

Abstract
Pulmonary edema associated with increased vascular permeability is a severe complication of Pseudomonas (P.) aeruginosa-induced acute lung injury. The mechanisms underlying P aeruginosa-induced vascular permeability are not well understood. In the present study, we investigated the role of neuronal Wiskott Aldrich syndrome protein (N-WASP) in modulating P aeruginosa-induced vascular permeability. Using lung microvascular endothelial and alveolar epithelial cells, we demonstrated that N-WASP downregulation attenuated P aeruginosa-induced actin stress fiber formation and prevented paracellular permeability. P aeruginosa-induced dissociation between VE-cadherin and β-catenin, but increased association between N-WASP and VE-cadherin, suggesting a role for N-WASP in promoting P aeruginosa-induced adherens junction rupture. P aeruginosa increased N-WASP-Y256 phosphorylation, which required the activation of Rho GTPase and focal adhesion kinase. Increased N-WASP-Y256 phosphorylation promotes N-WASP and integrin αVβ6 association as well as TGF-β-mediated permeability across alveolar epithelial cells. Inhibition of N-WASP-Y256 phosphorylation by N-WASP-Y256F overexpression blocked N-WASP effects in P aeruginosa-induced actin stress fiber formation and increased paracellular permeability. In vivo, N-WASP knockdown attenuated the development of pulmonary edema and improved survival in a mouse model of P aeruginosa pneumonia. Together, our data demonstrate that N-WASP plays an essential role in P aeruginosa-induced vascular permeability and pulmonary edema through the modulation of actin cytoskeleton dynamics.
Keywords: β-catenin, αvβ6, acute lung injury, small Rho GTPases, VE-cadherin
1 |. INTRODUCTION
Pseudomonas aeruginosa is one of the most common Gram-negative pathogens that causes pneumonia and accounts for high mortality associated with the development of acute lung injury (ALI) and pulmonary edema.1–3 Dysfunction of lung vascular endothelial (VE) and alveolar epithelial barriers is central to the formation of P aeruginosa-induced pulmonary edema. P aeruginosa utilizes various strategies to breach lung barrier functions, resulting in ALI complications such as inadequate alveolar fluid clearance, inadequate gas exchange, and development of protein-rich edema.1,4–16
Neuronal Wiskott-Aldrich syndrome protein (N-WASP) is a ubiquitously expressed scaffolding protein that plays an important role in cytoskeletal dynamics.4–9 N-WASP transmits upstream signals to the cellular machinery that is directly involved in the modulation of actin structures, such as induction of new actin polymerization, stabilization of newly formed actin filaments, and remodeling of actin cytoskeleton structures.17–22 We and others have previously demonstrated that the activation of small Rho family GTPases is responsible for actin stress fiber formation and increased paracellular permeability in lung endothelial cell monolayers induced by transforming growth factor beta-1 (TGF-β1) or other proinflammatory mediators including VE growth factor and interleukin 1.8,13–16,23,24 N-WASP is a demonstrated downstream signaling protein of small Rho family GTPases.17,18,25–27 In patients with ALI, transforming growth factor beta-1 (TGF-β1) expression and activation are elevated in bronchoalveolar lavage fluid.28 We have previously reported that N-WASP mediates lung vascular permeability in response to TGF-β1.29
N-WASP has three independent domains at the C terminus, collectively termed the verprolin homology, central, and acidic regions (VCA) domain.29,30 The middle region of N-WASP contains a GTPase-binding domain (GBD) and Rac interactive binding domain, which binds to small Rho GTPases and activates N-WASP.17,18,25–27 N-WASP activation unfolds N-WASP and allows VCA domain to bind downstream signaling proteins.29,30 The VCA domain is necessary to activate actin polymerization via an actin-related protein (Arp) 2/3-dependent mechanism.17,18,25–27 However, whether N-WASP plays a role in P aeruginosa-induced actin stress fiber formation and increased paracellular permeability in microvascular endothelial cells and alveolar epithelial cells remains unknown.
Here, we investigated the role of N-WASP in modulating P aeruginosa-induced paracellular permeability in lung microvascular endothelial and alveolar epithelial cells. Furthermore, we examined whether conditional depletion of N-WASP would significantly limit the development of pulmonary edema and prevent mortality in mice challenged with P aeruginosa. The results demonstrated that inhibition of N-WASP-mediated pathway(s) significantly inhibits the development of pulmonary edema and prevents mortality.
2 |. MATERIALS AND METHODS
2.1 |. Reagent and antibodies
TGF-β1 was obtained from R&D Systems (Minneapolis, MN, USA). Antibodies were purchased from: anti-N-WASP, anti-FAK, anti-phospho-Y256-N-WASP (Millipore, Billerica, MA, USA); anti-phospho-Y397-FAK (Cell Signaling Technology, Danvers, MA, USA); anti-GAPDH, anti-myc (Santa Cruz Biotechnology, Dallas, TX, USA); Alexa Fluor 488 phalloidin (Thermo Fisher Scientific, Waltham, MA, USA); Rho inhibitor I (Cytoskeleton, Inc, Denver, CO, USA); PF573228 (Millipore Sigma, Burlington, MA, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Thermo Fisher Scientific (Waltham, MA, USA).
2.2 |. Cells
Rat lung microvascular endothelial cells (RMVEC) were derived and cultured in DMEM supplemented with 10% fetal bovine serum and 100 U/ml penicillin/streptomycin.31,32 L2 rat lung alveolar epithelial cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in F-12K medium supplemented with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin per the manufacturer’s instructions. Human lung microvascular endothelial cells (HMVEC) and human lung epithelial cells (H441, from ATCC, Manassas, VA, USA) were cultured in DMEM/F12 supplemented with 10% fetal bovine serum and 100 U/mL penicillin/streptomycin.
2.3 |. Lentiviral and adenoviral vectors
Lentivirus expressing N-WASP short hairpin RNA (shRNA) or scrambled shRNA were generated as described previously.30 Replication-deficient adenoviral vectors were generated by the Adeno-X Expression System 2 in 293 cells followed by CsCl gradient centrifugation according to the manufacturer’s instructions (Takara Bio USA, Inc Mountain View, CA, USA).33–35 Cells were transfected as previously described30 and knockdown efficiency was examined 48–72 hours posttransfection by both Western blot analysis (for protein level) and quantitative RT-PCR (for mRNA level) as previously described.36,37
2.4 |. Western blotting
Western blotting was performed as previously described.35 Briefly, equivalent amounts of whole-cell lysates extracted from cells or mouse lungs were separated by SDS-PAGE, transferred to Immobilon-P PVDF membrane (Millipore Sigma, Burlington, MA, USA), followed by incubation with specific antibodies of interest, and developed with enhanced chemiluminescent detection system (Pharmacia Biotech, Inc, Piscataway, NJ, USA). The expression of GAPDH protein was used as a loading control to ensure equal protein loading.
2.5 |. Immunofluorescence microscopy
Immunofluorescence staining and microscopy were performed as previously described.38 In brief, cell monolayers grown on glass coverslips were treated as indicated and fixed in 3.7% paraformaldehyde solution in PBS and were permeabilized. Actin filaments were stained with Alexa Fluor 488-conjugated phalloidin (1:100) overnight at 4°C. Cell nuclei were stained with blue Hoechst fluorescence dye. Digital fluorescence images were obtained by a Nikon microscope (Nikon, Tokyo, Japan).
2.6 |. Measurement of active TGF-β1
Measurement of active TGF-β1 was described previously.29,39 In brief, cells were challenged with P aeruginosa and the culture medium was collected after 6 hours. Active TGF-β1 was measured by ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions as previously described.40
2.7 |. Measurement of transcellular resistance
Endothelial and epithelial barrier integrity was measured by an electric cell-substrate impedance sensing (ECIS) system (Applied Biophysics, Troy, NY, USA) as previously described.31 Briefly, RMVECs, HMVECs, human lung epithelial cells (H441), or rat lung alveolar epithelial cells (L2) were grown to confluence on ECIS 8W10E culture ware. Resistance values were used to evaluate the integrity of monolayers. Resistance was measured every 10 minutes for the duration of the experiments. Baseline resistances were monitored for 1 hour before the addition of P aeruginosa. Data were normalized to control readings, then averaged.
2.8 |. In vivo P aeruginosa-induced ALI model
All animal interventions were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Alabama at Birmingham. The wild-type strain of P aeruginosa (PAK strain) was a gift from Stephen Lory, PhD at Harvard Medical School. Preparation of P aeruginosa was previously described.41 The P aeruginosa suspension was diluted further with sterile phosphate-buffered saline (PBS) to obtain a final concentration of 2 × 107 CFU/mL. Counts were confirmed by serial dilution and growing on Luria-Bertani agar.
The P aeruginosa-induced ALI mouse model and administration of P aeruginosa were previously described.9 Saline with or without recombinant adenoviral vectors (50 μL, 108 plaque-forming units (PFU) were instilled intratracheally 1 day before P aeruginosa challenge as previously described.42 For P aeruginosa challenge, 8–12 weeks old mice were anesthetized and then instilled with PBS containing 107 CFU P aeruginosa into both lungs via the trachea. Mice were then allowed to recover and return to the cage. Mice were euthanized and extravascular plasma equivalents were calculated as previously described.43 N-WASP floxed (N-WASPflox/flox) mice were kindly provided by Dr Scott Snapper at Harvard University. For survival studies, N-WASPflox/flox mice were randomly grouped and each mouse was instilled with 107 CFU of P aeruginosa. Mice were checked every 6 hours up to 10 days after the instillation of P aeruginosa until death or survival at 10 days. Survival time was defined as the time from bacterial instillation to death.
