

Abstract
Background
Integrin αM (CD11b), which is encoded by the Integrin Subunit Alpha M (ITGAM) gene, is not only a surface marker of monocytes but also an essential adhesion molecule. In this study, we investigated the effect of CD11b on experimental abdominal aortic aneurysm and the potential underlying mechanisms.
Methods and Results
The incidence of abdominal aortic aneurysm was not significantly lower in ITGAM(‐/‐) mice than in control mice. Nevertheless, knockout of CD11b reduced the maximum abdominal aortic diameter, macrophage infiltration, matrix metalloproteinase‐9 expression, and elastin and collagen degradation. Additionally, lower expression of IL‐6 was found in both the peripheral blood and abdominal aortas of ITGAM(‐/‐) mice, indicating a biological correlation between CD11b and the inflammatory response in abdominal aortic aneurysm. In vitro, the number of ITGAM(‐/‐) bone marrow–derived macrophages (BMDMs) that adhered to endothelial cells was significantly lower than the number of wild‐type BMDMs. Moreover, the CD11b monoclonal antibody and CD11b agonist leukadherin‐1 decreased and increased the number of adherent wild‐type BMDMs, respectively. Through RNA sequencing, genes associated with leukocyte transendothelial migration were found to be downregulated in ITGAM(‐/‐) BMDMs. Furthermore, immunoprecipitation–mass spectrometry analysis predicted that the Akt pathway might be responsible for the impaired transmigratory ability of ITGAM(‐/‐) BMDMs. The reduced activation of Akt was then confirmed, and the Akt agonist SC79 partially rescued the transendothelial migratory function of ITGAM(‐/‐) BMDMs.
Conclusions
CD11b might promote the development and progression of abdominal aortic aneurysm by mediating the endothelial cells adhesion and transendothelial migration of circulating monocytes/macrophages.
Keywords: abdominal aortic aneurysm, adhesion molecule, inflammation, integrin αM, migration
Subject Categories: Aneurysm
Nonstandard Abbreviations and Acronyms
- BMDM
bone marrow‐derived macrophages
- CCL2
C‐C motif chemokine ligand 2
- DEG
differentially expressed genes
- EC
endothelial cell
- ITGAM
Integrin Subunit Alpha M
- MMP
matrix metalloproteinase
- MS
mass spectrometry
- RACK1
Receptor For Activated C Kinase 1
- TLR
Toll‐like receptor
- WT
wild type
Clinical Perspective
What Is New?
Deficiency of the integrin αM protects against experimental abdominal aortic aneurysm in a CaCl2‐induced mouse model.
A biological correlation between integrin αM and the inflammatory response exists in the experimental abdominal aortic aneurysm.
Integrin αM mediates the endothelial cells adhesion and transendothelial migration of bone marrow–derived macrophages.
What Are the Clinical Implications?
Integrin αM is an attractive therapeutic target in the medical treatment of abdominal aortic aneurysm.
Further studies are warranted on the translational implications of integrin αM‐specific agonist leukadherin‐1 in the development of experimental abdominal aortic aneurysm.
Abdominal aortic aneurysm (AAA) is a common and life‐threatening disease characterized by focal, permanent dilation and degradation of the aortic wall. In White people, population‐based studies have found a prevalence of AAA ranging from 1.2% to 3.3% in men older than 60 years. 1 Currently, open surgery and endovascular aortic repair are the first choices for the treatment of AAA, but these 2 established therapies are associated with high postoperative morbidity and high risk of requiring reintervention, respectively. 2 Thus, extensive research on the underlying molecular mechanism of AAA is warranted to develop effective medical therapies.
Pathologically, AAA is considered a chronic inflammatory response that includes the infiltration of macrophages into the media and adventitia, which is accompanied by the excessive secretion of proinflammatory cytokines and matrix metalloproteinases (MMPs). 3 , 4 Notably, the first step in the infiltration of macrophages is the establishment of adhesive interactions between circulating monocytes and vascular endothelial cells (ECs), and these interactions are mediated by 2 families of molecules named selectins and integrins. 5 Targeting these cell adhesion molecules may favor the repression of monocyte/macrophage recruitment and reduce the vascular inflammatory response. Ricci et al demonstrated that an anti‐CD18 (integrin β2) monoclonal antibody could ameliorate the exacerbation of AAA by reducing monocyte infiltration. 6 However, it remains unclear whether direct intervention targeting integrin‐induced monocyte adhesion could attenuate experimental AAA.
Mac‐1, as a member of integrins, is a heterodimeric protein composed of integrin αM (CD11b) and integrin β2. Of these molecules, CD11b is not only a surface marker of circulating monocytes but also mediates various cell functions, including adhesion, chemotaxis, and migration. 7 Moreover, CD11b contains an inserted domain that broadly contributes to the binding of Mac‐1 by many ligands, including intercellular adhesion molecule‐1 and fibrinogen. 8 Previously, several studies determined that the CD11b‐mediated interaction between monocytes and ECs promoted neurovascular injury, lupus nephritis, and vascular inflammation. 9 , 10 , 11 , 12 Samadzadeh et al found higher levels of CD11b on circulating monocytes from patients with AAA compared with those on circulating monocytes from control subjects. In vitro, monocytes isolated from patients with AAA showed increased adhesion and transmigration activities, which were also positively associated with AAA diameter. 13 Therefore, we hypothesized that depleting CD11b might limit monocyte/macrophage adhesion and transendothelial migration, reduce the inflammatory response, and promote vascular remodeling.
In the present study, we first detected the CD11b content in human abdominal aortic specimens. The expression of CD11b was also examined during the progression of AAA in mice. Then, we determined whether CD11b modulates proinflammatory mediator production and macrophage infiltration and polarization during the formation and development of AAA in vivo. Finally, by isolating bone marrow–derived macrophages (BMDMs) from Integrin Subunit Alpha M (ITGAM) (‐/‐) and wild‐type (WT) mice, we investigated the effect of CD11b on the cell viability, cell adhesion, and transendothelial migratory function of macrophages, as well as the underlying mechanisms.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Human Aortic Tissue Samples
According to a protocol approved by the Institutional Ethics Committees of Zhongshan Hospital, Fudan University (B2018‐040R), AAA tissue specimens were collected from 4 patients who underwent elective open surgery from March 2018 to September 2018. During the same time period, 4 nonaneurysmal abdominal aortic wall tissues were obtained from organ donors and used as controls. The clinical characteristics of the patients with AAA and organ donors are shown in Table S1. Written informed consent was obtained from all these patients and from the donors’ next‐of‐kin. Each aortic wall sample was divided into 2 portions: 1 portion was stored in liquid nitrogen for subsequent protein and nucleic acid extraction, and the other portion was fixed with 4% paraformaldehyde overnight and embedded in paraffin.
Experimental Animals
The animal study design was approved by the Institutional Animal Care and Use Committee at Fudan University (Figure S1). All the experimental procedures were conducted in accordance with the National Institutes of Health guidelines for laboratory animals, and were performed at the animal facility of Zhongshan Hospital Fudan University. Because of the poor susceptibility to AAA in female animals, only 8‐week‐old male mice were used for the establishment of the AAA animal model throughout this study. 14 ITGAM transgenic mice on a C57BL/6J background were generously provided by Dr Yiqin Shi. Genotyping was performed using polymerase chain reaction (PCR) according to the Jackson Laboratory’s protocol. All the mice were kept on a 12‐hour light/dark cycle and fed with normal chow.
CaCl2‐Induced AAA Model
The mice were anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg) before laparotomy. Then, the infrarenal abdominal aorta was exposed and isolated from the surrounding retroperitoneal tissues. The gross morphology of the abdominal aorta was recorded by video microscopy to measure the maximum diameters. Subsequently, gauze presoaked in 0.5 mol/L CaCl2 was used to wrap the dissected abdominal aorta for 15 minutes. The CaCl2 solution was replaced with a 0.9% NaCl solution in the sham group. The abdominal cavities were rinsed with sterile saline before suturing. After the procedure, the mice were allowed to recover on heated (37–40℃) pads. The mice were observed daily for sudden death and euthanized 6 weeks after the establishment of the model for serum and histological analysis.
Enzyme‐Linked Immunosorbent Assay
The concentrations of C‐C motif chemokine ligand 2 (CCL2), tumor necrosis factor‐α (TNF‐α), interleukin 6 (IL‐6), and interleukin 1β in the mouse serum were evaluated using ELISA kits according to the manufacturer’s instructions (GuangRui, Shanghai, China). All the samples were analyzed in triplicate.
Histological and Immunohistochemical Analysis
During follow‐up, necropsies were performed to determine the cause of death if the mice died suddenly. The survival of the mice is presented using Kaplan–Meier curves. Six weeks after CaCl2 stimulation, the mice were euthanized, followed by perfusion with PBS. The entire aorta from the thoracic aorta to the bifurcation of the iliac artery was carefully dissected under a microscope. The segment of the abdominal aorta was photographed, and the maximum diameter was recorded using Image‐Pro Plus software (Media Cybernetics, Silver Spring, MD). The occurrence of AAA was defined as a >50% increase in the diameter of the abdominal aorta compared with that before intervention.
