

Abstract
The field of protein design has grown enormously in the past few decades. In this review we discuss the minimalist approach to design of artificial enzymes, in which protein sequences are created with the minimum number of elements for folding and function. This method relies on identifying starting points in catalytically inert scaffolds for active site installation. The progress of the field from the original helical assemblies of the 1980s to the more complex structures of the present day is discussed, highlighting the variety of catalytic reactions which have been achieved using these methods. We outline the strengths and weaknesses of the minimalist approaches, describe representative design cases and put it in the general context of the de novo design of proteins.
Keywords: Protein design, catalysis, minimalism, enzymology, design approaches
Graphical Abstract
1.1. Introduction.
The field of protein design has grown enormously in the past few decades. A number of excellent reviews have been written describing the incredible achievements made both in term in practical applications and fundamental knowledge of protein function.1–4 However, as it is often the case with new and rapidly evolving fields, many definitions of what protein design actually encompasses are found in the literature. The protein part is clear – new proteins (polypeptides with well-defined secondary and tertiary structures) are to be made, but what does the word “design” really mean? It seems quite logical that every protein design problem begins with a purpose. Making large libraries of proteins with diverse sequences is easy, but what one wants is a protein with a predetermined property. Common examples of such properties include three-dimensional folds, catalytic function and target molecule binding. The next step is to determine how to achieve this purpose. We believe that most researchers would agree that methods that rely solely on screening or selection of a functional protein with a particular property from large libraries of proteins (e.g. phage/yeast/ribosome display and directed evolution) belong to the realm of protein engineering. Thus, design must include a substantial amount of a priori rational input. Indeed, if one wants to catalyze a chemical reaction, at least some knowledge of potential mechanisms is required, which in turn will define the types of residues to be chosen. Protein design combines both rational vision for the overall outcome and the process of its translation into a sequence of amino acids. The commonly found characterization of protein design as a reverse protein folding problem is therefore incomplete. What good is the ability to accurately create a protein that does not serve the intended purpose? Similar to Michelangelo’s David, which required both artistic vision as well as mastery of the necessary tools to carve it out from a piece of marble with natural defects, scientists come up with a broad strokes idea of what a particular protein function would require and then make a protein that satisfies that vision making adjustments along the way (Figure 1). As such, in addition to any practical value that the designed protein might have, the process itself serves a tool to test (and advance) understanding of protein function.
Figure 1.
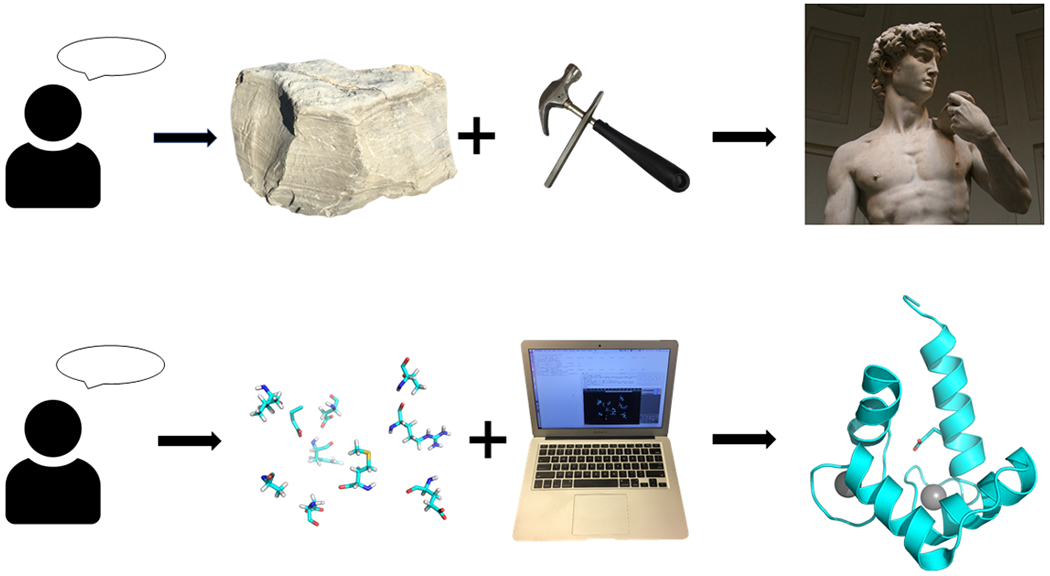
Protein design requires consideration of the correct method to apply to materials for optimal outcome.
To summarize, we define protein design as the rational creation of proteins with novel activity, behavior, or purpose, with the ultimate goal to advance understanding of protein function. There are several important conclusions to be drawn from this definition. First, protein design is inherently rational. This, of course, does not mean that no sampling of multiple possibilities is taking place – quite the opposite, but the overall concept of what the protein does and how it does this is decided in the very beginning. Second, the tools of protein design, while incredibly important, are secondary to and potentially independent of the rational input. Continuing with the David analogy, the statue can be either chiseled from stone or 3D printed with the same esthetically pleasing result.
Catalysis has been the Holy Grail of protein design since the days of Emil Fischer.5 The efficiency and selectivity of enzymes have inspired many generations of scientists to come up with approaches to create catalysts for reactions not found in nature, or to make new ways of catalyzing reactions for which enzymes are already known. Despite much effort, however, the problem of creating efficient protein catalysts a la carte is essentially unsolved.6 This is perhaps not surprising given the challenge of maintaining the intricate balance between substrate binding, transition state geometry, product dissociation, overall protein fold and dynamics all at once. Nonetheless, the progress in the field has led to the development of methods that at the very least can produce useful starting points for subsequent directed evolution.
1.2. Approaches to the design of protein catalysts.
As we have mentioned above, there has been tremendous progress in repurposing existing enzymes for new applications using directed evolution as well as other methods, the advances in this fields have been extensively reviewed elsewhere.7, 8 In this review we will focus only on protein catalysts that came from a non-natural protein/peptide scaffold OR a case where a protein fold that served as a basis for the design had no detectable catalytic activity of interest. For the purpose of this article we will refer to these protein catalysts as de novo designs. These situations provide the most realistic estimates of the ability to create enzymes from scratch and put understanding of enzymatic function to a real test.
Over three decades of work, quite a few successful examples of de novo designed protein catalysts have emerged. Despite large variability in their structures and the types of chemical reactions they catalyze, their design was accomplished using only several broadly defined approaches (Figure 2).
Figure 2.
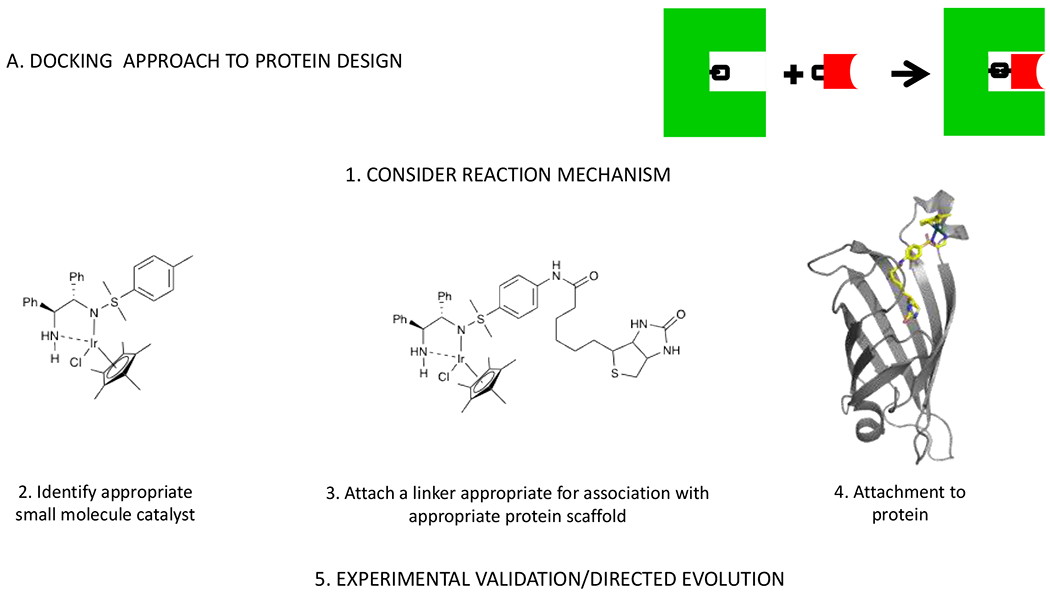
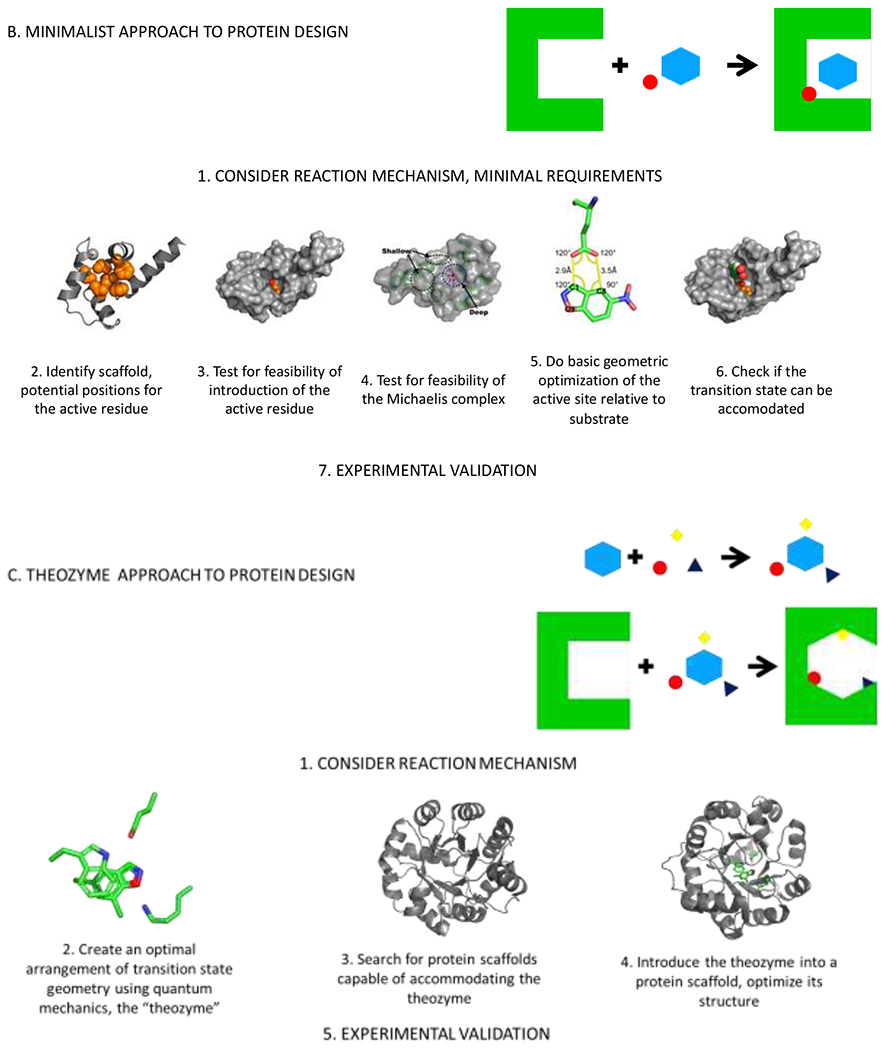
Schematic representation and representative cases of the A) docking approach with a representative example from ref. 16; B) minimalist approach with a representative example from ref. 17; and C) theozyme approach to design of protein catalysts with a representative example from ref. 18.
