

Abstract
Background
The menopausal transition is characterized by increased cardiovascular risk, weight gain, and increased adiposity for many women. The adipose‐derived secretory proteins adiponectin and leptin are associated with insulin resistance, metabolic syndrome, and cardiovascular disease but their role in subclinical atherosclerotic disease is unclear. This cross‐sectional study evaluated the associations of adiponectin and leptin with carotid artery intima‐media thickness, adventitial diameter, presence of carotid plaques, and brachial‐ankle pulse wave velocity (baPWV) in women aged 54 to 65 years.
Methods and Results
Participants were 1399 women from SWAN (Study of Women's Health Across the Nation), a community‐based study of women transitioning through menopause. Carotid ultrasound and baPWV measures were obtained at SWAN follow‐up visits 12 or 13, when 97% of participants were post‐menopausal. Adipokines were assayed from serum specimens obtained concurrently at these visits. Linear and logistic regression models were used to evaluate adiponectin or leptin, both log‐transformed attributable to skewness, in relationship to carotid artery intima‐media thickness, adventitial diameter, baPWV, and presence of carotid plaque. Covariates included age, race, study site, smoking, alcohol use, obesity, cardiovascular disease risk factors, and menopausal status. Lower levels of adiponectin were related to greater carotid artery intima‐media thickness, wider adventitial diameter, and faster baPWV; associations were attenuated after adjusting for cardiovascular disease risk factors. Higher levels of leptin were associated with greater carotid artery intima‐media thickness and wider adventitial diameter in minimally and fully adjusted models, and contrary to expectation, with slower baPWV, particularly among women with diabetes mellitus or obesity.
Conclusions
Adiponectin and leptin are 2 important inflammatory pathways that may contribute to adverse subclinical cardiovascular disease risk profiles in women at midlife.
Keywords: adiponectin, atherosclerosis, cardiovascular risk factors, leptin, menopause
Subject Categories: Cardiovascular Disease, Epidemiology, Risk Factors, Women
Nonstandard Abbreviations and Acronyms
- AD
adventitial diameter
- baPWV
brachial artery pulse wave velocity
- cIMT
carotid intimal‐medial thickness
- HMW
high molecular weight
- SHBG
sex hormone binding globulin
- SWAN
Study of Women's Health Across the Nation
Clinical Perspective
What Is New?
In a sample of nearly 1400 community‐dwelling post‐menopausal women without clinical cardiovascular disease, we observed that lower levels of adiponectin and higher levels of leptin were associated with greater subclinical cardiovascular disease.
Associations between subclinical cardiovascular disease and adiponectin were attenuated by known cardiovascular risk factors, but associations with leptin were mostly robust to risk factor adjustments.
What Are the Clinical Implications?
Our study findings suggest that inflammatory pathways are critical to understanding the increasing cardiovascular risk women experience as they age.
Adipokines, secretory proteins derived from adipose tissue, likely play a role in obesity‐related conditions, including insulin resistance, metabolic syndrome, and cardiovascular disease (CVD). 1 Adiponectin is a highly abundant anti‐inflammatory adipokine; leptin is proinflammatory, involved in metabolic regulation, energy balance, and autonomic nervous system functioning. Whether these adipokines play a significant role early in the atherosclerotic process is unclear; available evidence, though mixed, suggests these adipokines may be important to consider. Low adiponectin levels independently related to severity, extent, and pattern of atherosclerosis whereas high leptin related to severity and pattern of atherosclerosis in 1 case‐control study. 2 Lower adiponectin levels were independently correlated with greater carotid atherosclerosis in patients with chronic kidney disease (CKD) but not among patients without CKD in a Japanese study. 3 Among Asian Indian patients with coronary artery disease (CAD) and their unaffected family members (controls), lower adiponectin and higher leptin were related to higher odds of CAD and more adverse risk factor profiles, including higher levels of lipids, coagulation, and inflammatory markers. 4 In 2 studies from Taiwan, higher serum leptin levels were positively related to central arterial stiffness among middle‐aged and older patients with hypertension 5 and with documented CAD. 6 However, leptin was unrelated to arterial stiffness in a South African study of younger, healthy adults although it was inversely related to carotid wall thickness among men who were overweight. 7 Among healthy adults, circulating plasma levels of adiponectin were inversely associated with intima‐media thickness in men only 8 ; another study showed similar results in healthy middle‐aged women. 9
CVD risk increases after the menopausal transition. An increase in adipose tissue occurs at this time, with weight gain and increasing central adiposity commonly reported during the menopause transition. Whether concomitant changes in adipose‐derived adiponectin and leptin contribute to increasing CVD risk among women during midlife is unclear. Moreover, racial/ethnic differences in CVD risk and adiposity are evident in women during the menopausal transition, 10 , 11 , 12 , 13 although data on racial/ethnic differences in subclinical CVD are mixed. 14 , 15 A prior report from the SWAN (Study of Women's Health Across the Nation) study showed racial/ethnic differences in adiponectin and leptin levels. 16 Relative to White women, Black women had significantly higher leptin, and lower total and high molecular weight (HMW) adiponectin, whereas both Japanese and Chinese women had significantly lower total and HMW adiponectin, after adjusting for adiposity. The authors highlighted the need for additional research to determine if difference in adipokines contribute to observed CVD racial disparities.
Several reports in the literature have noted paradoxical or null associations of adiponectin and leptin with risk for coronary heart disease and/or total mortality, particularly in high‐risk individuals, eg, person with diabetes mellitus or Class III obesity. 17 , 18 , 19 Thus, the relationship of these pro‐ and anti‐inflammatory cytokines with CVD may vary across stages of the atherosclerotic disease process and in the face of metabolic dysregulation.
This study brings together these lines of research to evaluate the association of adiponectin and leptin with markers of subclinical CVD in a multi‐ethnic cohort of women, 97% of whom were post‐menopausal at the time of data collection. We investigated adiponectin and leptin in relationship to subclinical CVD, including markers of subclinical atherosclerosis (carotid artery intimal medial thickness [cIMT] and presence of carotid plaque), vascular remodeling (adventitial diameter [AD]), and arterial stiffness (brachial artery pulse wave velocity [baPWV]) in SWAN. We hypothesized that lower levels of adiponectin and higher levels of leptin would be associated with increased cIMT, greater AD, greater prevalence of carotid plaques and faster baPWV, all indicators of greater subclinical CVD.
Because of previously identified racial/ethnic differences in adiponectin and leptin in SWAN 19 and documented racial/ethnic differences in CVD risk, we examined potential differences in the hypothesized associations by race/ethnicity. Further, we investigated whether associations were independent of behavioral risk factors, traditional cardiovascular risk factors, and menopausal status, assessed by bleeding criteria and levels of estradiol and sex hormone binding globulin (SHBG).
Methods
Transparency and Reproducibility
SWAN provides access to public use data sets that include data from SWAN screening, baseline, and follow‐up visits (https://agingresearchbiobank.nia.nih.gov/). To preserve participant confidentiality, some, but not all, of the data used for this article are contained in the public use data sets. A link to the public use data sets also is located on the SWAN website: http://www.swanstudy.org/swan‐research/data‐access/. Investigators requiring assistance accessing the public use data set may contact the SWAN Coordinating Center by email at swanaccess@edc.pitt.edu.
Population
Participants were from SWAN, a multi‐ethnic, community‐based, longitudinal study of the menopausal transition among 3302 women enrolled at 7 US field sites (Boston, MA; Chicago, IL; Detroit, MI; Los Angeles, CA; Davis, CA; Newark, NJ; and Pittsburgh, PA) as previously described. 20 At baseline (1996–1997), eligible women were non‐pregnant, aged 42 to 52 years, had an intact uterus with at least 1 ovary, and at least 1 menstrual period and no use of hormone therapy in the preceding 3 months.
The current analysis includes eligible women who participated in the carotid ultrasound and baPWV exams during the 12th or 13th annual follow‐up clinic visit (n=1606). Women missing adipokine data (n=104) were excluded. Women with a history of prior myocardial infarction (n=103) also were excluded, given our focus on subclinical CVD; past diagnoses of other CVD conditions (eg, heart failure) were infrequent among participants and were not considered exclusionary. Thus, we identified a potential sample of 1399 with any subclinical CVD measure. Carotid plaque analyses included 1398 women as 1 participant had no plaque data available; poor quality carotid images for 29 participants limited analyses of cIMT and AD to 1370 participants; 1096 completed the baPWV assessment but 6 women had poor quality baPWV data resulting in 1090 participants for baPWV analyses. The Institutional Review Board at each SWAN site and the SWAN Data Coordinating Center approved the study protocol; all participants provided written informed consent.
