

Abstract
Background
Impairment of glycolytic metabolism is suggested to contribute to diabetic cardiomyopathy. In this study, we explored the roles of SIRT3 (Sirtuin 3) on cardiomyocyte glucose metabolism and cardiac function.
Methods and Results
Exposure of H9c2 cardiomyocyte cell lines to high glucose (HG) (30 mmol/L) resulted in a gradual decrease in SIRT3 and 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase isoform 3 (PFKFB3) expression together with increases in p53 acetylation and TP53‐induced glycolysis and apoptosis regulator (TIGAR) expression. Glycolysis was significantly reduced in the cardiomyocyte exposed to HG. Transfection with adenovirus‐SIRT3 significantly increased PFKFB3 expression and reduced HG‐induced p53 acetylation and TIGAR expression. Overexpression of SIRT3 rescued impaired glycolysis and attenuated HG–induced reactive oxygen species formation and apoptosis. Knockdown of TIGAR in cardiomyocytes by using siRNA significantly increased PFKFB3 expression and glycolysis under hyperglycemic conditions. This was accompanied by a significant suppression of HG–induced reactive oxygen species formation and apoptosis. In vivo, overexpression of SIRT3 by an intravenous jugular vein injection of adenovirus‐SIRT3 resulted in a significant reduction of p53 acetylation and TIGAR expression together with upregulation of PFKFB3 expression in the heart of diabetic db/db mice at day 14. Overexpression of SIRT3 further reduced reactive oxygen species formation and blunted microvascular rarefaction in the diabetic db/db mouse hearts. Overexpression of SIRT3 significantly blunted cardiac fibrosis and hypertrophy and improved cardiac function at day 14.
Conclusions
Our study demonstrated that SIRT3 attenuated diabetic cardiomyopathy via regulating p53 acetylation and TIGAR expression. Therefore, SIRT3 may be a novel target for abnormal energy metabolism in diabetes mellitus.
Keywords: diabetic cardiomyopathy, glycolysis, p53 acetylation, Sirtuin 3, TP53‐induced glycolysis and apoptosis regulator
Subject Categories: Angiogenesis, Metabolism
Nonstandard Abbreviations and Abbreviations
- Ad‐SIRT3
Adenovirus SIRT3
- EC
endothelial cell
- HG
high glucose
- NIH
National Institutes of Health
- PAEC
pig artery endothelial cells
- PFKFB3
6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase isoform 3
- PFU
plaque‐forming units
- ROS
reactive oxygen species
- SIRT3
sirtuin 3
- TIGAR
TP53‐induced glycolysis and apoptosis regulator
Clinical Perspective
What Is New?
Hyperglycemia induced p53 acetylation and an imbalanced TP53‐induced glycolysis and apoptosis regulator/6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase isoform 3 in favoring TP53‐induced glycolysis and apoptosis regulator, thus resulted in an impairment of glycolytic metabolism, which may contribute to diabetic cardiomyopathy.
In vitro, overexpression of sirtuin 3 blunted hyperglycemia‐induced impairments of glycolysis, oxidative stress, and apoptosis by suppression of p53 acetylation and TP53‐induced glycolysis and apoptosis regulator expression.
In vivo, overexpression of sirtuin 3 blunted p53‐acetylation, microvascular rarefaction, and cardiac dysfunction in db/db mice.
What Are the Clinical Implications?
This is the first time it has been demonstrated that sirtuin 3 modulates diabetic cardiomyopathy by a mechanism involving inhibition of p53 acetylation and TP53‐induced glycolysis and apoptosis regulator–mediated glycolytic metabolism.
Sirtuin 3–mediated metabolic reprogram may be a novel target for diabetic cardiomyopathy.
Diabetic cardiomyopathy is the main cause of heart failure, which contributes to higher morbidity and mortality in patients with diabetes mellitus. The prevalence of diabetic cardiomyopathy is increasing at an alarming rate 1 , 2 , 3 ; it is, therefore, urgent to identify potential therapeutic targets. Although multiple factors including oxidative stress, apoptosis, cardiac fibrosis, impaired angiogenesis, and glycolytic metabolism are considered to be the important pathophysiological factors, the underlying mechanisms of diabetic cardiomyopathy are still not fully understood.
SIRT3 (sirtuin 3) regulates cell metabolism through the deacetylation of its substrates. 4 , 5 Accumulative evidence highlighted the regulatory role of SIRT3 in cardiovascular and metabolic diseases such as heart failure, diabetes mellitus, and obesity. Activation of SIRT3 by resveratrol has been shown to improve cardiac function and attenuate cardiac fibrosis. 6 A study also showed that the expression and activity of SIRT3 were reduced in the diabetic hearts. 7 In contrast, a reduction of SIRT3 levels is associated with obesity‐induced microvascular rarefaction and cardiac dysfunction. 8 We have shown that overexpression of apelin with adenovirus‐apelin administration increased myocardial vascular density and attenuated ischemia‐induced cardiac dysfunction via the upregulation of SIRT3 in diabetes mellitus. 9 Furthermore, specific knockout of SIRT3 in endothelial cells resulted in an increase in oxidative phosphorylation and a reduction of glycolysis that was associated with impairment in angiogenesis and diastolic dysfunction. 10 , 11
TP53‐induced glycolysis and apoptosis regulator (TIGAR) is a novel downstream target gene of tumor suppressor p53. TIGAR was originally identified as a fructose 2, 6 bisphosphatase of the dual phosphofructose kinase/fructose 2, 6 bisphosphatase (PFKFB) family and regulates glycolysis and apoptosis. TIGAR is involved in various biological processes, including glycolytic metabolism, apoptosis, cell cycle, and cell death. 12 The p53 and its transcriptional target gene TIGAR are activated in the cardiomyocyte under hypoxia. 13 , 14 The expression of p53 is also elevated in the human failing heart. 15 , 16 So far, two roles of p53 accumulation in the failing heart have been demonstrated: (1) induction of cell cycle arrest that causes proliferative cells arrest in the G1 stage of the cell cycle to induce senescence; and (2) suppression of angiogenesis. 17 , 18 Elevation of p53 reduced capillary formation by inhibition of hypoxia‐inducible factor‐1α in the hypertrophic hearts. 19 Studies also show that global knockout of p53 in mice attenuates doxorubicin‐induced cardiac dysfunction. 20 , 21 Knockout of p53 in endothelial cells (ECs) reduces EC apoptosis and increases capillary density in the model of pressure overload–induced heart failure. 22 Knockout of TIGAR also has been shown to attenuate ischemic heart failure. 13 , 14 These studies provide a strong rationale for targeting p53 and TIGAR to ameliorate the microvascular dysfunction and heart failure. The p53 was deacetylated by histone deacetylases. Impairment of SIRT3 has been shown to promote acute kidney injury through elevated acetylation of p53. 23 So far, the direct link among SIRT3, p53 acetylation, and TIGAR in diabetic cardiomyopathy has not been investigated. In this study, we tested our hypothesis that SIRT3 regulates diabetic cardiomyopathy by a mechanism involving removal of p53 acetylation and downregulation of TIGAR, which modulates cardiomyocyte glycolytic metabolism.
METHODS
The authors declare that all supporting data are available within the article.
All protocols were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center (Protocol ID: 2018C) and were consistent with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85‐23, Revised 1996).
Experimental Mice
Male db+/− mice and diabetic db/db mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The experimental mice at the age of 10 to 12 months were divided into the following: (1) db+/− mice; (2) db/db mice+adenovirus‐β‐gal; and (3) db/db+adenovirus‐SIRT3 mice. Experimental mice received an intravenous jugular vein injection of adenovirus‐β‐gal or adenovirus‐SIRT3 (human adenovirus type 5 [dE1/E3], cytomegalovirus promoter) (1×109 plaque‐forming units [PFU]) (Vector Biolabs, Malvern, PA). After 14 days of adenovirus‐β‐gal or adenovirus‐SIRT3 administration, experimental mice were euthanized by cervical dislocation under anesthesia with isoflurane.
