

Abstract
Background
Recent accumulating evidence suggests that toll‐like receptor 9 (TLR9) is involved in the pathogenesis of cardiovascular diseases. However, its role in pulmonary hypertension remains uncertain. We hypothesized that TLR9 is involved in the development of pulmonary hypertension.
Methods and Results
A rat model of monocrotaline‐induced pulmonary hypertension was used to investigate the effects of TLR9 on hemodynamic parameters, vascular remodeling, and survival. Monocrotaline‐exposed rats significantly showed increases in plasma levels of mitochondrial DNA markers, which are recognized by TLR9, TLR9 activation in the lung, and interleukin‐6 mRNA level in the lung on day 14 after monocrotaline injection. Meanwhile, monocrotaline‐exposed rats showed elevated right ventricular systolic pressure, total pulmonary vascular resistance index and vascular remodeling, together with macrophage accumulation on day 21. In the preventive protocol, administration (days −3 to 21 after monocrotaline injection) of selective (E6446) or nonselective TLR9 inhibitor (chloroquine) significantly ameliorated the elevations of right ventricular systolic pressure and total pulmonary vascular resistance index as well as vascular remodeling and macrophage accumulation on day 21. These inhibitors also significantly reduced NF‐κB activation and interleukin‐6 mRNA levels to a similar extent. In the short‐term reversal protocol, E646 treatment (days 14–17 after monocrotaline injection) almost normalized NF‐κB activation and interleukin‐6 mRNA level, and reduced macrophage accumulation. In the prolonged reversal protocol, E6446 treatment (days 14–24 after monocrotaline injection) reversed total pulmonary vascular resistance index and vascular remodeling, and improved survival in monocrotaline‐exposed rats.
Conclusions
TLR9 is involved in the development of pulmonary hypertension concomitant via activation of the NF‐κB‒IL‐6 pathway. Inhibition of TLR9 may be a novel therapeutic strategy for pulmonary hypertension.
Keywords: interleukin‐6, perivascular inflammation, pulmonary hypertension, toll‐like receptor 9, vascular remodeling
Subject Categories: Pulmonary Biology, Inflammation
Nonstandard Abbreviations and Acronyms
- IL‐6
interleukin‐6
- mtDNA
mitochondrial DNA
- NF‐κB
nuclear factor κB
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary arterial smooth muscle cell
- PH
pulmonary hypertension
- TLR9
toll‐like receptor 9
Clinical Perspective
What Is New?
Better understanding of the pathogenesis of pulmonary hypertension (PH) and development of novel therapeutic strategies are needed.
Toll‐like receptor 9 is involved in the development of PH concomitant via activation of the NF‐κB‒IL‐6 pathway.
Inhibition of toll‐like receptor 9 may be a novel therapeutic strategy for PH.
What Are the Clinical Implications?
Given the lack of established treatments for perivascular inflammation in PH, treatment targeting toll‐like receptor 9 can be a novel strategy for PH.
Pulmonary hypertension (PH) is characterized by progressive increases in pulmonary vascular resistance and pressure, resulting in right ventricular failure. 1 Conventional pulmonary vasodilators including prostacyclin and its analogues, soluble guanylate cyclase stimulator, phosphodiesterase type 5 inhibitors, and/or endothelin receptor antagonists, have successfully delayed clinical aggravation of PH and improved survival. 2 However, the prognosis of advanced cases remains poor. Therefore, better understanding of the pathogenesis of PH and development of novel therapeutic strategies are needed.
Previous animal studies reported that inflammation precedes vascular remodeling in experimental PH, suggesting that altered immunity is a cause rather than a consequence of this disorder. However, the underlying mechanisms that initiate inflammatory and the immune processes involved in the progression of PH have not been fully elucidated. Recent study has revealed that cellular injury can release endogenous damaged mitochondrial DNA (mtDNA) that activates toll‐like receptors (TLRs) to induce inflammatory responses. 3 Recent reports also indicate that mtDNA production and the resultant inflammation via activation of TLR9 play an important role in the pathogenesis of cardiovascular diseases. 4 , 5 , 6 , 7 In mice with transverse aortic constriction, damaged mtDNA released by hemodynamic stress leads to TLR9‐mediated inflammatory responses that may induce left‐sided heart failure. 7 The circulating mtDNA activates TLR9, which contributes to elevate systemic blood pressure in spontaneous hypertensive rats. 6 More recent study indicates that TLR9 activation stimulates interleukin‐6 (IL‐6), a multifunctional pro‐inflammatory cytokine in rat pulmonary endothelial cells, causing proliferation and migration of rat pulmonary arterial smooth muscle cells (PASMCs). 8 However, very little is known about the role of this pathogenic mechanism in PH.
In the present study, we hypothesized that mtDNA might activate TLR9, and stimulate inflammatory cell accumulation and IL‐6 production in the lung, leading to PH progression. To test our hypothesis, we evaluated the plasma levels of mtDNA markers as well as TLR9 activation and IL‐6 level in lung during the course of development of PH in an experimental PH rat model, and examined whether chronic inhibition of TLR9 could attenuate PH accompanied by suppression of the augmented IL‐6 pathway in this model.
Methods
The data that support the findings of this study are available from the corresponding author on request.
Animal Experiments
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Kyushu University, Japan, and all animal procedures were performed in compliance with the principles of the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011).
Monocrotaline‐Induced PH Rat Model
Adult male Sprague‐Dawley rats weighing 200 to 250 g (Japan SLC, Hamamatsu, Japan) were given a single subcutaneous injection of 60 mg/kg of monocrotaline (Sigma‐Aldrich, Saint Louis, MO) to induce PH. 9 We used male rats because there is a more aggressive course of MCT‐induced PH in male rats compared with female rats. A total of 165 rats were used, including 41 for survival study, 72 for hemodynamic, histology, and immunohistochemistry studies, 52 for reverse transcription–polymerase chain reaction, and Western blot analysis.
Drugs
Monocrotaline (Sigma‐Aldrich, Saint Louis, MO) was dissolved in 1N HCl, neutralized with 1N NaOH, and diluted with distilled water to 20 mg/mL. E6446 provided by Eisai Inc. (São Paulo, SP, Brazil) 10 , 11 and chloroquine (Sigma‐Aldrich, Saint Louis, MO) were dissolved in drinking water and PBS, respectively. 12
Pharmacological Blockade of TLR9
To examine the effect of TLR9 on PAH in rats, we tested 2 types of TLR9 inhibitors, E6446 provided by Eisai Inc. (São Paulo, SP, Brazil) 10 , 11 and chloroquine (Sigma‐Aldrich, Saint Louis, MO). Chloroquine impairs acidification of lysosomes to inhibit the interaction between nonmethylated cytidine‐phosphate‐guanosine (CpG) DNA and TLR9. The metabolic product of chloroquine, hydroxychloroquine, is used as a treatment for malaria and systemic lupus erythematosus in humans. 13 , 14 E6446 directly inhibits the binding between nonmethylated CpG and TLR9, and inhibits TLR9 more selectively than other TLRs. As an orally active TLR9 inhibitor, its specificity is maintained in vivo (up to 20 mg/kg per day). 10 , 11 Thus, we consider that E6446 used at the dose of 10 mg/kg in the present study is a reasonably selective inhibitor for TLR9 in rats, as reported previously. 15
Hemodynamic Evaluation in Rats
Hemodynamic parameters were evaluated as described previously. 16 , 17 Briefly, a rat was anesthetized with isoflurane (1.5% in room air). An 18‐gauge BD Angiocath catheter (Becton Dickinson) with the tip at a 30‐degree angle was inserted into the right jugular vein and advanced into the right ventricle (RV) for measurement of RV systolic pressure (RVSP) as a marker of systolic pulmonary arterial pressure. 18 The catheter was connected to a fluid‐filled transducer (DX‐360, Nihon Kohden, Japan). A microtip P‐V catheter (FTH‐1912B8018, Transonic Inc., Ithaca, NY) was inserted into the right carotid artery and then advanced into the left ventricle (LV). The RVSP, left ventricle systolic pressure, heart rate, and cardiac output were continuously recorded using ML880/9 PowerLab 16/30 (AD Instruments, Dunedin, New Zealand), an ADVantage P‐V control unit (v 5.0) (FY097B, Transonic Inc.), and a dedicated laboratory computer system. Cardiac index (CI) was calculated by dividing cardiac output by body weight. The ratio of RVSP to CI (RVSP/CI) yielded a surrogate marker of total pulmonary vascular resistance index (TPRI). 16 , 19 After hemodynamic measurements, the rat was euthanized by exsanguination under isoflurane. The heart was removed for assessment of RV hypertrophy, and the lungs were collected for histological evaluation, immunoblot analysis, and reverse transcription–polymerase chain reaction. RV hypertrophy was expressed as a ratio of the RV weight to the LV weight plus septum weight (RV/LV+S). 16
Experimental Protocols
Protocol 1. Preventive Protocol
Chloroquine (50 mg/kg, intraperitoneal injection) or E6446 (10 mg/kg, oral administration in drinking water) was administered from 3 days before monocrotaline injection (day −3) through day 21 after monocrotaline injection, and rats were euthanized on day 21 in the following 4 groups; (1) age‐matched normal rats (normal), (2) monocrotaline alone, (3) monocrotaline with chloroquine, and (4) monocrotaline with E6446 (Figure 1).