Predictor Variables and Covariates
All SWAN participants underwent approximately annual study visits with interviewer‐administered questionnaires to ascertain medication use and current smoking status, physical measures, and a fasting blood draw. Measures of adiponectin and leptin and other cardiovascular and metabolic risk factors were obtained from these data (detailed below).
Adipokines
Leptin and adiponectin were measured from serum specimens corresponding to the same clinic visit at which carotid ultrasound and baPWV were assessed. Blood draws were done before 10 am in a fasted state for all SWAN participants, timed to the early follicular phase (days 2–5) for women who were still cycling, with cycle day of blood draw recorded as within or outside the targeted early follicular phase window. Among women who were no longer cycling (>97% of participants at visit 12 and 13), blood draws were completed within 90 days of the anniversary of their SWAN baseline visit. Assays for adiponectin and leptin were completed in duplicate using commercially available colorimetric enzyme‐linked immunosorbent assay kits per instructions from the manufacturer (Millipore, St. Charles, MO). The inter‐assay coefficient of variation percentage for adiponectin was 3.23±0.04 µg/mL and the coefficient of variation percentage for leptin was 7.38±0.09 ng/mL.
Covariates
Demographics, Health Behaviors, and History
Race or ethnicity and education were self‐reported at the SWAN baseline; education was classified as high school or less, some college/vocational, and college or more. Smoking status was categorized as current smoking (yes/no) based on self‐report of smoking at SWAN visit 12. Alcohol consumption was categorized as none/low (alcohol use less than once a month); moderate alcohol use (defined as greater than once per month and <2 times per week); and high alcohol use (defined as ≥2 times per week). Physical activity was assessed by self‐report of physical activity in 4 domains: occupation, household and caregiving, sports and exercise, and daily routine. 21 , 22 Women were considered to have diabetes mellitus if they self‐reported diabetes mellitus or had fasting glucose levels ≥126 mg/dL or reported any use of insulin/anti‐diabetic mellitus agents at ≥70% of study visits or for ≥3 consecutive visits.
Cardiovascular Biomarkers and Risk Factors
Data on blood pressure, anthropometric and biomarker assessments were obtained from the visit corresponding to the carotid ultrasound visit (visit 12/13). Systolic blood pressure (SBP) was measured manually per standard protocol in a seated position after at least a 5‐minute rest; the average of 2 sequential readings was used in the analyses. Height was measured by a stadiometer and weight was measured using calibrated scales; body mass index (BMI) was calculated as kg/m2. Triglycerides, high‐density lipoprotein cholesterol (HDL‐C) and direct low‐density lipoprotein cholesterol (LDL‐C) were analyzed via coupled enzymatic methods that used lipase, glycerol kinase, glycerol‐3‐phosphate oxidase, and peroxidase (for triglycerides) and cholesterol esterase, cholesterol oxidase, and peroxidase (for HDL‐C and direct LDL‐C). Serum insulin was measured by a 2‐site sandwich immunoassay using direct chemiluminescent technology. Glucose was measured using a 2‐step enzymatic reaction that uses hexokinase and glucose‐6‐phosphate dehydrogenase enzymes. Lipid, glucose, and insulin assays were performed on a Siemens ADVIA 2400 automated chemistry analyzer (Siemens Healthcare Diagnostics, Deerfield, IL) at the CLIA‐certified University of Michigan Pathology Laboratory.
Sex Hormones and Menopausal Status
Estradiol was measured by a modified, off‐line Automated Chemiluminescence System: 180 (E2‐6). Lower limit of detection range was 1 to 7 pg/mL. SHBG was evaluated by a 2‐site chemiluminescent immunoassay; limit of detection was between 1.9 and 3.2 nmol/L. Menopausal status at the year 12 and 13 visit was self‐reported and reclassified into 3 categories: pre‐menopausal/peri‐menopausal (menstrual period within 12 months), post‐menopausal (≥12 months of amenorrhea), and surgical menopausal (hysterectomy and/or bilateral oophorectomy).
Outcome Variables
Subclinical Cardiovascular Disease Measures
At each site, centrally trained and certified sonographers obtained carotid ultrasound images using a Terason t3000 Ultrasound System (Teratech Corp, Burlington, MA) equipped with a variable frequency 5 to 12 MHz linear array transducer. Two digitized images for later reading were obtained of the left and right distal common carotid artery (CCA) during end‐diastole. Using artery measurement system (AMS) semi‐automated edge detection software, 23 near and far wall CCA‐IMT measures were obtained by electronically tracing the lumen‐intima interface and media‐adventitia interface across a 1‐cm segment proximal to the carotid bulb. Averages of these measurements were recorded for all 4 images, with the mean of the average readings used in analyses. Common carotid artery AD was measured directly as the distance from the adventitial‐medial interface on the near wall to the medial‐adventitial interface on the far wall across the same CCA segments used for CCA‐IMT measurement. Carotid scan images were read centrally at the SWAN Ultrasound Reading Center (University of Pittsburgh Ultrasound Research Lab) by 2 trained readers without knowledge of or access to participant data. Reproducibility of IMT and AD measures was excellent with an intraclass correlation coefficient (ICC) between site sonographers ≥0.77 for IMT and >0.85 for AD, and the ICC between readers >0.90 for IMT and >0.80 for AD. Sonographers at each site evaluated the presence and extent of plaque in each of 5 segments of the left and right carotid artery (distal and proximal common carotid artery, carotid bulb, and proximal internal and external carotid arteries). 24 , 25 Plaque was defined as a distinct area protruding into the vessel lumen at least 50% thicker than the adjacent IMT. The presence (yes/no) of any plaque was used for analysis. For quality control, each site completed at least 20 repeat plaque assessments for total of 136 repeat scans across sites; the resulting Kappa coefficient (95% CI) of 0.7561 (95% CI, 0.64‒0.87) indicates good reliability. Identical scanning and reading protocols have been used in several studies. 25 , 26 , 27
Brachial‐ankle pulse wave velocity (baPWV) measures were automatically generated using a non‐invasive automated waveform analyzer (VP2000, Omron Co., Komaki, Japan). 28 This device provides a measure of mixed central and peripheral artery stiffness and ankle/brachial index by simultaneously recording baPWV, blood pressure, ECG and heart sounds. Following a 10‐minute rest in a supine position, occlusion and monitoring cuffs were placed around both upper arms and ankles, following standardized placement procedures. Arm cuffs were placed on bare arms or over light clothing, and ankle cuffs were placed on bare ankles. ECG electrodes were placed on both wrists and a phonocardiogram was placed on the left edge of the sternum. Cuffs were connected to a plethysmographic sensor that determines pulse volume waveforms and to an oscillometric pressure sensor that measures blood pressure. Volume waveforms for the arm (brachial artery) and ankle (tibial artery) were stored for a 10‐second sampling time with automatic gain analysis and quality adjustment. The baPWV was calculated as the path length between arterial sites divided by the time delay between the foot of the respective waveforms (ΔTba). The path length was calculated using height‐based formulas rather than the actual “above the body” distances which corrects for the opposite direction of blood flow by subtracting the heart‐to‐brachial distance from the heart‐to‐tibial distance, as seen in the following equations 29 : path length from the heart to the brachium (Lb)=0.2195×height of the patient (cm) −2.0734; path length from the heart to ankle (La)=0.8129×height of the patient (cm) +12.328. The baPWV was calculated by time‐phase analysis, for the right and left sides, using the following equation: (La−Lb)/(ΔTba). Left and right side baPWV values were averaged for use in analyses. Data were collected from 3 runs for each participant with values averaged.
Before the start of data collection, study sonographers from all SWAN sites were trained on the study protocol at the Ultrasound Research Laboratory at the University of Pittsburgh and monitored during the study period for reproducibility. This approach ensured accurate and consistent assessment of subclinical cardiovascular disease measures across study sites.