Cell Culture
Rat cardiomyocytes H9C2 (ATCC CRL1446) cell lines were grown in DMEM (Sigma‐Aldrich, St. Louis, MO) with the addition of 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 2 mmol/L glutamine, 104×diluted 10 000 U/mL penicillin and 10 mg/mL streptomycin (Sigma‐Aldrich).
Pig artery endothelial cells (PAECs) were obtained from the main pulmonary artery of 6‐ to 7‐month‐old pigs. PAECs were cultured in endothelial basal medium (EBM‐2, Clonetics, San Diego, CA) supplemented with EGM‐2 Endothelial Growth Single Quot Kit Supplement & Growth Factors (CC4176; Lonza, Basel, Switzerland).
Western Blot Analysis
Cultured cells or left ventricular samples were homogenized with an ice‐cold radioimmunoprecipitation assay buffer. Protein concentration was measured with the Bradford reagent (B6916; Sigma‐Aldrich). The polyvinylidene fluoride membranes were probed with antibodies specific to SIRT3 (Cell Signaling Technology, Beverly, MA) (1:2000), acetylated‐lysine (Cell Signaling Technology) (1:1000), p53 and acetylated‐p53 (K381) (Abcam, Cambridge, MA) (1:1000), TIGAR (Santa Cruz Biotechnology, Dallas, TX) (1:2000), cleaved Caspase‐3 (Cell Signaling Technology) (1:1000), vascular endothelial growth factor (VEGF) (1:1000), 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase isoform 3 (PFKFB3) (Abcam) (1:2000), β‐actin (Cell Signaling Technology), or β‐actin (Cell Signaling Technology) (1:3000). The membranes were then washed and incubated with an anti‐rabbit or anti‐mouse secondary antibody conjugated with horseradish peroxidase. Densitometries were analyzed using TINA 2.0 image analysis software (DesignSoft, Budapest, Hungary).
RNA Interference
H9C2 cells were transfected with 25 nmol/L TIGAR siRNA or p53 siRNA as well as control scramble siRNA (TriFecTaDsi DNA duplex; Integrated DNA Technologies, Coralville, IA) by using TransIT‐X2 Dynamic Delivery System (Integrated DNA Technologies) following the manufacturer's instruction. The transfection efficiency was detected by western blotting.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling Assay
In situ Dead End Colorimetric Apoptosis Detection System (Promega, Madison, WI) was used to detect apoptotic cells according to the manufacturer's instructions. For the apoptotic cell number, only fields which terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) positive cells were presented were counted. The number of TUNEL positive cells was quantified by measuring 6 microscopic fields using image‐analysis software (Image J, NIH, Bethesda, MD).
Dihydroethidium Staining
Equal numbers of cells were plated in 8‐well plates. After treatment, cells were rinsed with PBS and incubated with 0.5 mmol/L dihydroethidium in PBS for 10 minutes at room temperature in the dark. Similarly, left ventricular sections were prepared using a cryostat on microscope slides. Slides were rinsed with PBS and incubated with 0.5 mmol/L dihydroethidium in PBS for 15 minutes at room temperature in the dark. The relative density of dihydroethidium (red) fluorescence was quantified by measuring 6 random microscopic fields using image‐analysis software (Image J).
Metabolic Assays
Glycolysis was determined using the XFe24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) following the manufacturer's instruction. Briefly, cells were seeded in tissue culture–treated 24‐well plates (V7‐PS) at a density of 25 000 cells per well as determined by a pilot cell density assay. The next day, the cells were incubated in unbuffered assay medium supplemented with various substrates as described below, at 37°C in a non‐CO2 incubator for 1 hour before analysis. The unbuffered assay medium was supplemented with glutamine (2 mmol/L) only. The baseline of extracellular acidification rate was measured, followed by the sequential injection of the following compounds with indicated final concentration: glucose (10 mmol/L), oligomycin (1 μmol/L), and 2‐deoxyglucose (100 mmol/L). The levels of basal glycolysis, glycolytic reserve and glycolytic capacity were calculated from the raw data using the Seahorse report generator (Agilent Technologies, Santa Clara, CA).
Tube Formation Assay
PAECs (1×104 cells/well) were seeded into a 96‐well plate precoated with 40 μL reduced‐growth factor ECM gel (Life Technologies, New York, NY) and incubated for 6 hours. A tubelike structure was captured with an inverted phase‐contrast microscope (AMG, Life Technologies). The numbers of branching points and segments were quantified by Image J software with angiogenesis analyzer plug‐in (developed by Gilles Carpentier).
Wound Scratch Migration Assay
PAECs were seeded onto 24‐well plates. At near confluence, PAEC cultures were scratched with a 10‐μL pipette tip. Images of the scraped area were obtained for reference. Immediately following the scraping, the wells were washed with PBS and then incubated with medium overnight. After incubation for 16 hours, the cells migrating into the scraped area were measured. Images of the scraped area were taken and overlaid with the original reference image. The average distance migrated by cells at increasing times following scratching was calculated by comparison of the denuded areas remaining relative to the zero‐time point per each cell well.
Analysis of Myocardial Capillary and Arteriole Densities
Five‐micrometer sections of left ventricle were cut and incubated with fluorescein‐labeled Isolectin B4 (1:200; Molecular Probe, Invitrogen, Eugene, OR) and Cy3‐conjugated anti‐α smooth muscle actin (1:100; Sigma‐Aldrich). The number of capillaries (isolectin B4–positive EC) was counted and expressed as capillary density per square millimeter. Myocardial arteriole (smooth muscle actin–positive smooth muscle cells located in vascular walls) density was measured using Image J software.
Histology and Immunofluorescence Analysis
Cells were fixed with 10% formalin for 15 minutes at room temperature. After this, the cells were permeabilized with 0.2% Triton X‐100 in PBS for 10 minutes. Nonspecific binding was blocked through the incubation with 10% FCS in PBS for 30 minutes at 37°C in a humidified chamber. Cells were immunostained with SIRT3 primary antibodies (1:200) followed by incubation with second antibodies conjugated with fluorescein isothiocyanate or Cy3 (1:500). The area percentage of fluorescence intensity was quantified at 6 random microscopic fields using Image J software.
The left ventricle was embedded in frozen optimal cutting temperature compound (4583; Sakura Finetek, Torrance, CA) and 10‐µm frozen sections prepared. Sections were immunostained with troponin and SIRT3 primary antibodies (1:200) followed by incubation with second antibodies conjugated with fluorescein isothiocyanate or Cy3 (1:500). Photomicrographs were obtained with an Olympus BX51 microscope, a Q‐Color5 digital camera and a Q‐Capture Suite acquisition software (Olympus, Tokyo, Japan). For the measurement of myocardial fibrosis, frozen sections were stained with Masson's trichrome staining (blue) and hematoxylin and eosin (red). Fibrotic fraction was calculated as the percentage of blue‐stained area to total myocardial area.
Echocardiography
Transthoracic echocardiograms were performed on mice using a Vevo770 High‐Resolution In Vivo Micro‐Imaging System equipped with an RMV 710B scan head (Visual Sonics Inc., Toronto, ON, Canada). Mice were anesthetized by inhalation of 1.5% to 2% isoflurane mixed with 100% medical oxygen in an isolated chamber for induction. Anesthesia was maintained with 1% to 1.5% isoflurane to control the heart rate between 400 and 450 beats per minute. M‐mode cine loops were recorded and analyzed by High‐Frequency Ultrasound Imaging software (Visual Sonics Inc.) to assess myocardial parameters and cardiac functions of left ventricle, including left ventricular (LV) end‐systolic diameter, end‐diastolic diameter, LV end‐systolic volume, end‐diastolic volume, stroke volume, and cardiac output as well as ejection fraction and fractional shortening.
Fasting Glucose Levels
Blood glucose was measured at the end of the experiments. Blood was obtained from experimental mice by tail snip, and blood glucose levels were measured using the One Touch SureStep meter. Experimental mice were fasted overnight before collecting blood. Glucose levels were expressed as milligrams per deciliter.
