

Abstract
In this paper, a set of 3-methylquniazolinone derivatives were designed, synthesised, and studied the preliminary structure-activity relationship for antiproliferative activities. All target compounds performed significantly inhibitory effects against wild type epidermal growth factor receptor tyrosine kinase (EGFRwt-TK) and tumour cells (A431, A549, MCF-7, and NCI-H1975). In particular, compound 4d 3-fluoro-N-(4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)phenyl)benzamide showed higher antiproliferative activities against all tumour cells than Gefitinib (IC50 of 3.48, 2.55, 0.87 and 6.42 μM, respectively). Furthermore, compound 4d could induce apoptosis of MCF-7 cells and arrest in G2/M phase at the tested concentration. Molecular docking and ADMET studies showed that compound 4d could closely form many hydrogen bonds with EGFRwt-TK. Therefore, compound 4d is potential to develop as novel anti-cancer drug.
Keywords: Quinazolinone, EGFR, kinase inhibitor, structure-activity relationship, antiproliferative
1. Introduction
Epidermal growth factor receptor (EGFR) tyrosine kinase (TK), a receptor tyrosine kinase, plays an important role in the process of survival, proliferation, angiogenesis, tumour micro environment, and adhesion of several malignant tumors1. Overexpression or mutation of EGFR has been observed in many human tumour cells, such as nonsmall-cell lung cancer (NSCLC), breast cancer, and brain cancer2. Due to its crucial status related to cancer development, EGFR has been considered a potential drug target for many years3–5. Currently, a series of EGFR inhibitors were developed and approved into clinical phases, such as Gefitinib, Erlotinib, Icotinib, Afatinib, and Osimertinib6. However, with the continuous emergence of drug resistance in EGFR inhibitors in clinic, design and synthesis of novel EGFR inhibitors have become an urgent work for anti-tumour drug discovery7.
Quinazolone is the main skeleton of many natural products8,9, for example Allicin C, Cyclosporin F, Rutaecarpine, and (+)-Changshanine (Figure 1). These compounds have a wide range of biological activities, including antitumor, antibacterial, antiviral, and anti-inflammatory10–12. Therefore, many groups have developed new methods to synthesise quinazolone derivatives13. Patel et al. reported 2-phenylquinazolone derivatives as EGFR inhibitors in 201714 and recently inspired from natural alkaloid L-norephedrine, Ghorab et al. published a 3-substituted quniazolinones for EGFR inhibitors15. These results indicate that the development of novel quinazolone EGFR inhibitors has attracted extensive attentions.
Figure 1.

Natural products of quinazolinone and our strategy.
Our group has been focussed on the research of developing new anti-tumour drugs for several years16–19. We have synthesised lots of antiproliferative compounds such as benzodiazepines, 1,3,4-oxadiazoles, 1,3,4-thiadiazoles20, and pyrimidines21. Based on the broad activities of quinazolone and imperative need for EGFR inhibitors, we reported a series of 3-methylquinazolone derivatives for EGFR inhibitors in last year22. As shown in Figure 1, the IC50 value of compound A against EGFRwt reached to 47 nm. However, further studies showed that compound A was not stable in mice plasma (Figure S1). The result prompted us to seek more valuable 3-methylquniazolinone derivatives. To overcome the metabolic stability in our previous research, we herein describe the design, synthesis, and studies the preliminary structure-activity relationship (SAR) of new 3-methylquinazolinone derivatives as EGFR inhibitors.
2. Experimental section
All reagents were commercially available in Sigma-Aldrich with analytical purity. Melting points were tested in digital melting point analyser with micro-display window (uncorrected, Shanghai Microelectronics Technology Co. Ltd.). The 1H and 13 C NMR spectra were recorded on Bruker (Avance) 400 MHz and JEOL (Japan) 500 MHz NMR instrument with chemical reported as δ in CDCl3 and DMSO-d6, tetramethylsilane (TMS) as the internal standard. The high-resolution mass spectrometer (HRMS) was tested in TSQ 8000 high-resolution mass spectrometer and AB SCIEX X500R QTOF.
2.1. Synthesis
The synthetic route of compounds 4a–4g was shown in Scheme 1. The experimental methods and details were described as follows.
Scheme 1.

Synthetic route of compounds 4a-4g. Reagents and conditions: i) 2-chloroacetyl chloride, AcOH, reflux, 50% yield; ii) 4-hydroxybenzaldehyde, K2CO3, KI, DMF, rt, 68% yield; iii) 3-fluorophenylethanone, NaOH, EtOH, rt, 42% yield; iv) ethyl 4-hydroxybenzoate, K2CO3, KI, DMF, rt, 64% yield; v) NaOH, EtOH, rt, 58% yield; vi) 3-fluorobenzenamine, HATU, DIEA, DMF, rt, 38% yield; vii) triethyl phosphate, tetrabutylammonium bromide, 130 °C, then 3-fluorobenzaldehyde, NaH, THF, rt, 43% yield; viii) BBr3, DCM, 0–10 °C, 72% yield; ix) compound 2, K2CO3, KI, DMF, rt, 47% yield; x) 4-nitrophenol, K2CO3, KI, DMF, rt, 53% yield; xi) Pd/C, MeOH, H2, rt, 72% yield; xii–xv) corresponding acid or isocyanate, HATU, DIEA, DMF, rt, 30–37% yield.
2.1.1. 2-(Chloromethyl)-3-methylquinazolin-4(3H)-one (2)
2-Amino-N-methylbenzamide (1.5 g, 10 mmol) was dissolved in acetic acid (50 ml), and then 2-chloroacetyl chloride (3.36 g, 30 mmol) was added. The mixture was stirred for 6 h under refluxing. Then, the solution was evaporated in vacuo and neutralised with aqueous NaHCO3. The crude product was filtered and purified with recrystallisation from ethanol. The product was dried in vacuum to give a white solid. 1.04 g; 50% yield; 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 8.0 Hz, 1H), 7.80 − 7.75 (m, 1H), 7.68 − 7.69 (m, 1H), 7.50 − 7.54 (m, 1H), 4.65 (s, 2H), 3.76 (s, 3H). Spectral properties were in accordance with the literature23.
