


Keywords: diabetic hyperfiltration, dietary NaCl, glomerular capillary pressure, tubuloglomerular feedback, Na-glucose cotransporter
Abstract
Inhibitors of the main proximal tubular Na-glucose cotransporter (SGLT2) mitigate diabetic glomerular hyperfiltration and have been approved by the United States Food and Drug Administration for slowing the progression of diabetic kidney disease. It has been proposed that SGLT2 inhibitors improve hard renal outcomes by reducing glomerular capillary pressure (PGC) via a tubuloglomerular feedback (TGF) response to a decrease in proximal reabsorption (Jprox). However, the effect of SGLT2 inhibition on PGC has not been measured. Here, we studied the effects of acute SGLT2 blockade (ertugliflozin) on Jprox and glomerular hemodynamics in two-period micropuncture experiments using streptozotocin-induced diabetic rats fed high- or low-NaCl diets. PGC was measured by direct capillary puncture or computed from tubular stop-flow pressure (PSF). TGF is intact while measuring PGC directly but rendered inoperative when measuring PSF. Acute SGLT2 inhibitor reduced Jprox by ∼30%, reduced PGC by 5–8 mmHg, and reduced glomerular filtration rate (GFR) by ∼25% (all P < 0.0001) but had no effect on PSF. The decrease in PGC was larger with the low-NaCl diet (8 vs. 5 mmHg, P = 0.04) where PGC was higher to begin with (54 vs. 50 mmHg, P = 0.003). Greater decreases in PGC corresponded, unexpectedly, to lesser decreases in GFR (P = 0.04). In conclusion, these results confirm expectations that PGC would decline in response to acute SGLT2 inhibition and that a functioning TGF system is required for this. We infer a contribution of postglomerular vasorelaxation to the TGF responses where decreases in PGC were large and decreases in GFR were small.
NEW & NOTEWORTHY It has been theorized that Na-glucose cotransporter (SGLT2) blockade slows progression of diabetic kidney disease by reducing physical strain on the glomerulus. This is the first direct measurement of intraglomerular pressure during SGLT2 blockade. Findings confirmed that SGLT2 blockade does reduce glomerular capillary pressure, that this is mediated through tubuloglomerular feedback, and that the tubuloglomerular feedback response to SGLT2 blockade involves preglomerular vasoconstriction and postglomerular vasorelaxation.
INTRODUCTION
Na-glucose cotransporter (SGLT2) inhibitors reversibly reduce glomerular filtration rate (GFR) and slow the progression of diabetic kidney disease (1–4). SGLT2 inhibition is the first treatment to slow the progression toward end-stage renal disease for patients with diabetes since angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor blockers in the 1990s (5–7). The United States Food and Drug Administration has recently certified an SGLT2 inhibitor for the treatment of kidney disease (https://www.drugs.com/history/invokana.html).
Among the mechanisms that have been proposed to account for the renal-sparing effects of SGLT2 blockers, a theory based on renal hemodynamics is the most well developed. This theory begins with the assumption that diabetic hyperfiltration is a treatable stressor that puts the kidney at long-term risk (8) and invokes evidence that SGLT2 blockade mitigates hyperfiltration by reducing fluid and NaCl reabsorption by the proximal tubule, thereby evoking a tubuloglomerular feedback (TGF) response (9–11). An appealing feature of the theory is that using SGLT2 inhibitors to reduce hyperfiltration takes on the immediate upstream cause of hyperfiltration, which is a depressed TGF signal resulting from an SGLT2-dependent increase in NaCl and fluid reabsorption by the proximal tubule (10–16). In other words, inhibition of SGLT2 restores the glomerular hemodynamics to a more “natural” state.
Leading textbooks in the field of nephrology continue to describe a TGF effector mechanism that is restricted to the afferent arteriole (17). If this explanation is complete, then a TGF response elicited by SGLT2 blockade will reduce glomerular capillary pressure (PGC). However, this explanation of the TGF mechanism may not be adequate for all purposes (18). It is also commonly held that diabetic hyperfiltration implies elevated PGC and there are recent articles asserting that SGLT2 blockers improve renal outcomes by eliminating glomerular capillary hypertension (19–23). However, there are several published reports of diabetic hyperfiltration where PGC was not elevated (24–30), and there are no published measurements of PGC during SGLT2 inhibition.
Here, we report the results of micropuncture experiments specifically designed to test for effects of acute SGLT2 blockade on PGC in diabetic rats. These data confirm that SGLT2 blockade does reduce PGC, that the effect is magnified when rats are fed a low-NaCl diet, and that the effect requires a functioning TGF response.
METHODS
All animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with an institutional animal care and use committee-registered protocol.
Overview
Effects of acute SGLT2 blockade were studied by renal micropuncture in Wistar rats and Wistar-Froemter (MWF) rats with 3 wk of streptozotocin-induced diabetes. Animals were free fed with low- or high-NaCl rodent chow (Teklad) for 8–10 days before micropuncture. Data were obtained from each animal before and during SGLT2 blockade. SGLT2 blockade was achieved by intravenous bolus of ertugliflozin (Merck, 6 mg/kg over 10 min). In some animals, vehicle was given in lieu of ertugliflozin to provide a time control. Principal micropuncture data included direct and indirect measurements of pressure in glomerular capillaries, proximal tubules, peritubular capillaries, and Bowman's space as well as measurements of single-nephron GFR (SNGFR) and proximal tubular reabsorption.
Diabetes and Diets
Adult male Wistar rats (Harlan, Indianapolis, IN) and MWF rats (Veterans Affairs San Diego Healthcare System breeding colony) were made diabetic with streptozotocin (65 mg/kg as single intraperitoneal injection, Sigma). Thereafter, blood glucose was measured daily by a glucometer, and long-acting insulin (PZI, Blue Ridge Pharmaceuticals, Memphis, TN) was administered subcutaneously on a sliding scale with a blood glucose target of 15–20 mM. Rats were housed in pairs, given free access to tap water, and free fed rat chow (Teklad) containing 19% protein by weight and modified to contain either 0.12% NaCl (low NaCl) or 8% NaCl (high NaCl). Food and water consumption were recorded daily for each cage, and half of the total consumption was ascribed to each animal for the days that two animals were housed together.
Surgical Preparation and Micropuncture
Micropuncture experiments were performed after 3 wk of diabetes and 8–10 days on low- or high-NaCl diet. Rats that were housed together were studied on consecutive days. Animals were surgically prepared for micropuncture according to previously established protocols (31). Briefly, animals were anesthetized with Inactin (100 mg/kg intraperitoneal injection, Research Biochemicals, Natick, MA), and body temperature was maintained at 37°C on a servo-controlled heating table. The airway was maintained with a tracheostomy. Catheters were placed in the left jugular vein, left femoral artery, and urinary bladder. Arterial blood pressure was monitored throughout by an intra-arterial catheter. The left kidney was exposed through a flank incision, immobilized in a Lucite cup, and bathed with warm Ringer saline. The left ureter was cannulated for separate urine collection. Ringer saline containing [3H]inulin was infused at 1.5 mL/h as a marker of GFR. [3H]inulin was delivered at 80 µCi/h when protocols called for SNGFR to be measured and at 4 µCi/h otherwise. A separate syringe of Ringer saline was used to bring the total volume replacement to 3.5 mL/h in low NaCl-fed rats and 5 mL/h in high NaCl-fed rats. During SGLT2 blockade, the infusion rate of replacement fluid was increased by 2.5 mL/h and dextrose was added (4–8 g/dL) to stabilize blood glucose, which otherwise declines rapidly during SGLT2 blockade. At least 1 h was allowed for equilibration after surgery was completed before the initiation of micropuncture. Twenty minutes elapsed between administration of ertugliflozin and the start of the second period of micropuncture. Arterial pH, Pco2, Po2, glucose, hematocrit, Na, K, total protein, and radioactivity were determined before the start of micropuncture. At the end of each micropuncture period, additional blood samples were obtained to measure blood glucose concentration, hematocrit, total protein, and radioactivity. Urine collected during micropuncture was used to measure urine flow rate, whole kidney GFR, and electrolyte excretion.