2.9 |. Statistical analysis
Experiments were performed at least in triplicate and repeated three times. Results are shown as means ± SEM. The normal distribution was verified using the Kolmogorov-Smirnov test. For normally distributed data, a Student t-test was used to compare two experimental groups. Bonferroni correction, controlling for false positive error rate, was used to adjust for multiple comparisons. A Kaplan-Meier analysis followed by a log rank (Mantel-Cox) test was used to compare the survival between the two experimental groups of mice at 10 days. A P value of <.05 was considered statistically significant. All statistical comparison of means was bilateral (two-tailed tests).
3 |. RESULTS
3.1 |. N-WASP modulates P aeruginosa-induced permeability across lung microvascular endothelial monolayers
We have previously shown that N-WASP is an important modulator of lung microvascular endothelial permeability To study whether N-WASP modulates P aeruginosa-induced endothelial permeability, we downregulated N-WASP expression in rat microvascular endothelial cells (RMVECs) and evaluated changes in endothelial paracellular permeability after P aeruginosa infection. The downregulation of N-WASP expression was mediated by lentiviral shRNA as previously reported and confirmed at protein and mRNA levels.29 P aeruginosa infection decreased electrical resistance in RMVECs by 81% at 24 hours (Figure 1A,B, red). In contrast, the downregulation of N-WASP attenuated P aeruginosa-induced endothelial barrier disruption, reflected by only a 20% decrease in electrical resistance (Figure 1A,B, black). Smaller decreases in electrical resistance reflect increased barrier integrity and decreased paracellular permeability. These results demonstrate that the downregulation of N-WASP prevented P aeruginosa-induced paracellular permeability across RMVEC monolayers.
FIGURE 1.
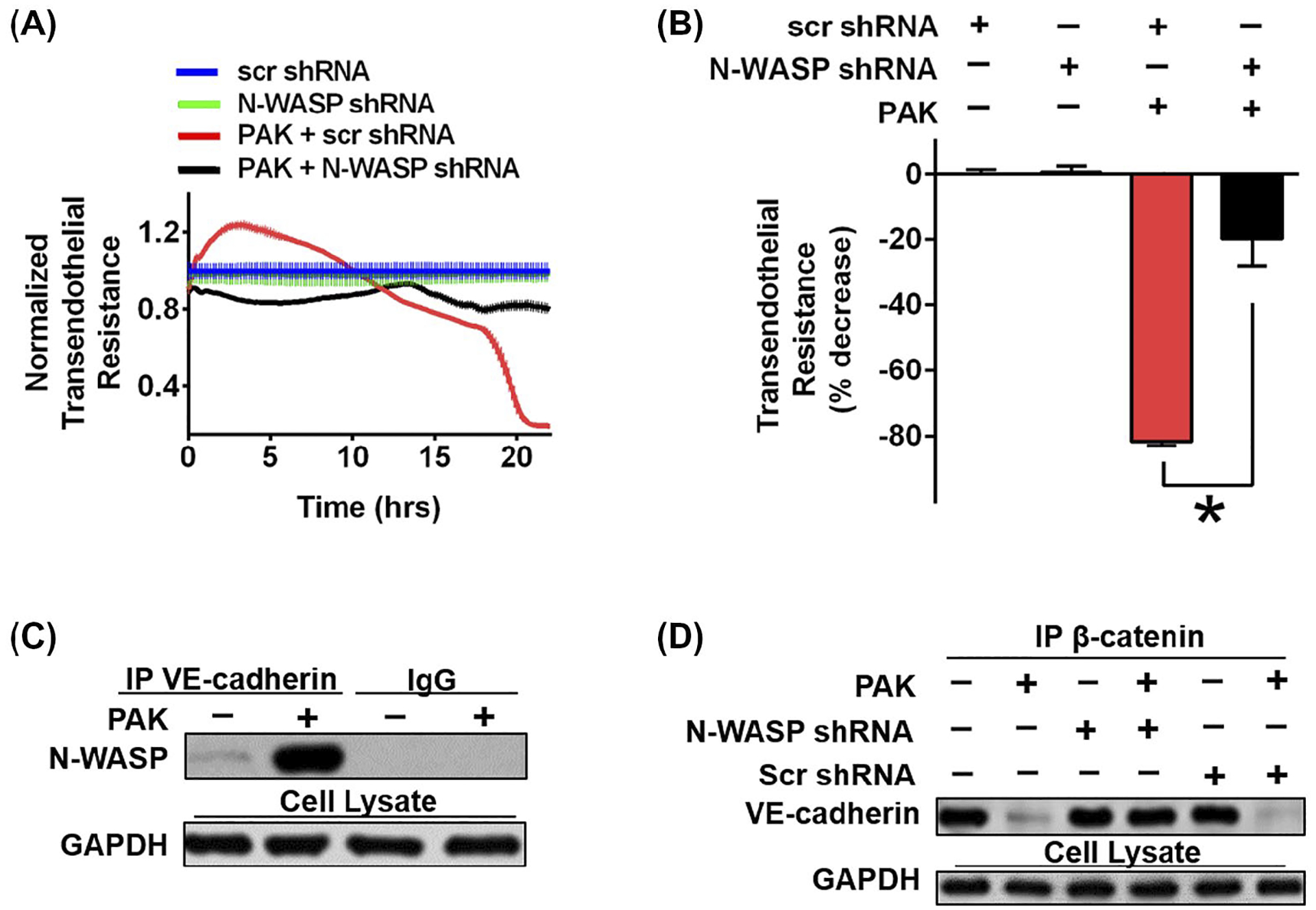
N-WASP downregulation significantly decreased P aeruginosa induced permeability and VE-cadherin dissociation. A, RMVECs were challenged with P aeruginosa infection (MOI = 40), followed by ECIS measurement with or without N-WASP downregulation. B, Resistance value was measured up to 24h post P aeruginosa stimulation. Data are presented as the percentage of decreased resistance normalized to control (n = 7–8). C, Representative blot of co-immunoprecipitation assays showing increased association of N-WASP and VE-cadherin in response to P aeruginosa in RMVECs. D, Representative blot of co-immunoprecipitation assays showing the downregulation of N-WASP inhibited P aeruginosa induced dissociation of VE-cadherin and β-catenin in RMVECs. Data are presented as means ± SEM. *P < .01
3.2 |. N-WASP modulates P aeruginosa-induced increased permeability through increased interaction with adherens junction protein VE-cadherin in RMVECs
To address whether N-WASP contributes to P aeruginosa-induced AJ disruption, we examined if N-WASP directly associates with VE-cadherin by performing co-immunoprecipitation (co-IP) of RMVEC lysates after P aeruginosa infection. There was a significant binding of N-WASP and VE-cadherin in RMVECs infected with P aeruginosa, but not in uninfected RMVECs (Figure 1C). This result indicates that N-WASP is distributed at cell-cell junctions in endothelial cells and directly interacts with VE-cadherin upon P aeruginosa infection.
To determine whether the increased association of VE-cadherin with N-WASP induced by P aeruginosa infection would disrupt the VE-cadherin and β-catenin complex, we performed a co-IP assay using RMVECs with or without N-WASP downregulation and P aeruginosa infection. P aeruginosa infected RMVECs had impaired binding of VE-cadherin and β-catenin compared to uninfected cells (Figure 1D lane 2 vs lane 1). The downregulation of N-WASP restored VE-cadherin and β-catenin association in P aeruginosa infected cells, suggesting that N-WASP plays a central role in P aeruginosa-induced disassembly of the VE-cadherin and β-catenin complex (Figure 1D, lane 4 vs 2). The downregulation of N-WASP alone, or scramble shRNA, have no effect on the cadherin and β-catenin binding in RMVECs without in P aeruginosa infection (Figure 1D, lane 3 and 5). Together, our findings demonstrate a central role of N-WASP in modulating P aeruginosa-induced paracellular permeability derangements in RMVECs. P aeruginosa infection increased the interaction between N-WASP and VE-cadherin, which further promoted the dissociation of the VE-cadherin and β-catenin complex, resulting in cell-cell junction destabilization and increased paracellular permeability.
3.3 |. N-WASP modulates P aeruginosa-induced permeability derangements in alveolar epithelial cell monolayers
To study the role of N-WASP in P aeruginosa-induced alveolar epithelial permeability, we downregulated N-WASP with lentiviral shRNA in rat alveolar type II epithelial cells (L2) followed by P aeruginosa infection (MOI = 40). Subsequently, continuous transepithelial resistance was measured by ECIS for 24h. P aeruginosa infected cells showed a decrease of 57% resistance at 24hr post P aeruginosa exposure. In contrast, N-WASP downregulation blocked P aeruginosa-induced paracellular permeability and significantly attenuated the resistance decrease (only a 10% drop in resistance) in L2 cells (Figure 2A). This result demonstrates that the downregulation of N-WASP protected barrier integrity from P aeruginosa infection in alveolar epithelial cell monolayers.