The harvested aorta was fixed in 4% paraformaldehyde for 24 hours, and the site of the maximal diameter was cut and embedded in paraffin. Histological analysis was performed in serial cross sections (5 μm each). Elastica Van Gieson staining and Masson trichrome staining were conducted to assess the elastin fragmentation and collagen content, respectively. 15 An established standard for the elastin degradation scale was adapted: 1 indicated <25% degradation; 2 indicated 25% to 50% degradation; 3 indicated 50% to 75% degradation; and 4 indicated >75% degradation. 16 The abundance of collagen in the aorta was quantified by the percentage of the positive area.
Serial paraffin sections were further subjected to immunohistochemical staining as described. 17 The primary antibodies used were F4/80 (ab111101; Abcam, Cambridge, UK), MMP2 (ab86607; Abcam), MMP9 (ab38898; Abcam), and rabbit IgG. A negative control group was established to confirm the specificity of antibody binding. Five random fields were imaged to count the number of F4/80‐positive macrophages. The expression of MMP2 and MMP9 was assessed by 2 independent investigators who were blinded to the study design according to the intensity and range of immunostaining. The mean value of 5 sections from each animal was determined (n=6).
Cell Isolation and Culture
BMDMs were isolated from 7‐week‐old WT and ITGAM(‐/‐) mice as previously described. 18 In brief, the femur and tibia bones were cut and flushed with PBS under aseptic conditions. The cell suspension was filtered through a 70‐μm cell strainer into a new tube, and incubated with ammonium‐chloride‐potassium lysis buffer for 5 minutes to eliminate red blood cells. The remaining bone marrow–derived cells were resuspended in complete RPMI medium, treated with macrophage colony‐stimulating factor (20 ng/mL), and then seeded in a 10‐cm petri dish. The media was changed every other day until day 7.
Primary mouse aortic ECs were obtained from aortic rings implanted in growth factor–reduced matrix with the endothelium facing down. 19 When a large number of ECs emerged from the aorta, the aortic segments were removed. The isolated cells were washed, incubated with neutral proteinase for 20 minutes at 37°C, and resuspended in EC growth medium. The harvested ECs were continually cultured in a dish coated with gelatin until they reached ≈90% confluence. Cells in passages 2 to 3 were used for the experiments.
Cell Adhesion Assay
Mature BMDMs were fluorescently labeled with 10 μmol/L calcein AM (Molecular Probes) for 30 minutes at 37℃ in the dark and washed twice with complete RPMI medium. Confluent primary mouse aortic ECs were pretreated with 1 μmol/L angiotensin II overnight, followed by coculture with 1 × 106 labeled BMDMs for 30 minutes. Then, the nonadherent cells were removed by washing with PBS 3 times. The adherent BMDMs were fixed with precooled 1% glutaraldehyde and observed under a fluorescence microscope. Five randomly selected fields were imaged and quantified by Image J software (National Institutes of Health). To block P selectin, the ECs were incubated with 10 μg/mL, 100 μg/mL, or 1000 μg/mL fucoidan or vehicle for 10 minutes after angiotensin II stimulation. For the CD11b inhibition and activation experiments, BMDMs were mixed with 20 μg/mL CD11b monoclonal antibody (ab133357, Abcam) and 10 μmol/L leukadherin‐1 (LA‐1) and incubated for 15 minutes and 2 hours, respectively, before they were added to the coated cells.
Cell Survival Assay
Cell apoptosis assays were performed using FITC‐Annexin V and propidium iodide apoptosis kits. 20 Briefly, 1 × 105 BMDMs were harvested and suspended in 100 μL of Annexin V binding buffer containing 10 μL of propidium iodide and 5 μL of Annexin V‐FITC. After incubation for 15 minutes at room temperature in the dark, 400 μL of Annexin V binding buffer was added. Then, the stained cells were analyzed by fluorescence‐activated cell sorting Calibur flow cytometer.
Cell viability was measured at 24‐hour intervals for 3 days using the Cell Counting Kit‐8. WT and ITGAM(‐/‐) BMDMs were seeded into a 96‐well plate at a density of 1 × 104 cells per well. After culturing for 22 hours, 46 hours, and 70 hours, 10 μL of Cell Counting Kit‐8 solution was added into each well. The BMDMs were further incubated for 2 hours in the dark. The cell viability rates were determined by measuring the absorbance at 450 nm using a microplate reader.
Cell Transendothelial Migration Assay
Primary mouse aortic ECs were precultured in monolayers on Matrigel‐coated Transwell inserts (8‐μm pore size; Corning Costar, Cambridge, MA). After adhesion, the ECs were treated with 20 ng/mL TNF‐α overnight and fixed with 1% paraformaldehyde. Then, 5 × 104 BMDMs were seeded on each Transwell insert, and 10 μmol/L CCL5 was added to the lower chamber. The entire setup was incubated for 16 hours, and the cells together with Matrigel in the upper chamber were gently removed by scrubbing. The transmigrated BMDMs on the lower side of the Transwell insert were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet (Beyotime, Shanghai, China), and counted in 5 random fields. For rescue experiment, BMDMs were pretreated with the Akt agonist SC‐79 (10 μmol/L) for 24 hours before transfer.
Western Blot
Human and mouse aortic tissues or BMDMs were lysed in radioimmunoprecipitation buffer. The protein concentrations were evaluated using a BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Equal amounts of total protein were separated using 10% SDS‐PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). After blocking, the membranes were probed with primary antibodies against CD11b (ab128797, Abcam), p53 (ab26, Abcam), caspase3 (ab1384, Abcam), Bax (ab32503, Abcam), Akt (4691, Cell Signaling Technology, Beverly, MA), p‐Akt (4060, Cell Signaling Technology), β‐actin (ab8226, Abcam), and GAPDH (ab9485, Abcam) at 4℃ overnight. Then, the membranes were incubated with the appropriate horseradish peroxidase–conjugated secondary antibodies. The protein bands were detected by a chemiluminescence detection kit, and β‐actin or GAPDH was used as a loading control. All the experiments were independently conducted in triplicate.
Quantitative Real‐Time PCR
Total RNA was extracted from the aortic tissues and BMDMs using TRIzol reagent (Invitrogen) following the manufacturer’s instructions. Equal amounts of total RNA were then converted to cDNA using the PrimeScript RT Reagent Kit (Takara Biotechnology, Otsu, Japan) and primer sequences specific for the examined molecules. Real‐time PCR was conducted with SYBR‑Green PCR Mix (Takara Biotechnology) using an ABI PRISM 7500 thermal cycler (Applied Biosystems, Foster City, CA). The relative expression of mRNA was determined using the 2‐ΔΔCt method with β‐actin as the control. The primer sequences are listed in Table S2.
Immunofluorescence Staining
Serial paraffin sections from human and mouse aortic tissues were deparaffinized and rehydrated as previously described. 21 Antigen retrieval was performed by boiling in 10 mmol/L Tris, 1 mmol/L, and 0.05% Tween20 retrieval buffer. The adherent BMDMs were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X‐100 in PBS for 10 minutes. After blocking with 1% bovine serum in PBS at room temperature for 1 hour, the sections or slides were incubated with diluted primary antibodies against CD11b (ab128797, Abcam), MOMA‐2 (MCA519GT, Bio‐Rad), F4/80 (ab111101, Abcam), iNOS (ab15323, Abcam), and CD206 (PA5‐101657, Invitrogen) at 4°C overnight. Secondary fluorescent antibodies (Alexa Fluor 488, green; Alexa Fluor 568, red) were added and incubated at room temperature in the dark. D4',6‐diamidino‐2‐phenylindole was used for the immunofluorescence staining of cellular nuclei. The fluorescence signals were visualized by a fluorescence laser scanning confocal microscope (Leica Imaging Systems, Cambridge, UK).
RNA Sequencing
WT and ITGAM (‐/‐) BMDMs were lysed in TRIzol for total RNA extraction. The quality of the RNA samples was analyzed using a 2100 Bioanalyzer (Agilent). Then, the samples were run on an Illumina HiSeq sequencer (Illumina, San Diego, CA) with 150‐bp paired‐end reads. Differentially expressed genes (DEGs) were identified using the Ballgown package in the R language environment. Only DEGs (P<0.05 and fold change ≥1.5) were considered for Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis to determine relevant biological significance.
