35911‐259146 and NC/K000225/1 to Dr Denning and NC/T001747/1 to Dr Gehmlich); Accelerator Award (AA/18/2/34218) to Institute of Cardiovascular Sciences at Birmingham; and Oxford British Heart Foundation Centre of Research Excellence (RE/13/1/30181).
Disclosures
None.
(J Am Heart Assoc. 2021;10:e019338. DOI: 10.1161/JAHA.120.019338.)
For Sources of Funding and Disclosures, see page 11.
Contributor Information
Chris Denning, Email: chris.denning@nottingham.ac.uk.
Davor Pavlovic, Email: d.pavlovic@bham.ac.uk.
References
- 1. GBD 2017 Causes of Death Collaborators . Global, regional, and national age‐sex‐specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1736–1788. DOI: 10.1016/S0140-6736(18)32203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gheorghe A, Griffiths U, Murphy A, Legido‐Quigley H, Lamptey P, Perel P. The economic burden of cardiovascular disease and hypertension in low‐ and middle‐income countries: a systematic review. BMC Public Health. 2018;18:975. DOI: 10.1186/s12889-018-5806-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903. DOI: 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li J, Philip JL, Xu X, Theccanat T, Razzaque MA, Akhter SA. β‐arrestins regulate human cardiac fibroblast transformation and collagen synthesis in adverse ventricular remodeling. J Mol Cell Cardiol. 2014;76:73–83. DOI: 10.1016/j.yjmcc.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eijgenraam TR, Sillje HHW, de Boer RA. Current understanding of fibrosis in genetic cardiomyopathies. Trends Cardiovasc Med. 2020;30:353–361. DOI: 10.1016/j.tcm.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 6. Ongstad E, Kohl P. Fibroblast‐myocyte coupling in the heart: potential relevance for therapeutic interventions. J Mol Cell Cardiol. 2016;91:238–246. DOI: 10.1016/j.yjmcc.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu T‐J, Ong JJC, Hwang C, Lee JJ, Fishbein MC, Czer L, Trento A, Blanche C, Kass RM, Mandel WJ, et al. Characteristics of wave fronts during ventricular fibrillation in human hearts with dilated cardiomyopathy: role of increased fibrosis in the generation of reentry. J Am Coll Cardiol. 1998;32:187–196. DOI: 10.1016/S0735-1097(98)00184-3. [DOI] [PubMed] [Google Scholar]
- 8. Azevedo PS, Polegato BF, Minicucci MF, Paiva SA, Zornoff LA. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. 2016;106:62–69. DOI: 10.5935/abc.20160005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci USA. 2005;102:437–442. DOI: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN. Arrhythmogenic consequences of myofibroblast‐myocyte coupling. Cardiovasc Res. 2012;93:242–251. DOI: 10.1093/cvr/cvr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end‐stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. DOI: 10.1161/01.CIR.89.1.151. [DOI] [PubMed] [Google Scholar]
- 12. Pick R, Janicki JS, Weber KT. Myocardial fibrosis in nonhuman primate with pressure overload hypertrophy. Am J Pathol. 1989;135:771–781. [PMC free article] [PubMed] [Google Scholar]
- 13. Vasquez C, Mohandas P, Louie KL, Benamer N, Bapat AC, Morley GE. Enhanced fibroblast‐myocyte interactions in response to cardiac injury. Circ Res. 2010;107:1011–1020. DOI: 10.1161/CIRCRESAHA.110.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang LI, Yu P, Zhou B, Song J, Li Z, Zhang M, Guo G, Wang Y, Chen X, Han L, et al. Single‐cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 2020;22:108–119. DOI: 10.1038/s41556-019-0446-7. [DOI] [PubMed] [Google Scholar]
- 15. Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol. 2007;293:H1883–H1891. DOI: 10.1152/ajpheart.00514.2007. [DOI] [PubMed] [Google Scholar]
- 16. Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, Akkad AD, Herndon CN, Arduini A, Papangeli I, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020;142:466–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou P, Pu WT. Recounting cardiac cellular composition. Circ Res. 2016;118:368–370. DOI: 10.1161/CIRCRESAHA.116.308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nag AC. Study of non‐muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 19. Vliegen HW, van der Laarse A, Cornelisse CJ, Eulderink F. Myocardial changes in pressure overload‐induced left ventricular hypertrophy: a study on tissue composition, polyploidization and multinucleation. Eur Heart J. 1991;12:488–494. DOI: 10.1093/oxfordjournals.eurheartj.a059928. [DOI] [PubMed] [Google Scholar]