FIGURE 2.
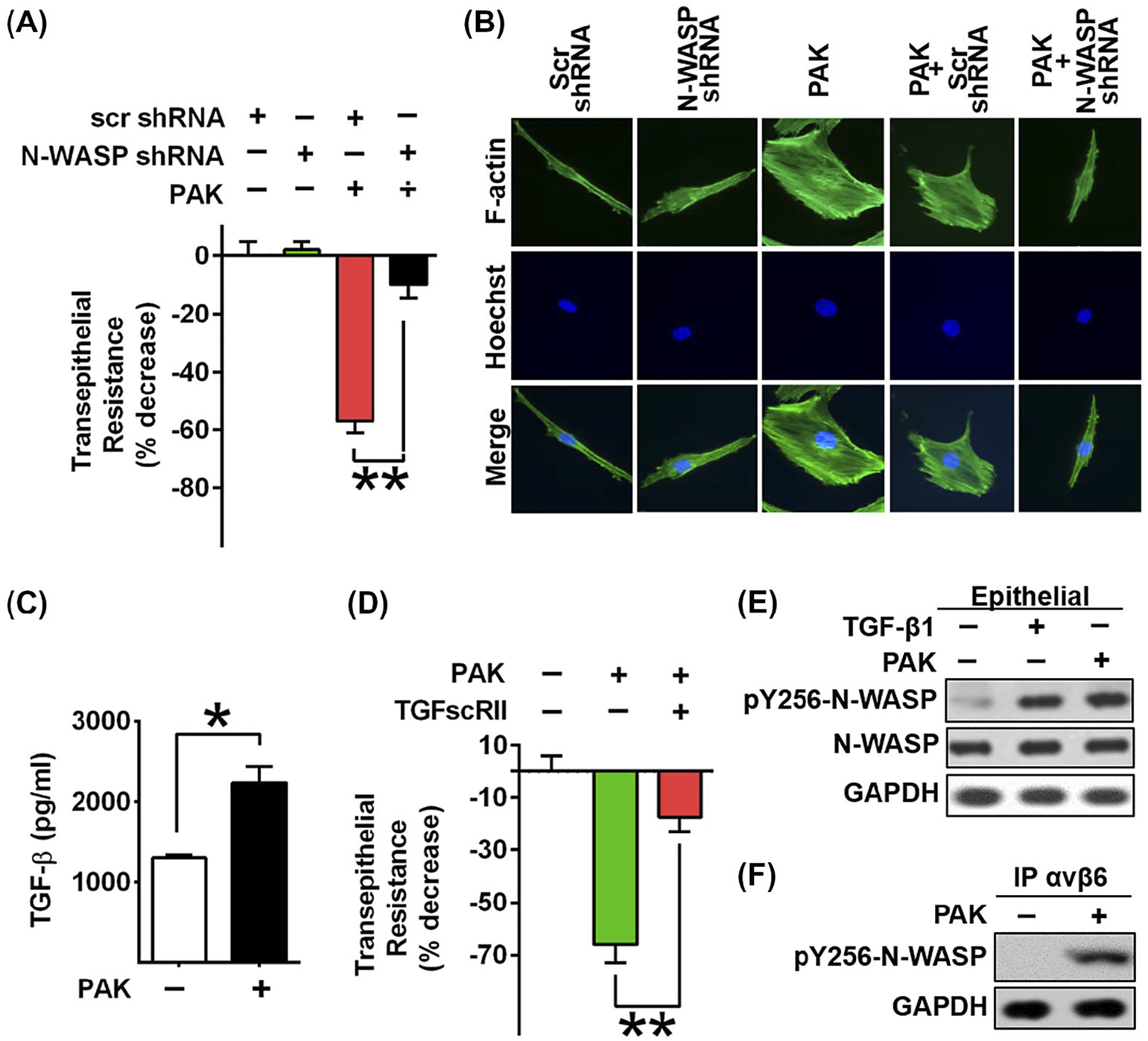
N-WASP downregulation inhibited P aeruginosa induced permeability and cytoskeleton rearrangement in epithelial cells. A, Lung epithelial cells (L2) were challenged with P aeruginosa infection, followed by the measurement of transepithelial resistance with or without N-WASP downregulation. Resistance value was measured 24 h post P aeruginosa stimulation. Data are presented as the percentage of decreased resistance normalized to control (n = 6–8). B, Representative images of actin stress fiber formation in L2 cells transduced with lentivirus targeting N-WASP or scramble shRNA followed by P aeruginosa treatment. Actin stress fibers are immunostained with phalloidin (green) and nuclei with Hoechst (blue). The downregulation of N-WASP blocked P aeruginosa induced actin stress fiber formation. C, ELISA assays showed increased active TGF-β1 production upon P aeruginosa stimulation in L2 cells. D, Blockade of TGF-β1 signaling pathway by treatment of L2 cells with a soluble chimeric TGF-β type II receptor (10 ng/mL) inhibited P aeruginosa induced epithelial paracellular permeability in L2 monolayers (n = 6–8). E, Representative blot shows increased phosphorylation of N-WAPS Y256 in response to TGF-β1 and P aeruginosa treatment in L2 cells. F, Western blot of co-immunoprecipitation assays showed that P aeruginosa induced the association of integrin αvβ6 with active form of N-WASP. Data are presented as means ± SEM. *P < .01 and **P < .001
To understand the role of N-WASP in modulating P aeruginosa-induced actin cytoskeleton dynamics, we examined the cytoskeletal changes by immunostaining F-actin in L2 monolayers with or without N-WASP downregulation or P aeruginosa infection. In non-infected cells, cortical ring structures were present around cell edges (Figure 2B, column 1) and no significant cytoskeletal change was observed in cells with N-WASP downregulation (Figure 2B, column 2). L2 cells with P aeruginosa infection showed disrupted cortical ring structures, with formation of pronounced thick and long actin stress fibers (Figure 2B, column 3). The downregulation of N-WASP blocked the P aeruginosa-induced prominent actin stress fiber formation (Figure 2B, column 5). These results demonstrate that N-WASP is essential for P aeruginosa-induced actin stress fiber formation in alveolar epithelial cells.
3.4 |. P aeruginosa induced increased TGF-β1 production, which promotes P aeruginosa-induced permeability across alveolar epithelial cell monolayers
Our previous study support that N-WASP modulates paracellular permeability in response to TGF-β1.29 To address whether N-WASP modulates P aeruginosa-induced paracellular permeability via TGF-β1-mediated pathway, we first measured the level of active TGF-β1 production in L2 cells with or without P aeruginosa infection. The production of active TGF-β1 was increased 1.7-fold in P aeruginosa-challenged cells compared to uninfected cells (Figure 2C). We next determined whether N-WASP-mediated paracellular permeability is secondary to P aeruginosa-induced TGF-β1 production. To test this, we pretreated L2 monolayers with a soluble chimeric TGF-β type II receptor (TGFscRII), and assessed transepithelial resistance across these monolayers in response to P aeruginosa infection. As shown in Figure 2D, treatment with P aeruginosa alone caused a significant decrease in resistance, whereas pretreatment with TGFscRII improved the resistance, suggesting that blocking TGF-β1 attenuated P aeruginosa-induced paracellular permeability. Furthermore, there was an increased phosphorylation of N-WASP Y256 in response to TGF-β1 in response to P aeruginosa treatment in L2 cells (Figure 2E). These results demonstrate that N-WASP modulates paracellular permeability by P aeruginosa-induced increases of TGF-β1 activation in alveolar epithelial cells.
3.5 |. P aeruginosa exposure induced interaction between N-WASP and αvβ6 integrin in alveolar epithelial cell monolayers
During ALI, αvβ6 expression is upregulated, resulting in increased TGF-β1 activation in lung epithelial cells.23,41 We hypothesize that N-WASP-mediated cytoskeleton derangements contribute to TGF-β1 activation by interacting with αvβ6 integrin. To test this hypothesis, we performed co-IP assay using L2 lysates with or without P aeruginosa infection. αvβ6 co-precipitated with tyrosine 256 phosphorylated N-WASP (Y256-N-WASP) only in P aeruginosa infected cells (Figure 2F). The findings support an increased interaction between activated N-WASP and αvβ6 integrin in response to P aeruginosa infection (Figure 2F). This result suggests that N-WASP directly participates in P aeruginosa-mediated upregulation of TGF-β1 activation through interaction with αvβ6 integrin.
3.6 |. N-WASP Y256 phosphorylation is a key step for P aeruginosa-induced disruption of the lung endothelial barrier integrity
We next examined whether exposure to P aeruginosa would cause the phosphorylation of N-WASP at Y256 in microvascular endothelial and alveolar epithelial cell monolayers. As shown in Figures 2E and 3A, the level of N-WASP Y256 phosphorylation was increased in both cell types infected with P aeruginosa. Interestingly, P aeruginosa-induced N-WASP Y256 phosphorylation was increased similarly when compared to that observed in cells after exposure to TGF-β1 in microvascular endothelial cells (Figure 3A), suggesting that TGF-β1 could have an important effect on N-WASP activation in response to P aeruginosa infection.