2.1.2. 4-((3-Methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)benzaldehyde (3)
p-Hydroxybenzaldehyde (610 mg, 5 mmol) was dissolved in DMF (20 ml), and then anhydrous K2CO3 (759 mg, 5.5 mmol) was added. After stirred for 15 min at room temperature, the mixture was added compound 2 (1.04 g, 5 mmol) and KI (913 mg, 5.5 mmol). The solution was stirred for the other 2 h, and poured into water (40 ml). The crude product was filtered and purified with recrystallisation from ethanol. The product was dried in vacuum to give a light-yellow solid. 1 g; 68% yield; 1H NMR (DMSO, 400 MHz), δ 9.90 (s, 1H), 8.16 (dd, J = 8.0, 1.0 Hz, 1H), 7.90 (d, J = 8.8 Hz, 2H), 7.82 (ddd, J = 8.4, 7.2, 1.6 Hz, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.60–7.52 (m, 1H), 7.32 (d, J = 8.8 Hz, 2H), 5.45 (s, 2H), 3.62 (s, 3H). Spectral properties were in accordance with the literature23.
2.1.3. (E)-2-((4–(3-(3-Fluorophenyl)-3-oxoprop-1-enyl)phenoxy)methyl)-3-methylquinazolin-4(3H)-one (4a)
1–(3-Fluorophenyl)ethanone (276 mg, 2 mmol) and 4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)benzaldehyde (588 mg, 2 mmol) were dissolved in EtOH (20 ml), and then 2 mol/l NaOH (1 ml) was added. The mixture was stirred for 6 h at room temperature. Then, the solution was neutralised with 2 mol/l HCl (1 ml). The crude product was filtered and purified by chromatography using PE/EtOAc = 1/1 as the fluent solvent. The product was dried in vacuum to give a white solid. 348 mg; 42% yield; m.p. 208–209 °C; 1H NMR (400 MHz, DMSO) δ 8.17 (d, J = 7.6 Hz, 1H), 8.02 − 7.75 (m, 7H), 7.67 − 7.52 (m, 4H), 7.22 (d, J = 8.4 Hz, 2H), 5.40 (s, 2H), 3.63 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 189.5, 161.8, 161.6, 160.5, 159.6, 152.7, 146.9, 145.1, 135.0, 134.5 (d, J = 8.5 Hz), 131.3, 130.9 (d, J = 3.0 Hz, 7H), 128.2, 127.9, 127.7, 126.8, 125.3 (d, J = 3.5 Hz), 124.2 (d, J = 3.5 Hz), 120.8, 117.1 (d, J = 22.0 Hz), 116.1, 69.0, 30.4; HRMS-ESI (m/z): [M + H]+ calcd for C25H19FN2O3, 415.1452; found, 415.1452.
2.1.4. Ethyl 4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)benzoate (5)
Ethyl 4-hydroxybenzoate (830 mg, 5 mmol) was dissolved in DMF (20 ml), and then anhydrous K2CO3 (759 mg, 5.5 mmol) was added. After stirred for 15 min at room temperature, the mixture was added compound 2 (1.04 g, 5 mmol) and KI (913 mg, 5.5 mmol). The solution was stirred for the other 2 h, and poured into water (40 ml). The crude product was filtered and purified with recrystallisation from ethanol. The product was dried in vacuum to give a white solid. 1.08 g; 64% yiled; m.p. 114–116 °C; 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 8.8 Hz, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.77 (t, J = 7.6 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.10 (d, J = 8.8 Hz, 2H), 5.25 (s, 2H), 4.35 (q, J = 7.2 Hz, 2H), 3.74 (s, 3H), 1.38 (t, J = 7.2 Hz, 3H); 13 C NMR (100 MHz, DMSO) δ 165.7, 162.0, 161.7, 152.5, 146.9, 134.9, 131.6, 127.8, 127.6 126.7, 123.4, 120.7, 115.5, 68.8, 60.9, 30.3, 14.7; HRMS-ESI (m/z): [M + H]+ calcd for C19H18N2O4, 339.1339; found, 339.1339.
2.1.5. 4-((3-Methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)benzoic acid (6)
Compound 5 (676 mg, 2 mmol) was dissolved in EtOH (10 ml), and then 2 mol/l NaOH (2 ml) was added. The mixture was stirred for 6 h at room temperature. Then, the solution was neutralised with 2 mol/l HCl (2 ml). The crude product was filtered and dried in vacuum to give a white solid. 360 mg; 58% yield; m.p. 97–99 °C; 1H NMR (400 MHz, DMSO) δ 12.69 (s, 1H), 8.16 (d, J = 7.6 Hz, 1H), 7.91 (d, J = 8.8 Hz, 2H), 7.83 (t, J = 8.0 Hz, 1H), 7.65 (d, J = 8.4 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.20 (d, J = 8.8 Hz, 2H), 5.39 (s, 2H), 3.61 (s, 3H); 13 C NMR (100 MHz, DMSO) δ 167.4, 161.7, 152.6, 146.9, 134.9, 131.8, 127.8, 127.6, 126.7, 124.3, 120.7, 115.3, 68.8, 30.3; HRMS-ESI (m/z): [M + H]+ calcd for C17H14N2O4, 311.1036; found, 311.1023.
2.1.6. N-(3-Fluorophenyl)-4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)benzamide (4 b)
Compound 6 (310 mg, 1 mmol) and 3-fluorobenzenamine (111 mg, 1 mmol) were dissolved in DMF (5 ml), and then HATU (o-(7-aza-1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate) (380 mg, 1 mmol) was added. After stirred for 30 min, the mixture was added DIEA (N,N-diisopropylethylamine) (129 mg, 1 mmol) and stirred for 12 h at room temperature. The solution was poured into water (20 ml). The final product was filtered and dried in vacuum to give a white solid. 153 mg; 38% yield; m.p. 196–197 °C; 1H NMR (400 MHz, DMSO) δ 10.28 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 8.8 Hz, 2H), 7.83 (t, J = 7.6 Hz, 1H), 7.74 (d, J = 12.0 Hz, 1H), 7.65 (d, J = 8.0 Hz, 1H), 7.56 (dd, J = 14.0, 7.6 Hz, 2H), 7.37 (dd, J = 15.6, 8.0 Hz, 1H), 7.26 (d, J = 8.8 Hz, 2H), 6.91 (td, J = 8.4, 2.4 Hz, 1H), 5.42 (s, 2H), 3.63 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 152.5, 150.8, 149.4 (d, J = 19.0 Hz), 148.8, 142.2, 137.5, 133.3 (d, J = 9.0 Hz), 128.0, 124.6 (d, J = 7.5 Hz), 124.1, 122.4 (d, J = 15.5 Hz), 122.1, 121.4, 116.6, 113.1, 112.2, 108.3 (d, J = 16.5 Hz), 105.9, 105.8, 75.1, 44.3; HRMS-ESI (m/z): [M + H]+ calcd for C23H18FN3O3, 404.1404; found, 404.1405.
