

Abstract
Background
The FHOD3 (formin homology 2 domain‐containing 3) gene has recently been identified as a causative gene of hypertrophic cardiomyopathy (HCM). However, the pathogenicity of FHOD3 variants remains to be evaluated. This study analyzed the spectrum of FHOD3 variants in a large HCM and control cohort, and explored its correlation with the disease.
Methods and Results
The genetic analysis of FHOD3 was performed using the whole exome sequencing data from 1000 patients with HCM and 761 controls without HCM. A total of 37 FHOD3 candidate variants were identified, including 25 missense variants and 2 truncating variants. In detail, there were 27 candidate variants detected in 33 (3.3%) patients with HCM, which was significantly higher than in the 12 controls (3.3% versus 1.6%; odds ratio, 2.13; P<0.05). On the basis of familial segregation, we identified one truncating variant (c.1286+2delT) as a causal variant in 4 patients. Furthermore, the FHOD3 candidate variant experienced significantly more risk of cardiovascular death and all‐cause death (adjusted hazard ratio [HR], 3.71; 95%, 1.32–8.59; P=0.016; and adjusted HR, 3.02; 95% CI, 1.09–6.85; P=0.035, respectively).
Conclusions
Our study suggests that FHOD3 is a causal gene for HCM, and that the presence of FHOD3 candidate variants is an independent risk for cardiovascular death and all‐cause death in HCM.
Keywords: cardiovascular events, formin homology 2 domain‐containing 3, genetic testing, hypertrophic cardiomyopathy
Subject Categories: Cardiomyopathy, Hypertrophy, Genetics
Nonstandard Abbreviations and Acronyms
- FHOD3
formin homology 2 domain‐containing 3
- HCM
hypertrophic cardiomyopathy
- SCD
sudden cardiac death
- VUS
variants of unknown significance
Clinical Perspective
What Is New?
The spectrum of FHOD3 (formin homology 2 domain‐containing 3) variants was analyzed in a large cohort consisting of patients with hypertrophic cardiomyopathy and controls.
The presence of FHOD3 candidate variants is an independent risk for cardiovascular death and all‐cause death in hypertrophic cardiomyopathy.
What Are the Clinical Implications?
In patients with hypertrophic cardiomyopathy, FHOD3 gene should be included for sequencing and interpretation into genetic testing as a causal gene.
Genetic testing for FHOD3 variants is important for the management and risk stratification of patients with hypertrophic cardiomyopathy and should be recommended in clinical practice.
Hypertrophic cardiomyopathy (HCM) is one of the most common cardiovascular disorders, and is classically regarded as an autosomal dominant mendelian disease. 1 Characterized by its clinical variability and genetic heterogeneity, HCM is a worldwide disease, with a prevalence of at least 1 in 500. 2 , 3 , 4 Since MYH7 was first identified as a gene associated with HCM, variants in 7 sarcomere genes (MYBPC3, TNNT2, TNNI3, MYL2, MYL3, TPM1, and ACTC1) have been reported to cause HCM. 5 , 6 However, causal variants located within these genes were not detected in around half of patients, suggesting that new disease‐associated genes remained to be discovered. 7 , 8 , 9
FHOD3 (formin homology 2 domain‐containing 3) protein, a myocardial formin that localizes to thin actin filaments, is encoded by the FHOD3 gene. 10 The formin homology‐2 domain of FHOD3 protein terminates filament extension through blocking capping protein from binding the actin filament end. 11 The role of FHOD3 in regulating sarcomere organization, myofibrillogenesis, and contractility in cardiomyocytes suggests that FHOD3 may be a potential candidate gene for HCM. 12 , 13 Previously, Wooten et al had reported FHOD3 variants were associated with HCM in the Tufts HCM Cohort by genome‐wide association study. 14 In another study, FHOD3 was considered a novel genetic cause of HCM in a European cohort, accounting for ≈1% to 2% of patients with HCM. 15 More recently, an in‐frame variant (NM_001281740, c.1578_1580del, p.Ser527del) of FHOD3 was identified as a causal variant in a Chinese family with HCM. 16 However, the pathogenicity of FHOD3 variants has not been evaluated systemically. Herein, we analyzed the spectrum of FHOD3 variants in a large cohort with HCM, and established a correlation of FHOD3 variants with clinical manifestations.
Methods
Because of privacy, the data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure.
Patient and Control Cohorts
From 2012 to 2018, 1039 patients with HCM and 823 controls without HCM were recruited through Fuwai Hospital, Chinese Academy of Medical Sciences, Beijing, China. HCM was defined by a maximal ventricular wall thickness ≥15 mm that was not solely explained by abnormal loading conditions. Patients with lesser degrees of maximal ventricular wall thickness (13–14 mm) were diagnosed as having HCM if they had a family history of disease.
Clinical evaluation was performed in all patients, including history of disease, systematic clinical examinations, and pedigree investigation. HCM was excluded in all of the controls by the lack of primary left ventricular hypertrophy in echocardiography.
The study was approved by the Ethics Committees of Fuwai Hospital, and complies with the principles of the Declaration of Helsinki. Written informed consent was obtained from all patients.