Statistical Analysis
Data were presented as mean±SD. The assumption of normality in both comparison groups was determined by normality test. Statistical significance was determined using 2‐tailed independent Student t test for comparisons between the 2 groups, 1‐way or 2‐way ANOVA, followed by Tukey's post hoc test for multiple comparisons using Prism 8.1.1 software (GraphPad Software, La Jolla, CA). P<0.05 was considered statistically significant.
RESULTS
HG Reduced SIRT3 Expression and Upregulated p53 Acetylation and TIGAR Expression
To explore the effects of HG on the SIRT3 expression and p53 acetylation, the time‐dependent response was examined in cardiomyocyte H9C2 cell lines under hyperglycemic conditions. Exposure of cardiomyocyte cell lines to HG (30 mmol/L) for various time up to 72 hours led to a gradual downregulation of SIRT3 expression. This was accompanied by increases in p53, p53 acetylation, TIGAR, and caspase‐3 expression. Exposure of H9c2 to HG further led to reductions of PFKFB3 and VEGF expression (Figure 1A). To exclude the possible role of HG‐induced high osmolarity in the culture medium, H9c2 was exposed to mannitol (30 mmol/L) for 72 hours. Exposure of H9c2 cell lines to mannitol (30 mmol/L) had no effects on the levels of SIRT3, p53, p53 acetylation, PFKFB3, and TIGAR expression (Figure S1).
Figure 1. SIRT3 overexpression inhibited HG‐induced activation of p53/TIGAR axis and modulate cardiomyocyte metabolism.
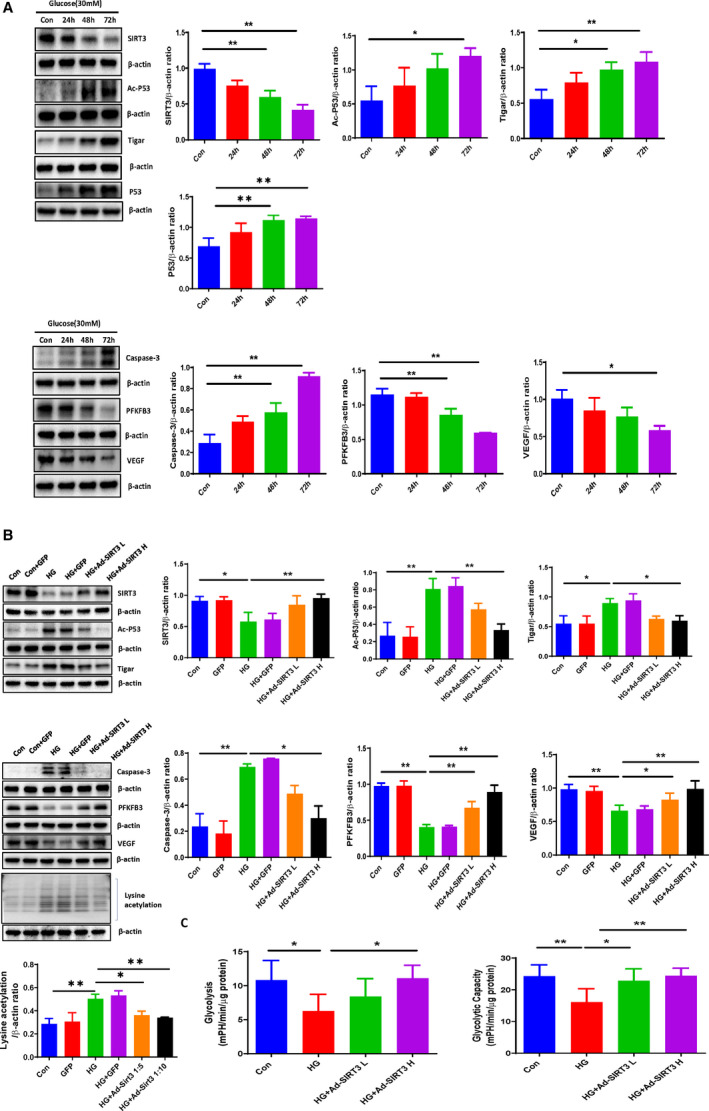
A, Western blot analysis demonstrating that exposure of H9c2 cells to HG (30 mmol/L) for 24, 48, and 72 hours resulted in a gradual increase in expression of p53, Ac‐p53, TIGAR, and caspase‐3, but a significant reduction of SIRT3, PFKFB3, and VEGF expression. B, Western blot analysis confirming that adenovirus‐SIRT3 treatment significantly attenuated the HG‐induced Ac‐p53, lysine acetylation, TIGAR, Caspase‐3 expression, but upregulated SIRT3, PFKFB3, and VEGF expression in H9c2 cells under HG conditions. All data represent mean±SD (n=3 per group, *P<0.05 and **P<0.01). C, Ad‐SIRT3 enhanced glycolysis and glycolytic capacity compared with HG treatment group. All data represent mean±SD (n=5, *P<0.05 and **P<0.01). Ac‐p53 indicates acetyl‐p53; Ad‐SIRT3, adenovirus SIRT3; caspase‐3, cleaved caspase‐3; HG, high glucose; p53, tumor suppressor p53; PFKFB3, 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase isoform 3; SIRT3, sirtuin 3; TIGAR, TP53‐induced glycolysis and apoptosis regulator; and VEGF, vascular endothelial growth factor.
SIRT3 Blunted HG‐Induced p53 Acetylation and TIGAR Expression in Cardiomyocytes
To test the potential role of SIRT3 in the HG‐induced p53 acetylation, the human SIRT3 adenoviral vectors (adenovirus‐SIRT3) were utilized to overexpress SIRT3 in cardiomyocytes. H9c2 cells were infected by adenovirus‐SIRT3 at 0.5×106 PFU/mL (low dose) or 1×106 PFU/mL (high dose) for 48 hours. Adenovirus‐SIRT3 treatment led to a dose‐dependent increase in SIRT3 expression in H9c2 cells (Figure S2). Moreover, adenovirus‐SIRT3 treatment significantly reduced the HG‐induced p53 acetylation and lysine acetylation as well as expression of TIGAR and caspase‐3. Adenovirus‐SIRT3 treatment also significantly upregulated PFKFB3 and VEGF expression under HG conditions (Figure 1B). Transfection of H9c2 cells with adenovirus—green fluorescent protein had little effects as compared with control or HG exposure (Figure 1B).
SIRT3 Enhanced Cardiomyocyte Glycolysis Under HG Conditions
Since TIGAR was downregulated while PFKFB3 was upregulated after adenovirus‐‐SIRT3 treatment, we next examined whether SIRT3 regulated glycolysis in H9c2 cells under HG conditions by using the Seahorse analyzer. Exposure of H9c2 cells to HG led to a significant reduction of glycolysis and glycolytic capacity. Overexpression of SIRT3 significantly improved HG‐induced impairments of glycolysis and glycolytic capacity in H9c2 cells (Figure 1C).
SIRT3 Ameliorated the HG‐Induced ROS Formation and Apoptosis
Exposure of H9c2 cells to HG for 72 hours significantly increased the intracellular formation of ROS and apoptosis (Figure 2A and 2B). Overexpression of SIRT3 significantly attenuated HG‐induced ROS formation and apoptosis in H9c2 cells (Figure 2A and 2B).
Figure 2. SIRT3 ameliorated the HG‐induced ROS formation and apoptosis, and promoted angiogenesis in EC cocultured with cardiomyocyte conditioned media.