FIGURE 3.
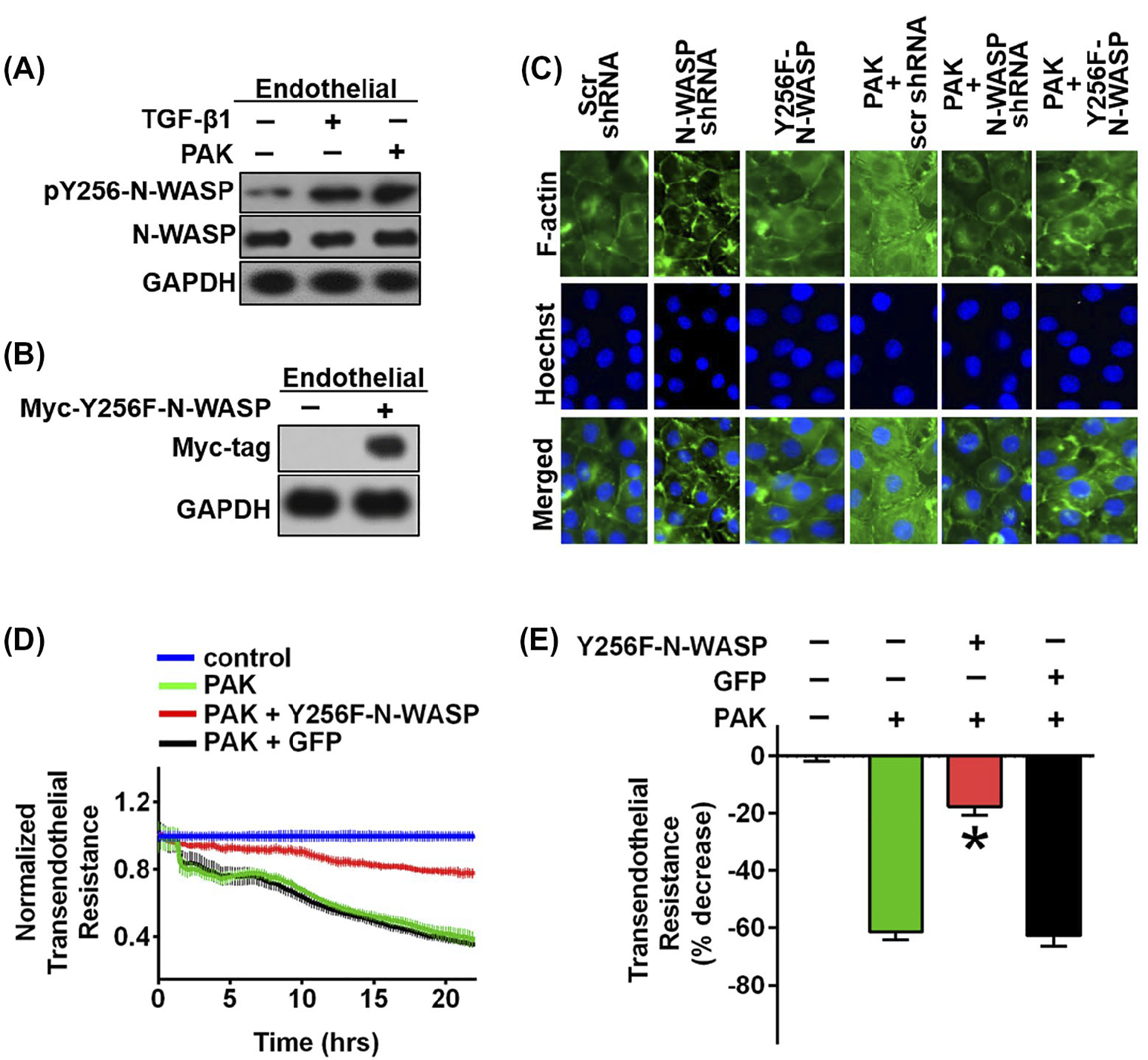
Phosphorylation of N-WASP Y256 is required for P aeruginosa induced permeability and actin stress fiber formation in RMVECs. A, Increased phosphorylation of N-WASP Y256 in response to TGF-β1 and P aeruginosa treatment in RMVEC cells. B, Forced expression of the dominant negative N-WASP mutant (myc-Y256F-N-WASP), a non-phosphorylatable mutant, in RMVEC cells. C, Expression of N-WASP mutant, myc-Y256F-N-WASP (Y256F-N-WASP), inhibited P aeruginosa induced stress fiber formation in RMVEC cells. Actin stress fibers are immunostained with phalloidin (green) and nuclei with Hoechst (blue). Representative images are showed. D, Expression of N-WASP mutant (Y256F-N-WASP) blocked P aeruginosa-induced endothelial permeability. ECIS measurement of RMVEC monolayers with or without myc-Y256F expression in response to P aeruginosa challenge (MOI = 40), electrical resistance was measured 24 h post P aeruginosa stimulation. E, Resistance value was calculated based on Figure D, 24 h post P aeruginosa stimulation. Data are presented as percentage of decreased resistance normalized to control (n = 6–8). Data are presented as means ± SEM. *P < .01
Next, if N-WASP plays a critical role in P aeruginosa-induced lung vascular permeability, blocking its active form should compromise the formation of actin stress fibers. To block the endogenous unfolding of N-WASP, we overexpressed a dominant negative myc-tagged N-WASP mutant (tyrosine-to-phenylalanine mutation, myc-Y256F-N-WASP) by adenoviral vectors (Figure 3B). Generation and usage of N-WASP mutant adenoviral vector was described previously.30 Y256F-N-WASP acts as dominant-negative that competes and blocks the phosphorylation of Y256-N-WASP. Compared to P aeruginosa infected cells (Figure 3C, column 4), cells with N-WASP downregulation or Y256F-N-WASP overexpression showed less stress fiber formation in response to P aeruginosa (Figure 3C, columns 5–6), suggesting that Y256-N-WASP phosphorylation is necessary for P aeruginosa-induced actin cytoskeleton derangement. Furthermore, as expected, cells overexpressing Y256F-N-WASP mutant showed significant improvement in resistance in response to P aeruginosa (Figure 3D, red line) and about 80% in resistance at 24hr poststimulation (Figure 3E, red bar). In contrast, cells treated with P aeruginosa alone or with green fluorescent protein (GFP) overexpression showed a continuing decrease in barrier resistance over time (Figure 3D, green and black line) with a decrease of about 60% resistance at 24hrs poststimulation (Figure 3E, green and black bars). Taken together, these results demonstrate that N-WASP plays an essential role, by regulating the GBD switch, in mediating actin cytoskeletal dynamics and lung permeability in P aeruginosa infection.
3.7 |. P aeruginosa-induced N-WASP activation is dependent upon Rho GTPase and FAK activation
To test if Rho GTPase is required in P aeruginosa-induced N-WASP Y256 phosphorylation, we pharmacologically blocked Rho activity by pretreating RMVEC monolayers with pan Rho inhibitor (1μg/ml) before P aeruginosa exposure. As shown in Figure 4A, P aeruginosa-induced Rho activation and N-WASP Y256 phosphorylation were inhibited by Rho inhibitor. This result demonstrates that N-WASP Y256 phosphorylation induced by P aeruginosa requires an upstream active Rho GTPase signaling.
FIGURE 4.
