

Abstract
A chiral phosphoric acid-catalyzed kinetic resolution and desymmetrization of para-quinols operating via oxa-Michael addition was developed and subsequently subjected to mechanistic study. Good to excellent s-factors/enantioselectivities were obtained over a broad range of substrates. Kinetic studies were performed, and DFT studies favor a hydrogen bonding activation mode. The mechanistic studies provide insights to previously reported chiral anion phase transfer reactions involving chiral phosphate catalysts in combination with boronic acids.
Keywords: asymmetric catalysis, chiral phosphoric acid, reaction mechanisms, kinetics, directed reactivity, oxa-Michael addition
Chiral Anion Phase Transfer (CAPT) Catalysis has proven to be a powerful strategy for achieving asymmetric induction.[1,2] A central hypothesis in this catalytic manifold is the use of suitable H-bonding directing groups which have empirically proven crucial to obtain high selectivity in these transformations.[1h] Along these lines, our group recently reported the fluorination of allylic alcohols via the in situ generation of boronic acid-derived directing groups (Figure 1A).[3] This work was in turn inspired by a 2008 report from Falck and co-workers, in which a formal enantioselective oxa-Michael addition of hydroxide was achieved by employing a bifunctional organo-catalyst, affording cyclization of a boronic acid monoester intermediate (Figure 1B).[4]
Figure 1.
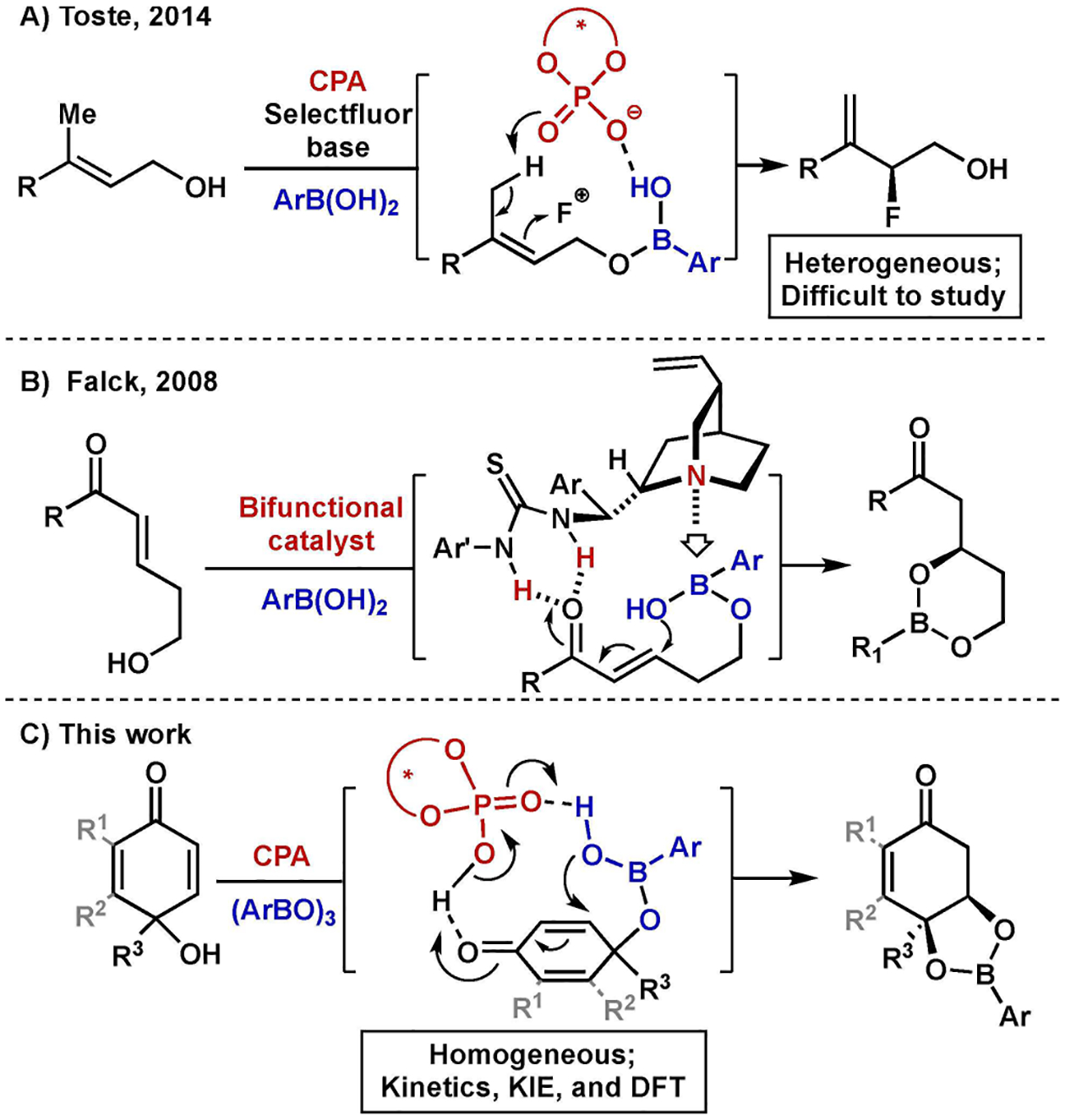
A) Boronic acid monoester as an in stiu directing group for CAPT enantioselective fluorination of allylic alcohols (CPA=chiral phosphoric acid); B) Oxa-Michael addition of boronic acid monoester promoted by push/pull-type catalyst; C) Kinetic resolution of para-quinols via complexation of in situ directing group and phosphate.
