

Abstract
IMPORTANCE
While older children and adults with achromatopsia have been studied, less is known of young children with achromatopsia.
OBJECTIVES
To characterize the macular and foveal architecture of patients with achromatopsia during early childhood with handheld spectral-domain optical coherence tomographic imaging and to make phenotype-genotype correlations.
DESIGN, SETTING, AND PARTICIPANTS
Comparative case series of 9 patients with achromatopsia and 9 age-matched control participants at a tertiary ophthalmology referral center.
MAIN OUTCOMES AND MEASURES
Patients underwent complete ocular examination, full-field electroretinography, handheld spectral-domain optical coherence tomographic imaging, and screening for genetic mutations.
RESULTS
The mean (SD) age of the patients with achromatopsia was 4.2 (2.4) years, and the mean (SD) age of the control participants was 4.0 (2.1) years. Cone-driven responses to photopic single-flash or 30-Hz stimuli were nonrecordable in 7 patients and severely attenuated in 2. Rod-driven responses to dim scotopic single-flash stimuli were normal in 7 patients and mildly subnormal in 2. Six patients (67%) had foveal ellipsoid zone disruption, of which 1 had a hyporeflective zone. Four patients (44%) had foveal hypoplasia. The average total retinal thicknesses of the macula and fovea in the patients with achromatopsia were 14% and 17% thinner than in the control participants (P < .001 and P = .001), which was mostly due to the outer retina that was 18% and 26% thinner than in control participants (both P < .001), respectively. Genetic testing revealed a common homozygous mutation in CNGB3 in 5 patients with complete achromatopsia and heterozygous mutations in CNGA3 in 2 patients with incomplete achromatopsia. The youngest and worst-affected patient harbored compound heterozygous mutations in CNGB3 and a single mutation in CNGA3.
CONCLUSIONS AND RELEVANCE
In early childhood, there is a spectrum of foveal pathology that is milder than reported in older individuals with achromatopsia, which suggests the need for early therapeutic intervention. Neither age alone nor genotype alone predicts the degree of photoreceptor loss or preservation. Thus, in anticipation of future gene therapy trials in humans, we propose that handheld spectral-domain optical coherence tomography is an important tool for the early assessment and stratification of macular architecture in young children with achromatopsia.
Achromatopsia is an autosomal recessive disorder characterized by reduced visual acuity, nystagmus, increased sensitivity to light, impaired color discrimination that corresponds to an absence or dysfunction of all 3 cone classes, normal-appearing fundus, and severely attenuated or nonrecordable cone-driven responses with normal or mildly subnormal rod-driven responses on full-field electroretinography (ffERG).1 Most patients with achromatopsia have a total loss of cone function (complete type), but some have residual cone function (incomplete type) and better visual potential.2 Postmortem histopathologic studies in patients with achromatopsia have confirmed that the cones in the macula have abnormal morphology and extensively reduced numbers.3–6 The molecular genetic etiology of achromatopsia is due to mutations in the following 5 genes that affect the cone photoreceptor transduction pathway: α3 or β3 subunit of the cone cyclic nucleotide-gated (CNG) ion channel (CNGA3 or CNGB3, respectively),7–9 α2 subunit of the cone guanine nucleotide-binding protein G(t) (GNAT2),10 α subunit of the cone cyclic guanosine monophosphate-specific 3′,5′-cyclic phosphodiesterase 6C (PDE6C),11 and γ subunit of the cone cyclic guanosine monophosphate-specific 3′,5′-cyclic phosphodiesterase 6H (PDE6H).12 There is currently no proven treatment for patients with congenital achromatopsia, but the use of recombinant adeno-associated virus-mediated gene replacement therapy has been promising in mouse and dog models of achromatopsia.13–15 In preparation for translation to human clinical trials, it will be important to be able to identify the best candidates who still have intact retinal architecture and may best benefit from genetic therapy as well as to develop objective tools for monitoring safety and efficacy.
Initial studies using lower-resolution time-domain optical coherence tomography (OCT) have shown some general changes in macular thickness,16,17 but subsequent higher-resolution spectral-domain (SD) OCT devices have revealed changes in foveal architecture that suggest achromatopsia is a progressive disease that worsens with age.18–21 Additional studies are required to determine whether retinal architecture is indeed more preserved at an earlier age, but only co-operative adults and older children are able to be imaged on conventional SD-OCT systems. Handheld SD-OCT imaging of infants and young children under anesthesia is capable of yielding high-quality images comparable to conventional SD-OCT.22–27 In this study, we used a handheld SD-OCT protocol to assess the macular architecture of young children with achromatopsia and correlated the findings with genotype and age to identify criteria that may be helpful in preparation for future genetic therapy clinical trials.
Methods
Participants
This study followed the tenets of the Declaration of Helsinki and was approved by the institutional review board at Oregon Health & Science University, Portland. Written informed consent was obtained from all patients with achromatopsia and control participants. Nine patients (5 boys and 4 girls) with achromatopsia were included in the study, and patients 7 and 8 were siblings. Nine control participants (4 boys and 5 girls) were included in the study. The mean (SD) age was 4.2 (2.4) years with a range of 0.8 to 7.8 years for the patients with achromatopsia and 4.0 (2.1) years with a range of 1.3 to 7.0 years for the control participants. Using a propofol sedation protocol as previously described,28 patients had a complete ocular examination, ffERG, color fundus photography, handheld SD-OCT, and blood draw for genetic testing. The clinical diagnosis of achromatopsia was based on a history of congenital decreased vision, nystagmus, photophobia, a severely attenuated or absent cone response with a normal or mildly subnormal rod response on ffERG, and otherwise normal findings on ophthalmic examination. Exclusion criteria for all study patients included any other vision-limiting ocular disorder such as media opacities, chorioretinal lesions, or optic nerve disease. Additional criteria for study eyes in control participants with strabismus included no history of amblyopia or developmental delay.