Genotyping
Targeted capture was performed using the Agilent Sure SelectXT Human All Exon V6 kit, followed by 2×150‐bp paired‐end sequencing on the Illumina NovaSeq platform using manufacturer's protocol. A total of 25 patients and 12 controls were removed with excess missing rates or excess heterozygosity, both defined by 1.5× interquartile range above the third quartile. Moreover, 14 patients and 50 controls were excluded with the mean identical‐by‐descent sharing >0.125 with any other individuals, which was estimated after linkage disequilibrium pruning using PLINK. 17 The sequencing achieved a mean coverage of 142× with >99.9% of targeted regions in the FHOD3 gene. Variants detected in FHOD3 were described according to the Human Genome Variation Society recommendations 18 and were annotated according to the longest transcript of FHOD3 gene (NM_001281740.1). A modified classification scheme, based on the criteria of American College of Medical Genetics and Genomics, was constructed for FHOD3 variant classification (Table S1). 19 The pathogenicity of detected variants was classified as pathogenic, likely pathogenic, variants of unknown significance (VUS), likely benign, or benign (Table S2). FHOD3 variants of pathogenic, likely pathogenic, and VUS status were defined as candidate variants. All FHOD3 candidate variants were verified by Sanger sequencing, and the primers for PCR amplification are listed in Table S3.
Variants in 8 sarcomere disease genes (MYH7, MYBPC3, TNNT2, TNNI3, MYL2, MYL3, TPM1, and ACTC1) were classified following the American College of Medical Genetics and Genomics guideline. 19 Patients were divided as FHOD3 HCM and non‐FHOD3 HCM, including genotype‐positive HCM and genotype‐negative HCM based on whether patients carried any pathogenic, likely pathogenic, or VUS variants of sarcomere genes or not.
Pedigree Analysis
Patients with HCM with FHOD3 candidate variants underwent pedigree analysis. Each family member underwent 12‐lead electrocardiography and echocardiography to assess his/her cardiac condition. Candidate variants found in probands were tested among family members using Sanger capillary sequencing. Two‐point logarithm of the odds scores were calculated using the PARAMLINK package for R software 20 with a parametric linkage model of autosomal dominance, θ=0, phenocopy rate=0.005, and 2 different penetrance values: 0.80 and 0.95.
Follow‐Up and End Points
Follow‐up was performed by a clinic visit or telephone interview for all patients with HCM until December 2018. The primary end point was cardiovascular death, defined as death caused by cardiogenic or vascular causes, including sudden cardiac death (SCD), heart failure (HF)–related death, and stroke‐related death. The secondary end point was all‐cause death. SCD was defined as witnessed sudden and unexpected death with or without documented ventricular fibrillation within 1 hour of new symptoms or nocturnal deaths with no antecedent history of worsening symptoms. HF‐related death was defined as death proceeded by HF or heart transplantation in the end stage of HF.
Statistical Analysis
All statistical analysis was performed with SPSS version 24.0 software (SPSS, Chicago, IL) unless otherwise specified. Categorical variables are presented as number (percentage), and continuous variables are presented as mean±SD. The Pearson χ2 test or the Fisher exact test was used for comparing categorical variables. The independent‐sample t test was used for continuous variable comparisons, and the Mann‐Whitney U test was used for abnormally distributed variables. Survival curves were constructed according to the Kaplan‐Meier method, and comparisons were performed using the log‐rank test. Cox regression with Firth penalized maximum likelihood models was used to calculate the hazard ratio (HR) and 95% CI to estimate the effect of candidate variants on end points using R software version 3.4.3 (R Core Team, Vienna, Austria) with the “coxphf” package (https://cran.r‐project.org/web/packages/coxphf/coxphf.pdf). The characteristics with a P<0.05 in univariable analysis were chosen for the multivariable model, including left ventricular end‐diastolic dimension, family history of SCD, maximal ventricular wall thickness, and left atrial diameter. P values are 2 sided and considered significant when <0.05.
Results
FHOD3 Candidate Variants in the Study Population
There were a total of 1000 patients with HCM and 761 controls without HCM included in the final analysis of this study. The characteristics of the study population at enrollment are summarized in Table S4. The participants consisted of 1000 patients with HCM and 761 controls. The cohort with HCM was 64.5% men, with a median age of 47.9±14.6 years. There was no significant difference in sex and age between patients with HCM and controls.
A total of 37 FHOD3 candidate variants were identified, including 25 missense variants and 2 truncating variants (Table 1). 21 , 22 , 23 In detail, 14 (37.8%) variants were reported in the Exome Aggregation Consortium database or the Genome Aggregation database, whereas 23 (62.2%) variants were first detected in patients with HCM or the total population. In our cohort, 27 candidate variants were detected in 33 (3.3%) patients with HCM (Table 1). Comparatively, 12 variants were found in 12 (1.6%) controls, which represented a significant difference (odds ratio [OR], 2.13; P<0.05). Furthermore, 6 variants were clustered in an exclusively cardiac isoform domain of the protein (amino acids 400–574), which maintained the 2 truncating variants (Figure S1). Notably, all these 6 variants were only detected in 9 patients with HCM.
Table 1.