Figure 1. Experimental protocols.
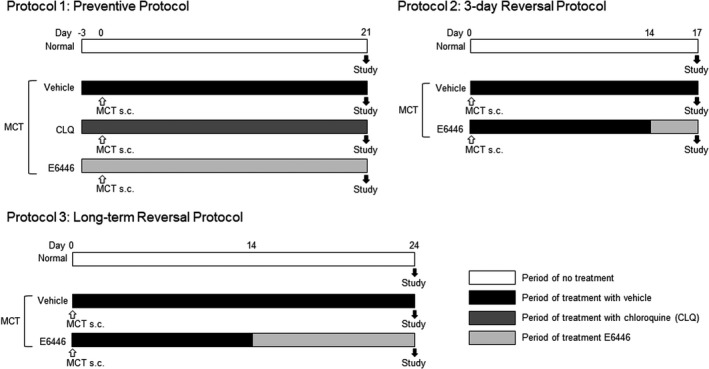
The protocol 1 aims to examine the preventive effect of a toll‐like receptor 9 (TLR9) inhibitor, E6446 on pulmonary hypertension (PH) in monocrotaline (MCT)‐exposed rats. The protocol 2 aims to examine the short‐term effect of TLR9 inhibition on inflammatory responses in the established PH in MCT‐exposed rats. The protocol 3 aims to examine the therapeutic effect of E6446 on the established PH in MCT‐exposed rats. CLQ: chloroquine; MCT: monocrotaline; and TLR9: toll‐like receptor 9.
Protocol 2. Three‐Day Reversal Protocol (Short‐Term Administration)
E6446 (10 mg/kg, drinking water) was administered from day 14 to day 17 after monocrotaline injection, and rats were euthanized on day 17 in 3 groups; (1) age‐matched normal rats (normal), (2) monocrotaline alone, and (3) monocrotaline with E6446. This protocol is used to examine a short‐term effect of TLR9 inhibition on inflammatory responses in monocrotaline‐exposed rats.
Protocol 3. Long‐Term Reversal Protocol (Prolonged Administration)
E6446 (10 mg/kg, drinking water) was administered from day 14 to day 24 after monocrotaline injection, and rats were euthanized on day 24 in 3 groups: (1) age‐matched normal rats (normal), (2) monocrotaline alone, and (3) monocrotaline with E6446. This protocol is used to examine the therapeutic effect of a TLR9 inhibitor, E6446 on the established PH in monocrotaline‐exposed rats.
Survival Study
In the preventive protocol, survival of rats was followed until day 35 after monocrotaline injection in monocrotaline alone and monocrotaline with E6446 groups. In the reversal protocol, survival was followed until day 28 after monocrotaline injection in those groups.
Mitochondrial DNA Isolation and Purification
To investigate the increase of mitochondrial DNA (mtDNA) in plasma, 6 rats were injected with monocrotaline on day 0, and plasma samples were collected after euthanization on days 1, 7, and 14. As controls, rats were injected with PBS on day 0 and euthanized on day 1. Arterial blood was collected from the carotid artery in tubes containing EDTA. Blood was immediately centrifuged at 1500g for 15 minutes at 4°C. Plasma was then stored in multiple aliquots at −80°C until analysis. Circulating DNA was extracted from plasma samples and purified using the Plasma/Serum Cell‐Free Circulating DNA Purification Mini Kit (Norgen Biotek Corp., Canada). Mitochondrial DNA was amplified and quantified using SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad, Hercules, CA). The primers (Invitrogen, Grand Island, NY) used to amplify mtDNA were: NADH dehydrogenase subunit 4 (ND4) (forward 5’‐CTC CGC AAC AGA ACT AAT‐3’ and reverse 5’‐GTT GAG TGT TCC TAT TGA GT‐3’), cytochrome c oxidase subunit 2 (COX2) (forward 5’‐GCT GTC ATT CTT ATT CTA A‐3’ and reverse 5’‐GGA TTA TGT AGG AGT CAA‐3’) and ATPase 6 (ATP6) (forward 5’‐CGA AAC TAT CAG CCT ATT‐3’ and reverse 5’‐AGT AGA AGT AGA ATA ATA AAT GTA A‐3’). Primer sequences have no significant homology with DNA found in any bacterial species published on BLAST. Real‐time results were presented as the inverse of cycle threshold for gene amplification. 20
Histopathological Analysis
After catheterization, the rat was euthanized by exsanguination under overdose of isoflurane (2%–5% in room air) and an intraperitoneal injection of a lethal dose of pentobarbital sodium (30 mg/kg), and then the lungs were removed. The left lobe of the lungs was inflated with PBS containing 1% formalin plus 0.5% agarose via the trachea at a constant pressure of 20 cm H2O, and was then fixed in 10% formalin neutral buffer solution overnight. After embedding in paraffin, 5‐μm‐thick slices obtained at the level of the hilum were subjected to Elastin van Gieson staining and immunohistochemical staining. 16
Medial Wall Thickness
Small pulmonary arteries with outer diameters of 50 to 100 µm were analyzed. They were used for the evaluation of the medial wall thickness. 21 , 22 The medial wall thickness index was obtained using the following formula: medial wall thickness =(outer diameter−inner diameter)/outer diameter. At least 10 arteries were analyzed, and the mean medial wall thickness was obtained for each animal. The analysis was performed in a blinded manner.
Immunohistochemical Analysis
All sections were incubated with primary antibody reactive to smooth muscle actin (1:500; M085101; Agilent DAKO, Santa Clara, CA), Ki67 (1:400; RM‐9106; Thermo Scientific K.K., Tokyo, Japan), CD68 (1:100; ab31630; Abcam), nuclear factor kappa B (NF‐κB; 1:200; MAB3026; Merck KGaA, Darmstadt, Germany), and TLR9 polyclonal antibody (1:200; PA5‐20203; Thermo Scientific K.K., Tokyo, Japan) at 4°C overnight. Sections were then incubated with biotinylated secondary antibody before horseradish peroxidase–labeled streptavidin. 4
Muscularization in Small Pulmonary Arteries
Pulmonary arteries with outer diameters of 50 µm or less were analyzed. 23 Nonmuscular, partially muscular, and fully muscular (FM) arterioles were defined by positive staining of α‐smooth muscle actin in <25%, 25% to 75%, and >75%, respectively, of the circumference of the arteriole. At least 70 arterioles were evaluated for each specimen in a blinded manner.
Cell Proliferation in Media of Pulmonary Arteries
Proliferative cells were detected by immunostaining by anti‐Ki67 antibody. 16 The number of Ki67‐positive cells in the media of pulmonary arteries in 15 random fields was counted at a magnification of ×400.
Macrophage Accumulation
Macrophages were detected by immunostaining by anti‐CD68 antibody. 24 The number of CD68‐positive macrophages in 15 random fields was counted at a magnification of ×400.
NF‐κB Activity
The redox‐sensitive transcription factor, nuclear factor κB (NF‐κB), is known as one of the key inflammatory markers involved in the downstream of the TLR9 pathway. 25 Immunohistochemical staining of NF‐κB was performed using the Vectastain Universal Quick kit (Vector Laboratories). 16 The αp65 mAb of NF‐κB, which recognizes an epitope on the p65 subunit that is masked by the bound inhibitor of κB, detects activated NF‐κB. The numbers of NF‐κB‒positive cells were counted in 20 random fields at a magnification of ×400. The analysis was performed in a blinded manner.