Coimmunoprecipitation and Mass Spectrometry
Total proteins were extracted from BMDMs via ice‐cold radioimmunoprecipitation buffer. After centrifugation, the supernatant was incubated with anti‐CD11b antibodies and protein A/G beads at 4°C overnight. The control immunoprecipitation was carried out by incubation with nonimmune rabbit polyclonal antibodies. The immunoprecipitated samples were washed 3 times with ice‐cold radioimmunoprecipitation buffer. Before electrophoresis, the samples were boiled and resolved in SDS‐PAGE loading buffer.
For the mass spectrometry (MS) assay, gel lanes were sliced into small pieces and washed twice. 22 Then, the gel pieces were digested using 25 mmol/L ammonium bicarbonate, and the resulting peptides were extracted and dried. The peptides were dissolved and detected by liquid chromatography coupled with tandem MS (ekspertTMnanoLC; AB Sciex TripleTOF 5600‐plus). The MS raw data were converted to Proteinpilot software and searched against a decoy UniProt mouse protein database. The peptides were accepted as accurate if the CI was ≥95% and there was at least 1 unique peptide. A list of potentially interacting proteins was generated after discarding the control IP proteins. The STRING database was used to search for bait proteins and known interactors.
Statistical Analysis
All the statistical analyses were conducted using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA) or SPSS 23.0 software (IBM, Armonk, NY). The data are presented as the proportions or means±SEMs. Based on the distribution of the experimental data, Student t test, Fisher exact test, and nonparametric Mann–Whitney U test were used for comparisons between 2 groups. For comparisons among ≥3 groups, 1‐way or 2‐way ANOVA and the Kruskal–Wallis test followed by a post hoc Bonferroni test were used. A P<0.05 (2‐tailed) was considered statistically significant.
Results
CD11b is Upregulated in Human and Mouse AAA Tissues
To explore the role of CD11b in human aortic aneurysms, we first investigated the expression of CD11b in AAA and control aortic tissues. Immunofluorescence analysis revealed that CD11b expression was significantly higher in the human AAA tissues than in the control tissues (P<0.01, Figure 1A). Similarly, Western blot analysis also confirmed the upregulation of CD11b in the human AAA tissues (P<0.05, Figure 1B). Moreover, CD11b mRNA expression was markedly higher in the human AAA tissues than in the control tissues (P<0.05, Figure 1C). We further induced the formation of aneurysm in WT mice via CaCl2. Western blot analysis showed that CD11b expression progressively increased with time, especially from 2 to 6 weeks (Figure 1D). These findings indicated that CD11b might be associated with the development of AAA in humans and in the present experimental model.
Figure 1. CD11b is upregulated in human AAA tissues and a CaCl2‐induced AAA model.
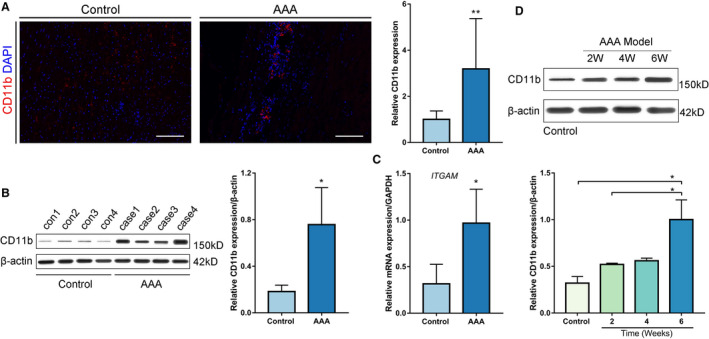
A, Representative confocal immunofluorescence staining of CD11b and DAPI in human AAA tissues and control tissues. Scale bars=100 μm. Quantification of CD11b based on the integral optical density in AAA (n=4) and control (n=4) tissues, **P<0.01 vs control. B, WB and densitometric analysis of CD11b protein expression in human AAA samples (n=4) and control samples (n=4), *P<0.05 vs control. C, Quantitative real‐time polymerase chain reaction for CD11b mRNA expression in human AAA tissues (n=4) and nonaneurysmal abdominal aortic tissues (n=4). *P<0.05 vs control. D, WB and densitometric analyses of CD11b protein expression in abdominal aortic tissues from control wild‐type mice, and from mice 2 weeks, 4 weeks, and 6 weeks after AAA establishment, *P<0.05 vs control or 2 weeks. AAA indicates abdominal aortic aneurysm; DAPI, 4',6‐diamidino‐2‐phenylindole; ITGAM, integrin subunit alpha M; and WB, Western blot.
Depletion of ITGAM Attenuates the Development of Experimental AAA
To further determine the role of CD11b in AAA, we induced experimental AAA in WT and ITGAM knockout mice. As shown in Figure 2A, the infrarenal abdominal aortas obviously expanded in the WT mice 6 weeks after CaCl2 treatment, whereas ITGAM deficiency abated the expansion of the abdominal aortas. Two ITGAM(‐/‐) mice died suddenly at 18 and 25 days following the construction of AAA model, respectively. We excluded the possibility of aortic rupture because of the absence of blood clots in the retroperitoneal cavity. The survival rates were not significantly different between the WT and ITGAM(‐/‐) mice (Figure 2B). Similarly, the incidence of AAA formation also failed to show a significant difference between the WT and ITGAM(‐/‐) mice (73.3% versus 33.3%, P=0.066; Figure 2C). However, compared with the WT mice, the ITGAM(‐/‐) mice exhibited a significant reduction in the maximum diameter of the abdominal aorta (P<0.01, Figure 2D). Moreover, H&E staining showed that the depletion of ITGAM effectively protected against a decrease in the media thicknesses (Figure 2E). Elastica Van Gieson and Masson trichrome staining, as shown in Figure 2E through 2G, demonstrated that ITGAM deficiency also inhibited the elastin and collagen degradation associated with AAA formation, respectively. Furthermore, serum analysis (Figure 2H through 2K) showed that the expression of the proinflammatory cytokines CCL2, TNF‐α, and IL‐6 in the WT and ITGAM(‐/‐) mice was significantly higher than that in the sham‐treated mice, but there was no significant difference in interleukin 1β expression among the 3 groups. Compared with the WT mice, the ITGAM(‐/‐) mice had lower levels of IL‐6 in the peripheral blood. Overall, ITGAM deficiency attenuated AAA development, vascular structural injury, and the inflammatory response, suggesting a causal role of CD11b in AAA.
Figure 2. ITGAM deficiency attenuates the development of experimental AAA, vascular structural injury, and the inflammatory response.
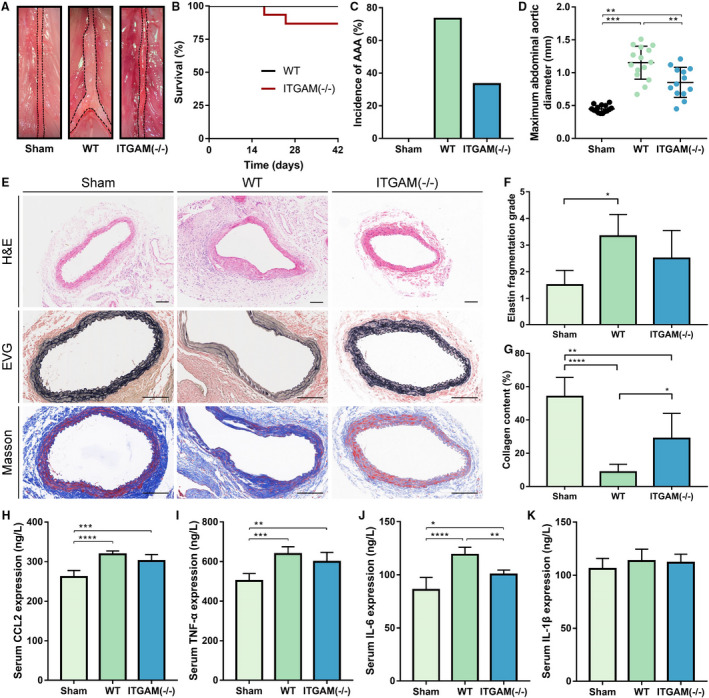
A, Representative photographs of the infrarenal abdominal aortas in saline‐ or CaCl2‐treated WT and ITGAM(‐/‐) male mice. The black dotted lines indicate the edge of abdominal aortas. B, Kaplan–Meier survival curves of WT (n=15) and ITGAM(‐/‐) mice (n=15). No significant difference was found regarding the survival rate. C, Incidence of AAA formation in WT and ITGAM(‐/‐) mice in response to CaCl2. D, Maximal abdominal aortic diameter of WT (n=15) and ITGAM(‐/‐) mice (n=13) 6 weeks after being exposed to CaCl2 and the sham‐treated mice (n=15). The data represent the mean±SEM. **P<0.01, ***P<0.001. E, Representative H&E, EVG, and Masson trichrome staining of the abdominal aortic tissues from the sham‐treated, WT, and ITGAM(‐/‐) mice. Scale bar=100 μm. F and G, Quantification of elastin fragmentation and collagen deposition in the experimental groups (n=6), *P<0.05, **P<0.01, ****P<0.0001. H through K, Serum CCL2, TNF‐α, IL‐6, and IL‐1β levels in the experimental groups (n=6 for each group) detected by ELISA, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. AAA indicates abdominal aortic aneurysm; EVG, Elastica Van Gieson; H&E, hematoxylin and eosin; ITGAM, integrin subunit alpha M; and WT, wild‐type.