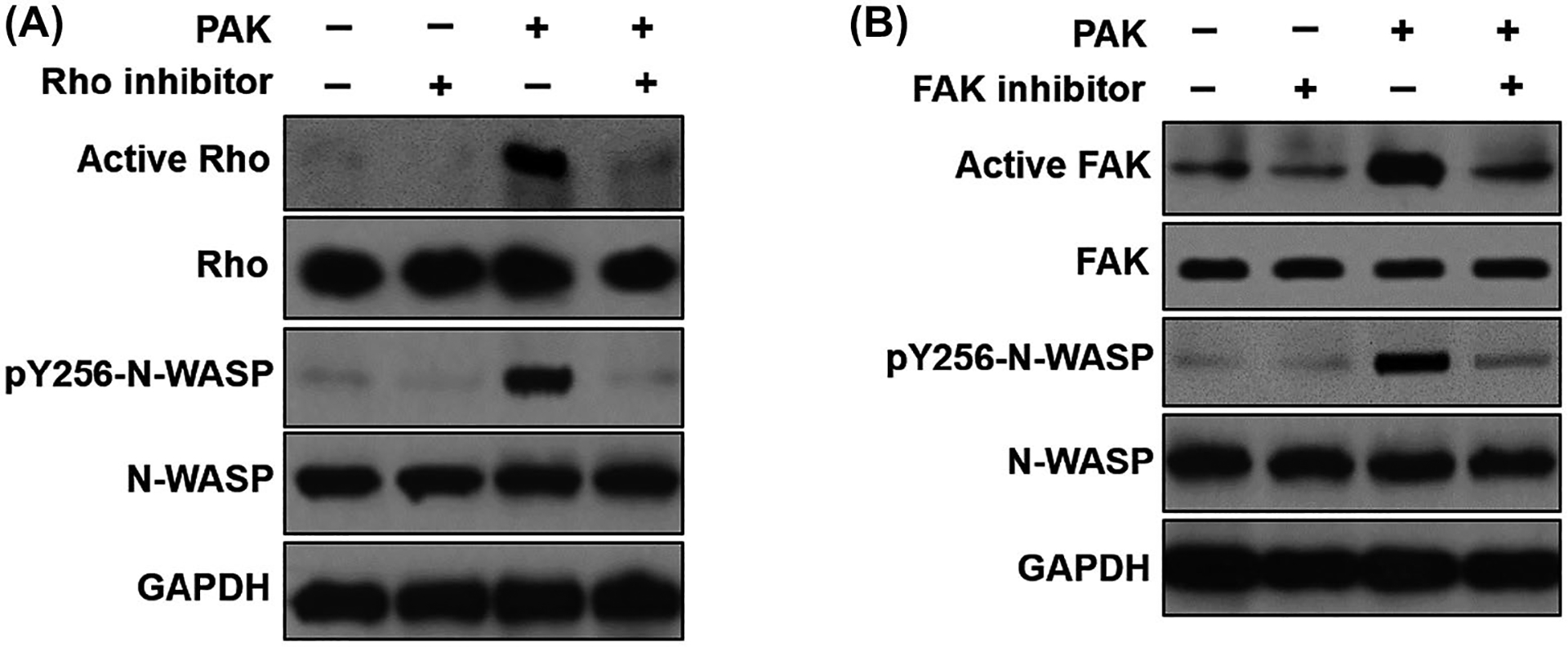
P aeruginosa induced the phosphorylation of Y256-N-WASP requires activation of upstream effectors Rho and FAK. A, Western blot showed that the pharmacological inhibition of Rho with Rho inhibitor (1 μg/mL pan Rho inhibitor) blocked P aeruginosa induced N-WASP activation in RMVECs. B, Western blot showed that pharmacological inhibition of FAK with FAK inhibitor (1 μM) blocked P aeruginosa induced N-WASP activation in RMVECs
We have previously demonstrated that focal adhesion kinase (FAK) as an essential mediator of TGF-β1-induced N-WASP Y256 phosphorylation in fibroblast and lung microvascular endothelial cells.29,30 To explore whether FAK is necessary for P aeruginosa-induced N-WASP Y256 phosphorylation, RMVECs were pretreated with vehicle or FAK inhibitor (PF573228, 1 μM) before P aeruginosa exposure. As shown in Figure 4B, P aeruginosa-induced FAK activation (phosphorylation of Y397) and downstream N-WASP Y256 phosphorylation. Pretreatment of cells with FAK inhibitor significantly inhibited P aeruginosa-induced FAK activation and N-WASP Y256 phosphorylation (Figure 4B). The data demonstrate that N-WASP Y256 phosphorylation induced by P aeruginosa requires FAK activation in the upstream signaling. Taken together, these data indicate that the activation of upstream effector Rho and FAK signaling is necessary for N-WASP Y256 phosphorylation induced by P aeruginosa.
3.8 |. N-WASP modulates P aeruginosa induced paracellular permeability in human lung microvascular endothelial and human alveolar epithelial cell monolayers
Next, we downregulated N-WASP expression by lentiviral shRNA in human lung microvascular endothelial (HMVECs) and human alveolar epithelial (H441) cells and examined whether it would attenuate P aeruginosa induced paracellular permeability. N-WASP expression was efficiently knocked down in both human endothelial and epithelial cells (Figure 5A,B; #2 shRNA achieving the maximal knockdown). We then evaluated the paracellular permeability in both HMVECs and H441 cells with or without N-WASP downregulation or P aeruginosa infection. In N-WASP downregulated HMVEC cells, P aeruginosa infection induced a significantly less drop in paracellular resistance (Figure 5C,E, black), with about 18% decrease comparing to about 60% decrease in control cells (Figure 5C,E, red). Similarly, in N-WASP downregulated H441 cells, P aeruginosa induced only about 40% drop in paracellular resistance (Figure 5D,F, black) when compared to about 75% decrease in control cells (Figure 5D,F, red). These results confirm the role of N-WASP in mediating P aeruginosa-induced permeability in human lung microvascular endothelial and alveolar epithelial monolayers.
FIGURE 5.
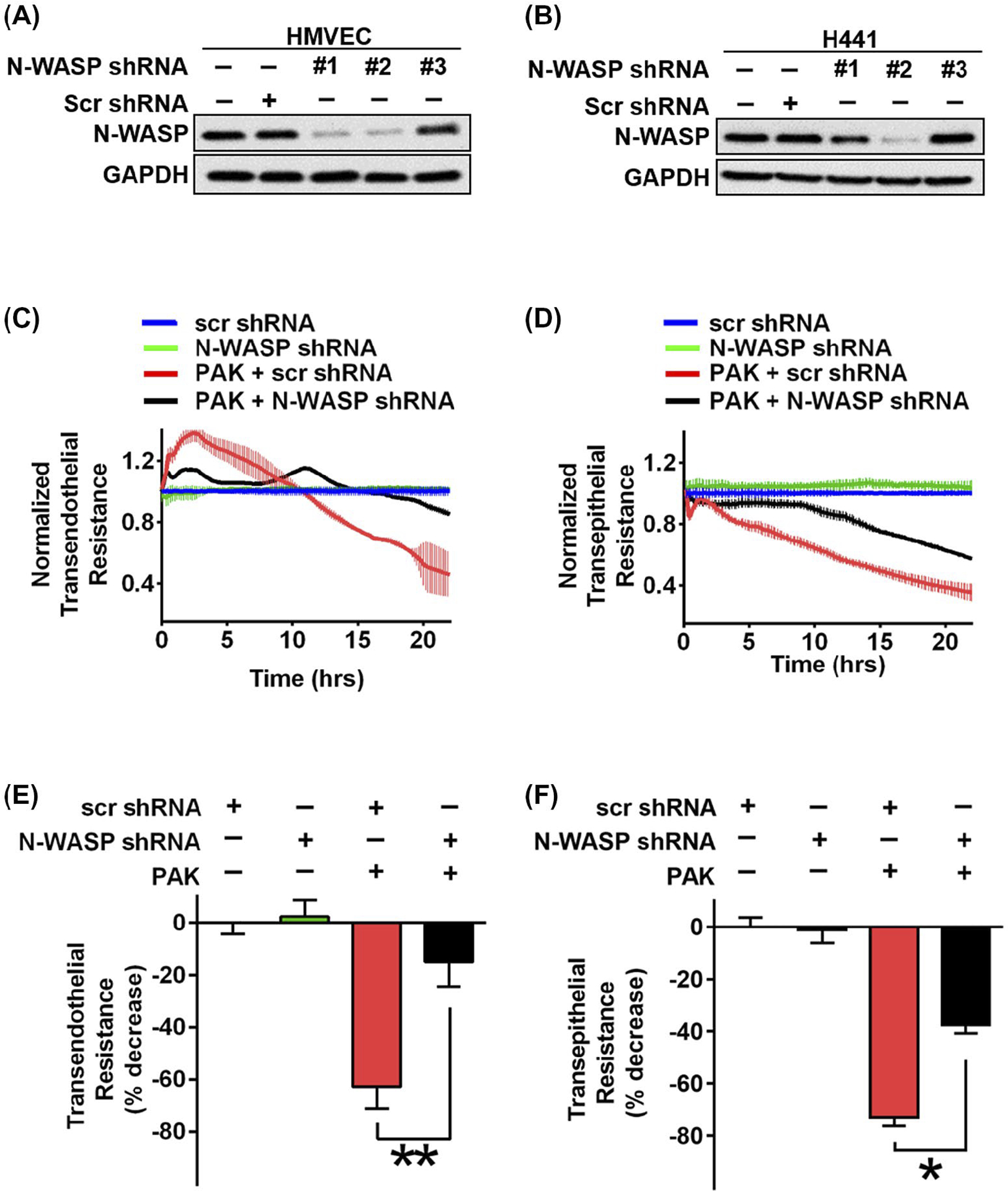
Downregulation of N-WASP blocked P aeruginosa induced permeability in human microvascular endothelial and human alveolar epithelial cells. A and B, Western blot showed the lentivirus-mediated knockdown effects of N-WASP in human microvascular endothelial cells (HMVECs) in panel A and a human alveolar epithelial cell line (H441) in panel B by three different lentiviral shRNA constructs. The #2 lentiviral vector had the optimal efficiency and used for all following experiments. C and D, Measurement of monolayer resistance across HMVEC (C) and H441 monolayers (D) with or without N-WASP downregulation in response to P aeruginosa stimulation (MOI = 40). Electrical resistance was measured as described above up to 24 h post P aeruginosa stimulation. E and F, Resistance value was calculated based on panels C and D, in HMVEC cells (E) and H441 cells (F) with or without N-WASP downregulation. Data are presented as the percentage of decreased resistance normalized to control (n = 7–8). *P < .01, **P < .001
3.9 |. Conditional deletion of N-WASP protects mice from P aeruginosa-induced pulmonary edema
To understand whether N-WASP plays a role in P aeruginosa-induced pulmonary edema, N-WASP floxed (N-WASPflox/flox) mice were intratracheally instilled with recombinant adenoviral Cre vectors (Ad-Cre, 50 μL, 108 PFU).35,42 Adenoviral vector with GFP (Ad-GFP, 50 μL, 108 PFU) was used as control. For P aeruginosa challenge, mice were intratracheally instilled with 107 CFU P aeruginosa as described previously.9 Pulmonary edema was evaluated by the abnormal accumulation of extravascular pulmonary edema (EVPE) in the lungs. P aeruginosa challenge induced excessive lung water and pulmonary edema in control mice (instilled with Ad-GFP). In comparison to mice instilled with Ad-GFP, P aeruginosa challenge induced a significant less lung water and pulmonary edema in mice instilled with Ad-Cre (Figure 6A). As a result, survival is significantly improved in mice instilled with Ad-Cre when compared with mice instilled with Ad-GFP in response to P aeruginosa challenge (Figure 6B, P < .01). These results clearly support that conditional deletion of N-WASP has protected mice from the development of pulmonary edema, and has improved mouse survival in a P aeruginosa-induced pneumonia model.