Reverse Transcription–Polymerase Chain Reaction Analysis
Total RNAs were extracted from whole lung using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and mRNA expression level was determined by reverse transcription–polymerase chain reaction. 24 The ReverTra Ace qPCR Kit (Toyobo, Tokyo, Japan) and SYBR Premix Ex Taq (TaKaRa, Tokyo, Japan) were used for reverse transcription and amplification, respectively. We purchased polymerase chain reaction (PCR) primers and probes from TaKaRa. A 10‐μL RT reaction mixture containing 200 ng of total RNA, oligo dT primer, random primer, and Moloney murine leukemia virus reverse transcriptase was subjected to transverse transcription. An aliquot of RT product was diluted with water, and 25 ng of the cDNA was subjected to real‐time PCR analysis using SYBR Premix Ex TaqII and 7500 real‐time PCR system (Applied biosystems). The thermal cycle consisted of an initial denaturation at 95°C for 30 seconds followed by 40 cycles of denaturation at 95°C for 5 seconds and annealing at 60°C for 34 seconds. We analyzed the melting curve of the PCR product at the end of the real‐time PCR analysis and identified the single peak of the melting profile. Electrophoresis confirmed that each PCR product showed a single band with the expected molecular size. We used the ΔΔCt method to analyze the fluorescence data using 18s as an internal control. The primers that were used to amplify mRNA were interleukin‐6 (Il‐6; forward 5’‐ATT GTA TGA ACA GCG ATG ATG CAC‐3’ and reverse 5’ ‐CCA GGT AGA AAC GGA ACT CCA GA‐3’) and 18s (forward 5’‐AAG TTT CAG CAC ATC CTG CGA GTA‐3’ and reverse 5’‐TTG GTG AGG TCA ATG TCT GCT TTC‐3’). 24
Western Blotting
The lungs were frozen at −80°C and subsequently homogenized in 50 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 0.5% (vol/vol) Nonidet P‐40, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 0.5 mmol/L Na3VO4, 10 µg/mL leupeptin, 10 µg/mL aprotinin, 5 µmol/L microcystin‐LR, 10 µmol/L calpain inhibitor, and 10 µmol/L 4‐aminidophenylmethane sulfonyl fluoride. 26 The protein concentration of the lysate was determined using a Coomassie protein assay kit (Pierce, Rockford, IL) with bovine serum albumin as standard. Equal amounts of total proteins (10 µg) were separated on 10% (wt/vol) polyacrylamide gels for SDS‐PAGE and transferred to a polyvinylidene difluoride membrane (0.2 µm pore size; Bio‐Rad, Hercules, CA). The membranes were blocked with 5% (wt/vol) skim milk in 20 mmol/L Tris‐HCl, pH 7.5, 150 mmol/L NaCl, and 0.05% (vol/vol) Tween 20 (Tween 20 containing Tris‐buffered saline) for 1 hour at room temperature. The membranes were then incubated overnight at 4°C with primary antibodies: anti‐TLR9 (1:5000, Abcam, Cambridge, UK, ab134368), 6 phospho‐STAT3 (1: 3000, Cell Signaling Technology, Denver, MA, #9131), 27 , 28 anti‐STAT3 antibody (1:5000, Cell Signaling Technology, Denver, CO, #124H6), 27 and β‐actin (1:5000, Santa Cruz Biotechnology, Dallas, TX, #sc‐47778), followed by 1‐hour incubation with secondary antibodies conjugated to horseradish peroxidase (1 5000). The immune complexes were detected using an ECL select detection kit (GE Healthcare, Buckinghamshire, UK). Light emission was detected and analyzed with VersaDoc 5000 and the computer program Quantity One (Bio‐Rad). We measured the following protein levels: phospho‐STAT3, STAT3, TLR9, and β‐actin. The optical density of the phospho‐STAT3 band was normalized to that of the corresponding STAT3 band. 8 The optical density of the TLR9 band was normalized to that of the corresponding β‐actin band.
Human Lung Specimens
Lung specimens were obtained from autopsy material of a patient with severe PAH. Informed consent to use the tissue for research purposes had previously been obtained. The tissue was fixed in 10% formalin and paraffin‐embedded after routine processing. Slides were cut at 5 µm and stained with Elastin van Gieson staining and immunohistochemical staining.
Evaluation of Long‐Term Effect of TLR9 Inhibition on PH in SU5416/Hypoxia/Normoxia‐Exposed PH Model of Rats
Adult male Sprague‐Dawley rats weighing 180 to 220 g were injected subcutaneously with SU5416 (20 mg/kg) and exposed to hypoxia (10% O2) for 3 weeks. 16 They were returned to normoxia (21% O2) for an additional 5 weeks (total 8 weeks after SU5416 injection). E6446 (10 mg/kg, drinking water) was administered from week 3 to week 8 after SU5416 injection, and rats were euthanized on week 8 in 3 groups; (1) age‐matched normal rats (normal), (2) SU5416/hypoxia/normoxia alone, and (3) SU5416/hypoxia/normoxia with E6446. This protocol is used to examine the therapeutic effect of a TLR9 inhibitor, E6446, on the established PH in SU5416/hypoxia/normoxia‐exposed rats.
Statistical Analysis
Values are expressed as mean±SEM. ANOVA with Bonferroni post hoc test was used for comparisons among the experimental groups. The survival curves were analyzed by the Kaplan‐Meier method, and the statistical differences were analyzed by a log‐rank test. Differences were considered significant at P<0.05.
Results
Inflammatory Responses in Monocrotaline‐Exposed Rats
Circulating mitochondrial DNAs, ND4, ATP6, and COX2 in plasma samples increased significantly on days 7 and 14 (ND4) or day 14 (ATP6, COX2) in monocrotaline‐exposed rats compared with normal control rats injected with PBS (Figure 2A). TLR9, a receptor for mitochondrial DNAs, was expressed predominantly in endothelial cells, which were also positive for NF‐κB, in the monocrotaline‐exposed rats 3 weeks after monocrotaline injection (Figure S1). The similar localization was observed in the specimens of a patient with severe PAH (Figure S1). In monocrotaline‐exposed rats, the protein level of cleaved C‐terminal fragment of TLR9, an indicator of TLR9 activation, in the lung increased significantly on days 7 and 14, compared with control rats (Figure 2B). The mRNA levels of Il‐6 in lung increased significantly on day 14 after monocrotaline injection (Figure 2C). Concomitantly, immunostaining indicated that the number of CD68‐positive macrophages increased significantly in areas surrounding small pulmonary arteries on day 14 in monocrotaline‐exposed rats (Figure 2D).
Figure 2. Quantification of mitochondrial DNA genes, TLR9 activation, Il‐6 and macrophage accumulation in normal control and MCT‐exposed rats.
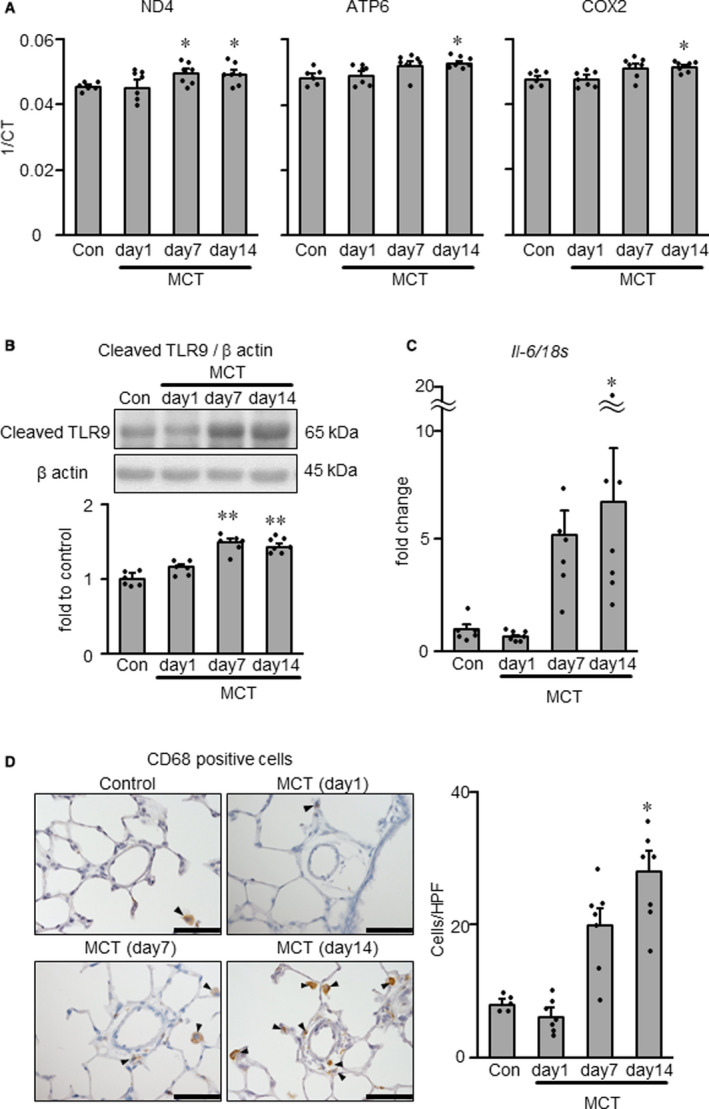
A, NADH dehydrogenase subunit 4 (ND4, left), ATP synthase 6 (ATP6, middle) and cytochrome C oxidase subunit II (COX2, right) in plasma of normal and MCT‐exposed rats at days 1, 7, and 14 after MCT injection, as measured by real‐time PCR. 1/CT denotes the reciprocal of the count where the sequence is detected, and is a direct function of gene concentration. B, Protein expression level of cleaved TLR9 in whole lung measured by Western blotting. C, Il‐6 mRNA expression level in whole lung measured by RT–PCR. D, Representative photomicrographs (left) and counts (right) of CD68‐positive macrophages (arrowheads) in the lung. Scale bars indicate 50 μm. Con indicates normal rats injected with PBS; and MCT, monocrotaline. N=5–7. *P<0.05 and **P<0.01 vs Con., according to a 1‐way ANOVA followed by Bonferroni post hoc test. HPF, high‐power field; RT‐PCR, reverse transcriptase polymerase chain reaction; and TLR9, toll‐like receptor 9.