In our own report,[3] we hypothesized that the boronic acid forms an intermediate hemiester which serves as the corresponding directing group. Because the CAPT fluorination chemistry requires the use of heterogeneous conditions, support for this hypothesis derived from the observation of hemiester formation under conditions lacking the insoluble components, with no test for kinetic relevance of the intermediate possible.[5] In order to gain more mechanistic insight, we sought a homogeneous model reaction to enable kinetic analysis. By examining oxa-Michael addition rather than fluorination, the insoluble reagent could be excluded. As such, we developed a chiral phosphoric acid-catalyzed kinetic resolution /desymmetrization of quinol derivatives (Figure 1C).[6–9] The homogeneity of the reaction mixtures allowed us to examine the kinetic relevance of alcohol boronic acid condensation. These studies have enabled the gathering of some new insights into the nature of reactions involving boronic acid-directed chiral phosphoric acid catalysis.
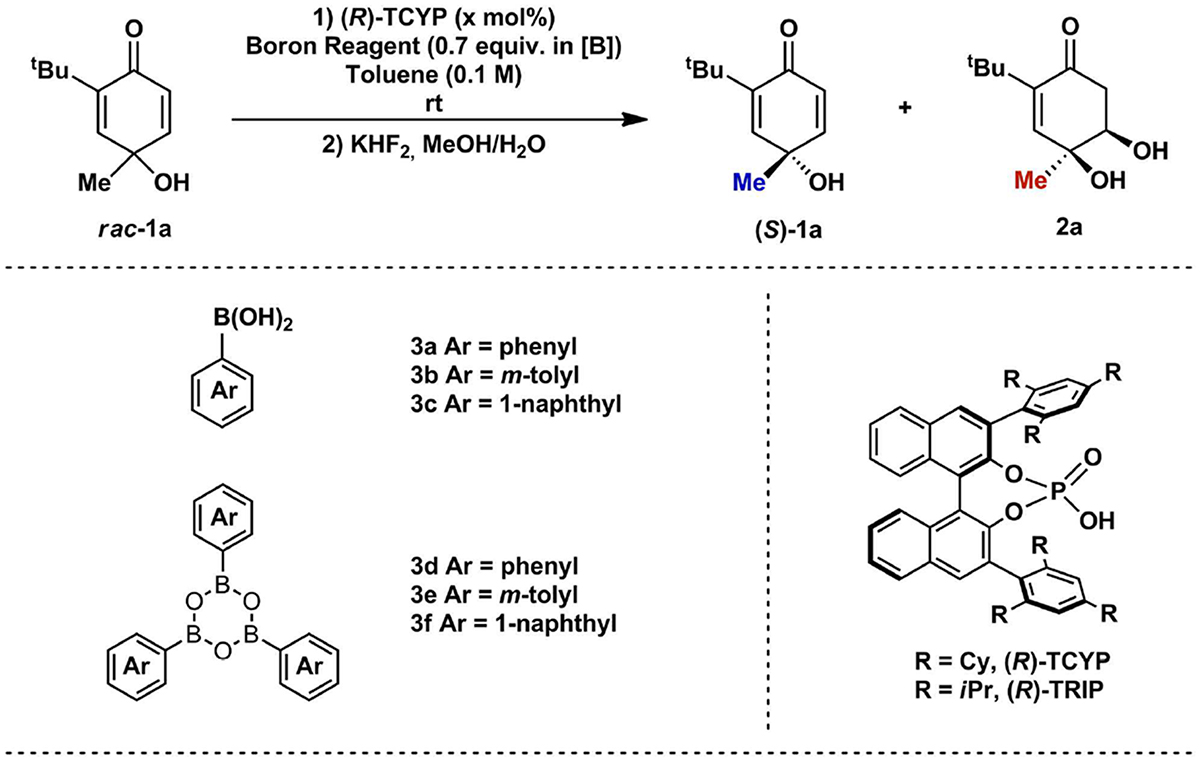
We began our investigation by examining the reaction of quinol rac-1a with phenylboronic acid (Table 1). Routine optimization identified (R)-TCYP as the optimal catalyst (Table 1, entry 6) (replacing (R)-TCYP with (R)-TRIP gives s=21)[10] and toluene as the optimal solvent for kinetic resolution.[10,11] Various arylboronic acids were examined, culminating in the identification of 1-naphthylboronic acid as the optimal boron reagent, affording a significant increase in selectivity (s=23). A key finding was the discovery that implementation of 1-naphthyl boroxine in place of its boronic acid analogue decreased the required reaction time and afforded an improved s factor of 27 in the presence of only 2 mol% catalyst at room temperature for 24 h. This trend of increased enantioselectivity and increased rate held across a variety of boronic acid/boroxine pairs, including phenylboroxine and m-tolylboroxine (Table 1).