A servo-nulling micro-pressure pipette apparatus (IPM, San Diego, CA) was used to measure pressures in proximal tubules, peritubular capillaries, glomerular capillaries, and Bowman's space, as previously described (28, 32). Pressures were sampled at 120 Hz, digitized, recorded on a desktop computer, and then downsampled to 1 Hz for analysis. MWF rats are uniquely suited for glomerular hemodynamic studies because they have glomeruli on the kidney surface, which are necessary for direct measurement of pressures in glomerular capillaries and Bowman’s space. In Wistar rats, PGC must be assessed indirectly, by the "stop-flow method," where an obstructing wax block is inserted in a proximal tubule and the stop-flow pressure (PSF) is measured upstream of the obstruction. Because there is no filtration in the obstructed nephron, protein concentration throughout the glomerular capillary is the same as that in systemic blood so PGC becomes the sum of PSF and oncotic pressure of systemic blood: PGC = PSF + π, where π (mmHg) is calculated from blood protein concentration c (g/dL) according to the Landis-Pappenheimer equation, π = 1.74c + 0.28c2 (33). Advantages to the stop-flow method are that it does not require surface glomeruli and it is technically easier to perform than direct capillary micropuncture. A disadvantage to the stop-flow method is that it renders TGF inoperative while PSF is being measured.
SNGFR and late proximal flow were measured in timed collections from late proximal tubules in Wistar rats. Net proximal fluid reabsorption (Jprox) is the difference between SNGFR and late proximal flow. Proximal tubular pressure was monitored during some of the collections.
Statistical Analysis
Results are expressed as means ± SE produced by least-squares ANOVA. ANOVA was done with proprietary software (Systat, Evanston, IL) and included design for repeated measures and analysis of covariance, as appropriate. For micropuncture experiments where there was more than one data point per experimental period, the individual experiment was included as a factor in the ANOVA. Data more than 3 SDs from the mean were excluded from the analysis. Effects with α < 0.05 were termed “significant.”
RESULTS
Baseline Metrics in Diabetic Rats Fed High-NaCl or Low-NaCl Diets
Control period data were obtained from 38 animals, and a full two periods of data were obtained from 35 animals. Thirty of these were administered ertugliflozin during the second period and five were administered vehicle to serve as time controls. Baseline values for body weight, mean arterial blood pressure, blood glucose, hematocrit, arterial blood gases, plasma electrolyte and protein concentrations, GFR, urine flow rate, and urine electrolyte excretion rates are shown in Table 1. In Wistar rats, GFR tended to be higher (P = 0.08) while hematocrit (P = 0.01) and baseline NaCl excretion were lower than in MWF rats. Rats fed high NaCl tended toward lower GFR (P = 0.09), lower arterial Po2 (P = 0.06), and a higher rate of NaCl excretion (P = 0.03) compared with rats fed low NaCl. Average body weight, baseline arterial blood pressure, blood glucose concentration, arterial CO2, plasma electrolyte concentrations, urine flow rate, and urine K excretion rate were not different between the rat strains and were unaffected by the prescribed dietary NaCl.
Table 1.
Baseline whole-animal metrics in Wistar-Froemter and Wistar rats fed high-NaCl or low-NaCl
| Wistar-Froemter Rats |
Wistar Rats |
P Values |
||||
|---|---|---|---|---|---|---|
| High NaCl | Low NaCl | High NaCl | Low NaCl | Diet | Strain | |
| n | 14 | 8 | 6 | 10 | ||
| Body weight, g | 315 ± 12 | 326 ± 15 | 330 ± 18 | 371 ± 14 | ||
| Mean arterial blood pressure, mmHg | 105 ± 3 | 99 ± 4 | 97 ± 5 | 115 ± 4 | ||
| Blood glucose, mg/dL | 411 ± 27 | 344 ± 39 | 407 ± 45 | 358 ± 35 | ||
| Hematocrit, % | 46 ± 1 | 47 ± 1 | 50 ± 1 | 50 ± 1 | 0.01 | |
| Arterial pH | 7.41 ± 0.01 | 7.47 ± 0.02 | 7.44 ± 0.1 | 7.43 ± 0.01 | ||
| Arterial Po2, mmHg | 73 ± 2 | 77 ± 5 | 72 ± 3 | 82 ± 3 | 0.06 | |
| Arterial total CO2, mM | 32 ± 1 | 31 ± 1 | 32 ± 1 | 31 ± 1 | ||
| Plasma Na, mM | 141 ± 1 | 140 ± 4 | 138 ± 1 | 140 ± 1 | ||
| Plasma K, mM | 4.0 ± 0.1 | 3.9 ± 0.2 | 4.1 ± 0.2 | 4.2 ± 0.1 | ||
| Plasma protein, g/dL | 7.4 ± 0.2 | 6.3 ± 0.2 | 6.5 ± 0.2 | 6.8 ± 0.1 | ||
| Glomerular filtration rate, mL/min | 3.9 ± 0.2 | 3.9 ± 0.3 | 3.9 ± 0.4 | 4.9 ± 0.3 | 0.09 | 0.08 |
| Urine flow, µL/min | 51 ± 6 | 36 ± 8 | 36 ± 10 | 40 ± 7 | ||
| Urine K, µeq/min | 2.5 ± 0.3 | 2.6 ± 0.4 | 2.0 ± 0.4 | 2.1 ± 0.4 | ||
| Urine Na, µeq/min | 4.9 ± 0.5 | 1.3 ± 0.7 | 0.8 ± 0.7 | 1.2 ± 0.6 | 0.02 | <0.01 |
| Urine Cl, µeq/min | 5.6 ± 0.5 | 2.4 ± 0.7 | 1.9 ± 0.8 | 1.9 ± 0.7 | 0.03 | <0.01 |
Values are shown as means ± SE. P values were generated by least-squares ANOVA.
Baseline Micropuncture Results
Baseline micropuncture results are shown in Table 2. SNGFR and Jprox were determined from [3H]inulin clearance and the volume of fluid in timed collections from late proximal tubules in Wistar rats on high-NaCl and low-NaCl diets. The purpose of making proximal tubular collections was to confirm that SGLT2 blockade reduces proximal reabsorption. Control period collections included 63 late proximal tubular fluid collections with SNGFR ranging from 20 to 76 nL/min and fractional reabsorption ranging from 8% to 78%. Average SNGFR was ∼10% higher in animals fed the low-NaCl diet, but the effect of diet was not statistically significant. After adjustment for SNGFR, Jprox averaged ∼10% higher in rats fed low-NaCl diet, but the effect of diet on Jprox was not statistically significant. The method of correcting Jprox for SNGFR is discussed later where we describe effects of SGLT2 blockade on Jprox.
Table 2.
Baseline micropuncture data
| n | Wistar-Froemter rats |
Wistar rats |
P Value |
||||
|---|---|---|---|---|---|---|---|
| High NaCl | Low NaCl | High NaCl | Low NaCl | Diet | Strain | ||
| Tubular fluid collections | |||||||
| Single-nephron glomerular filtration rate, nL/min | 63 | 40.2 ± 2.5 | 44.6 ± 2.1 | 0.2 | |||
| Proximal fluid reabsorption, nL/min | 63 | 16.9 ± 1.3† | 17.8 ± 1.1† | ||||
| TF/P inulin | 63 | 1.7 ± 0.1 | 1.9 ± 0.1 | 0.2 | |||
| Micropressures, mmHg | |||||||
| Glomerular capillary | 231 | 48.7 ± 0.5 | 53.9 ± 0.6 | 47.7 ± 0.8* | 54.2 ± 0.7* | <0.0001 | |
| Free-flow tubule | 294 | 13.2 ± 0.3 | 13.2 ± 0.3 | 13.5 ± 0.3 | 11.8 ± 0.4 | <0.01 | |
| Transcapillary pressure difference | 231 | 35.6 ± 0.6 | 40.7 ± 0.5 | 34.2 ± 0.7* | 42.4 ± 0.7* | <0.0001 | |
| Peritubular capillary | 175 | 12.9 ± 0.3 | 11.4 ± 0.4 | 14.9 ± 0.4 | 14.5 ± 0.4 | 0.02 | <0.001 |
Values are means ± SE. Significance was calculated from ANOVA from individual data with date of experiment as a factor in ANOVA. *Calculated from the sum of stop-flow pressure and plasma oncotic pressure. †Adjusted for covariate single-nephron glomerular filtration rate. Context is provided for the tubular fluid results in the discussion in Conditioning Effect of Dietary NaCl.