Preventive Effects of Long‐Term Treatment With TLR9 Inhibitors on Hemodynamics and Survival in Monocrotaline‐Exposed Rats
In the monocrotaline group, RVSP (Figure 3A) and TPRI (Figure 3C) increased significantly while CI (Figure 3B) decreased significantly compared with the normal control group. The RV/LV+S weight ratio also increased significantly in the monocrotaline group compared with the normal control group (Figure 3D). The mean systemic arterial pressure and heart rate in the monocrotaline group did not differ significantly from those in the normal control group (data not shown). When treatment with chloroquine or E6446 was started 3 days before monocrotaline injection (protocol 1, Figure 1), chloroquine and E6446 both significantly ameliorated the increases in RVSP, TPRI, and RV/LV+S in monocrotaline‐exposed rats (Figure 3A, 3C, and 3D). Chloroquine and E4666 had no effects on CI (Figure 3B).
Figure 3. Preventive effects of long‐term treatment with toll‐like receptor 9 inhibitors on hemodynamic parameters and survival in MCT‐exposed rats.
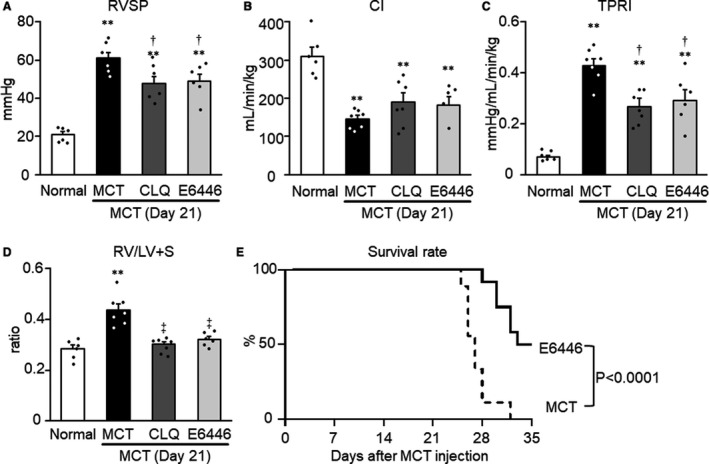
Rats were given a subcutaneous injection of MCT on day 0. Some MCT‐injected rats were treated with CLQ or E6446 from 3 days before to 21 days after MCT injection. Normal: normal controls. A, RVSP. B, CI. C, TPRI. D, right ventricular hypertrophy (RV/LV+S: weight ratio of RV free wall to the sum of LV free wall and septum) were measured. Data are expressed as mean±SEM. N=6 to 7. **P<0.01 vs Normal. † P<0.05 and ‡ P<0.01 vs MCT, based on a 1‐way ANOVA followed by Bonferroni post hoc test. E, Survival curves are shown for MCT‐exposed rats not treated (MCT, n=9) and those treated with E6446 (10 mg/kg per day, oral, n=12) from the day of MCT injection. The statistical differences were analyzed by a log‐rank test. CI indicates cardiac index; CLQ, indicates chloroquine; MCT, monocrotaline; RV/LV+S, ratio of the right ventricle weight to the left ventricle weight plus septum weight; RVSP, right ventricular systolic pressure; and TPRI, total pulmonary vascular resistance index.
All rats in the monocrotaline group died within 32 days after the injection of monocrotaline (Figure 3E). Autopsies of all deceased rats demonstrated severe RV hypertrophy and a significant degree of pleural effusion, ascites, and hepatic congestion. These manifestations indicated the existence of RV failure at the time of death. Thus, severe pulmonary hypertension appeared to be the main cause of death in these animals. Survival was significantly improved by administration of E6446 compared with untreated monocrotaline‐exposed rats (Figure 3E).
Preventive Effects of TLR9 Inhibitors on Pulmonary Artery Remodeling in Monocrotaline‐Induced Rats With PH
Medial wall hypertrophy in muscular arteries, a manifestation of vascular remodeling in PH, was evaluated in small arteries with diameters of 50 to 100 µm. The medial wall thickness was significantly increased on day 21 in the monocrotaline group compared with the normal control group (Figure 4A). Administration of chloroquine or E6446 significantly ameliorated thickening of the medial wall of pulmonary arteries in treated monocrotaline‐exposed rats compared with untreated monocrotaline‐exposed rats (Figure 4A).
Figure 4. Preventive effects of long‐term treatment with TLR9 inhibitors on pulmonary artery remodeling in MCT‐exposed rats.
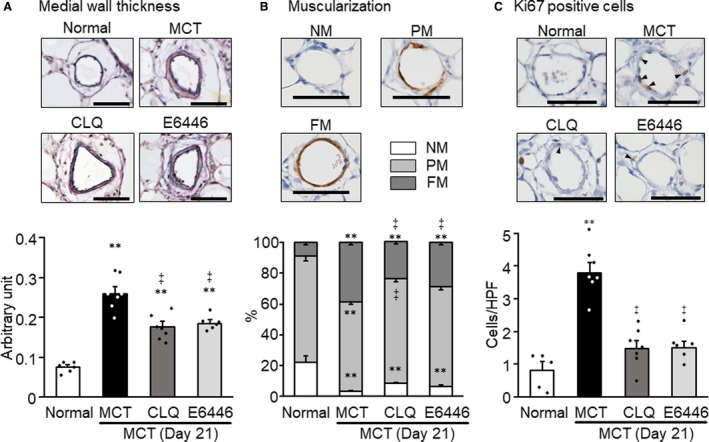
Rats were given a subcutaneous injection of MCT on day 0. Some MCT‐injected rats were treated with CLQ or E6446 from 3 days before to 21 days after MCT injection. Normal: normal controls. A, Representative photomicrographs of Verhoeff‐van Gieson stained pulmonary arteries with outer diameters of 50 to 100 µm, and comparison of medial wall thickness in 4 groups. B, Representative photomicrographs of pulmonary arterioles with outer diameters of 50 µm or less immunostained for β‐smooth muscle actin showing NM, PM, and TM patterns, and comparison in 4 groups. C, Representative photomicrographs of Ki67‐positive cells (arrowheads) in medial layer of pulmonary arteries with outer diameters of 50 µm or less, and comparison of counts in 4 groups. Bars indicate 50 µm. Data are expressed as mean±SEM. N=6 to 7. **P<0.01 vs Normal. ‡ P<0.01 vs MCT, according to a 1‐way ANOVA followed by Bonferroni post hoc test. CLQ indicates chloroquine; FM, fully muscular; HPF, high‐power field; MCT, monocrotaline; NM, nonmuscular; PM, partially muscular; TM, totally muscular; and TLR9, toll‐like receptor 9.
Muscularization of the arterioles, another manifestation of vascular remodeling in PH, was evaluated in arterioles with diameters of 50 µm or less. Immunohistochemical detection of α‐smooth muscle actin and quantitative evaluation of the degree of positive staining relative to the circumferential length revealed a significant increase in the percentage of partially and totally muscularized arterioles in the monocrotaline group on day 21 after monocrotaline injection (Figure 4B). Treatment with either chloroquine or E6446 significantly ameliorated the muscularization of pulmonary arterioles in monocrotaline‐exposed rats (Figure 4B). The number of Ki67‐positive, thus proliferative, cells in the media of muscular arteries increased in the monocrotaline group compared with the normal group (Figure 4C). Treatment with chloroquine or E6446 ameliorated the increase in the Ki67‐positive cells (Figure 4C).