Table 1.
Key Influence of Boron Reagent Hydration State on Reaction Selectivity.[a]
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Boron reagent | (R)-TCYP loading | Time (h) | Conversion (%)[b] | (S)-1a ee1 (%)[c] | 2a ee2 (%)[c] | s |
| 1 | 3a | 5.0 mol% | 48 | 58 | 71 | 54 | 6 |
| 2 | 3b | 5.0 mol% | 48 | 64 | 86 | 54 | 7 |
| 3 | 3c | 5.0 mol% | 48 | 54 | 90 | 78 | 23 |
| 4 | 3d | 2.0 mol% | 24 | 51 | 70 | 73 | 10 |
| 5 | 3e | 2.0 mol% | 24 | 45 | 62 | 78 | 14 |
| 6 | 3f | 2.0 mol% | 24 | 53 | 90 | 84 | 27 |
Reaction conditions: rac-1a (0.2 mmol), (R)-TCYP (2 or 5 mol%), Boronic acid or Boroxine (0.7 equiv.), solvent (2.0 mL), rt for 24 or 48 h;
Determined by 1H-NMR using internal standard;
Determined by chiral phase HPLC; Selectivity factor (s) was calculated according ln[(1-C)(1 eeSM)/1-C](1+eeSM)].[10]
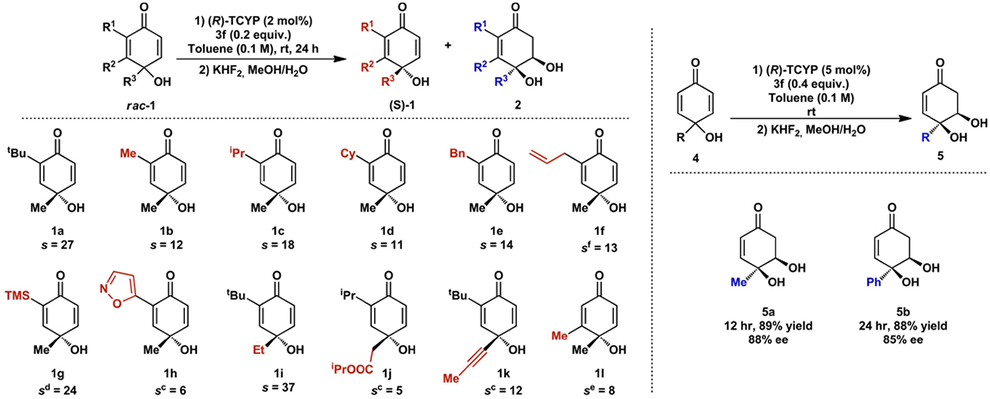
With optimized conditions in hand, we next explored the scope of the kinetic resolution (Table 2). Various α-substitutents (R1), β-substituents (R2), and γ-substituents were tolerated with varying levels of selectivity, including various alkyl, unsaturated, and heteroaromatic groups. The protocol was also found to apply well in the desymmetrization of prochiral p-quinols (Table 2). Two representative substrates were examined under conditions similar to the optimized kinetic resolution protocol. Gratifyingly, the reaction proceeded smoothly to yield the corresponding diols in 88% and 85% ee, respectively.
Table 2.
|
Isolated yield. Absolute configuration assigned by analogy to that of 1g, which was determined to be (S) by single-crystal X-ray diffraction.
Selectivity factor (s) was calculated according ln[(1-C)(1 eeSM)/1-C](1+eeSM)]. For isolated yields of kinetic resolution, see Supporting Information.
5.0 mol% of catalyst was used.
Reaction time variation: 12 h.
Reaction time variation: 18 h.
Reaction time variation: 20 h.
As noted above, the homogeneity of these reactions enabled a closer examination of the mechanism. Initially, we were drawn to the observation that the boroxine hydration state affects both rate of the reaction and its enantioselectivity, suggesting that formation of boronic acid esters of the substrate may be relevant. We began our investigation by examining association between the boroxine and the substrate (4a) by 1H-NMR spectroscopy.[12] However, no discrete observable new species were formed, and no detectable changes in the spectra were found as the concentration of each species was varied.
This lack of observable association between the substrate and boroxine was surprising given our previous observation of quantitative condensation of boroxine with primary allylic alcohols.[5] In order to gain further insight, a series of kinetic experiments were performed. Unfortunately, kinetics conducted under typical reaction stoichiometries were found at extended reaction times to deviate from simple integer-order behavior.[10,13] To simplify the analysis, the method of initial rates was employed, along with flooding conditions in one or more substrates. Under flooding in boroxine, an order in (R)-TRIP deviating from linearity was observed, with reasonable fits obtained to either ½ order or saturation kinetics (Figure 2A). In order to distinguish these possibilities, the diffusion constants of both (R)-TRIP and the O-methylated analogue were measured by DOSY (diffusion ordered spectroscopy), affording nearly indistinguishable diffusion constants.[10] These experiments indicated that the phosphoric acid catalyst is likely monomeric under the reaction conditions, excluding the possibility of ½ order.[14]
Figure 2. A).