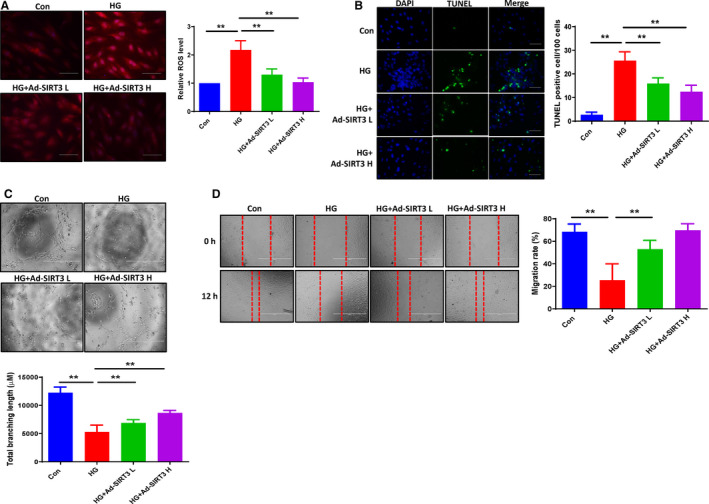
A, Quantitative analysis of ROS formation by dihydroethidium staining revealing that exposure of H9c2 cells HG (30 mmol/L) for 72 hours increased the intracellular ROS formation, whereas adenovirus‐SIRT3 ameliorated the HG‐induced ROS formation. All data represent mean±SD (n=5 per group, **P<0.01). B, Representative images and quantification of TUNEL+ cells in cultured H9c2 cells. Cell apoptosis was significantly increased in HG exposed group, but adenovirus‐SIRT3 decreased the HG‐induced TUNEL+ cells numbers in H9c2 cells (green, ×20). All data represent mean±SD (n=5 per group, **P<0.01). C, Adenovirus‐SIRT3 significantly increased total branching length compared with HG media treatment. All data represent mean±SD (n=4 per group, **P<0.01). D, Migration of PAECs was assessed using a scratch wound assay. Cocultured ECs with adenovirus‐SIRT3 media significant increased EC migration rate. All data represent mean±SD (n=5, **P<0.01). Ad‐SIRT3 indicates adenovirus SIRT3; HG, high glucose; PAECs, pig artery endothelial cells; ROS, reactive oxygen species; and TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
SIRT3 Promoted Angiogenesis in ECs Cocultured With Cardiomyocyte Conditioned Media
Using ECs cocultured with cardiomyocyte conditioned media, we investigated the paracrine role of SIRT3 in H9c2 cells on EC angiogenesis. In the first set of experiments, H9c2 cells were exposed to HG for 72 hours. We then removed the HG media and replaced it with DMEM for 24 hours. The DMEM was collected and cocultured with PAECs. As shown in Figure 2C, cocultures of ECs with these media led to a significant impairment of tube formation as evidenced by the reduced branching length. In cocultures with cardiomyocyte conditioned media, EC migration was also significantly decreased (Figure 2D). In the SIRT3 overexpression experiments, H9C2 cells were treated with adenovirus‐‐SIRT3 in HG media for 48 hours, then removed the adenovirus‐SIRT3 and HG media and replaced it with the DMEM for 24 hours. The DMEM was collected and used to coculture PAECs. Cocultured ECs with adenovirus‐SIRT3 DMEM significantly increased EC tube formation (Figure 2C). Similarly, EC migration rate was significantly increased in cocultures with adenovirus‐SIRT3 cardiomyocyte media (Figure 2D).
Knockdown of TIGAR Improved Cardiomyocyte Glycolysis
H9c2 cells were transfected by using TransIT‐X2 Dynamic Delivery System and 25 nmol/L siRNA was used to knock down the p53 gene. Transfection with p53 siRNA dramatically reduced p53 expression in H9c2 cell lines, while control scramble siRNA had little effect on p53 expression (Figure 3A). Knockdown of p53 by siRNA‐p53 significantly reduced HG‐induced TIGAR and caspase‐3 expression. Furthermore, siRNA‐p53 treatment ameliorated HG‐induced downregulation of PFKFB3 expression (Figure 3B).
Figure 3. Knockdown of TIGAR improved cardiomyocyte metabolism under HG conditions.
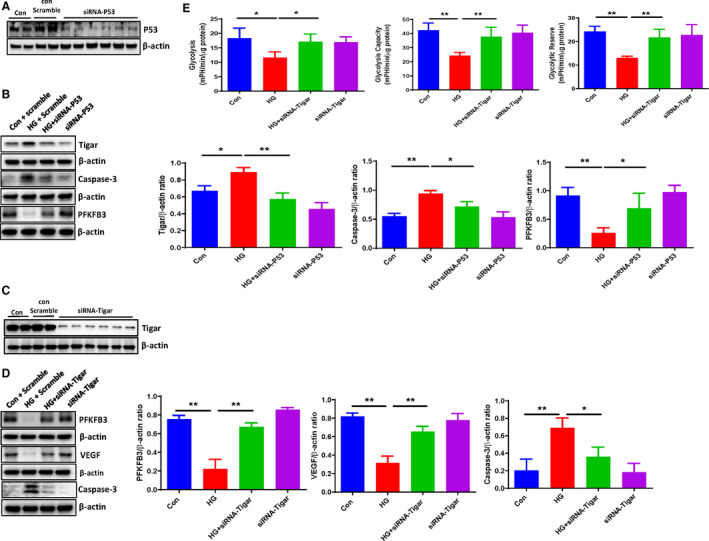
A, Western blot analysis demonstrating that transfection with p53 siRNA resulted in a knockdown of p53 expression in H9c2 cell lines. B, Western blot analysis demonstrating that siRNA‐p53 significantly reduced the HG‐induced expression of TIGAR and Caspase‐3, but increased the expression of PFKFB3 compared with the control H9c2 cells. C, Western blot analysis demonstrating that transfection with TIGAR siRNA led to a knockdown of TIGAR expression in H9c2 cell lines. D, Western blot analysis demonstrating that knockdown of TIGAR ameliorated the HG‐induced caspase‐3 expression, but increased PFKFB3 and VEGF expression compared with the control H9c2 cells. All data represent mean±SD (n=3 per group, *P<0.05 and **P<0.01). E, Knockdown of TIGAR rescued HG‐induced impairments of glycolysis, glycolytic capacity, and glycolytic reserve in H9c2 cell lines. All data represent mean±SD (n=4, *P<0.05 and **P<0.01). Caspase‐3 indicates cleaved caspase‐3; HG, high glucose; p53, tumor suppressor p53; PFKFB3, 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase isoform 3; TIGAR, TP53‐induced glycolysis and apoptosis regulator; and VEGF, vascular endothelial growth factor.
To investigate the HG‐induced activation of TIGAR and its downstream signal pathway, TIGAR was silenced by siRNA. Similarly, transfection with TIGAR siRNA significantly reduced TIGAR expression in H9c2 cell lines, while control scramble siRNA had little effect on TIGAR expression (Figure 3C). Furthermore, knockdown of TIGAR by TIGAR siRNA treatment significantly blunted HG‐induced downregulation of PFKFB3 and VEGF expression. Similarly, knockdown of TIGAR significantly attenuated HG‐induced caspase‐3 expression (Figure 3D). Measurement of glycolysis showed that knockdown of TIGAR resulted in a significant improvement of glycolysis, glycolytic capacity, and glycolytic reserve in H9c2 cells under HG conditions (Figure 3E).
Knockdown of TIGAR Attenuated HG‐Induced ROS Formation and Apoptosis
Exposure of H9c2 cells to HG (30 mmol/L) for 72 hours significantly increased the intracellular ROS levels and apoptosis. Knockdown of TIGAR significantly reduced HG‐induced ROS formation (Figure 4A). Knockdown of TIGAR also significantly suppressed HG‐induced apoptosis in H9c2 cells (Figure 4B).
Figure 4. Knockdown of TIGAR ameliorated the HG‐induced ROS formation and apoptosis, promoted angiogenesis in ECs cocultured with cardiomyocyte conditioned media.