Full-Field ERG
Full-field ERGs were obtained binocularly using Burian-Allen bipolar contact lens electrodes (Hansen Ophthalmic Development Laboratory), proparacaine hydrochloride, 0.5%, topical anesthetic, and a Ganzfeld stimulator according to the International Society for Clinical Electrophysiology of Vision guidelines.29 All 30-Hz flicker responses were filtered with discrete Fourier transform for a fundamental frequency.
Handheld SD-OCT
Handheld SD-OCT images were obtained using the Envisu R2200-HR handheld SD-OCT device (Bioptigen). Volume scans of the macula (nominal lateral scan length of 10 mm, 1000 a-scans/b-scan, 230 b-scans) were obtained, followed by macular horizontal linear scan stacks (nominal lateral scan length of 7-9 mm, 900 a-scans/b-scan, 145 b-scans/stack) through the fovea. In this young cohort, axial length measurement was not possible owing to time and technical constraints during the sedation protocol. The recordings from each eye were judged qualitatively for image stability and signal to noise ratio. For each patient, the retinal findings were similar in each eye, and the eye with the better recording was chosen for representative data.
Image Processing and Segmentation
Individual frames from the SD-OCT scans were registered with the Stack Reg Rigid Body plug-in30 and averaged as a z-stack using ImageJ version 1.47e (National Institutes of Health). An SD-OCT image segmentation program was created in IGOR Pro version 6.22A (WaveMetrics Inc) and used to manually segment the following layers: total retina (TR), retinal nerve fiber layer, retinal ganglion cell plus inner plexiform layer (RGC+), inner nuclear layer (INL), outer nuclear layer (ONL), inner segment (IS), outer segment plus retinal pigment epithelium (OS+), and the photoreceptor layer from the Bruch membrane to the INL/outer plexiform layer interface (REC+). The standard method of obtaining OCT scans perpendicular to the macula does not visualize the obliquely oriented Henle fibers,31,32 so the ONL was measured from the outer limiting membrane to the INL/outer plexiform layer interface. Because the interdigitation zone (previously known as the cone outer segment tip33) was not always visualized in patients with achromatopsia, the outer segment layer was analyzed in combination with the retinal pigment epithelium (OS+). For each of the 8 retinal layers defined in this study, the average thickness of the macular (central 6 mm), foveal (central 1.5 mm), and extrafoveal (central 1.5-6 mm) areas were calculated. This was performed for each patient and control participant and shown in a scatterplot. The segmentation profile of each retinal layer for each patient with achromatopsia was aligned to the foveal center and plotted against the mean retinal layer thickness ± 2 SDs for the control group.
Molecular Analysis
Genetic mutation screening was performed by the Casey Eye Institute Molecular Diagnostics Laboratory, Oregon Health & Science University, except for 1 patient whose sample was tested by the University of Colorado DNA Diagnostic Laboratory, University of Colorado, Aurora. At the Casey Eye Institute Molecular Diagnostics Laboratory, DNA was isolated from whole blood, amplified by polymerase chain reaction, and used for next-generation sequencing to screen for mutations in 5 genes encoding CNGA3, CNGB3, GNAT2, PDE6C, and PDE6H. Thereafter, Sanger sequencing was used to confirm the identified mutations.
Statistical Analysis
Statistical analysis was performed using Excel (Microsoft Corp) and Igor Pro version 6.22A (WaveMetrics Inc). Group differences between the patients with achromatopsia and control participants were analyzed for significance using 2-tailed Mann-Whitney U test and Bonferroni correction for multiple comparisons (α = .05/8 = .006).
Results
Clinical Examination
All patients with achromatopsia had a history of nystagmus and photophobia since birth. Seven patients were white and 2 were Hispanic (patients 2 and 3). Visual acuity was able to be quantified in the 6 patients older than 3 years, which ranged from 20/100 to 20/400 (Table). Cycloplegic retinoscopic refraction revealed hyperopia with a range of +1.00 to +5.00 spherical equivalent (SE) in 7 patients and mild myopia with a range of −0.50 to −1.00 SE in 2 patients. The mean SE of the patients with achromatopsia was not significantly different from the control participants (mean [SD], 2.36 [2.03] vs 2.25 [1.15] diopters, respectively; P = .45). Fundus examination showed either mild foveal changes or normal-appearing macula (Table).
Table.