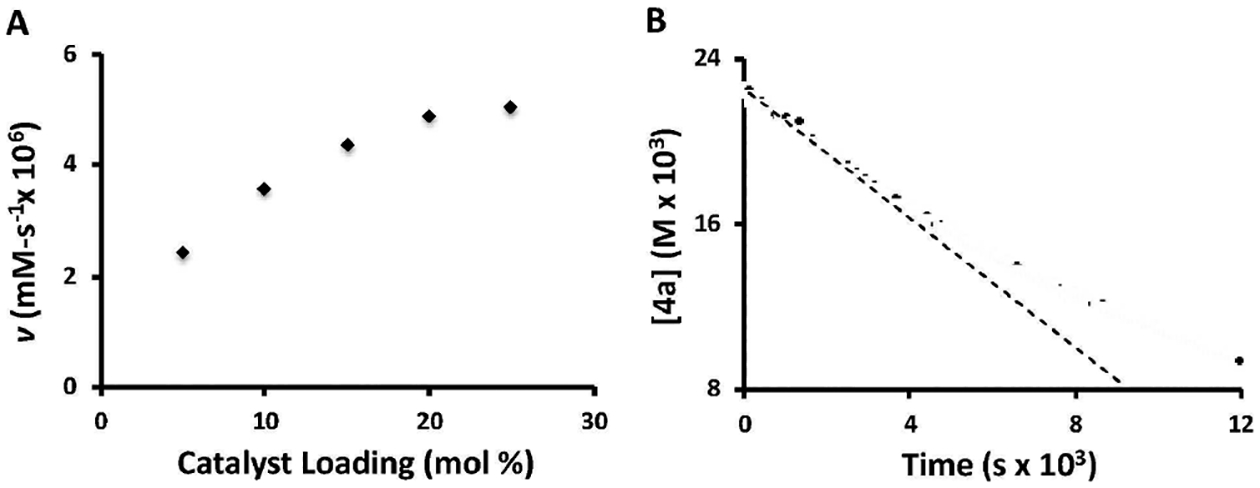
Initial rate-(R)-TRIP catalyst loading plot; B) Reaction plot of [4a] under saturation of 3e.
For the remaining components, under the pseudo-zero order conditions, first-order dependence on the substrate concentration (Figure 2B) was observed. The boroxine exhibited kinetic behaviour deviating from first order which could be modelled as first-order dependence on the concentration of monomeric arylboron.[10] Taken together, this kinetic behavior indicates a preequilibrium to form boronic acid ester before the involvement of chiral phosphoric acid.
The 1H-NMR spectroscopy of the mixtures of TRIP and boroxine 3e reveals a concentration dependent shift in the resonances of the phosphoric acid, with partial decoalesence of the signals achieved upon cooling to 217 K.[10,16,17] A DFT study at the B3LYP-D3/def2-TZVPP/SMD(toluene)//B3LYP/6–31G(d) level of theory suggests that the most likely candidate for the associated complex is the mixed phosphoric/boronic anhydride (vide infra).[10] This association complex is potentially kinetically relevant.[15]
Finally, KIE experiments were conducted using 4a labelled at the vinylic C H bonds. A separate pot experiment showed a clear inverse secondary KIE (Scheme 1), indicating that oxa-Michael addition is the rate-limiting step, either concerted with catalyst association or as a discrete second step following reversible catalyst association.[10,18,19] Initial Michael addition followed by esterification is likely inconsistent with the exclusively observed syn diastereoselectivity and the observation that methylation of the hydroxyl group of 4a shuts down reactivity altogether, with no conversion observed over 72 h.[10]
Scheme 1.