Candidate Variants of FHOD3 Detected in Patients With HCM and Controls
| Transcript Effect (NM_ 001281740.1) | Protein (NP_001268669.1) | Type | Variant Classification* | SNP | FHOD3 Domain | CADD | SIFT | Polyphen | GnomAD MAF% | ExAC MAF% | In‐House MAF% | Subject Identifier (Phenotype) | Sarcomere Gene Variants |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.274C>T | p.Arg92Trp | Missense | VUS | rs759696197 | GBD/FH3 | 27.1 | 0.005 | 0.967 | 0.0032 | 0.0051 | 0.0284 | H8267 (HCM) | MYH7, p.Arg453Cys |
| c.562C>T | p.Arg188Cys | Missense | VUS | rs143579901 | GBD/FH3 | 24.2 | 0.001 | 0.015 | 0.0032 | 0 | 0.0284 | H7105 (HCM) | None |
| c.646G>A | p.Val216Ile | Missense | VUS | rs551483382 | GBD/FH3 | 26.6 | 0.004 | 0.758 | 0.0084 | 0.0084 | 0.0284 | H7191 (HCM) | MYH7, p.Glu930Lys |
| c.776C>T | p.Thr259Met | Missense | VUS | rs770013602 | GBD/FH3 | 26.2 | 0.008 | 0.414 | 0.0096 | 0.0165 | 0.0284 | HT065 (HCM) | None |
| c.796A>G | p.Met266Val | Missense | VUS | Novel | GBD/FH3 | 24.1 | 0.187 | 0.21 | 0 | 0 | 0.0284 | A073 (control) | None |
| c.1007G>A | p.Ser336Asn | Missense | VUS | Novel | GBD/FH3 | 13.15 | NA | NA | 0 | 0 | 0.0284 | H8138 (HCM) | None |
| c.1063C>T | p.Arg355Trp | Missense | VUS | Novel | GBD/FH3 | 25.9 | 0.001 | 0.471 | 0 | 0 | 0.0284 | S285 (HCM) | None |
| c.1097C>T | p.Ser366Leu | Missense | VUS | rs747730516 | GBD/FH3 | 27.4 | 0.001 | 0.982 | 0.0032 | 0.0041 | 0.0568 | H7597 (HCM) | None |
| H8282 (HCM) | None | ||||||||||||
| c.1157C>T | p.Pro386Leu | Missense | VUS | Novel | GBD/FH3 | 24.7 | NA | NA | 0 | 0 | 0.0284 | Y398 (control) | None |
| c.1189C>T | p.Arg397Cys | Missense | VUS | rs760874847 | GBD/FH3 | 20.7 | 0.091 | 0.001 | 0 | 0 | 0.0568 | S101 (HCM) | MYBPC3,p.Gln469Ter |
| H7177 (HCM) | MYH7, p.Arg1045His | ||||||||||||
| c.1286+2delT | NA | Spicing | Pathogenic | Novel | Ex | 0 | 0 | 0.1136 | H8306 (HCM) | None | |||
| H7571 (HCM) | None | ||||||||||||
| H7147 (HCM) | None | ||||||||||||
| H7104 (HCM) | None | ||||||||||||
| c.1309C>T | p.Gln437Ter | Nonsense | LP | Novel | Ex | 35 | NA | NA | 0 | 0 | 0.0284 | H7115 (HCM) | None |
| c.1552G>A | p.Val518Met | Missense | VUS | Novel | Ex | 25.7 | NA | NA | 0 | 0 | 0.0284 | H1260 (HCM) | MYBPC3, p.Asp1091Gly |
| c.1580C>T | p.Ser527Phe | Missense | VUS | Novel | Ex | 25.5 | NA | NA | 0 | 0 | 0.0284 | H8136 (HCM) | MYH7, p.Leu832Phe |
| c.1640A>C | p.Glu547Ala | Missense | VUS | Novel | Ex | 23.7 | NA | NA | 0 | 0 | 0.0284 | H8194 (HCM) | MYBPC3, p.Glu611fs |
| c.1703G>T | p.Arg568Leu | Missense | VUS | Novel | Ex | 20.9 | NA | NA | 0 | 0 | 0.0284 | H7327 (HCM) | None |
| c.1733T>A | p.Phe578Tyr | Missense | VUS | Novel | 19.2 | NA | NA | 0 | 0 | 0.0284 | Y5332 (control) | None | |
| c.1924G>A | p.Glu642Lys | Missense | VUS | Novel | CC | 26.2 | 0.005 | 0.979 | 0 | 0 | 0.0284 | S099 (HCM) | MYH7, p.Gly823Glu |
| c.2077C>T | p.Arg693Trp | Missense | VUS | rs533572045 | DID | 29.1 | 0.0 | 0.292 | 0.0064 | 0 | 0.0284 | Y2722 (control) | None |
| c.2078G>A | p.Arg693Gln | Missense | VUS | rs148866621 | DID | 23.2 | 0.083 | 0.001 | 0.0096 | 0.0087 | 0.0284 | H7520 (HCM) | MYBPC3,p.Glu542Lys |
| c.2090G>A | p.Arg697Gln | Missense | VUS | rs553341694 | DID | 23.2 | 0.006 | 0.811 | 0.0065 | 0.0095 | 0.0284 | H8315 (HCM) | None |
| c.2429G>T | p.Gly810Val | Missense | VUS | Novel | DID | 22.7 | 0.02 | 0.022 | 0 | 0 | 0.0284 | H1341 (HCM) | None |
| c.2584G>A | p.Asp862Asn | Missense | VUS | rs544119818 | DID | 21.1 | 0.671 | 0.002 | 0.0032 | 0.0043 | 0.0568 | H8819 (HCM) | None |
| HT020 (HCM) | MYH7, p.Arg663His | ||||||||||||