Statistical Analysis
Variables were examined for normality with skewed variables transformed as needed for analyses. Adiponectin, leptin, glucose, HDL‐C, triglyceride, and SHBG were log‐transformed. Estradiol was square‐root transformed. The cIMT, AD, and baPWV measures were slightly skewed; thus, log‐transformed, and untransformed measures were used in regression models. Results were similar; therefore, the untransformed models are presented for ease of interpretation. Descriptive statistics (mean and SD or median and interquartile range values for continuous variables and counts and percentages for dichotomous or categorical variables) were used to summarize individual characteristics or risk factors by quartile of serum levels of adipokines. Analysis of variance F‐test and Chi‐square test were performed to test differences of continuous and categorical variables across quartiles of adipokines, respectively. Linear and logistic regression models were used to test for trends of continuous and categorical variables across quartiles of adipokines, respectively. Correlations of covariates, adipokines, and subclinical cardiovascular measures were examined to identify potential collinearity. BMI was highly correlated with adiponectin (r, −0.33, P<0.0001) and leptin (r, 0.75, P<0.0001); thus, we dichotomized BMI for analyses, with BMI values ≥30 noted as obese, and <30 as non‐obese. Linear regression models were used with cIMT, AD, and baPWV as the dependent variables in separate models with adiponectin and leptin as predictors. With each outcome, the first model adjusted for age, race, and study site; the second model added covariates for smoking and alcohol consumption; and the third model added dichotomous BMI as a covariate. We also performed sensitivity analyses using either waist:hip ratio or the calculated BMI residuals instead of obesity status as a covariate; results were similar to what is reported herein with obesity status as the covariate for adiposity. The final model included all covariates in the third model plus other potential confounders that were significant at P<0.1. Established health and CVD risk factors were candidate covariates for the final model and included SBP, obesity, glucose, HDL‐C, LDL‐C, triglycerides, physical activity, use of anti‐hypertensives and lipid‐lowering medications, SHBG, estradiol, diabetes mellitus status, and menopausal status, classified as surgical menopause, postmenopause, or combined pre‐/peri‐menopause (referent). Serum glucose level is highly correlated with diabetes mellitus status, so these 2 variables were tested in separate models. Interactions of race and adipokines, diabetes mellitus, and adipokines, as well as obesity status and adipokines were tested after the final model was determined. Additional interactions of SBP with leptin and with obesity were tested in the baPWV model. Subgroup analyses were performed if interactions were statistically significant. SAS version 9.3 (SAS Institute, Cary, NC) was used for the analyses. All tests were 2‐sided with α=0.05.
Results
The study sample was multi‐ethnic, including Black (30.0%), Chinese (12.7%), Hispanic (6.6%), and White (50.7%) women. Participants were predominantly post‐menopausal (96.9%), non‐smokers (90.9%), and, on average, aged 60 years at the time of the carotid exam visit. Fourteen percent reported a history of diabetes mellitus, 40% were currently on antihypertensive medications (65% ever use), 35.8% on lipid‐lowering medications, and 4.5% reported currently using hormone replacement therapy.
Participants' demographic and cardiovascular risk characteristics by quartile of adiponectin and leptin are summarized in Tables 1 and 2. Black women were more likely to be in the lower quartiles of adiponectin and higher quartiles of leptin while White women were more likely to be in the higher quartiles of adiponectin and Chinese women in the lowest quartile of leptin. Overall, lower adiponectin and higher leptin quartiles were associated with adverse CVD risk profiles, indicated by measures of adiposity, cardiometabolic health, and subclinical CVD. The box plots in Figure 1 graphically depict patterns of observed relationships for cIMT, AD, and baPWV across quartiles of adiponectin (Figure 1A through 1C) and quartiles of leptin (Figure 1D through 1F), adjusted for age, race, and study site. CIs (depicted by error bars) are generally wide, yet distributions of cIMT, AD, and baPWV mean values across quartiles of adiponectin from low (Quartile 1) to high (Quartile 4) show a graded, inverse pattern for each outcome. Across quartiles of leptin, graded patterns are seen for cIMT and AD, but not for baPWV, with the highest mean cIMT and AD values among women in the highest leptin quartile.
Table 1.
Sample Characteristics by Quartiles of Adiponectin: Study of Women's Health Across the Nation Visit 12/13
| Quartile 1 (Low) | Quartile 2 | Quartile 3 | Quartile 4 (High) | No. | P Value | P‐for‐Trend | |
|---|---|---|---|---|---|---|---|
| No. (total subjects) | 352 | 352 | 353 | 342 | 1399 | ||
| Race/Ethnicity, n(%) | |||||||
| Black | 180 (51.1) | 127 (36.1) | 72 (20.4) | 42 (12.3) | 421 | <0.0001 | <0.0001 |
| Chinese | 40 (11.4) | 44 (12.5) | 53 (15.0) | 40 (11.7) | 177 | ||
| Hispanic | 38 (10.8) | 18 (5.1) | 19 (5.4) | 17 (5.0) | 92 | ||
| White | 94 (26.7) | 163 (46.3) | 209 (59.2) | 243 (71.0) | 709 | ||
| Age, y, mean (SD) | 59.3 (2.6) | 59.6 (2.8) | 59.7 (2.8) | 59.7 (2.6) | 1399 | 0.132 | 0.028 |
| Diabetes mellitus, n (%) | |||||||
| Yes | 96 (27.3) | 50 (14.2) | 19 (5.4) | 26 (7.6) | 191 | <0.0001 | <0.0001 |
| No | 256 (72.7) | 302 (85.8) | 334 (94.6) | 316 (92.4) | 1208 | ||
| Current smoker, n (%) | |||||||
| Yes | 55 (15.8) | 28 (8.0) | 26 (7.5) | 17 (5.0) | 126 | <0.0001 | <0.0001 |
| No | 294 (84.2) | 320 (92.0) | 322 (92.5) | 324 (95.0) | 1260 | ||
| Alcohol use, n (%) | |||||||
| High (2+/wk) | 39 (11.3) | 59 (17.1) | 95 (27.5) | 117 (34.8) | 310 | <0.0001 | <0.0001 |
| Moderate | 92 (26.7) | 85 (24.6) | 89 (25.7) | 83 (24.7) | 349 | ||
| Low (<1/mo) | 213 (61.9) | 201 (58.3) | 162 (46.8) | 136 (40.5) | 712 | ||
| Menopause status, n (%) | |||||||
| Surgical menopause | 40 (11.4) | 40 (11.4) | 24 (6.8) | 24 (7.0) | 128 | 0.045 | 0.006 |
| Post‐menopause | 298 (85.1) | 298 (84.9) | 318 (90.6) | 313 (91.5) | 1227 | ||
| Pre‐ or peri‐menopause | 12 (3.4) | 13 (3.7) | 9 (2.6) | 5 (1.5) | 39 | ||
| Current use of anti‐hypertensives, n (%) | |||||||
| Yes | 218 (74.7) | 195 (71.4) | 138 (54.8) | 122 (55.7) | 673 | <0.0001 | <0.0001 |
| No | 74 (25.3) | 78 (28.6) | 114 (45.2) | 97 (44.3) | 363 | ||
| Current use of lipid‐lowering drug, n (%) | |||||||
| Yes | 152 (43.8) | 141 (40.3) | 112 (31.9) | 91 (26.9) | 496 | <0.0001 | <0.0001 |
| No | 195 (56.2) | 209 (59.7) | 239 (68.1) | 247 (73.1) | 890 | ||
| Current use of hormone therapy, n (%) | |||||||
| Yes | 14 (4.0) | 15 (4.3) | 12 (3.4) | 22 (6.43) | 63 | 0.237 | 0.198 |
| No | 338 (96.0) | 337 (95.7) | 341 (96.6) | 320 (93.6) | 1336 | ||
| BMI, kg/m2, means (SD) | 33.2 (6.9) | 31.4 (7.2) | 28.9 (6.9) | 26.7 (6.3) | 1377 | <0.0001 | <0.0001 |
| Obesity (BMI ≥30 kg/m2), n (%) | |||||||
| Yes | 219 (62.2) | 180 (51.1) | 128 (36.3) | 76 (22.2) | 603 | <0.0001 | <0.0001 |
| No | 133 (37.8) | 172 (48.9) | 225 (63.7) | 266 (77.8) | 796 | ||
| Systolic blood pressure, mm Hg, means (SD) | 128.5 (19.2) | 122.7 (15.8) | 119.6 (16.1) | 118.4 (16.4) | 1384 | <0.0001 | <0.0001 |
| Fasting glucose, mg/dL, median (IQR) | 99 (91–114) | 93 (88–104) | 91 (85–97) | 88 (83–96) | 1387 | <0.0001 | <0.0001 |
| Triglycerides, mg/dL, median (IQR) | 121 (89–163) | 112 (81–157) | 96 (73–126) | 81 (63–110) | 1381 | <0.0001 | <0.0001 |
| HDL‐C, mg/dL, median (IQR) | 51 (45–58) | 56 (48–64) | 63 (54.5–8) | 70 (62–82) | 1392 | <0.0001 | <0.0001 |
| LDL‐C, mg/dL, means (SD) | 116.0 (32.7) | 120.0 (32.0) | 122.1 (29.8) | 121.7 (32.0) | 1382 | 0.047 | 0.013 |
| SHBG, nmol/L, median (IQR) | 44.0 (30.5–57.4) | 48.3 (35.8–67.9) | 61.9 (44.3–84.8) | 84.0 (63.5–112.2) | 1399 | <0.0001 | <0.0001 |
| Estradiol, pg/mL, median (IQR) | 21.5 (13.5–29.8) | 18.8 (11.0–26.7) | 19.8 (11.2–27.2) | 16.1 (9.7–23.3) | 1397 | <0.0001 | <0.0001 |
| Average CCA, mm, median (IQR) | 0.81 (0.74–0.9) | 0.78 (0.72–0.86) | 0.77 (0.7–0.83) | 0.76 (0.69–0.84) | 1363 | <0.0001 | <0.0001 |
| Max CCA, mm, median (IQR) | 0.95 (0.87–1.04) | 0.91 (0.84–1.01) | 0.88 (0.81–0.97) | 0.88 (0.8–0.96) | 1368 | <0.0001 | <0.0001 |
| AD, mm, median (IQR) | 7.33 (6.88–7.77) | 7.14 (6.73–7.61) | 7.1 (6.65–7.48) | 6.99 (6.63–7.4) | 1366 | <0.0001 | <0.0001 |
| Plaque, n (%) | |||||||
| Yes | 157 (44.7) | 141 (40.0) | 140 (39.7) | 155 (45.3) | 593 | 0.278 | 0.922 |
| no | 194 (55.3) | 211 (60.0) | 213 (60.3) | 187 (54.7) | 805 | ||
| baPWV, cm/s, median (IQR) | 1540.0 (1358.2–1692.4) | 1462 (1301.9–1636.2) | 1404.8 (1276.2–1517.2) | 1417.7 (1301.9–1570.2) | 1090 | <0.0001 | <0.0001 |
The column labeled "P value" shows P values for overall differences between groups; the column labeled "P‐for‐trend" shows P values from analyses testing for linear trends across groups. To obtain the P‐for‐trend values, linear regression analysis was performed for continuous risk factor variables, logistic regression analysis was performed for binary risk factor variables, and proportional odds logistic regression was performed for categorical variables. AD indicates adventitial diameter; baPWV, brachial artery pulse wave velocity; BMI, body mass index; CCA, common carotid artery; HDL‐C, high density lipoprotein cholesterol; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; and SHBG, sex hormone binding globulin.