Preventive Effects of TLR9 Inhibitors on Inflammatory Responses in Monocrotaline‐Induced Rats With PH
The infiltration of CD68‐positive macrophages and NF‐κB‐positive cells increased in the areas surrounding small pulmonary arteries in the monocrotaline group, compared with the normal group. Treatment with chloroquine or E6446 significantly reduced the accumulation of CD68‐positive macrophages and NF‐κB‐positive cells in the monocrotaline‐exposed rats (Figure 5A and 5B). The mRNA levels of Il‐6 increased significantly in the monocrotaline group, while this increase was significantly and almost completely inhibited by treatment with chloroquine or E6446 (Figure 5C). The level of phospho‐STAT3, a major downstream molecule of IL‐6, increased in lungs of monocrotaline‐exposed rats compared with normal rats. Treatment with E6446 significantly ameliorated the activation of STAT3 compared with the monocrotaline group (Figure 5D).
Figure 5. Preventive effects of long‐term treatment with TLR9 inhibitors on perivascular inflammation in MCT‐exposed rats.
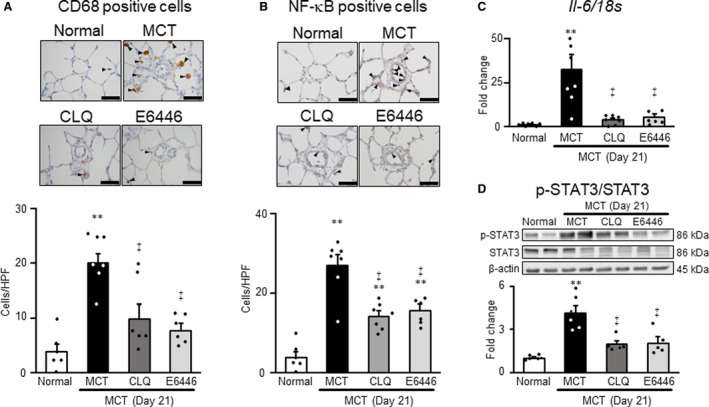
Rats were given a subcutaneous injection of MCT on day 0. Some MCT‐injected rats were treated with CLQ or E6446 from 3 days before to 21 days after MCT injection. Normal: normal controls. A and B, Representative photomicrographs of CD68‐positive macrophages and NF‐κB‒positive cells (arrowheads) in medial layer of pulmonary arteries with outer diameters of 50 µm or less, and comparisons of counts in 4 groups. Scale bars indicate 50 µm. C, The mRNA expression levels of Il‐6 in lung measured by RT‐PCR. D, Representative photographs of Western blots of phospho‐STAT3, STAT3, and β‐actin in lung (upper) and summary of the protein levels of phospho‐STAT3/STAT3 in lung (lower). Data are expressed as mean±SEM. N=6 to 7. **P<0.01 vs Normal. ‡ P<0.01 vs MCT, according to a 1‐way ANOVA followed by Bonferroni post hoc test. CLQ indicates chloroquine; HPF, high‐power field; MCT, monocrotaline; NF‐κB, nuclear factor κB; RT‐PCR, reverse transcriptase polymerase chain reaction; and TLR9, toll‐like receptor 9.
Reversal Effects of Short‐Term (3‐Day) Treatment With a TLR9 Inhibitor on Perivascular Inflammation in Established Monocrotaline‐Induced Rat Model With PH
Previous studies demonstrated that monocrotaline‐exposed rats showed significant pulmonary hypertension on day 14. 29 , 30 The numbers of CD68‐positive macrophages and NF‐κB‐positive cells increased in the areas surrounding small pulmonary arteries in the monocrotaline group compared with the normal group (Figure 6A and 6B). Short‐term treatment (from day 14 to day 17) with E6446 (3‐day reversal protocol, protocol 2, Figure 1) significantly reduced CD68‐positive macrophages and NF‐κB–positive cells in the lung of monocrotaline‐exposed rats (Figure 6A and 6B). However, short‐term treatment with E6446 had no significant effect on the medial wall thickness and RVSP (data not shown). The level of Il‐6 mRNA in the lung increased significantly in the monocrotaline group compared with the normal group (Figure 6C). Short‐term treatment with E6446 almost completely normalized the level of Il‐6 mRNA expression in the lung of monocrotaline‐exposed rats (Figure 6C). E6446 also significantly inhibited the activation of STAT3 compared with the monocrotaline group (Figure 6D).
Figure 6. Reversal effects of short‐term (3‐day) treatment with TLR9 inhibitors on perivascular inflammation in rats with established MCT‐induced PH.
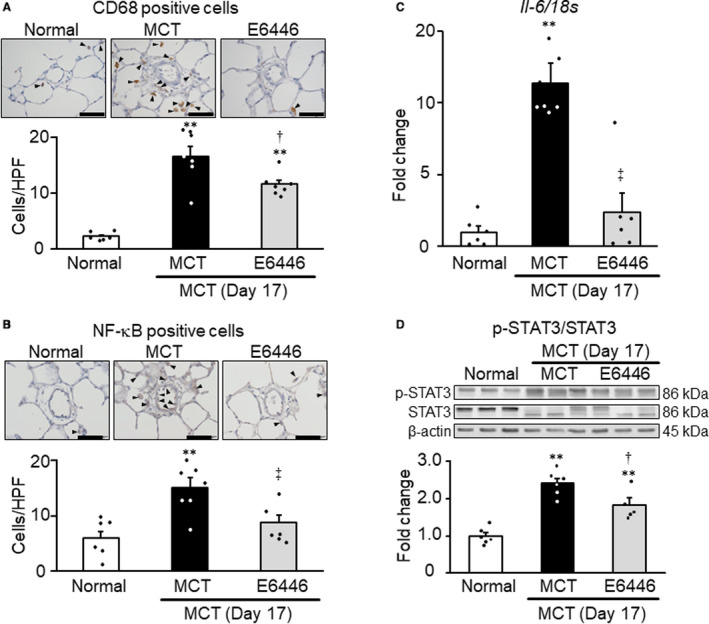
Rats were given a subcutaneous injection of MCT on day 0. Some MCT‐injected rats were treated with E6446 from days 14 to 17 after MCT injection. Normal: normal controls. A and B, Representative photomicrographs of CD68‐positive macrophages and NF‐κB‒positive cells (arrowheads) in medial layer of pulmonary arteries with outer diameters of 50 µm or less, and comparisons of counts in 4 groups. Scale bars indicate 50 µm. C, The mRNA expression levels of Il‐6 in lung measured by RT‐PCR. D, Representative photographs of Western blots of phospho‐STAT3, STAT3, and β‐actin in lung (upper) and summary of the protein levels of phospho‐STAT3/STAT3 in lung (lower). Data are expressed as mean±SEM. N=6 to 7. **P<0.01 vs normal. † P<0.05 and ‡ P<0.01 vs MCT, according to a 1‐way ANOVA followed by Bonferroni post hoc test. HPF indicates high‐power field; MCT, monocrotaline; NF‐κB, nuclear factor κB; PH, pulmonary hypertension; RT‐PCR, reverse transcriptase polymerase chain reaction; and TLR9, toll‐like receptor 9.
Reversal Effect of Prolonged Treatment With a TLR Inhibitor on Pulmonary Hypertension, PA Remodeling, and Survival in Established Monocrotaline‐Induced Rat Model With PH
Treatment with E6446 from day 14 to day 24 after monocrotaline injection (10‐day reversal protocol, protocol 3, Figure 1) reduced the increases of TPRI, medial wall thickness, and percentage of fully muscularized vessels on day 24, compared with the monocrotaline group (Figure 7C, 7E, and 7F), whereas E6446 treatment had no significant effect on RVSP, CI, and RV/LV+S ratio (Figure 7A, 7B, and 7D). In addition, E6446 treatment significantly improved survival of the monocrotaline‐exposed rats (Figure 7G).
Figure 7. Reversal effect of prolonged (10‐day) treatment with a selective TLR inhibitor on pulmonary hypertension, PA remodeling, and survival in rats with MCT‐induced PH.