| c.2746T>G | p.Ser916Ala | Missense | VUS | Novel | DID | 20.8 | 0.091 | 0.711 | 0 | 0 | 0.0568 | H1424 (HCM) | None |
| T903 (control) | None | ||||||||||||
| c.2824G>C | p.Glu942Gln | Missense | VUS | rs779000457 | DID | 27.1 | 0.016 | 0.986 | 0.0025 | 0.0035 | 0.0284 | H8818 (HCM) | MYBPC3, p.Tyr842Ter |
| c.2837G>A | p.Ser946Asn | Missense | VUS | Novel | DID | 15.46 | 0.247 | 0.058 | 0 | 0 | 0.0284 | S122 (HCM) | MYH7, p.Ile263Thr |
| c.2954C>A | p.Ala985Asp | Missense | VUS | Novel | DID | 25.9 | 0.003 | 0.996 | 0 | 0 | 0.0284 | B014 (control) | None |
| c.3005A>T | p.Glu1002Val | Missense | VUS | Novel | DID | 26.9 | 0.003 | 0.408 | 0 | 0 | 0.0284 | H1253 (HCM) | None |
| c.3412T>C | p.Ser1138Pro | Missense | VUS | Novel | FH2 | 23.1 | 0.071 | 0.101 | 0 | 0 | 0.0284 | HT033 (HCM) | None |
| c.3478G>A | p.Ala1160Thr | Missense | VUS | rs746707013 | FH2 | 24.6 | 0.004 | 0.777 | 0.0032 | 0 | 0.0284 | Y3909 (control) | None |
| c.3587C>G | p.Thr1196Arg | Missense | VUS | Novel | FH2 | 25.2 | 0.01 | 0.999 | 0 | 0 | 0.0284 | Y6233 (control) | None |
| c.3624G>C | p.Gln1208His | Missense | VUS | Novel | FH2 | 24.2 | 0.001 | 0.909 | 0 | 0 | 0.0284 | H8258 (HCM) | None |
| c.3796A>G | p.Ile1266Val | Missense | VUS | Novel | FH2 | 25.2 | 0.276 | 0.028 | 0 | 0 | 0.0284 | H8911 (HCM) | MYH7, p.Lys1485Arg |
| c.4270T>A | p.Tyr1424Asn | Missense | VUS | rs753641918 | FH2 | 25.1 | 0.314 | 0.003 | 0.0012 | 0.0017 | 0.0568 | H8004 (HCM) | None |
| T303 (control) | None | ||||||||||||
| c.4586C>T | p.Pro1529Leu | Missense | VUS | Novel | 18.4 | 0.342 | 0.052 | 0 | 0 | 0.0284 | Y1615 (control) | None | |
| c.4702C>T | p.Arg1568Cys | Missense | VUS | rs770836110 | DAD | 28.1 | 0.187 | 0.917 | 0.0016 | 0.0008 | 0.0284 | A2015 (control) | None |
| c.4787T>C | p.Leu1596Ser | Missense | VUS | Novel | DAD | 24.1 | 0.0 | 0.994 | 0 | 0 | 0.0284 | Y4609 (control) | None |
CADD indicates combined annotation‐dependent depletion score (phred; v1.3; August 2015) 21 ; CC, coiled‐coiled; DAD, diaphanous autoregulation domain; DID, diaphanous autoinhibitory domain; Ex, exclusively cardiac isoform; ExAC, Exome Aggregation Consortium (http://exac.broadinstitute.org); FH2, formin homology 2 domain; FHOD3, formin homology 2 domain‐containing 3; GBD/FH3, GTPase‐binding domain/formin homology 3 domain; GnomAD, Genome Aggregation (https://gnomad.broadinstitute.org); HCM, hypertrophic cardiomyopathy; LP, likely pathogenic; MAF, minor allele frequency; NA, not available; Polyphen, polymorphism phenotyping v2 (September 2014) 23 ; SIFT, sorting intolerant from tolerant (computed from ENSEMBL 55; September 2014) 22 ; SNP, single nucleotide polymorphism; and VUS, variants of uncertain significance.
Determined according to criteria in Table S1.
Segregation Study
Pedigree analysis was performed in 6 of 33 patients with FHOD3 candidate variants (Figure 1). Of these, 4 families included at least a second subject with HCM. Among these family members, all 6 affected patients were FHOD3 candidate variant carriers (Figure 1).
Figure 1. Cosegregation of FHOD3 (formin homology 2 domain‐containing 3) candidate variants with hypertrophic cardiomyopathy.
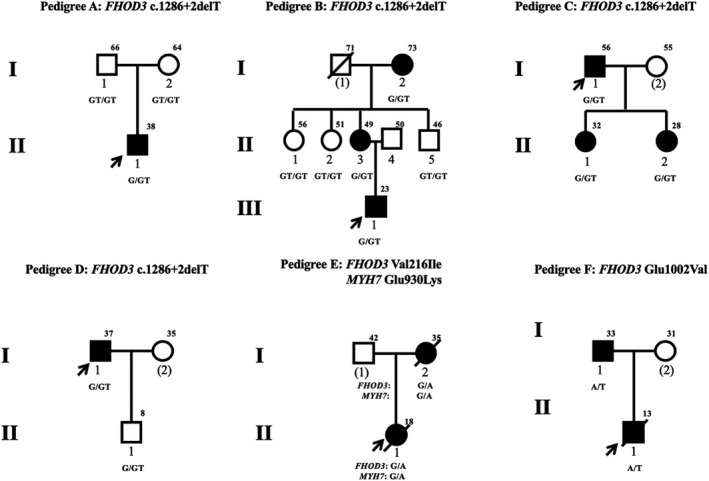
Individuals affected by left ventricular hypertrophy are indicated by black symbols. Unfilled symbols represent individuals without ventricular hypertrophy. Arrows indicate the probands. Circles represent women; squares represent men. Symbols with a slash through them indicate deceased subjects. The current age or the age at death is indicated to the upper right of each symbol. Numbers in parentheses indicate individuals without DNA available.