Pressures were measured in glomerular capillaries, Bowman’s space, peritubular capillaries, and proximal tubules under free-flow and stop-flow conditions. Baseline results are shown in Table 2. Free-flow pressure and pressure in Bowman’s space were interchangeable for nephrons with superficial glomeruli, where both pressure in Bowman’s space and free-flow pressure can be measured, so free-flow pressure results are reported. PGCs were computed for each PSF by adding to it the oncotic pressure of systemic plasma. The transcapillary pressure difference (ΔP) is the difference between PGC and free-flow tubular pressure. Group mean values for PGC and ΔP obtained by direct measurement in MWF rats differed by <1 mmHg from those computed from PSFs in Wistar rats. PGC and ΔP were 5–8 mmHg higher in rats fed the low-NaCl diet (P < 0.0001) regardless of whether they were obtained by direct capillary micropuncture or computed from PSF.
Average free-flow pressure was 0.8 mmHg higher in animals fed the high-NaCl diet (P < 0.01 where diet, rat strain, and individual experiments were factors in the ANOVA).
Peritubular capillary pressure was slightly higher in those fed high NaCl versus low NaCl and in Wistar versus MWF rats. There was a drift in peritubular capillary pressure between first and second periods, which was the same in rats that received placebo as in those that received SGLT2 blocker, which casts doubts on the physiological importance of the baseline differences, statistical significance notwithstanding.
Effects of Acute SGLT2 Blockade on GFR and Urinary Electrolytes
Acute SGLT2 blockade with ertugliflozin reduced GFR by ∼20% (P < 0.00001) while increasing urine flow rate by ∼3-fold (P < 0.00001) and increasing electrolyte excretion by ∼1.5-fold (P = 0.002; Fig. 1). These effects were not different between rat strains so results from the two rat strains are pooled for presentation. The effects of SGLT2 blockade on GFR, urine flow rate, and K excretion were not affected by dietary NaCl, whereas the increases in Na and Cl excretion during SGLT2 blockade were greater in rats fed the low-NaCl diet (P < 0.03). The concentration ratio of urinary electrolyte cations (Na + K) to anions (Cl) increased during SGLT2 blockade (P = 0.02), which implies that acute SGLT2 blockade either increased the excretion of a non-Cl anion or decreased excretion of a nonelectrolyte cation. The impact of ertugliflozin on urine flow rate was independent of dietary NaCl, rat strain, blood glucose, or the effect of SGLT2 blockade on GFR.
Figure 1.
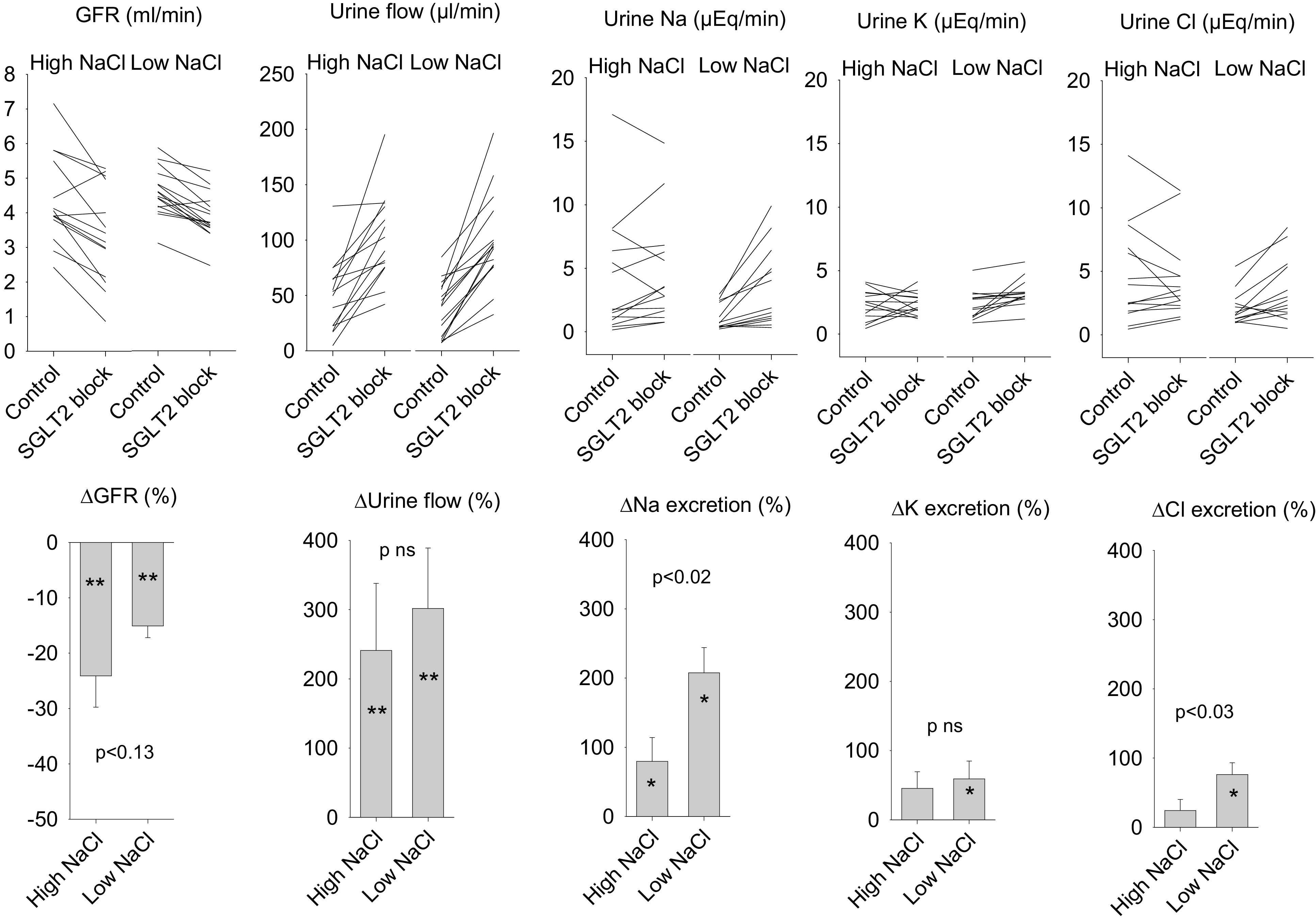
Effects of Na-glucose cotransporter (SGLT2) inhibitor on glomerular filtration rate (GFR), urine flow rate, and urine electrolyte excretion in diabetic rats fed high-NaCl or low-NaCl diets. The line graphs show values for individual animals. The bar graphs show group means ± SE for the effects of SGLT2 inhibitor. SGLT2 inhibitor reduced GFR and was diuretic and natriuretic (P < 0.001). P values in the bar graphs refer to the differential response to SGLT2 inhibitor between animals fed high-NaCl or low-NaCl diets. *P < 0.02 and **P < 0.001 for effect of SGLT2 blocker in animals on the designated diet.