Historically, the first protein catalysts were created by incorporating a large cofactor (exclusively metal-containing) capable of promoting catalysis on its own into a protein to gain a substantial increase in activity and/or selectivity.9 The major advantages of this docking approach are simplicity, high levels of activity and low computational cost. Activities on par with those of natural enzymes can be achieved.10
In an approach we term minimalistic, very few (often one) residues/cofactors are introduced into a protein (whether it is based on a de novo designed scaffold or not) without extensive modeling, with the aim to establish the feasibility of catalysis rather than trying to create the optimal environment. The advantages of this approach include generality (many different types of residues and protein scaffolds can be sampled) and low computational and experimental cost. Moreover, large libraries of different proteins can be designed and experimentally characterized.
Separately, increased computational power allowed for the development of a theozyme approach, where multiple interactions of the substrate with the active site residues are modelled to achieve optimal geometry for the transition state first.11 The resultant active site arrangement is then fitted in a range of protein scaffolds identified as suitable, followed by optimization of the newly introduced interactions in the context of the designed protein.
While these approaches are fundamentally different in the way catalytic activity is achieved, the tools used, and lessons learned can be combined to achieve the desired goal. The progress in designing enzymes using the theozyme2, 12–14 and the docking approaches15 have been comprehensively reviewed recently, thus we will focus this review on the minimalist approach to design proteins with catalytic activity.
2.1. The minimalist approach.
The minimalist approach,19 originally described by DeGrado as ”sequences that are simpler than their natural counterparts but, nevertheless, retain sufficient complexity for folding and function”20 follows the evolutionary path. In nature, a low level of promiscuous activity present in the vast majority of enzymes is improved stepwise to ultimately reach extremely high catalytic efficiencies. The difficulty is to achieve the levels of activity that are significant enough for the evolution (directed or otherwise) to begin. Fortunately, even the fairly limited chemical diversity of proteinogenic amino acids provides a sufficient set of functionalities for a single mutation to offer enough reactivity for subsequent directed evolution. This approach requires very little computation and is very general. It is often applied in a hierarchal manner, where elements required to optimize or increase activity are built in as needed from the initial simplified model.21 In this review we will outline the main features of the minimalist approach and compare it to the other methods of creating protein catalysts.
2.2. Single amino acid mutations can impart high catalytic activity onto a protein scaffold.
Early attempts to replicate enzymes were intrinsically minimalist: one residue that can feasibly promote catalysis was introduced into a nascent hydrophobic cavity in a de novo designed helical scaffolds. In one of the earliest examples of minimalist design of catalysts Johnsson et. al. reported a lysine-containing helical bundle of 14-mers which was able to catalyze the decarboxylation of oxaloacetate to pyruvate, 3–4 orders of magnitude faster than their control amine catalyzed reaction.22
Baltzer and co-workers designed KO-42, a polypeptide which folded into a helix-loop-helix hairpin and dimerized.23 The histidine residues introduced onto the surface of the engineered polypeptide catalyzed the hydrolysis of p-nitrophenyl esters and follow second-order kinetics in water or water-trifluoroethanol mixtures, three orders of magnitude above the rate of the same reaction catalyzed by 4-methylimidazole. Attempts to understand the mechanism of KO-42 generated the JN-42, MN-42 and MKNR peptides, which either reduced the histidine content of the peptides or added glutamine, arginine or lysine mutations to stabilize the transition state.24 The mutants demonstrated reduced p-nitrophenyl ester hydrolysis rates but showed an enantiomeric preference, with D-norleucine p-nitrophenyl ester being hydrolyzed faster than the L-enantiomer. The arginine modifications were extended to a two histidine four arginine mutant which was capable of hydrolyzing phosphodiesters such as uridine 3–2,2,2-trichloroethylphosphate,25 with a second order rate constant of 4.2 × 10−4 M−1 s−1, and the introduction of tyrosine residues close to the active site increased the activity further.26
Not limited to ester hydrolysis, the KO-42 family of peptides was utilized for the conversion of aldimine into ketamine, a model reaction to emulate the biosynthesis of amino acids.27 This transamination reaction relies on aspartic acid as the donor and pyridoxal phosphate as the acceptor. Cognizant of the rate limiting proton transfer step required, Baltzer and co-workers employed histidine residues to facilitate the transamination, along with positively charged residues to stabilize the negatively charged substituents present in the aldimine. Designs T-4 and T-16 had millimolar affinity to the target aldimines and catalyzed their conversion to ketamine with rates three orders of magnitude higher than the control imidazole (Initial rates of 1.4–2.2 × 10−6 s−1 respectively for the two designs, compared with 9.2 × 10−10 s−1 for imidazole).
2.3. The minimalist approach is suitable for installing catalytic activity into a diverse set of protein architectures.
While de novo designed scaffolds represent the purest case of protein design, there are many well-characterized natural proteins with design potential. The main advantage of these proteins is access to a highly structurally and functionally diverse set of protein folds. However, care should be taken to ensure that no catalytic activity is present in the original scaffold. The use of stable proteins for initial starting points allows for the modification of the protein without undue concern for the stability of the mutants. This builds on elements of rational design but does not require extensive consideration of all elements of the protein. A drawback with this approach is that it requires a substrate-binding, often hydrophobic, cavity. At the same time the typical hydrophobic core of a protein is well packed to prevent interactions with water, so there are few protein structures with cavities within their hydrophobic core. Cavities can be introduced by replacing bulky hydrophobic side chains with smaller amino acids (i.e. Leu/Phe to Gly) but this often leads to significant destabilization of the protein.28 However, those proteins that already possess a cavity before any mutation are also very stable, which makes them ideal targets for modification, as this can compensate for any instability caused by the introduction of noncomplimentary amino acids within the cavity.
We have shown the utility of this approach using Calmodulin (CaM), a highly stable, allosterically regulated calcium binding messenger protein with no known catalytic activity, as a scaffold.28 Kemp elimination (Table 1) has become a de facto benchmark reaction for testing different approaches to protein design.6 It is simple, it goes through a single transition state and until very recently has been considered to be completely unnatural.29–31 The minimal requirement for Kemp eliminase activity is an appropriately placed carboxylate in a hydrophobic pocket that fits the desired substrate. In a three-step approach, the hydrophobic pocket of CaM was computationally scanned to identify a site where a catalytic carboxylate (Glu or Asp) could be accommodated (Figure 2) using Monte Carlo protocols and CHARMM minimization.32 Then the substrate was docked into the protein with AutoDock to ensure a properly positioned Michaelis complex.33 Finally, a group of super rotamers was constructed by computationally linking the side chain carboxylate oxygen in the standard rotamers of Glu and Asp to C1 of the substrate in a geometry predicted to be optimal for the reaction (C-O distance of 2.9 Å, N(substrate)C1(substrate)Oδ(Asp/Glu)Cδ(Asp/Glu) dihedral angle of 180°) to ensure the of the transition state feasibility. The only position to satisfy these requirements was F92. The resulting protein, nicknamed AlleyCat (for ALLostEricallY Controlled cATalyst) created by a single F92E mutation showed activity comparable to the first catalytic antibodies as well as Kemp eliminases designed using different approaches (5.8 M−1 s−1 vs 1.4–5,500 M−1 s−1).18, 34, 35 The catalytic activity of the protein was further improved using directed evolution; after seven rounds of mutations, a 220-fold increase in kcat/KM was achieved.36 Importantly, both the original AlleyCat and its evolved version AlleyCat7 preserve CaM allosteric regulation and are only active in the presence of calcium.