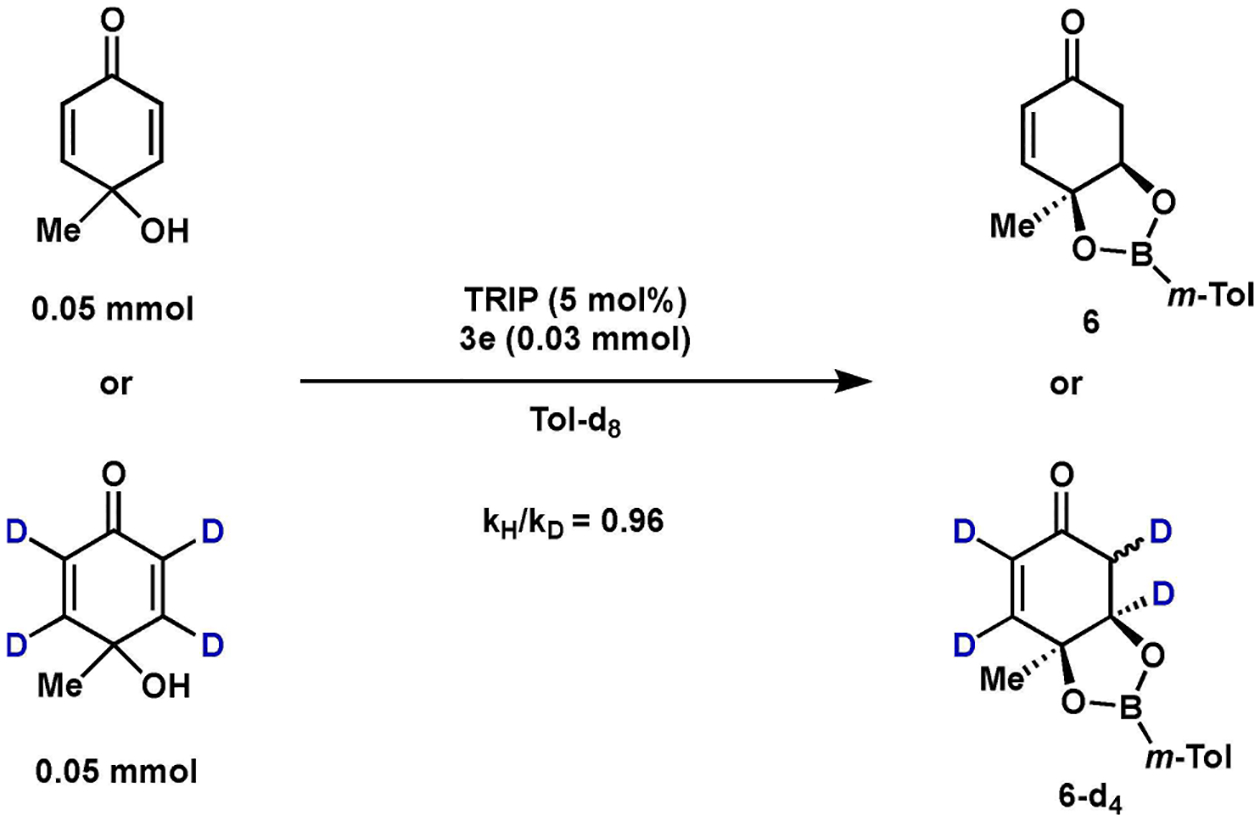
Separate-pot kinetic isotope effect.
As a whole, these data are consistent with a mechanism involving two roles for the boroxine: productive association with the substrate, and delete-rious association with the catalyst (though we cannot conclusively rule out the possibility that the mixed phosphoric boronic anhydride is on-cycle).
A DFT study conducted at the B3LYP-D3/def2-TZVPP/SMD(toluene)/ /B3LYP/6-31G(d) level of theory located four transition structures for the TRIP-catalyzed reaction. Importantly, formation of the observed major enantiomer is correctly predicted. Our proposed mechanism for the overall reaction, considering all of the collected data, is shown in Scheme 2. The lowest-energy computed transition state for oxa-Michael addition is shown, as well as the computationally suggested structure for the mixed anhydride. Based on this mechanism, we propose that the boronic acid/boroxine effect on enantioselectivity arises from a background reaction catalyzed by the boronic acid (further exacerbated by catalyst inhibition by boron), whereas the rate enhancement is a function of the equilibrium for substrate esterification.
Scheme 2.
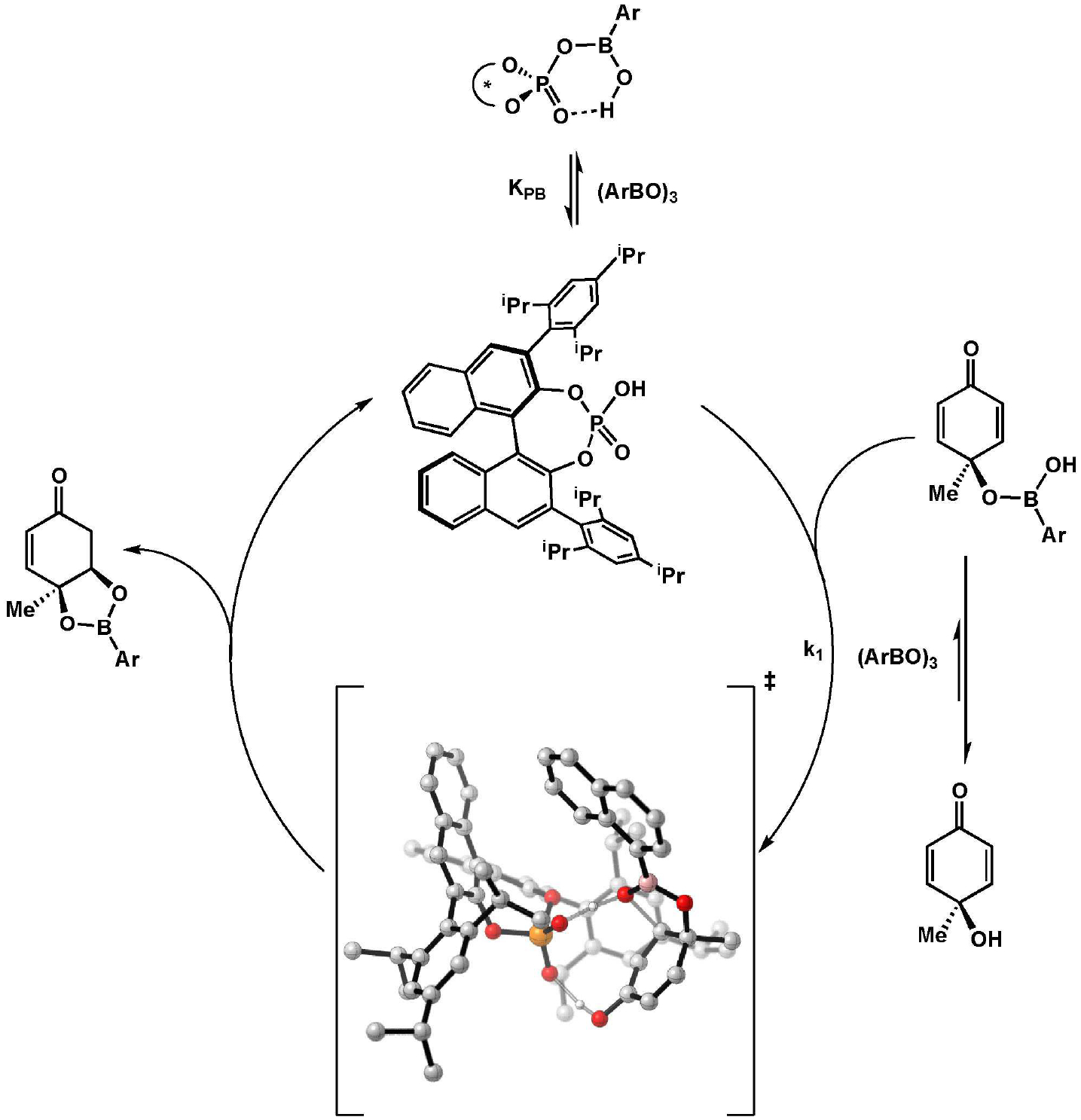
Proposed mechanism with calculated transition state.
In conclusion, a chiral phosphoric acid-catalyzed kinetic resolution (or desymmetrization) of para-quinols was developed. This method provides the access to enantioenriched para-quinols and corresponding diols. Kinetic experiments are in line with the canon of indirect experimental evidence we have garnered over the course of our studies on the role of H-bonding in CAPT catalysis – namely, that appropriately situated H-bonding directing groups, including in situ formed boronic acid esters, orient the chiral phosphate, enabling high enantiocontrol.[1h,20] Further-more, preliminary evidence supporting formation of mixed anhydrides between boronic and phosphoric acids was garnered, suggesting a potentially under-appreciated role for these species. We anticipate that new asymmetric transformations will be enabled by the knowledge garnered herein concerning the dynamics of this potent reagent pair.