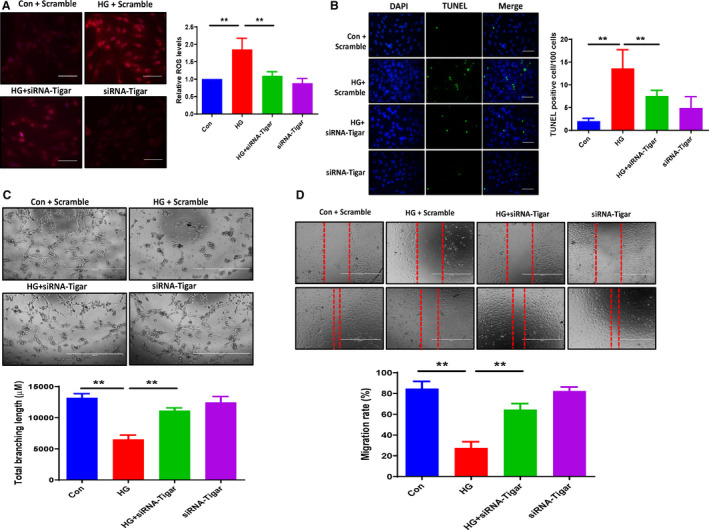
A, Dihydroethidium staining revealing that knockdown of TIGAR ameliorated the HG‐induced ROS formation. All data represent mean±SD (n=3 per group, **P<0.01). B, Representative images and quantification of TUNEL+ cells. Knockdown of TIGAR decreased the HG‐induced TUNEL+ cells numbers (green, ×20). All data represent mean±SD (n=5 per group, **P<0.01). C, Knockdown of TIGAR significantly increased total branching length compared with HG media treatment. All data represent mean±SD (n=3 per group, **P<0.01). D, Migration of PAECs was assessed using a scratch wound assay. Migration in distance was significantly decreased in the HG media treatment group, while knockdown of TIGAR significantly increased PAEC migration rate compared with HG media treatment. All data represent mean±SD (n=5 per group; **P<0.01). HG indicates high glucose; PAEC, pig artery endothelial cells; ROS, reactive oxygen species; TIGAR, TP53‐induced glycolysis and apoptosis regulator; and TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Knockdown of TIGAR Increased Angiogenesis in ECs Cocultured With Cardiomyocyte Conditioned Media
To test whether knockdown of TIGAR in cardiomyocytes improved angiogenesis in ECs, H9c2 cells were exposed to HG for 72 hours and transfected with siRNA‐TIGAR for 48 hours; then we removed the medium and replaced it with the DMEM for 24 hours. The DMEM was collected and cocultured with PAECs. In ECs cocultured with cardiomyocyte conditioned media, knockdown of TIGAR in cardiomyocytes resulted in a significant increase in tube formation in ECs (Figure 4C). Also, PAEC migration rate was significantly increased in cocultures with siRNA‐TIGAR cardiomyocyte conditioned media (Figure 4D).
Overexpression of SIRT3 Suppressed p53 Acetylation in Diabetic db/db Mice
Systemic delivery of adenovirus‐SIRT3 (1×109 PFU) led to a significant increase in SIRT3 expression in the hearts of db/db mice compared with db/db mice receiving adenovirus‐β‐gal (1×109 PFU) at day 14 (Figure 5A). Immunohistochemical analysis revealed that SIRT3 was colocalized with troponin on cardiomyocytes and isolectin B4 on ECs in the adenovirus‐SIRT3–treated db/db mouse hearts (Figure S3). Systemic administration of adenovirus‐SIRT3 had little effects on body weight and glucose levels in diabetic db/db mice, but significantly reduced heart weight (0.172±0.001 g versus 0.148±0.017 g; P<0.05; Table 1).
Figure 5. SIRT3 gene therapy reduced protein acetylation, suppressed TIGAR expression, and reduced microvascular rarefaction in diabetic db/db mice.
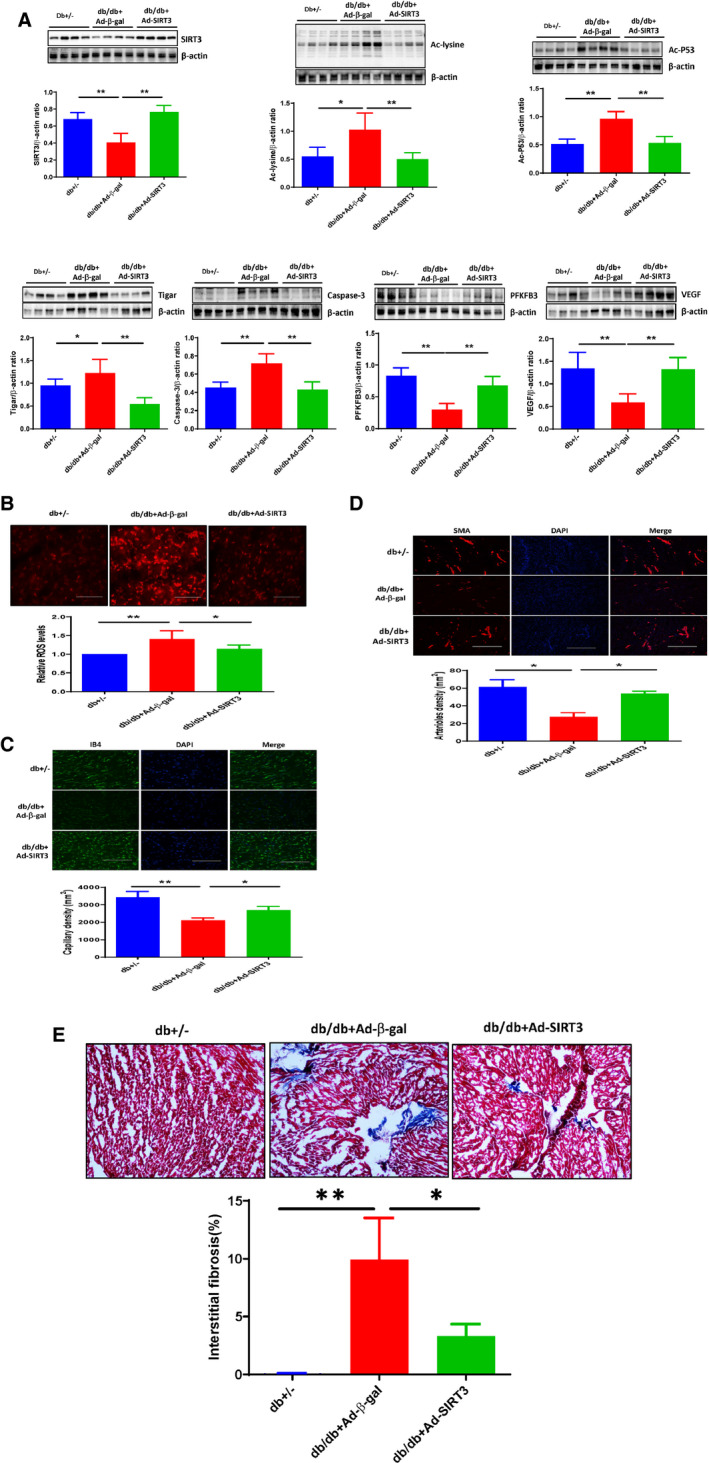
A, Western blot analysis demonstrating that systemic administration of adenovirus‐SIRT3 (1×109 PFU) resulted in overexpression of SIRT3 in the db/db mouse hearts compared with db/db mice receiving adenovirus‐β‐gal (1×109 PFU). Overexpression of SIRT3 significantly increased the expression of PFKFB3 and VEGF, but reduced the expression of Ac‐lysine, Ac‐p53, TIGAR, caspase‐3 compared with db/db mice+Ad‐β‐gal. B, Representative images of dihydroethidium staining demonstrating that Overexpression of SIRT3 in db/db mice exhibited a significant reduced ROS levels in the hearts. C, Representative images and quantitative analysis showing that treatment with adenovirus‐SIRT3 significantly increased capillary density in db/db mice by isolectin B4 staining (green, ×10). D, Representative images and quantitative analysis showing that treatment with Ad‐SIRT3 significantly increased myocardial arteriole density in db/db mice by SMA staining (red, ×10). All data represent mean±SD (n=4 per group, *P<0.05 and **P<0.01). E, Representative images and quantitative analysis showing that treatment with Ad‐SIRT3 significantly reduced myocardial fibrosis in db/db mice by Masson's trichrome staining (blue, ×10). All data represent mean±SD (n=4 per group, *P<0.05 and **P<0.01). Ad‐SIRT3 indicates adenovirus SIRT3; Caspase‐3, cleaved caspase‐3; p53, tumor suppressor p53; PFKFB3, 6‐phosphofructo‐2‐kinase/fructose‐2, 6‐bisphosphatase isoform 3; ROS, reactive oxygen species; SIRT3, sirtuin 3; SMA, smooth muscle actin; TIGAR, TP53‐induced glycolysis and apoptosis regulator; and VEGF, vascular endothelial growth factor.
