

Abstract
(–)-Lomaiviticin A (1) is a genotoxic C2-symmetric metabolite that arises from the formal dimerization of two bis(glycosylated) diazotetrahydrobenzo[b]fluorenes. Here we present a synthesis of the monomer 17 and its coupling to form (2S,2’S)-lomaiviticin A (4), an unnatural diastereomer of 1. (2S,2’S)-Lomaiviticin A (4) is significantly less genotoxic, a result we attribute to changes in the orientation of the diazofluorene and carbohydrate residues, relative to 1. These data bring the importance of the configuration of the conjoining bond to light and place the total synthesis of 1 itself within reach.
Graphical Abstract

Introduction.
(–)-Lomaiviticin A (1, Fig. 1a) is a C2-symmetric cytotoxic bacterial metabolite1 that possesses half-maximal inhibitory potencies (IC50s) in the nanomolar–picomolar range against cultured human cancer cell lines.2 (–)-Lomaiviticin A (1) itself is isolated in low yield (~0.5–1 mg/L) from the producing organism1a but is accessible in one step by diazo transfer to the more abundant (~60 mg/L) C1-symmetric isolate (–)-lomaiviticin C (2).1b The cytotoxicity of (–)-lomaiviticin A (1) derives from the induction of double-strand breaks (DSBs) in DNA.3,2g The mechanism of DNA cleavage involves sequential 1,7-nucleophilic addition to each diazo residue within (–)-lomaiviticin A (1),3g followed by fragmentation with loss of dinitrogen and the formation of sp2 carbon-centered radicals.4 These radicals, in turn, effect hydrogen atom abstraction from each DNA strand, ultimately leading to DSBs (Scheme S1 depicts the cleavage pathway graphically). Consistent with this mechanism, both diazotetrahydrobenzo[b]fluorene (diazofluorene) residues of (–)-lomaiviticin A (1) penetrate the DNA duplex on binding (Fig. 1b).3e This binding completely disrupts base pairing; consequently, (–)-lomaiviticin A (1) displays a preference for AT-rich regions. The aminosugar residues of (–)-lomaiviticin A (1) stabilize the complex by an electrostatic interaction and preorganize the molecule in its binding conformation wherein the diazofluorene residues overlay each other (the bound conformation of 1 is calculated as <1 kcal/mol higher in energy than the free molecule). This preorganization serves to minimize non-bonded interactions between the carbohydrates on opposing diazofluorene residues. Evidence suggests it also enhances the electrophilicity of (–)-lomaiviticin A (1) via a through-space interaction between the diazofluorenes.3g
Figure 1.

a. Structures of (–)-lomaiviticin A (1), (–)-lomaiviticin C (2), (–)-lomaiviticin aglycon (3), and (2S,2’S)-lomaiviticin A (4). b. View of (–)-lomaiviticin A (1; pale blue) bound to d(GCTATAGC)2. The diazo nitrogen and carbon atoms are shown in royal blue and magenta, respectively. The light and dark red spheres correspond to A6H4′ and A14H1′ which are the aliphatic DNA hydrogen atoms closest to each diazo carbon (4.2 Å).
We reported a synthesis of (–)-lomaiviticin aglycon (3) that proceeded via the stereoselective oxidative coupling of two monomeric diazofluorenes to form the conjoining carbon–carbon bond (blue in 1–4).5,6 Here we describe the application of this strategy to glycosylated monomeric diazofluorenes, ultimately providing access to (2S,2’S)-lomaiviticin A (4), an unnatural diastereomer of (–)-lomaiviticin A (1).7 (2S,2’S)-Lomaiviticin A (4) is non-toxic at concentrations where (–)-lomaiviticin A (1) induces extensive cell kill, and does not damage DNA in vitro or in tissue culture. This work reveals the gentotoxicity of (–)-lomaiviticin A (1) as critically dependent upon the stereochemistry of the bridging carbon–carbon bond and provides a foundation for the synthesis of (–)-lomaiviticin A (1) itself.