Effect of Acute SGLT2 Blockade on SNGFR and Proximal Fluid Reabsorption
SNGFR and Jprox were analyzed for 135 late proximal tubular fluid collections. SNGFR tended to be less in animals fed high NaCl (39.7 ± 1.6 versus 43.8 ± 1.5 nL/min, P = 0.07) and during SGLT2 blockade (40.0 ± 1.6 versus 43.5 ± 1.6 nL/min, P = 0.1). Jprox declined markedly during acute SGLT2 blockade, regardless of dietary NaCl. This is illustrated in Fig. 2. As expected, SNGFR was the major determinant of Jprox. Therefore, to properly test for direct effects of SGLT2 blockade on Jprox, it is necessary to correct for variations in Jprox that result from differences in SNGFR. In the present case, inspection of the raw data revealed Jprox to be a concave upward function of SNGFR (Fig. 2). This implied that standard analysis of covariance, which assumes the effect of SNGFR to be linear, is not the best way to control for effects of SNGFR on Jprox. Instead, we modeled Jprox as an exponential function of SNGFR and then computed the differences between measured and predicted Jprox associated with the two diets and the two experimental periods and tested them for significance by ANOVA. The logistical best fit was Jprox = 5.08 + 0.00099 × SNGFR2.36733. The model attributed ∼75% of the raw variance in Jprox to differences in SNGFR (r2 = 0.87). The 95% confidence interval for the exponent extended from 1.95 to 2.79. The curve is concave upward if the exponent exceeds unity. Therefore, it is established that Jprox was a concave upward function of SNGFR for these data, with a high degree of certainty.
Figure 2.

Proximal fluid reabsorption (Jprox) versus single-nephron glomerular filtration rate (SNGFR) before and during Na-glucose cotransporter (SGLT2) blockade (n = 135 tubular fluid collections). Data were combined from rats fed high-NaCl and low-NaCl diets. The exponential curve, Jprox = 5.08 + 0.00099 × SNGFR2.36733 was a logistical fit to the raw data. The effect of SNGFR accounted for 75% of the total variance in Jprox. The effect of SGLT2 blockade accounted for 70% of the remaining variance in Jprox after controlling for SNGFR. The bar graph shows mean ± SE values for proximal reabsorption before and during SGLT2 blockade where Jprox′ is the value obtained for proximal reabsorption after adjusting for SNGFR.
SGLT2 blockade reduced Jprox by ∼30% (P < 0.00001). The effect of SGLT2 blockade explained ∼70% of the variance in Jprox that remained after accounting for the effect of SNGFR. The combined effects of SNGFR and SGLT2 blockade accounted for over 90% of the variance in Jprox. This indicates that SGLT2 exerted a major influence on proximal reabsorption. After adjusting for SNGFR, prescribed dietary NaCl was not a significant predictor of Jprox in these experiments. This is addressed further in the discussion.
Effects of Acute SGLT2 Blockade on Intrarenal Pressures
PGC was measured by direct micropuncture of 105 glomerular capillaries before SGLT2 blockade and in 128 glomerular capillaries during SGLT2 blockade. PGC averaged 6.7 mmHg less during SGLT2 blockade (P < 0.00001). The average decline in PGC during SGLT2 blockade was greater in rats fed the low-NaCl diet (9 vs. 5 mmHg, P = 0.01) in which PGC was ∼5 mmHg higher to begin with.
Average PGC drifted ∼3 mmHg higher during the second experimental period in animals given vehicle in lieu of the SGLT2 blocker (n = 62, P = 0.03).
PSF was measured in 80 tubules before SGLT2 blockade and 83 tubules during SGLT2 blockade. Unlike PGC, which was lower during SGLT2 blockade, average PSF was ∼1.5 mmHg higher during SGLT2 blockade than during the prior control period (P = 0.03). The effect of SGLT2 blocker on PSF was similar to the effect of vehicle on PGC.
There was a positive association between PGC and blood pressure between animals. After accounting for diet and SGLT2 blockade, differences in blood pressure accounted for ∼10% of the residual variance in PGC, and PGC increased by ∼0.2 mmHg per 1-mmHg increase in blood pressure (P < 0.001; Fig. 3). Within individual experiments and within individual PGC recordings, there was too little variability in blood pressure to evaluate the relationship between PGC and blood pressure over short timescales.
Figure 3.

Relationship of glomerular capillary pressure (PGC) to arterial blood pressure (BP). Each point represents PGC and BP averaged over a measurement. The median duration of the measurements was 99 s. Regression lines are shown with 95% confidence intervals. After controlling for dietary NaCl and Na-glucose cotransporter (SGLT2) blockade, most animals fit within a distribution of BP and PGC where a 1-mmHg increase in BP associated with an ∼0.25-mmHg increase in PGC. This analysis was sensitive to outliers such as those depicted with x’s in the bottom left graph.
The effects of acute SGLT2 blockade on ΔP measured in surface glomeruli for each animal are shown in Fig. 4 along with the relationship between ΔP and GFR and the relationship between changes in ΔP and changes in GFR associated with acute SGLT2 blockade. ΔP was a poor predictor of GFR overall (Pearson correlation, r = −0.14 for the control period and r = 0.18 during SGLT2 blockade). However, whereas SGLT2 blockade led to decreases in both GFR and ΔP, these changes in GFR and ΔP were inversely correlated (P = 0.04). In other words, a larger decrease in ΔP was associated with a smaller decrease in GFR (Fig. 4C). This has implications for the mechanism whereby TGF activation reduces PGC (vide infra).
Figure 4.
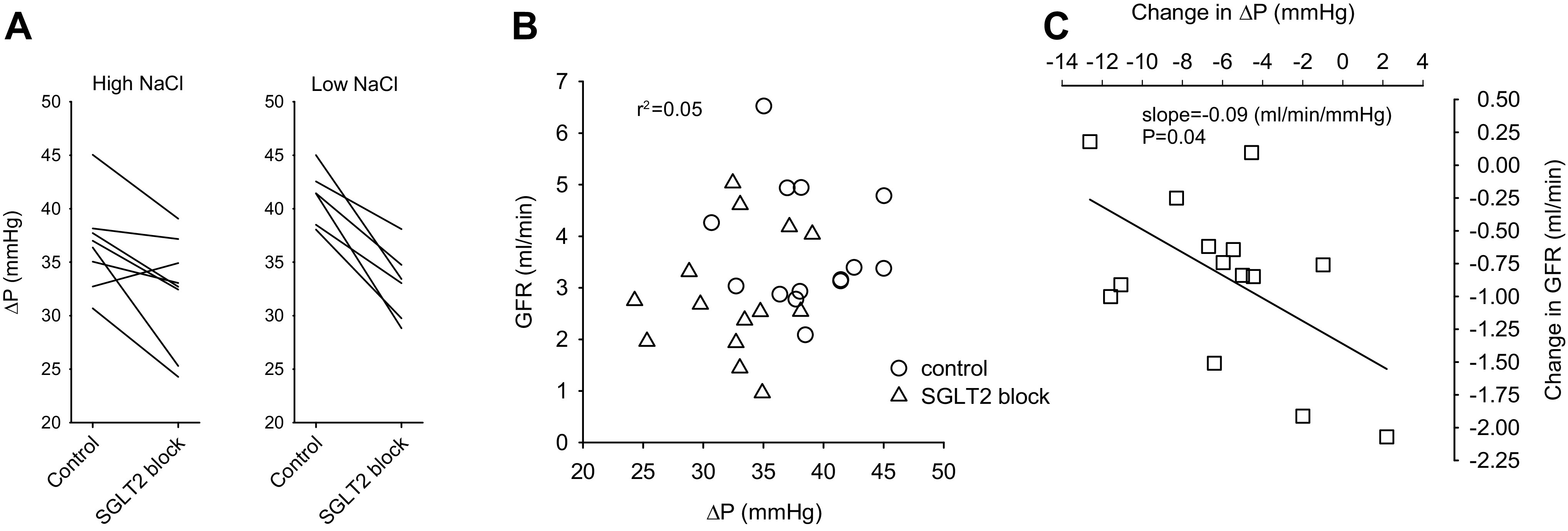
Effects of acute Na-glucose cotransporter (SGLT2) blockade on the glomerular filtration rate (GFR) and transcapillary pressure difference (ΔP) for individual animals where ΔP was measured directly in surface glomeruli. The values shown for ΔP were averaged from several glomeruli in that animal. A: ΔP before and during SGLT2 blockade shown separately for high-NaCl and low-NaCl diets. B: demonstration that GFR and ΔP were uncorrelated. Data for both diets were combined in this graph. C: corresponding changes in GFR and ΔP in response to SGLT2 blockade. SGLT2 blockade decreased both GFR and ΔP. However, greater decreases in ΔP were associated with lesser decreases in GFR (P = 0.04). This cannot be explained by isolated changes in preglomerular resistance (see discussion).