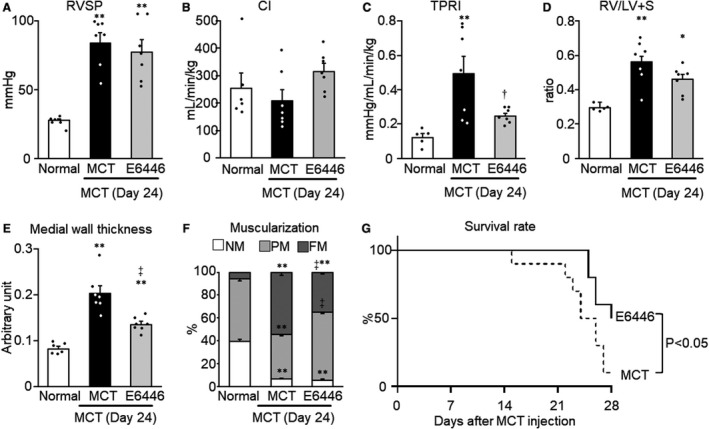
Rats were given a subcutaneous injection of MCT on day 0. Some MCT‐injected rats were treated with E6446 from days 14 to 24 after MCT injection. Normal: normal controls. A, RVSP. B, CI. C, TPRI. D, Right ventricular hypertrophy. E, Medial wall thickness of pulmonary arteries with outer diameters of 50 to 100 µm. F, Muscularization of pulmonary arteries with outer diameters of 50 µm or less. Data are expressed as mean±SEM. N=6 to 7. *P<0.05 and **P<0.01 vs Normal. † P<0.05 and ‡ P<0.01 vs MCT, according to a 1‐way ANOVA followed by Bonferroni post hoc test. G, Survival curves (day 14 to day 28 after MCT injection) are shown for MCT‐exposed rats not treated (MCT, n=10) and those treated with E6446 (10 mg/kg per day, oral, n=10). The statistical differences were analyzed by a log‐rank test. CI indicates cardiac index; FM, fully muscular; MCT, monocrotaline; NM, nonmuscular; PA, pulmonary artery; PH, pulmonary hypertension; PM, partially muscular; RV/LV+S RV/LV+S, right ventricular hypertrophy (weight ratio of RV free wall to the sum of LV free wall and septum); RVSP, right ventricular systolic pressure; TLR9, toll‐like receptor 9; and TPRI, total pulmonary vascular resistance index.
Effect of Prolonged Treatment With a TLR Inhibitor on Pulmonary Hypertension in Established SU5416/Hypoxia/Normoxia‐Induced Rat Model With PH
In order to generalize the findings of the monocrotaline‐exposed PH model, a SU5416/hypoxia/normoxia‐exposed PH rat model was utilized to examine the therapeutic effect of E6446 (Figure 8A). The treatment with E6446 from week 3 through week 8 after SU5416 injection had no significant effect on RVSP, CI, TPRI, and RV/LV+S ratio (Figure 8B through 8E).
Figure 8. Treatment with a selective TLR inhibitor on hemodynamic parameters and RV hypertrophy in rats with SU5416/hypoxia/normoxia–induced PH rats.
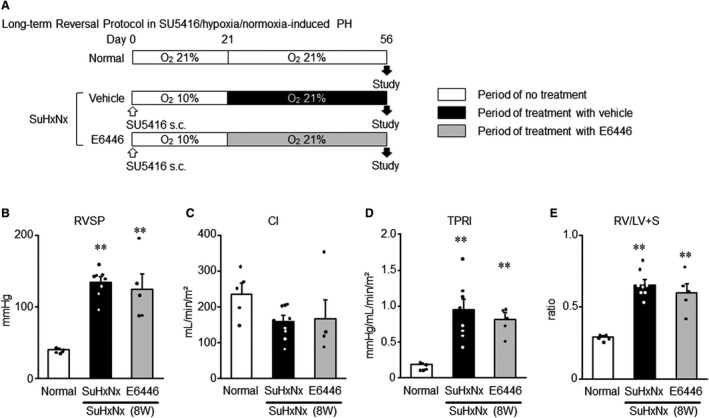
A, Experimental protocol. Rats received a single subcutaneous injection of SU5416 (20 mg/kg weight), and were housed in hypoxic condition (10% O2) for 3 weeks and then normoxic condition for 5 weeks. Some SU5416/hypoxia/normoxia–exposed rats were treated with E6446 (10 mg/kg per day, oral) from week 3 through week 8 after SU5416 injection according to a protocol similar to the reversal protocol (protocol 3) used for the MCT‐exposed rats. Normal: normal controls. SU5416/hypoxia/normoxia: SuHxNx. B, RVSP. C, CI. D, TPRI. E, Right ventricular hypertrophy (RV/LV+S). Data are expressed as mean±SEM. N=5 to 8. **P<0.01 vs normal, based on a 1‐way ANOVA followed by Bonferroni post hoc test. CI indicates cardiac index; LV, left ventricle; MCT, monocrotaline; PH, pulmonary hypertension; RV, right ventricle; RV/LV+S, weight ratio of RV free wall to the sum of LV free wall and septum; RVSP, right ventricular systolic pressure; TLR9, toll‐like receptor 9; and TPRI, total pulmonary vascular resistance index.
Discussion
The novel findings of the present study are the following: (1) TLR9 activation was involved in the development of PH induced by monocrotaline injection in rats; and (2) long‐term inhibition of TLR9 in monocrotaline‐exposed rats not only ameliorated but also reversed the pathophysiological findings in PH, reduced Il‐6 mRNA level, and prolonged survival. These results indicate that TLR9 inhibition is a potential novel therapeutic strategy for PH.
In the present study, 2 types of TLR9 inhibitors—chloroquine and E6446—were studied. Chloroquine impairs acidification of lysosomes, thereby inhibiting the interaction between nonmethylated CpG DNA and TLR9, and chloroquine has high selectivity for TLR9 among other TLRs (IC50 for TLR9, 7, and 4 are 0.08, 2.78, and >30 mmol/L, respectively). 10 , 11 On the other hand, E6446 directly inhibits the binding between nonmethylated CpG DNA and TLR9. Although E6446 inhibits TLR7 and TLR9 signaling by accumulating inside cells and binding to nucleic acids, it can inhibit TLR9 more selectively than TLR7 (IC50 for TLR9, TLR7, and TLR4 are 0.01, 1.78, and 10.58 mmol/L, respectively). 10 , 11 Previous in vivo study demonstrated that administration of 20 mg/kg of E6446 for 5 days completely inhibited the increase of Il‐6 mRNA level in response to a TLR9 agonist (CpG ODN1668) but not to synthetic nonnucleic acid agonists of TLR7 (CL097 and R848) or lipopolysaccharide in the spleen cells harvested from mice. 10 Another study also demonstrated that 20 or 60 mg/kg per day of E6446 markedly and dose‐dependently inhibited an increase in serum IL‐6 level 2 hours after challenge with CpG ODN1668 in mice. 10 Considering that relatively lower doses of E6446 (10 mg/kg per day) as well as chloroquine (50 mg/kg per day) similarly and almost completely inhibited the increase of Il‐6 mRNA level in the lung of monocrotaline‐exposed rats, it is conceivable that these inhibitors prevented the development of PH by inhibiting TLR9 signaling.
When TLR9 senses its specific agonist of subcellular CpG DNA, TLR9 first translocates from endoplasmic reticulum to the endosome, and subsequently binds to the agonist in the endosome. 31 Then, TLR9 is cleaved in the endosome into N‐ and C‐terminal fragments. The C‐terminal fragment is the active form of TLR9 and interacts with MyD88 to induce cytokine production. 32 In the present study, we found an increased protein level of C‐terminal TLR9 fragment in the lungs concomitant with increased circulating mtDNAs on days 7 and 14 after monocrotaline injection, suggesting that mtDNAs activate TLR9 during the progression of PH. Considering that mtDNAs did not increase to a significant level at the earliest time point (day 1) when monocrotaline and its metabolites in blood should reach peak concentrations, we speculate that monocrotaline and its metabolites may not directly increase circulating mtDNAs. In a previous study, pressure‐overload toward LV induced deposition of mtDNA that co‐localized with TLR9 in autolysosomes in mice on day 2 after transverse aortic constriction. 7 We demonstrated that TLR9 is expressed in pulmonary artery endothelial cells of monocrotaline‐exposed rats and human PAH (Figure S1). We have also reported that hemodynamic stress plays a critical role in the development and maintenance of NF‐κB‒IL‐6‒mediated perivascular inflammation and vascular remodeling in rats with PH. 24 It is therefore possible that hemodynamic stress induces mtDNA‐mediated TLR9 activation in pulmonary artery endothelial cells and that such activation of TLR9 may be responsible for the initiation and development of PH. Further study is needed to investigate how hemodynamic stress induces mtDNA production in the lung and which cell types are responsible for TLR9 activation in PH.