The synthesis of (–)-lomaiviticin A (1) presents several challenges not found in the aglycon 3. The 2,6-dideoxygenated α-oleandrose residues of 1 are susceptible to hydrolysis under mildly acidic conditions.8,1b Control of stereochemistry in both glycosylation steps is particularly challenging owing to the absence of a C2 substituent in either sugar.9 The steric congestion about the tertiary, secondary vicinal diol was expected to slow the rates of glycosylation, particularly following introduction of the first sugar. Moreover, while the β-linked-2,4,6-trideoxy-4-aminosugar (N,N-dimethyl-β-pyrrolosamine) residue is stable toward acidic hydrolysis (due to preferential N-protonation), we found in exploratory studies that the basic amine was incompatible with certain steps in the sequence. For example, intermediates bearing both halonaphthoquinone and N,N-dimethylamine residues (analogous to 15, Scheme 2) underwent spontaneous N-demethylation, potentially via intramolecular electron transfer. Thus, it was necessary to mask the amine as a carbamate, a modification which led to intractable NMR spectra (owing to rotamer isomerization on the NMR time scale), and a separate carbamate cleavage step was required to confidently characterize the products of each transformation (see Supporting Information). Finally, the α-oleandrose residue has a greater propensity for β-elimination relative to the tertiary alcohol in the aglycon 3, which was stabilized via a cyclic acetal.5,6n In light of these challenges, the recent synthesis of the fully deprotected monomeric unit of (–)-lomaiviticin A (1) by Nicolaou and co-workers is an especially notable milestone.6q We approached these challenges by developing the sequence below using a deaminated derivative, and then adapting that chemistry to the substrates shown.10
Scheme 2.

Synthesis of the monomeric bis(glycosylated) diazofluorene 17.
Results and Discussion.
Our synthesis began with the diol 5, which is accessible in four steps, 55% yield, and 91% ee from 3-ethylphenol (Scheme 1).5,6n Konigs–Knorr glycosylation11 using the bromide 6 (generated in situ from the corresponding acetate) provided the β-glycoside 7 in 44% yield (based on the donor 6) and with 5.2:1 β:α selectivity (LC/MS analysis). Other common donors, such as the corresponding glycosyl fluoride, imidate, or phosphinate, provided lower selectivity (<2:1 β:α) and the separation of these mixtures was not possible on preparative scales. Slow addition of the Schmidt imidate 812 (1h) to a solution of tri-iso-propylsilyl trifluoromethanesulfonate, 2,6-lutidine, and 7 provided the bis(glycoside) 9 in 64% yield (based on 7) and with >20:1 α-selectivity (LC/MS analysis). The alternate glycosylation sequence (introduction of a β-pyrrolosamine donor to a substrate bearing the α-oleandrose residue) was not feasible.
Scheme 1.

Synthesis of the bis(glycoside) 12.
Removal of the p-(methoxy)benzyl protecting groups (2,3- dichoro-5,6-dicyano-1,4-benzoquinone, DDQ, 94%) followed by benzoylation (benzoyl chloride, pyridine, 88%) proceeded smoothly to furnish the bis(benzoate) ester 11. We found that 11 constituted the last intermediate in the sequence that was stable toward the conditions (diethylamine, 24 °C) required to remove the 9-fluorenylmethyl (Fmoc) carbamate. Accordingly, it was exchanged at this point with a trifluoroacetamide by treatment with diethylamine, followed by trifluoroacetylation (trifluoroacetic anhydride, pyridine, 87% overall). NMR spectra of the trifluoroacetamide derivatives revealed two distinct rotameric isomers, which facilitated characterization. However, while the trifluoroacetamide corresponding to 6 could be employed in the first glycosylation, the rotameric mixture of products was found to equilibrate on silica gel, which complicated purification on preparative scales.
The enone 12 was elaborated to the monomeric diazofluorene 17 by the pathway shown in Scheme 2. Silicon-promoted, copper-catalyzed 1,4-addition of trimethylsilylmethyl magnesium chloride, followed by oxidation of the unpurified enoxysilane intermediate (not shown) with DDQ provided the γ-silylenone 13 (85%, two steps). Addition of tris(diethylamino)sulfonium trimethyldifluorosilicate [TASF(Et)] to a solution of the γ-silylenone 13 and the naphthoquinone derivative 145 generated the fragment coupling product 15 (84%). Cyclization of 15 to the cyclopentadiene of the target was achieved by heating with palladium bis(chloroacetate) [Pd(CLA)2] in the present of polymer-supported triphenylphosphine (PS-PPh3) and silver carbonate, to provide the hydroxyfulvene 16. Use of palladium acetate or trifluoroacetate in the cyclization5,6n led to low recovery of product (25–30%). The hydroxyfulvene 16 was unstable toward 1,6-elimination of the aminosugar residue upon attempted purification.3a Consequently, the unpurified product was treated directly with trifluoromethanesulfonyl azide to afford the monomeric bis(glycosylated) diazofluorene 17, which was stable toward chromatographic purification (44%, two steps).