The novel truncating variant (c.1286+2delT) classified as likely pathogenic was detected in 4 patients, of whom none carried a probable pathogenic mutation in 8 sarcomere genes. In pedigree A, the variant was a de novo mutation because the cardiac structure was normally in both parents of the proband without this variant (Figure 1A). The cosegregation of HCM and the variant is shown in pedigree B (Figure 1B). The grandmother (I‐1) and mother (II‐3) of the proband are variant carriers, and present left ventricular hypertrophy. All 3 uncles without this variant have a normal phenotype. In addition, the variant is also detected in 2 daughters of the proband in pedigree C, who show cardiac hypertrophy (Figure 1C). No other patients were found in pedigree D, although the son of the proband, an 8‐year‐old boy, carried the variant but had a normal echocardiogram and 12‐lead electrocardiography (Figure 1D). The combined logarithm of the odds score of the variant was 1.44 in family linkage analysis of pedigrees B through D (Table S5). In general, the variant was defined as pathogenic variant and causative of disease onset in these patients.
The Val216Ile variant in FHOD3 (rs551483382) was detected in 1 patient, but was also present in 10 (0.0084%) individuals in the Exome Aggregation Consortium database, and classified as VUS (Table 1). The proband also carried the Glu930Lys variant in MYH7 (rs397516171), which was classified as pathogenic. Pedigree analysis showed that both rs551483382 and rs397516171 are present in the mother of the proband (I‐2) diagnosed as having HCM (Figure 1E). The pathogenicity of FHOD3 Val216Ile has not been determined.
The Glu1002Val variant, classified as VUS, was novel and not present in Exome Aggregation Consortium or Genome Aggregation database (Table S2). There was no probable sarcomere pathogenic mutation detected in the patient with the Glu1002Val variant. The father of the patient who presented with left ventricular hypertrophy also carried FHOD3 Glu1002Val variant, but whole exome sequencing found no sarcomere pathogenic mutation. We assumed that Glu1002Val variant might be the causal variant in this family, but the available evidence is insufficient to determine its pathogenicity.
Characteristics of Patients With FHOD3 Candidate Variants
Similar clinical and echocardiographic characteristics were observed between patients with HCM with or without FHOD3 candidate variants (Table 2). Patients with FHOD3 variants presented a similar probability of outflow tract obstruction as patients without FHOD3 variants. In addition, there was no difference of maximal ventricular wall thickness between patients with or without FHOD3 candidate variants.
Table 2.
Demographic and Clinical Characteristics of Patients With HCM With or Without FHOD3 Candidate Variants
| Variable | FHOD3 Variant Carriers | Noncarriers | P Value* |
|---|---|---|---|
| Sample size | 33 | 967 | |
| Age at enrollment, y | 45.2±17.2 | 48.0±14.5 | 0.282 |
| Age at diagnosis, y | 40.0±14.5 | 43.5±14.6 | 0.179 |
| Men, n (%) | 24 (72.7) | 622 (64.3) | 0.321 |
| BMI, kg/m2 | 24.6±2.8 | 25.6±3.7 | 0.133 |
| Family history of SCD, n (%) | 4 (12.1) | 112 (11.6) | 0.847 |
| Echocardiography | |||
| MVT, mm | 23.6±6.9 | 22.6±5.8 | 0.340 |
| Left atrium, mm | 43.0±6.8 | 41.7±7.2 | 0.300 |
| LVEDD, mm | 45.1±7.7 | 44.0±6.3 | 0.327 |
| Ejection fraction, % | 66.3±11.3 | 67.6±8.1 | 0.372 |
| Outflow tract obstruction, n (%) | 19 (57.6) | 544 (56.3) | 0.881 |
Continuous variables are presented as mean±SD; the categorical variable sex was presented as number (percentage). BMI indicates body mass index; FHOD3, formin homology 2 domain‐containing 3; HCM, hypertrophic cardiomyopathy; LVEDD, left ventricular end‐diastolic diameter; MVT, maximal left ventricular wall thickness; and SCD, sudden cardiac death.
Continuous variables were compared by Student t test; the categorical variables were compared by χ2 test.
The mean age of patients with FHOD3 candidate variants at diagnosis was 45.2±17.2 years, and 24 (72.7%) patients were men. The clinical manifestations of male and female patients with FHOD3 candidate variants were similar (Table S6).