- 20. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom S, Szewczykowska M, Jackowska T, dos Remedios C, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;161:1566–1575. DOI: 10.1016/j.cell.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 21. Litviňuková M, Talavera‐López C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. Cells of the adult human heart. Nature. 2020;588:466–472. DOI: 10.1038/s41586-020-2797-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagaraju CK, Robinson EL, Abdesselem M, Trenson S, Dries E, Gilbert G, Janssens S, Van Cleemput J, Rega F, Meyns B, et al. Myofibroblast phenotype and reverisbility of fibrosis in patients with end‐stage heart failure. J Am Coll Cardiol. 2019;73:2267–2282. [DOI] [PubMed] [Google Scholar]
- 23. Sridhar S, Vandersickel N, Panfilov AV. Effect of myocyte‐fibroblast coupling on the onset of pathological dynamics in a model of ventricular tissue. Sci Rep. 2017;7:40985. DOI: 10.1038/srep40985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Furtado MB, Nim HT, Boyd SE, Rosenthal NA. View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development. 2016;143:387–397. DOI: 10.1242/dev.120576. [DOI] [PubMed] [Google Scholar]
- 25. Ackers‐Johnson M, Li PY, Holmes AP, O'Brien SM, Pavlovic D, Foo RS. A simplified, langendorff‐free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ Res. 2016;119:909–920. DOI: 10.1161/CIRCRESAHA.116.309202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kohl P, Gourdie RG. Fibroblast‐myocyte electrotonic coupling: does it occur in native cardiac tissue? J Mol Cell Cardiol. 2014;70:37–46. DOI: 10.1016/j.yjmcc.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15. DOI: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction‐from repair and remodeling to regeneration. Cell Tissue Res. 2016;365:563–581. DOI: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruiz‐Villalba A, Simon AM, Pogontke C, Castillo MI, Abizanda G, Pelacho B, Sanchez‐Dominguez R, Segovia JC, Prosper F, Perez‐Pomares JM. Interacting resident epicardium‐derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J Am Coll Cardiol. 2015;65:2057–2066. DOI: 10.1016/j.jacc.2015.03.520. [DOI] [PubMed] [Google Scholar]
- 30. Eghbali M, Czaja MJ, Zeydel M, Weiner FR, Zern MA, Seifter S, Blumenfeld OO. Collagen chain mRNAs in isolated heart cells from young and adult rats. J Mol Cell Cardiol. 1988;20:267–276. DOI: 10.1016/S0022-2828(88)80059-2. [DOI] [PubMed] [Google Scholar]
- 31. Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann N Y Acad Sci. 2006;1080:76–84. DOI: 10.1196/annals.1380.007. [DOI] [PubMed] [Google Scholar]
- 32. Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell. 2017;28:1871–1882. DOI: 10.1091/mbc.e17-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagaraju CK, Dries E, Gilbert G, Abdesselem M, Wang N, Amoni M, Driesen RB, Sipido KR. Myofibroblast modulation of cardiac myocyte structure and function. Sci Rep. 2019;9:8879. DOI: 10.1038/s41598-019-45078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, Lacraz GPA, Huibers MMH, van Oudenaarden A, van Rooij E. Single‐cell sequencing of the healthy and diseased heart reveals cytoskeleton‐associated protein 4 as a new modulator of fibroblasts activation. Circulation. 2018;138:166–180. DOI: 10.1161/CIRCULATIONAHA.117.030742. [DOI] [PubMed] [Google Scholar]
- 35. Nguyen DT, Nagarajan N, Zorlutuna P. Effect of substrate stiffness on mechanical coupling and force propagation at the infarct boundary. Biophys J. 2018;115:1966–1980. DOI: 10.1016/j.bpj.2018.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emig R, Zgierski‐Johnston CM, Beyersdorf F, Rylski B, Ravens U, Weber W, Kohl P, Horner M, Peyronnet R. Human atrial fibroblast adaptation to heterogeneities in substrate stiffness. Front Physiol. 2019;10:1526. DOI: 10.3389/fphys.2019.01526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kollmannsberger P, Bidan CM, Dunlop JWC, Fratzl P, Vogel V. Tensile forces drive a reversible fibroblast‐to‐myofibroblast transition during tissue growth in engineered clefts. Sci Adv. 2018;4:eaao4881. DOI: 10.1126/sciadv.aao4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ren J, Yang M, Qi G, Zheng J, Jia L, Cheng J, Tian C, Li H, Lin X, Du J. Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by angiotensin II infusion. Am J Hypertens. 2011;24:701–707. DOI: 10.1038/ajh.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Van Linthout S, Miteva K, Tschope C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. 2014;102:258–269. DOI: 10.1093/cvr/cvu062. [DOI] [PubMed] [Google Scholar]