The synthesis of (2S,2’S)-lomaiviticin A (4) was completed by the pathway shown in Scheme 3. Thus, soft enolization of the monomeric diazofluorene 17 (trimethylsilyl trifluoromethanesulfonate, 2,6-lutidine) at −78 °C formed a β-alkoxy enoxysilane intermediate (23, Fig. 2c). Following aqueous workup, the enoxysilane was dissolved in acetonitrile, cooled to −30 °C, and treated with ceric ammonium nitrate (CAN) and 2,6-di-tert-butylpyridine. Under these conditions two diastereomeric dimeric products were formed, in a ratio of ~4:1 (LC analysis). The thermal instability of the products (decomposition was observed on warming to 60 °C) and their sensitivity to bases and nucleophiles, coupled with the presence of four amide rotamers and a molecular mass exceeding 2000, rendered NMR and MS analysis of these mixtures indeterminate. The relative stereochemistry of the major diastereomer was established by its successful elaboration to (2S,2’S)-lomaiviticin A (4). For reasons that remain unclear, manganese tris(hexafluoroacetylacetonate) [Mn(hfacac)3], which was singularly successful in mediating the synthesis of the aglycon 3,5,6n was not a competent oxidant in this context.
Scheme 3.
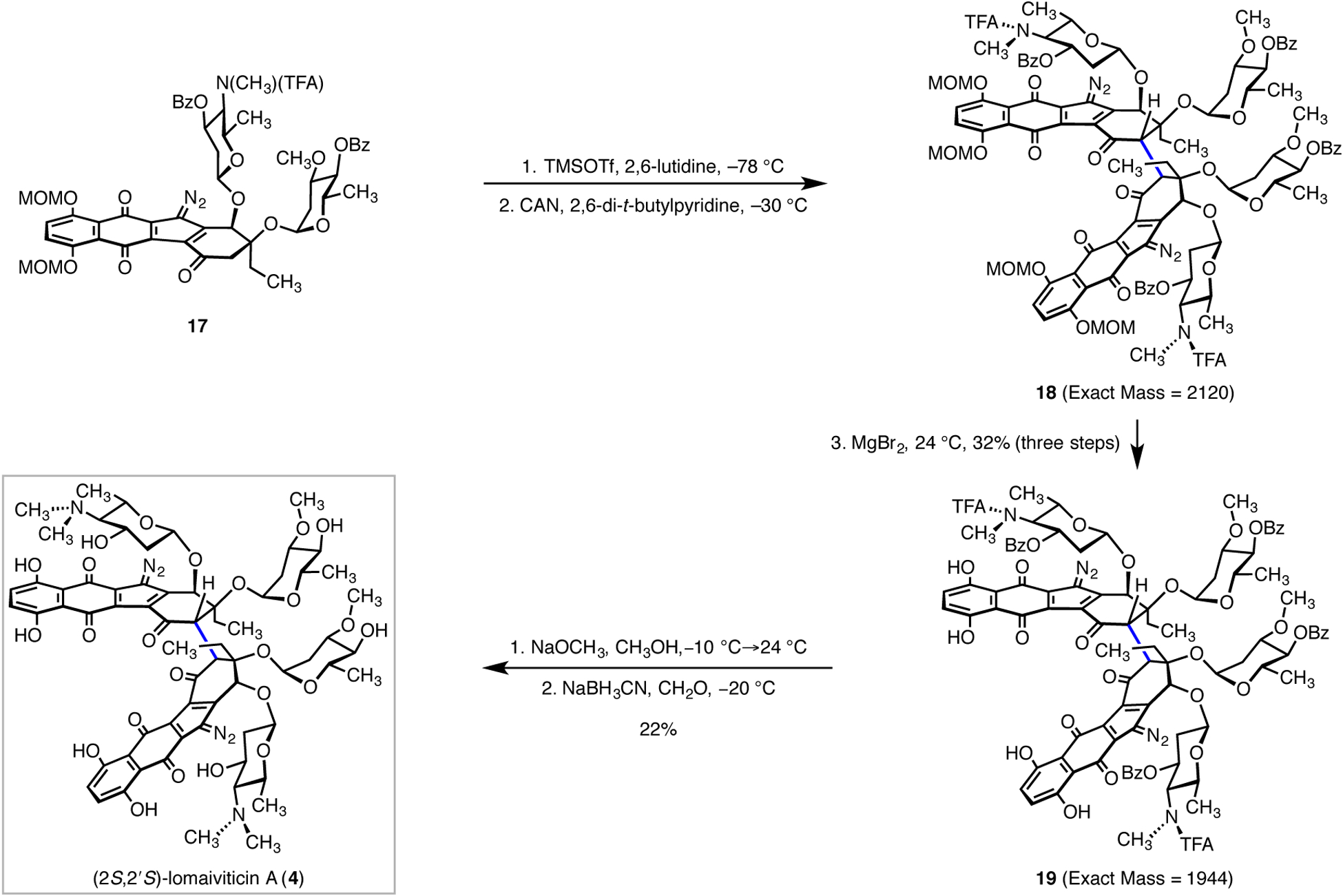
Conversion of the monomeric bis(glycosylated) diazofluorene 17 to (2S,2’S)-lomaiviticin A (4).