Experimental Section
For details of instruments used and the general experimental procedures, see the Supporting Information.
General Procedure of Kinetic Resolution of Para-Quinols
The rac-1 (0.20 mmol), 1-naphthylboroxine (0.04 mmol) and (R)-TCYP (2.0 mol%) were added to a vial containing a stir bar. The vial was twined with Teflon tape, toluene (2.0 ml) was added using a syringe, and the vial was fitted with a cap. The reaction mixture was stirred at room temperature for the indicated time. To the mixture was added MeOH/H2O (1/1,2.0 mL), and then KHF2 (10 eqiv.) was added and the mixture was stirred vigorously for 5–30 min until the boronic ester was removed as evident from TLC analysis. The aqueous phase was separated and collected, and the organic phase was evaporated and then re-dissolved in n-hexanes. The mixture was then washed MeOH/H2O (1/1) twice and the aqueous phase was combined. The combined aqueous layers were then evaporated and purified by column chromatography carefully over silica gel affording the desired chiral p-quinols and diols. The products were further purified by preparative TLC as necessary.
Crystallographic Data for (S)-1g
CCDC-1950552 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
Acknowledgements
We gratefully acknowledge the National Institute of Health (R35GM118190) for financial support of this work. We thank Dr. Andrew Neel, Dr. David Kaphan, and Dr. Richard Thorn-bury for helpful discussions, Dr. Cynthia Hong for X-ray crystallographic collection and analysis, Dr. Hasan Celik for assistance with NMR experiments and Dr. Miao Zhang for assistance with HR-MS data. DFT studies were conducted at the Molecular Graphics and Computation Facility, College of Chemistry, University of California, Berkeley (NSF CHE-0840505).