A total of 64 subjects were lost to follow‐up, including 1 patient with an FHOD3 variant and 63 without. During the follow‐up period of 2.6±1.6 years (2411 patient‐years), 41 patients reached the primary end point, including 5 FHOD3 candidate variant carriers and 36 noncarriers. In detail, all 5 (15.6%) FHOD3 candidate variant carriers died of SCD, and 36 (4.0%) patients without FHOD3 variants died of cardiovascular death, including 20 (2.2%) of SCD, 12 (1.3%) of HF‐related death, and 5 (0.6%) of stroke‐related death. Univariate analysis showed that the risk of cardiovascular deaths was significantly higher in patients with FHOD3 candidate variants than those without (15.6% versus 4.0%; HR, 4.09; 95% CI, 1.48–9.22; P=0.009) (Table 3). Moreover, the FHOD3 candidate variant was associated with the risk of SCD (15.6% versus 2.2%; HR, 7.24; 95% CI, 2.54–17.36; P<0.001) (Table S7). Figure 2 displays Kaplan‐Meier survival curves of freedom of cardiovascular death and SCD. Multivariate analysis showed the patients with FHOD3 candidate variants experienced a significantly higher risk of cardiovascular death and SCD than those without (adjusted HR, 3.71; 95% CI, 1.32–8.59; P=0.016; and adjusted HR, 6.79; 95% CI, 2.26–17.35; P=0.001, respectively) (Table 3 and Table S7).
Table 3.
Univariable and Multivariable Cox Regression Analysis of the Association Between FHOD3 Candidate Variants and Cardiovascular Death in Patients With HCM
| Variants | Crude HR (95% CI) | Crude P Value | Adjusted HR (95% CI) | Adjusted P Value |
|---|---|---|---|---|
| FHOD3 variants | 4.086 (1.480–9.221) | 0.009 | 3.707 (1.320–8.594) | 0.016 |
| LVEDD | 1.066 (1.025–1.103) | 0.002 | 1.071 (1.025–1.113) | 0.003 |
| Family history of SCD | 2.258 (1.035–4.473) | 0.041 | 2.276 (1.032–4.580) | 0.042 |
| MVT | 1.034 (1.002–1.214) | 0.042 | 1.046 (0.9991–1.101) | 0.102 |
| Left atrial diameter | 1.047 (1.006–1.086) | 0.025 | 1.020 (0.977–1.062) | 0.361 |
FHOD3 indicates formin homology 2 domain‐containing 3; HCM, hypertrophic cardiomyopathy; HR, hazard ratio; LVEDD, left ventricular end‐diastolic diameter; MVT, maximal left ventricular wall thickness; and SCD, sudden cardiac death.
Figure 2. Cumulative Kaplan‐Meier analysis showing that FHOD3 (formin homology 2 domain‐containing 3) candidate variants were associated with a higher risk of cardiovascular death (A), sudden cardiac death (B), and all‐cause death (C).
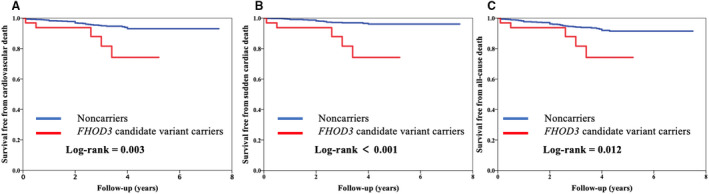
P values were calculated using the log‐rank test.
A total of 49 patients reached the secondary end point, including 5 (15.6%) patients with candidate variants and 44 (4.9%) without those variants. A total of 36 patients died of cardiovascular diseases, and another 8 patients without FHOD3 variants died of cancer or accidents. Patients with FHOD3 candidate variants had a higher risk of all‐cause death (HR, 3.35; 95% CI, 1.22–7.46; P=0.022) (Table S8). Kaplan‐Meier survival curves of freedom of all‐cause death are displayed in Figure 2C. Multivariate analysis showed that the FHOD3 candidate variant remained an independent predictor of all‐cause death (adjusted HR, 3.02; 95% CI, 1.09–6.85; P=0.035) (Table S8).
Among 986 non‐FHOD3 variant carriers, a total of 482 patients were included in non‐FHOD3 genotype‐positive group, carrying sarcomere gene mutations. Except for 29 patients who were lost to follow‐up, there were 18 subjects reaching primary outcome, containing 12 patients who died of SCD, and 4 subjects reaching secondary outcome. Kaplan‐Meier survival curves of primary and secondary outcomes were constructed for comparison between the prognosis of FHOD3 variant carriers and non‐FHOD3 genotype‐positive carriers. The results suggested FHOD3 patients with HCM had a higher risk of reverse outcomes than non‐FHOD3 genotype‐positive patients (Figure S2A through S2C).
Discussion
FHOD3 is highly expressed in the heart, and plays an important role in maintaining normal cardiac function. 24 , 25 As a regulator of actin assembly in cardiac sarcomeres, the FHOD3 mutants Ile1127Ala and Lys1273Asp are defective in actin binding. 12 , 13 Matsuyama et al reported that aberrantly mislocalized FHOD3 was deleterious, and contributed to the pathogenesis of MYBPC3 (cardiac myosin‐binding protein C)‐related cardiomyopathy by failing to directly interact with MYBPC3. 26 Recently, FHOD3 mutations were shown to be associated with heart diseases; for example, the Tyr1249Asn variant was identified to cause a dilated cardiomyopathy family by interfering with actin filament assembly. 27 Moreover, as some FHOD3 variants account for HCM cases, FHOD3 is also regarded as causative of HCM. 15 In our study, the FHOD3 gene was screened in a large Chinese cohort, and a total of 37 FHOD3 candidate variants were detected in patients with HCM or controls. The OR of FHOD3 candidate variants in patients with HCM was 2.13 with respect to controls, which is comparable with that reported by Ochoa et al. 15 Candidate variants of FHOD3 gene detected in population databases or controls indicated that not all FHOD3 candidate variants cause the HCM phenotype. Therefore, the pathogenicity of FHOD3 variants remains to be validated by segregation and functional assays. 9 Under the pedigree analysis, the combined logarithm of the odds score of truncating variant c.1286+2delT reached 1.44 as evidence for suggestive linkage. 28 Moreover, this variant was the de novo mutation in pedigree A, as strong support for the pathogenicity of this variant, according to the American College of Medical Genetics and Genomics guideline. 19 Therefore, the variant was identified as a causal variant for HCM. However, other candidate variants were not evaluated because of the absence of second patients or small size of families. Considering the caveat of variable expressivity and probability of incomplete penetrance, the pathogenicity of these variants remains to be determined.