Table 2.
Sample Characteristics by Quartiles of Leptin: Study of Women's Health Across the Nation Visit 12/13
| Quartile 1 (Low) | Quartile 2 | Quartile 3 | Quartile 4 (High) | No. | P Value | P‐for‐Trend | ||
|---|---|---|---|---|---|---|---|---|
| No. (total subjects) | 349 | 350 | 350 | 350 | 1399 | |||
| Race/Ethnicity, n (%) | ||||||||
| Black | 46 (13.2) | 93 (26.6) | 133 (3.08) | 149 (42.8) | 421 | <0.0001 | 0.0002 | |
| Chinese | 115 (33.0) | 40 (11.4) | 18 (5.1) | 4 (1.1) | 177 | |||
| Hispanic | 13 (3.7) | 27 (7.7) | 23 (6.6) | 29 (8.3) | 92 | |||
| White | 175 (50.1) | 190 (54.3) | 176 (50.3) | 168 (48.0) | 709 | |||
| Age, y, mean (SD) | 59.7 (2.6) | 59.6 (2.7) | 59.6 (2.7) | 59.4 (2.8) | 1399 | 0.611 | 0.212 | |
| Diabetes mellitus, n (%) | ||||||||
| Yes | 19 (5.4) | 43 (12.3) | 55 (15.7) | 74 (21.1) | 191 | <0.0001 | <0.0001 | |
| No | 330 (94.6) | 307 (87.7) | 295 (84.3) | 276 (78.9) | 1208 | |||
| Current smoker, n (%) | ||||||||
| Yes | 37 (10.6) | 34 (9.7) | 33 (9.5) | 22 (6.4) | 126 | 0.24 | 0.064 | |
| No | 311 (89.4) | 315 (90.3) | 313 (90.5) | 321 (93.6) | 1260 | |||
| Alcohol use, n (%) | ||||||||
| High (2+/wk) | 90 (26.2) | 102 (29.7) | 79 (23.2) | 39 (11.4) | 310 | <0.0001 | <0.0001 | |
| Moderate | 79 (23.0) | 85 (24.7) | 94 (27.6) | 91 (26.6) | 349 | |||
| <1/mo | 175 (50.9) | 157 (45.6) | 168 (49.3) | 212 (62.0) | 712 | |||
| Menopause status, n (%) | ||||||||
| Surgical menopause | 21 (6.0) | 28 (8.1) | 36 (10.3) | 43 (12.3) | 128 | 0.0037 | <0.0001 | |
| Post‐menopause | 324 (92.8) | 312 (89.9) | 301 (86.2) | 290 (83.1) | 1227 | |||
| Pre‐ and peri‐menopause | 4 (1.1) | 7 (2.0) | 12 (3.4) | 16 (4.6) | 39 | |||
| Current use of anti‐hypertensives, n (%) | ||||||||
| Yes | 99 (44.4) | 150 (62.8) | 179 (66.8) | 245 (80.1) | 673 | <0.0001 | <0.0001 | |
| No | 124 (55.6) | 89 (37.2) | 89 (33.2) | 61 (19.9) | 363 | |||
| Current use of lipid‐lowering drug, n (%) | ||||||||
| Yes | 93 (27.0) | 118 (34.1) | 119 (34.3) | 166 (47.7) | 496 | <0.0001 | <0.0001 | |
| No | 252 (73.0) | 228 (65.9) | 228 (65.7) | 182 (52.3) | 890 | |||
| Current use of hormone therapy, n (%) | ||||||||
| Yes | 21 (6.0) | 16 (4.6) | 12 (3.4) | 14 (4.0) | 63 | 0.390 | 0.149 | |
| No | 328 (94.0) | 334 (95.4) | 338 (96.6) | 336 (96.0) | 1336 | |||
| BMI, kg/m2, n (%) | 23.2 (3.2) | 27.4 (3.9) | 31.8 (4.9) | 38.0 (6.6) | 1377 | <0.0001 | <0.0001 | |
| Obesity (BMI ≥30 kg/m2, n (%) | ||||||||
| Yes | 10 (2.9) | 72 (20.6) | 217 (62.0) | 304 (86.9) | 603 | <0.0001 | <0.0001 | |
| No | 339 (97.1) | 278 (79.4) | 133 (38.0) | 46 (13.1) | 796 | |||
| Systolic blood pressure, mm Hg, means (SD) | 115.3 (17.3) | 122.7 (17.1) | 123.7 (15.4) | 127.6 (17.3) | 1384 | <0.0001 | <0.0001 | |
| Fasting glucose, mg/dL, median (IQR) | 89 (83–94) | 92 (86–99.5) | 94 (88–102) | 97 (89–110) | 1387 | <0.0001 | <0.0001 | |
| Triglycerides, mg/dL, median (IQR) | 85 (65–114) | 102 (76–143) | 108 (79–149) | 111 (81–147) | 1381 | <0.0001 | <0.0001 | |
| HDL‐C, mg/dL, median (IQR) | 68 (57–81) | 59 (51–72) | 56 (49–68) | 55 (47–63) | 1392 | <0.0001 | <0.0001 | |
| LDL‐C, mg/dL, mean (SD) | 119.3 (32.6) | 121.2 (30.7) | 121.2 (31.7) | 118.1 (31.7) | 1382 | 0.501 | 0.655 | |
| SHBG, nmol/L, median (IQR) | 80.2 (56–108.1) | 60.1 (42.9–8) | 52.0 (37.2–68.7) | 45.2 (33.3–63.8) | 1399 | <0.0001 | <0.0001 | |
| Estradiol, pg/mL, median (IQR) | 16.2 (8.4–22.3) | 18.0 (10.9–25.7) | 19.0 (12.7–27.7) | 23.6 (14.9–32.7) | 1397 | <0.0001 | <0.0001 | |
| Average CCA, mm, median (IQR) | 0.73 (0.68–0.81) | 0.78 (0.71–0.87) | 0.79 (0.73–0.87) | 0.81 (0.74–0.89) | 1363 | <0.0001 | <0.0001 | |
| Max CCA, mm, median (IQR) | 0.86 (0.79–0.93) | 0.91 (0.83–1.01) | 0.92 (0.85–1.01) | 0.95 (0.87–1.03) | 1368 | <0.0001 | <0.0001 | |
| AD, mm, median (IQR) | 6.96 (6.6–7.32) | 7.06 (6.65–7.54) | 7.22 (6.77–7.65) | 7.31 (6.9–7.79) | 1366 | <0.0001 | <0.0001 | |
| Plaque, n (%) | ||||||||
| Yes | 146 (41.8) | 146 (41.7) | 151 (43.1) | 150 (43.0) | 593 | 0.971 | 0.681 | |
| no | 203 (58.2) | 204 (58.3) | 199 (56.9) | 199 (57.0) | 805 | |||
| baPWV, cm/s, median (IQR) | 1387.0 (1195.8–1431.1) | 1428.6 (1319.5–1596.8) | 1437.5 (1310.5–1623.9) | 1453.6 (1313.1–1644.0) | 822 | 0.669 | 0.698 | |
The column labeled "P value" shows P values for overall differences between groups; the column labeled "P‐for‐trend" shows P values from analyses testing for linear trends across groups. To obtain the P‐for‐trend values, linear regression analysis was performed for continuous risk factor variables, logistic regression analysis was performed for binary risk factor variables, and proportional odds logistic regression was performed for categorical variables. AD indicates adventitial diameter; baPWV, brachial artery pulse wave velocity; BMI, body mass index; CCA, common carotid artery; HDL‐C, high density lipoprotein cholesterol; IQR, interquartile range; LDL‐C, low‐density lipoprotein cholesterol; and SHBG, sex hormone binding globulin.