Per-animal effects of acute SGLT2 blockade on PSF are shown in Fig. 5. SGLT2 blockade did not reduce PSF, and changes in PSF during acute SGLT2 blockade did not correlate with changes in GFR.
Figure 5.
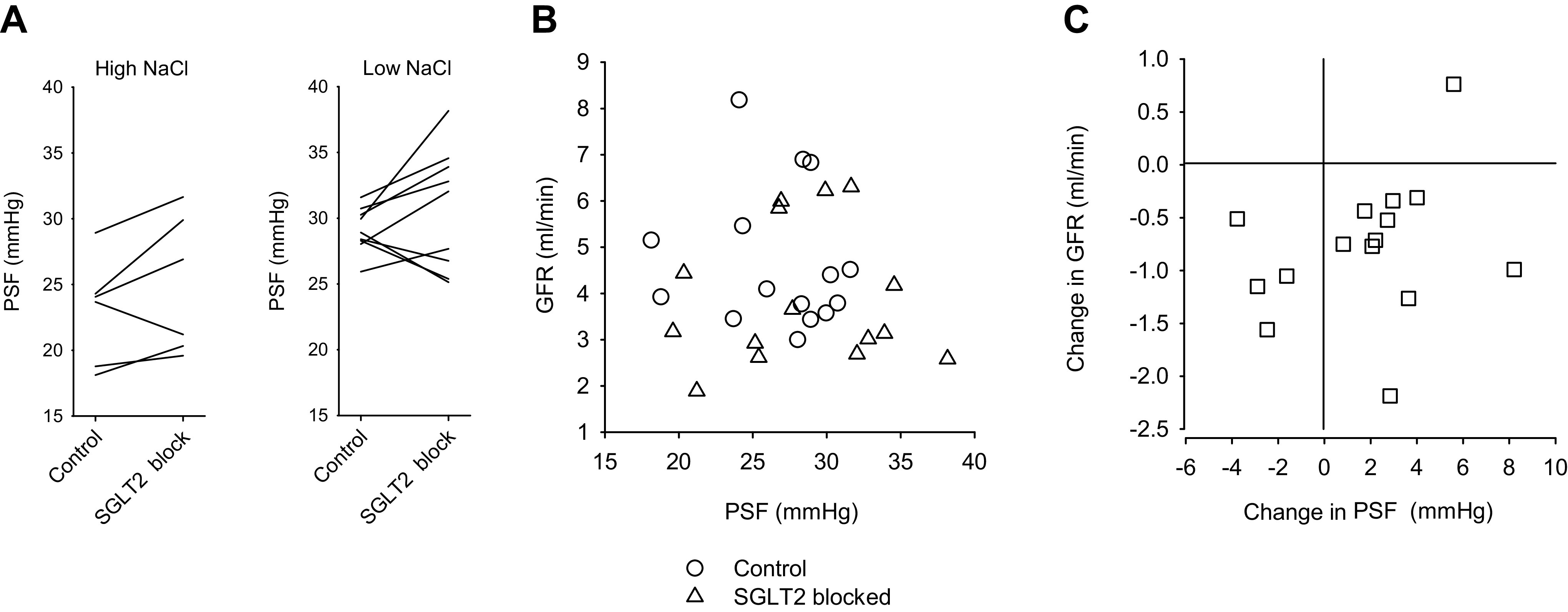
Effects of acute Na-glucose cotransporter (SGLT2) blockade on glomerular filtration rate (GFR) and tubular stop-flow pressure (PSF) for individual animals. Values shown for PSF were averaged from several nephrons in that animal. A: PSF before and during SGLT2 blockade shown separately for high-NaCl and low-NaCl diets. B: demonstration that GFR and PSF were uncorrelated. Data for both diets were combined in this graph. C: corresponding changes in GFR and PSF in response to SGLT2 blockade. SGLT2 blockade reduced GFR but did not reduce PSF. The effects of acute SGLT2 blockade on GFR and PSF were uncorrelated. The PGC-lowering effect of SGLT2 inhibitor shown in Fig. 4 was not reflected in the PSF response. From the difference between ΔP and PSF responses, we infer that SGLT2 blockade reduces ΔP by eliciting a tubuloglomerular feedback response.
DISCUSSION
SGLT2 Glomerular Hemodynamics and TGF
Whole kidney GFR emerges from glomerular hemodynamics, but glomerular hemodynamics cannot be deduced from whole kidney GFR. The primary goal of these experiments was to test for effects of acute SGLT2 blockade on intraglomerular pressures, which can only be determined using micropuncture. There is interest and published speculation that salutary effects of SGLT2 blockade on the diabetic kidney are mediated through lower PGC. However, the effect of SGLT2 blockade on PGC had not been confirmed with measurements until now. The main new finding was a major decline in PGC (and ΔP) during acute SGLT2 inhibition when PGC was measured directly but not when PGC (or ΔP) was estimated by the tubular stop-flow method. A crucial difference between PGC as measured directly versus PGC obtained by the stop-flow method is that the stop-flow method renders TGF inoperative. Hence, we infer that the effect of acute SGLT2 blockade on PGC is mediated through TGF. This is consistent with expectation based on two previous micropuncture studies in diabetic rats where SGLT1/2 blockade applied to individual nephrons (11) or SGLT2 blockade applied systemically (10) depressed Jprox, increased delivery of Na and Cl to the macula densa, activated TGF, and reduced SNGFR. All existing micropuncture data obtained during SGLT2 blockade are consistent with the theory that SGLT2 inhibitors reduce GFR by activating TGF.
Implications for the TGF Effector Mechanism
An unexpected finding in the current study was that, whereas SGLT2 blockade consistently reduced both ΔP and GFR, greater declines in ΔP were associated with lesser declines in GFR. This has implications for the TGF effector mechanism, which is a broader topic in kidney physiology that we now discuss.
It is not disputed that TGF impinges on the afferent arteriole. However, if it were true that the entire TGF response occurs in the afferent arteriole, the existence of a TGF-mediated decrease in GFR would imply a corresponding decrease in PGC, which could be determined mathematically. Therefore, accepting that TGF mediates the effect of SGLT2 blockade on GFR would render PGC measurements superfluous. The impression that TGF-mediated changes in GFR and ΔP imply each other is understandable because micropuncture experiments are usually designed to measure the effect of an imposed stimulus to the macula densa on SNGFR or on PSF, but not on both at once. However, when the effort was made to simultaneously record changes in SNGFR and PGC within the physiological range of TGF inputs, SNGFR was found to be most sensitive over a lower range of inputs than was PGC (18). This could not be explained by a TGF mechanism that only acts on the afferent arteriole. We extended this finding using perturbation analysis and showed that the SNGFR response is sensitive to inputs encompassing the ambient tubular flow and that PGC is most sensitive to perturbations over a higher range of inputs where the SNGFR response is saturated (34). This implied that typical TGF responses from the shoulder of the GFR curve through the TGF operating point involve parallel changes in resistances upstream and downstream of the glomerulus whereas responses from the elbow of the GFR curve and beyond include decreased resistance in the efferent arteriole. Reducing resistance to flow along the efferent arteriole allows SNGFR to remain stable because the effect of declining PGC is offset by increasing glomerular blood flow. It has been noted that both upstream vasoconstriction and downstream vasorelaxation caused by TGF could be explained by adenosine derived from the macula densa acting on constrictor A1 and dilatory A2 adenosine receptors, which are preferentially expressed upstream and downstream, respectively (35, 36). The transition point to efferent dilation in such a system could be flexible and sensitive to manipulation by changing the differential expression of A1 and A2 receptors or by various modulators of the segmental responses. In fact, there are situations where the SNGFR response to a TGF stimulus remains intact while the PSF response disappears altogether (37, 38). The current data provide a new example of a TGF response that cannot be fully accounted for by changes in resistance upstream of the glomerulus. In this way, the current findings complement prior observations on the nuances of the TGF response. These concepts are shown graphically in Fig. 6.