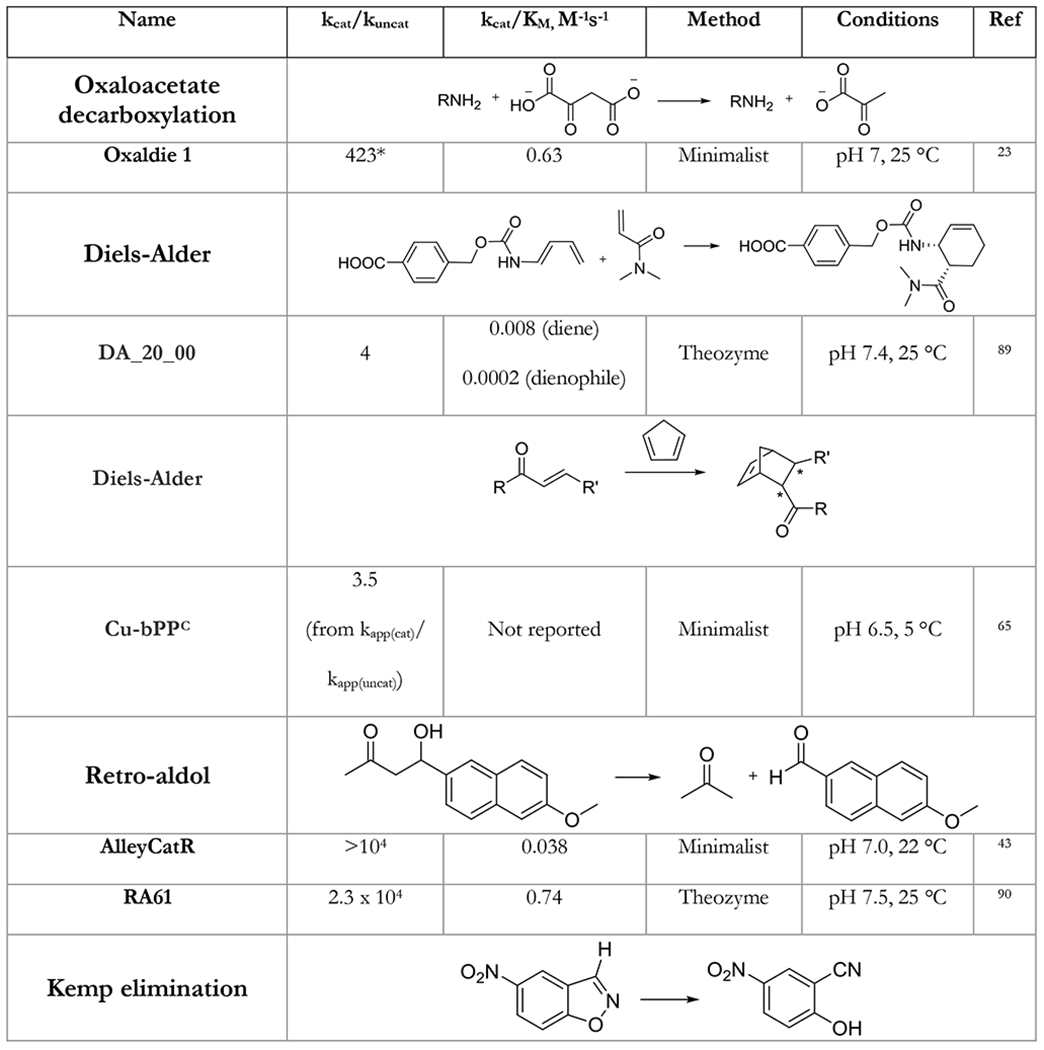
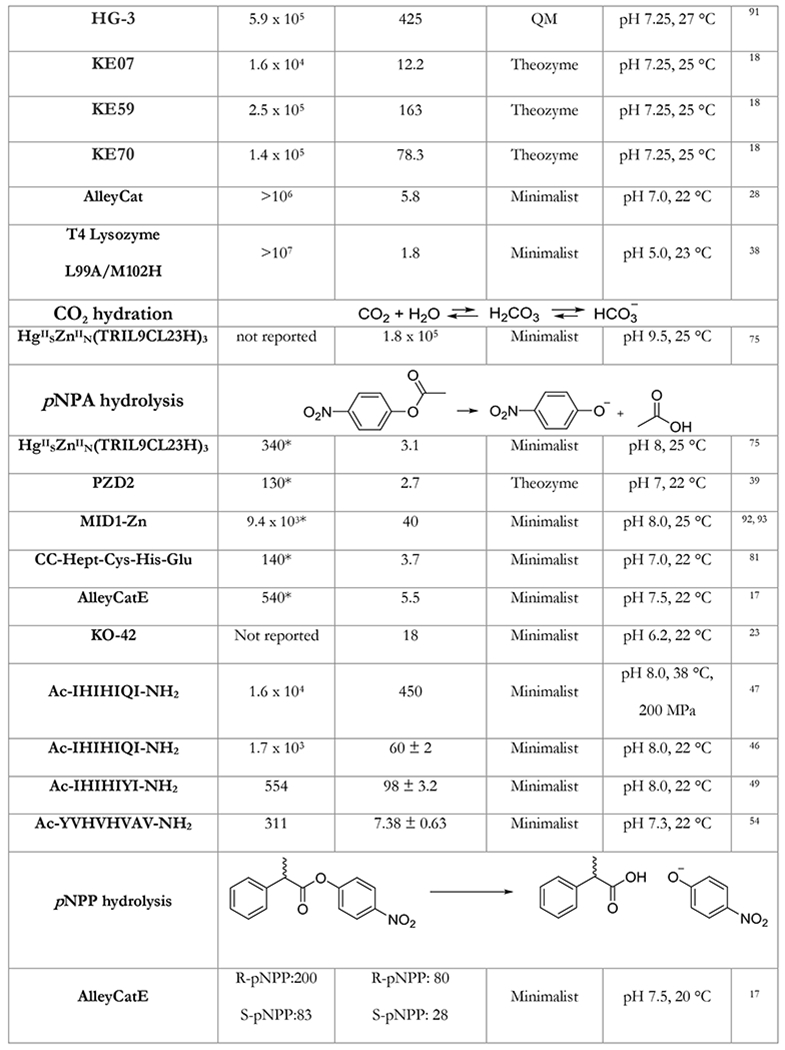
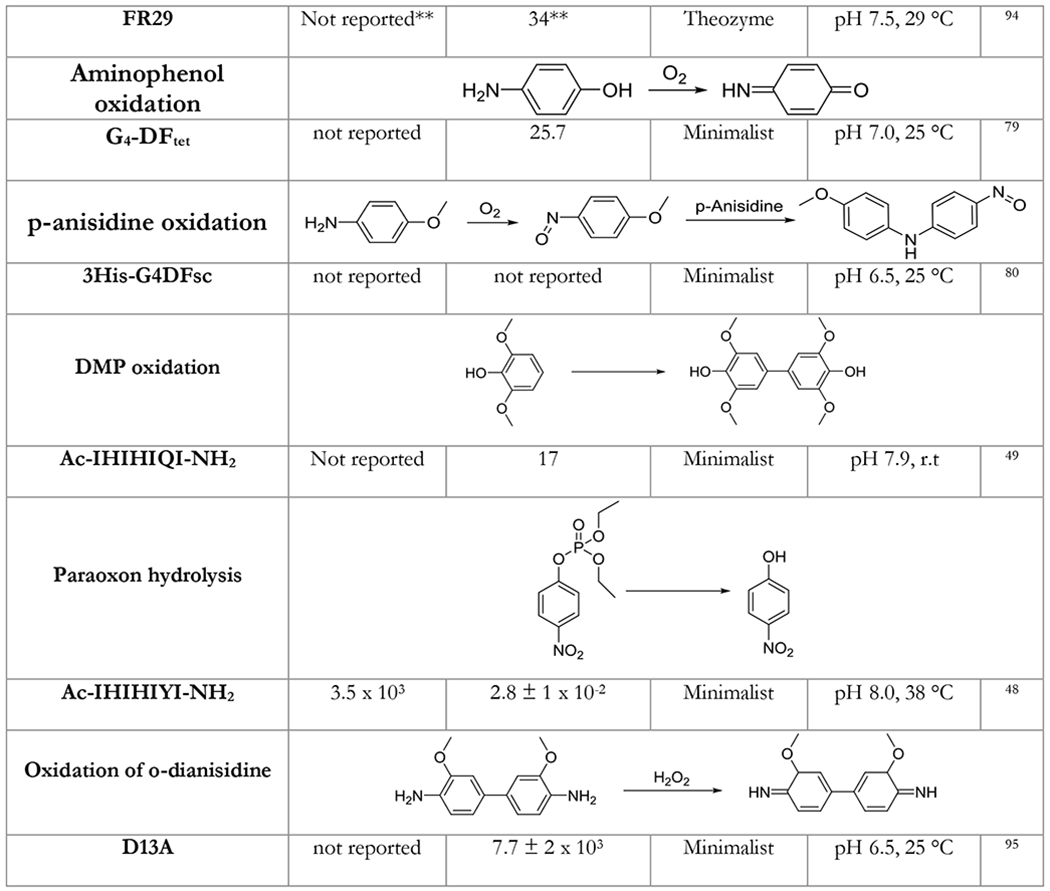
Table 1.