Summary of Study Cohort
| Patient No./Sex/Age, y | OD, OS | Fundus Appearance | Full-Field ERG | SD-OCT | Gene Mutation | |||
|---|---|---|---|---|---|---|---|---|
| VA | Ret, SE | Scotopic | Photopic | FH | Foveal EZ | |||
| 1/M/0.8 | FF, FF | +5.00, +3.50 | Foveal pigment mottling | Normal | NR | Yes | Complete loss | CNGA3 heterozygous p.D260N:c.778G>A; CNGB3 heterozygous c.1148delC, p.R403Q:c.1208G>A |
| 2/F/1.3 | FF, FF | +3.25, +3.00 | Normal | Mildly reduced | Severely reduced and prolonged | No | Disrupted | CNGA3 heterozygous c.940-942delATC, p.R661S:c.1981C>A |
| 3/M/2.8 | FF, FF | +3.50, +3.50 | Normal | Mildly reduced and prolonged | NR | Yes | Disrupted | Declined testing |
| 4/M/3.5 | 20/400, 20/400 | +4.25, +3.75 | Normal | Normal | NR | No | Normal | CNGB3 homozygous c.1148delC |
| 5/M/4.6 | 20/100, 20/100 | −1.00, −1.00 | No FLR | Normal | NR | Yes | Intact but flat | CNGB3 homozygous c.1148delC |
| 6/F/4.9 | 20/200, 20/200 | +2.50, +1.25 | No FLR | Normal | Severely reduced and prolonged | No | Disrupted with hyporeflective zone | CNGA3 heterozygous p.Y357C:c.1070A>G, p.T565M:c.1694C>T |
| 7/M/5.8 | 20/400, 20/400 | +3.00, +2.75 | Normal | Normal | NR | No | Disrupted | CNGB3 homozygous c.1148delC |
| 8/F/6.7 | 20/200, 20/150 | +1.50, +1.00 | No FLR | Normal | NR | No | Disrupted | CNGB3 homozygous c.1148delC |
| 9/F/7.8 | 20/150, 20/200 | −0.50, −0.50 | No FLR | Normal | NR | Yes | Intact but flat | CNGB3 homozygous c.1148delC |
Abbreviations: ERG, electroretinogram; EZ, ellipsoid zone; FF, fixes and follows; FH, foveal hypoplasia; FLR, foveal light reflex; NR, nonrecordable; OD, right eye; OS, left eye; Ret, retinoscopic refraction; SD-OCT, spectral-domain optical coherence tomography; SE, spherical equivalent; VA, visual acuity.
Genetic Analysis
Genetic testing revealed that 5 patients (patients 4, 5, and 7-9) had the same homozygous c.1148delC frameshift mutation in CNGB3 that has been commonly reported7,9 (Table). Two patients (patients 2 and 6) had heterozygous mutations in CNGA3. Patient 2 had 1 reported mutation, c.940-942delATC,8 and a second novel mutation, c.1981C>A, with a low allele frequency of 0.001 and high PolyPhen-2 score of 1.0. The latter mutation results in an amino acid substitution of serine in place of arginine at residue 661 on CNGA3. Patient 6 had 2 reported mutations, c.1070A>G34 and c.1694C>T,8 which together have been reported to be associated with oligocone trichromacy.34 Patient 1 had 1 reported mutation in CNGA3, c.778G>A,8 as well as 2 reported mutations in CNGB3, c.1148delC9 and c.1208G>A,35 which together have been associated with cone dystrophy.35 Patient 3 declined genetic testing.
ffERG and Photoreceptor Function
The dark-adapted isolated rod response was normal in 7 patients with achromatopsia and mildly abnormal in 2 (patients 2 and 3) (Figure 1 and Table). The light-adapted single-flash and 30-Hz cone responses were nonrecordable in 7 patients (patients 1, 3-5, and 7-9), questionably detectable in patient 2, and severely abnormal but recordable in patient 6.
Figure 1. Representative Full-Field Electroretinographic Photopic and Scotopic Recordings From Patients 1, 2, 4, and 6, Showing the Spectrum of Cone-Driven and Rod-Driven Responses in the Cohort.
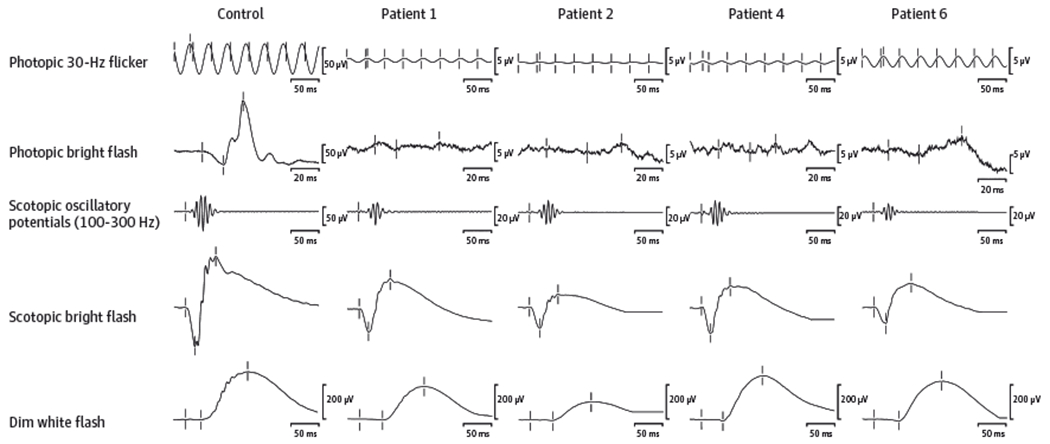
The amplitude scales for photopic responses and scotopic oscillatory potentials are different between the patients and the control participant.
Handheld SD-OCT and Macular Morphology
Six patients with achromatopsia (67%) had disruption of the foveal ellipsoid zone (EZ; previously known as the IS/outer segment junction36) that ranged from a complete loss (patient 1) to a foveal hyporeflective zone (FHZ) (patient 6) and milder changes (patients 2,3,7, and 8) (Figure 2 and Table). Two patients (patients 5 and 9) had an EZ that appeared intact but lacked the prominent foveolar outer segment thickening and EZ peak that are expected in normal foveal morphology. Only 1 patient (patient 4) had a normal-appearing foveal EZ. While the interdigitation zone was easily distinguished in all control participants, it was either absent or much more subtle in patients with achromatopsia, especially at the fovea. Four patients (44%) (patients 1,3,5, and 9) had foveal hypoplasia with a retained inner plexiform layer, INL, and outer plexiform layer across the fovea.
Figure 2. Spectral-Domain Optical Coherence Tomographic Images of Patients With Achromatopsia and a Representative Control Participant Aged 1.3 Years.