Figure 1. Distributions of carotid artery intima‐medial thickening, adventitial diameter, and brachial artery pulse wave velocity across quartiles of adiponectin and leptin: SWAN (Study of Women's Health Across the Nation).
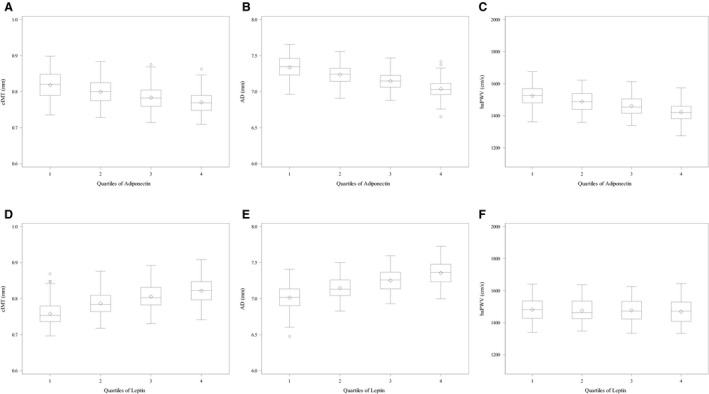
A and D, Means and distribution of carotid artery intima‐medial thickening by quartiles of adiponectin and leptin, respectively, adjusted for age, race and study site. B and E, Means and distribution of adventitial diameter by quartiles of adiponectin and leptin, respectively, adjusted for age, race, and study site. C and F, means and distribution of brachial artery pulse wave velocity by quartiles of adiponectin and leptin, respectively, adjusted for age, race and study site. AD indicates adventitial diameter; baPWV, brachial artery pulse wave velocity; and cIMT, carotid artery intima‐medial thickening.
Adiponectin Levels and Subclinical CVD
Every 1 unit lower log‐transformed serum adiponectin related to a 0.0159 mm (P=0.0013) thicker cIMT after adjusting for age, race, and study site (top half of Table 3, Model 1). The association remained statistically significant after adjusting for smoking and alcohol consumption (Model 2), but not after adjusting for obesity status (Model 3); adding covariates related to CVD risk, and menopausal status eliminated the observed relationship of adiponectin with cIMT (Model 4). For every 1 unit lower log‐transformed serum adiponectin, AD was 0.141 mm wider (P<0.001) adjusting for age, site, and race (middle columns, Table 3, Model 1) and health behaviors (Model 2). The association remained statistically significant after adjusting for obesity status (Model 3) but reduced to null after adjusting for other CVD risk factors and menopausal status (Model 4). Adjusting for age, site, and race, lower serum adiponectin was significantly associated with faster baPWV (β=−41.21, P<0.001), an association that persisted after adjusting for smoking, alcohol consumption, and obesity status (right columns, Table 3, Models 1–3) but not after adjusting for other covariates (Model 4). Including diabetes mellitus status in the model in place of fasting glucose did not alter the association between adiponectin and any of the subclinical vascular measures (data not shown). There was no statistically significant modification by race, diabetes mellitus status, or obesity (data not shown). Adiponectin levels were unrelated to presence of carotid plaques (data not shown).
Table 3.
Associations of Adiponectin and Leptin With Markers of Subclinical CVD in the Study of Women's Health Across the Nation
| No. | cIMT | No. | AD | No. | baPWV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | SE | P Value | R 2 | Estimate | SE | P Value | R 2 | Estimate | SE | P Value | R 2 | ||||
| Adiponectin | |||||||||||||||
| Model 1 | 1344 | −0.016 | 0.005 | 0.001 | 0.108 | 1337 | −0.141 | 0.029 | <0.001 | 0.072 | 1056 | −41.213 | 11.255 | <0.001 | 0.101 |
| Model 2 | 1344 | −0.013 | 0.005 | 0.007 | 0.115 | 1337 | −0.127 | 0.029 | <0.001 | 0.079 | 1056 | −38.630 | 11.410 | <0.001 | 0.105 |
| Model 3 | 1344 | −0.007 | 0.005 | 0.195 | 0.131 | 1337 | −0.070 | 0.029 | 0.017 | 0.114 | 1056 | −36.198 | 11.694 | 0.002 | 0.106 |
| Model 4* | 1326 | 0.002 | 0.005 | 0.649 | 0.168 | 1323 | 0.035 | 0.032 | 0.281 | 0.204 | 1015 | −11.240 | 10.927 | 0.304 | 0.302 |
| Leptin | |||||||||||||||
| Model 1 | 1323 | 0.020 | 0.004 | <0.001 | 0.123 | 1326 | 0.156 | 0.021 | <0.001 | 0.093 | 1047 | −4.487 | 8.617 | 0.603 | 0.090 |
| Model 2 | 1323 | 0.020 | 0.004 | <0.001 | 0.134 | 1326 | 0.165 | 0.021 | <0.001 | 0.106 | 1047 | −3.691 | 8.874 | 0.678 | 0.095 |
| Model 3 | 1323 | 0.012 | 0.005 | 0.006 | 0.136 | 1326 | 0.100 | 0.026 | <0.001 | 0.119 | 1047 | −16.456 | 10.614 | 0.121 | 0.099 |
| Model 4* | 1315 | 0.008 | 0.004 | 0.091 | 0.171 | 1318 | 0.057 | 0.025 | 0.021 | 0.202 | 1006 | −33.237 | 9.568 | <0.001 | 0.309 |
Both adioponectin and leptin were log‐transformed for analyses; thus, estimates shown represent the difference in carotid artery intima‐medial thickening, adventitial diameter, and brachial artery pulse wave velocity per 1‐unit increase in adiponectin (top rows) or leptin (bottom rows), respectively. Model 1 covariates: age, race/ethnicity, and study site. Model 2 covariates: Model 1+smoking, and alcohol consumption. Model 3 covariates: Model 2+obesity status (body mass index <30 vs ≥30). Model 4 covariates: Model 3+systolic blood pressure, and fasting glucose for all outcomes. AD indicates adventitial diameter; baPWV, brachial artery pulse wave velocity; and cIMT, carotid artery intima‐medial thickening.
Menopausal status is an added covariate in Model 4 for carotid artery intima‐medial thickening and adventitial diameter outcomes (for both the adiponectin and leptin models); high‐density lipoprotein cholesterol is an added covariate for Model 4 for adventitial diameter for just the adiponectin model; and low‐density lipoprotein cholesterol is a covariate in both Model 4s with brachial artery pulse wave velocity as the outcome.
Leptin Levels and Subclinical CVD
After adjusting for age, site, and race, every 1‐log increment of leptin was associated with 0.0197 mm thicker cIMT (P<0.0001; see left columns, lower half of Table 3, Model 1). This association remained statistically significant after adjusting for smoking and alcohol consumption (Model 2) and obesity status (Model 3) but not for the other covariates (Model 4). AD was 0.156 mm wider for every 1‐unit increase of log‐transformed leptin after adjusting for age and race (P<0.001) (middle columns Table 3, Model 1), and remained statistically significant after covariate adjustment, albeit somewhat attenuated (Models 2–4). Higher concentrations of leptin were associated with slower baPWV across models (right columns, lower half of Table 3, Models 1–4) but the association only became statistically significant after SBP, fasting glucose, and LDL‐C were added as covariates (ie, Model 4). The coefficient for leptin is ≈8 times larger in Model 4 (β=−33.237; P=0.0005) than in Model 1 (β=−4.487; ns). The R 2 value of Model 4 is 0.309, compared with <0.10 for each other model. The leptin‐baPWV association appears to be especially impacted by fasting glucose and systolic blood pressure (both P<0.0001) but less so by LDL‐C (P=0.09), all of which were positively related to baPWV (data not shown). Calculating the change in baPWV for each 1‐SD increase in these covariates shows that baPWV was 41.7 cm/s faster per 1‐SD of fasting glucose (SD, 27.7 mg/dL), 110.6 cm/s faster for each 1‐SD increase in SBP (SD, 17.4 mm Hg), and 10.6 cm/s faster for each 1‐SD higher LDL‐C value (SD, 32.2 mg/dL) (data not shown). Leptin is positively associated with fasting glucose and with SBP, as indicated in Table 2 where participants in the top quartile of leptin have the highest median value of glucose (P‐for‐trend<0.0001) and highest mean SBP (P‐for‐trend<0.0001). Using diabetes mellitus status in any of the fully adjusted leptin models in place of fasting glucose did not alter any of the associations observed between leptin and the subclinical CVD measures (data not shown). We observed significant effect modification by diabetes mellitus status in the leptin and baPWV model (βleptin×diabetes mellitus=−70.77, SE=28.35, P=0.013); stratified models showed that leptin was related to slower baPWV in women with and without diabetes mellitus, but a stronger association was seen among women with diabetes mellitus. This was the case with or without fasting glucose included as a covariate. We also explored effect modification by obesity status (βleptin×obese=−55.49, SE=20.76, P=0.008). Analysis stratified by BMI (<30 versus ≥30) showed that leptin was related to slower baPWV in women in both BMI groups, but a stronger relationship was seen among women with obesity (data not shown). In contrast, no evidence of effect modification by diabetes mellitus status was observed in either the IMT or AD models (data not shown). There were no significant leptin×race interactions for any outcomes, and leptin levels were unrelated to presence of carotid plaques (data not shown).