Figure 2.

a. Dimerization of the enoxysilane 20 using manganese tris(hexafluoroacetylacetonate) provides the (2R,2’R)-coupling product selectively. The selectivity was attributed to contact ion pairing with the reduced oxidant, as in 21. b. The silyl ether product 22 was formed with 1:1–3:2 selectivity relative to the (2S,2’R) coupling product, suggesting the stereoselectivity in the dimerization of 17 does not derive from simple non-bonded interactions. c. The stereochemical outcome in the oxidative coupling of 23 (to form 18, Scheme 3) is attributed to neighboring group participation by the oleandrose residue, which shields the si face of the monomer and (potentially) promotes the final oxidation step following addition of the radical cation to the re face of the remaining enoxysilane.
In a carefully-optimized sequence, the methoxymethyl ether substituents were removed by exposure of the product mixture to magnesium bromide in ether–tetrahydrofuran at 24 °C, to provide the tetraphenol 19 (32% from 17). The mass of the tetraphenol 19 (1944) was within the range of our mass spectrometry detector, which facilitated optimization of the following steps. Treatment of 19 with sodium methoxide in methanol served to remove the benzoate and trifluoroacetamide protecting groups. Attempts to cleave the trifluoroacetamide from derivatives bearing an O-benzyl substituent were unsuccessful, suggesting this transformation proceeds by two-fold removal of the benzoate ester, N-to-O migration of the trifluoroacetate substituent, and methanolysis of the resulting trifluoroacetate ester. Attempts to remove the ester and amide protecting groups from 18 under basic conditions resulted in decomposition. Presumably, one or more of the phenol substituents in 19 is ionized on treatment with sodium methoxide, rendering the diazo residues less electrophilic. Finally, installation of the remaining N-methyl substituents was achieved by treatment with aqueous formaldehyde and sodium cyanoborohydride, to provide (2S,2’S)- lomaiviticin A (4; 22% from 19). The minor diastereomer was separated and identified as the C1-symmetric coupling product (1H NMR analysis), but could not be isolated in quantities sufficient for complete characterization.
The C2-symmetry of (2S,2’S)-lomaiviticin A (4) was evident upon NMR analysis (see Supporting Information), and the material was distinct from natural (–)-lomaiviticin A (1) by NMR, LC/MS, and TLC analysis, supporting the stereochemical assignment depicted. In our prior studies we successfully prepared (–)-lomaiviticin aglycon (3) by oxidative coupling of the exo-mesityl monomeric diazofluorene 20 using [Mn(hfacac)3] as oxidant (Fig. 2a). In that study the acetal was used strategically to bias the stereoselectivity toward the desired (2R,2′R) configuration.5,6n We reasoned that formation of the contact ion pair 21, wherein the reduced oxidant associates closely with less-hindered re face of the radical cation intermediate, directs addition to a second enoxysilane to the si face.13
To a first approximation, the stereoselectivity in the oxidative coupling of 17 may seem to derive from simple steric considerations. However the bis(silyl ether) 22 was consistently obtained as a 1:1–3:2 mixture of the (2S,2’S)- and (2S,2’R)-coupling products by dimerization of the corresponding enoxysilane (Fig. 2b). Based on these data we hypothesize that the stereoselectivity in the oxidative coupling of 17 arises from the greater charge separation between the radical cation 24 and the nitrate counterion, which may in turn promote neighboring group participation by the adjacent oleandrose residue (Fig. 2c). The resulting dative interaction shields the si face of the radical cation, promoting addition of the remaining enoxysilane to the re face (leading to 18). The facial selectivity in addition to the remaining enoxysilane is not well-understood but if the addition step were reversible, the oleandrose may promote the second (final) oxidation via a similar interaction. This mechanistic model provides a basis to develop strategies to control selectivity in the coupling, which are ongoing.