FIGURE 6.
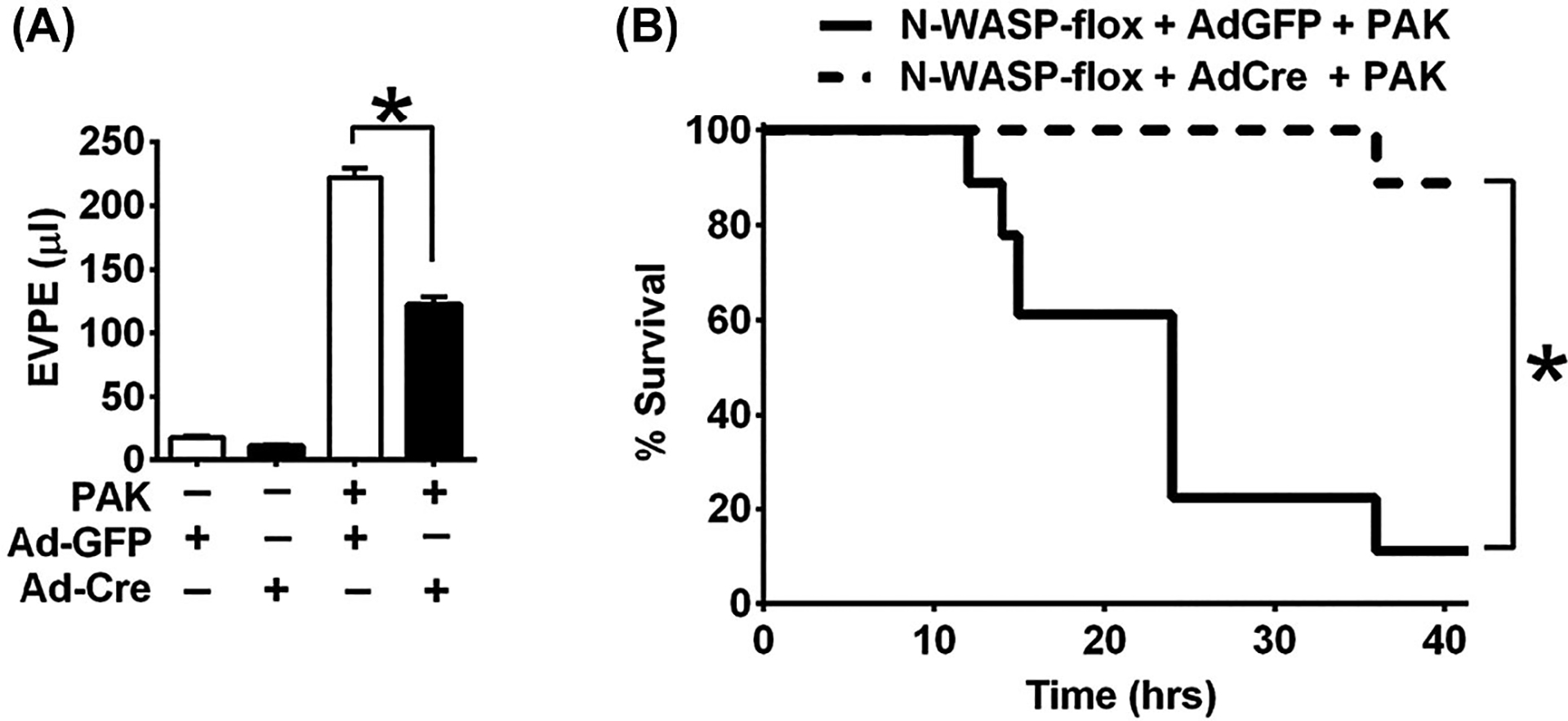
Conditional deletion of N-WASP protected against the development of pulmonary edema and improved survival in mice challenged with P aeruginosa. A, N-WASP floxed (N-WASPflox/flox) mice were instilled with adenoviral vector encoding Cre Recombinase (Ad-Cre) or adenoviral vector encoding control green fluorescent protein (Ad-GFP) (50 μL, 108 PFU), challenged with P aeruginosa (107 CFU), and followed by measurement of extravascular pulmonary edema (EVPE) 6 h post P aeruginosa challenge (n = 9–10 mice per group). The volume of EVPE in Ad-Cre instilled mice is significantly reduced when compared to mice instilled with control Ad-GFP in response to P aeruginosa challenge. B, Survival analysis showed improved survival in Ad-Cre instilled mice post P aeruginosa infection, when compared to the mortality in control Ad-GFP instilled mice (n = 18 mice per group). *P < .01
4 |. DISCUSSION
P aeruginosa is a common opportunistic pathogen that causes pneumonia in critically ill patients, leading to high mortality associated with the development of ALI and pulmonary edema secondary to increased permeability across the lung endothelial and alveolar epithelial barriers. Thus, dysfunction of lung vascular endothelial and alveolar epithelial barrier function is central to the pathology of pulmonary edema. N-WASP is a ubiquitously expressed multifunctional scaffolding protein. N-WASP is directly involved in the formation of newly synthesized actin filaments and thus serves as an important regulator of actin cytoskeletal dynamics. In this study, we investigated the role of N-WASP in P aeruginosa-induced increased vascular permeability across lung microvascular endothelial and alveolar epithelial cell monolayers and in a P aeruginosa-induced ALI mouse model. Our data indicate that the downregulation of N-WASP blocked P aeruginosa-induced permeability across monolayers of lung microvascular endothelial and alveolar epithelial cells. Conditional depletion of N-WASP significantly limited the development of pulmonary edema and prevented mortality in mice challenged with P aeruginosa. These findings support an important role of N-WASP and N-WASP-mediated pathway(s) in the development of pulmonary edema in response to P aeruginosa challenge.
Lung vascular endothelial and alveolar epithelial barriers play a fundamental role in the pathology of pulmonary edema during P aeruginosa pneumonia.5,6,44 Our data demonstrate that the downregulation of N-WASP blocked P aeruginosa-induced detrimental cytoskeletal dynamics and de novo actin stress fiber formation in monolayers of lung microvascular endothelial and alveolar epithelial cells. VE-cadherin is a major component of AJs that plays a role in maintaining vascular endothelial barrier function. Disruption of VE-cadherin by blocking antibody, genetic deletion or pathogenic protease can induce increased vascular permeability.45–47 Loss of VE-cadherin has been reported in P aeruginosa infected cells and is associated with adherens junction (AJ) rupture.47 Whether N-WASP contributes to P aeruginosa-induced AJ disruption is unclear. Our results suggest that an association of N-WASP and VE-cadherin is increased in P aeruginosa-treated microvascular endothelial cells. N-WASP also promotes P aeruginosa-induced actin stress fiber formation that further enhances cellular mechanical tensions and results in distorted cell-cell junction functions. Lung microvascular endothelium is a tightly controlled barrier.48 VE-cadherin plays a central role in the modulation of endothelial permeability. VE-cadherin is anchored to actin cytoskeleton via its cytoplasmic tail, linking the functions of adherens junctions to actin-cytoskeleton-mediated cell retraction. Indeed, our result suggests that P aeruginosa promotes the recruitment of N-WASP to VE-cadherin-dependent adherens junction and subsequently induces dynamics and dysfunctional of VE-cadherin-dependent adherens junctions. In line with our result, others have also observed the disruption of endothelial barrier function in N-WASP depleted cells.49
The nature of N-WASP as a scaffolding protein enables N-WASP to bridge the actin cytoskeleton and cell junction organizations. Cells with N-WASP downregulation have been reported to present misshaped cell junctions.50 β-catenin links VE-cadherin complex to the actin cytoskeleton and thus plays an important role in stabilizing endothelial cell-cell contacts and barrier integrity. VE-cadherin is anchored to the actin cytoskeleton by forming a complex with β-catenin. β-catenin deficient mouse embryos display abnormal AJs and vascular fragility.51 Our data suggest that P aeruginosa increases N-WASP and VE-cadherin interplay and association, promotes VE-cadherin and β-catenin dissociation, and induces stress fiber formation in an N-WASP-dependent manner. These events result in increased changes in actin cytoskeletal dynamics and cellular mechanical tensions that disrupt endothelial barrier integrity. Our results suggest a novel mechanism that N-WASP promotes P aeruginosa-induced cellular cytoskeletal dynamics and mechanical tensions by coordinating dynamics of actin cytoskeleton and VE-cadherin at adherens junctions, eventually leading to the subsequent barrier dysfunction.