Discussion
This study examined the relationship of adiponectin and leptin, 2 important adipose‐derived secretory proteins, with several indicators of subclinical CVD among nearly 1400 middle‐aged and older community‐dwelling women. Consistent with our hypotheses, women with lower levels of adiponectin or higher levels of leptin showed greater atherosclerotic burden, as indicated by greater cIMT and greater AD. Notably, adiposity (BMI), SBP, and high fasting glucose levels, recognized risk factors for atherosclerotic vascular disease, were important covariates in our models, accounting for some though not all of the observed associations between adipokines and subclinical atherosclerosis. For example, the associations of adiponectin with cIMT and AD were weakened when BMI was included in the analytic models and largely explained by adjustment or other CVD risk factors. In contrast, associations between leptin and these subclinical CVD measures remained significant (or nearly so) after adjusting for BMI and other CVD risk factors. Our observed pattern of findings highlights the importance of proinflammatory processes in atherogenesis.
Prior studies investigating the role of adiponectin in subclinical atherosclerosis produced mixed results. Lo et al 9 reported that adiponectin was inversely related to mean carotid IMT in analyses adjusted for age, race, pack‐years of smoking, and indicators of subcutaneous and visceral fat in a sample of ethnically diverse, younger (mean age=40 years) community‐dwelling women. Lipids, blood pressure, and indicators of glucose metabolism were not included as covariates in their multivariable model so it is unknown if further adjustment for these CVD risk factors would have impacted their observed relationship between adiponectin and cIMT. Significant cross‐sectional associations between lower levels of adiponectin and greater cIMT also have been reported in adolescents with obesity in analyses adjusted for age, sex, and CVD risk factors 29 and in middle‐aged White adults at high risk for, but without clinical manifestation of atherosclerosis 30 ; in the latter study, associations were stronger in men than women, particularly in fully adjusted models with CVD risk factors, C‐reactive protein, and history of diabetes mellitus. Another study of healthy adults in their 60s observed a relationship between circulating adiponectin and mean IMT in men but not in women. 8 Studies of patients with CAD 2 and CKD 3 reported that lower adiponectin levels were independently associated with greater carotid atherosclerosis, although the association was not evident in controls without those conditions. Lower adiponectin and higher leptin were related to greater odds of CAD and more adverse cardiovascular risk factor profiles in a sample of South Asian patients with CAD and their unaffected family members who were study controls selected from the Indian Atherosclerosis Research Study. 4 However, another study of South Asian and White patients with angiographically documented CAD residing in the United Kingdom found no association between adiponectin levels and extent or severity of CAD. 31 Our work shows potentially important relations of adiponectin and leptin to subclinical atherosclerosis among midlife women without established CVD, but additionally underscores the importance of comprehensive consideration of key CVD risk factors in these relationships.
Our analyses evaluating adipokines in relationship to arterial stiffness, measured by baPWV, revealed mixed findings. Women with lower adiponectin levels showed greater arterial stiffness (ie, faster baPWV). This inverse association was attenuated and no longer statistically significant with SBP, fasting glucose, and LDL‐C included as covariates. Other cross‐sectional studies similarly have found that lower levels of adiponectin are associated with arterial stiffness, in hypertensive populations and among healthy inviduals. 32 Few longitudinal studies have evaluated adiponectin in relationship to changes in arterial stiffness; one population‐based study of men without CVD 33 and another study of patients with hypertension 34 reported that lower baseline levels of adiponectin were related to increased arterial stiffness over time. In our data, leptin was associated with slower baPWV only in the fully adjusted model, with the addition of covariates for SBP and especially fasting glucose influencing this relationship; contrary to expectations, in this model, higher levels of leptin were associated with less arterial stiffness. Further analyses revealed this relationship was strongest among women with obesity or diabetes mellitus, although still evident among women without obesity or diabetes mellitus and with or without fasting glucose included as a covariate. The extant literature, though neither extensive nor consistent, suggests that higher levels of leptin may contribute to vascular stiffness and hypertension. 32 Thus, it is not clear why the pattern we observed emerged in our data. The interactions we identified suggest that obesity may be particularly important in helping to understand our findings. We accounted for obesity status using BMI and as we noted, sensitivity analyses with either BMI residuals or waist‐to‐hip ratio showed the same findings. It is possible that the BMI‐leptin relationship may not be fully accounted for in how we analyzed our data. However, though leptin is strongly related to BMI in our data, BMI is only weakly associated with baPWV (rs=0.0556, P=0.059). It could be that the relationship of leptin with arterial stiffness varies depending on whether the specific pulse wave velocity measure that is obtained captures vascular stiffening in central or peripheral artery beds. Our study measured baPWV, a composite indicator of central and peripheral artery stiffness. The studies by Tsai and colleagues 5 , 6 showed that leptin was positively related to central arterial stiffness, as measured by carotid‐femoral pulse wave velocity, in patients with hypertension or with CAD. However, this same measure of arterial stiffness was unrelated to leptin in healthy young adults in 1 study 7 and inversely related with leptin in a recent report from the Framingham Third Generation cohort. 35 Yet both central and peripheral artery stiffness are associated with glucose dysregulation and greater cardiovascular risk. 36 , 37 , 38 It is clear that further research is needed to better understand the complex interrelationships among leptin, arterial stiffness, glucose regulation, and adiposity among post‐menopausal women.
In our sample of mostly post‐menopausal women, there was little impact of menopausal status, estradiol, or SHBG on the observed relationships between the adipokines and subclinical CVD. Menopausal status, established by bleeding pattern criteria, met our criteria (P<0.1) for inclusion as a covariate in 4 multivariable models evaluating the associations of adiponectin and leptin with cIMT and AD (Model 4s in Table 3); however, it was not a statistically significant covariate in any of the fully adjusted models. The patterns of association of SHBG and estradiol with the adipokines were similar to the pattern observed with menopausal status. SHBG values were increasingly higher and estradiol values were increasingly lower from the bottom to top quartiles of adiponectin (Table 1), with a reverse pattern noted across quartiles of leptin (Table 2), which is consistent with prior research. 39 However, neither measure met criteria for inclusion in the final, fully adjusted models for either adipokine.
Study Limitations and Strengths
This study has limitations; data are cross‐sectional so we cannot determine temporal relationships or whether changes in leptin or adiponectin over time affect progression of IMT, arterial stiffness, or the other subclinical CVD measures. Additional data collection in SWAN at visit 15 included repeat assessments of several markers of subclinical CVD in a subset of participants and we also have ongoing adjudication of cardiovascular events, so we will be able to examine longitudinal relationships of adipokines and cardiovascular risk in future analyses. Results are not generalizable to women younger or older than the age range of our participants or from other racial/ethnic backgrounds, or to men. We used total adiponectin rather than HMW adiponectin in analyses, which might be interpreted as a limitation. HMW adiponectin is more biologically active than total adiponectin and increases with improved insulin sensitivity so may be a better measurement than total adiponectin for insulin sensitivity and CVD 40 ; however, levels of venous total and HMW adiponectin levels are highly correlated in humans and declines in both forms of adiponectin are associated with type 2 diabetes mellitus and an increased CVD risk. 41
Our study also has important strengths. SWAN includes a well‐measured and comprehensive set of cardiovascular risk factors, and our study design enabled us to examine associations of adipokines with several indicators of subclinical CVD among women before onset of clinical manifestations of CVD.