Figure 6.

Glomerular hemodynamic effects of Na-glucose cotransporter (SGLT2) inhibitor superimposed on a model of tubuloglomerular feedback (TGF), which incorporates effects on pre- and postglomerular resistances. Idealized TGF curves are shown for glomerular filtration rate (GFR) and PGC. The PGC curve lies to the right of the GFR curve owing to differences in the way that pre- and postglomerular resistances react across the range of inputs, as shown in the bottom portion of the figure. Within a group of nephrons, the position of operating points along the respective TGF curves will form a distribution. The figure shows the effects on GFR and PGC when identical increases in macula densa delivery are imposed by SGLT2 inhibitor for two nephrons drawn from this distribution (nephrons A and B). For nephron A, the TGF response to SGLT2 blocker included a large decrease in GFR and small decrease in PGC. For nephron B, there was a smaller decline in GFR and a larger decline in PGC. The inverse relationship between decreases in whole kidney GFR and decreases in average PGC for that animal is expected if animals with the smaller decreases in GFR yet larger decreases in PGC had more nephrons operating near the elbow of their respective TGF curves and vice versa.
PGC in Diabetes
The current data also invite review of the role played by PGC in diabetes, which is an older subject. It is frequently asserted that diabetic hyperfiltration implies glomerular capillary hypertension. This impression emerged from iconic micropuncture studies published in the 1980s where certain diabetic rats manifest markedly elevated PGC, which could be normalized by reducing dietary protein or by blocking the formation of angiotensin II (24, 30, 39). However, those micropuncture studies associating diabetes with glomerular capillary hypertension are outnumbered by studies in which PGC was normal in diabetes (24–30, 39–42). Furthermore, maneuvers that reduced or prevented glomerular capillary hypertension in those iconic studies did not reverse or prevent hyperfiltration (24, 30, 39). Hence, the assertion that diabetic hyperfiltration implies glomerular capillary hypertension is not supported by the majority of micropuncture data. Differences in ΔP and GFR between diabetic and nondiabetic controls for 20 published micropuncture series are shown in Fig. 7. Diabetic hyperfiltration was present in 13 of 20 series across 8 published papers (24–30, 39). Among the 13 groups of hyperfiltering diabetic rats, mean ΔP was elevated in 5 groups across 4 papers (24, 30, 39, 43) and normal in the other 8 groups. For 7 groups of diabetic rats without hyperfiltration across 6 papers (30, 40–42, 44, 45), ΔP was elevated in 3 groups (41, 44, 45) and normal in the other 4 groups. Among these published series, diabetes conferred a 40% probability of glomerular capillary hypertension, which was no more likely to occur when there was hyperfiltration. The current study was not designed to study the effect diabetes on PGC, but the baseline PGC of ∼50 mmHg in the current study is typical among nondiabetic rats we have studied for other reasons in the past (28, 32, 46, 47). Although glomerular capillary hypertension may not be the norm in diabetes, there are only two validated treatments for diabetic nephropathy (renin-angiotensin-aldosterone system blockers and SGLT2 inhibitors), and the most obvious thing renin-angiotensin-aldosterone system blockers and SGLT2 inhibitors have in common is that they stereotypically reduce PGC.
Figure 7.

Ratio of diabetic to control glomerular filtration rate (GFR) and transcapillary pressure difference (ΔP) for each of 20 published series where this comparison was possible. Ratios are shown instead of absolute changes because some studies reported GFR and others reported single-nephron GFR (SNGFR). Confidence intervals for this figure are SEs computed from reported variances and sample sizes and the following equation for computing variance of a ratio: The blue curve was generated by the Brenner-Deen model and depicts the relative changes in GFR and ΔP where renal plasma flow and ultrafiltration coefficient are held constant starting from the base case where ΔP = 35 mmHg, plasma flow = 150 nL/min, plasma protein concentration = 5.6 g/dL, and ultrafiltration coefficient = 0.06 nL/s/mmHg. The cases shown in red are series in which effects of diabetes on GFR cannot be explained by associated changes in ΔP.
Conditioning Effect of Dietary NaCl
The current experiments were performed in diabetic rats fed low-NaCl or high-NaCl diets. This allowed us to gauge the impact of a variable commonly linked to hemodynamics (dietary NaCl) on the glomerular hemodynamic response to SGLT2 blockade. We found that the decrease in PGC, the increases in Na and Cl excretion, and the increase in urinary anion gap during SGLT2 blockade were all greater in animals fed the low-NaCl diet. It has recently come to light that SGLT2 and Na/H exchanger isoform 3 (NHE3) reinforce each other’s actions in the proximal tubule (48–50). Because the basal activity of NHE3 is expected to increase on a low-NaCl diet, the greater impacts of SGLT2 blockade on urinary electrolytes and glomerular hemodynamics in animals given low-NaCl diet could be manifestations of a given fraction of NHE3 activity being susceptible to the SGLT2 blocker.
According to the foregoing argument, dietary NaCl should have predicted Jprox. However, the 10% lower Jprox in rats fed high NaCl was not statistically significant. We have previously published micropuncture data from experiments that were more carefully designed to examine the effect of dietary NaCl on Jprox. In those experiments, Jprox was reduced by high-NaCl diet, leading to increased TGF activation and a paradoxical effect of dietary NaCl on GFR (51). In those experiments, we perfused Henle’s loop during micropuncture, which made TGF a tool for manipulating SNGFR so we could interpolate to Jprox at the natural TGF operating point. In the present case, all collections were done in the absence of a TGF signal. Due to intrinsic properties of glomerulotubular balance, an increase in SNGFR should cause Jprox to increase whereas fractional reabsorption should decline (31). Thus, the simple approach of making all Jprox measurements in the absence of a TGF signal will overestimate what fractional reabsorption would be at the TGF operating point to an unknown degree. The simple approach was required to leave enough time to measure Jprox and micropressures in the same experiment, and it sufficed to prove that ertugliflozin is a potent proximal diuretic, which was the main reason for measuring Jprox.
Measuring PGC on low-NaCl and high-NaCl diets provided an opportunity to examine the role of PGC in the “salt paradox,” which is a term coined in reference to a reciprocal effect of dietary NaCl on GFR in diabetes. The salt paradox has been confirmed in human and experimental diabetes (16, 52–55). The salt paradox could have implications for dietary prescribing in diabetes because the usual admonition to minimize dietary NaCl could be counterproductive in diabetes if low-NaCl diet exacerbates hyperfiltration or raises PGC. The potential for dietary salt to mitigate hyperfiltration and to reduce PGC provide a convenient explanation for the striking association of long-term renal survival with higher NaCl intakes in the FinnDiane study (56, 57).
Heterogeneity in GFR
One factor that obscured effects that dietary NaCl may have on GFR and Jprox in these experiments was heterogeneity in baseline GFR, especially among those prescribed high NaCl (see Fig. 1). Heterogeneity is generally undesirable in physiology experiments because it reduces statistical power to detect differences between groups. In the two-period design where each animal served as its own control, we were able to quantify various effects of acute SGLT2 blockade within narrow limits, despite a wide range of baseline GFR. The accident of variable baseline GFR allowed us to show that acute SGLT2 blockade reduces ΔP and GFR in both hyperfiltering and nonhyperfiltering diabetic kidneys. Another feature that was brought out by high variability in baseline GFR is the concave upward relationship between Jprox and SNGFR (see Fig. 2). As previously mentioned, this is contrary to what is expected when the relationship between Jprox and SNGFR is dominated by glomerulotubular balance. However, a concave upward relationship between Jprox and SNGFR is expected if the relationship is dominated by primary differences in Jprox, which affect GFR by signaling through TGF. The curve is concave upward because the TGF response is saturable. This is the same phenomenon reported by Pruijm et al., who compared hyperfiltering to nonhyperfiltering humans with early diabetes and discovered that those with higher GFR had lower lithium clearance (13). These findings are consistent with the “tubular hypothesis” of glomerular filtration in diabetes (58, 59).