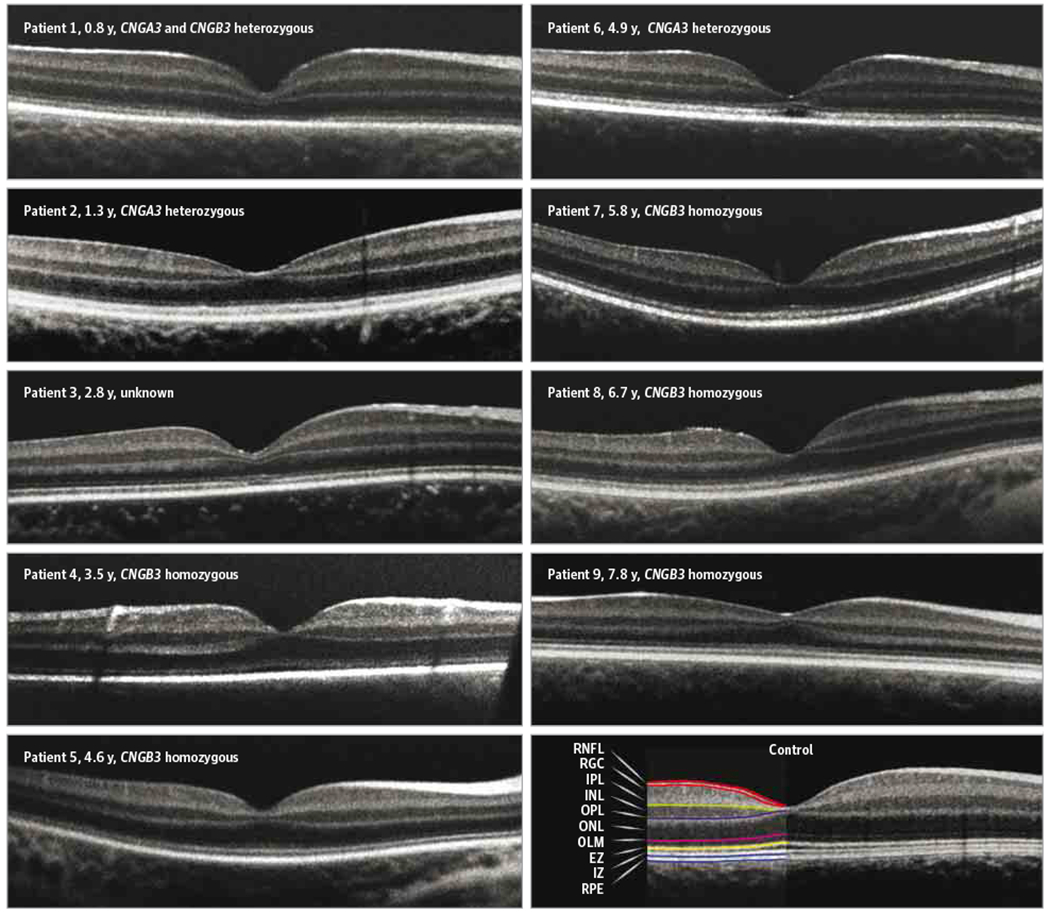
The ages and genotypes are indicated for each patient with achromatopsia. In this young cohort, axial length measurement was not possible owing to time and technical constraints during the sedation protocol, so a common scale bar was not applied. EZ indicates ellipsoid zone; INL, inner nuclear layer; IPL, inner plexiform layer; IZ, interdigitation zone; OLM, outer limiting membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; RGC, retinal ganglion cell; RNFL, retinal nerve fiber layer; and RPE, retinal pigment epithelial layer.
Segmentation and quantification of the SD-OCT linear scans showed that the total retinal thickness of the macula (central 6 mm) was on average 38.8 μm (14%) thinner in the patients with achromatopsia than the control participants (mean [SD] thickness, 247.7 [13.7] vs 286.5 [9.9] μm, respectively; P < .001) (Figure 3). Analysis of the different retinal layers revealed that this difference was mostly due to a decrease in the REC+, which was on average 29.5 μm (18%) thinner in the patients with achromatopsia compared with the control participants (mean [SD] thickness, 131.9 [9.6] vs 161.5 [6.4] μm, respectively, P < .001). The mean (SD) thicknesses of the component layers of the outer retina were all thinner in the patients with achromatopsia than the control participants: ONL (69.6 [8.2] vs 86.5 [5.1] μm; P < .001), IS (21.3 [1.5] vs 27.0 [2.0] μm; P < .001), and OS+ (41.1 [3.4] vs 47.9 [2.5] μm; P < .001). The mean (SD) thickness of the RGC+ layer was significantly thinner (11%) in patients with achromatopsia than the control participants (59.6 [4.4] vs 66.9 [3.3] μm; P = .002). The mean thicknesses of the macular retinal nerve fiber layer and INL were not significantly different between the achromatopsia and control groups.
Figure 3. Mean Retinal Layer Thickness Scatterplots.