2.1.7. (E)-1-(4-Methoxystyryl)-3-fluorobenzene (8)
1-(Chloromethyl)-4-methoxybenzene (1.56 g, 10 mmol) was dissolved in triethyl phosphate (50 ml), and then tetrabutylammonium bromide (322 mg, 1 mmol) was added. The mixture was stirred at 130 °C under argon for overnight. After evaporated under vacuum, the solution was added to the solution of 3-fluorobenzaldehyde (1.24 g, 10 mmol) in anhydrous THF (50 ml). The mixture was added NaH (600 mg, 15 mmol, 60% in oil) at 0–10 °C and stirred for 12 h at room temperature. The solution was added to water (100 ml) and extracted with EtOAc (100 ml × 2), and dried over MgSO4 for 6 h. The crude product was evaporated and purified with flash chromatography using PE/EtOAc = 1/1 as the fluent solvent. The product was dried in vacuum to give a light solid. 980 mg; 43% yield; 1H NMR (400 MHz, CDCl3) δ 7.47 − 7.42 (m, 2H), 7.28 (dd, J = 8.0, 6.0 Hz, 1H), 7.24 − 7.16 (m, 2H), 7.05 (d, J = 16.4 Hz, 1H), 6.94 − 6.87 (m, 4H), 3.83 (s, 3H). Spectral properties were in accordance with the literature24.
2.1.8. (E)-4-(3-Fluorostyryl)phenol (9)
Compound 8 (1.14 g, 5 mmol) was dissolved in DCM (10 ml), and then 1 mol/l BBr3 (10 ml) in THF was added at 0–10 °C. The mixture was stirred at room temperature for 6 h. The solution was evaporated under vacuum. The crude product was filtered and purified with recrystallisation from ethanol. The product was dried in vacuum to give a white solid. 770 mg; 72% yield; 1H NMR (400 MHz, CDCl3) δ 7.43 − 7.39 (m, 2H), 7.30 (td, J = 8.0, 6.0 Hz, 1H), 7.24 − 7.16 (m, 2H), 7.05 (d, J = 16.4 Hz, 1H), 6.95 − 6.89 (m, 2H), 6.86 − 6.82 (m, 2H), 4.83 (s, 1H). Spectral properties were in accordance with the literature25.
2.1.9. (E)-2-((4-(3-Fluorostyryl)phenoxy)methyl)-3-methylquinazolin-4(3H)-one (4c)
Compound 9 (214 mg, 1 mmol) was dissolved in DMF (5 ml), and then anhydrous K2CO3 (152 mg, 1.1 mmol) was added. After stirred for 15 min at room temperature, the mixture was added compound 2 (208 g, 1 mmol) and KI (183 mg, 1.1 mmol). The solution was stirred for the other 2 h, and poured into water (40 ml). The crude product was filtered and purified with recrystallisation from ethanol. The product was dried in vacuum to give a white solid. 181 mg; 47% yield; m.p. 160–162 °C; 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 8.0 Hz, 1H), 7.79 − 7.70 (m, 2H), 7.54 − 7.45 (m, 3H), 7.29 (dd, J = 8.0, 6.0 Hz, 1H), 7.20 (dd, J = 20.0, 9.2 Hz, 2H), 7.05 (dd, J = 12.8, 6.4 Hz, 3H), 6.93 (dd, J = 9.2, 7.2 Hz, 2H), 5.21 (s, 2H), 3.75 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 151.3, 149.7, 149.5, 146.5, 142.4, 137.6, 132.4 (d, J = 6.5 Hz), 128.0, 124.8 (d, J = 6.5 Hz), 124.6, 124.0, 122.8, 122.2 (d, J = 19.5 Hz), 121.4, 120.7, 118.5, 116.6, 112.7, 111.51 (d, J = 17.0 Hz), 110.2 (d, J = 17.0 Hz), 75.4, 44.4; HRMS-ESI (m/z): [M + H]+ calcd for C24H19FN2O2, 387.1503; found, 387.1499.
2.1.10. 3-Methyl-2-((4-nitrophenoxy)methyl)quinazolin-4(3H)-one (10)
4-Nitrophenol (695 mg, 5 mmol) was dissolved in DMF (20 ml), and then anhydrous K2CO3 (759 mg, 5.5 mmol) was added. After stirred for 15 min at room temperature, the mixture was added compound 2 (1.04 g, 5 mmol) and KI (915 mg, 5.5 mmol). The solution was stirred for the other 2 h, and poured into water (80 ml). The crude product was filtered and purified with recrystallisation from ethanol. The product was dried in vacuum to give a yellow solid. 824 mg; 53% yield; m.p. 185–186 °C; 1H NMR (400 MHz, CDCl3) δ 8.32 (dd, J = 8.0, 1.2 Hz, 1H), 8.27 − 8.23 (m, 2H), 7.81 (ddd, J = 8.4, 7.2, 1.6 Hz, 1H), 7.74 (dd, J = 8.0, 0.8 Hz, 1H), 7.56 (ddd, J = 8.0, 7.2, 1.2 Hz, 1H), 7.23 − 7.19 (m, 2H), 5.33 (s, 2H), 3.77 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 150.9, 149.4, 141.8, 137.5, 133.5, 128.0, 122.3, 122.1, 121.4, 121.0, 116.6, 112.9, 75.0, 44.1; HRMS-ESI (m/z): [M + H]+ calcd for C16H13N3O4, 312.0978; found, 312.0982.
2.1.11. 2-((4-Aminophenoxy)methyl)-3-methylquinazolin-4(3H)-one (11)
Compound 10 (622 mg, 2 mmol) was dissolved in MeOH (10 ml), and then Pd/C (62 mg, 10%) was added. The mixture was stirred at room temperature under hydrogen atmosphere for overnight. The solution was filtered with celite and the filtration was evaporated under vacuum. The crude product was purified with recrystallisation from MeOH. The product was dried in vacuum to give a light-yellow solid. 405 mg; 72% yield; m.p. 165–166 °C; 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 8.8 Hz, 1H), 7.78 − 7.68 (m, 2H), 7.53 − 7.48 (m, 1H), 6.90 − 6.85 (m, 2H), 6.67 − 6.62 (m, 2H), 5.09 (s, 2H), 3.75 (s, 3H), 3.49 (s, 2H); 13 C NMR (125 MHz, DMSO) δ 149.5, 142.8, 139.4, 137.6, 135.2, 128.0, 122.3, 122.1, 121.4, 116.6, 113.4, 112.2, 76.6, 44.6; HRMS-ESI (m/z): [M + H]+ calcd for C16H15N3O2, 282.1236; found, 282.1241.