A table listing the kinetic parameters of protein catalysts designed using the minimalist approach. Representative examples of protein catalysts designed by other approaches are given for comparison.
 |
 |
 |
- rate enhancement (kcat/kuncat) values were calculated based on the data from ref 85
- only k2/KS value reported
Merski and Shoichet applied a similar approach in designing a Kemp eliminase from T4 lysozyme, a protein that shows no Kemp elimination activity. Rather than focusing on naturally occurring protein cavities, they took inspiration from the synthetic cavitands introduced by Cram and Lehn.37 A fully apolar hydrophobic pocket was first generated within the protein to accommodate the substrate, and the introduction of a single histidine residue into the newly generated active site resulted into a 100-fold increase in Kemp elimination activity.38
The minimalistic strategy of introducing a single highly reactive functional group in the protein scaffold is not limited to Kemp elimination. In an effort to create a de novo designed esterase, both enantiomers of a model substrate (p-nitrophenyl-(2-phenyl)-propanoate, pNPP) was selected as a substrate and both were docked into calmodulin’s C-terminal domain using AutoDock Vina to ensure that the substrate fits well in the hydrophobic pocket.17, 33 Next, residues in close proximity to the docked substrate were computationally mutated to histidine, a residue shown to promote ester hydrolysis,39 using Rosetta,40 followed by pNPP docking into the model of the resulting mutants. The docking poses were evaluated for suitable distances and geometry of the nucleophilic attack of the histidine nitrogens on the carbonyl group of the substrate. Only a single mutation in the C-terminal domain of CaM M144H satisfied the latter requirement and the resulting protein, AlleyCatE (E stands for esterase) was used for further studies. AlleyCatE catalyzed pNPP hydrolysis with efficiency that rivals that of catalytic antibodies in this reaction and was further improved by 40% using a single round of saturation mutagenesis. In agreement with the design, AlleyCatE showed kinetic preference for hydrolyzing the R-enantiomer. CaM M144H, a version of AlleyCatE, that is based on the full length CaM shows abilitiy to selectively transfer acyl groups onto calmodulin-binding targets.41
To test whether a minimalist approach can be utilized to facilitate more complex transformations, that proceed through multiple transition states, we modeled potential accommodation of the Michaelis complex formed by methodol in a hydrophobic cavity of the C-terminal domain of calmodulin using AutoDock Vina.42 Seven residues had direct Van der Waals contacts with the complex, and all these positions tolerated computational mutation to lysine. Screening of the mutants demonstrated retroaldol activity in all cases after a single lysine mutation, in some cases being high enough to display fluorescence in the crude cell lysate.43
By the very nature of minimalist protein design, the catalysts produced using these methods can be quite promiscuous. The AlleyCat family of Kemp eliminases can promote ring opening in related compounds, such as those found in leflunomide, opening the path for using libraries of designed enzymes as a starting point for creating enzymes with different functionalities.44
2.4. The minimalist approach is suitable for designing large functional supramolecular assemblies.
The minimalist approach can be applied to both the structural design of supramolecular assemblies and the installation of catalytic activity using basic physico-chemical considerations. The broader field of catalytic peptide assemblies has been comprehensively reviewed very recently,45 thus we’ll only discuss few representative cases here and put them into the overall context of design.
Since the major goal of minimalist design is to obtain some level of initial activity rather than accurately create a structure with highest possible activity, very simple approaches such as creating a peptide structures with hydrophobic residues (e.g. Leu, Ile, Val) alternating with hydrophilic residues, including those that can tightly bind metal ions, can lead to formation of functional catalytic assemblies (Figure 3). Amyloid-like fibrils formed by peptides with as few as seven amino acid residues promote hydrolysis of p-nitrophenyl esters with rates which rival those of naturally occurring hydrolases when compared by weight.46 Amyloid-like materials are extremely robust over a range of temperatures and pressures, highlighting their tolerance to adverse conditions and suggesting they are potentially useful for the development of catalysis beyond the tolerance of more sensitive protein enzymes.47 After the initial discovery of high catalytic activity in de novo designed catalytic amyloids, different groups greatly expanded the sequence space of the peptides, substrate scope and the repertoire of the reactions catalyzed.48–50, Further, Lynn and co-workers have demonstrated that amyloid assemblies can start from simple peptide self-assembly and form structures capable of enantioselective chemical reactions.51 Small changes in the individual residues, such as exchanging lysine for ornithine, changed the selectivity of the system.
Figure 3.
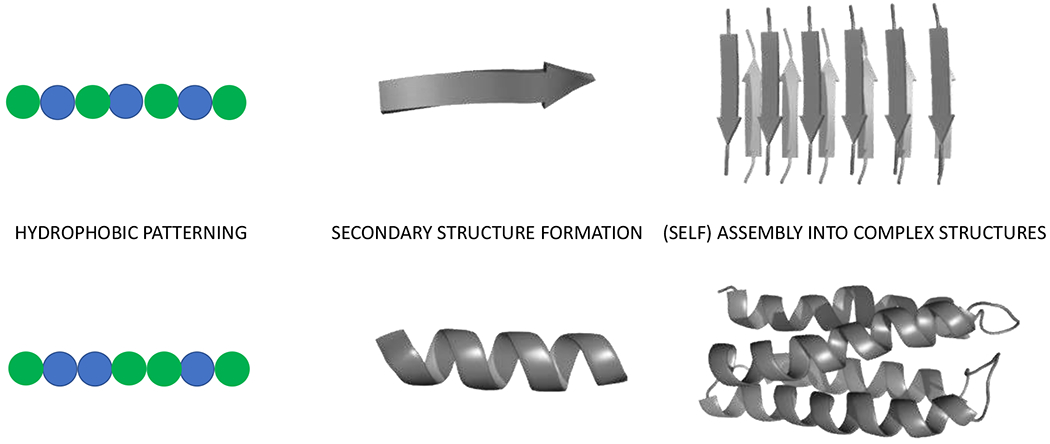
Hierarchical approach to create secondary structure elements by hydrophobic patterning (green circles – hydrophobic residues, blue circles – polar residues) that can self-assemble or be assembled in complex structures. This patterning can be applied to the formation of large libraries which can be screened for activity.
One of the difficulties in the rational improvement of these assemblies is the characterization of the active fibril,52, 53 so there is an element of high throughput screening in all catalyst designs. Friedmann et. al. designed libraries of amyloid catalysts capable of p-nitrophenyl acetate hydrolysis, with sequence variation from those previously reported.54 The broad structural similarity of the amyloid fibrils seems counterintuitive when considering the range of catalytic activities available from relatively small changes to the same structure but is consistent with observations that small differences in amyloid structure can propagate to generate large changes in the properties of the macromolecular assembly.55
2.5. The minimalist approach can be easily adapted to use unnatural amino acids.
Functionalities offering the ability to promote chemical reactions beyond the repertoire of the proteinogenic residues can be directly introduced. Moreover, a wide range of unnatural amino acids can also now be expressed in proteins, allowing them to be used in the design of large proteins, rather than restricting them to incorporation in peptides.56, 57 In one of the earliest examples of this approach, Roy and Imperiali introduced an pyridoxamine-containing unnatural amino acid into peptide S, one of the two fragments of RNase A generated by subtilisin-promoted proteolysis.58 This engineered mutant promotes the conversion of L-alanine to pyruvate 18-fold faster as compared to the blank when non-covalently associated with Protein S, the other protelolytic fragment of bovine RNase A. More recently, Liu and co-workers demonstrated that introduction of a single selenocysteine residue into the allosteric domain of adenylate kinase in a minimalist manner results in a creation of an enzyme with an antioxidative effect.59 A subsequent phenylalanine-arginine mutation improved the activity of the catalyst by 20%. The resulting allosterically modulated selenoenzyme has demonstrated activity at a subcellular level, offering potential in vivo applications.
The introduction of unnatural amino acid residues is particularly productive for binding metal ions, as a single functionality capable of effective caption of metals can be easily introduced. The design of various metalloproteins using unnatural amino acids has been recently comprehensively reviewed,60–62 so we’ll focus here on a representative case to illustrate the approach.
The Diels-Alder reaction has incredibly high synthetic value. Until very recently,63 no natural enzymes capable of promoting this transformation were known. At the same time, fundamentally this transformation requires fairly little – strong Lewis acid alone is sufficient for catalysis.64 Incorporating a Lewis acid functionality into a protein would allow for increased stereoselectivity, and thus design of various Diels-Alderases was subject of many studies. Designing enzymes for abiological reactions is challenging, but an important step in the development of the protein design field. The easiest approach to introducing a Lewis acid center into a protein core capable of operating under mild conditions in water is through use a metal ion (most frequently Cu(II)). Pyridylalanine residues can be introduced into bovine pancreatic polypeptide which can then catalyze Diels-Alder cycloadditions.65 This Lewis acid behavior can be extended to catalyse Michael additions and Friedel-Crafts alkylations, with conversions and enantiomeric excess of up to 98%.65–67 These scaffolds, which feature high stability, and availability of large (often hydrophobic) cavities for the substrate to associate with, are a common theme in minimalist protein design, as they are excellent tabulae rasae to design enzymes with.
2.6. Minimalist approaches to protein design using metal ions.
Metal ions have been utilized for introducing catalytic properties into proteins since the very early days of protein design. Indeed, metal ions provide a variety of different inherent catalytic functionalities (especially when used in complex cofactors, such as metalloporphyrins) thus greatly simplifying designer’s task. Excellent reviews exist on this topic,68–70 so we will briefly put in the context of the overall design approaches. With few notable exceptions,71 nearly all examples of designed metalloproteins are inherently minimalist – indeed a single catalytic metal center is introduced into a protein with minimal optimization of its properties (at most limited to redesign of the coordination sphere). While metal ions can be readily introduced using unnatural amino acids, as was discussed in more detail above, employing only the twenty proteinogenic amino acids offers access to easy and inexpensive production of proteins in large quantities, and allows the use of the incredible amount of structural information from the PDB to guide the design and thus presents an attractive target in advancing protein design principles.