The synthesis of (2S,2’S)-lomaiviticin A (4) provides an opportunity to directly evaluate the influence of the C2–C2’ bond stereochemistry on cytotoxicity. We anticipated that (2S,2’S)-lomaiviticin A (4) might possess cellular potencies comparable to (–)-lomaiviticin A (1), as the diazofluorenes were expected to be capable of adopting similar orientations in 1 and 4. Surprisingly, we found that (2S,2’S)-lomaiviticin A (4) was non-toxic to K562 cells at concentrations up to 5 nM. By comparison, (–)-lomaiviticin A (1) decreased cell viability by >90% at the same concentration (Fig. 3a). Additionally, (2S,2’S)-lomaiviticin A (4) did not damage DNA in a cell-free plasmid cleavage assay at concentrations up to 1 mM (Fig. 3b). By comparison, treatment with 500 nM (–)-lomaiviticin A (1) resulted in transformation of >60% of the plasmid to nicked (Form II) and linearized (Form III) DNA, as expected (first lane, Fig. 3b). Consistent with these results, (2S,2’S)-lomaiviticin A (4) did not activate the DNA damage response in K562 cells at concentrations as high as 50 nM, as determined by immunofluorescence imaging of the DNA repair factors γH2AX14 and 53BP115 (Fig. 3c). (–)-Lomaiviticin A (1) induced activation of the DNA damage response at 0.5 nM concentration.
Figure 3.

a. Cytotoxicity of (–)-lomaiviticin A (1) and (2S,2’S)-lomaiviticin A (4) against the K562 leukemia cell line. Increasing concentrations of (–)-lomaiviticin A (1; 0⟶5 nM) reduced cell viability by >80% while (2S,2’S)-lomaiviticin A (4) was non-toxic at 5 nM concentration. Cell viability was determined using the CellTiter-Glo assay. b. Plasmid DNA cleavage assay employing (–)-lomaiviticin A (1) or (2S,2’S)-lomaiviticin A (4). (–)-Lomaiviticin A (1) extensively damaged DNA at 500 nM concentration while (2S,2’S)-lomaiviticin A (4) did not generate detectable levels of DNA damage at up to 1 mM concentration in the presence or absence of DTT. c. Immunofluorescence imaging of the DNA DSB repair factors γH2AX and 53BP1 in K562 cells treated with (–)-lomaiviticin A (1) or (2S,2[isp]’S)-lomaiviticin A (4). (–)-Treatment with 0.5 nM (–)-lomaiviticin A (1) led to activation of the DNA DSB response while (2S,2’S)-lomaiviticin A (4) did not activate the DNA DSB damage response at 50 nM concentration. Immunological detection was performed using a primary antibody (rabbit polyclonal anti-53BP1 antibody and mouse monoclonal anti-phospho-histone H2AX (SER139) antibody and visualized with Alexa 488 (goat-anti-mouse IgG) and Alexa 594 (goat-anti-rabbit IgG)). Mounting medium contained 4’,6-diamidino-2-phenylindole (DAPI) to visualize nuclear DNA. Full imaging data are shown in Fig. S1
Molecular modeling was conducted to elucidate conformational dissimilarities between (–)-lomaiviticin A (1) and (2S,2’S)-lomaiviticin A (4). A molecular dynamics search was conducted on both structures, and the geometry of the lowest energy conformation was optimized by DFT (ωB97X-D/6–311G*; Fig. 4).16 While the diazofluorenes of (–)-lomaiviticin A (1) are nearly parallel (Δ = 1.1°), the diazofluorenes of (2S,2’S)-lomaiviticin A (4) are 18.5° from parallel (Fig. 4a). Additionally, one can measure the angle between the nitrogen atoms and C2 (or C2’) to place the proximity of the aminosugars to the bis(diazofluorene core) on quantitative footing (shown as a yellow line in Fig. 4b). We find that these angles are 142° and 174° for (–)-lomaiviticin A (1) and (2S,2’S)-lomaiviticin A (4), respectively. Thus, the aminosugar residues of (2S,2’S)-lomaiviticin A (4) are nearly perpendicular with respect to the core of the molecule (this is also evident in Fig. 4a).
Figure 4.

Optimized (wB97X-D/6–311G*) structures of (–)-lomaiviticin A (1) and (2S,2’S)-lomaiviticin A (4). a. Front view. b. Side view. The naphthoquinone and cyclopentadiene rings are shown in yellow. The N-methyl substituents of the aminosugar are shown in magenta. Hydrogen atoms are omitted for clarity.