2.1.12. 3-Fluoro-N-(4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)phenyl)benzamide (4d)
Compound 11 (281 mg, 1 mmol) and 3-fluorobenzoic acid (140 mg, 1 mmol) were dissolved in DMF (5 ml), and then HATU (o-(7-aza-1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate) (380 mg, 1 mmol) was added. After stirred for 30 min, the mixture was added DIEA (N,N-diisopropylethylamine) (129 mg, 1 mmol) and stirred for 12 h at room temperature. The solution was poured into water (20 ml). The final product was filtered and dried in vacuum to give a white solid. 149 mg; 37% yield; m.p. 228–229 °C; 1H NMR (400 MHz, DMSO) δ 10.23 (s, 1H), 8.17 (dd, J = 8.0, 1.2 Hz, 1H), 7.86 − 7.79 (m, 2H), 7.77 − 7.73 (m, 1H), 7.71 − 7.67 (m, 3H), 7.57 (dt, J = 2.0, 1.6 Hz, 2H), 7.44 (td, J = 8.4, 2.4 Hz, 1H), 7.15 − 7.10 (m, 2H), 5.29 (s, 2H), 3.64 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 151.4, 150.7, 149.5, 149.2, 143.6, 142.5, 137.6, 130.2 (d, J = 5.5 Hz), 128.0, 126.7, 124.9 (d, J = 6.5 Hz), 122.2 (d, J = 17.0 Hz), 121.4, 119.5, 118.0, 116.6, 115.1 (d, J = 16.5 Hz), 112.5, 111.9 (d, J = 18.0 Hz), 75.6, 44.4; HRMS-ESI (m/z): [M + Na]+ calcd for C23H17FN3NaO3, 426.1224; found, 426.1226.
2.1.13. 3,4-Difluoro-N-(4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)phenyl)benzamide (4e)
Compound 11 (281 mg, 1 mmol) and 3,4-difluorobenzoic acid (158 mg, 1 mmol) were dissolved in DMF (5 ml), and then HATU (o-(7-aza-1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate) (380 mg, 1 mmol) was added. After stirred for 30 min, the mixture was added DIEA (N,N-diisopropylethylamine) (129 mg, 1 mmol) and stirred for 12 h at room temperature. The solution was poured into water (20 ml). The final product was filtered and dried in vacuum to give a white solid. 139 mg; 33% yield; m.p. 248–249 °C; 1H NMR (400 MHz, DMSO) δ 10.23 (s, 1H), 8.16 (dd, J = 8.0, 1.2 Hz, 1H), 8.05 − 7.98 (m, 1H), 7.84 (dd, J = 12.0, 4.8 Hz, 2H), 7.70 − 7.55 (m, 5H), 7.13 (d, J = 9.2 Hz, 2H), 5.28 (s, 2H), 3.63 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 152.1, 150.7, 149.5, 143.7, 142.5, 137.6, 128.0, 126.6, 126.2 (d, J = 6.0 Hz), 122.2 (d, J = 18.0 Hz), 121.4, 120.5 (d, J = 2.0 Hz), 118.0, 116.6, 114.6, 114.5, 114.1, 114.0, 112.5, 75.6, 44.4; HRMS-ESI (m/z): [M + Na]+ calcd for C23H16F2N3NaO3, 444.1130; found, 444.1126.
2.1.14. (E)-3-(3,4-Difluorophenyl)-N-(4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)phenyl)acrylamide (4f)
Compound 11 (281 mg, 1 mmol) and (E)-3–(3,4-difluorophenyl)acrylic acid (184 mg, 1 mmol) were dissolved in DMF (5 ml), and then HATU (o-(7-aza-1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate) (380 mg, 1 mmol) was added. After stirred for 30 min, the mixture was added DIEA (N,N-diisopropylethylamine) (129 mg, 1 mmol) and stirred for 12 h at room temperature. The solution was poured into water (20 ml). The final product was filtered and dried in vacuum to give a white solid. 134 mg; 30% yield; m.p. 235–236 °C; 1H NMR (400 MHz, DMSO) δ 10.16 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.86 − 7.82 (m, 1H), 7.75 − 7.63 (m, 4H), 7.59 − 7.49 (m, 4H), 7.11 (d, J = 9.1 Hz, 2H), 6.78 (d, J = 15.7 Hz, 1H), 5.27 (s, 2H), 3.63 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 150.6, 149.5, 143.4, 142.5, 137.6, 130.5, 128.0, 127.0, 126.6 (d, J = 5.0 Hz), 122.3, 122.1, 121.4, 120.2 (d, J = 2.5 Hz), 119.4, 118.2, 116.9, 116.6, 114.9 (d, J = 14.0 Hz), 113.6 (d, J = 14.0 Hz), 113.3, 112.6, 75.7, 44.5; HRMS-ESI (m/z): [M + Na]+ calcd for C25H18F2N3NaO3, 470.1287; found, 470.1288.
2.1.15. 1-(3-Fluorophenyl)-3-(4-((3-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methoxy)phenyl)urea (4 g)
Compound 11 (281 mg, 1 mmol) was dissolved in anhydrous THF (5 ml), and then 1-fluoro-3-isocyanatobenzene (137 mg, 1 mmol) was added. After stirred for 30 min, the mixture was added DIEA (129 mg, 1 mmol) and stirred for 12 h at room temperature. The solution was poured into water (20 ml). The crude product was purified with recrystallisation from MeOH. The product was dried in vacuum to give a white solid. 142 mg; 34% yield; m.p. 195–196 °C; 1H NMR (400 MHz, DMSO) δ 9.13 (s, 1H), 8.84 (s, 1H), 8.16 (dd, J = 8.0, 1.2 Hz, 1H), 7.87 − 7.80 (m, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.48 (dt, J = 12.0, 2.4 Hz, 1H), 7.39 (d, J = 9.2 Hz, 2H), 7.28 (dd, J = 15.2, 8.0 Hz, 1H), 7.12 (dd, J = 8.4, 0.8 Hz, 1H), 7.06 (d, J = 9.2 Hz, 2H), 6.75 (td, J = 8.4, 2.4 Hz, 1H), 5.24 (s, 2H), 3.63 (s, 3H); 13 C NMR (125 MHz, DMSO) δ 151.1, 149.5 (d, J = 10.0 Hz), 142.6, 142.5, 137.6, 134.0 (d, J = 9.5 Hz), 128.0, 127.3, 124.6 (d, J = 8.0 Hz), 122.3, 122.2, 121.4, 116.6, 116.4, 112.7, 111.4, 106.6 (d, J = 17.0 Hz), 104.2, 104.0, 75.8, 44.5; HRMS-ESI (m/z): [M + Na]+ calcd for C23H18FN4NaO3, 441.1333; found, 441.1333.