In addition to the inherent catalytic abilities metal ions provide, they can be used to drive protein folding/assembly into a particular conformation.72 Pecoraro et al. modified the previously developed trimeric α3D scaffold by introducing cysteines in the core of the bundle.73, 74 This created a binding site for Cd(II), Hg(II) and Pb(II). Such stable metal coordination allowed for introduction of an additional tris(His) site for bind Zn(II) binding into the same metalloprotein. The resulting bimetallic construct promoted efficient pNPA (paranitrophenyl acetate) hydrolysis and CO2 hydration (Table 1).75 The related peptide TRIL2BH was designed using similar principles, binds copper (II) ions and possesses nitrite reductase activity.76, 77
DeGrado and co-workers designed a Due Ferri (DF) family of four-helical bundles using a genetic algorithm together with the CVFF forcefield for energy minimization (Figure 4).78 The original DF protein assembles as an antiparallel A2B2 heterotetramer in the presence of iron(II) to catalyse oxidation of 4-aminophenol by dioxygen.79 The antiparallel geometry of the bundle allowed for introduction of loops to link the helices into a single chain protein DFsc. DFsc is no longer subject to the constraints imposed by symmetry, and site directed mutagenesis within the active site as well as in the second-sphere coordination environment yielded an efficient N-hydroxylase for arylamines.80
Figure 4.
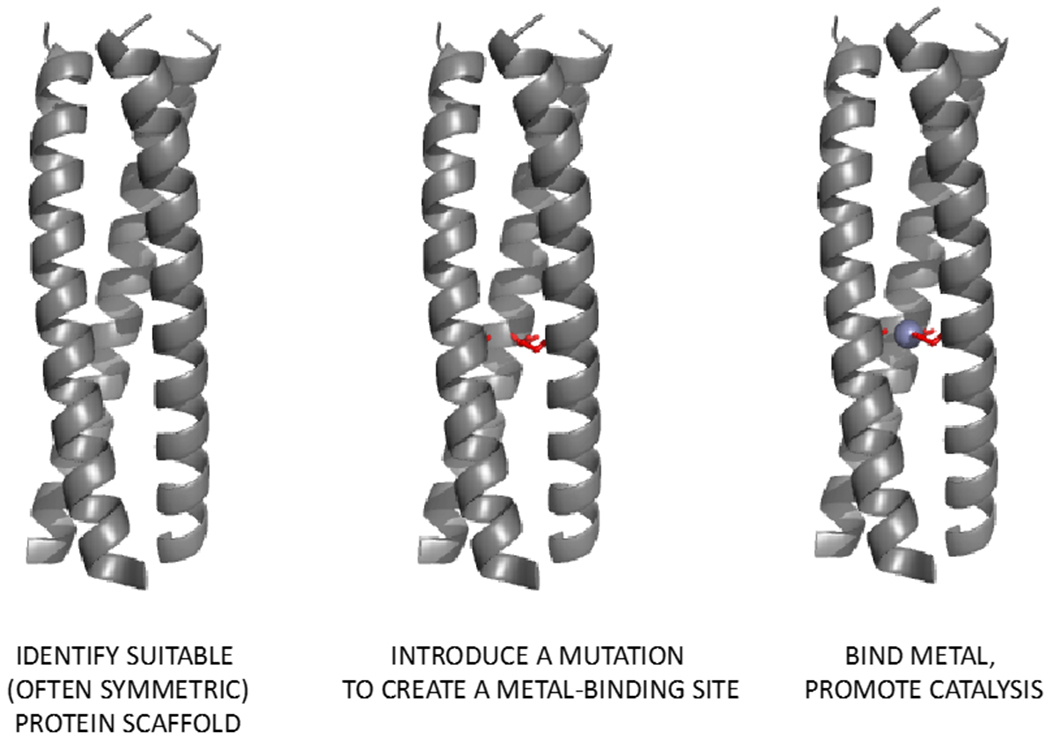
Schematic representative of the general minimalist approach to creation of metalloproteins using the trimeric coiled coils.
2.7. Multiple amino acids can be simultaneously introduced to create synergistic interactions and subsequently high activity.
To test whether new catalytic activity can be installed in a de novo designed scaffold, Woolfson and co-workers introduced a catalytic triad comprised of Cys, His and Glu in the a and d positions of each helix in CC-Hept (Figure 5).81 This highly stable protein assembly with no natural analogs was designed in a modular fashion using a toolbox of tectons,82–84 Importantly, the positioning of the triad members was not extensively optimized, they were simply arranged in close proximity to each other. The stabilizing effects of the coiled coil assembly overcame the destabilization caused by the incorporation of the catalytic residues within the hydrophobic core of the barrel. While the single and double mutants had little or no hydrolytic activity, the final design, CC-Hept-Cys-His-Glu facilitated p-nitrophenyl acetate (pNPA) hydrolysis. The resulting catalyst created by simply placing of several residues in a stable hydrophobic cavity of a coiled coil bundle is comparable to the best examples of esterases created by other approaches (kcat = 0.05 s−1 vs 0.05–4.2 s−1).
Figure 5.
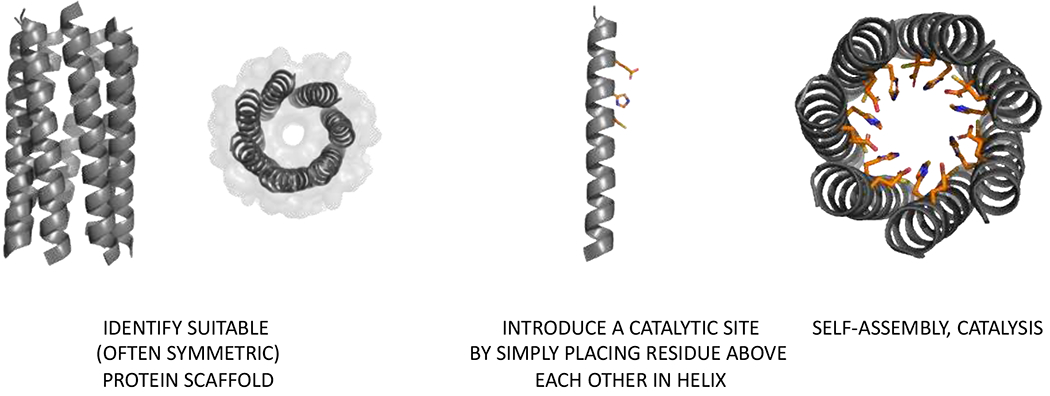
Minimalist design of a catalytic triad in a helical coiled coil bundle CC-Hept. Catalytic residues are arranged in close proximity in a peptide that upon self-assembly from an efficient p-nitrophenol esterase (PDB code 5EZC).
2.8. The minimalist approach is well suited to effectively designing large libraries of protein candidates.
Hecht and co-workers have pioneered an approach in which simple hydrophobic patterning can be used to generate a large variety of different well-folded and stable proteins.85, 86 Recently they reported a novel helical bundle enzymatic protein, which sustains the growth of E. coli in minimal media.87 This novel enzyme identified from a large library of candidates hydrolyzes the naturally occurring ferric L-enterobactin siderophore with efficiency sufficient to promote bacterial survival. The enzyme’s active site does not rely on a metal center, nor the Ser-His-Glu triad utilized by naturally occurring enzymes of the same reaction. Rather, a glutamate residue interacts with the τ-NH of an adjacent histidine, priming it for the base-catalyzed hydrolysis of the ester carbonyl of FeEnt. This shows the applicability of protein design for synthetic biology applications.
2.9. The catalysts designed by the minimalist approach have high catalytic efficiency.
In the table below, we summarize kinetic parameters reported for the de novo designed protein catalysts using minimalist approach and compare them to enzymes designed by other approaches. Catalytic efficiency (kcat/KM) has been commonly used as a yardstick for measuring success. One important caveat to keep in mind is that improving catalytic efficiency solely by decreasing the KM can only go so far.88 Indeed, if an enzyme’s KM is less that the practically used concentration of the substrate, improving it further has no practical utility whatsoever in the observed reaction rates. The ratio of the turnover number and the background rate of the reaction kcat/kuncat provides a much more realistic measure of design success.
While direct comparison of the activities of catalysts designed by different approaches should be taken with a grain of salt, Table 1 provides an instructive insight into capabilities of various methods. Kemp elimination and p-nitrophenol ester hydrolysis were extensively used as model reactions for various design methodologies shedding light on their performance. As can be seen from Table 1, the minimalist approach is on average performing just as well as the theozyme method in terms of activity of designs (especially in terms of kcat). Importantly, the predictive power of the minimalist approach is unrivaled – in addition to much lower computational cost most minimalist designs were active (in some cases simple computation was able to pinpoint the most active configuration with 100% accuracy).
3-. Conclusions.
The field of protein design has come a long way in the in the past decades. Many examples of efficient, selective and robust enzyme-like de novo designed protein catalysts have been reported. Subsequent directed evolution has led to further improvement in catalytic efficiency to reach the levels of activity observed in naturally occurring enzymes. In addition to its inherent practical utility, protein design has greatly advanced our understanding of the fundamental principles that drive enzymatic catalysis. At the same time, there are still plenty of challenges ahead. First, with the exception of the docking approach where the desired chemical activity is available in the beginning, the progress in the field is limited to promoting model chemical reactions. Second, despite the availability of tools to accurately model protein structure, the current approaches produce catalysts with fairly modest chemical activities. This is especially disconcerting given today’s impressive tools to accurately design proteins with predetermined structures.96 Indeed, while the design tools have greatly evolved, the very same principles of minimalism and modelling ideal transition states are still used to this day.19, 97 The first scenario follows evolutionary pathways and thus are likely to be very general but is not always practical in the absence of truly high-throughput selection or screening assays to evolve the original designs. The second scenario does not account for the requirement for all the steps in the enzymatic catalysis to be appropriately tuned in energy.