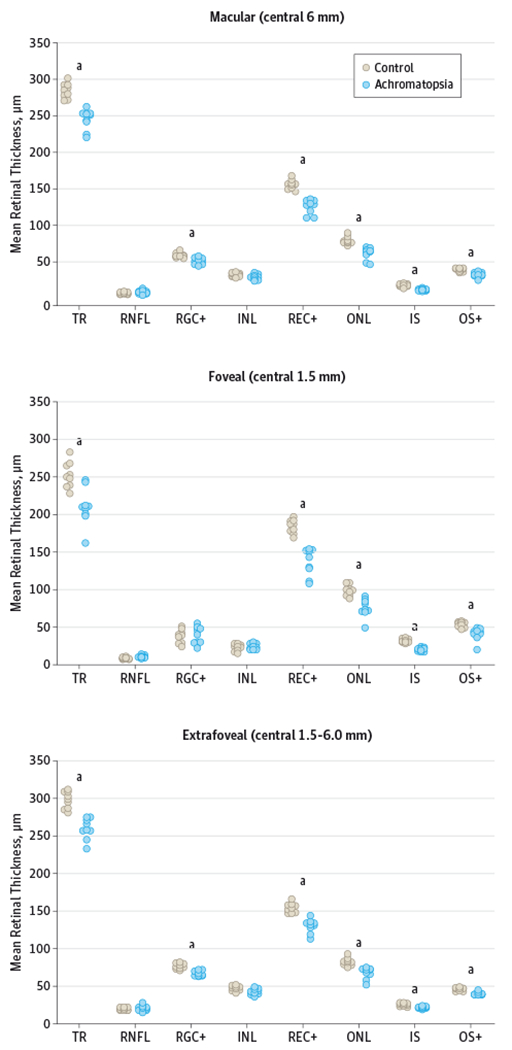
INL indicates inner nuclear layer; IS, inner segment layer; ONL, outer nuclear layer; OS+, outer segment plus retinal pigment epithelium; REC+, photoreceptor layer from Bruch membrane to INL/outer plexiform interface; RGC+, retinal ganglion cell plus inner plexiform layer; RNFL, retinal nerve fiber layer; and TR, total retina. aP < .006.
Analysis of the foveal area (central 1.5 mm) showed similar findings as in the macular analysis, except there was even greater thinning of the outer retinal layers (Figure 3). The mean (SD) total retinal thickness of the fovea was 42.2 μm (17%) thinner in the patients with achromatopsia than in the control participants (210.2 [24.8] vs 252.4 [17.2] μm; P = .001). Foveal thinning was mainly due to the REC+, which was 47.1 μm (26%) thinner in the patients with achromatopsia than in the control participants (mean [SD] thickness, 136.7 [18.0] vs 183.8 [9.2] μm; P < .001). The average amount of thinning in the TR was less than the amount of thinning in the REC+ because of the additional contribution of retained inner retinal layers to TR in patients with achromatopsia with foveal hypoplasia. The mean (SD) thicknesses of the component layers of the outer retina in the fovea were all thinner in the patients with achromatopsia than the control participants: ONL (75.4 [12.5] vs 98.5 [7.3] μm; P < .001), IS (20.3 [2.5] vs 31.8 [2.7] μm; P < .001), and OS+ (41.0 [8.6] vs 53.5 [3.8] μm; P < .001). Analysis of the extrafoveal area (central 1.5-6.0 mm) showed the same significant results as in the macular analysis for TR (P < .001), RGC+ (P < .001), REC+ (P < .001), ONL (P < .001), IS (P = .002), and OS+ (P < .001) (Figure 3).
The distribution of the segmentation profiles was distinctly different between the achromatopsia and control groups, except for the INL (Figure 4). The patients with achromatopsia had diffuse thinning of the macular TR, REC+, ONL, IS, and OS+ layers. Three patients with the worst foveal EZ pathology (patients 1, 3, and 6) also had further relative thinning of the foveolar REC+ and ONL, which caused a bimodal distribution of the foveolar contour of the REC+ and ONL plots among the patients with achromatopsia. For the OS+ traces, patient 1 had complete loss of the central foveal EZ, which is seen in Figure 4 as a wide central trough. For the foveal thickness scatterplots, patient 1 was an outlier for thinnest OS+, while patient 6 with an FHZ was an outlier for the thinnest ONL and TR; both patients were outliers for the thinnest foveal REC+ (Figure 3). The 2 outliers for thickest foveal TR were patients 4 and 5, who also had the thickest foveal REC+ and ONL.
Figure 4. Retinal Layer Thicknesses of Patients With Achromatopsia Plotted Against the Control Mean ± 2 SDs.
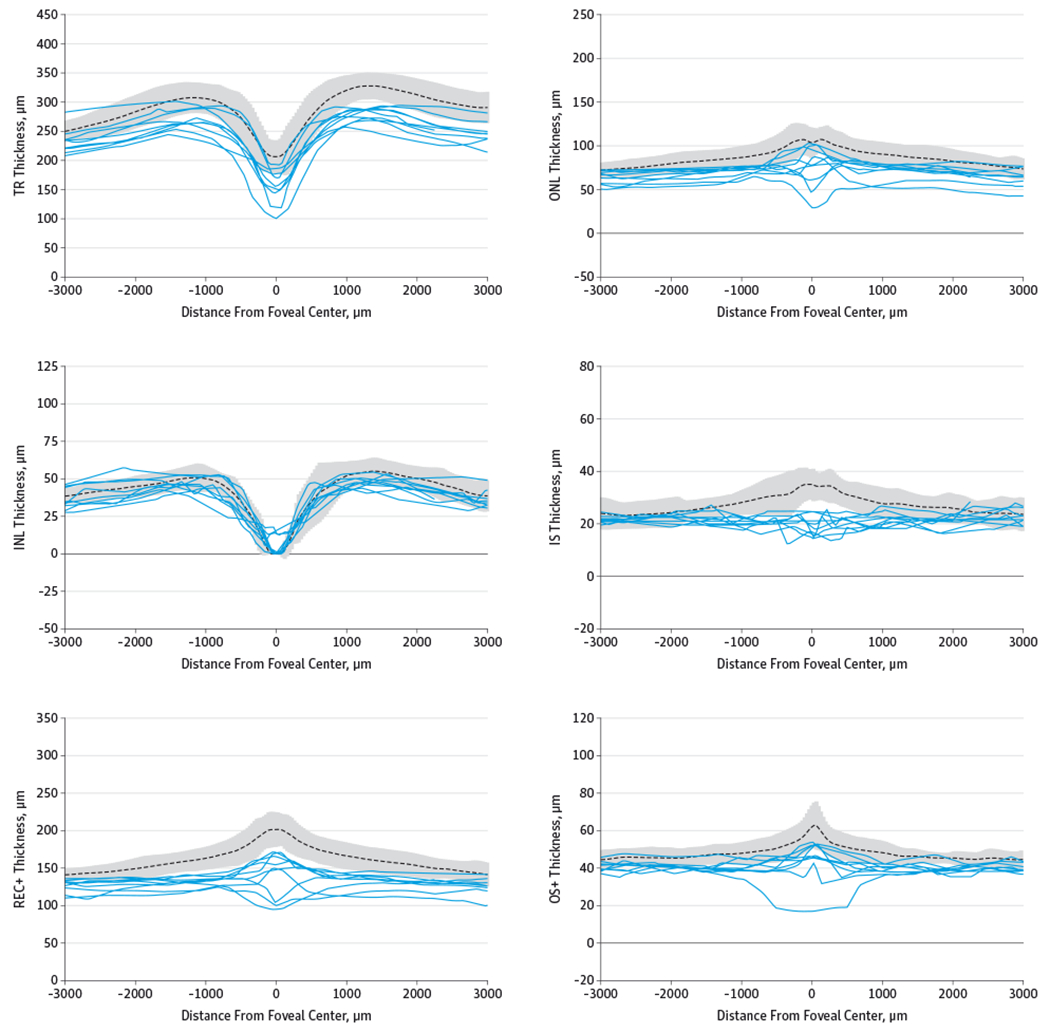