Depletion of ITGAM Ameliorates the Infiltration and Polarization of Macrophage
Macrophage infiltration plays an important role in inflammatory pathology during AAA formation. 4 , 23 Thus, we asked whether the ITGAM deficiency‐induced reduction in AAA was attributed to the regulation of macrophage recruitment. Then, we characterized the macrophages infiltrating the AAA lesions in WT and ITGAM(‐/‐) mice using immunohistochemistry. Reduced immunostaining of F4/80‐positive macrophages was observed within the medial region of the abdominal aortas in the ITGAM(‐/‐) mice (Figure 3A and 3B). Notably, macrophage‐derived MMPs and proinflammatory mediators have been reported to mediate AAA development. 15 , 24 As expected, the MMP9 immunostaining in the aortic walls of the ITGAM(‐/‐) mice was significantly decreased compared with that in the aortic walls of the WT mice following CaCl2 induction (Figure 3A, 3C and 3D). However, there was no obvious difference in the MMP2 expression in the aortic sections from the WT and ITGAM(‐/‐) mice in response to CaCl2. Moreover, the changes in MMP9 and MMP2 mRNA expression were consistent with the immunohistochemistry results (Figure 3E and 3F). In addition, quantitative reverse transcriptase PCR verified the upregulation of CCL2, TNF‐α, and IL‐6 in the aortic walls of the CaCl2‐treated WT mice (Figure 3G through 3I), while the aortas of the CaCl2‐induced ITGAM(‐/‐) mice exhibited reduced transcription of CCL2 and IL‐6 compared with those of the CaCl2‐treated WT mice. In contrast, the expression of interleukin 1β was not markedly different among the 3 groups (Figure 3J). These findings indicated that ITGAM deficiency could reduce recruitment of macrophages from the peripheral blood into the aortic wall as well as the concomitant proinflammatory cytokine expression. Additionally, macrophages in inflammatory tissues are classified into the classically activated M1 type and alternatively activated M2 type. 25 Immunofluorescence staining showed that iNOS‐positive M1 macrophages, rather than CD206‐positive M2 macrophages, infiltrated into the aortic walls of the CaCl2‐induced WT mice, facilitating AAA formation. However, in the CaCl2‐treated ITGAM(‐/‐) mice, the numbers of infiltrating M1 macrophages were reduced, whereas the numbers of M2 macrophages were increased (Figure 3K and 3L).
Figure 3. ITGAM deficiency ameliorates macrophage infiltration and polarization.
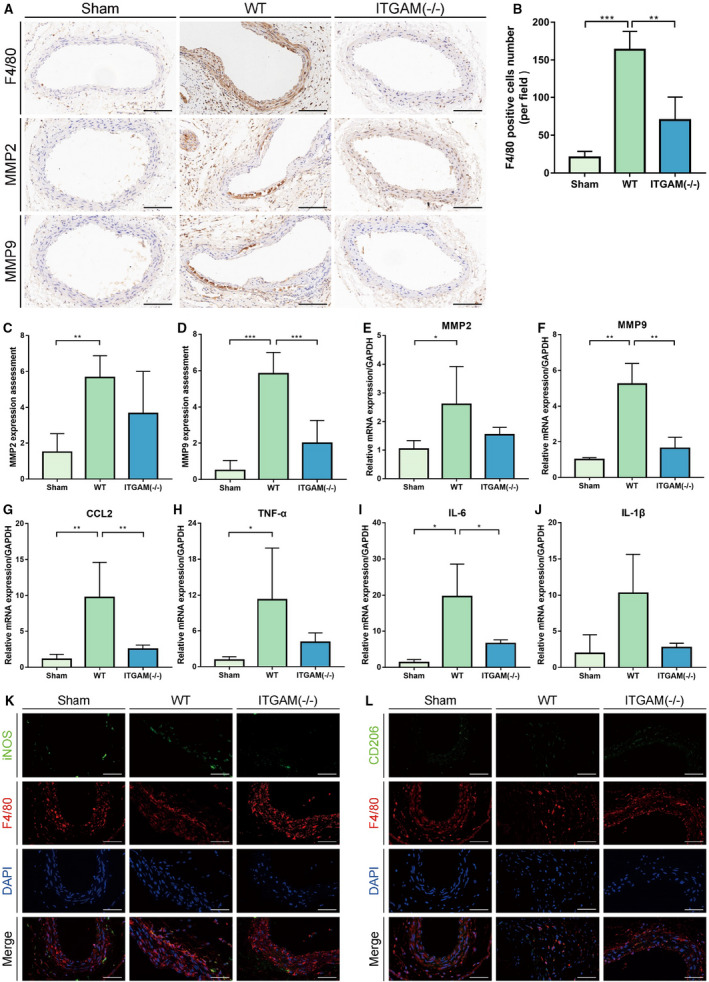
A, Representative images of immunohistochemical staining for F4/80, MMP2, and MMP9 in the aortic tissues of saline‐ or CaCl2‐treated WT or ITGAM(‐/‐) mice. Scale bar=100 μm. B, Quantification of F4/80‐positive macrophages in the infrarenal abdominal aortas of the mice from the 3 experimental groups under a microscope (n=6 for each group), **P<0.01, ***P<0.001. C and D, Quantification of MMP2 and MMP9 immunostaining in the abdominal aortic walls of the experimental mice (n=6), **P<0.01, ***P<0.001. E through J, Relative MMP2, MMP9, CCL2, TNF‐α, IL‐6, and IL‐1β mRNA expression in the abdominal aortic walls of the experimental mice (n=3), *P<0.05, **P<0.01. K and L, Representative images of dual immunofluorescence staining of iNOS (green), CD206 (green), F4/80 (red), and DAPI (blue) in the abdominal aortas of WT and ITGAM(‐/‐) mice 6 weeks after exposure to CaCl2. Scale bars=50 μm. DAPI indicates 4',6‐diamidino‐2‐phenylindole; IL‐6, interleukin 6; IL‐1β, interleukin 1β; iNOS, inducible nitric oxide synthase; ITGAM, integrin subunit alpha M; MMP, matrix metalloproteinase; TNF‐α, tumor necrosis factor α; and WT, wild‐type.
CD11b Mediates Macrophage Adhesion
The first step of macrophage infiltration is the interaction between ECs and circulating monocytes/macrophages, and this step includes macrophage adhesion to and transmigration across ECs. To commence, we investigated whether CD11b mediates macrophage adhesion. As shown by WT BMDM and EC coculture experiments in vitro, a CD11b‐neutralizing antibody significantly inhibited BMDM adhesion to angiotensin II–stimulated ECs, while the CD11b agonist LA‐1 markedly promoted the attachment of BMDMs (Figure 4A). Furthermore, we isolated primary BMDMs from WT and ITGAM(‐/‐) mice, and verified that the number of adhesive ITGAM(‐/‐) BMDMs was significantly lower than that of adhesive WT BMDMs. These findings indicated that CD11b was involved in macrophage adhesion (Figure 4B). Notably, the initial tethering and rolling of macrophages rely on the P‐selectin of ECs, which is followed by the CD11b‐dependent firm adhesion of macrophages. 26 It remains unclear whether the simultaneous blockade of P‐selectin and CD11b has a synergistic effect on the inhibition of macrophage adhesion. Fucoidan is a well‐established sulfated polysaccharide that exhibits high affinity for the P‑selectin on activated ECs and can abolish the selectin‑dependent recruitment of leukocytes. 27 In this study, we also confirmed that fucoidan limited WT BMDM adhesion to angiotensin II‐stimulated ECs in a dose‐dependent manner. However, the number of adhesive ITGAM(‐/‐) BMDMs failed to show a significant decrease after treatment with different concentrations of fucoidan compared with vehicle (Figure 4B and 4C).
Figure 4. CD11b induces macrophage adhesion.