These conformational differences provide an explanation for the substantial decrease in genotoxiticy of (2S,2’S)-lomaiviticin A (4). (–)-Lomaiviticin A (1) binds d(GCTATAGC)2 by penetration of both diazofluorene residues into the duplex in a nearly parallel arrangement (Fig. 1b). This penetration completely disrupts base pairing about the central binding region and induces a bend in the DNA. Prior studies established that the bound conformation of (–)-lomaiviticin A (1) is <1 kcal/mol higher in energy than its conformation in solution; thus, only minor conformational changes occur when (–)-lomaiviticin A (1) associates with DNA.3e By comparison, the large deviation from parallel found in the aromatic residues of (2S,2’S)-lomaiviticin A (4) suggests the molecule would have to access a higher energy conformation (to obtain a near-parallel arrangement) or induce a greater distortion in the DNA duplex to obtain the same mode of binding. Additionally, the aminosugar residues of (–)-lomaiviticin A (1) associate (<4.3 Å) with the phosphate residues spanning the disrupted base pairs. This electrostatic interaction undoubtedly helps to overcome the enthalpic penalty associated with disruption of the duplex and may be the primary driving force for association. The translocation of the aminosugar residues of (2S,2’S)-lomaiviticin A (4) further from the diazofluorenes would substantially diminish this effect. Finally, we have previously established that the electrophilicity of (–)-lomaiviticin A (1) is enhanced by a through-space interaction between the diazofluorenes.3g The distortion of the diazofluorenes of (2S,2’S)-lomaiviticin A (4) may decrease the reactivity of the molecule.
Conclusion.
We have described a synthesis of (2S,2’S)-lomaiviticin A (4), an unnatural diastereomer of the antiproliferative bacterial metabolite (–)-lomaiviticin A (1). The synthesis proceeds via oxidative coupling of the monomeric diazofluorene 17. Cytotoxicity and genotoxicity studies have established that the (2R,2’R) configuration of the bridging carbon atoms in (–)-lomaiviticin A (1) is required for potent activity. The change in configuration of this bond in (2S,2’S)-lomaiviticin A (4) prevents the diazofluorene residues from obtaining a parallel arrangement and results in positioning of the aminosugars further from the diazofluorenes, ultimately impeding DNA cleavage. This work establishes an efficient route to bis(glycosylated) monomeric diazofluorenes such as 17 and points toward the protecting group and end-game strategies that might be suitable to prepare (–)-lomaiviticin A (1) itself. Thus, the obstacles posed by (–)-lomaiviticin A (1) synthesis have been reduced to the single (admittedly significant) challenge of controlling stereochemistry in the oxidative coupling.
Supplementary Material
ACKNOWLEDGMENT
Financial support from the National Institutes of Health (R35-GM131913) and Yale University is gratefully acknowledged. Z.L. dedicates his work in the present paper to Professor Henry N. C. Wong on the occasion of his 70th birthday.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting Information: Detailed experimental procedures and characterization data for all new compounds (PDF)
REFERENCES
- 1.Isolation of (–)-lomaiviticin A (1): [Google Scholar]; (a) He H; Ding WD; Bernan VS; Richardson AD; Ireland CM; Greenstein M; Ellestad GA; Carter GT Lomaiviticins A and B, Potent Antitumor Antibiotics from Micromonospora lomaivitiensis. J. Am. Chem. Soc 2001, 123, 5362. For elucidation of the absolute stereochemistry of 1, see: [DOI] [PubMed] [Google Scholar]; (b) Woo CM; Beizer NE; Janso JE; Herzon SB Isolation of Lomaiviticins C–E. Transformation of Lomaiviticin C to Lomaiviticin A, Complete Structure Elucidation of Lomaiviticin A, and Structure–Activity Analyses. J. Am. Chem. Soc 2012, 134, 15285. [DOI] [PubMed] [Google Scholar]