Retinal layer thicknesses are shown for patients with achromatopsia (solid lines) plotted against the control mean (dotted line) ± 2 SDs (gray area). INL indicates inner nuclear layer; IS, inner segment layer; ONL, outer nuclear layer; OS+, outer segment plus retinal pigment epithelial layer; REC+, photoreceptor layer from Bruch membrane to INL/outer plexiform interface; and TR, total retina. For distance from foveal center, −3000 is in the direction of temporal retina and +3000 is nasal retina.
Discussion
Studies of macular morphology in older patients with achromatopsia using conventional SD-OCT systems showed that foveal EZ disturbance (70%-100%) and foveal hypoplasia (50%-80%) were common earlier in life with subsequent age-dependent development of FHZ (54%-60%) and ONL thinning.18–21 Using our handheld SD-OCT protocol, we show for the first time, to our knowledge, that foveal EZ disturbance (71%) and foveal hypoplasia (43%) were also common in early childhood, while FHZ was found in only 1 patient (14%). Taken together, these data indicate that some disease characteristics such as foveal hypoplasia appear early and persist into adulthood, but other signs such as FHZ tend to be associated with older age. Neither foveal hypoplasia nor FHZ is specific to achromatopsia, and it is thought that while foveal hypoplasia is a product of abnormal macular development, an FHZ is the result of an imbalance between the rate of cellular debris accumulation and the efficiency of apoptotic cell clearance and phagocytosis.18–21 Foveal EZ disturbance was common in early childhood but relatively mild in most patients, which is encouraging for future genetic therapy trials. However, the patient with the most severe foveolar EZ loss was also the youngest patient in our cohort (patient 1; age 0.8 year), serving as a reminder that age alone is not completely predictive of foveal pathology in achromatopsia.
Further segmentation analysis of the SD-OCT data showed that the average macular and foveal thicknesses of the patients with achromatopsia were thinner than in the control participants in our cohort, which is in agreement with previous reports.3,5,18–20 Macular thinning in early childhood was mainly due to attenuation of the foveal outer retina, which suggests either that cone photoreceptor atrophy begins very early in life or that shortened photoreceptors are a congenital characteristic of abnormal cone morphology in individuals with achromatopsia. Specifically, the 3 patients with the worst foveal EZ disruption (patients 1, 3, and 6) had additional relative thinning of the foveolar photoreceptor cell body layer, which suggests that these patients may not benefit as greatly from early intervention with genetic therapy, at least for preservation or restoration of visual function. In contrast, patients with milder foveal EZ pathology did not have additional relative foveolar photoreceptor cell body atrophy. The 2 patients (patients 4 and 5) with the most normal-appearing foveal EZ architecture also had the most preserved foveal photoreceptor cell body layer, which suggests that they would likely benefit the most from future gene therapy clinical trials. Thus, these results underscore the utility of handheld SD-OCT as an objective tool for assessing young children with achromatopsia.