The current experiments were done in male rats. We originally demonstrated the salt paradox in female (54) and male (55) rats. Adjusting for sex did not affect the results of Pruijm et al., who examined the tubular hypothesis in humans (13).
CONCLUSIONS
Acute SGLT2 inhibition reduces PGC in moderately hyperglycemic rats by way of TGF. This effect persists on high- or low-NaCl diets and does not require background hyperfiltration or background glomerular capillary hypertension. The TGF response elicited by acute SGLT2 inhibition includes a variable mixture of preglomerular vasoconstriction and postglomerular vasorelaxation such that the decrease in PGC can exceed expectation based on the associated decrease in GFR.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK112042, University of Alabama at Birmingham/University of California at San Diego O'Brien Center for Acute Kidney Injury Research Grant P30DK079337, and an Investigator Initiated Grant from Merck.
DISCLOSURES
S.C.T. received non-salary funds by investigator-initiated grant from Merck. V.V. has grant funding from Astra-Zeneca, Boehringer Ingelheim, and Janssen.
AUTHOR CONTRIBUTIONS
S.C.T. and V.V. conceived and designed research; S.C.T. performed experiments; S.C.T. analyzed data; S.C.T. and V.V. interpreted results of experiments; S.C.T. and V.V. prepared figures; S.C.T. drafted manuscript; S.C.T. and V.V. edited and revised manuscript; S.C.T. and V.V. approved final version of manuscript.
ACKNOWLEDGEMENTS
Technical assistance was provided by Ser Khang and Hai Pham.
REFERENCES
- 1.Neal B, Perkovic V, Mahaffey KW, de ZD, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377: 644–657, 2017. doi: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 2.Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, Cannon CP, Capuano G, Chu PL, de Zeeuw D, Greene T, Levin A, Pollock C, Wheeler DC, Yavin Y, Zhang H, Zinman B, Meininger G, Brenner BM, Mahaffey KW. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 380: 2295–2306, 2019. doi: 10.1056/NEJMoa1811744. [DOI] [PubMed] [Google Scholar]
- 3.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von EM, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 375: 323–334, 2016. doi: 10.1056/NEJMoa1515920. [DOI] [PubMed] [Google Scholar]
- 4.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE; EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373: 2117–2128, 2015. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 5.Brenner BM, Cooper ME, de ZD, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 6.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329: 1456–1462, 1993. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 7.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345: 851–860, 2001. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 8.Mogensen CE. Early glomerular hyperfiltration in insulin-dependent diabetics and late nephropathy. Scand J Clin Lab Invest 46: 201–206, 1986. doi: 10.3109/00365518609083660. [DOI] [PubMed] [Google Scholar]
- 9.Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, Fagan NM, Woerle HJ, Johansen OE, Broedl UC, von EM. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 129: 587–597, 2014. doi: 10.1161/CIRCULATIONAHA.113.005081. [DOI] [PubMed] [Google Scholar]
- 10.Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, Singh P. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol 302: R75–R83, 2012. doi: 10.1152/ajpregu.00357.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 10: 2569–2576, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Pollock CA, Lawrence JR, Field MJ. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am J Physiol Renal Physiol 260: F946–F952, 1991. doi: 10.1152/ajprenal.1991.260.6.F946. [DOI] [PubMed] [Google Scholar]
- 13.Pruijm M, Wuerzner G, Maillard M, Bovet P, Renaud C, Bochud M, Burnier M. Glomerular hyperfiltration and increased proximal sodium reabsorption in subjects with type 2 diabetes or impaired fasting glucose in a population of the African region. Nephrol Dial Transplant 25: 2225–2231, 2010. doi: 10.1093/ndt/gfq008. [DOI] [PubMed] [Google Scholar]
- 14.Vallon V. The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 300: R1009–R1022, 2011. doi: 10.1152/ajpregu.00809.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vallon V, Blantz RC, Thomson S. Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am J Physiol Renal Physiol 269: F876–F883, 1995. doi: 10.1152/ajprenal.1995.269.6.F876. [DOI] [PubMed] [Google Scholar]
- 16.Vallon V, Schroth J, Satriano J, Blantz RC, Thomson SC, Rieg T. Adenosine A(1) receptors determine glomerular hyperfiltration and the salt paradox in early streptozotocin diabetes mellitus. Nephron Physiol 111: p30–38, 2009. doi: 10.1159/000208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navar LG, Maddox DA, Munger KA. The renal circulations and glomerular filtration. In: Brenner & Rector’s The Kidney. Elsevier, 2020, p. 80–112. [Google Scholar]
- 18.Schnermann J, Briggs JP. Single nephron comparison of the effect of loop of Henle flow on filtration rate and pressure in control and angiotensin II-infused rats. Miner Electrolyte Metab 15: 103–107, 1989. [PubMed] [Google Scholar]
- 19.Barutta F, Bernardi S, Gargiulo G, Durazzo M, Gruden G. SGLT2 inhibition to address the unmet needs in diabetic nephropathy. Diabetes Metab Res Rev 35: e3171, 2019. doi: 10.1002/dmrr.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherney D, Lund SS, Perkins BA, Groop PH, Cooper ME, Kaspers S, Pfarr E, Woerle HJ, von Eynatten M. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia 59: 1860–1870, 2016. doi: 10.1007/s00125-016-4008-2. [DOI] [PubMed] [Google Scholar]
- 21.Kimura G. Importance of inhibiting sodium-glucose cotransporter and its compelling indication in type 2 diabetes: pathophysiological hypothesis. J Am Soc Hypertens 10: 271–278, 2016. doi: 10.1016/j.jash.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Perico N, Ruggenenti P, Remuzzi G. ACE and SGLT2 inhibitors: the future for non-diabetic and diabetic proteinuric renal disease. Curr Opin Pharmacol 33: 34–40, 2017. doi: 10.1016/j.coph.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Vallianou NG, Geladari E, Kazazis CE. SGLT-2 inhibitors: Their pleiotropic properties. Diabetes Metab Syndr 11: 311–315, 2017. doi: 10.1016/j.dsx.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Hostetter TH, Troy JL, Brenner BM. Glomerular hemodynamics in experimental diabetes mellitus. Kidney Int 19: 410–415, 1981. doi: 10.1038/ki.1981.33. [DOI] [PubMed] [Google Scholar]
- 25.Jensen PK, Christiansen JS, Steven K, Parving HH. Renal function in streptozotocin-diabetic rats. Diabetologia 21: 409–414, 1981. [PubMed] [Google Scholar]
- 26.Michels LD, Davidman M, Keane WF. Determinants of glomerular filtration and plasma flow in experimental diabetic rats. J Lab Clin Med 98: 869–885, 1981. [PubMed] [Google Scholar]
- 27.O'Donnell MP, Kasiske BL, Daniels FX, Keane WF. Effects of nephron loss on glomerular hemodynamics and morphology in diabetic rats. Diabetes 35: 1011–1015, 1986. doi: 10.2337/diab.35.9.1011. [DOI] [PubMed] [Google Scholar]