- 40. Melendez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56:225–231. DOI: 10.1161/HYPERTENSIONAHA.109.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang J, Chen H, Seth A, McCulloch CA. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2003;285:H1871–H1881. DOI: 10.1152/ajpheart.00387.2003. [DOI] [PubMed] [Google Scholar]
- 42. Penke LR, Peters‐Golden M. Molecular determinants of mesenchymal cell activation in fibroproliferative diseases. Cell Mol Life Sci. 2019;76:4179–4201. DOI: 10.1007/s00018-019-03212-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ten Tusscher KH, Panfilov AV. Influence of diffuse fibrosis on wave propagation in human ventricular tissue. Europace. 2007;9(suppl 6):vi38–vi45. DOI: 10.1093/europace/eum206. [DOI] [PubMed] [Google Scholar]
- 44. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al. Fibroblast‐specific TGF‐beta‐Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor‐beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180–189. [DOI] [PubMed] [Google Scholar]
- 46. Scharenberg MA, Pippenger BE, Sack R, Zingg D, Ferralli J, Schenk S, Martin I, Chiquet‐Ehrismann R. TGF‐beta‐induced differentiation into myofibroblasts involves specific regulation of two MKL1 isoforms. J Cell Sci. 2014;127:1079–1091. [DOI] [PubMed] [Google Scholar]
- 47. Hecker L, Jagirdar R, Jin T, Thannickal VJ. Reversible differentiation of myofibroblasts by MyoD. Exp Cell Res. 2011;317:1914–1921. DOI: 10.1016/j.yexcr.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light‐mediated reduction in substrate modulus. PLoS One. 2012;7:e39969. DOI: 10.1371/journal.pone.0039969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor‐beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure‐overloaded rats. Circulation. 2002;106:130–135. [DOI] [PubMed] [Google Scholar]
- 50. Arora PD, Narani N, McCulloch CA. The compliance of collagen gels regulates transforming growth factor‐beta induction of alpha‐smooth muscle actin in fibroblasts. Am J Pathol. 1999;154:871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schroedl NA, Hartzell CR. Myocytes and fibroblasts exhibit functional synergism in mixed cultures of neonatal rat heart cells. J Cell Physiol. 1983;117:326–332. DOI: 10.1002/jcp.1041170307. [DOI] [PubMed] [Google Scholar]
- 52. Suzuki T, Tsuruda A, Katoh S, Kubodera A, Mitsui Y. Purification of endothelin from a conditioned medium of cardiac fibroblastic cells using beating rate assay of myocytes cultured in a serum‐free medium. J Mol Cell Cardiol. 1997;29:2087–2093. DOI: 10.1006/jmcc.1997.0443. [DOI] [PubMed] [Google Scholar]
- 53. Miragoli M, Gaudesius G, Rohr S. Electrotonic modulation of cardiac impulse conduction by myofibroblasts. Circ Res. 2006;98:801–810. DOI: 10.1161/01.RES.0000214537.44195.a3. [DOI] [PubMed] [Google Scholar]
- 54. Grove D, Zak R, Nair KG, Aschenbrenner V. Biochemical correlates of cardiac hypertrophy, IV: observations on the cellular organization of growth during myocardial hypertrophy in the rat. Circ Res. 1969;25:473–485. DOI: 10.1161/01.RES.25.4.473. [DOI] [PubMed] [Google Scholar]
- 55. Quinn TA, Camelliti P, Rog‐Zielinska EA, Siedlecka U, Poggioli T, O'Toole ET, Knopfel T, Kohl P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc Natl Acad Sci USA. 2016;113:14852–14857. DOI: 10.1073/pnas.1611184114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. 2004;94:828–835. DOI: 10.1161/01.RES.0000122382.19400.14. [DOI] [PubMed] [Google Scholar]
- 57. He K, Shi X, Zhang X, Dang S, Ma X, Liu F, Xu M, Lv Z, Han D, Fang X, et al. Long‐distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc Res. 2011;92:39–47. DOI: 10.1093/cvr/cvr189. [DOI] [PubMed] [Google Scholar]
- 58. Guo Y, Zeng Q‐C, Zhang C‐Q, Zhang X‐Z, Li R‐X, Wu J‐M, Guan J, Liu LU, Zhang X‐C, Li J‐Y, et al. Extracellular matrix of mechanically stretched cardiac fibroblasts improves viability and metabolic activity of ventricular cells. Int J Med Sci. 2013;10:1837–1845. DOI: 10.7150/ijms.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vozzi C, Dupont E, Coppen SR, Yeh HI, Severs NJ. Chamber‐related differences in connexin expression in the human heart. J Mol Cell Cardiol. 1999;31:991–1003. DOI: 10.1006/jmcc.1999.0937. [DOI] [PubMed] [Google Scholar]