We also assessed for genotype-phenotype correlations in our young cohort, even though no clear correlations have been found thus far in conventional SD-OCT studies of older patients with achromatopsia.18,19 Five patients (patients 4,5, and 7-9) with complete achromatopsia all had the most common mutation in CNGB3, homozygous c.1148delC, which results in complete loss of function of the CNG β subunit due to a 1-base pair frameshift mutation that eliminates all protein sequences downstream of Thr383, including the pore, S6 transmembrane, and cyclic guanosine monophosphate binding domains.9 Even though all CNG β subunits are predicted to be dysfunctional in these 5 patients, they all had relatively mild foveal pathology and thicker retinal layers compared with the rest of the cohort. The 2 patients with heterozygous mutations in CNGA3 had evidence of incomplete achromatopsia on ffERG (patient 6 definite, patient 2 suspect) (Figure 1). A correlation of incomplete achromatopsia in patients with mutations in CNGA3 is supported by in vitro studies that demonstrate the ability of wild-type CNG β3 subunits to partially rescue the effect of defective mutant α3 subunits.37 Patient 2 had 1 previously reported mutation associated with complete achromatopsia that causes a deletion in isoleucine at the 312 residue in the S5 transmembrane domain8 and had 1 novel mutation corresponding to an Arg661Ser missense mutation that is likely disease causing but with relatively mild effect owing to its distal location toward the carboxyl end of the CNG α subunit. Patient 6 had the same heterozygous mutations in CNGA3 that have been associated with oligocone trichromacy,34 which is a rare and mild variant of achromatopsia with normal color vision. Thus, it is curious that patient 6 had an FHZ, which is a phenotype associated with later-stage disease. Interestingly, the patient with the worst foveal pathology (patient 1) was the only individual to have mutations in 2 achromatopsia genes, CNGB3 and CNGA3. In this patient, the c.778G>A mutation in CNGA3 has been associated with complete achromatopsia,8 while the combined c.1148delC and c.1208G>A mutations in CNGB3 have been associated with an autosomal recessive progressive cone dystrophy.35 While one CNGB3 allele results in a complete loss of function, the other CNGB3 allele has an Arg403Gln mutation, which produces a loss of charge in the pore domain of the β subunit. The likely result is CNG channels with limited ability to transfer cations, restricting but not completely eliminating cone function. However, patient 1 presented with complete loss of photopic ffERG at a very early age, which is unusual for progressive cone dystrophy and more consistent with complete achromatopsia. Thus, it is possible that the additional Asp260Asn mutation in 1CNGA3 allele located in the S3 transmembrane domain of the α subunit may exacerbate alterations in subunit interaction, complex assembly, and/or plasma membrane targeting to a degree that there are no functional CNG channels in cone photoreceptors. Overall, these genotype-phenotype studies showed that CNGB3 homozygous c.1148delC was associated with milder foveal pathology, but these cases also demonstrate that neither the predicted severity of genotype mutations nor the degree of photopic ffERG attenuation always correlated with foveal pathology in achromatopsia.
These data add to the cumulative evidence that supports earlier intervention to better preserve retinal architecture and visual potential in patients. Indeed, it has been shown that recombinant adeno-associated virus-mediated gene replacement therapy in models of achromatopsia produced the best results in the youngest animals,14 which further underscores the importance of assessing patients with achromatopsia with SD-OCT in early childhood as we have done here. Hopefully, gene therapy would not only stabilize retinal architecture and prevent progression of abnormal foveal morphology but also reduce photophobia and improve visual acuity. It is likely that clinical benefit would vary, but it is possible that among the patients with good foveal architecture, those with worse visual acuity may have the most to gain from treatment.
The major limitation of this study was the lack of axial length measurement owing to prior limitations of our sedation protocol. However, any disparity in image size due to the lack of axial length correction between the study groups likely would have been minimal considering that the refractive error of the patients with achromatopsia was similar to that of the control participants. Thus, axial length correction in this study would be unlikely to change our relative comparisons and conclusions. As with all clinical studies of rare diseases, our study was limited by small sample size, but differences between the study groups were large enough to be statistically significant. Multicenter large-scale studies of patients with achromatopsia in early childhood will be required to confirm our findings and further examine genotype-phenotype correlations.
Conclusions
This is the first study, to our knowledge, to show that the spectrum of foveal pathology in early childhood is milder than reported in older individuals with achromatopsia, which supports the need for early therapeutic intervention. We have also shown that age, genotype, and ffERG findings were not fully predictive of the degree of photoreceptor loss. Therefore, we propose that handheld SD-OCT is an important and objective tool for early assessment and stratification of young patients for potential therapeutic intervention, particularly as gene therapy for achromatopsia transitions to human trials.
Acknowledgments
Funding/Support: This work was supported by grants CF-CL-0612-0583-OHSU (Dr Yang) and CD-CL-0808-0469-OHSU (Dr Pennesi) from the Foundation Fighting Blindness, grant 1K08 EY0231186-01 from the National Institutes of Health (Dr Pennesi), Research to Prevent Blindness (Dr Pennesi), Medical Research Foundation (Dr Pennesi), Collins Medical Trust (Dr Pennesi), unrestricted grant C-CL-0711-0534-OHSU01 from the Foundation Fighting Blindness (Casey Eye Institute), and an unrestricted grant from Research to Prevent Blindness (Casey Eye Institute).
Role of the Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Conflict of Interest Disclosures: Dr Wen is a consultant to QLT, Inc. Dr Weleber is a consultant to Novartis, Pfizer, and Wellstat; is a member of the scientific advisory board for Applied Genetic Technologies Corp; and serves on the scientific advisory board for the Foundation Fighting Blindness (the relationship has been reviewed and managed by Oregon Health & Science University). No other disclosures were reported.