2.2. In vitro EGFRwt-TK assay
Enzymatic inhibition of synthesised compounds against wild type EGFR tyrosine kinase was determined with enzyme-linked immunosorbent assays (ELISAs) as our previous methods. The recombinant EGFRwt-TK and Antiphosphotyrosine mouse mAb were purchased from PTM Bio. The IC50 value was determined from a sigmoid dose − response curve using Graph-Pad Prism (GraphPad Software, San Diego, CA).
2.3. In vitro activity assay at cell level
2.3.1. Cell culture
A431 (Human epidermoid carcinoma cell line) cell, A549 (Human non-small cell lung cancer cell line) cell, MCF-7 (Human breast adenocarcinoma cell line) cell, and NCI-H1975 (Human non-small cell lung cancer cell line) cell were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences. NRK-52E (Normal rat kidney cell line) cell was donated by Guizhou Medical University. All cell lines were maintained in RPMI 1640 or DMEM complete medium.
2.3.2. Cytotoxicity evaluation (MTT assay)
In vitro cytotoxicity of compounds 4a–4g against four cancer cell lines (A431, A549, MCF-7, and NCI-H1975) and normal rat kidney cells (NRK 52E) was determined by MTT assay as our previous report. Gefitinib were used as positive controls. The IC50 value was determined from a sigmoid dose − response curve using Graph-Pad Prism (GraphPad Software, San Diego, CA).
2.3.3. Cell apoptosis analysis
The apoptosis of tumour cells MCF-7 treated by different concentrations of compound 4d, was measured with Annexin V - FITC/PI apoptosis detection kit (Solarbio, Beijing, China), according to instructions of kit, and detected by BD Accuri C6 flow cytometry (American BD Corporation Shanghai Co. Ltd.)
2.3.4. Cell cycle analysis
The distribution of cell cycle for MCF-7 treated by different concentrations of compound 4d, was measured with Annexin V - FITC/PI cell cycle detection kit (Solarbio, Beijing, China), according to instructions of kit, and detected by BD Accuri C6 flow cytometry (American BD Corporation Shanghai Co. Ltd.)
2.4. Molecular docking
X-ray crystal structures of EGFR in both “active” (PDB entry 1M17) and “inactive” (PDB entry 4HJO) states were used for identifying candidate binding modes. The possible binding modes of compound A, compounds 4a–4g, Gefitinib with EGFR were predicted by molecular docking with Sybyl X-2.0 software from Tripos Inc, St. Louis, MI.
2.5. Admet studies
The absorption, distribution, metabolism, elimination, and toxicity (ADMET) parameters of compounds 4a–4g and Gefitinib were calculated in and calculated in CHARMM Force Field of Discovery Studio 2.5 Software (Accelrys Inc., San Diego, CA). The data of ADMET included Solubility, Absorption, Cytochrome P450 (CYP2D6), Hepatotoxiciy, Plasma protein binding (PPB), Blood brain barrier permeability (BBB), and water partition coefficients for the unionised species (AlogP98)26.
3. Results and discussions
3.1. Chemistry
All target compounds (4a–4g) were synthesised and confirmed based on 1H-NMR, 13 C-NMR and HRMS. The synthetic route was shown in Scheme 1. At first, compound 2 and 3 were prepared under the described conditions in literature23. And then, compound 3 experienced in aldol-condensation with 3-fluorophenylethanone to give the product 4a. Subsequently, compound 3 reacted with ethyl 4-hydroxybenzoate under the condition of K2CO3/KI/DMF to give the compound 5 with 64% yield. After hydrolysis in the presence of NaOH, compound 6 was obtained with 58% yield and formed amide 4 b with 3-fluorobenzenamine. Next, compound 9 was prepared with the reported method and reacted with compound 2 to give styrylquniazolinone 4c. At last, treatment 2 with 4-nitrophenol and reduce the nitro-group to give the intermediate 11. Compound 11 reacted with corresponding acid or isocyanate to obtain the final targets 4d–4g. The synthetic details were described in experimental section and the spectra can be found in the supplementary material.
3.2. In vitro EGFR kinase inhibitory activity and antitumor activity of target quinazolinone derivatives
With the compounds 4a–4g in hand, the activity against EGFRwt-TK was tested with ELISA assay27. As shown in Table 1, when the enamine bond of compound A (IC50 of 0.047 μM) was substituted with ketene group (Table 1, 4a), vinyl group (Table 1, 4c), and amide bond (Table 1, 4b and 4d), the IC50 values of compounds 4a–d to EGFRwt-TK were 2.71 μM, 0.2 μM, 1.63 μM and 0.053, respectively. Fortunately, compound 4d reached in the similar activity with compound A. Compared to 4d, 3,4-difluoro substitution on the phenyl ring (Table 1, 4e) decreased the activity. Meanwhile, the extended amide bond (Table 1, 4f) and urea bond (Table 1, 4g) also significantly weaken the activity.
Table 1.
IC50 values for EGFRwt-TK. 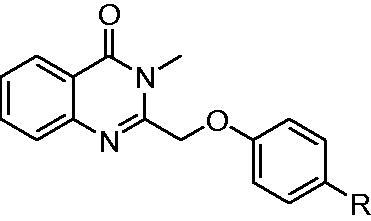
| Entry | Comp. | R | EGFRwt-TK IC50 (μM) |
|---|---|---|---|
| 1 | 4a | 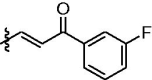 |
2.71 ± 0.22 |
| 2 | 4b | 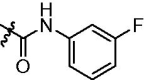 |
1.63 ± 0.15 |
| 3 | 4c | 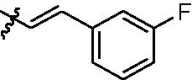 |
0.20 ± 0.041 |
| 4 | 4d | 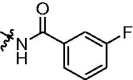 |
0.053 ± 0.008 |
| 5 | 4e | 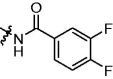 |
0.061 ± 0.007 |
| 6 | 4f | 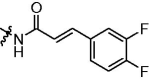 |
0.38 ± 0.075 |
| 7 | 4g | |
0.71 ± 0.093 |
| 8 | A | 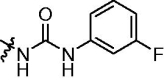 |
0.047 ± 0.004 |
| Gefitinib | 0.0061 ± 0.0003 | ||
aThe values are mean ± SD of three replicates.