Table 1.
Measurement of Body Weight, Glucose Levels and Heart Weight
| Db+/− Mice (n=6) | Db/db+Adenovirus‐β‐gal (n=7) | Db/db+Adenovirus‐SIRT3 (n=5) | P Value | |
|---|---|---|---|---|
| Body weight, g | 35.8±2.4 | 69.7±5.9* | 65.9±3.7* | |
| Heart weight, g | 0.154±0.011 | 0.172±0.001* | 0.148±0.017 † | <0.05 |
| Glucose levels, mg/dL | 89±18 | 243±142* | 268±157* |
All data represent mean±SD. One‐way ANOVA, followed by Tukey's post hoc test.
P<0.05 vs db+/−.
P<0.05 vs Db/db+Ad‐β‐gal.
Western blot analysis showed that overexpression of SIRT3 significantly reduced total lysine acetylation and p53 acetylation levels, and attenuated TIGAR and caspase‐3 expression in the hearts of db/db mice at day 14. Overexpression of SIRT3 significantly increased the expression of PFKFB3 and VEGF as compared with db/db mice+adenovirus‐β‐gal (Figure 5A).
Overexpression of SIRT3 Blunted Microvascular Rarefaction in the Hearts of Diabetic db/db Mice
The db/db mice had a significant increase in ROS levels in the hearts as compared with that of db+/− mice. Overexpression of SIRT3 in db/db mice led to a significant reduction of ROS formation in the heart (Figure 5B).
Microvascular rarefaction was determined by measuring capillary (isolectin B4) and arteriole (smooth muscle actin) densities. There was a significant decrease in capillary density in the hearts of db/db mice. Immunostaining study showed that capillary density was significantly increased in adenovirus‐SIRT3‐treated db/db mice compared with db/db mice+adenovirus‐β‐gal (Figure 5C). Similarly, arteriole density was decreased in db/db mice, this reduction was reversed by treating with adenovirus‐SIRT3 (Figure 5D). Treatment with adenovirus‐SIRT3 further significantly reduced myocardial fibrosis in db/db mice (Figure 5E).
Echocardiographic analysis showed that diabetic db/db mice had an impaired cardiac function as evidenced by an increased LV end‐systolic diameter (LVSD)/LV end‐diastolic diameter (LVDD) and LV end‐systolic volume (LVSV)/‐LV end‐diastolic volume (LVDV), and a reduced ejection fraction (EF%) and ejection shortening (FS%) as compared with db+/− mice. Overexpression of SIRT3 led to a significant improvement of ejection fraction and fractional shortening with a reduction of LV end‐systolic diameter (2.88±0.02 mm versus 2.58±0.037 mm, P<0.05) and LV end‐systolic volume (31.85±0.531 versus 27.57±1.11, P<0.05) in db/db mice (Figure 6A, Table 2). Overexpression of SIRT3 also significantly suppressed cardiac hypertrophy in db/db mice (98±3 mg/cm versus 84±10 mg/cm; P<0.05; Figure 6B).
Figure 6. SIRT3 gene therapy attenuated cardiac hypertrophy and improved cardiac function in db/db mice.
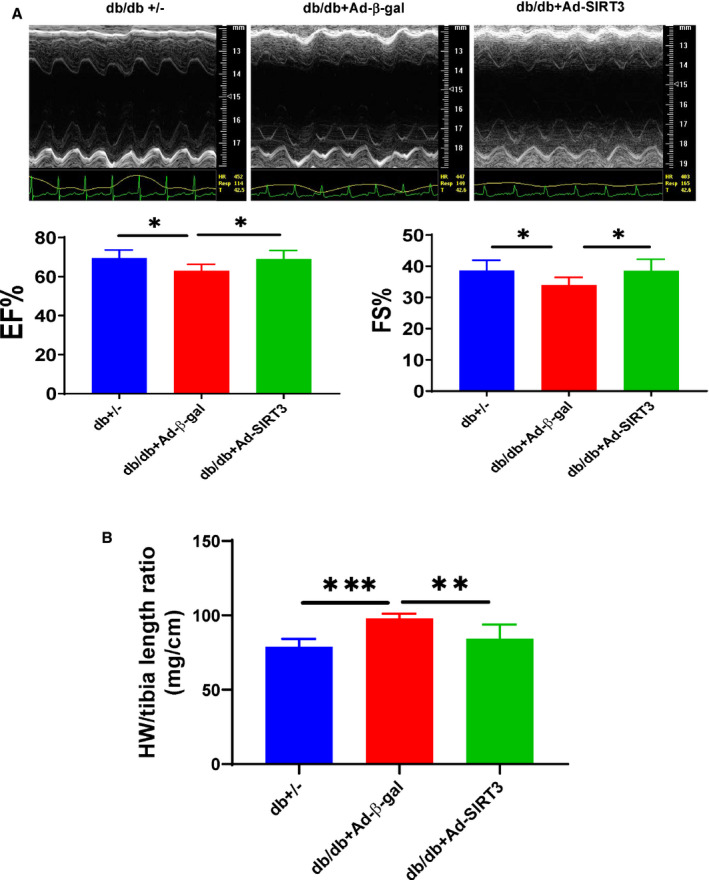
A and B, Overexpression of SIRT3 led to a significant improvement of EF% and FS% with reduction of heart weight (HW/tibia length ratio) in db/db mice. All data represent mean±SD (n=5–8 mice, *P<0.05, **P<0.01, and ***P<0.001). Ad‐SIRT3 indicates adenovirus SIRT3; EF%, ejection fraction; and FS%, fractional shortening.
Table 2.
Measurement of Heart Rate and Cardiac Function by Echocardiography
| Db+/− Mice (n=5) | Db/db+Adenovirus‐β‐gal (n=8) | Db/db+Adenovirus‐SIRT3 (n=5) | P Value | |
|---|---|---|---|---|
| Heart rate, bpm | 485±30 | 425±60 | 435±50 | |
| LVSD, mm | 2.37±0.033 | 2.88±0.02* | 2.58±0.037 † | <0.05 |
| LVDD, mm | 3.86±0.026 | 4.36±0.032* | 4.19±0.32* | |
| LVSV, µL | 19.89±0.685 | 31.85±0.531* | 27.57±1.11 † | <0.05 |
| LVDV, µL | 64.73±1.06 | 86.26±1.47* | 84.9±1.803* | |
| Stroke volume, µL | 44.84±0.61 | 54.41±1.233* | 57.33±0.888* | |
| Cardiac output, mL/min | 19.76±0.165 | 23.59±0.539* | 25.21±0.356* |
All data represent mean±SD. One‐way ANOVA, followed by Tukey's post‐hoc test. LVDD indicates left ventricular end‐diastolic diameter; LVDV, left ventricular end‐diastolic volume; LVSD, left ventricular end‐systolic diameter; and LVSV, left ventricular end‐systolic volume.
P<0.05 vs db+/−.
P<0.05 vs Db/db+Ad‐β‐gal.
DISCUSSION
In this study, we demonstrated that overexpression of SIRT3 alleviated diabetic cardiomyopathy by a mechanism involving inhibition of p53 acetylation and TIGAR expression. In vitro, overexpression of SIRT3 in cardiomyocytes blunted the HG‐induced impairment of glycolysis and increases in oxidative stress and apoptosis by targeting p53 acetylation‐mediated TIGAR expression. In vivo, overexpression of SIRT3 resulted in suppression of p53 acetylation and improvements of capillary density and cardiac function in db/db mice. Our data for the first time demonstrated that SIRT3 modulates diabetic cardiomyopathy by a mechanism involving inhibition of p53 acetylation and TIGAR‐mediated cardiomyocyte glycolytic metabolism.