Phosphorylation of Y256 on N-WASP is critical in maintaining N-WASP in its active status. We have previously shown that phosphorylation of Y256 on N-WASP is necessary for TGF-β1-induced actin cytoskeletal dynamics and vascular permeability.29 In this study, we found that overexpression of N-WASP mutant (Y256F), a dominant-negative mutation that is unable to be phosphorylated, blocked P aeruginosa-induced cytoskeletal dynamics and paracellular permeability. Further, we showed that the activation of N-WASP upstream effectors, RhoA and FAK, is necessary for P aeruginosa-induced phosphorylation of N-WASP-Y256. This may due to the effect of active small Rho GTPases on releasing N-WASP from the inactive status (autoinhibited conformation) and subsequent Y256 phosphorylation through FAK kinase. Our findings are in line with the previous report in fibroblasts that TGF-β1 induced FAK activation is a critical upstream event required for N-WASP activation. FAK and its signaling partners are known to play important roles in promoting lung vascular permeability.29,52–56 Our findings in this study explained, as least in part, how FAK modulates vascular permeability in response to P aeruginosa, by mediating downstream N-WASP activation and associated actin cytoskeletal reorganizations in lung microvascular endothelial monolayers. We have reported that increased epithelial permeability is attributable to RhoA-αvβ6-mediated TGF-β1 activation in response to pro-inflammatory factor interleukin-1β during ALI.23,57 TGF-β1 plays a critical role in mediating epithelial and endothelial permeability and development of pulmonary edema.39,49,50 αvβ6 integrin is associated with inactive TGF-β1. Our data show an increased association of N-WASP with αvβ6 integrin, and increased TGF-β1 activation, in P aeruginosa-treated epithelial cells. The data suggest that N-WASP-mediated actin stress fiber dynamics likely increases cell contractility, and facilitates the releasing of active TGF-β1 from latency-associated peptide. Indeed, our current findings suggest that P aeruginosa promotes the recruitment of activated N-WASP to αvβ6 in alveolar epithelial cells and promote permeability.
In this study, we investigated the role of N-WASP in modulating the barrier functions of lung vascular endothelium and alveolar epithelium in a P aeruginosa induced mouse ALI model. Our data demonstrate that N-WASP is required for the development of pulmonary edema in vivo. The development of pulmonary edema is significantly attenuated, while survival is significantly improved, in mice with conditional N-WASP deletion in response to P aeruginosa challenge.
P aeruginosa uses multiple toxins to increase vascular permeability. We have previously reported that ExoS and ExoT from the Type III secretion system increase in vivo and in vitro lung vascular permeability via the activation of RhoA and thus may play an important role in the activation of N-WASP.8,9 Furthermore, ExoY, another toxin from the type III secretion system, also increases lung vascular permeability via its nucleotidyl cyclase activity.10–12 Its type III secretion system disrupts alveolar epithelial carrier and increase alveolar epithelial permeability.4,58 P aeruginosa virulence factors can also weaken cell repair ability and disrupt tight junction integrity, resulting in increased alveolar epithelial permeability.35 In addition to the Type III secretion system, P aeruginosa has several other toxins that can affect the integrity of the alveolar epithelial barrier. Among these toxins, the lectin LecB, targets epithelial adherens junctions, inducing β-catenin degradation.59 Exotoxin A, secreted by the Type II secretion system, inhibits host protein synthesis by ADP-ribosylating elongation factor-2.60 Another toxin from the Type II secretion system, LasB Protease, increases epithelial permeability via the disruption of intercellular tight junctions by down-regulating the expression of several tight junction proteins.61 Finally, a pore-forming toxin named Exolysin, was identified in a subset of P aeruginosa outlier strains lacking the Type III secretion system. Exolysin causes E-cadherin cleavage and intercellular junction disruption via pore formation that triggers a massive Ca2+ influx into the cytosol.62 Ubiquitin E3 ligase HECTD2 and Activated protein C pathways also have been reported to regulate epithelial permeability during P aeruginosa infection.63,64
In summary, we demonstrate the important role of N-WASP in modulating vascular permeability through actin cytoskeletal derangement and modulation of VE-cadherin and αvβ6 function in response to P aeruginosa. Phosphorylation of N-WASP, mediated by upstream effector Rho GTPase and FAK, is an essential step that is required for N-WASP to modulate actin cytoskeletal dynamics. In mice challenged with P aeruginosa, conditional deletion of N-WASP significantly limits the development of pulmonary edema and protects mice from edema associated death. Currently, efforts in drug discovery focus primarily on defined target binding sites with enzymatic activities. Autoregulatory domains, such as N-WASP autoinhibition domain or N-WASP-Y256, may present attractive alternative targets for the search of novel inhibitors against P aeruginosa-induced pulmonary edema. Our investigation of the role of N-WASP in mediating lung vascular permeability and pulmonary edema development provides evidence to support the development of therapeutic drugs targeting the involved signaling pathways.
ACKNOWLEDGMENTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) grants R01HL143017 and R01HL127338, and FAMRI to Q. Ding, J. Pittet, and B. Wagener, a Mentored Research Training Grant from the Foundation for Anesthesia Education and Research to B. Wagener, and the American Heart Association postdoctoral fellowship to P. Che.
Funding information
HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI), Grant/Award Number: R01HL143017 and R01HL127338; Flight Attendant Medical Research Institute (FAMRI); Foundation for Anesthesia Education and Research (FAER); American Heart Association (AHA)
Abbreviations:
- AJ
adherens junction
- ALI
acute lung injury
- Arp
actin-related protein
- CFU
colony-forming unit
- ECIS
electric cell-substrate impedance sensing
- EVPE
extravascular pulmonary equivalents
- FAK
focal adhesion kinase
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GBD
GTPase-binding domain
- GFP
green fluorescent protein
- HMVEC
human microvascular endothelial cells
- N-WASP
neuronal Wiskott-Aldrich syndrome protein
- PAGE
polyacrylamide gel electrophoresis
- PFU
plaque-forming unit
- RMVEC
rat lung microvascular endothelial cells
- RT-PCR
reverse transcriptase-polymerase chain reaction
- SDS
sodium dodecyl sulfate
- shRNA
short hairpin RNA
- TGF-β1
transforming growth factor beta-1
- VCA
verprolin homology (V), central (C), and acidic (A) regions of N-WASP
- VE
vascular endothelial
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. [DOI] [PubMed] [Google Scholar]
- 2.Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. [DOI] [PubMed] [Google Scholar]
- 3.Rello J, Rue M, Jubert P, et al. Survival in patients with nosocomial pneumonia: impact of the severity of illness and the etiologic agent. Crit Care Med. 1997;25:1862–1867. [DOI] [PubMed] [Google Scholar]
- 4.Sawa T, Shimizu M, Moriyama K, Wiener-Kronish JP. Association between Pseudomonas aeruginosa type III secretion, antibiotic resistance, and clinical outcome: a review. Crit Care. 2014;18:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiener-Kronish JP, Sakuma T, Kudoh I, et al. Alveolar epithelial injury and pleural empyema in acute P. aeruginosa pneumonia in anesthetized rabbits. J Appl Physiol (1985). 1993;75:1661–1669. [DOI] [PubMed] [Google Scholar]
- 6.Sayner SL, Frank DW, King J, Chen H, VandeWaa J, Stevens T. Paradoxical cAMP-induced lung endothelial hyperpermeability revealed by Pseudomonas aeruginosa ExoY. Circ Res. 2004;95:196–203. [DOI] [PubMed] [Google Scholar]
- 7.Liu A, Park JH, Zhang X, et al. Therapeutic effects of hyaluronic acid in bacterial pneumonia in ex vivo perfused human lungs. Am J Respir Crit Care Med. 2019;200:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganter MT, Roux J, Su G, et al. Role of small GTPases and alphavbeta5 integrin in Pseudomonas aeruginosa-induced increase in lung endothelial permeability. Am J Respir Cell Mol Biol. 2009;40:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carles M, Lafargue M, Goolaerts A, et al. Critical role of the small GTPase RhoA in the development of pulmonary edema induced by Pseudomonas aeruginosa in mice. Anesthesiology. 2010;113:1134–1143. [DOI] [PubMed] [Google Scholar]
- 10.Kloth C, Schirmer B, Munder A, Stelzer T, Rothschuh J, Seifert R. The role of Pseudomonas aeruginosa ExoY in an acute mouse lung infection model. Toxins (Basel). 2018;10:E185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balczon R, Prasain N, Ochoa C, et al. Pseudomonas aeruginosa exotoxin Y-mediated tau hyperphosphorylation impairs microtubule assembly in pulmonary microvascular endothelial cells. PLoS ONE. 2013;8:e74343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochoa CD, Alexeyev M, Pastukh V, Balczon R, Stevens T. Pseudomonas aeruginosa exotoxin Y is a promiscuous cyclase that increases endothelial tau phosphorylation and permeability. J Biol Chem. 2012;287:25407–25418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duluc L, Wojciak-Stothard B. Rho GTPases in the regulation of pulmonary vascular barrier function. Cell Tissue Res. 2014;355:675–685. [DOI] [PubMed] [Google Scholar]