To investigate the anticancer activity of the synthesised compounds, four EGFR over-expressed human tumour cell lines derived from human epidermoid cancer (A431), non-small cell lung cancer (A549), breast cancer (MCF-7), and non-small cell lung cancer (NCI-H1975) were used to evaluate the antiproliferative activities by methyl thiazolyl tetrazolium colorimetric assay (MTT). Gefitinib, a clinical drug targeted EGFR was employed as positive control. As shown in Table 2, against A431 cells compounds 4d and 4e (IC50 values of 3.48 μM and 3.53 μM) were more potent than Gefitinib (4.45 μM), while compounds 4a, 4 b, 4c, 4f, and 4 g (IC50 values of 9.32, 8.72, 6.79, 8.76 μM and 8.34 μM, respectively). Against A549 cells compounds 4c–4f (with IC50 values of 4.26, 2.55, 2.69, 8.46, and 7.31 μM, respectively) were more potent than Gefitinib (8.47 μM). Against NCI-H1975 cells compounds 4c–4f showed higher inhibitor activities than Gefitinib. Against MCF-7 cells compounds 4d–4f (with IC50 values of 0.87, 0.96, and 2.84 μM, respectively) were more efficient than Gefitinib (5.89 μM).
Table 2.
IC50 values for human cancer cell linesa.
| Comp. | R | IC50 (μM) |
|||
|---|---|---|---|---|---|
| A431 | A549 | MCF-7 | NCI-H1975 | ||
| 4a | 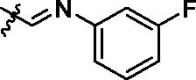 |
9.32 ± 0.57 | 8.63 ± 0.41 | 10.58 ± 1.03 | 13.68 ± 1.61 |
| 4b | 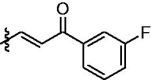 |
8.72 ± 0.46 | 9.08 ± 0.69 | 9.64 ± 0.88 | 12.87 ± 1.44 |
| 4c |  |
6.79 ± 0.72 | 4.26 ± 0.57 | 6.04 ± 0.73 | 6.44 ± 0.85 |
| 4d | 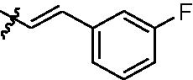 |
3.48 ± 0.49 | 2.55 ± 0.26 | 0.87 ± 0.14 | 6.42 ± 0.47 |
| 4e | 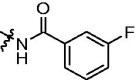 |
3.53 ± 0.34 | 2.69 ± 0.32 | 0.96 ± 0.21 | 7.18 ± 0.49 |
| 4f | 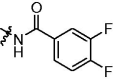 |
8.76 ± 0.74 | 8.46 ± 0.67 | 2.84 ± 0.22 | 9.33 ± 0.78 |
| 4g | 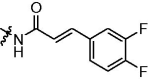 |
8.35 ± 0.46 | 7.31 ± 0.59 | 10.74 ± 2.07 | 10.19 ± 1.03 |
| A |  |
17.71 ± 2.08 | 17.86 ± 2.11 | 14.09 ± 1.85 | 19.18 ± 3.02 |
| Gefitinib | 4.45 ± 0.25 | 8.47 ± 1.06 | 5.89 ± 0.72 | 9.71 ± 1.96 | |
aThe values are mean ± SD of three replicates.
Based on the result of EGFRwt-TK and four tumour cell assays, we found that the similar structure–activity relationship (SAR) was observed in cell lines and EGFR-TKs. The amide group (4d and 4e) instead of enamine was more potent than the ketene group (4a), vinyl group (4c), urea group (4 g) and other groups (4 b and 4f). Besides, compound A showed lower inhibitor activities against four tumour cell lines than the compounds 4a–4g. The reason may be due to the lower stability of compound A in cells. Generally, compound 4d was the best in the synthesised compounds and particularly showed much higher inhibitor activities than Gefitinib against four tumour cells. These data indicate that 4d was worth to further investigation.
3.3. In vitro cytotoxicity of 3-methyl quinazolinone derivatives on normal cells
The cytotoxicity study of compounds 4a–4g was first evaluated by MTT colorimetric assays to normal rat kidney cell line (NRK-52E) at different concentrations. There was no inhibition of all compounds including Gefitinib at 10 μM. Therefore, the concentration was accelerated to 100 μM. As shown in Figure 2, all compounds were similar with Gefitinib and not more than 25% against NRK-52E at 100 μM. This indicate that the synthesised compounds 4a–4g were low cytotoxicity and potent for development for drugs.
Figure 2.

In vitro cytotoxicity of compounds 4a-g and Gefitinib on NRK-52E cell.
3.4. Effects of compound 4d on cell apoptosis of MCF-7 cell line
Apoptosis of tumour cell is the basic mechanism of tissue homeostasis, which is regarded as the efficient method to eliminate the excess cells28. Most of the anti-cancer drugs perform as a well inducing apoptosis for cancer cells29. As shown in Figure 3, compound 4d was used to investigate the mechanism of inhibition of MCF-7 cell proliferation. Flow cytometry analysis of MCF-7 treated with 4d at 5 and 10 µM for 48 h demonstrated a prominent increase in apoptotic cells with a dose-dependent fashion. The trend of compound 4d showed that early apoptosis rate was priority over late apoptosis. Compared compound 4d with Gefitinib, the results in Figure 3(B) showed that 4d significantly performed better ability to induce early apoptosis at the same concentration. Particularly, the ratio of apoptotic cells for compound 4d reached 28.14% (early) and 41.36% (late) even at 5 µM.
Figure 3.

Compound 4d induced cell apoptosis in Annexin V-FITC assay. (A) Density plot were obtained by flow cytometry, Gefitinib was used as reference drug. (B) Total apoptotic cells (%) at various concentrations of 4d and Gefitinib.