- 60. McArthur L, Chilton L, Smith GL, Nicklin SA. Electrical consequences of cardiac myocyte: fibroblast coupling. Biochem Soc Trans. 2015;43:513–518. DOI: 10.1042/BST20150035. [DOI] [PubMed] [Google Scholar]
- 61. Rackauskas M, Verselis VK, Bukauskas FF. Permeability of homotypic and heterotypic gap junction channels formed of cardiac connexins mCx30.2, Cx40, Cx43, and Cx45. Am J Physiol Heart Circ Physiol. 2007;293:H1729–H1736. DOI: 10.1152/ajpheart.00234.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rackauskas M, Kreuzberg MM, Pranevicius M, Willecke K, Verselis VK, Bukauskas FF. Gating properties of heterotypic gap junction channels formed of connexins 40, 43, and 45. Biophys J. 2007;92:1952–1965. DOI: 10.1529/biophysj.106.099358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nagibin V, Egan Benova T, Viczenczova C, Szeiffova Bacova B, Dovinova I, Barancik M, Tribulova N. Ageing related down‐regulation of myocardial connexin‐43 and up‐regulation of MMP‐2 may predict propensity to atrial fibrillation in experimental animals. Physiol Res. 2016;65(suppl 1):S91–S100. DOI: 10.33549/physiolres.933389. [DOI] [PubMed] [Google Scholar]
- 64. Watanabe M, Ichinose S, Sunamori M. Age‐related changes in gap junctional protein of the rat heart. Exp Clin Cardiol. 2004;9:130–132. [PMC free article] [PubMed] [Google Scholar]
- 65. Yan J, Thomson JK, Zhao W, Wu X, Gao X, DeMarco D, Kong W, Tong M, Sun J, Bakhos M, et al. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J Mol Cell Cardiol. 2018;114:105–115. DOI: 10.1016/j.yjmcc.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanchez J, Gomez JF, Martinez‐Mateu L, Romero L, Saiz J, Trenor B. Heterogeneous effects of fibroblast‐myocyte coupling in different regions of the human atria under conditions of atrial fibrillation. Front Physiol. 2019;10:847. DOI: 10.3389/fphys.2019.00847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang Y, Kanter EM, Yamada KA. Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovasc Pathol. 2010;19:e233–e240. DOI: 10.1016/j.carpath.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, Mains IM, Mingoia JT, Flack EC, Lindsey ML. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39:699–707. DOI: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 69. Peters NS, Green CR, Poole‐Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–875. DOI: 10.1161/01.CIR.88.3.864. [DOI] [PubMed] [Google Scholar]
- 70. Blyszczuk P, Zuppinger C, Costa A, Nurzynska D, Di Meglio FD, Stellato M, Agarkova I, Smith GL, Distler O, Kania G. Activated cardiac fibroblasts control contraction of human fibrotic cardiac microtissues by a beta‐adrenoreceptor‐dependent mechanism. Cells. 2020;9:1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gerdes HH, Carvalho RN. Intercellular transfer mediated by tunneling nanotubes. Curr Opin Cell Biol. 2008;20:470–475. DOI: 10.1016/j.ceb.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 72. Pellman J, Zhang J, Sheikh F. Myocyte‐fibroblast communication in cardiac fibrosis and arrhythmias: mechanisms and model systems. J Mol Cell Cardiol. 2016;94:22–31. DOI: 10.1016/j.yjmcc.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cartledge JE, Kane C, Dias P, Tesfom M, Clarke L, Mckee B, Al Ayoubi S, Chester A, Yacoub MH, Camelliti P, et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc Res. 2015;105:260–270. DOI: 10.1093/cvr/cvu264. [DOI] [PubMed] [Google Scholar]
- 74. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium: fibrosis and renin‐angiotensin‐aldosterone system. Circulation. 1991;83:1849–1865. DOI: 10.1161/01.CIR.83.6.1849. [DOI] [PubMed] [Google Scholar]
- 75. LaFramboise WA, Scalise D, Stoodley P, Graner SR, Guthrie RD, Magovern JA, Becich MJ. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol. 2007;292:C1799–C1808. DOI: 10.1152/ajpcell.00166.2006. [DOI] [PubMed] [Google Scholar]
- 76. Kaur K, Zarzoso M, Ponce‐Balbuena D, Guerrero‐Serna G, Hou L, Musa H, Jalife J. TGF‐beta1, released by myofibroblasts, differentially regulates transcription and function of sodium and potassium channels in adult rat ventricular myocytes. PLoS One. 2013;8:e55391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, Field LJ. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor‐beta(1) transgene in the heart. Circ Res. 2000;86:571–579. [DOI] [PubMed] [Google Scholar]