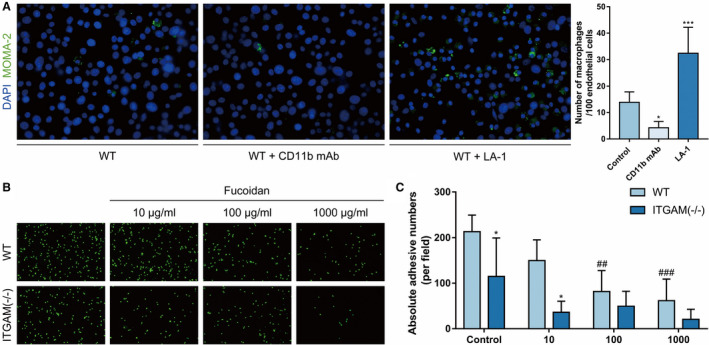
A, Representative images and quantification of WT BMDM adhesion to primary mouse aortic ECs in the absence or presence of CD11b monoclonal antibody or the CD11b agonist LA‐1. The ECs were stimulated with 1 μmol/L angiotensin II overnight before coculture. n=6, *P<0.05 vs control, ***P<0.001 vs control. B, Representative images of WT and ITGAM(‐/‐) BMDM adhesion to primary ECs, which were activated and incubated with 10 μg/mL, 100 μg/mL, or 1000 μg/mL fucoidan or vehicle before coculture. C, Quantification of WT and ITGAM(‐/‐) BMDM adhesion to activated primary ECs under different conditions. n=5, *P<0.05 vs WT BMDMs under the same conditions, ##P<0.01 vs control wild type BMDMs, ###P<0.001 vs control WT BMDMs. BMDMs indicates bone marrow–derived macrophages; DAPI, 4',6‐diamidino‐2‐phenylindole; ECs, endothelial cells; ITGAM, integrin subunit alpha M; LA‐1, leukadherin‐1; MOMA, monocyte/macrophage and WT, wild‐type.
CD11b Does Not Affect Macrophage Survival
In vivo, the survival of circulating monocytes/macrophages also potentially affects the infiltration of macrophage. Flow cytometry was performed to assess the apoptosis of WT and ITGAM(‐/‐) BMDMs. Neither the early apoptosis nor the late apoptosis rates were significantly different between the WT and ITGAM(‐/‐) BMDMs (Figure 5A). We further detected the expression of apoptosis‐related proteins, including p53, caspase3, and Bax, using Western blot. The results confirmed that there was no significant difference in apoptosis‐related protein expression between the 2 groups (Figure 5B). Additionally, the cell counting assays also suggested that the viability of the ITGAM(‐/‐) BMDMs was not significantly different from that of the WT BMDMs (Figure 5C).
Figure 5. CD11b does not affect macrophage survival.
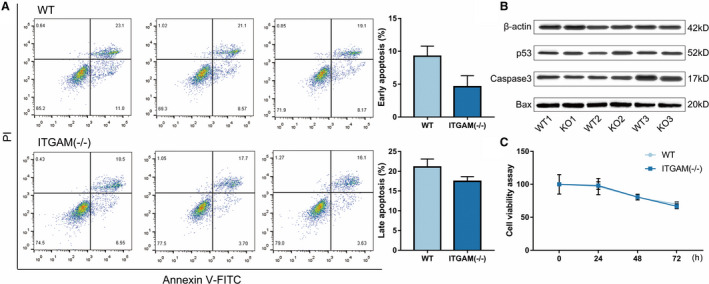
A, Primary BMDMs from WT and ITGAM(‐/‐) mice were collected and stained with FITC‐Annexin V and PI. The early and late cell apoptosis rates were measured by flow cytometry. No significant difference in the apoptosis rate was observed between the 2 groups (n=3). B, The expression of apoptosis‐related proteins, including p53, caspase3, and Bax, in WT and ITGAM(‐/‐) BMDMs was measured by Western blot. C, A cell viability assay was performed in WT and ITGAM(‐/‐) BMDMs using the Cell Counting Kit‐8. No significant difference in the absorbance (OD 450 nm) was found between the 2 groups (n=3). BMDMs indicates bone marrow–derived macrophages; ITGAM, integrin subunit alpha M; FITC, fluorescein isothiocyanate conjugated; PI, propidium iodide and WT, wild‐type.
CD11b Regulates Macrophage Transendothelial Migration
To further determine the effect of CD11b on macrophage functions, we performed RNA sequencing of 3‐paired BMDM samples from WT and ITGAM(‐/‐) mice. After preprocessing, a total of 11 376 genes were identified. Of these genes, 102 DEGs, consisting of 54 significantly upregulated DEGs and 48 significantly downregulated DEGs, were retrieved for the subsequent bioinformatics analysis (Figure 6A). The expression patterns of the top 10 upregulated and downregulated DEGs are shown in Figure 6B. Moreover, Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses showed that the downregulated genes were mainly enriched in leukocyte transendothelial migration (Figure 6C), while the upregulated genes were enriched in the Toll‐like receptor (TLR) signaling pathway (Figure 6D). Then, we isolated another 3‐paired BMDM samples from WT and ITGAM(‐/‐) mice and verified the DEGs involved in the enriched pathways (CCL3, CCL4, CCR5, CYBB, and GNB4) and several proinflammatory mediators (CCL2, IL1B, IL6, and TNF‐α) using quantitative reverse transcriptase PCR. The changes in the mRNA expression levels of CCL3, CCL4, CCR5, and GNB4 were validated in ITGAM(‐/‐) and WT BMDMs (Figure 6E).
Figure 6. CD11b regulates macrophage transendothelial migration.
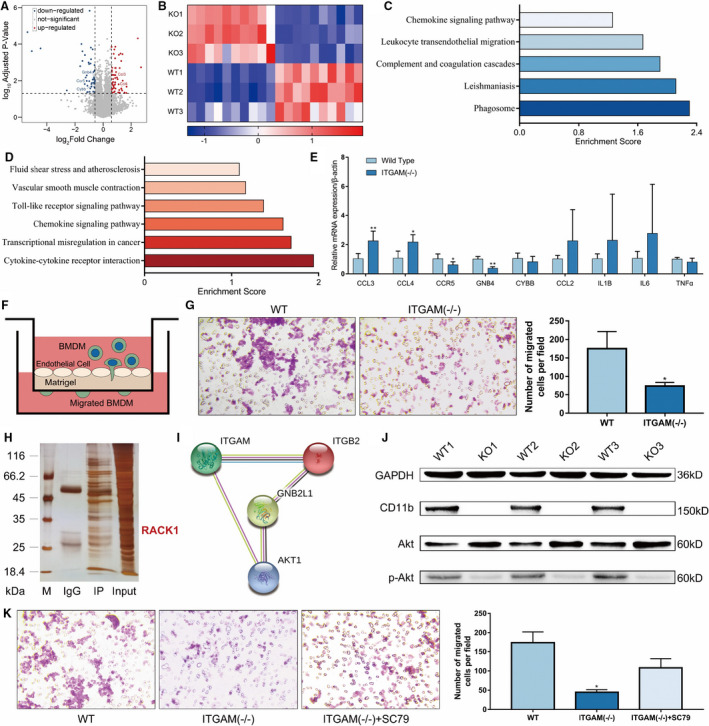
A, Volcano plots show the ‐lg (adjusted P value) against the log2 (fold change) for each gene. The black horizontal and vertical dashed lines indicate the filtering criteria (|log2 (fold change)| >1.5 and P <0.05). The red dots indicate upregulated DEGs, and the blue dots indicate downregulated DEGs. B, Heat maps of the top 10 upregulated and downregulated DEGs between WT and ITGAM(‐/‐) BMDMs. C, Pathway enrichment analysis of downregulated DEGs in ITGAM(‐/‐) BMDMs, P<0.05. D, Pathway enrichment analysis of upregulated DEGs in ITGAM(‐/‐) BMDMs, P<0.05. E, Quantification of CCL3, CCL4, CCR5, GNB4, CYBB, CCL2, IL‐1β, IL‐6, and TNF‐α mRNA expression in WT and ITGAM(‐/‐) BMDMs, *P<0.05 vs WT BMDMs, **P<0.01 vs WT BMDMs. F, Schematic experimental design of BMDM transendothelial migration, BMDMs seeded in the Transwell insert transmigrated across the endothelial cells and adhered to the lower side of the upper chamber. G, Representative images and quantification of WT and ITGAM(‐/‐) BMDMs that had migrated through the endothelial cell layer. The data represent the mean±SEM of 3 independent experiments, *P<0.05 vs WT BMDMs. H, MS assays of CD11b‐interacting proteins. Whole‐cell lysates of WT BMDMs were immunoprecipitated with anti‐flag protein A/G or control IgG. The immunoprecipitated products were separated and silver stained. MS identified an interaction between CD11b and RACK1 in WT BMDMs via the differential bands. I, The protein–protein interaction network showed that CD18 (ITGB2) was a bridging protein between CD11b (ITGAM) and RACK1 (GNB2L1), and Akt was a common protein downstream of RACK1 and CD11b. J, Western blot of CD11b, Akt, and phosphorylated Akt expression in WT and ITGAM(‐/‐) BMDMs (n=3). K, Representative images and quantification of WT and ITGAM(‐/‐) BMDMs that had migrated through the endothelial cell layer in the absence or presence of the Akt agonist SC79. The data represent the mean±SEM of 3 independent experiments, *P<0.05 vs WT BMDMs. BMDMs indicates bone marrow–derived macrophages; CCL, C‐C motif chemokine ligand; DEGs, differentially expressed genes; ITGAM, integrin subunit alpha M; MS, mass spectrometry; TNF‐α, tumor necrosis factor α; and WT, wild‐type.