Emerging evidence suggests that SIRT3 may have a protective role against diabetic cardiomyopathy. 24 , 25 However, the underlying mechanisms of SIRT3 in diabetic cardiomyopathy remain unclear. SIRT3 is a protein deacetylase through the deacetylation of a variety of transcriptional factors. One of the targets of SIRT3 is transcriptional factor p53. 26 SIRT3 has been shown to deacetylate and promote p53 degradation in PTEN‐defective non–small cell lung cancer. 27 Our previous study revealed that deficiency of SIRT3 increased p53 acetylation in the mouse hearts. 28 Under normal conditions, levels of p53 protein were maintained low because of its short half‐life through the ubiquitin‐proteasome degradation pathway. In the absence of SIRT3, p53 may be acetylated and accumulated, which caused its activation and stabilization. In the present study, we found that SIRT3 expression was reduced, while p53 acetylation was increased in the hearts of db/db mice. Hyperglycemia is considered as one of the major factors contributing to diabetic cardiomyopathy. 29 To test the direct link between SIRT3 and p53 acetylation, cardiomyocyte cell line H9c2 cells were exposed to HG conditions. Under HG conditions, SIRT3 levels were reduced, which led to p53 acetylation. Overexpression of SIRT3 significantly attenuated the HG‐induced p53 acetylation, further suggesting a regulatory role of SIRT3 on p53 acetylation under hyperglycemia.
Hyperglycemia‐induced ROS and apoptosis in cardiomyocytes have been contributed to diabetic cardiomyopathy. 30 In the present study, we demonstrated the involvement of SIRT3 in mediating hyperglycemia‐induced ROS and apoptosis in cardiomyocytes. Overexpression of SIRT3 ameliorated the HG‐induced ROS formation and apoptosis. Our data also indicate that the p53/TIGAR axis appeared to be a direct downstream signal pathway of SIRT3 since overexpression of SIRT3 resulted in reductions of p53 acetylation and TIGAR expression. Moreover, knockdown of p53 significantly inhibited HG‐induced TIGAR expression. Furthermore, specific knockdown of TIGAR reduced HG‐induced ROS levels, apoptosis, and promoted EC angiogenesis. These data suggest that SIRT3 may alleviate HG‐induced cardiomyopathy by a mechanism involving inhibition of p53/TIGAR signaling pathway.
We have shown that knockout of SIRT3 disrupts glucose transport from endothelial cells to cardiomyocytes, reduces cardiomyocyte glucose utilization and glycolysis, and sensitizes pressure overload‐induced heart failure. 31 SIRT3 may regulate cardiomyocyte glucose availability and glycolysis, which governs the function of the heart. 31 PFKFB3 and TIGAR are the key regulators for phosphofructokinase and glycolysis. Accumulating evidence reveals that balanced PFKFB3/TIGAR is critical for glycolysis and cell survival. A study showed an upregulation of TIGAR levels in response to PFKFB3 knockdown in HeLa cells. 32 Furthermore, TIGAR was overexpressed in multiple human leukemia cell lines and knockdown of TIGAR activated glycolysis via the upregulation of PFKFB3 in human leukemia cells. 33 Glycolysis is essential to cardiac metabolism and impaired glycolysis has contributed to diabetic cardiomyopathy. 34 Metformin, which abrogated the downregulation of hexokinase and phosphofructokinase in the hearts of streptozotocin‐induced diabetic mice, exhibited protection to diabetic hearts. 35 In the present study, exposure of cardiomyocytes to HG caused an imbalanced PFKFB3/TIGAR in favoring TIGAR. This was accompanied by significant reductions of glycolysis, glycolysis capacity, and glycolytic reserve. In contrast, knockdown of TIGAR upregulated PFKFB3 expression and improved glycolysis in H9c2 cells. These results indicated that hyperglycemia may disrupt glycolysis by activation of p53, which leads to an imbalanced TIGAR/PFKFB3 in favoring TIGAR. This notion was further validated by that the overexpression of SIRT3 rebalanced PFKFB3/TIGAR and enhanced glycolysis under hyperglycemic conditions.
In vivo, we demonstrated that SIRT3 expression was significantly reduced in the hearts of db/db mice. Overexpression of SIRT3 inhibited p53 acetylation and rebalanced PFKFB3/TIGAR expression in the hearts of db/db mice. SIRT3 overexpression also reduced myocardial ROS formation. SIRT3 overexpression further blunted myocardial fibrosis and myocardial microvascular rarefaction in diabetes mellitus. Most importantly, overexpression of SIRT3 ameliorated cardiac hypertrophy and diabetic cardiac dysfunction with a significant improvement of fractional shortening and ejection fraction in db/db mice. These in vivo results further confirmed the protective role of SIRT3 in diabetic cardiomyopathy.
Our data suggest a novel role of SIRT3 in diabetes mellitus–mediated metabolism reprogramming and cardiac remodeling; however, this study had some limitations. First, we did not examine how SIRT3 affects p53 acetylation. Although our data showed an imbalanced of TIGAR/PFKFB3 in the hearts of diabetic db/db mice, we did not actually measure the levels of glycolytic enzyme activity and glycolysis. Further studies are warranted to further explore the potential role of an imbalanced of TIGAR/PFKFB3 on cardiac insulin resistance in the diabetic cardiomyopathy. In addition, our study in vivo only focused on SIRT3 overexpression on diabetic cardiomyopathy, whereas the physiologic role of adenovirus‐SIRT3 on nondiabetic db+/− mice is lack. Further studies are warranted to address these questions.
In conclusion, our present study suggested that overexpression of SIRT3 alleviated diabetic cardiomyopathy by a mechanism involving inhibition of the p53/TIGAR and in favor of the PFKFB3 signaling pathway, which may improve glycolysis under hyperglycemia. Results from the present studies provided the foundation for exploitation of the regulation of the SIRT3 and p53/TIGAR axis, especially a targeted reduction in TIGAR and the glycolytic metabolism pathway, to ameliorate or reverse the diabetic cardiomyopathy.
Sources of Funding
This work was supported by the National Institutes of Health grant 2R01HL102042 and University of Mississippi Medical Center Intramural Research Support Program to J.X. Chen.
Disclosures
None.
Supporting information
Figures S1–S3
(J Am Heart Assoc. 2021;10:e018913. DOI: 10.1161/JAHA.120.018913.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018913
For Sources of Funding and Disclosures, see pages 13 and 14.