- 78. Lijnen P, Petrov V, Rumilla K, Fagard R. Transforming growth factor‐beta 1 promotes contraction of collagen gel by cardiac fibroblasts through their differentiation into myofibroblasts. Methods Find Exp Clin Pharmacol. 2003;25:79–86. DOI: 10.1358/mf.2003.25.2.723680. [DOI] [PubMed] [Google Scholar]
- 79. Parichatikanond W, Luangmonkong T, Mangmool S, Kurose H. Therapeutic targets for the treatment of cardiac fibrosis and cancer: focusing on TGF‐beta signaling. Front Cardiovasc Med. 2020;7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Thompson NL, Bazoberry F, Speir EH, Casscells W, Ferrans VJ, Flanders KC, Kondaiah P, Geiser AG, Sporn MB. Transforming growth factor beta‐1 in acute myocardial infarction in rats. Growth Factors. 1988;1:91–99. DOI: 10.3109/08977198809000251. [DOI] [PubMed] [Google Scholar]
- 81. Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non‐myocyte proliferation and requires Tgf‐beta. J Clin Invest. 2010;120:3520–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF‐beta 1 and endothelin‐1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. [DOI] [PubMed] [Google Scholar]
- 83. Rahmutula D, Marcus GM, Wilson EE, Ding CH, Xiao Y, Paquet AC, Barbeau R, Barczak AJ, Erle DJ, Olgin JE. Molecular basis of selective atrial fibrosis due to overexpression of transforming growth factor‐beta1. Cardiovasc Res. 2013;99:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thompson SA, Copeland CR, Reich DH, Tung L. Mechanical coupling between myofibroblasts and cardiomyocytes slows electric conduction in fibrotic cell monolayers. Circulation. 2011;123:2083–2093. DOI: 10.1161/CIRCULATIONAHA.110.015057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Davies L, Jin J, Shen W, Tsui H, Shi Y, Wang Y, Zhang Y, Hao G, Wu J, Chen SI, et al. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J Am Heart Assoc. 2014;3:e000340. DOI: 10.1161/JAHA.113.000340 10.1161/JAHA.113.000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Dzialo E, Tkacz K, Blyszczuk P. Crosstalk between the TGF‐beta and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim Pol. 2018;65:341–349. [DOI] [PubMed] [Google Scholar]
- 87. Blyszczuk P, Muller‐Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, Glatz K, Basler K, Luscher TF, Distler O, et al. Transforming growth factor‐beta‐dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J. 2017;38:1413–1425. [DOI] [PubMed] [Google Scholar]
- 88. Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, Regan JN, Rojas M, Willis M, Leask A, et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012;31:429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E, et al. Cardiac fibroblast glycogen synthase kinase‐3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation. 2014;130:419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cao L, Chen Y, Lu L, Liu Y, Wang Y, Fan J, Yin Y. Angiotensin II upregulates fibroblast‐myofibroblast transition through Cx43‐dependent CaMKII and TGF‐beta1 signaling in neonatal rat cardiac fibroblasts. Acta Biochim Biophys Sin (Shanghai). 2018;50:843–852. [DOI] [PubMed] [Google Scholar]
- 91. Ceccato TL, Starbuck RB, Hall JK, Walker CJ, Brown TE, Killgore JP, Anseth KS, Leinwand LA. Defining the cardiac fibroblast secretome in a fibrotic microenvironment. J Am Heart Assoc. 2020;9:e017025. DOI: 10.1161/JAHA.120.017025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rikard SM, Athey TL, Nelson AR, Christiansen SLM, Lee JJ, Holmes JW, Peirce SM, Saucerman JJ. Multiscale coupling of an agent‐based model of tissue fibrosis and a logic‐based model of intracellular signaling. Front Physiol. 2019;10:1481. DOI: 10.3389/fphys.2019.01481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fukamizu A, Takahashi S, Seo MS, Tada M, Tanimoto K, Uehara S, Murakami K. Structure and expression of the human angiotensinogen gene: identification of a unique and highly active promoter. J Biol Chem. 1990;265:7576–7582. [PubMed] [Google Scholar]
- 94. Yang G, Merrill DC, Thompson MW, Robillard JE, Sigmund CD. Functional expression of the human angiotensinogen gene in transgenic mice. J Biol Chem. 1994;269:32497–32502. [PubMed] [Google Scholar]