3.5. Effects of compound 4d on cell cycle of MCF-7 cell line
Next, we wished to determine whether 4d-induced decrease of EGFR phosphorylation would result in cell cycle arrest in MCF-7 cell lines, compared with Gefitinib. As shown in Figure 4, cell cycle analysis was tested in flow cytometry. MCF-7 cell lines were treated with compound 4d at 5 and 10 µM. Against MCF-7 cells at 5 µM, the control consisted of 6.06% G2 phase cells, compound 4d increased to 10.38% and Gefitinib also reached to 11.58%. However, at 10 μM of tested samples, G2/M phase decreased and S phase increased, this might be that compound 4d and Gefitinib undergo significant apoptosis. These results indicate that compound 4d and Gefitinib could arrest MCF-7 cells in G2/M phase at the appropriate concentration.
Figure 4.

Cell cycle distribution of compound 4d and Gefitinib against MCF-7 was studied by flow cytometry. (A) Profiles were obtained by FACS. The percentages for different phases of cell cycle were illustrated in the histogram. (B) MCF-7 cells were cultured in the presence of different concentrations of 4d (5 μM and 10 μM) or Gefitinib (5 μM and 10 μM) for 48 h, harvested, fixed, and labelled with PI, then analysed by FACS. Percentage of cells in G0/G1, S and G2/M phases are indicated.
3.6. Molecular docking studies
To explain the structural activity relationship of EGFR with our compounds, the possible binding modes were performed in molecular docking studies30. Former researches showed that the “active” or “inactive” state of EGFR-TK has different preference of ligand binding profile, so X-ray crystal structures of EGFR in both “active” (PDB entry 1M17) and “inactive” (PDB entry 4HJO) states were used for identifying candidate binding modes31.
As shown in Tables S1 and S2, the predicted score of all compounds 4a–4g with inactive state of EGFR (PDB: 4HJO) is much higher than the score of active state of EGFR (PDB: 1M7), which suggests that the compounds 4a–4g are likely to bind with EGFR in inactive state. By contrast, the predicted binding model for Gefitinib in two states are similar, which is consistent with our previous work. More interestingly, as shown in Figure 5(A,C), all the compounds 4a–4g were employed in the same binding configuration with EGFR active state or inactive state. Take compound 4d as example, in Figure 5(B) for active state, simply formed one hydrogen bond with Met769 during the docking situation. However, in Figure 5(D) for inactive state, quinazolinone ring of compound 4d formed stable hydrogen bonds with Lys692 and Lys704. Meanwhile, amide group and fluorine atom of 4d also formed hydrogen bonds with Thr766, Thr830, and Lys721 respectively. These results indicate that the synthesised compounds 4a–4g are particularly likely to be potential EGFR inhibitors against inactive state.
Figure 5.

Docking structures of compounds 4a-4g. (A) Different binding configurations of compounds 4a-4g with EGFR (PDB: 1M17). (B) The 3 D model of compound 4d bound to EGFR (PDB: 1M17). (C) Different binding configurations of compounds 4a-4g with EGFR (PDB: 4HJO). (D) The 3 D model of compound 4d bound to EGFR (PDB: 4HJO).
3.7. Admet and stability studies
The ADMET are essential aspects for drug candidates32. Computer aided ADMET studies have been employed in the early stage of drug discovery. Therefore, the ADMET properties of compounds 4a–4g were calculated through Discovery Studio 2.5 software (Accelrys Inc., San Diego, CA). As shown in Table 3, the levels of solubility and absorption were the same with compounds 4a–4g and Gefitinib. CYP2D6 is non-inhibitor of cytochrome P450 enzyme for predicting drug toxicity. The compounds 4a, 4f and 4 g were less toxic than Gefitinib, and the others were similar with Gefitinib. Meanwhile, the calculated log p values of compound 4 b, 4d, 4e and 4 g were less than Gefitinib (4.20). Particularly, compound 4d reached the lowest log p (3.52) and performed highly oral bioavailability. These data are consistent with the structure-activity relationships.
Table 3.
ADMET properties of compounds 4a–g.
| Comp. | Solubility | Absorption | CYP2D6 | Hepatotoxicity | PPB | BBB | AlogP98 |
|---|---|---|---|---|---|---|---|
| 4a | 2 | 0 | 0 | 1 | 2 | 1 | 4.61 |
| 4b | 2 | 0 | 1 | 1 | 2 | 2 | 3.53 |
| 4c | 2 | 0 | 1 | 1 | 2 | 1 | 4.73 |
| 4d | 2 | 0 | 1 | 1 | 2 | 2 | 3.52 |
| 4e | 2 | 0 | 1 | 1 | 2 | 2 | 3.73 |
| 4f | 2 | 0 | 0 | 1 | 2 | 1 | 4.20 |
| 4g | 2 | 0 | 0 | 1 | 2 | 2 | 3.63 |
| Gefitinib | 2 | 0 | 1 | 0 | 1 | 1 | 4.20 |
At last, we investigated the stability of compound 4d in mice plasma. The results exhibited that compound 4d was stable with purity higher than 98% after incubation with Sprague Dawley (SD) mice plasma (Figure S2). All the data suggest that compound 4d was a good candidate for developing new anti-cancer drug.
4. Conclusions
To solve the issues of metabolic stability in our previous research of EGFR inhibitors, seven 3-methylquinazolinone derivatives were designed and synthesised. After evaluated activities of these compounds in enzyme and cell level, we found that compounds 4d and 4e were performed well antiproliferative activities. Furthermore, the best compound 4d displayed IC50 value of 53 nM against EGFRwt-TK activity and IC50 value of 0.87 μM against MCF-7 cells. Analysis of apoptosis and cell cycle for MCF-7 cells indicate that compound 4d could induce apoptosis and arrest in G2/M phase at tested concentration. At last, molecular docking, ADMET, and stability studies suggested that compound 4d was closely formed hydrogen bonds with EGFRwt-TK and potential to develop new anti-cancer drug.