The c.1286+2delT variant is located within the exclusively cardiac isoform domain, which is expressed in the heart but spiced out in the kidney and brain. 29 In a previous study, the FHOD3 isoform without an exclusively cardiac isoform domain could not localize to the sarcomere, indicating that this domain is paramount in FHOD3 localization. 12 Notably, all 6 variants in the exclusively cardiac isoform domain were only found in patients with HCM in our study, further suggesting that this domain is crucial to HCM. The FHOD3 protein also contains 3 formin homology domains, a GTPase binding domain, a diaphanous autoinhibitory domain, a coiled‐coil domain, and a diaphanous autoregulation domain. 30 , 31 The interaction of diaphanous autoinhibitory domain and diaphanous autoregulation domain was reported to be responsible for FHOD3 dimerization. 32 Half of the pathogenic variants identified by Ochoa et al were clustered in the coiled‐coil domain, indicating that this is associated with HCM. 15 However, only Glu642Lys from the coiled‐coil domain was detected in a patient with HCM in our study, whereas we did not determine the pathogenicity of variants in the diaphanous autoinhibitory domain or diaphanous autoregulation domain. Thus, the function of FHOD3 domains remains unclear and to be explored.
The genotype‐phenotype correlation between mutations and prognosis in patients with HCM has varied in studies. 33 , 34 , 35 Phenotypes were found to be similar in patients with MYH7 or MYBPC3 mutations, 36 whereas patients with variants in genes encoding thin myofilament proteins presented with milder hypertrophy but a higher risk of systolic dysfunction. 37 Our study revealed the FHOD3 candidate variants to be an independent predictor for cardiovascular death and all‐cause death. The FHOD3 variant carriers showed worse prognosis than non‐FHOD3 carriers or non‐FHOD3 genotype‐positive carriers, indicating the FHOD3 variants should be considered in the management and risk stratification of patients with HCM. Ochoa et al described the sex difference that female carriers were diagnosed 10 years later than male carriers. Nevertheless, the sex difference did not reproduce in our study, which might be as a result of the difference of ethnicity or particular variants.
Our study has some limitations. First, the FHOD3 sequence was limited within the exome region. Second, the pathogenicity of most of the candidate variants requires confirmation by further study; therefore, our finding underestimates the effect of the FHOD3 gene in HCM.
In conclusion, our study screened the FHOD3 gene in a large Chinese cohort, which consisted of 1000 patients with HCM and 761 controls without HCM, identifying a total of 37 FHOD3 candidate variants. Among these variants, the truncating variant c.1286+2delT was identified as a causal variant in 4 patients. Thus, we verified the FHOD3 gene was a causal gene for HCM. Finally, we found that FHOD3 candidate variants increased the risk of cardiovascular death and all‐cause death, suggesting that they should be included in the management and risk stratification of patients with HCM.
Sources of Funding
This work was supported by grants from the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Sciences (CAMS‐I2M and 2016‐I2M‐1‐015) and National Natural Science Foundation of China (81870286).
Disclosures
None.
Supporting information
Tables S1–S8
Figures S1–S2
Acknowledgments
We thank all the patients who are participating in this study and their families.
(J Am Heart Assoc. 2021;10:e018236. DOI: 10.1161/JAHA.120.018236.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018236
For Sources of Funding and Disclosures, see page 9.
Contributor Information
Yubao Zou, Email: zouyb1973@sina.com.
Lei Song, Email: songlqd@126.com.