On the basis of this function enrichment analysis, we used a Transwell system (Figure 6F) and found that the number of transendothelial migrated ITGAM(‐/‐) BMDMs was significantly lower than that of transendothelial migrated WT BMDMs (P<0.05, Figure 6G). To understand the molecular mechanisms by which CD11b induces transendothelial migration, we detected potential CD11b interacting proteins in stable CD11b‐expressing BMDMs by coimmunoprecipitation and MS analysis. The proteomic analysis identified 92 unique proteins that interact with CD11b, including receptor for activated C kinase 1 (RACK1) (Figure 6H). However, a protein–protein interaction network revealed that CD11b might bind to RACK1 via CD18 (integrin β2), and it might not directly bind to RACK1 (Figure 6I). Notably, as a common protein downstream of RACK1 and CD11b, Akt has been proven to be associated with macrophage transendothelial migration. 28 Western blot analysis confirmed that knocking out CD11b could reduce the level of phosphorylated Akt in the BMDMs (Figure 6J). Moreover, pretreatment with the Akt agonist SC79 partly rescued the impaired transendothelial migration of the ITGAM(‐/‐) BMDMs (Figure 6K).
Discussion
In the current study, we determined that CD11b, a member of integrins, is upregulated in human and experimental AAA tissues. In vivo, ITGAM deficiency obviously attenuates AAA development and ameliorates macrophage infiltration and polarization, therefore limiting the inflammatory response and extracellular matrix degradation in the aortic wall. Mechanistically, ITGAM deficiency reduces the adhesion of macrophages to ECs and transmigration across EC layers by inhibiting the expression of CCR5 and activation of the Akt pathway in vitro (Figure 7). Notably, while both of the mRNA and protein expression levels of MMP9 and MMP2 were reported, further studies could correlate these end points with their activity. Besides inflammation, CD11b has also been proven to mediate the interaction between monocytes and platelets, thereby promoting thrombosis. 29 , 30 Intraluminal thrombus is a well‐established risk factor for the exacerbation of AAA. 31 , 32 Overall, our study confirms a pathogenic role of CD11b in AAA progression other than its role as a biomarker of monocytes. CD11b might be a potential therapeutic target in the medical treatment of AAA.
Figure 7. Proposed mechanism of CD11b‐mediated macrophage adhesion and transendothelial migration.
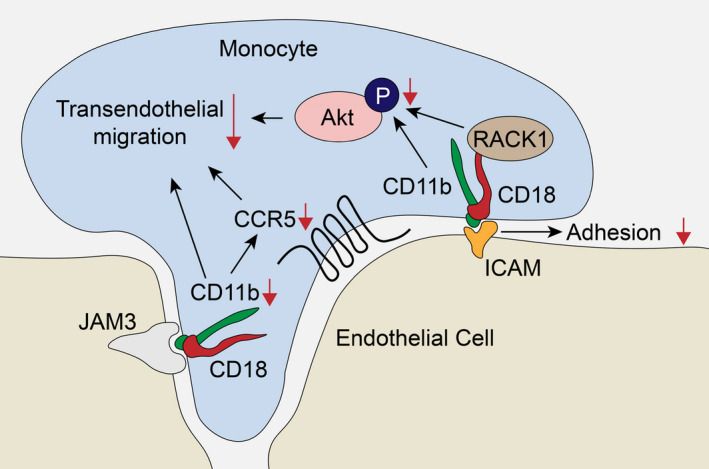
(1) Monocyte‐derived CD11b specifically recognizes ICAM on endothelial cells, helping monocytes adhere to endothelial cells and further transmigrate into the subendothelial space. (2) ITGAM deficiency impairs the macrophage transendothelial migratory function by limiting the expression of CCR5, inhibiting the phosphorylation of Akt via RACK1, and reducing the interaction with JAM3. ICAM indicates intercellular adhesion molecule; ITGAM, integrin subunit alpha M; and RACK1, receptor for activated C kinase 1.
Over the past decades, circulating monocyte mobilization and infiltration into the aortic wall have been demonstrated to participate in all stages of AAA formation. 4 Various studies have attempted to limit macrophage accumulation at sites of vascular injury by blocking chemokine signaling axes, such as CCL2/CCR2 and CXCL12/CXCR4. 33 , 34 However, chemokine pathways are abundant in vivo, of which a broad inhibitor is warranted to enable effectively targeting monocytes chemotaxis in AAA. In this study, we focused on the interactions between monocytes and ECs, including tethering, rolling, adhesion, and transendothelial migration. To commence, the adhesion of monocytes to ECs mainly depends on selectins and integrins. 5 Hannawa et al reported that P‐selectin deficiency attenuated AAA formation in the elastase aortic perfusion model because of a blunting of the inflammatory response. Here, we found that in vitro neutralization of P‐selectin on ECs using fucoidan and ITGAM deficiency could reduce macrophage adhesion. Unexpectedly, ECs pretreated with fucoidan failed to show a significant decrease in the number of adhesive ITGAM(‐/‐) BMDMs. We hypothesized that CD11b‐mediated firm adhesion might play a leading role in the interaction between ECs and monocytes, and this adhesion might overpower the effect of P‐selectin‐induced rolling adhesion. Notably, the binding of P‐selectin to P‐selectin glycoprotein ligand 1 can activate αLβ2 integrin, but does not increase the affinity of leukocytes. 35
To further explore the role of CD11b in macrophage function, we performed RNA sequencing in primary BMDMs from WT and ITGAM(‐/‐) mice. As a result, the downregulated genes in the ITGAM(‐/‐) BMDMs were enriched in leukocyte transendothelial migration, and this finding was confirmed by in vitro Transwell experiments. Previously, several studies showed that the transendothelial migration of macrophages is regulated by the aforementioned adhesive molecules, chemokine axes, and migratory properties. 5 Here, we verified the downregulation of CCR5 in ITGAM(‐/‐) BMDMs using quantitative reverse transcriptase PCR. CCR5 plays a crucial role in the migration and recruitment of monocytes/macrophages in vascular diseases. 36 Blockade of CCR5 has been demonstrated to attenuate macrophage accumulation and infiltration into atherosclerotic arteries. 37 However, the potential mechanism by which CCR5 is reduced in ITGAM(‐/‐) BMDMs remains unclear. Furthermore, a Mac‐1‐neutralizing antibody or Akt knockout has been shown to significantly inhibit the migration of BMDMs. 28 Through coimmunoprecipitation–MS and bioinformatics analyses, we found that CD11b potentially interacts with RACK1 via CD18, and regulates the activation of the downstream Akt pathway. Nevertheless, pretreatment with the Akt agonist SC79 partially rescued the transendothelial migration of ITGAM(‐/‐) BMDMs, suggesting that CD11b might mediate the migratory properties of macrophages via the Akt pathway. Moreover, Cui et al investigated the distinct migratory properties of M1, M2, and resident macrophages in a 3‐dimensional environment and determined that CD11b knockout decreases the migration of M2 macrophages. 38 This decreased migration may result in the retention of ITGAM(‐/‐) M2‐polarized macrophages in the aortic wall and exert anti‐inflammatory effects.
Recently, 2 studies have shown that CD11b can inhibit the expression of MyD88 and the activation of NF‐κB, thus negatively regulating the TLR‐dependent immune response in macrophages. 39 , 40 Here, we also found that ITGAM(‐/‐) BMDMs contained upregulated CCL3 and CCL4, which are the downstream of the TLR pathway; these results indicate that ITGAM deficiency might enhance the activation of the TLR pathway. Additionally, Yao et al also confirmed that CD11b interacts with TLR4 and subsequently induces its endocytosis, which partially inhibits the interaction of lipopolysaccharide with TLR4 and the subsequent inflammatory response. 41 Interestingly, TLR4 was not detected in our coimmunoprecipitation–MS analysis, which might be because of the transmembrane protein properties of both TLR4 and CD11b.
In fact, other β2 integrins, including αLβ2 (CD11a/CD18), αXβ2 (CD11c/CD18), and αDβ2 (CD11d/CD18), also play an important role in chronic vascular inflammation. 42 , 43 Nie et al reported that in rats with diet‐induced hypercholesterolemia, an anti‐CD11a monoclonal antibody could reduce the number of macrophages adhering to the intima by 31% compared with the control. 44 Similarly, depletion of CD11c‐positive dendritic cells abrogates inflammatory cell recruitment and reduces neutrophil elastase activity, thus limiting AAA growth. 45 Moreover, Aziz et al demonstrated the enhanced migration of CD11d‐deficient M1 macrophages and proved the contribution of CD11d to the retention of macrophages at inflammatory sites during atherogenesis. 46 Overall, CD18‐blocking antibodies significantly decreased neutrophil adhesion and transmigration, thereby ameliorating AAA development in rats. 6 To the best of our knowledge, CD11b is the last subset of CD11 families, whose role in vascular diseases has not been previously determined.