Supplementary Material
Funding Statement
This work was financially supported by GZU (Guizhou University) Found for Newly Enrolled Talent ([2019]15), GZU (Guizhou University) Found for Cultivation ([2019]65), and the State Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medical University [Grant number FAMP202005K]; Guizhou Science and Technology Platform Talents [QKHRCPT [2019]5106].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Ayati A, Moghimi S, Salarinejad S, et al. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg Chem 2020;99:103811. [DOI] [PubMed] [Google Scholar]
- 2.Roskoski R. Jr., Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res 2020;152:104609. [DOI] [PubMed] [Google Scholar]
- 3.Sigismund S, Avanzato D, Lanzetti L.. Emerging functions of the EGFR in cancer. Mol Oncol 2018;12:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu X, Mao L, Xu W, et al. AC0010, an irreversible EGFR inhibitor selectively targeting mutated EGFR and overcoming T790M-induced resistance in animal models and lung cancer patients. Mol Cancer Ther 2016;15:2586–97. [DOI] [PubMed] [Google Scholar]
- 5.Lu X, Yu L, Zhang Z, et al. Targeting EGFR(L858R/T790M) and EGFR(L858R/T790M/C797S) resistance mutations in NSCLC: current developments in medicinal chemistry. Med Res Rev 2018;38:1550–81. [DOI] [PubMed] [Google Scholar]
- 6.Lee DH. Treatments for EGFR-mutant non-small cell lung cancer (NSCLC): the road to a success, paved with failures. Pharmacol Ther 2017;174:1–21. [DOI] [PubMed] [Google Scholar]
- 7.Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016;534:129–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mhaske SB, Argade NP.. The chemistry of recently isolated naturally occurring quinazolinone alkaloids. Tetrahedron 2006;62:9787–826. [Google Scholar]
- 9.Auti PS, George G, Paul AT.. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv 2020;10:41353–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shang XF, Morris-Natschke SL, Yang GZ, et al. Biologically active quinoline and quinazoline alkaloids part II. Med Res Rev 2018;38:1614–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shang XF, Morris-Natschke SL, Liu YQ, et al. Biologically active quinoline and quinazoline alkaloids part I. Med Res Rev 2018;38:775–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zayed MF, Rateb HS, Ahmed S, et al. Quinazolinone-amino acid hybrids as dual inhibitors of EGFR kinase and tubulin polymerization. Molecules 2018;23:1699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong X-F, Guo X-Y, Gu Z-Y, et al. Silver(i)-catalyzed selective hydroalkoxylation of C2-alkynyl quinazolinones to synthesize quinazolinone-fused eight-membered N,O-heterocycles. Org Chem Front 2020;7:2055–62. [Google Scholar]
- 14.Patel HM, Pawara R, Ansari A, et al. Design and synthesis of quinazolinones as EGFR inhibitors to overcome EGFR resistance obstacle. Bioorg Med Chem 2017;25:2713–23. [DOI] [PubMed] [Google Scholar]
- 15.Ghorab MM, Abdel-Kader MS, Alqahtani AS, et al. Synthesis of some quinazolinones inspired from the natural alkaloid L-norephedrine as EGFR inhibitors and radiosensitizers. J Enzyme Inhib Med Chem 2021;36:218–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan L, Che X, Bai X, et al. Syntheses of novel diaryl[d,f] [1,3]diazepines via one-pot Suzuki coupling followed by direct ring closure with carboxylic acids. Mol Divers 2012;16:489–501. [DOI] [PubMed] [Google Scholar]
- 17.Yan L, Wang H, Chen Y, et al. Synthesis and structure-activity relationship study of diaryl[d,f][1,3]diazepines as potential anti-cancer agents. Mol Divers 2018;22:323–33. [DOI] [PubMed] [Google Scholar]
- 18.Yan L, Li Y, Deng M, et al. Design, synthesis and biological activities of compounds containing 1,3,4-oxadiazole or 1,3,4-thiadiazole. Chin J Org Chem 2020;40:731–9. [Google Scholar]
- 19.Yan L, Le Y, Chen D, et al. Synthesis and evaluation of diaminopyrimidine derivatives as dual inhibitors of EGFR and SRC for antitumor treatment. Heterocycles 2020;100:418–28. [Google Scholar]
- 20.Yan L, Deng M, Chen A, et al. Synthesis of N-pyrimidin[1,3,4]oxadiazoles and N-pyrimidin[1,3,4]-thiadiazoles from 1,3,4-oxadiazol-2-amines and 1,3,4-thiadiazol-2-amines via Pd-catalyzed heteroarylamination. Tetrahedron Lett 2019;60:1359–62. [Google Scholar]
- 21.Le Y, Zhang Y, Wang Q, et al. Microwave-assisted synthesis of phenylpyrimidine derivatives via Suzuki-Miyaura reactions in water. Tetrahedron Lett 2021;68:152903. [Google Scholar]
- 22.Le Y, Gan Y, Fu Y, et al. Design, synthesis and in vitro biological evaluation of quinazolinone derivatives as EGFR inhibitors for antitumor treatment. J Enzyme Inhib Med Chem 2020;35:555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shao L-H, Fan S-L, Meng Y-F, et al. Design, synthesis, biological activities and 3D-QSAR studies of quinazolinone derivatives containing hydrazone structural units. New J Chem 2021;45:4626–31. [Google Scholar]
- 24.Altarejos J, Sucunza D, Vaquero JJ, et al. Practical solvent-free microwave-assisted hydroboration of alkynes. Eur J Org Chem 2020;2020:3024–9. [Google Scholar]
- 25.Kuramochi T, Kakefuda A, Yamada H, et al. Synthesis and structure-activity relationships of phenoxypyridine derivatives as novel inhibitors of the sodium-calcium exchanger. Bioorg Med Chem 2004;12:5039–56. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Yan L, Cai J, et al. Development of novel theranostic agents for in vivo amyloid imaging and protective effects on human neuroblastoma cells. Eur J Med Chem 2019;181:111585. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Chen L, Xu H, et al. 6,7-Dimorpholinoalkoxy quinazoline derivatives as potent EGFR inhibitors with enhanced antiproliferative activities against tumor cells. Eur J Med Chem 2018;147:77–89. [DOI] [PubMed] [Google Scholar]
- 28.D'Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int 2019;43:582–92. [DOI] [PubMed] [Google Scholar]
- 29.Xie Z, Wu K, Wang Y, et al. Discovery of 4,6-pyrimidinediamine derivatives as novel dual EGFR/FGFR inhibitors aimed EGFR/FGFR1-positive NSCLC. Eur J Med Chem 2020;187:111943. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Chen L, Li X, et al. Novel 4-arylaminoquinazolines bearing N,N-diethyl(aminoethyl)amino moiety with antitumour activity as EGFR(wt)-TK inhibitor. J Enzyme Inhib Med Chem 2019;34:1668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park JH, Liu Y, Lemmon MA, et al. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem J 2012;448:417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia CY, Li JY, Hao GF, et al. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov Today 2020;25:248–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.