References
- 1. Stratmann B, Gawlowski T, Tschoepe D. Diabetic cardiomyopathy–to take a long story serious. Herz. 2010;35:161–168.DOI: 10.1007/s00059-010-3336-0. [DOI] [PubMed] [Google Scholar]
- 2. Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. DOI: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 3. Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two‐faced disease with restrictive and dilated phenotypes. Eur Heart J. 2015;36:1718–1727, 1727a–1727c. DOI: 10.1093/eurheartj/ehv134. [DOI] [PubMed] [Google Scholar]
- 4. Maimaitiming A, Xiao K, Hu C, Chen J, Yang XH, Zhou DH, Gao LP, Dong XP, Shi Q. Aberrant decrease of the endogenous sirt3 and increases of acetylated proteins in scrapie‐infected cell line smb‐s15 and in the brains of experimental mice. ACS Chem Neurosci. 2019;10:4293–4302. DOI: 10.1021/acschemneuro.9b00341. [DOI] [PubMed] [Google Scholar]
- 5. Yang W, Nagasawa K, Münch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS, Zaganjor E, et al. Mitochondrial sirtuin network reveals dynamic sirt3‐dependent deacetylation in response to membrane depolarization. Cell. 2016;167:985–1000.e21. DOI: 10.1016/j.cell.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen T, Li J, Liu J, Li N, Wang S, Liu H, Zeng M, Zhang Y, Bu P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF‐beta/Smad3 pathway. Am J Physiol Heart Circ Physiol. 2015;308:H424–H434. DOI: 10.1152/ajpheart.00454.2014. [DOI] [PubMed] [Google Scholar]
- 7. Bagul PK, Katare PB, Bugga P, Dinda AK, Banerjee SK. SIRT‐3 modulation by resveratrol improves mitochondrial oxidative phosphorylation in diabetic heart through deacetylation of TFAM. Cells. 2018;7:235. DOI: 10.3390/cells7120235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zeng H, Vaka VR, He X, Booz GW, Chen JX. High‐fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847–1856. DOI: 10.1111/jcmm.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeng H, He X, Hou X, Li L, Chen JX. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol. 2014;306:H585–H597. DOI: 10.1152/ajpheart.00821.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He X, Zeng H, Chen JX. Ablation of SIRT3 causes coronary microvascular dysfunction and impairs cardiac recovery post myocardial ischemia. Int J Cardiol. 2016;215:349–357. DOI: 10.1016/j.ijcard.2016.04.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He X, Zeng H, Chen ST, Roman RJ, Aschner JL, Didion S, Chen JX. Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J Mol Cell Cardiol. 2017;112:104–113. DOI: 10.1016/j.yjmcc.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geng J, Yuan X, Wei M, Wu J, Qin ZH. The diverse role of TIGAR in cellular homeostasis and cancer. Free Radic Res. 2018;52:1240–1249. DOI: 10.1080/10715762.2018.1489133. [DOI] [PubMed] [Google Scholar]
- 13. Hoshino A, Matoba S, Iwai‐Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, et al. P53‐TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol. 2012;52:175–184. DOI: 10.1016/j.yjmcc.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 14. Kimata M, Matoba S, Iwai‐Kanai E, Nakamura H, Hoshino A, Nakaoka M, Katamura M, Okawa Y, Mita Y, Okigaki M, et al. p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress. Am J Physiol Heart Circ Physiol. 2010;299:H1908–H1916. DOI: 10.1152/ajpheart.00250.2010. [DOI] [PubMed] [Google Scholar]
- 15. Birks EJ, Latif N, Enesa K, Folkvang T, Luong LA, Sarathchandra P, Khan M, Ovaa H, Terracciano CM, Barton PJ Jr, et al. Elevated p53 expression is associated with dysregulation of the ubiquitin‐proteasome system in dilated cardiomyopathy. Cardiovasc Res. 2008;79:472–480. DOI: 10.1093/cvr/cvn083. [DOI] [PubMed] [Google Scholar]
- 16. Song H, Conte JV Jr, Foster AH, McLaughlin JS, Wei C. Increased p53 protein expression in human failing myocardium. J Heart Lung Transplant. 1999;18:744–749. DOI: 10.1016/S1053-2498(98)00039-4. [DOI] [PubMed] [Google Scholar]
- 17. Oka T, Morita H, Komuro I. Novel molecular mechanisms and regeneration therapy for heart failure. J Mol Cell Cardiol. 2016;92:46–51. DOI: 10.1016/j.yjmcc.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 18. Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114:565–571. DOI: 10.1161/CIRCRESAHA.114.300507. [DOI] [PubMed] [Google Scholar]
- 19. Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, et al. p53‐induced inhibition of hif‐1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. DOI: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 20. Nithipongvanitch R, Ittarat W, Velez JM, Zhao R, St Clair DK, Oberley TD. Evidence for p53 as guardian of the cardiomyocyte mitochondrial genome following acute adriamycin treatment. J Histochem Cytochem. 2007;55:629–639. DOI: 10.1369/jhc.6A7146.2007. [DOI] [PubMed] [Google Scholar]
- 21. Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM. Targeted disruption of p53 attenuates doxorubicin‐induced cardiac toxicity in mice. Mol Cell Biochem. 2005;273:25–32. DOI: 10.1007/s11010-005-5905-8. [DOI] [PubMed] [Google Scholar]
- 22. Gogiraju R, Xu X, Bochenek ML, Steinbrecher JH, Lehnart SE, Wenzel P, Kessel M, Zeisberg EM, Dobbelstein M, Schafer K. Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc. 2015;4:e001770. DOI: 10.1161/JAHA.115.001770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ouyang J, Zeng Z, Fang H, Li F, Zhang X, Tan W. SIRT3 inactivation promotes acute kidney injury through elevated acetylation of SOD2 and p53. J Surg Res. 2019;233:221–230. DOI: 10.1016/j.jss.2018.07.019. [DOI] [PubMed] [Google Scholar]
- 24. Yu W, Gao B, Li NA, Wang J, Qiu C, Zhang G, Liu M, Zhang R, Li C, Ji G, et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: role of Foxo3A‐parkin‐mediated mitophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1973–1983. DOI: 10.1016/j.bbadis.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 25. Li Y, Ma Y, Song L, Yu L, Zhang L, Zhang Y, Xing Y, Yin Y, Ma H. SIRT3 deficiency exacerbates p53/parkinmediated mitophagy inhibition and promotes mitochondrial dysfunction: implication for aged hearts. Int J Mol Med. 2018;41:3517–3526. DOI: 10.3892/ijmm.2018.3555. [DOI] [PubMed] [Google Scholar]
- 26. Chen J, Wang A, Chen Q. SirT3 and p53 deacetylation in aging and cancer. J Cell Physiol. 2017;232:2308–2311. DOI: 10.1002/jcp.25669. [DOI] [PubMed] [Google Scholar]
- 27. Xiong Y, Wang L, Wang S, Wang M, Zhao J, Zhang Z, Li X, Jia L, Han Y. SIRT3 deacetylates and promotes degradation of P53 in PTEN‐defective non‐small cell lung cancer. J Cancer Res Clin Oncol. 2018;144:189–198. DOI: 10.1007/s00432-017-2537-9. [DOI] [PubMed] [Google Scholar]
- 28. Zeng H, Chen JX. Sirtuin 3, endothelial metabolic reprogramming, and heart failure with preserved ejection fraction. J Cardiovasc Pharmacol. 2019;74:315–323. DOI: 10.1097/FJC.0000000000000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jia G, Whaley‐Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia‐ and insulin‐resistance‐induced heart disease. Diabetologia. 2018;61:21–28. DOI: 10.1007/s00125-017-4390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen X, Qian J, Wang L, Li J, Zhao Y, Han J, Khan Z, Chen X, Wang J, Liang G. Kaempferol attenuates hyperglycemia‐induced cardiac injuries by inhibiting inflammatory responses and oxidative stress. Endocrine. 2018;60:83–94. DOI: 10.1007/s12020-018-1525-4. [DOI] [PubMed] [Google Scholar]
- 31. Zeng H, He X, Chen JX. Endothelial sirtuin 3 dictates glucose transport to cardiomyocyte and sensitizes pressure overload‐induced heart failure. J Am Heart Assoc. 2020;9:e015895. DOI: 10.1161/JAHA.120.015895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon‐Molas H, Calvo‐Vidal MN, Castano E, Rodriguez‐Garcia A, Navarro‐Sabate A, Bartrons R, Manzano A. Akt mediates TIGAR induction in HeLa cells following PFKFB3 inhibition. FEBS Lett. 2016;590:2915–2926. DOI: 10.1002/1873-3468.12338. [DOI] [PubMed] [Google Scholar]
- 33. Qian S, Li J, Hong M, Zhu YU, Zhao H, Xie Y, Huang J, Lian Y, Li Y, Wang S, et al. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. J Hematol Oncol. 2016;9:128. DOI: 10.1186/s13045-016-0360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sowton AP, Griffin JL, Murray AJ. Metabolic profiling of the diabetic heart: toward a richer picture. Front Physiol. 2019;10:639. DOI: 10.3389/fphys.2019.00639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Da Silva D, Ausina P, Alencar EM, Coelho WS, Zancan P, Sola‐Penna M. Metformin reverses hexokinase and phosphofructokinase downregulation and intracellular distribution in the heart of diabetic mice. IUBMB Life. 2012;64:766–774. DOI: 10.1002/iub.1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3