REFERENCES
- 1.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004;88 (2):291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pokorny J, Smith VC, Pinckers AJ, Cozijnsen M. Classification of complete and incomplete autosomal recessive achromatopsia. Graefes Arch Clin Exp Ophthalmol. 1982;219(3):121–130. [DOI] [PubMed] [Google Scholar]
- 3.Falls HF, Wolter JR, Alpern M. Typical total monochromacy: a histological and psychophysical study. Arch Ophthalmol. 1965;74(5):610–616. [DOI] [PubMed] [Google Scholar]
- 4.Glickstein M, Heath GG. Receptors in the monochromat eye. Vision Res. 1975;15(6):633–636. [DOI] [PubMed] [Google Scholar]
- 5.Harrison R, Hoefnagel D, Hayward JN. Congenital total color blindness: a clinicopathological report. Arch Ophthalmol. 1960;64:685–692. [DOI] [PubMed] [Google Scholar]
- 6.Larsen H Demonstration mikroskopischer praparate von einem monochromatischen Auge. Klin Monatsbl Augenheilkd. 1921;67:301–302. [Google Scholar]
- 7.Kohl S, Baumann B, Broghammer M, et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet. 2000;9(14):2107–2116. [DOI] [PubMed] [Google Scholar]
- 8.Wissinger B, Gamer D, Jägle H, et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet. 2001;69(4):722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sundin OH, Yang JM, Li Y, et al. Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet. 2000;25(3):289–293. [DOI] [PubMed] [Google Scholar]
- 10.Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet. 2002;71(2): 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thiadens AA, den Hollander AI, Roosing S, et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet. 2009;85(2):240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohl S, Coppieters F, Meire F, et al. ; European Retinal Disease Consortium. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet. 2012;91(3):527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alexander JJ, Umino Y, Everhart D, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007;13(6):685–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvalho LS, Xu J, Pearson RA, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. 2011;20(16): 3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komáromy AM, Alexander JJ, Rowlan JS, et al. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19(13):2581–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishiguchi KM, Sandberg MA, Gorji N, Berson EL, Dryja TP. Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum Mutat. 2005;25(3):248–258. [DOI] [PubMed] [Google Scholar]
- 17.Varsányi B, Somfai GM, Lesch B, Vámos R, Farkas A. Optical coherence tomography of the macula in congenital achromatopsia. Invest Ophthalmol Vis Sci. 2007;48(5):2249–2253. [DOI] [PubMed] [Google Scholar]
- 18.Genead MA, Fishman GA, Rha J, et al. Photoreceptor structure and function in patients with congenital achromatopsia. Invest Ophthalmol Vis Sci. 2011;52(10):7298–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiadens AA, Somervuo V, van den Born LI, et al. Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2010;51(11):5952–5957. [DOI] [PubMed] [Google Scholar]
- 20.Thomas MG, Kumar A, Kohl S, Proudlock FA, Gottlob I. High-resolution in vivo imaging in achromatopsia. Ophthalmology. 2011;118(5):882–887. [DOI] [PubMed] [Google Scholar]
- 21.Thomas MG, McLean RJ, Kohl S, Sheth V, Gottlob I. Early signs of longitudinal progressive cone photoreceptor degeneration in achromatopsia. Br J Ophthalmol. 2012;96(9):1232–1236. [DOI] [PubMed] [Google Scholar]
- 22.Chavala SH, Farsiu S, Maldonado R, Wallace DK, Freedman SF, Toth CA. Insights into advanced retinopathy of prematurity using handheld spectral domain optical coherence tomography imaging. Ophthalmology. 2009;116(12):2448–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong GT, Farsiu S, Freedman SF, et al. Abnormal foveal morphology in ocular albinism imaged with spectral-domain optical coherence tomography. Arch Ophthalmol. 2009;127(1):37–44. [DOI] [PubMed] [Google Scholar]
- 24.Gerth C, Zawadzki RJ, Héon E, Werner JS. High-resolution retinal imaging in young children using a handheld scanner and Fourier-domain optical coherence tomography. J AAPOS. 2009;13 (1):72–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muni RH, Kohly RP, Sohn EH, Lee TC. Hand-held spectral domain optical coherence tomography finding in shaken-baby syndrome. Retina. 2010;30(4)(suppl):S45–S50. [DOI] [PubMed] [Google Scholar]
- 26.Rootman DB, Gonzalez E, Mallipatna A, et al. Hand-held high-resolution spectral domain optical coherence tomography in retinoblastoma: clinical and morphologic considerations. Br J Ophthalmol. 2013;97(1):59–65. [DOI] [PubMed] [Google Scholar]
- 27.Scott AW, Farsiu S, Enyedi LB, Wallace DK, Toth CA. Imaging the infant retina with a hand-held spectral-domain optical coherence tomography device. Am J Ophthalmol. 2009;147(2):364–373, e2. [DOI] [PubMed] [Google Scholar]
- 28.Lalwani K, Tompkins BD, Burnes K, Krahmer MR, Pennesi ME, Weleber RG. The “dark” side of sedation: 12 years of office-based pediatric deep sedation for electroretinography in the dark. Paediatr Anaesth. 2011;21(1):65–71. [DOI] [PubMed] [Google Scholar]
- 29.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118(1):69–77. [DOI] [PubMed] [Google Scholar]
- 30.Thévenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans Image Process. 1998;7(1):27–41. [DOI] [PubMed] [Google Scholar]
- 31.Lujan BJ, Roorda A, Knighton RW, Carroll J. Revealing Henle’s fiber layer using spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2011;52(3):1486–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otani T, Yamaguchi Y, Kishi S. Improved visualization of Henle fiber layer by changing the measurement beam angle on optical coherence tomography. Retina. 2011;31(3):497–501. [DOI] [PubMed] [Google Scholar]
- 33.Lima LH, Sallum JM, Spaide RF. Outer retina analysis by optical coherence tomography in cone-rod dystrophy patients. Retina. 2013;33(9): 1877–1880. [DOI] [PubMed] [Google Scholar]
- 34.Vincent A, Wright T, Billingsley G, Westall C, Héon E. Oligocone trichromacy is part of the spectrum of CNGA3-related cone system disorders. Ophthalmic Genet. 2011;32(2):107–113. [DOI] [PubMed] [Google Scholar]
- 35.Michaelides M, Aligianis IA, Ainsworth JR, et al. Progressive cone dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis Sci. 2004;45(6):1975–1982. [DOI] [PubMed] [Google Scholar]
- 36.Spaide RF, Curcio CA. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: literature review and model. Retina. 2011;31(8):1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tränkner D, Jägle H, Kohl S, et al. Molecular basis of an inherited form of incomplete achromatopsia. J Neurosci. 2004;24(1):138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]