Currently, the CD11b‐specific agonist LA‐1 has been proven to promote the endocytosis of TLR4 and inhibit the lipopolysaccharide‐induced inflammatory response in macrophages. 41 In vitro, LA‐1 enhances the binding of CD11b to intercellular adhesion molecule‐1 via the formation of long membrane tethers, abates the detachment force, and reduces the transendothelial migration of macrophages. 47 , 48 In vivo, various studies confirmed that LA‐1 could attenuate hyperoxia‐induced lung injury and vascular injury by inhibiting macrophage infiltration. 47 , 49 In addition to its effects on monocytes/macrophages, Roberts et al determined that LA‐1 pretreatment also reduces the secretion of the proinflammatory cytokines interferon‐γ, TNF, and CCL4 by monokine‐stimulated NK cells. 50 Moreover, LA‐1 does not induce global conformational changes in CD11b/CD18, while CD11b‐activating antibodies cause significant CD11b/CD18 macro clustering and ligand‐mimetic outside‐in signaling. 51 Thus, LA‐1 has great potential in limiting the development of AAA.
Conclusions
In summary, CD11b mediates the adhesion and transendothelial migration of macrophages via the regulation of CCR5 expression and the Akt pathway, and further promotes macrophage infiltration without affecting their apoptosis and proliferation. ITGAM deficiency attenuates AAA development and ameliorates macrophage infiltration and polarization in vivo, thereby indicating that CD11b may hold promise as a therapeutic target for the treatment of AAA.
Sources of Funding
The present research was sponsored by the National Natural Science Foundation of China (grant numbers 81870342, 81870476).
Disclosures
None.
Supporting information
Tables S1–S2
Figure S1
(J Am Heart Assoc. 2021;10:e019900. DOI: 10.1161/JAHA.120.019900.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.019900
For Sources of Funding and Disclosures, see page 13.
References
- 1. Force USPST , Owens DK, Davidson KW, Krist AH, Barry MJ, Cabana M, Caughey AB, Doubeni CA, Epling JW, Kubik M, et al. Screening for abdominal aortic aneurysm: US preventive services task force recommendation statement. JAMA. 2019;322:2211–2218. DOI: 10.1001/jama.2019.18928. [DOI] [PubMed] [Google Scholar]
- 2. Kent KC. Clinical practice. Abdominal aortic aneurysms. N Engl J Med. 2014;371:2101–2108. DOI: 10.1056/NEJMcp1401430. [DOI] [PubMed] [Google Scholar]
- 3. Golledge J. Abdominal aortic aneurysm: update on pathogenesis and medical treatments. Nat Rev Cardiol. 2019;16:225–242. DOI: 10.1038/s41569-018-0114-9. [DOI] [PubMed] [Google Scholar]
- 4. Raffort J, Lareyre F, Clement M, Hassen‐Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. 2017;14:457–471. DOI: 10.1038/nrcardio.2017.52. [DOI] [PubMed] [Google Scholar]
- 5. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. DOI: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 6. Ricci MA, Strindberg G, Slaiby JM, Guibord R, Bergersen LJ, Nichols P, Hendley ED, Pilcher DB. Anti‐CD 18 monoclonal antibody slows experimental aortic aneurysm expansion. J Vasc Surg. 1996;23:301–307. DOI: 10.1016/S0741-5214(96)70274-4. [DOI] [PubMed] [Google Scholar]
- 7. Solovjov DA, Pluskota E, Plow EF. Distinct roles for the alpha and beta subunits in the functions of integrin alphambeta2. J Biol Chem. 2005;280:1336–1345. [DOI] [PubMed] [Google Scholar]
- 8. Zhou L, Lee DH, Plescia J, Lau CY, Altieri DC. Differential ligand binding specificities of recombinant CD11b/CD18 integrin I‐domain. J Biol Chem. 1994;269:17075–17079. DOI: 10.1016/S0021-9258(17)32522-X. [DOI] [PubMed] [Google Scholar]
- 9. Lee SJ, Baek SE, Jang MA, Kim CD. SIRT1 inhibits monocyte adhesion to the vascular endothelium by suppressing Mac‐1 expression on monocytes. Exp Mol Med. 2019;51:1–12. DOI: 10.1038/s12276-019-0239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buffone A Jr, Anderson NR, Hammer DA. Human neutrophils will crawl upstream on ICAM‐1 if Mac‐1 is blocked. Biophys J. 2019;117:1393–1404. DOI: 10.1016/j.bpj.2019.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H, Hong L‐J, Huang J‐Y, Jiang Q, Tao R‐R, Tan C, Lu N‐N, Wang C‐K, Ahmed MM, Lu Y‐M, et al. P2RX7 sensitizes Mac‐1/ICAM‐1‐dependent leukocyte‐endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. 2015;25:674–690. DOI: 10.1038/cr.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan SQ, Khan I, Gupta V. CD11b activity modulates pathogenesis of lupus nephritis. Front Med (Lausanne). 2018;5:52. DOI: 10.3389/fmed.2018.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Samadzadeh KM, Chun KC, Nguyen AT, Baker PM, Bains S, Lee ES. Monocyte activity is linked with abdominal aortic aneurysm diameter. J Surg Res. 2014;190:328–334. DOI: 10.1016/j.jss.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 14. Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies–statement from ATVB council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. DOI: 10.1161/ATVBAHA.117.309524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma N, Dev R, Belenchia AM, Aroor AR, Whaley‐Connell A, Pulakat L, Hans CP. Deficiency of il12p40 (interleukin 12 p40) promotes ang ii (angiotensin ii)‐induced abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2019;39:212–223. DOI: 10.1161/ATVBAHA.118.311969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Satoh K, Nigro P, Matoba T, O'Dell MR, Cui Z, Shi XI, Mohan A, Yan C, Abe J‐I, Illig KA, et al. Cyclophilin A enhances vascular oxidative stress and the development of angiotensin II‐induced aortic aneurysms. Nat Med. 2009;15:649–656. DOI: 10.1038/nm.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhong L, He X, Si X, Wang H, Li B, Hu Y, Li M, Chen X, Liao W, Liao Y, et al. SM22alpha (smooth muscle 22alpha) prevents aortic aneurysm formation by inhibiting smooth muscle cell phenotypic switching through suppressing reactive oxygen species/NF‐kappab (nuclear factor‐kappab). Arterioscler Thromb Vasc Biol. 2019;39:e10–e25. DOI: 10.1161/ATVBAHA.118.311917. [DOI] [PubMed] [Google Scholar]
- 18. Assouvie A, Daley‐Bauer LP, Rousselet G. Growing murine bone marrow‐derived macrophages. Methods Mol Biol. 2018;1784:29–33. DOI: 10.1007/978-1-4939-7837-3_3. [DOI] [PubMed] [Google Scholar]
- 19. Wang JM, Chen AF, Zhang K. Isolation and primary culture of mouse aortic endothelial cells. J Vis Exp. 2016;118: 52965. DOI: 10.3791/52965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ. Quantitation of apoptosis and necrosis by Annexin V binding, propidium iodide uptake, and flow cytometry. Cold Spring Harb Protoc. 2016;2016.953–957. 10.1101/pdb.prot087288. [DOI] [PubMed] [Google Scholar]
- 21. Hadi T, Boytard L, Silvestro M, Alebrahim D, Jacob S, Feinstein J, Barone K, Spiro W, Hutchison S, Simon R, et al. Macrophage‐derived netrin‐1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat Commun. 2018;9:5022. DOI: 10.1038/s41467-018-07495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maccarrone G, Bonfiglio JJ, Silberstein S, Turck CW, Martins‐de‐Souza D. Characterization of a protein interactome by co‐immunoprecipitation and shotgun mass spectrometry. Methods Mol Biol. 2017;1546:223–234. DOI: 10.1007/978-1-4939-6730-8_19. [DOI] [PubMed] [Google Scholar]
- 23. Ma D, Zheng B, Suzuki T, Zhang R, Jiang C, Bai D, Yin W, Yang Z, Zhang X, Hou L, et al. Inhibition of KLF5‐Myo9b‐RhoA pathway‐mediated podosome formation in macrophages ameliorates abdominal aortic aneurysm. Circ Res. 2017;120:799–815. DOI: 10.1161/CIRCRESAHA.116.310367. [DOI] [PubMed] [Google Scholar]
- 24. Kusters PJH, Seijkens TTP, Beckers L, Lievens D, Winkels H, de Waard V, Duijvestijn A, Lindquist Liljeqvist M, Roy J, Daugherty A, et al. CD40L deficiency protects against aneurysm formation. Arterioscler Thromb Vasc Biol. 2018;38:1076–1085. DOI: 10.1161/ATVBAHA.117.310640. [DOI] [PubMed] [Google Scholar]