Regarding the effects of TLR9 inhibitors on mtDNA and TLR9 activation, the treatment with chloroquine or E6446 according to Protocol 1–preventive protocol had no significant effect on the levels of mtDNAs on days 21 in monocrotaline‐exposed PH rats (Figure S2A). The previous reports demonstrated that the hemodynamic stress or other pathological stresses trigger the release of mtDNA. 3 , 4 , 6 , 7 , 8 , 15 The released mtDNA in turn activate TLR9 and its downstream signaling. Therefore, it is conceivable that mtDNA release is an upstream event, but not downstream event, of TLR9 signaling. In addition, the treatment with chloroquine or E6446 according to the preventive protocol (protocol 1) and the 3‐day reversal protocol (protocol 2) had no significant effect on the levels of cleaved TLR9 in the lungs of monocrotaline‐exposed PH rats (Figure S2B). As mentioned above, the cleavage and activation of TLR9 occur in the endosome after translocating from endoplasmic reticulum upon insult of subcellular CpG DNA, such as mtDNA. The C‐terminal fragment, which represents an active form of TLR9, then binds to mtDNA, and interacts with downstream signaling molecules such as MyD88 to induce inflammatory responses including cytokine production. Chloroquine and E6446 are considered to inhibit the interaction between the agonist and cleaved TLR9, thereby inhibiting the TLR9 function. 11 Chloroquine also inhibits TLR9 signaling by inhibiting acidification of endosome, which is a prerequisite for the TLR9 cleavage. These results are therefore consistent with such proposed mechanism of action of chloroquine and E6446.
IL‐6 was increased in the lung of PH rodent models and in the blood of patients with severe PH. 29 , 33 , 34 The level of IL‐6 was shown to correlate with the prognosis of patients with PAH. 34 In previous studies, lung‐specific Il‐6‒expressing transgenic mice displayed spontaneous PH under normoxia condition and developed greatly exaggerated hypoxia‐induced PH, 35 and RVSP was reduced in Il‐6–deficient mice compared with wild‐type mice subjected to 3‐week exposure to hypoxia. 36 Based on these findings, the concept that IL‐6 is a key cytokine in the development of PH has been widely accepted. In the present study, not only preventive but also 3‐day reversal treatment with TLR9 inhibitor almost completely normalized the elevated Il‐6 mRNA level in the lung of monocrotaline‐exposed rats. These findings suggest that the attenuation of the IL‐6 pathway is involved in the therapeutic effect of TLR9 inhibitor on PH. However, further study using IL‐6 receptor antagonist or Il‐6–deficient mice is required to verify this hypothesis.
Previous in vitro studies demonstrated that activated NF‐κB induced IL‐6 production, which in turn phosphorylated STAT3 and activated the transcription factor Krüppel‐like factor 5 in control PASMCs, 27 and that inhibition of STAT3 abrogated Krüppel‐like factor 5 activation and reduced proliferation of PAH‐PASMCs. 27 In response to CpG DNA, TLR9 induces NF‐κB activation in mouse pulmonary artery endothelial cells. 37 As shown in Figure S1, both TLR9 and NF‐κB were co‐expressed predominantly on the single endothelial layers of remodeled pulmonary arteries in the monocrotaline‐exposed rat and human PAH. It is therefore speculated that TLR9 activation, probably in pulmonary artery endothelial cells, may stimulate proliferation of PASMCs via NF‐κB‒IL‐6‒mediated STAT3 activation in PH. This speculation is partly supported by our finding that TLR9 inhibition decreased the number of Ki67‐positive cells in the medial wall layer and muscularization of pulmonary artery in monocrotaline‐exposed rats. It is possible that TLR9 inhibitor may suppress proliferation of PASMCs, thereby reducing pulmonary vascular resistance in monocrotaline‐exposed rats.
The conventional monocrotaline‐induced PH model used in the present study exhibits only medial hypertrophy and muscularization of pulmonary arteries as manifestations of vascular remodeling, while this model does not show occlusive neointimal or plexiform lesions that are characteristic of the advanced lesions seen in patients with PAH. 38 , 39 The present study showed no significant effect of E6446 on hemodynamic and RV hypertrophy in a second PH model, SU5416/hypoxia/normoxia‐exposed PH rats, which closely mimics the pathophysiology and pulmonary arteriopathy of human PAH. 16 The differences in the therapeutic effects of E6446 on monocrotaline‐ and SU5416/hypoxia/normoxia‐induced PH suggest that the contribution of TLR9 to the pathophysiology of PH differs depending on the model of PH and that it also depends on the progression of PAH and varies during the time course of development of PH. Multiple mechanisms are considered to contribute to the development and/or maintenance of complex pulmonary arteriopathy. Our data suggest that TLR9 does not play a critical role at least in the established stage of SU5416/hypoxia/normoxia‐induced PH. However, the temporal change in the contribution of TLR9 to pathophysiology of SU5416/hypoxia/normoxia‐induced PH thus remains to be elucidated. Furthermore, the present study is limited in the ability to suggest the role of TLR9 in the development of advanced lesions seen in patients with PAH. The contribution of mtDNA and TLR9 to the development and progression of human PAH still remains largely unknown. Therefore, it is still difficult to extrapolate the observations we obtained with a rat PH model to human PH. When the contribution of mtDNA and TLR9 to the pathophysiology of human PH is elucidated, our observations of the present study are useful for promoting the development of novel therapeutic strategies for the treatment of the patients with PH by targeting the TLR9. The therapeutic effect of E6446 should be further investigated in PAH patients.
Conclusion
In conclusion, we demonstrated that TLR9 contributed to the development of PH in monocrotaline‐induced rats with PH. Given the lack of established treatments for perivascular inflammation in PH, treatment targeting TLR9 can be a novel strategy for PH. To translate our present findings to clinical medicine, we need to investigate whether TLR9 is activated in patients with PH, and whether TLR9 inhibition by E6446 effectively and safely improves PH in those patients.
Sources of Funding
This work was supported by supported by JSPS KAKENHI Grant (17K09591 and 20K08425) and Japan Agency for Medical Research and Development (18ek0109371h0001).
Disclosures
Abe worked in a department endowed by Actelion Pharmaceuticals Japan, and received a research grant from Mochida Pharmaceutical Co. H. Tsutsui received honoraria from Daiichi Sankyo, Inc., Otsuka Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co. Ltd., Mitsubishi Tanabe Pharma Corporation, Boehringer Ingelheim Japan, Inc., Novartis Pharma K.K., Bayer Yakuhin, Ltd., Bristol‐Myers Squibb KK, and Astellas Pharma Inc., and research funds from Actelion Pharmaceuticals Japan, Daiichi Sankyo, Inc., and Astellas Pharma Inc. The remaining authors have no disclosures to report.
Supporting information
Figures S1–S2
Acknowledgments
We thank Akiko Ando for technical assistance. We appreciate the technical assistance from The Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences.
Author contributions: Abe designed the study and wrote the manuscript. Ishikawa, Takana‐Ishikawa, Yoshida, Watanabe, and Imakiire performed the experiments and data collection and analysis. K. Hosokawa, Hirano, K. Hirano, and Tsutsui wrote and revised the manuscript with input from all authors.
(J Am Heart Assoc. 2021;10:e019247. DOI: 10.1161/JAHA.120.019247.)
For Sources of Funding and Disclosures, see page 14.