- 28.Slomowitz LA, Peterson OW, Thomson SC. Converting enzyme inhibition and the glomerular hemodynamic response to glycine in diabetic rats. J Am Soc Nephrol 10: 1447–1454, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Tucker BJ, Anderson CM, Thies RS, Collins RC, Blantz RC. Glomerular hemodynamic alterations during acute hyperinsulinemia in normal and diabetic rats. Kidney Int 42: 1160–1168, 1992. doi: 10.1038/ki.1992.400. [DOI] [PubMed] [Google Scholar]
- 30.Zatz R, Meyer TW, Rennke HG, Brenner BM. Predominance of hemodynamic rather than metabolic factors in the pathogenesis of diabetic glomerulopathy. Proc Natl Acad Sci USA 82: 5963–5967, 1985. doi: 10.1073/pnas.82.17.5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomson SC, Deng A, Bao D, Satriano J, Blantz RC, Vallon V. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest 107: 217–224, 2001. doi: 10.1172/JCI10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomson SC, Tucker BJ, Gabbai F, Blantz RC. Functional effects on glomerular hemodynamics of short-term chronic cyclosporine in male rats. J Clin Invest 83: 960–969, 1989. doi: 10.1172/JCI113982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landis EM, Pappenheimer JR. Exchange of substances through the capillary walls. In: Handbook of Physiology, edited by Orloff J, Berliner R.W.. Washington DC: American Physiological Society, 1964, pp 961–1034. [Google Scholar]
- 34.Thomson S, Vallon V, Blantz RC. Asymmetry of tubuloglomerular feedback effector mechanism with respect to ambient tubular flow. Am J Physiol Renal Physiol 271: F1123–F1130, 1996. doi: 10.1152/ajprenal.1996.271.6.F1123. [DOI] [PubMed] [Google Scholar]
- 35.Ren Y, Garvin JL, Carretero OA. Efferent arteriole tubuloglomerular feedback in the renal nephron. Kidney Int 59: 222–229, 2001. doi: 10.1046/j.1523-1755.2001.00482.x. [DOI] [PubMed] [Google Scholar]
- 36.Vallon V, Mühlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev 86: 901–940, 2006. doi: 10.1152/physrev.00031.2005. [DOI] [PubMed] [Google Scholar]
- 37.Huang WC, Bell PD, Harvey D, Mitchell KD, Navar LG. Angiotensin influences on tubuloglomerular feedback mechanism in hypertensive rats. Kidney Int 34: 631–637, 1988. doi: 10.1038/ki.1988.227. [DOI] [PubMed] [Google Scholar]
- 38.Persson AE, Gushwa LC, Blantz RC. Feedback pressure-flow responses in normal and angiotensin-prostaglandin-blocked rats. Am J Physiol Renal Physiol 247: F925–931, 1984. doi: 10.1152/ajprenal.1984.247.6.F925. [DOI] [PubMed] [Google Scholar]
- 39.Zatz R, Dunn BR, Meyer TW, Anderson S, Rennke HG, Brenner BM. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest 77: 1925–1930, 1986. doi: 10.1172/JCI112521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Nicola L, Blantz RC, Gabbai FB. Renal functional reserve in the early stage of experimental diabetes. Diabetes 41: 267–273, 1992. doi: 10.2337/diabetes.41.3.267. [DOI] [PubMed] [Google Scholar]
- 41.Jensen PK, Christiansen JS, Steven K, Parving HH. Strict metabolic control and renal function in the streptozotocin diabetic rat. Kidney Int 31: 47–51, 1987. doi: 10.1038/ki.1987.7. [DOI] [PubMed] [Google Scholar]
- 42.Michels LD, O'Donnell MP, Keane WF. Glomerular hemodynamic and structural correlations in long-term experimental diabetic rats. J Lab Clin Med 103: 840–847, 1984. [PubMed] [Google Scholar]
- 43.Bank N, Klose R, Aynedjian HS, Nguyen D, Sablay LB. Evidence against increased glomerular pressure initiating diabetic nephropathy. Kidney Int 31: 898–905, 1987. doi: 10.1038/ki.1987.83. [DOI] [PubMed] [Google Scholar]
- 44.Jensen PK, Steven K, Blaehr H, Christiansen JS, Parving HH. Effects of indomethacin on glomerular hemodynamics in experimental diabetes. Kidney Int 29: 490–495, 1986. doi: 10.1038/ki.1986.26. [DOI] [PubMed] [Google Scholar]
- 45.Remuzzi A, Fassi A, Sangalli F, Malanchini B, Mohamed EI, Bertani T, Remuzzi G. Prevention of renal injury in diabetic MWF rats by angiotensin II antagonism. Exp Nephrol 6: 28–38, 1998. doi: 10.1159/000020502. [DOI] [PubMed] [Google Scholar]
- 46.Thomson SC, Gabbai FB, Tucker BJ, Blantz RC. Interaction between alpha 2-adrenergic and angiotensin II systems in the control of glomerular hemodynamics as assessed by renal micropuncture in the rat. J Clin Invest 90: 604–611, 1992. doi: 10.1172/JCI115899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomson SC, Tucker BJ, Gabbai FB, Blantz RC. Glomerular hemodynamics and alpha 2-adrenoreceptor stimulation: the role of renal nerves. Am J Physiol Renal Physiol 258: F21–F27, 1990. doi: 10.1152/ajprenal.1990.258.1.F21. [DOI] [PubMed] [Google Scholar]
- 48.Onishi A, Fu Y, Darshi M, Crespo-Masip M, Huang W, Song P, Patel R, Kim YC, Nespoux J, Freeman B, Soleimani M, Thomson SC, Sharma K, Vallon V. Effect of renal tubule-specific knockdown of the Na(+)/H(+) exchanger NHE3 in Akita diabetic mice. Am J Physiol Renal Physiol 317: F419–F434, 2019. doi: 10.1152/ajprenal.00497.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, Song P, Freeman B, Kim YC, Soleimani M, Sharma K, Thomson SC, Vallon V. A role for the tubular Na(+)-H(+)-exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 319: F712–F728, 2020. doi: 10.1152/ajprenal.00264.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 25: 2028–2039, 2014. doi: 10.1681/ASN.2013060588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vallon V, Huang DY, Deng A, Richter K, Blantz RC, Thomson S. Salt-sensitivity of proximal reabsorption alters macula densa salt and explains the paradoxical effect of dietary salt on glomerular filtration rate in diabetes mellitus. J Am Soc Nephrol 13: 1865–1871, 2002. doi: 10.1097/01.asn.0000016441.41118.57. [DOI] [PubMed] [Google Scholar]
- 52.Lau C, Sudbury I, Thomson M, Howard PL, Magil AB, Cupples WA. Salt-resistant blood pressure and salt-sensitive renal autoregulation in chronic streptozotocin diabetes. Am J Physiol Regul Integr Comp Physiol 296: R1761–R1770, 2009. doi: 10.1152/ajpregu.90731.2008. [DOI] [PubMed] [Google Scholar]
- 53.Miller JA. Renal responses to sodium restriction in patients with early diabetes mellitus. J Am Soc Nephrol 8: 749–755, 1997. [DOI] [PubMed] [Google Scholar]
- 54.Vallon V, Kirschenmann D, Wead LM, Lortie MJ, Satriano J, Blantz RC, Thomson SC. Effect of chronic salt loading on kidney function in early and established diabetes mellitus in rats. J Lab Clin Med 130: 76–82, 1997. doi: 10.1016/s0022-2143(97)90061-5. [DOI] [PubMed] [Google Scholar]
- 55.Vallon V, Wead LM, Blantz RC. Renal hemodynamics and plasma and kidney angiotensin II in established diabetes mellitus in rats: effect of sodium and salt restriction. J Am Soc Nephrol 5: 1761–1767, 1995. [DOI] [PubMed] [Google Scholar]
- 56.Thomas MC, Moran J, Forsblom C, Harjutsalo V, Thorn L, Ahola A, Wadén J, Tolonen N, Saraheimo M, Gordin D, Groop PH, FinnDiane Study Group. The association between dietary sodium intake, ESRD, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 34: 861–866, 2011. doi: 10.2337/dc10-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vallon V, Thomson SC. Anomalous role for dietary salt in diabetes mellitus? Nat Rev Endocrinol 7: 377–378, 2011. doi: 10.1038/nrendo.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomson SC, Vallon V, Blantz RC. Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am J Physiol Renal Physiol 286: F8–15, 2004. doi: 10.1152/ajprenal.00208.2003. [DOI] [PubMed] [Google Scholar]
- 59.Vallon V, Thomson SC. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat Rev Nephrol 16: 317–336, 2020. doi: 10.1038/s41581-020-0256-y. [DOI] [PMC free article] [PubMed] [Google Scholar]