- 25. Murray P, Allen J, Biswas S, Fisher E, Gilroy D, Goerdt S, Gordon S, Hamilton J, Ivashkiv L, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. DOI: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of PSGL‐1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100:485–491. DOI: 10.1172/JCI119556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Piqueras L, Kubes P, Alvarez A, O'Connor E, Issekutz AC, Esplugues JV, Sanz MJ. Angiotensin II induces leukocyte‐endothelial cell interactions in vivo via AT(1) and AT(2) receptor‐mediated P‐selectin upregulation. Circulation. 2000;102:2118–2123. DOI: 10.1161/01.cir.102.17.2118. [DOI] [PubMed] [Google Scholar]
- 28. Jin SY, Kim EK, Ha JM, Lee DH, Kim JS, Kim IY, Song SH, Shin HK, Kim CD, Bae SS. Insulin regulates monocyte trans‐endothelial migration through surface expression of macrophage‐1 antigen. Biochim Biophys Acta. 2014;1842:1539–1548. DOI: 10.1016/j.bbadis.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 29. Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, Soloviev DA, Bledzka K, Ustinov V, Zhu L, Qin J, et al. Leukocyte integrin mac‐1 regulates thrombosis via interaction with platelet gpibalpha. Nat Commun. 2017;8:15559. DOI: 10.1038/ncomms15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamad OA, Mitroulis I, Fromell K, Kozarcanin H, Chavakis T, Ricklin D, Lambris JD, Ekdahl KN, Nilsson B. Contact activation of C3 enables tethering between activated platelets and polymorphonuclear leukocytes via CD11b/CD18. Thromb Haemost. 2015;114:1207–1217. DOI: 10.1160/TH15-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Domonkos A, Staffa R, Kubicek L. Effect of intraluminal thrombus on growth rate of abdominal aortic aneurysms. Int Angiol. 2019;38:39–45. DOI: 10.23736/S0392-9590.18.04006-3. [DOI] [PubMed] [Google Scholar]
- 32. Wiernicki I, Parafiniuk M, Kolasa‐Wolosiuk A, Gutowska I, Kazimierczak A, Clark J, Baranowska‐Bosiacka I, Szumilowicz P, Gutowski P. Relationship between aortic wall oxidative stress/proteolytic enzyme expression and intraluminal thrombus thickness indicates a novel pathomechanism in the progression of human abdominal aortic aneurysm. FASEB J. 2019;33:885–895. DOI: 10.1096/fj.201800633R. [DOI] [PubMed] [Google Scholar]
- 33. Nie H, Wang HX, Tian C, Ren HL, Li FD, Wang CY, Li HH, Zheng YH. Chemokine (C‐X‐C motif) receptor 2 blockade by SB265610 inhibited angiotensin II‐induced abdominal aortic aneurysm in Apo E(‐/‐) mice. Heart Vessels. 2019;34:875–882. DOI: 10.1007/s00380-018-1301-7. [DOI] [PubMed] [Google Scholar]
- 34. Michineau S, Franck G, Wagner‐Ballon O, Dai J, Allaire E, Gervais M. Chemokine (C‐X‐C Motif) receptor 4 blockade by AMD3100 inhibits experimental abdominal aortic aneurysm expansion through anti‐inflammatory effects. Arterioscler Thromb Vasc Biol. 2014;34:1747–1755. DOI: 10.1161/ATVBAHA.114.303913. [DOI] [PubMed] [Google Scholar]
- 35. Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E‐ or P‐selectin induces the extended but not high‐affinity conformation of LFA‐1 in neutrophils. Blood. 2010;116:617–624. DOI: 10.1182/blood-2010-01-266122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mateo T, Naim Abu Nabah Y, Abu Taha M, Mata M, Cerdá‐Nicolás M, Proudfoot AEI, Stahl RAK, Issekutz AC, Cortijo J, Morcillo EJ, et al. Angiotensin II‐induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J Immunol. 2006;176:5577–5586. DOI: 10.4049/jimmunol.176.9.5577. [DOI] [PubMed] [Google Scholar]
- 37. Combadiere C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. DOI: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- 38. Cui K, Ardell CL, Podolnikova NP, Yakubenko VP. Distinct migratory properties of M1, M2, and resident macrophages are regulated by alphadbeta2 and alphambeta2 integrin‐mediated adhesion. Front Immunol. 2018;9:2650. DOI: 10.3389/fimmu.2018.02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR‐triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl‐b. Nat Immunol. 2010;11:734–742. DOI: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 40. Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, Kumar V, Armstrong AR, Carmona‐Rivera C, Dorschner JM, et al. CD11b activation suppresses TLR‐dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest. 2017;127:1271–1283. DOI: 10.1172/JCI88442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yao X, Dong G, Zhu Y, Yan F, Zhang H, Ma Q, Fu X, Li X, Zhang QingQing, Zhang J, et al. Leukadherin‐1‐mediated activation of CD11b inhibits LPS‐induced pro‐inflammatory response in macrophages and protects mice against endotoxic shock by blocking LPS‐TLR4 interaction. Front Immunol. 2019;10:215. DOI: 10.3389/fimmu.2019.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, Fury W, Bai YU, Wagner S, Li D, et al. Role of interleukin‐1 signaling in a mouse model of kawasaki disease‐associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2016;36:886–897. DOI: 10.1161/ATVBAHA.115.307072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kawamura A, Miura S, Murayama T, Iwata A, Zhang B, Nishikawa H, Tsuchiya Y, Matsuo K, Tsuji E, Saku K. Increased expression of monocyte CD11a and intracellular adhesion molecule‐1 in patients with initial atherosclerotic coronary stenosis. Circ J. 2004;68:6–10. DOI: 10.1253/circj.68.6. [DOI] [PubMed] [Google Scholar]
- 44. Nie Q, Fan J, Haraoka S, Shimokama T, Watanabe T. Inhibition of mononuclear cell recruitment in aortic intima by treatment with anti‐ICAM‐1 and anti‐LFA‐1 monoclonal antibodies in hypercholesterolemic rats: implications of the ICAM‐1 and LFA‐1 pathway in atherogenesis. Lab Invest. 1997;77:469–482. [PubMed] [Google Scholar]
- 45. Krishna SM, Moran CS, Jose RJ, Lazzaroni S, Huynh P, Golledge J. Depletion of CD11c+ dendritic cells in apolipoprotein E‐deficient mice limits angiotensin II‐induced abdominal aortic aneurysm formation and growth. Clin Sci (Lond). 2019;133:2203–2215. DOI: 10.1042/CS20190924. [DOI] [PubMed] [Google Scholar]
- 46. Aziz MH, Cui K, Das M, Brown KE, Ardell CL, Febbraio M, Pluskota E, Han J, Wu H, Ballantyne CM, et al. The upregulation of integrin alphadbeta2 (CD11d/CD18) on inflammatory macrophages promotes macrophage retention in vascular lesions and development of atherosclerosis. J Immunol. 2017;198:4855–4867. DOI: 10.4049/jimmunol.1602175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, Barth CJ, Lugo G, Donnelly M, Nayer A, et al. Small molecule‐mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal. 2011;4:ra57. DOI: 10.1126/scisignal.2001811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Celik E, Faridi MH, Kumar V, Deep S, Moy VT, Gupta V. Agonist leukadherin‐1 increases cd11b/cd18‐dependent adhesion via membrane tethers. Biophys J. 2013;105:2517–2527. DOI: 10.1016/j.bpj.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jagarapu J, Kelchtermans J, Rong M, Chen S, Hehre D, Hummler S, Faridi MH, Gupta V, Wu S. Efficacy of leukadherin‐1 in the prevention of hyperoxia‐induced lung injury in neonatal rats. Am J Respir Cell Mol Biol. 2015;53:793–801. DOI: 10.1165/rcmb.2014-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roberts AL, Furnrohr BG, Vyse TJ, Rhodes B. The complement receptor 3 (CD11b/CD18) agonist leukadherin‐1 suppresses human innate inflammatory signalling. Clin Exp Immunol. 2016;185:361–371. DOI: 10.1111/cei.12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Faridi MH, Altintas MM, Gomez C, Duque JC, Vazquez‐Padron RI, Gupta V. Small molecule agonists of integrin CD11b/CD18 do not induce global conformational changes and are significantly better than activating antibodies in reducing vascular injury. Biochim Biophys Acta. 2013;1830:3696–3710. DOI: 10.1016/j.bbagen.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hannawa KK, Cho BS, Sinha I, Roelofs KJ, Myers DD, Wakefield TJ, Stanley JC, Henke PK, Upchurch GR, Jr . Attenuation of experimental aortic aneurysm formation in p‐selectin knockout mice. Ann N Y Acad Sci. 2006;1085: 353–359. 10.1196/annals.1383.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figure S1