Arguably, the most successful de novo designed protein catalyst to date is Kemp eliminase reported by Hilvert, Mayo and coworkers.98 It is quite instructive to look at its development. The original design HG1 showed no activity and it took two rounds of rational development using X-ray crystal structural information and MD simulations to create HG3, which was subsequently improved using state-of-the-art directed evolution techniques to achieve incredibly high turnover (kcat of 700 s−1). Similarly, evolved variants of other Kemp eliminases show very high activity17, 99 and it is conceivable that given additional rounds of directed evolution they could be evolved further. Recently, Hilvert, Kuhlman and co-workers have shown even a very simple, effectively minimalist metallopeptide MID1 with nascent hydrolytic activity can be evolved to attain extremely high activity and stereoselectivity (kcat/KM 9.8 × 105).93 This suggests that from a practical standpoint the problem of catalyst design for the reactions suitable for high throughput screening is solved. This is certainly not the case for most of chemical reactions, including those of high practical importance, and better methods for designing catalysts for these reactions are needed. This is especially important from the standpoint of better understanding of the principles of enzymatic function. Moreover, resorting to improvement of activity by directed evolution assumes defeat in the rational prediction of the optimal arrangement of functional groups for catalysis.
The decades of work in the fields allowed for testing of various concepts and produced several important lessons. First, it is quite clear that focus of the transition state alone is not sufficient for creating efficient catalysts. High relative stabilization of the transition state geometry can lead to product/substrate inhibition (as in the case of catalytic antibodies) and low turnover (theozyme-based catalysts). Second, it is apparent from the location and identities of the mutations identified in the course of directed evolution that consideration of the active site alone without accounting for dynamics of the protein as a whole leads to potentially low activity, especially when simultaneously introducing multiple residues into the protein scaffold to achieve catalysis. Finally, are we there yet in terms of predicting the optimal arrangement of the functional groups for catalysis in the first place? Consideration of local electrostatics, vibrational modes, etc. can help improve the target design model further. The inadequacy of a single step, “small molecule-like” approach to enzyme design has been recognized for quite some time6 and computational approaches to simultaneously consider multiple states along the enzymatic path have been proposed.12 Undoubtedly, they will lead to improved designs and better understanding of the mechanism of enzymatic activity, especially with the development of more powerful and inexpensive computational tools.
The principal approaches to design of protein catalysts have distinct features. The docking approach is highly efficient in improving enantioselectivity and activity of existing small molecule catalysts, especially when coupled with directed evolution.100 The method is very broadly applicable, but its inherent disadvantage is the requirement of high initial activity. The theozyme approach succeeds in achieving high kcat/KM values of the designed catalysts but suffers from high cost and somewhat low success rate, perhaps contributing to the declining number of its examples in recent years. This approach is arguably best for practical testing of enzymological concepts. The minimalist approach is very computationally and experimentally inexpensive, allows for easy modeling of all steps along the Michaelis path. The initial catalytic efficiency is modest (although kcat values are comparable to those of other approaches) but can be improved by directed evolution. Its applicability to complex multistep transformation remains to be established. Recent state-of the-art examples of protein design combine the elements of all of these methods and studies are likely to continue with this trend.
Acknowledgements
This work was supported by the NIH (grant GM119634), the NSF (grants 1332349), the Nappi Family Award and the Alexander von Humboldt Foundation.
5- References
- 1.Nanda V; Koder RL, Designing Artificial Enzymes by Intuition and Computation. Nat Chem, 2010, 2, 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiss G; Celebi-Ölcum N; Moretti R; Baker D; Houk KN, Computational Enzyme Design. Angew. Chem. Int. Ed , 2013, 52, 5700–5725. [DOI] [PubMed] [Google Scholar]
- 3.Kries H; Blomberg R; Hilvert D, De Novo Enzymes by Computational Design. Curr. Opin. Chem. Biol, 2013, 17, 221–228. [DOI] [PubMed] [Google Scholar]
- 4.Peacock AFA, Recent Advances in Designed Coiled Coils and Helical Bundles with Inorganic Prosthetic Groups - from Structural to Functional Applications. Curr. Opin. Chem. Biol, 2016, 31, 160–165. [DOI] [PubMed] [Google Scholar]
- 5.Fruton JS, Contrasts in Scientific Style. Emil Fischer and Franz Hofmeister: Their Research Groups and Their Theory of Protein Structure. Proc. Am. Philos. Soc, 1985, 129, 313–370. [PubMed] [Google Scholar]
- 6.Korendovych IV; DeGrado WF, Catalytic Efficiency of Designed Catalytic Proteins. Curr. Opin. Struct. Biol, 2014, 27, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.García-Guevara F; Avelar M; Ayala M; Segovia L, Computational Tools Applied to Enzyme Design − a Review. In Biocatalysis, 2016; Vol. 1, p 109. [Google Scholar]
- 8.Damborsky J; Brezovsky J, Computational Tools for Designing and Engineering Biocatalysts. Curr. Opin. Chem. Biol, 2009, 13, 26–34. [DOI] [PubMed] [Google Scholar]
- 9.Wilson ME; Whitesides GM, Conversion of a Protein to a Homogeneous Asymmetric Hydrogenation Catalyst by Site-Specific Modification with a Diphosphinerhodium(I) Moiety. J. Am. Chem. Soc, 1978, 100, 306–307. [Google Scholar]
- 10.Watkins DW; Jenkins JMX; Grayson KJ; Wood N; Steventon JW; Le Vay KK; Goodwin MI; Mullen AS; Bailey HJ; Crump MP; MacMillan F; Mulholland AJ; Cameron G; Sessions RB; Mann S; Anderson JLR, Construction and in Vivo Assembly of a Catalytically Proficient and Hyperthermostable De Novo Enzyme. Nat. Commun, 2017, 8, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tantillo DJ; Chen J; Houk KN, Theozymes and Compuzymes: Theoretical Models for Biological Catalysis. Curr. Opin. Chem. Biol, 1998, 2, 743–750. [DOI] [PubMed] [Google Scholar]
- 12.St-Jacques AD; Gagnon O; Chica RA, Chapter 4 Computational Enzyme Design: Successes, Challenges, and Future Directions. In Modern Biocatalysis: Advances Towards Synthetic Biological Systems, The Royal Society of Chemistry: 2018; pp 88–116. [Google Scholar]
- 13.Khare SD; Fleishman SJ, Emerging Themes in the Computational Design of Novel Enzymes and Protein-Protein Interfaces. FEBS letters, 2013, 587, 1147–54. [DOI] [PubMed] [Google Scholar]
- 14.Vaissier Welborn V; Head-Gordon T, Computational Design of Synthetic Enzymes. Chem. Rev, 2018, 119, 6613–6630. [DOI] [PubMed] [Google Scholar]
- 15.Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR, Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev, 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- 16.Fujieda N; Schätti J; Stuttfeld E; Ohkubo K; Maier T; Fukuzumi S; Ward TR, Enzyme Repurposing of a Hydrolase as an Emergent Peroxidase Upon Metal Binding. Chem. Sci, 2015, 6, 4060–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moroz YS; Dunston TT; Makhlynets OV; Moroz OV; Wu Y; Yoon JH; Olsen AB; McLaughlin JM; Mack KL; Gosavi PM; van Nuland NAJ; Korendovych IV, New Tricks for Old Proteins: Single Mutations in a Nonenzymatic Protein Give Rise to Various Enzymatic Activities. J. Am. Chem. Soc, 2015, 137, 14905–14911. [DOI] [PubMed] [Google Scholar]
- 18.Röthlisberger D; Khersonsky O; Wollacott AM; Jiang L; DeChancie J; Betker J; Gallaher JL; Althoff EA; Zanghellini A; Dym O; Albeck S; Houk KN; Tawfik DS; Baker D, Kemp Elimination Catalysts by Computational Enzyme Design. Nature, 2008, 453, 190–195. [DOI] [PubMed] [Google Scholar]
- 19.DeGrado WF; Wasserman ZR; Lear JD, Protein Design, a Minimalist Approach. Science, 1989, 243, 622–628. [DOI] [PubMed] [Google Scholar]
- 20.DeGrado WF; Summa CM; Pavone V; Nastri F; Lombardi A, De Novo Design and Structural Characterization of Proteins and Metalloproteins. Annu. Rev. Biochem, 1999, 68, 779–819. [DOI] [PubMed] [Google Scholar]
- 21.Bryson JW; Betz SF; Lu HS; Suich DJ; Zhou HX; Neil KT; DeGrado WF, Protein Design: A Hierarchic Approach. Science, 1995, 270, 935–941. [DOI] [PubMed] [Google Scholar]