- 95. Toto RD, Rinner S, Ram CV. ACE inhibitors and target organ protection: an expanded role for these antihypertensive agents? Postgrad Med. 2004;116:11–16, 19–22, 48. [DOI] [PubMed] [Google Scholar]
- 96. Schieffer B, Wirger A, Meybrunn M, Seitz S, Holtz J, Riede UN, Drexler H. Comparative effects of chronic angiotensin‐converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation. 1994;89:2273–2282. DOI: 10.1161/01.CIR.89.5.2273. [DOI] [PubMed] [Google Scholar]
- 97. Sun Y, Zhang JQ, Zhang J, Ramires FJ. Angiotensin II, transforming growth factor‐beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–1569. [DOI] [PubMed] [Google Scholar]
- 98. Sopel MJ, Rosin NL, Lee TD, Legare JF. Myocardial fibrosis in response to angiotensin II is preceded by the recruitment of mesenchymal progenitor cells. Lab Invest. 2011;91:565–578. DOI: 10.1038/labinvest.2010.190. [DOI] [PubMed] [Google Scholar]
- 99. Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin‐6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol. 2005;204:428–436. DOI: 10.1002/jcp.20307. [DOI] [PubMed] [Google Scholar]
- 100. Sadoshima J, Izumo S. Molecular characterization of angiotensin II–induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts: critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–423. DOI: 10.1161/01.RES.73.3.413. [DOI] [PubMed] [Google Scholar]
- 101. Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, Takahashi T, Makino S, Kato T, Ogawa S. Interleukin‐6 family of cytokines mediate angiotensin II‐induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem. 2000;275:29717–29723. DOI: 10.1074/jbc.M003128200. [DOI] [PubMed] [Google Scholar]
- 102. Brilla CG, Funck RC, Rupp H. Lisinopril‐mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–1393. DOI: 10.1161/01.CIR.102.12.1388. [DOI] [PubMed] [Google Scholar]
- 103. Arcone R, Pucci P, Zappacosta F, Fontaine V, Malorni A, Marino G, Ciliberto G. Single‐step purification and structural characterization of human interleukin‐6 produced in Escherichia coli from a T7 RNA polymerase expression vector. Eur J Biochem. 1991;198:541–547. [DOI] [PubMed] [Google Scholar]
- 104. Banerjee I, Fuseler JW, Intwala AR, Baudino TA. IL‐6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. Am J Physiol Heart Circ Physiol. 2009;296:H1694–H1704. DOI: 10.1152/ajpheart.00908.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fontes JA, Rose NR, Cihakova D. The varying faces of IL‐6: from cardiac protection to cardiac failure. Cytokine. 2015;74:62–68. DOI: 10.1016/j.cyto.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal‐transducing receptor component for interleukin 6‐related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995;92:4862–4866. DOI: 10.1073/pnas.92.11.4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ancey C, Corbi P, Froger J, Delwail A, Wijdenes J, Gascan H, Potreau D, Lecron JC. Secretion of IL‐6, IL‐11 and LIF by human cardiomyocytes in primary culture. Cytokine. 2002;18:199–205. DOI: 10.1006/cyto.2002.1033. [DOI] [PubMed] [Google Scholar]
- 108. Ma F, Li Y, Jia L, Han Y, Cheng J, Li H, Qi Y, Du J. Macrophage‐stimulated cardiac fibroblast production of IL‐6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One. 2012;7:e35144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. McArthur L, Riddell A, Chilton L, Smith GL, Nicklin SA. Regulation of connexin 43 by interleukin 1beta in adult rat cardiac fibroblasts and effects in an adult rat cardiac myocyte: fibroblast co‐culture model. Heliyon. 2020;6:e03031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Akhtar R, Sherratt MJ, Cruickshank JK, Derby B. Characterizing the elastic properties of tissues. Mater Today (Kidlington). 2011;14:96–105. DOI: 10.1016/S1369-7021(11)70059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hinz B. Matrix mechanics and regulation of the fibroblast phenotype. Periodontol 2000. 2013;63:14–28. DOI: 10.1111/prd.12030. [DOI] [PubMed] [Google Scholar]
- 112. Bhana B, Iyer RK, Chen WL, Zhao R, Sider KL, Likhitpanichkul M, Simmons CA, Radisic M. Influence of substrate stiffness on the phenotype of heart cells. Biotechnol Bioeng. 2010;105:1148–1160. DOI: 10.1002/bit.22647. [DOI] [PubMed] [Google Scholar]