- 2.For reviews, see: [Google Scholar]; (a) Gould SJ. Biosynthesis of the Kinamycins. Chem. Rev 1997, 97, 2499. [DOI] [PubMed] [Google Scholar]; (b) Marco-Contelles J; Molina MT Naturally Occurring Diazo Compounds: The Kinamycins. Curr. Org. Chem 2003, 7, 1433. [Google Scholar]; (c) Arya DP. Diazo and Diazonium DNA Cleavage Agents: Studies on Model Systems and Natural Product Mechanisms of Action. Top. Heterocycl. Chem 2006, 2, 129. [Google Scholar]; (d) Nawrat CC; Moody CJ Natural Products Containing a Diazo Group. Nat. Prod. Rep 2011, 28, 1426. [DOI] [PubMed] [Google Scholar]; (e) Herzon SB. The Kinamycins. In Total Synthesis of Natural Products. At the Frontiers of Organic Chemistry; Li JJ, Corey EJ, Eds.; Springer-Verlag: Berlin Heidelberg, 2012; pp 39. [Google Scholar]; (f) Herzon SB; Woo CM The Diazofluorene Antitumor Antibiotics: Structural Elucidation, Biosynthetic, Synthetic, and Chemical Biological Studies. Nat. Prod. Rep 2012, 29, 87. [DOI] [PubMed] [Google Scholar]; (g) Herzon SB The Mechanism of Action of (–)-Lomaiviticin A. Acc. Chem. Res 2017, 50, 2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Mulcahy SP; Woo CM; Ding WD; Ellestad GA; Herzon SB Characterization of a Reductively-Activated Elimination Pathway Relevant to the Biological Chemistry of the Kinamycins and Lomaiviticins. Chem. Sci 2012, 3, 1070. [Google Scholar]; (b) Colis LC; Woo CM; Hegan DC; Li Z; Glazer PM; Herzon SB The Cytotoxicity of (–)-Lomaiviticin A Arises from Induction of Double-strand Breaks in DNA. Nat. Chem 2014, 6, 504. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Woo CM; Ranjan N; Arya DP; Herzon SB Analysis of Diazofluorene DNA Binding and Damaging Activity: DNA Cleavage by a Synthetic Monomeric Diazofluorene. Angew. Chem., Int. Ed 2014, 53, 9325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Colis LC; Hegan DC; Kaneko M; Glazer PM; Herzon SB Mechanism of Action Studies of Lomaiviticin A and the Monomeric Lomaiviticin Aglycon. Selective and Potent Activity Toward DNA Double-strand Break Repair-deficient Cell Lines. J. Am. Chem. Soc 2015, 137, 5741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Woo CM; Li Z; Paulson EK; Herzon SB Structural Basis for DNA Cleavage by the Potent Antiproliferative Agent (–)-Lomaiviticin A. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 2851. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Colis LC; Herzon SB Synergistic Potentiation of (−)-Lomaiviticin A Cytotoxicity by the ATR Inhibitor VE-821. Bioorg. Med. Chem. Lett 2016, 26, 3122. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Xue M; Herzon SB Mechanism of Nucleophilic Activation of (−)-Lomaiviticin A. J. Am. Chem. Soc 2016, 138, 15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For the first proposal that the diazo residue of (–)-lomaiviticin A (1) may be electrophilic, see: [Google Scholar]; (a) Laufer RS; Dmitrienko GI Diazo Group Electrophilicity in Kinamycins and Lomaiviticin A: Potential Insights into the Molecular Mechanism of Antibacterial and Antitumor Activity. J. Am. Chem. Soc 2002, 124, 1854. For the first proposal that vinyl radicals form from 1, see: [DOI] [PubMed] [Google Scholar]; (b) Feldman KS; Eastman KJ A Proposal for the Mechanism-of-Action of Diazoparaquinone Natural Products. J. Am. Chem. Soc 2005, 127, 15344. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Feldman KS; Eastman KJ Studies on the Mechanism of Action of Prekinamycin, a Member of the Diazoparaquinone Family of Natural Products: Evidence for Both sp2 Radical and Orthoquinonemethide Intermediates. J. Am. Chem. Soc 2006, 128, 12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herzon SB; Lu L; Woo CM; Gholap SL 11-Step Enantioselective Synthesis of (–)-Lomaiviticin Aglycon. J. Am. Chem. Soc 2011, 133, 7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For synthetic studies towards (–)-lomaiviticin A (1), see: [Google Scholar]; (a) Freed JD Toward a Synthesis of the Lomaiviticins. Ph.D. Thesis, Harvard University, 2005. [Google Scholar]; (b) Nicolaou KC; Denton RM; Lenzen A; Edmonds DJ; Li A; Milburn RR; Harrison ST Stereocontrolled Synthesis of Model Core Systems of Lomaiviticins A and B. Angew. Chem., Int. Ed 2006, 45, 2076. [DOI] [PubMed] [Google Scholar]; (c) Morris WJ; Shair MD Stereoselective Synthesis of 2-Deoxy-β-glycosides Using Anomeric O-Alkylation/Arylation. Org. Lett 2008, 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Krygowski ES; Murphy-Benenato K; Shair MD Enantioselective Synthesis of the Central Ring System of Lomaiviticin A in the Form of an Unusually Stable Cyclic Hydrate. Angew. Chem., Int. Ed 2008, 47, 1680. [DOI] [PubMed] [Google Scholar]; (e) Zhang W; Baranczak A; Sulikowski GA Stereocontrolled Assembly of the C3/C3’ Dideoxy Core of Lomaiviticin A/B and Congeners. Org. Lett 2008, 10, 1939. [DOI] [PubMed] [Google Scholar]; (f) Gholap SL; Woo CM; Ravikumar PC; Herzon SB Synthesis of the Fully Glycosylated Cyclohexenone Core of Lomaiviticin A. Org. Lett 2009, 11, 4322. [DOI] [PubMed] [Google Scholar]; (g) Nicolaou KC; Nold AL; Li H Synthesis of the Monomeric Unit of the Lomaiviticin Aglycon. Angew. Chem., Int. Ed 2009, 48, 5860. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Morris WJ; Shair MD Synthesis of the N-(tert-Butyloxycarbonyl)-O-triisopropylsilyl-D-pyrrolosamine Glycal of Lomaiviticins A and B via Epimerization of L-Threonine. Tetrahedron Lett. 2010, 51, 4310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Lee HG; Ahn JY; Lee AS; Shair MD Enantioselective Synthesis of the Lomaiviticin Aglycon Full Carbon Skeleton Reveals Remarkable Remote Substituent Effects During the Dimerization Event. Chem. –Eur. J 2010, 16, 13058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Scully SS; Porco JA Asymmetric Total Synthesis of the Epoxykinamycin FL-120 B′. Angew. Chem., Int. Ed 2011, 50, 9722. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Baranczak A; Sulikowski GA Synthetic Studies Directed toward Dideoxy Lomaiviticinone Lead to Unexpected 1,2- Oxazepine and Isoxazole Formation. Org. Lett 2012, 14, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Scully SS; Porco JA Studies Toward the Synthesis of the Epoxykinamycin FL-120B′: Discovery of a Decarbonylative Photocyclization. Org. Lett 2012, 14, 2646. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Feldman KS; Selfridge BR Enantioselective Synthesis of the ent-Lomaiviticin A Bicyclic Core. Org. Lett 2012, 14, 5484. [DOI] [PubMed] [Google Scholar]; (n) Woo CM; Gholap SL; Lu L; Kaneko M; Li Z; Ravikumar PC; Herzon SB Development of Enantioselective Synthetic Routes to (–)-Kinamycin F and (–)-Lomaiviticin Aglycon. J. Am. Chem. Soc 2012, 134, 17262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Lee AS; Shair MD Synthesis of the C4-Epi-Lomaiviticin B Core Reveals Subtle Stereoelectronic Effects. Org. Lett 2013, 15, 2390. [DOI] [PubMed] [Google Scholar]; (p) Feldman KS; Selfridge BR Synthesis Studies on the Lomaiviticin a Aglycone Core: Development of a Divergent, Two-directional Strategy. J. Org. Chem 2013, 78, 4499. [DOI] [PubMed] [Google Scholar]; (q) Nicolaou KC; Chen Q; Li R; Anami Y; Tsuchikama K Total Synthesis of the Monomeric Unit of Lomaiviticin A. J. Am. Chem. Soc 2020, 142, 20201. [DOI] [PubMed] [Google Scholar]; (r) Rose JA; Mahapatra S; Li X; Wang C; Chen L; Swick SM; Herzon SB Synthesis of the bis(cyclohexenone) core of (−)-lomaiviticin A. Chem. Sci 2020, 11, 7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The work described herein was taken in part from the dissertation of Dr. Miho Kaneko, Yale University, 2015. [Google Scholar]
- 8.Overend WG; Rees CW; Sequeira JS 675. Reactions at Position 1 of Carbohydrates. Part III. The Acid-catalysed Hydrolysis of Glycosides. J. Chem. Soc 1962, 3429. [Google Scholar]
- 9.For a recent review of 2-deoxyglycoside synthesis, see:; Bennett CS; Galan MC Methods for 2-Deoxyglycoside Synthesis. Chem. Rev 2018, 118, 7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Full details of the development of this sequence will be disclosed elsewhere. [Google Scholar]
- 11.Kaneko M; Herzon SB Scope and Limitations of 2-Deoxy and 2,6-Dideoxyglycosyl Bromides as Donors for the Synthesis of β−2-Deoxy and β−2,6-Dideoxyglycosides. Org. Lett 2014, 16, 2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For a review, see:; Zhu X; Schmidt RR New Principles for Glycoside-Bond Formation. Angew. Chem., Int. Ed 2009, 48, 1900. [DOI] [PubMed] [Google Scholar]
- 13.The basis for the facial selectivity in addition to the remaining enoxysilane remains unknown. [Google Scholar]
- 14.For a review of γH2AX in DSB repair, see:; Yuan J; Adamski R; Chen J Focus on Histone Variant H2AX: To Be or Not to Be. FEBS Lett. 2010, 584, 3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For a review of 53BP1 in DSB repair, see:; Panier S; Boulton SJ Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol 2014, 15, 7. [DOI] [PubMed] [Google Scholar]
- 16.All calculations were carried out using Spartan 18, Wavefunction, Inc., Irvine, CA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.