Footnotes
Supporting information for this article is available on the WWW under https://doi.org/10.1002/adsc.201900816
References
- [1].a) Rauniyar V, Lackner AD, Hamilton GL, Toste FD, Science 2011, 334, 1681–1684; [DOI] [PubMed] [Google Scholar]; b) Shunatona HP, Früh N, Wang Y-M, Rauniyar V, Toste FD, Angew. Chem. Int. Ed 2013, 52, 7724–7727; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 7878–7881; [Google Scholar]; c) Lackner AD, Samant AV, Toste FD, J. Am. Chem. Soc 2013, 135, 14090–14093; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Phipps RJ, Hiramatsu K, Toste FD,J. Am. Chem. Soc 2012, 134, 8376–8379; [DOI] [PubMed] [Google Scholar]; e) Phipps RJ, Toste FD, J. Am. Chem. Soc 2013, 135, 1268– 1271; [DOI] [PubMed] [Google Scholar]; f) Neel AJ, Hehn JP, Tripet PF, Toste FD, J. Am. Chem. Soc 2013, 135, 14044–14047; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wang Y-M, Wu J, Hoong C, Rauniyar V, Toste FD, J. Am. Chem. Soc 2012, 134, 12928–12931; [DOI] [PubMed] [Google Scholar]; h) Wu J, Wang Y-M, Drljevic A, Rauniyar V, Phipps RJ, Toste FD, Proc. Natl. Acad. Sci. USA 2013, 110, 13729–13733; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Yang X, Phipps RJ, Toste FD, J. Am. Chem. Soc 2014, 136, 5225–5228; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Nelson HM, Reisberg SH, Shunatona HP, Patel JS, Toste FD, Angew. Chem. Int. Ed 2014, 53, 5600–5603; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 5706–5709; [Google Scholar]; k) Nelson HM, Patel JS, Shunatona HP, Toste FD, Chem. Sci 2015, 6, 170–173; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Nelson HM, Williams BD, Miró J, Toste FD, J. Am. Chem. Soc 2015, 137, 3213–3216; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Milo A, Neel AJ, Toste FD, Sigman MS, Science 2015, 347, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].For other recent reports on CAPT catalysis, see:; a) Shen Z, Pan X, Lai Y, Hu J, Wan X, Li X, Zhang H, Xie W, Chem. Sci 2015, 6, 6986–6990; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Romanov-Michailidis F, Guénée L, Alexakis A, Angew. Chem. Int. Ed 2013, 52, 9266–9270; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 9436–9440; [Google Scholar]; c) Xie W, Jiang G, Liu H, Hu J, Pan X,Zhang H, Wan X, Lai Y, Ma D, Angew. Chem. Int. Ed 2013, 52, 12924–12927; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 13162–13165; [Google Scholar]; d) Yang Z, He Y, Toste FD, J. Am. Chem. Soc 2016, 138, 9775–9778; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yamamoto E,Hilton MJ, Orlandi M, Saini V, Toste FD, Sigman MS, J. Am. Chem. Soc 2016, 138, 15877–15880; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Avila CM, Patel JS, Reddi Y, Saito M, Nelson HM, Shunatona HP, Sigman MS, Sunoj RB,Toste FD, Angew. Chem. Int. Ed 2017, 56, 5806–5811; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 5900–5905; [Google Scholar]; g) Biswas S, Kubota K, Orlandi M, Turberg M, Miles DH, Sigman MS, Toste FD, Angew. Chem. Int. Ed 2018, 57, 589–593; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 598–602; [Google Scholar]; h) Ye B,Zhao J, Zhao K, McKenna JM, Toste FD, J. Am. Chem. Soc 2018, 140, 8350–8356; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Coelho JAS, Matsumoto A, Orlandi M, Hilton MJ, Sigman MS,Toste FD, Chem. Sci 2018, 9, 7153–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zi W, Wang Y-M, Toste FD, J. Am. Chem. Soc 2014, 136, 12864–12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li DR, Murugan A Falck AJR, J. Am. Chem. Soc 2008, 130, 46–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Neel AJ, Milo A, Sigman MS, Toste FD, J. Am. Chem. Soc 2016, 138, 3863–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For para-quinols in complex bioactive molecules and natural products:; a) Wang X, Porco JA Jr., Angew. Chem. Int. Ed 2005, 44, 3067–3071; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005, 117, 3127–3131; [Google Scholar]; b) Baldwin JE, Adlington RM, Sham VW-W, Marquez R, Bulger PG, Tetrahedron 2005, 61, 2353–2363; [Google Scholar]; c) Patil AD, Freyer AJ,Killmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK, Tetrahedron 1997, 53, 5047–5060; [Google Scholar]; d) Berry JM, Bradshaw TD, Fichtner I, Ren R, Schwalbe CH, Wells G, Chew E-H, Stevens MFG, Westwell AD, J. Med. Chem 2005, 48, 639–644; [DOI] [PubMed] [Google Scholar]; e) Stork G,La Clair JJ, Spargo P, Nargund RP, Totah N, J. Am. Chem. Soc 1996, 118, 5304–5305; [Google Scholar]; f) Mejorado LH,Pettus TRR, J. Am. Chem. Soc 2006, 128, 15625–15631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For recent reports on preparation of para-quinols, see; a) Carreño MC, González-López M, Urbano A, Angew. Chem. Int. Ed 2006, 45, 2737–2741; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2006, 118, 2803–2807; [Google Scholar]; b) Yakura T, Omoto M,Yamauchi Y, Tian Y, Ozono A, Tetrahedron 2010, 66, 5833–5840. [Google Scholar]