References
- 1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. DOI: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 2. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study: coronary artery risk development in (young) adults. Circulation. 1995;92:785–789. DOI: 10.1161/01.CIR.92.4.785. [DOI] [PubMed] [Google Scholar]
- 3. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. DOI: 10.1016/j.jacc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 4. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. DOI: 10.1161/CIR.0b013e318223e230. [DOI] [PubMed] [Google Scholar]
- 5. Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, Levi T, Donis‐Keller H, Seidman JG, Seidman CE. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–1378. DOI: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 6. Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60:705–715. DOI: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- 7. Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. 2019;12:e002460. DOI: 10.1161/CIRCGEN.119.002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sen‐Chowdhry S, Jacoby D, Moon JC, McKenna WJ. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat Rev Cardiol. 2016;13:651–675. DOI: 10.1038/nrcardio.2016.140. [DOI] [PubMed] [Google Scholar]
- 9. Walsh R, Buchan R, Wilk A, John S, Felkin LE, Thomson KL, Chiaw TH, Loong CCW, Pua CJ, Raphael C, et al. Defining the genetic architecture of hypertrophic cardiomyopathy: re‐evaluating the role of non‐sarcomeric genes. Eur Heart J. 2017;38:3461–3468. DOI: 10.1093/eurheartj/ehw603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iskratsch T, Lange S, Dwyer J, Kho AL, dos Remedios C, Ehler E. Formin follows function: a muscle‐specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. J Cell Biol. 2010;191:1159–1172. DOI: 10.1083/jcb.201005060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK. Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature. 2005;433:488–494. DOI: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- 12. Taniguchi K, Takeya R, Suetsugu S, Kan OM, Narusawa M, Shiose A, Tominaga R, Sumimoto H. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. J Biol Chem. 2009;284:29873–29881. DOI: 10.1074/jbc.M109.059303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kan OM, Takeya R, Abe T, Kitajima N, Nishida M, Tominaga R, Kurose H, Sumimoto H. Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis. Biol Open. 2012;1:889–896. DOI: 10.1242/bio.20121370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wooten EC, Hebl VB, Wolf MJ, Greytak SR, Orr NM, Draper I, Calvino JE, Kapur NK, Maron MS, Kullo IJ, et al. Formin homology 2 domain containing 3 variants associated with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2013;6:10–18. 10.1161/CIRCGENETICS.112.965277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ochoa JP, Sabater‐Molina M, García‐Pinilla JM, Mogensen J, Restrepo‐Córdoba A, Palomino‐Doza J, Villacorta E, Martinez‐Moreno M, Ramos‐Maqueda J, Zorio E, et al. Formin homology 2 domain containing 3 (FHOD3) is a genetic basis for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2018;72:2457–2467. DOI: 10.1016/j.jacc.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 16. Huang S, Pu T, Wei W, Xu R, Wu Y. Exome sequencing identifies a FHOD3 p. S527del mutation in a Chinese family with hypertrophic cardiomyopathy. J Gene Med. 2020;22:e3146. DOI: 10.1002/jgm.3146. [DOI] [PubMed] [Google Scholar]
- 17. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559–575. DOI: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan‐Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569. DOI: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- 19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Egeland T, Pinto N, Vigeland MD. A general approach to power calculation for relationship testing. Forensic Sci Int Genet. 2014;9:186–190. DOI: 10.1016/j.fsigen.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 21. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. DOI: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. DOI: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 23. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. DOI: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ushijima T, Fujimoto N, Matsuyama S, Kan OM, Kiyonari H, Shioi G, Kage Y, Yamasaki S, Takeya R, Sumimoto H. The actin‐organizing formin protein Fhod3 is required for postnatal development and functional maintenance of the adult heart in mice. J Biol Chem. 2018;293:148–162. DOI: 10.1074/jbc.M117.813931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katoh M, Katoh M. Identification and characterization of human FHOD3 gene in silico. Int J Mol Med. 2004;13:615–620. DOI: 10.3892/ijmm.13.4.615. [DOI] [PubMed] [Google Scholar]
- 26. Matsuyama S, Kage Y, Fujimoto N, Ushijima T, Tsuruda T, Kitamura K, Shiose A, Asada Y, Sumimoto H, Takeya R. Interaction between cardiac myosin‐binding protein C and formin Fhod3. Proc Natl Acad Sci USA. 2018;115:E4386–E4395. DOI: 10.1073/pnas.1716498115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Arimura T, Takeya R, Ishikawa T, Yamano T, Matsuo A, Tatsumi T, Nomura T, Sumimoto H, Kimura A. Dilated cardiomyopathy‐associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ J. 2013;77:2990–2996. DOI: 10.1253/circj.CJ-13-0255. [DOI] [PubMed] [Google Scholar]
- 28. Nyholt DR. All LODs are not created equal. Am J Hum Genet. 2000;67:282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanaya H, Takeya R, Takeuchi K, Watanabe N, Jing N, Sumimoto H. Fhos2, a novel formin‐related actin‐organizing protein, probably associates with the nestin intermediate filament. Genes Cells. 2005;10:665–678. DOI: 10.1111/j.1365-2443.2005.00867.x. [DOI] [PubMed] [Google Scholar]
- 30. Iskratsch T, Reijntjes S, Dwyer J, Toselli P, Degano IR, Dominguez I, Ehler E. Two distinct phosphorylation events govern the function of muscle FHOD3. Cell Mol Life Sci. 2013;70:893–908. DOI: 10.1007/s00018-012-1154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schonichen A, Geyer M. Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim Biophys Acta. 2010;1803:152–163. DOI: 10.1016/j.bbamcr.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 32. Kuhn S, Geyer M. Formins as effector proteins of Rho GTPases. Small GTPases. 2014;5:e29513. DOI: 10.4161/sgtp.29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A, Spirito P, Matsumori A, Moravec CS, Seidman JG, et al. Mutations in the genes for cardiac troponin T and alpha‐tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064. [DOI] [PubMed] [Google Scholar]
- 34. Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1108–1114. DOI: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 35. Marian AJ, Mares A Jr, Kelly DP, Yu QT, Abchee AB, Hill R, Roberts R. Sudden cardiac death in hypertrophic cardiomyopathy: variability in phenotypic expression of beta‐myosin heavy chain mutations. Eur Heart J. 1995;16:368–376. DOI: 10.1093/oxfordjournals.eurheartj.a060920. [DOI] [PubMed] [Google Scholar]
- 36. Lopes LR, Rahman MS, Elliott PM. A systematic review and meta‐analysis of genotype‐phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart. 2013;99:1800–1811. DOI: 10.1136/heartjnl-2013-303939. [DOI] [PubMed] [Google Scholar]
- 37. Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin‐filament gene mutations. J Am Coll Cardiol. 2014;64:2589–2600. DOI: 10.1016/j.jacc.2014.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S8
Figures S1–S2