- 113. Fomovsky GM, Holmes JW. Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am J Physiol Heart Circ Physiol. 2010;298:H221–H228. DOI: 10.1152/ajpheart.00495.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wilson CL, Hayward SL, Kidambi S. Astrogliosis in a dish: substrate stiffness induces astrogliosis in primary rat astrocytes. RSC Adv. 2016;6:34447–34457. DOI: 10.1039/C5RA25916A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kamkin A, Kiseleva I, Wagner KD, Scholz H. Mechano‐electric feedback in the heart: evidence from intracellular microelectrode recordings on multicellular preparations and single cells from healthy and diseased tissue. In: Kamkin A, Kiseleva I, eds. Mechanosensitivity in Cells and Tissues. Moscow, Russia: Academia; 2005. [PubMed] [Google Scholar]
- 116. Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodels cell‐to‐cell communication in cultured myocytes. Circ Res. 2000;87:316–322. DOI: 10.1161/01.RES.87.4.316. [DOI] [PubMed] [Google Scholar]
- 117. Estigoy CB, Ponten F, Odeberg J, Herbert B, Guilhaus M, Charleston M, Ho JWK, Cameron D, Dos Remedios CG. Intercalated discs: multiple proteins perform multiple functions in non‐failing and failing human hearts. Biophys Rev. 2009;1:43. DOI: 10.1007/s12551-008-0007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Gutstein DE, Liu FY, Meyers MB, Choo A, Fishman GI. The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci. 2003;116:875–885. DOI: 10.1242/jcs.00258. [DOI] [PubMed] [Google Scholar]
- 119. Yamada K, Green KG, Samarel AM, Saffitz JE. Distinct pathways regulate expression of cardiac electrical and mechanical junction proteins in response to stretch. Circ Res. 2005;97:346–353. DOI: 10.1161/01.RES.0000178788.76568.8a. [DOI] [PubMed] [Google Scholar]
- 120. Engler AJ, Carag‐Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, Sanger JW, Sanger JM, Discher DE. Embryonic cardiomyocytes beat best on a matrix with heart‐like elasticity: scar‐like rigidity inhibits beating. J Cell Sci. 2008;121:3794–3802. DOI: 10.1242/jcs.029678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Thompson SA, Blazeski A, Copeland CR, Cohen DM, Chen CS, Reich DM, Tung L. Acute slowing of cardiac conduction in response to myofibroblast coupling to cardiomyocytes through N‐cadherin. J Mol Cell Cardiol. 2014;68:29–37. DOI: 10.1016/j.yjmcc.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wessels A, Sedmera D. Developmental anatomy of the heart: a tale of mice and man. Physiol Genomics. 2003;15:165–176. DOI: 10.1152/physiolgenomics.00033.2003. [DOI] [PubMed] [Google Scholar]
- 123. Camacho P, Fan H, Liu Z, He JQ. Small mammalian animal models of heart disease. Am J Cardiovasc Dis. 2016;6:70–80. [PMC free article] [PubMed] [Google Scholar]
- 124. Goversen B, van der Heyden MAG, van Veen TAB, de Boer TP. The immature electrophysiological phenotype of iPSC‐CMs still hampers in vitro drug screening: special focus on IK1. Pharmacol Ther. 2018;183:127–136. DOI: 10.1016/j.pharmthera.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 125. Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–860. DOI: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Machiraju P, Greenway SC. Current methods for the maturation of induced pluripotent stem cell‐derived cardiomyocytes. World J Stem Cells. 2019;11:33–43. DOI: 10.4252/wjsc.v11.i1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Mosqueira D, Mannhardt I, Bhagwan JR, Lis‐Slimak K, Katili P, Scott E, Hassan M, Prondzynski M, Harmer SC, Tinker A, et al. CRISPR/Cas9 editing in human pluripotent stem cell‐cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur Heart J. 2018;39:3879–3892. DOI: 10.1093/eurheartj/ehy249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Buikema JW, Lee S, Goodyer WR, Maas RG, Chirikian O, Li G, Miao YI, Paige SL, Lee D, Wu H, et al. Wnt activation and reduced cell‐cell contact synergistically induce massive expansion of functional human iPSC‐derived cardiomyocytes. Cell Stem Cell. 2020;27:50–63.e5. DOI: 10.1016/j.stem.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC, Pabon L, et al. Fatty acids enhance the maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cell Rep. 2019;13:657–668. DOI: 10.1016/j.stemcr.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Abilez OJ, Tzatzalos E, Yang H, Zhao M‐T, Jung G, Zöllner AM, Tiburcy M, Riegler J, Matsa E, Shukla P, et al. Passive stretch induces structural and functional maturation of engineered heart muscle as predicted by computational modeling. Stem Cells. 