- [8].For enantioselective synthesis of para-quinols:; a) Volp KA, Harned AM, Chem. Commun 2013, 49, 3001–3003; [DOI] [PubMed] [Google Scholar]; For select reports for synthesis of chiral para-quinol derivatives, see; b) Wu W-T, Zhang L, You S-L, Chem. Soc. Rev 2016, 45, 1570–1580; [DOI] [PubMed] [Google Scholar]; c) Gu Q,Rong Z-Q, Zheng C, You S-L, J. Am. Chem. Soc 2010, 132, 4056–4057; [DOI] [PubMed] [Google Scholar]; d) Rubush DM, Rovis T, Synlett 2014, 25, 713–717; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Rubush DM, Morges MA, Rose BJ, Thamm DH, Rovis T, J. Am. Chem. Soc 2012, 134, 13554–13557; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Vo NT, Pace RD, O’Hara F, Gaunt MJ, J. Am. Chem. Soc 2008, 130, 404–405; [DOI] [PubMed] [Google Scholar]; g) Corbett MT, Johnson JS, Chem. Sci 2013, 4, 2828–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For recent reports on kinetic resolution of alcohols and 1,2-diols catalyzed by chiral phosphoric acid, see:; a) Yamanaka T, Kondoh A, Terada M, J. Am. Chem. Soc 2015, 137, 1048–1051; [DOI] [PubMed] [Google Scholar]; b) Kim JH, Čorić I, Palumbo C, List B, J. Am. Chem. Soc 2015, 137, 1778–1781; [DOI] [PubMed] [Google Scholar]; c) Xie C, Bai D, Huang S-H, Jia X, Hong R, Asian J Org. Chem 2014, 3, 277–280; [Google Scholar]; d) Harada S, Kuwano S, Yamaoka Y, Yamada K, Takasu K, Angew. Chem. Int. Ed 2013, 52, 10227–10230; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 10417–10420; [Google Scholar]; e) Fujii K, Mitsudo K, Mandai H, Suga S, Adv. Synth. Catal 2017, 359, 2778–2788; [Google Scholar]; f) Pawliczek M, Hashimoto T, Maruoka K, Chem. Sci 2018, 9, 1231–1235; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Liu B, Yan J, Huang R, Wang W, Jin Z, Zanoni G, Zheng P, Yang S,Chi YR, Org. Lett 2018, 20, 3447–3450. [DOI] [PubMed] [Google Scholar]
- [10]. For detailed results, see Supporting Information.
- [11].Rauniyar V, Wang ZJ, Burks HE, Toste FD, J. Am. Chem. Soc 2011, 133, 8486–8489. [DOI] [PubMed] [Google Scholar]
- [12]. m-tolylboroxine (3e) and (R)-TRIP were employed in place of the 1-naphthyl analogue (3f) and (R)-TCYP to maintain homogeneity across a wider range of concentrations and temperatures.
- [13]. We are unable to conclusively distinguish between product inhibition and catalyst deactivation due to our inability to isolate the product boronic ester without contaminating diol and/or boronic acid. In either case, initial rate kinetics obviate any confounding rate influence.
- [14].For dimerization of phosphoric acids:; a) Monaco MR,Fazzi D, Tsuji N, Leutzsch M, Liao S, Thiel W, List B, J. Am. Chem. Soc 2016, 138, 14740–14749; [DOI] [PubMed] [Google Scholar]; b) De Ford J, Chu F, Anslyn EV, Tetrahedron Lett 1996. 37, 1925–1928; [Google Scholar]; c) Peppard DF, Ferraro JR, Mason GW, J. Inorg. Nucl. Chem 1958, 7, 231–244; [Google Scholar]; For a recent study of dimerization in hydrogen-bond-donor catalysts, see:; d) Ford DD, Lehnerr D, Kennedy CR,Jacobsen EN, J. Am. Chem. Soc 2016. 138, 7860–7863; [DOI] [PMC free article] [PubMed] [Google Scholar]; For a full derivation of the relevant rate laws and discussion of the assumptions involved, see the supporting information.
- [15]. For a full derivation of the relevant rate laws and discussion of the assumptions involved, see the supporting information.
- [16].Thordarson P, Chem. Soc. Rev 2011, 40, 1305–1323. [DOI] [PubMed] [Google Scholar]
- [17].For a discussion on the interaction between phosphoric acids and boronic acids, see:; Martínez-Aguirre MA,Yatsimirsky AK, J. Org. Chem 2015, 80, 4985–4993. [DOI] [PubMed] [Google Scholar]
- [18].In principle, a post-rate-determining equilibration between parent and deuterated substrate prior to the enantiodetermining step is possible, but the observation of a separate-pot KIE indicates that C O bond formation occurs before the turnover-limiting step. For a discussion on KIE interpretation, see:; Simmons EM, Hartwig JF, Angew. Chem. Int. Ed 2012, 51, 3066–3072; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2012, 124, 3120–3126. [Google Scholar]
- [19]. These two possibilities are kinetically indistinguishable. Another possibility involves reversible oxa-Michael addition and rate-determining enolate protonation. See the Supporting information for details.
- [20]. Indeed, in the limit of protonation of the carbonyl, the quinone electrophile can be seen as directly analogous to previous examples of CAPT catalysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.