- 22.Johnsson K; Allemann RK; Widmer H; Benner SA, Synthesis, Structure and Activity of Artificial, Rationally Designed Catalytic Polypeptides. Nature, 1993, 365, 530–532. [DOI] [PubMed] [Google Scholar]
- 23.Broo KS; Brive L; Ahlberg P; Baltzer L, Catalysis of Hydrolysis and Transesterification Reactions of P-Nitrophenyl Esters by a Designed Helix-Loop-Helix Dimer. J. Am. Chem. Soc, 1997, 119, 11362–11372. [Google Scholar]
- 24.Broo KS; Brive L; Sott RS; Baltzer L, Site-Selective Control of the Reactivity of Surface-Exposed Histidine Residues in Designed Four-Helix-Bundle Catalysts. Fold Des, 1998, 3, 303–312. [DOI] [PubMed] [Google Scholar]
- 25.Razkin J; Nilsson H; Baltzer L, Catalysis of the Cleavage of Uridine 3’−2,2,2-Trichloroethylphosphate by a Designed Helix−Loop−Helix Motif Peptide. J. Am. Chem. Soc, 2007, 129, 14752–14758. [DOI] [PubMed] [Google Scholar]
- 26.Razkin J; Lindgren J; Nilsson H; Baltzer L, Enhanced Complexity and Catalytic Efficiency in the Hydrolysis of Phosphate Diesters by Rationally Designed Helix-Loop-Helix Motifs. ChemBioChem, 2008, 9, 1975–1984. [DOI] [PubMed] [Google Scholar]
- 27.Allert M; Baltzer L, Noncovalent Binding of a Reaction Intermediate by a Designed Helix-Loop-Helix Motif-Implications for Catalyst Design. ChemBioChem, 2003, 4, 306–318. [DOI] [PubMed] [Google Scholar]
- 28.Korendovych IV; Kulp DW; Wu Y; Cheng H; Roder H; DeGrado WF, Design of a Switchable Eliminase. Proc. Natl. Acad. Sci. U.S.A, 2011, 108, 6823–6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li AT; Wang BJ; Ilie A; Dubey KD; Bange G; Korendovych IV; Shaik S; Reetz MT, A Redox-Mediated Kemp Eliminase. Nat. Commun, 2017, 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khersonsky O; Malitsky S; Rogachev I; Tawfik DS, Role of Chemistry Versus Substrate Binding in Recruiting Promiscuous Enzyme Functions. Biochemistry, 2011, 50, 2683–2690. [DOI] [PubMed] [Google Scholar]
- 31.Yufeng M; Richard M; Yasuhisa A, Kemp Elimination Catalyzed by Naturally Occurring Aldoxime Dehydratases. ChemBioChem, 2017, 18, 451–454. [DOI] [PubMed] [Google Scholar]
- 32.Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M, Charmm: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem, 1983, 4, 187–217. [Google Scholar]
- 33.Goodsell DS; Olson AJ, Automated Docking of Substrates to Proteins by Simulated Annealing. Proteins, 1990, 8, 195–202. [DOI] [PubMed] [Google Scholar]
- 34.Thorn SN; Daniels RG; Auditor M-TM; Hilvert D, Large Rate Accelerations in Antibody Catalysis by Strategic Use of Haptenic Charge. Nature, 1995, 373, 228–230. [DOI] [PubMed] [Google Scholar]
- 35.Genre-Grandpierre A; Tellier C; Loirat M-J; Blanchard D; Hodgson DRW; Hollfelder F; Kirby AJ, Catalysis of the Kemp Elimination by Antibodies Elicited against a Cationic Hapten. Bioorg. Med. Chem. Lett, 1997, 7, 2497–2502. [Google Scholar]
- 36.Moroz OV; Moroz YS; Wu Y; Olsen AB; Cheng H; MacK KL; McLaughlin JM; Raymond EA; Zhezherya K; Roder H; Korendovych IV, A Single Mutation in a Regulatory Protein Produces Evolvable Allosterically Regulated Catalyst of Nonnatural Reaction. Angew. Chem. Int. Ed, 2013, 52, 6246–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cram DJ, The Design of Molecular Hosts, Guests, and Their Complexes (Nobel Lecture). Angew. Chem. Int. Ed, 1988, 27, 1009–1020. [PubMed] [Google Scholar]
- 38.Merski M; Shoichet BK, Engineering a Model Protein Cavity to Catalyze the Kemp Elimination. Proc. Natl. Acad. Sci. U.S.A. , 2012, 109, 16179–16183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolon DN; Mayo SL, Enzyme-Like Proteins by Computational Design. Proc. Natl. Acad. Sci. U. S. A, 2001, 98, 14274–14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman K; Renfrew PD; Smith CA; Sheffler W; Davis IW; Cooper S; Treuille A; Mandell DJ; Richter F; Ban YE; Fleishman SJ; Corn JE; Kim DE; Lyskov S; Berrondo M; Mentzer S; Popovic Z; Havranek JJ; Karanicolas J; Das R; Meiler J; Kortemme T; Gray JJ; Kuhlman B; Baker D; Bradley P, Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. Methods Enzymol., 2011, 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gosavi PM; Jayachandran M; Rempillo JJL; Zozulia O; Makhlynets OV; Korendovych IV, A Designed Enzyme Promotes Selective Post-Translational Acylation. ChemBioChem, 2018, 19, 1605–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trott O; Olson AJ, Autodock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem, 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raymond EA; Mack KL; Yoon JH; Moroz OV; Moroz YS; Korendovych IV, Design of an Allosterically Regulated Retroaldolase. Protein Sci, 2015, 24, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caselle EA; Yoon JH; Bhattacharya S; Rempillo JJL; Lengyel Z; D’Souza A; Moroz YS; Tolbert PL; Volkov AN; Forconi M; Castañeda CA; Makhlynets OV; Korendovych IV, Kemp Eliminases of the Alleycat Family Possess High Substrate Promiscuity. ChemCatChem, 2019, 11, 1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zozulia O; Dolan MA; Korendovych IV, Catalytic Peptide Assemblies. Chem. Soc. Rev, 2018, 47, 3621–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rufo CM; Moroz YS; Moroz OV; Stöhr J; Smith TA; Hu X; DeGrado WF; Korendovych IV, Short Peptides Self-Assemble to Produce Catalytic Amyloids. Nat. Chem, 2014, 6, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luong TQ; Erwin N; Neumann M; Schmidt A; Loos C; Schmidt V; Fändrich M; Winter R, Hydrostatic Pressure Increases the Catalytic Activity of Amyloid Fibril Enzymes. Angew. Chem. Int. Ed, 2016, 55, 12412–12416. [DOI] [PubMed] [Google Scholar]
- 48.Makhlynets OV; Gosavi PM; Korendovych IV, Short Self-Assembling Peptides Are Able to Bind to Copper and Activate Oxygen. Angew. Chem. Int. Ed, 2016, 55, 9017–9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lengyel Z; Rufo CM; Moroz YS; Makhlynets OV; Korendovych IV, Copper-Containing Catalytic Amyloids Promote Phosphoester Hydrolysis and Tandem Reactions. ACS Catal., 2018, 8, 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Al-Garawi ZS; McIntosh BA; Neill-Hall D; Hatimy AA; Sweet SM; Bagley MC; Serpell LC, The Amyloid Architecture Provides a Scaffold for Enzyme-Like Catalysts. Nanoscale, 2017, 9, 10773–10783. [DOI] [PubMed] [Google Scholar]
- 51.Omosun TO; Hsieh M-C; Childers WS; Das D; Mehta AK; Anthony NR; Pan T; Grover MA; Berland KM; Lynn DG, Catalytic Diversity in Self-Propagating Peptide Assemblies. Nat. Chem, 2017, 9, 805–809. [DOI] [PubMed] [Google Scholar]
- 52.Lee M; Wang T; Makhlynets OV; Wu Y; Polizzi NF; Wu H; Gosavi PM; Stöhr J; Korendovych IV; DeGrado WF; Hong M, Zinc-Binding Structure of a Catalytic Amyloid from Solid-State Nmr. Proc. Natl. Acad. Sci. U. S. A, 2017, 114, 6191–6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song R; Wu X; Xue B; Yang Y; Huang W; Zeng G; Wang J; Li W; Cao Y; Wang W; Lu J; Dong H, Principles Governing Catalytic Activity of Self-Assembled Short Peptides. J. Am. Chem. Soc, 2019, 141, 223–231. [DOI] [PubMed] [Google Scholar]
- 54.Friedmann MP; Torbeev V; Zelenay V; Sobol A; Greenwald J; Riek R, Towards Prebiotic Catalytic Amyloids Using High Throughput Screening. PLoS One, 2015, 10, e0143948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seuring C; Verasdonck J; Ringler P; Cadalbert R; Stahlberg H; Böckmann A; Meier BH; Riek R, Amyloid Fibril Polymorphism: Almost Identical on the Atomic Level, Mesoscopically Very Different. J. Phys. Chem. B, 2017, 121, 1783–1792. [DOI] [PubMed] [Google Scholar]
- 56.Liu CC; Schultz PG, Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem, 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- 57.Dumas A; Lercher L; Spicer CD; Davis BG, Designing Logical Codon Reassignment – Expanding the Chemistry in Biology. Chem. Sci, 2015, 6, 50–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Roy RS; Imperiali B, Pyridoxamine–Amino Acid Chimeras in Semisynthetic Aminotransferase Mimics. Prot. Eng, 1997, 10, 691–698. [DOI] [PubMed] [Google Scholar]
- 59.Pan T; Liu Y; Si C; Bai Y; Qiao S; Zhao L; Xu J; Dong Z; Luo Q; Liu J, Construction of Atp-Switched Allosteric Antioxidant Selenoenzyme. ACS Catal., 2017, 1875–1879. [Google Scholar]
- 60.Yu Y; Hu C; Xia L; Wang J, Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal., 2018, 8, 1851–1863. [Google Scholar]
- 61.Hu C; Chan SI; Sawyer EB; Yu Y; Wang J, Metalloprotein Design Using Genetic Code Expansion. Chem. Soc. Rev, 2014, 43, 6498–6510. [DOI] [PubMed] [Google Scholar]
- 62.Lewis JC, Metallopeptide Catalysts and Artificial Metalloenzymes Containing Unnatural Amino Acids. Curr. Opin. Chem. Biol, 2015, 25, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Byrne MJ; Lees NR; Han L-C; van der Kamp MW; Mulholland AJ; Stach JEM; Willis CL; Race PR, The Catalytic Mechanism of a Natural Diels–Alderase Revealed in Molecular Detail. J. Am. Chem. Soc, 2016, 138, 6095–6098. [DOI] [PubMed] [Google Scholar]