- 14.Lu Q, Harrington EO, Jackson H, Morin N, Shannon C, Rounds S. Transforming growth factor-beta1-induced endothelial barrier dysfunction involves Smad2-dependent p38 activation and subsequent RhoA activation. J Appl Physiol 1985. 2006;101:375–384. [DOI] [PubMed] [Google Scholar]
- 15.Chen XL, Nam JO, Jean C, et al. VEGF-induced vascular permeability is mediated by FAK. Dev Cell. 2012;22:146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weis S, Shintani S, Weber A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14:7–12. [DOI] [PubMed] [Google Scholar]
- 18.Rohatgi R, Ma L, Miki H, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. [DOI] [PubMed] [Google Scholar]
- 19.Sturge J, Hamelin J, Jones GE. N-WASP activation by a beta1-integrin-dependent mechanism supports PI3K-independent chemotaxis stimulated by urokinase-type plasminogen activator. J Cell Sci. 2002;115:699–711. [DOI] [PubMed] [Google Scholar]
- 20.Snapper SB, Meelu P, Nguyen D, et al. WASP deficiency leads to global defects of directed leukocyte migration in vitro and in vivo. J Leukoc Biol. 2005;77:993–998. [DOI] [PubMed] [Google Scholar]
- 21.Suetsugu S, Hattori M, Miki H, et al. Sustained activation of N-WASP through phosphorylation is essential for neurite extension. Dev Cell. 2002;3:645–658. [DOI] [PubMed] [Google Scholar]
- 22.Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8:37–48. [DOI] [PubMed] [Google Scholar]
- 23.Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, Pittet JF. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma 2008;64:1211–1217; discussion 1217. [DOI] [PubMed] [Google Scholar]
- 24.Birukova AA, Cokic I, Moldobaeva N, Birukov KG. Paxillin is involved in the differential regulation of endothelial barrier by HGF and VEGF. Am J Respir Cell Mol Biol. 2009;40:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. [DOI] [PubMed] [Google Scholar]
- 26.Dominguez R. Actin filament nucleation and elongation factors–structure-function relationships. Crit Rev Biochem Mol Biol. 2009;44:351–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–1809. [DOI] [PubMed] [Google Scholar]
- 28.Fahy RJ, Lichtenberger F, McKeegan CB, Nuovo GJ, Marsh CB, Wewers MD. The acute respiratory distress syndrome: a role for transforming growth factor-beta 1. Am J Respir Cell Mol Biol. 2003;28:499–503. [DOI] [PubMed] [Google Scholar]
- 29.Wagener BM, Hu M, Zheng A, et al. Neuronal Wiskott-Aldrich syndrome protein regulates TGF-beta1-mediated lung vascular permeability. FASEB J. 2016;30:2557–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai GQ, Chou CF, Hu M, et al. Neuronal Wiskott-Aldrich syndrome protein (N-WASP) is critical for formation of alpha-smooth muscle actin filaments during myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2012;303:L692–L702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jian MY, Alexeyev MF, Wolkowicz PE, Zmijewski JW, Creighton JR. Metformin-stimulated AMPK-alpha1 promotes microvascular repair in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2013;305:L844–L855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Creighton J, Jian M, Sayner S, Alexeyev M, Insel PA. Adenosine monophosphate-activated kinase alpha1 promotes endothelial barrier repair. FASEB J. 2011;25:3356–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18:2571–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai GQ, Zheng A, Tang Q, et al. Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp Cell Res. 2010;316:1600–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding Q, Gladson CL, Wu H, Hayasaka H, Olman MA. Focal adhesion kinase (FAK)-related non-kinase inhibits myofibroblast differentiation through differential MAPK activation in a FAK-dependent manner. J Biol Chem. 2008;283:26839–26849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding Q, Stewart J Jr, Olman MA, Klobe MR, Gladson CL. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem. 2003;278:39882–39891. [DOI] [PubMed] [Google Scholar]
- 37.Che P, Yang Y, Han X, et al. S100A4 promotes pancreatic cancer progression through a dual signaling pathway mediated by Src and focal adhesion kinase. Sci Rep. 2015;5:8453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding Q, Stewart J Jr, Prince CW, et al. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 2002;62: 5336–5343. [DOI] [PubMed] [Google Scholar]
- 39.Bechara RI, Brown LA, Roman J, Joshi PC, Guidot DM. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;15:188–194. [DOI] [PubMed] [Google Scholar]
- 40.Bechara RI, Brown LA, Roman J, Joshi PC, Guidot DM. Transforming growth factor beta1 expression and activation is increased in the alcoholic rat lung. Am J Respir Crit Care Med. 2004;170:188–194. [DOI] [PubMed] [Google Scholar]
- 41.Song Y, Pittet JF, Huang X, et al. Role of integrin alphav beta6 in acute lung injury induced by Pseudomonas aeruginosa. Infect Immun. 2008;76:2325–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding Q, Cai GQ, Hu M, et al. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am J Pathol. 2013;182:1572–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hijazi MH, MacIntyre NR. Advances in infection control: ventilator-associated pneumonia. Semin Respir Crit Care Med. 2000;21:245–262. [DOI] [PubMed] [Google Scholar]
- 44.Stevens T, Garcia JG, Shasby DM, Bhattacharya J, Malik AB. Mechanisms regulating endothelial cell barrier function. Am J Physiol Lung Cell Mol Physiol. 2000;279:L419–L422. [DOI] [PubMed] [Google Scholar]
- 45.Carmeliet P, Lampugnani MG, Moons L, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. [DOI] [PubMed] [Google Scholar]
- 46.Corada M, Mariotti M, Thurston G, et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Golovkine G, Faudry E, Bouillot S, Voulhoux R, Attree I, Huber P. VE-cadherin cleavage by LasB protease from Pseudomonas aeruginosa facilitates type III secretion system toxicity in endothelial cells. PLoS Pathog. 2014;10:e1003939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leligdowicz A, Chun LF, Jauregui A, et al. Human pulmonary endothelial cell permeability after exposure to LPS-stimulated leukocyte supernatants derived from patients with early sepsis. Am J Physiol Lung Cell Mol Physiol. 2018;315:L638–L644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mooren OL, Kim J, Li J, Cooper JA. Role of N-WASP in endothelial monolayer formation and integrity. J Biol Chem. 2015;290:18796–18805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otani T, Ichii T, Aono S, Takeichi M. Cdc42 GEF Tuba regulates the junctional configuration of simple epithelial cells. J Cell Biol. 2006;175:135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cattelino A, Liebner S, Gallini R, et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J Cell Biol. 2003;162:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganter MT, Roux J, Miyazawa B, et al. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. 2008;102:804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pittet JF, Koh H, Fang X, et al. HMGB1 accelerates alveolar epithelial repair via an IL-1beta- and alphavbeta6 integrin-dependent activation of TGF-beta1. PLoS ONE. 2013;8:e63907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt TT, Tauseef M, Yue L, et al. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am J Physiol Lung Cell Mol Physiol. 2013;305:L291–L300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu Q, Sakhatskyy P, Grinnell K, et al. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol. 2011;301:L847–L857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su G, Hodnett M, Wu N, et al. Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 2007;36:377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munger JS, Huang X, Kawakatsu H, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. [DOI] [PubMed] [Google Scholar]
- 58.Geiser TK, Kazmierczak BI, Garrity-Ryan LK, Matthay MA, Engel JN. Pseudomonas aeruginosa ExoT inhibits in vitro lung epithelial wound repair. Cell Microbiol. 2001;3:223–236. [DOI] [PubMed] [Google Scholar]
- 59.Cott C, Thuenauer R, Landi A, et al. Pseudomonas aeruginosa lectin LecB inhibits tissue repair processes by triggering beta-catenin degradation. Biochim Biophys Acta. 2016;1863:1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bleves S, Viarre V, Salacha R, Michel GP, Filloux A, Voulhoux R. Protein secretion systems in Pseudomonas aeruginosa: a wealth of pathogenic weapons. Int J Med Microbiol. 2010;300:534–543. [DOI] [PubMed] [Google Scholar]
- 61.Beaufort N, Corvazier E, Hervieu A, et al. The thermolysin-like metalloproteinase and virulence factor LasB from pathogenic Pseudomonas aeruginosa induces anoikis of human vascular cells. Cell Microbiol. 2011;13:1149–1167. [DOI] [PubMed] [Google Scholar]
- 62.Reboud E, Bouillot S, Patot S, Beganton B, Attree I, Huber P. Pseudomonas aeruginosa ExlA and Serratia marcescens ShlA trigger cadherin cleavage by promoting calcium influx and ADAM10 activation. PLoS Pathog. 2017;13:e1006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Coon TA, McKelvey AC, Lear T, et al. The proinflammatory role of HECTD2 in innate immunity and experimental lung injury. Sci Transl Med. 2015;7:295ra109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bir N, Lafargue M, Howard M, et al. Cytoprotective-selective activated protein C attenuates Pseudomonas aeruginosa-induced lung injury in mice. Am J Respir Cell Mol Biol. 2011;45:632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]