Conclusions
We observed a consistent association of greater subclinical atherosclerosis with lower levels of adiponectin and higher levels of leptin, 2 important inflammatory pathways that may underlie relationships between adiposity and CVD risk. Associations for leptin are mostly robust to risk factor adjustment whereas associations for adiponectin can be explained by known CVD risk factors. The data also show that low adiponectin is associated with greater arterial stiffness in our sample, with a surprising trend toward less arterial stiffness associated with higher leptin particularly in women with diabetes mellitus or obesity. Data are cross‐sectional only; future investigations should evaluate whether adverse changes in these adipocyte‐derived hormones contribute to a worsening of subclinical atherosclerotic vascular disease over time.
Sources of Funding
The SWAN study has grant support from the National Institutes of Health, Department of Health and Human Services (DHHS), through the National Institute on Aging, the National Institute of Nursing Research and the National Institutes of Health Office of Research on Women's Health (Grants U01NR004061; U01AG012505, U01AG012535, U01AG012531, U01AG012539, U01AG012546, U01AG012553, U01AG012554, U01AG012495). The Chicago site of SWAN also is supported by the Charles J. and Margaret Roberts Trust. This publication also was supported in part by the National Center for Research Resources and the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through University of California San Francisco (UCSF)‐Clinical and Translational Science Institute (CTSI) Grant Number UL1 RR024131 and UMN‐CTSI Grant Number UL1 TR002494, and by the Program in Health Disparities Research and the Applied Clinical Research Program at the University of Minnesota. The contents of this article are solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Aging, National Institute of Nursing Research, Office of Research on Women's Health, National Center for Research Resources, NCATS, or the National Institutes of Health. None of the funders had any involvement in the study design, the collection, analysis, and interpretation of data, in writing this article, or in the decision to submit this article for publication.
Disclosures
None.
Acknowledgments
We thank the study staff at each site and all the women who participated in the SWAN study). SWAN: Clinical Centers: University of Michigan, Ann Arbor—Siobán Harlow, Principal Investigator (PI) 2011 to present, MaryFran Sowers, PI 1994 to 2011; Massachusetts General Hospital, Boston, MA—Joel Finkelstein, PI 1999 to present; Robert Neer, PI 1994 to 1999; Rush University, Rush University Medical Center, Chicago, IL—Howard Kravitz, PI 2009 to present; Lynda Powell, PI 1994–2009; University of California, Davis/Kaiser—Ellen Gold, PI; University of California, Los Angeles, CA—Gail Greendale, PI; Albert Einstein College of Medicine, Bronx, NY—Carol Derby, PI 2011 to present, Rachel Wildman, PI 2010 to 2011; Nanette Santoro, PI 2004 to 2010; University of Medicine and Dentistry—New Jersey Medical School, Newark, NJ—Gerson Weiss, PI 1994 to 2004; and the University of Pittsburgh, Pittsburgh, PA—Karen Matthews, PI.
National Institutes of Health Program Office: National Institute on Aging, Bethesda, MD—Chhanda Dutta 2016 to present; Winifred Rossi 2012–2016; Sherry Sherman 1994–2012; Marcia Ory 1994 to 2001; National Institute of Nursing Research, Bethesda, MD—Program Officers.
Central Laboratory: University of Michigan, Ann Arbor—Daniel McConnell (Central Ligand Assay Satellite Services).
Mancuso Laboratory: University of Michigan, Ann Arbor—Peter Mancuso (Adipokine Assay Laboratory).
Coordinating Center: University of Pittsburgh, Pittsburgh, PA—Maria Mori Brooks, PI 2012 to present; Kim Sutton‐Tyrrell, PI 2001 to 2012; New England Research Institutes, Watertown, MA—Sonja McKinlay, PI 1995 to 2001.
Steering Committee: Susan Johnson, Current Chair; Chris Gallagher, Former Chair.
(J Am Heart Assoc. 2021;10:e019173. DOI: 10.1161/JAHA.120.019173.)
For Sources of Funding and Disclosures, see page 13.
References
- 1. Mattu HS, Randeva HS. Role of adipokines in cardiovascular disease. J Endocrinol. 2013;216:T17–T36. DOI: 10.1530/JOE-12-0232. [DOI] [PubMed] [Google Scholar]
- 2. Hasan‐Ali H, Abd El‐Mottaleb NA, Hamed HB, Abd‐Elsayed A. Serum adiponectin and leptin as predictors of the presence and degree of coronary atherosclerosis. Coron Artery Dis. 2011;22:264–269. DOI: 10.1097/MCA.0b013e3283452431. [DOI] [PubMed] [Google Scholar]
- 3. Hayashi M, Shibata R, Takahashi H, Ishii H, Aoyama T, Kasuga H, Yamada S, Ohashi K, Maruyama S, Matsuo S, et al. Association of adiponectin with carotid arteriosclerosis in predialysis chronic kidney disease. Am J Nephrol. 2011;34:249–255. DOI: 10.1159/000330178. [DOI] [PubMed] [Google Scholar]
- 4. Shanker J, Rao VS, Ravindran V, Dhanalakshmi B, Hebbagodi S, Kakkar VV. Relationship of adiponectin and leptin to coronary artery disease, classical cardiovascular risk factors and atherothrombotic biomarkers in the IARS cohort. Thromb Haemost. 2012;108:769–780. DOI: 10.1160/TH12-04-0263. [DOI] [PubMed] [Google Scholar]
- 5. Tsai JP, Hsu BG, Lee CJ, Hsieh YH, Chen YC, Wang JH. Serum leptin is a predictor for central arterial stiffness in hypertensive patients. Nephrology. 2017;22:783–789. DOI: 10.1111/nep.12859. [DOI] [PubMed] [Google Scholar]
- 6. Tsai JP, Wang JH, Chen ML, Yang CF, Chen YC, Hsu BG. Association of serum leptin levels with central arterial stiffness in coronary artery disease patients. BMC Cardiovasc Disord. 2016;16:80. DOI: 10.1186/s12872-016-0268-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahiante BO, Smith W, Lammertyn L, Schutte AE. Leptin and the vasculature in young adults: the African‐PREDICT study. Eur J Clin Invest. 2019;49:e13039. DOI: 10.1111/eci.13039. [DOI] [PubMed] [Google Scholar]
- 8. Nilsson PM, Engström G, Hedblad B, Frystyk J, Persson MM, Berglund G, Flyvbjerg A. Plasma adiponectin levels in relation to carotid intima media thickness and markers of insulin resistance. Arterioscler Thromb Vasc Biol. 2006;26:2758–2762. DOI: 10.1161/01.ATV.0000249638.01416.4b. [DOI] [PubMed] [Google Scholar]
- 9. Lo J, Dolan SE, Kanter JR, Hemphill LC, Connelly JM, Lees RS, Grinspoon SK. Effects of obesity, body composition, and adiponectin on carotid intima‐media thickness in healthy women. J Clin Endocrinol Metab. 2006;91:1677–1682. DOI: 10.1210/jc.2005-2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sowers M, Zheng H, Tomey K, Karvonen‐Gutierrez C, Jannausch M, Li X, Yosef M, Symons J. Changes in body composition in women over six years at midlife: ovarian and chronological aging. J Clin Endocrinol Metab. 2007;92:895–901. DOI: 10.1210/jc.2006-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perry AC, Martin L. Race differences in obesity and its relationship to the sex hormone milieu. Horm Mol Biol Clin Investig. 2014;19:151–161. [DOI] [PubMed] [Google Scholar]
- 12. Im EO, Ham OK, Chee E, Chee W. Racial/ethnic differences in cardiovascular symptoms in four major racial/ethnic groups of midlife women: a secondary analysis. Women Health. 2015;55:525–547. DOI: 10.1080/03630242.2015.1022813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gurka MJ, Vishnu A, Santen RJ, DeBoer MD. Progression of metabolic syndrome severity during the menopausal transition. J Am Heart Assoc. 2016;5:e003609. DOI: 10.1161/JAHA.116.003609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim C, Diez‐Roux AV, Nettleton JA, Polak JF, Post WS, Siscovick DS, Watson KE, Vahratian AM. Sex differences in subclinical atherosclerosis by race/ethnicity in the Multi‐Ethnic Study of Atherosclerosis. Am J Epidemiol. 2011;174:165–172. DOI: 10.1093/aje/kwr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hao G, Wang X, Treiber FA, Davis H, Leverett S, Su S, Kapuku G. Growth of carotid intima‐media thickness in black and white young adults. J Am Heart Assoc. 2016;5:e004147. DOI: 10.1161/JAHA.116.004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khan U, Wang D, Sowers MFR, Mancuso P, Everson‐Rose SA, Scherer PE, Wildman RP. Race‐ethnic differences in adipokine levels: the Study of Women’s Health Across the Nation (SWAN). Metabolism. 2012;61:1261–1269. DOI: 10.1016/j.metabol.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lawlor DA, Davey Smith G, Ebrahim S, Thompson C, Sattar N. Plasma adiponectin levels are associated with insulin resistance, but do not predict future risk of coronary heart disease in women. J Clin Endocrinol Metab. 2005;90:5677–5678. DOI: 10.1210/jc.2005-0825. [DOI] [PubMed] [Google Scholar]