- 64.Reetz MT, Artificial Metalloenzymes as Catalysts in Stereoselective Diels–Alder Reactions. Chem. Rec, 2012, 12, 391–406. [DOI] [PubMed] [Google Scholar]
- 65.Coquière D; Bos J; Beld J; Roelfes G, Enantioselective Artificial Metalloenzymes Based on a Bovine Pancreatic Polypeptide Scaffold. Angew. Chem. Int. Ed, 2009, 48, 5159–5162. [DOI] [PubMed] [Google Scholar]
- 66.Drienovská I; Rioz-Martínez A; Draksharapu A; Roelfes G, Novel Artificial Metalloenzymes by in Vivo Incorporation of Metal-Binding Unnatural Amino Acids. Chem. Sci, 2015, 6, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bersellini M; Roelfes G, Multidrug Resistance Regulators (Mdrs) as Scaffolds for the Design of Artificial Metalloenzymes. Org. Biomol. Chem, 2017, 15, 3069–3073. [DOI] [PubMed] [Google Scholar]
- 68.Plegaria JS; Pecoraro VL, Sculpting Metal-Binding Environments in De Novo Designed Three-Helix Bundles. Isr. J. Chem, 2015, 55, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peacock AFA, Incorporating Metals into De Novo Proteins. Curr. Opin. Chem. Biol, 2013, 17, 934–939. [DOI] [PubMed] [Google Scholar]
- 70.Schneider JP; Lombardi A; DeGrado WF, Analysis and Design of Three-Stranded Coiled Coils and Three-Helix Bundles. Fold Des, 1998, 3, R29–40. [DOI] [PubMed] [Google Scholar]
- 71.Khare SD; Kipnis Y; Greisen P Jr.; Takeuchi R; Ashani Y; Goldsmith M; Song Y; Gallaher JL; Silman I; Leader H; Sussman JL; Stoddard BL; Tawfik DS; Baker D, Computational Redesign of a Mononuclear Zinc Metalloenzyme for Organophosphate Hydrolysis. Nat. Chem. Biol, 2012, 8, 294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Handel TM; Williams SA; DeGrado WF, Metal Ion-Dependent Modulation of the Dynamics of a Designed Protein. Science, 1993, 261, 879–885. [DOI] [PubMed] [Google Scholar]
- 73.Bryson JW; Desjarlais JR; Handel TM; DeGrado WF, From Coiled Coils to Small Globular Proteins: Design of a Native-Like Three-Helix Bundle. Protein Sci., 1998, 7, 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chakraborty S; Kravitz JY; Thulstrup PW; Hemmingsen L; DeGrado WF; Pecoraro VL, Design of a Three-Helix Bundle Capable of Binding Heavy Metals in a Triscysteine Environment. Angew. Chem. Int. Ed, 2011, 50, 2049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zastrow ML; Peacock AFA; Stuckey JA; Pecoraro VL, Hydrolytic Catalysis and Structural Stabilization in a Designed Metalloprotein. Nat. Chem, 2011, 4, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tegoni M; Yu F; Bersellini M; Penner-Hahn JE; Pecoraro VL, Designing a Functional Type 2 Copper Center That Has Nitrite Reductase Activity within Α-Helical Coiled Coils. Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 21234–21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu F; Penner-Hahn JE; Pecoraro VL, De Novo-Designed Metallopeptides with Type 2 Copper Centers: Modulation of Reduction Potentials and Nitrite Reductase Activities. J. Am. Chem. Soc, 2013, 135, 18096–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Summa CM; Rosenblatt MM; Hong J-K; Lear JD; DeGrado WF, Computational De Novo Design, and Characterization of an A2b2 Diiron Protein. J. Mol. Biol, 2002, 321, 923–938. [DOI] [PubMed] [Google Scholar]
- 79.Kaplan J; DeGrado WF, De Novo Design of Catalytic Proteins. Proc. Natl. Acad. Sc.i U.S.A, 2004, 101, 11566–11570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reig AJ; Pires MM; Snyder RA; Wu Y; Jo H; Kulp DW; Butch SE; Calhoun JR; Szyperski T; Solomon EI; DeGrado WF, Alteration of the Oxygen-Dependent Reactivity of De Novo Due Ferri Proteins. Nat. Chem, 2012, 4, 900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Burton AJ; Thomson AR; Dawson WM; Brady RL; Woolfson DN, Installing Hydrolytic Activity into a Completely De Novo Protein Framework. Nat. Chem, 2016, 8, 837–844. [DOI] [PubMed] [Google Scholar]
- 82.Fletcher JM; Boyle AL; Bruning M; Bartlett GJ; Vincent TL; Zaccai NR; Armstrong CT; Bromley EH; Booth PJ; Brady RL; Thomson AR; Woolfson DN, A Basis Set of De Novo Coiled-Coil Peptide Oligomers for Rational Protein Design and Synthetic Biology. ACS Synth Biol, 2012, 1, 240–250. [DOI] [PubMed] [Google Scholar]
- 83.Thomson AR; Wood CW; Burton AJ; Bartlett GJ; Sessions RB; Brady RL; Woolfson DN, Computational Design of Water-Soluble Α-Helical Barrels. Science, 2014, 346, 485–488. [DOI] [PubMed] [Google Scholar]
- 84.Fletcher JM; Harniman RL; Barnes FRH; Boyle AL; Collins A; Mantell J; Sharp TH; Antognozzi M; Booth PJ; Linden N; Miles MJ; Sessions RB; Verkade P; Woolfson DN, Self-Assembling Cages from Coiled-Coil Peptide Modules. Science, 2013, 340, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei Y; Hecht MH, Enzyme-Like Proteins from an Unselected Library of Designed Amino Acid Sequences. Protein Eng Des Sel, 2004, 17, 67–75. [DOI] [PubMed] [Google Scholar]
- 86.Fisher MA; McKinley KL; Bradley LH; Viola SR; Hecht MH, De Novo Designed Proteins from a Library of Artificial Sequences Function in Escherichia Coli and Enable Cell Growth. PLoS One, 2011, 6, e15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Donnelly AE; Murphy GS; Digianantonio KM; Hecht MH, A De Novo Enzyme Catalyzes a Life-Sustaining Reaction in Escherichia Coli. Nat. Chem. Biol, 2018, 14, 253–255. [DOI] [PubMed] [Google Scholar]
- 88.Eisenthal R; Danson MJ; Hough DW, Catalytic Efficiency and Kcat/Km: A Useful Comparator? Trends Biotechnol, 2007, 25, 247–249. [DOI] [PubMed] [Google Scholar]
- 89.Preiswerk N; Beck T; Schulz JD; Milovník P; Mayer C; Siegel JB; Baker D; Hilvert D, Impact of Scaffold Rigidity on the Design and Evolution of an Artificial Diels-Alderase. Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 8013–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jiang L; Althoff EA; Clemente FR; Doyle L; Röthlisberger D; Zanghellini A; Gallaher JL; Betker JL; Tanaka F; Barbas CF; Hilvert D; Houk KN; Stoddard BL; Baker D, De Novo Computational Design of Retro-Aldol Enzymes. Science, 2008, 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Privett HK; Kiss G; Lee TM; Blomberg R; Chica RA; Thomas LM; Hilvert D; Houk KN; Mayo SL, Iterative Approach to Computational Enzyme Design. Proc. Natl. Acad. Sci. U.S.A, 2012, 109, 3790–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Der BS; Edwards DR; Kuhlman B, Catalysis by a De Novo Zinc-Mediated Protein Interface: Implications for Natural Enzyme Evolution and Rational Enzyme Engineering. Biochemistry, 2012, 51, 3933–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Studer S; Hansen DA; Pianowski ZL; Mittl PRE; Debon A; Guffy SL; Der BS; Kuhlman B; Hilvert D, Evolution of a Highly Active and Enantiospecific Metalloenzyme from Short Peptides. Science, 2018, 362, 1285–1288. [DOI] [PubMed] [Google Scholar]
- 94.Richter F; Blomberg R; Khare SD; Kiss G; Kuzin AP; Smith AJT; Gallaher J; Pianowski Z; Helgeson RC; Grjasnow A; Xiao R; Seetharaman J; Su M; Vorobiev S; Lew S; Forouhar F; Kornhaber GJ; Hunt JF; Montelione GT; Tong L; Houk KN; Hilvert D; Baker D, Computational Design of Catalytic Dyads and Oxyanion Holes for Ester Hydrolysis. J. Am. Chem. Soc, 2012, 134, 16197–16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fujieda N; Schätti J; Stuttfeld E; Ohkubo K; Maier T; Fukuzumi S; Ward TR, Enzyme Repurposing of a Hydrolase as an Emergent Peroxidase Upon Metal Binding. Chem. Sci, 2015, 6, 4060–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huang P-S; Feldmeier K; Parmeggiani F; Fernandez Velasco DA; Höcker B; Baker D, De Novo Design of a Four-Fold Symmetric TIM-Barrel Protein with Atomic-Level Accuracy. Nat. Chem. Biol, 2016, 12, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pollack SJ; Jacobs JW; Schultz PG, Selective Chemical Catalysis by an Antibody Science, 1986, 234, 1570–1573. [DOI] [PubMed] [Google Scholar]
- 98.Blomberg R; Kries H; Pinkas DM; Mittl PRE; Grutter MG; Privett HK; Mayo SL; Hilvert D, Precision Is Essential for Efficient Catalysis in an Evolved Kemp Eliminase. Nature, 2013, 503, 418–421. [DOI] [PubMed] [Google Scholar]
- 99.Khersonsky O; Kiss G; Röthlisberger D.; Dym O.; Albeck S.; Houk KN; Baker D; Tawfik DS, Bridging the Gaps in Design Methodologies by Evolutionary Optimization of the Stability and Proficiency of Designed Kemp Eliminase Ke59. Proc. Natl. Acad. Sc.i U.S.A, 2012, 109, 10358–10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang H; Swartz AM; Park HJ; Srivastava P; Ellis-Guardiola K; Upp DM; Lee G; Belsare K; Gu Y; Zhang C; Moellering RE; Lewis JC, Evolving Artificial Metalloenzymes Via Random Mutagenesis. Nat. Chem, 2018, 10, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]