2018;36:265–277. DOI: 10.1002/stem.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Rodriguez AG, Han SJ, Regnier M, Sniadecki NJ. Substrate stiffness increases twitch power of neonatal cardiomyocytes in correlation with changes in myofibril structure and intracellular calcium. Biophys J. 2011;101:2455–2464. DOI: 10.1016/j.bpj.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mihic A, Li J, Miyagi Y, Gagliardi M, Li SH, Zu J, Weisel RD, Keller G, Li RK. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell‐derived cardiomyocytes. Biomaterials. 2014;35:2798–2808. DOI: 10.1016/j.biomaterials.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 133. Zhang H, Tian L, Shen M, Tu C, Wu H, Gu M, Paik DT, Wu JC. Generation of quiescent cardiac fibroblasts from human induced pluripotent stem cells for in vitro modeling of cardiac fibrosis. Circ Res. 2019;125:552–566. DOI: 10.1161/CIRCRESAHA.119.315491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhang J, Tao R, Campbell KF, Carvalho JL, Ruiz EC, Kim GC, Schmuck EG, Raval AN, da Rocha AM, Herron TJ, et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat Commun. 2019;10:2238. DOI: 10.1038/s41467-019-09831-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wülfers EM, Seemann G, Courties G, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522.e20. DOI: 10.1016/j.cell.2017.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chen K, Bai H, Arzigian M, Gao YX, Bao J, Wu WS, Shen WF, Wu L, Wang ZZ. Endothelial cells regulate cardiomyocyte development from embryonic stem cells. J Cell Biochem. 2010;111:29–39. DOI: 10.1002/jcb.22680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Yoshida S, Miyagawa S, Fukushima S, Kawamura T, Kashiyama N, Ohashi F, Toyofuku T, Toda K, Sawa Y. Maturation of human induced pluripotent stem cell‐derived cardiomyocytes by soluble factors from human mesenchymal stem cells. Mol Ther. 2018;26:2681–2695. DOI: 10.1016/j.ymthe.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Giacomelli E, Bellin M, Sala L, van Meer BJ, Tertoolen LG, Orlova VV, Mummery CL. Three‐dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co‐differentiated from human pluripotent stem cells. Development. 2017;144:1008–1017. DOI: 10.1242/dev.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Giacomelli E, Meraviglia V, Campostrini G, Cochrane A, Cao XU, van Helden RWJ, Krotenberg Garcia A, Mircea M, Kostidis S, Davis RP, et al. Human‐iPSC‐derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non‐cardiomyocyte contributions to heart disease. Cell Stem Cell. 2020;26:862–879.e11. DOI: 10.1016/j.stem.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gluck JM, Herren AW, Yechikov S, Kao HKJ, Khan A, Phinney BS, Chiamvimonvat N, Chan JW, Lieu DK. Biochemical and biomechanical properties of the pacemaking sinoatrial node extracellular matrix are distinct from contractile left ventricular matrix. PLoS One. 2017;12:e0185125. DOI: 10.1371/journal.pone.0185125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Baudino TA, McFadden A, Fix C, Hastings J, Price R, Borg TK. Cell patterning: interaction of cardiac myocytes and fibroblasts in three‐dimensional culture. Microsc Microanal. 2008;14:117–125. DOI: 10.1017/S1431927608080021. [DOI] [PubMed] [Google Scholar]
- 142. Ronaldson‐Bouchard K, Yeager K, Teles D, Chen T, Ma S, Song L, Morikawa K, Wobma HM, Vasciaveo A, Ruiz EC, et al. Engineering of human cardiac muscle electromechanically matured to an adult‐like phenotype. Nat Protoc. 2019;14:2781–2817. DOI: 10.1038/s41596-019-0189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Lee MO, Jung KB, Jo SJ, Hyun SA, Moon KS, Seo JW, Kim SH, Son MY. Modelling cardiac fibrosis using three‐dimensional cardiac microtissues derived from human embryonic stem cells. J Biol Eng. 2019;13:15. DOI: 10.1186/s13036-019-0139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. DOI: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 145. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore‐Morris T, Scott B, Tsukamoto H, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109:9448–9453. DOI: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Kulkarni AB, Karlsson S. Transforming growth factor‐beta 1 knockout mice: a mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol. 1993;143:3–9. [PMC free article] [PubMed] [Google Scholar]