References
- 1. Seferian A, Simonneau G. Therapies for pulmonary arterial hypertension: where are we today, where do we go tomorrow? Eur Respir Rev. 2013;22:217–226. DOI: 10.1183/09059180.00001713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Humbert M, Sitbon O, Chaouat A, Bertocchi Michèle, Habib G, Gressin V, Yaïci A, Weitzenblum E, Cordier J‐F, Chabot François, et al. Survival in patients with idiopathic, familial, and anorexigen‐associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. DOI: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. DOI: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garcia‐Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, Shlomchik MJ, Coffman RL, Candia A, Mehal WZ. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 2016;126:859–864. DOI: 10.1172/JCI83885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Han SJ, Li H, Kim M, Shlomchik MJ, Lee HT. Kidney proximal tubular TLR9 exacerbates ischemic acute kidney injury. J Immunol. 2018;201:1073–1085. DOI: 10.4049/jimmunol.1800211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCarthy CG, Wenceslau CF, Goulopoulou S, Ogbi S, Baban B, Sullivan JC, Matsumoto T, Webb RC. Circulating mitochondrial DNA and Toll‐like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc Res. 2015;107:119–130. DOI: 10.1093/cvr/cvv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. DOI: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loomis Z, Eigenberger P, Redinius K, Lisk C, Karoor V, Nozik‐Grayck E, Ferguson SK, Hassell K, Nuss R, Stenmark K, et al. Hemoglobin induced cell trauma indirectly influences endothelial TLR9 activity resulting in pulmonary vascular smooth muscle cell activation. PLoS One. 2017;12:e0171219. DOI: 10.1371/journal.pone.0171219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kolettis T, Vlahos AP, Louka M, Hatzistergos KE, Baltogiannis GG, Agelaki MM, Mitsi A, Malamou‐Mitsi V. Characterisation of a rat model of pulmonary arterial hypertension. Hellenic J Cardiol. 2007;48:206–210. [PubMed] [Google Scholar]
- 10. Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, Zheng W, Ataíde MA, de Oliveira RB, Golenbock DT, et al. Therapeutical targeting of nucleic acid‐sensing Toll‐like receptors prevents experimental cerebral malaria. Proc Natl Acad Sci USA. 2011;108:3689–3694. DOI: 10.1073/pnas.1015406108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, Genest M, Yang H, Shaffer C, Zhao Y, et al. Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol. 2014;85:429–440. DOI: 10.1124/mol.113.089821. [DOI] [PubMed] [Google Scholar]
- 12. Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine prevents progression of experimental pulmonary hypertension via inhibition of autophagy and lysosomal bone morphogenetic protein type II receptor degradation. Circ Res. 2013;112:1159–1170. DOI: 10.1161/CIRCRESAHA.111.300483. [DOI] [PubMed] [Google Scholar]
- 13. Lafyatis R, York M, Marshak‐Rothstein A. Antimalarial agents: closing the gate on Toll‐like receptors? Arthritis Rheum. 2006;54:3068–3070. DOI: 10.1002/art.22157. [DOI] [PubMed] [Google Scholar]
- 14. Willis R, Seif AM, McGwin G Jr, Martinez‐Martinez LA, González EB, Dang N, Papalardo E, Liu J, Vilá LM, Reveille JD, et al. Effect of hydroxychloroquine treatment on pro‐inflammatory cytokines and disease activity in SLE patients: data from LUMINA (LXXV), a multiethnic US cohort. Lupus. 2012;21:830–835. DOI: 10.1177/0961203312437270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshida K, Abe K, Ishikawa M, Saku K, Shinoda‐Sakamoto M, Ishikawa T, Watanabe T, Oka M, Sunagawa K, Tsutsui H. Inhibition of TLR9‐NF‐kappaB‐mediated sterile inflammation improves pressure overload‐induced right ventricular dysfunction in rats. Cardiovasc Res. 2019;115:658–668. DOI: 10.1093/cvr/cvy209. [DOI] [PubMed] [Google Scholar]
- 16. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. DOI: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 17. Toba M, Alzoubi A, O'Neill KD, Gairhe S, Matsumoto Y, Oshima K, Abe K, Oka M, McMurtry IF. Temporal hemodynamic and histological progression in Sugen5416/hypoxia/normoxia‐exposed pulmonary arterial hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;306:H243–H250. DOI: 10.1152/ajpheart.00728.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwenke DO, Pearson JT, Sonobe T, Ishibashi‐Ueda H, Shimouchi A, Kangawa K, Umetani K, Shirai M. Role of Rho‐kinase signaling and endothelial dysfunction in modulating blood flow distribution in pulmonary hypertension. J Appl Physiol (1985). 2011;110:901–908. DOI: 10.1152/japplphysiol.01318.2010. [DOI] [PubMed] [Google Scholar]
- 19. Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, Kass DA. Measurement of cardiac function using pressure‐volume conductance catheter technique in mice and rats. Nat Protoc. 2008;3:1422–1434. DOI: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuck JL, Obiako BO, Gorodnya OM, Pastukh VM, Kua J, Simmons JD, Gillespie MN. Mitochondrial DNA damage‐associated molecular patterns mediate a feed‐forward cycle of bacteria‐induced vascular injury in perfused rat lungs. Am J Physiol Lung Cell Mol Physiol. 2015;308:L1078–L1085. DOI: 10.1152/ajplung.00015.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guignabert C, Phan C, Seferian A, Huertas A, Tu LY, Thuillet R, Sattler C, Le Hiress M, Tamura Y, Jutant E‐M, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126:3207–3218. DOI: 10.1172/JCI86249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tamura Y, Phan C, Tu L, Le Hiress M, Thuillet R, Jutant EM, Fadel E, Savale L, Huertas A, Humbert M, et al. Ectopic upregulation of membrane‐bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. 2018;128:1956–1970. DOI: 10.1172/JCI96462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuwabara Y, Tanaka‐Ishikawa M, Abe K, Hirano M, Hirooka Y, Tsutsui H, Sunagawa K, Hirano K. Proteinase‐activated receptor 1 antagonism ameliorates experimental pulmonary hypertension. Cardiovasc Res. 2019;115:1357–1368. DOI: 10.1093/cvr/cvy284. [DOI] [PubMed] [Google Scholar]
- 24. Abe K, Shinoda M, Tanaka M, Kuwabara Y, Yoshida K, Hirooka Y, McMurtry IF, Oka M, Sunagawa K. Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc Res. 2016;111:16–25. DOI: 10.1093/cvr/cvw070. [DOI] [PubMed] [Google Scholar]
- 25. Bliksoen M, Mariero LH, Torp MK, Baysa A, Ytrehus K, Haugen F, Seljeflot I, Vaage J, Valen G, Stenslokken KO. Extracellular mtDNA activates NF‐kappaB via toll‐like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol. 2016;111:42. DOI: 10.1007/s00395-016-0553-6. [DOI] [PubMed] [Google Scholar]
- 26. Tanaka M, Abe K, Oka M, Saku K, Yoshida K, Ishikawa T, McMurtry IF, Sunagawa K, Hoka S, Tsutsui H. Inhibition of nitric oxide synthase unmasks vigorous vasoconstriction in established pulmonary arterial hypertension. Physiol Rep. 2017;5:e13537. DOI: 10.14814/phy2.13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Courboulin A, Tremblay VL, Barrier M, Meloche J, Jacob MH, Chapolard M, Bisserier M, Paulin R, Lambert C, Provencher S, et al. Kruppel‐like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir Res. 2011;12:128. DOI: 10.1186/1465-9921-12-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Cote J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription‐3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation. 2011;123:1205–1215. DOI: 10.1161/CIRCULATIONAHA.110.963314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, Hamon M, Adnot S, Eddahibi S. Serotonin transporter inhibition prevents and reverses monocrotaline‐induced pulmonary hypertension in rats. Circulation. 2005;111:2812–2819. DOI: 10.1161/CIRCULATIONAHA.104.524926. [DOI] [PubMed] [Google Scholar]
- 30. Miyauchi T, Yorikane R, Sakai S, Sakurai T, Okada M, Nishikibe M, Yano M, Yamaguchi I, Sugishita Y, Goto K. Contribution of endogenous endothelin‐1 to the progression of cardiopulmonary alterations in rats with monocrotaline‐induced pulmonary hypertension. Circ Res. 1993;73:887–897. DOI: 10.1161/01.RES.73.5.887. [DOI] [PubMed] [Google Scholar]
- 31. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. DOI: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 32. Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll‐like receptor 9. Nat Immunol. 2008;9:1407–1414. DOI: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Otsuki S, Sawada H, Yodoya N, Shinohara T, Kato T, Ohashi H, Zhang E, Imanaka‐Yoshida K, Shimpo H, Maruyama K, et al. Potential contribution of phenotypically modulated smooth muscle cells and related inflammation in the development of experimental obstructive pulmonary vasculopathy in rats. PLoS One. 2015;10:e0118655. DOI: 10.1371/journal.pone.0118655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. DOI: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 35. Steiner MK, Syrkina OL, Kolliputi N, Mark EJ, Hales CA, Waxman AB. Interleukin‐6 overexpression induces pulmonary hypertension. Circ Res. 2009;104:236–244, 228p following 244. DOI: 10.1161/CIRCRESAHA.108.182014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Savale L, Tu L, Rideau D, Izziki M, Maitre B, Adnot S, Eddahibi S. Impact of interleukin‐6 on hypoxia‐induced pulmonary hypertension and lung inflammation in mice. Respir Res. 2009;10:6. DOI: 10.1186/1465-9921-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li J, Ma Z, Tang Z‐L, Stevens T, Pitt B, Li S. Song Li. CpG DNA‐mediated immune response in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L552–L558. DOI: 10.1152/ajplung.00436.2003. [DOI] [PubMed] [Google Scholar]
- 38. Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:25s–32s. DOI: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 39. Meyrick B. The pathology of pulmonary artery hypertension. Clin Chest Med. 2001;22:393–404, vii. DOI: 10.1016/S0272-5231(05)70279-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S2