- 18. Kizer JR, Benkeser D, Arnold AM, Mukamal KJ, Ix JH, Zieman SJ, Siscovick DS, Tracy RP, Mantzoros CS, deFilippi CR, et al. Associations of total and high‐molecular‐weight adiponectin with all‐cause and cardiovascular mortality in older persons: the Cardiovascular Health Study. Circulation. 2012;126:2951–2961. DOI: 10.1161/CIRCULATIONAHA.112.135202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sook Lee E, Park SS, Kim E, Sook Yoon Y, Ahn HY, Park CY, Ho Yun Y, Woo OHS. Association between adiponectin levels and coronary heart disease and mortality: a systematic review and meta‐analysis. Int J Epidemiol. 2013;42:1029–1039. DOI: 10.1093/ije/dyt087. [DOI] [PubMed] [Google Scholar]
- 20. Sowers MF, Crawford S, Sternfeld B, Morganstein D, Gold EB, Greendale GA, Evans D, Neer R, Matthews K, Sherman S, et al. SWAN: A multicenter, multiethnic, community‐based cohort study of women and the menopausal transition. In: Lobo R, Kelsey J, Marcus R, eds. Menopause: Biology and Pathobiology. San Diego, CA: Academic Press; 2000:175–188. [Google Scholar]
- 21. Baecke JAH, Burema J, Fritjers JER. A short questionnaire for the measurement of habitual physical activity in epidemiological studies. Am J Clin Nutr. 1982;36:936–942. DOI: 10.1093/ajcn/36.5.936. [DOI] [PubMed] [Google Scholar]
- 22. Sternfeld B, Ainsworth BA, Quesenberry CP Jr. Physical activity patterns in a diverse population of women. Prev Med. 1999;28:313–323. DOI: 10.1006/pmed.1998.0470. [DOI] [PubMed] [Google Scholar]
- 23. Wendelhag I, Gustavsson T, Suurkula M, Berglund G, Wikstrand J. Ultrasound measurement of wall thickness in the carotid artery: fundamental principles and description of a computerized analysing system. Clin Physiol. 1991;11:565–577. DOI: 10.1111/j.1475-097X.1991.tb00676.x. [DOI] [PubMed] [Google Scholar]
- 24. Thompson T, Sutton‐Tyrrell K, Wildman RP, Kao A, Fitzgerald SG, Shook B, Tracy RP, Kuller LH, Brockwell S, Manzi S. Progression of carotid intima‐media thickness and plaque in women with systemic lupus erythematosus. Arthritis Rheum. 2008;58:835–842. DOI: 10.1002/art.23196. [DOI] [PubMed] [Google Scholar]
- 25. Thurston RC, El Khoudary SR, Derby CA, Barinas‐Mitchell EJ, Lewis T, McClure C, Matthews KA. Low socioeconomic status over 12 years and subclinical cardiovascular disease: the Study of Women’s Health Across the Nation. Stroke. 2014;45:954–960. DOI: 10.1161/STROKEAHA.113.004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sutton‐Tyrrell K, Kuller LH, Matthews KA, Holubkov R, Patel A, Edmundowicz D, Newman A. Subclinical atherosclerosis in multiple vascular beds: an index of atherosclerotic burden evaluated in postmenopausal women. Atherosclerosis. 2002;160:407–416. DOI: 10.1016/S0021-9150(01)00591-3. [DOI] [PubMed] [Google Scholar]
- 27. Choo J, Ueshima H, Jang Y, Sutton‐Tyrrell K, El‐Saed A, Kadowaki T, Takamiya T, Okamura T, Ueno Y, Nakamura Y, et al. Difference in carotid intima‐media thickness between Korean and Japanese men. Ann Epidemiol. 2008;18:310–315. DOI: 10.1016/j.annepidem.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yamashina A, Tomiyama H, Takeda K, Tsuda H, Arai T, Hirose K, Koji Y, Hori S, Yamamoto Y. Validity, reproducibility, and clinical significance of noninvasive brachial‐ankle pulse wave velocity measurement. Hypertens Res. 2002;25:359–364. DOI: 10.1291/hypres.25.359. [DOI] [PubMed] [Google Scholar]
- 29. Pilz S, Horejsi R, Möller R, Almer G, Scharnagl H, Stojakovic T, Dimitrova R, Weihrauch G, Borkenstein M, Maerz W, et al. Early atherosclerosis in obese juveniles is associated with low serum levels of adiponectin. J Clin Endocrinol Metab. 2005;90:4792–4796. DOI: 10.1210/jc.2005-0167. [DOI] [PubMed] [Google Scholar]
- 30. Iglseder B, Mackevics V, Stadlmayer A, Tasch G, Ladurner G, Paulweber B. Plasma adiponectin levels and sonographic phenotypes of subclinical carotid artery atherosclerosis: data from the SAPHIR Study. Stroke. 2005;36:2577–2582. DOI: 10.1161/01.STR.0000190834.00284.fd. [DOI] [PubMed] [Google Scholar]
- 31. Lim HS, Tayebjee MH, Tan KT, Patel JV, Macfadyen RJ, Lip GY. Serum adiponectin in coronary heart disease: ethnic differences and relation to coronary artery disease severity. Heart. 2005;91:1605–1606. DOI: 10.1136/hrt.2004.047803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabbatini AR, Fontana V, Laurent S, Moreno H. An update on the role of adipokines in arterial stiffness and hypertension. J Hypertens. 2015;33:435–444. DOI: 10.1097/HJH.0000000000000444. [DOI] [PubMed] [Google Scholar]
- 33. El Khoudary SR, Barinas‐Mitchell E, White J, Sutton‐Tyrrell K, Kuller LH, Curb JD, Shin C, Ueshima H, Masaki K, Evans RW, et al. Adiponectin, systolic blood pressure, and alcohol consumption are associated with more aortic stiffness progression among apparently healthy men. Atherosclerosis. 2012;225:475–480. DOI: 10.1016/j.atherosclerosis.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Youn JC, Kim C, Park S, Lee SH, Kang SM, Choi D, Son NH, Shin DJ, Jang Y. Adiponectin and progression of arterial stiffness in hypertensive patients. Int J Cardiol. 2013;163:316–319. DOI: 10.1016/j.ijcard.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 35. Zachariah JP, Hwang S, Hamburg NM, Benjamin EJ, Larson MG, Levy D, Vita JA, Sullivan LM, Mitchell GF, Vasan RS. Circulating adipokines and vascular function: cross‐sectional associations in a community‐based cohort. Hypertension. 2016;67:294–300. DOI: 10.1161/HYPERTENSIONAHA.115.05949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Henry RMA, Kostense PJ, Spijkerman AMW, Dekker JM, Nijpels G, Heine RJ, Kamp O, Westerhof N, Bouter LM, Stehouwer CD; Hoorn Study . Arterial stiffness increases with deteriorating glucose tolerance status. the Hoorn Study. Circulation. 2003;107:2089–2095. DOI: 10.1161/01.CIR.0000065222.34933.FC. [DOI] [PubMed] [Google Scholar]
- 37. Schram MT, Henry RMA, van Dijk RAJM, Kostense PJ, Dekker JM, Nijpels G, Heine RJ, Bouter LM, Westerhof N, Stehouwer CD. Increased central artery stiffness in impaired glucose metabolism and type 2 diabetes: the Hoorn Study. Hypertension. 2004;43:176–181. DOI: 10.1161/01.HYP.0000111829.46090.92. [DOI] [PubMed] [Google Scholar]
- 38. Shin JY, Lee HR, Lee DC. Increased arterial stiffness in healthy subjects with high‐normal glucose levels and in subjects with pre‐diabetes. Cardiovasc Diabetol. 2011;10:30. DOI: 10.1186/1475-2840-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Karim R, Stanczyk FZ, Brinton RD, Rettberg J, Hodis HN, Mack WJ. Association of endogenous sex hormones with adipokines and ghrelin in postmenopausal women. J Clin Endocrinol Metab. 2015;100:508–515. DOI: 10.1210/jc.2014-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horáková D, Azeem K, Benešová R, Pastucha D, Horák V, Dumbrovská L, Martínek A, Novotný D, Švagera Z, Hobzová M, et al. Total and high molecular weight adiponectin levels and prediction of cardiovascular risk in diabetic patients. Int J Endocrinol. 2015;2015:545068. DOI: 10.1155/2015/545068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, et al. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione‐mediated improvement in insulin sensitivity. J Biol Chem. 2004;279:12152–12162. DOI: 10.1074/jbc.M311113200. [DOI] [PubMed] [Google Scholar]