

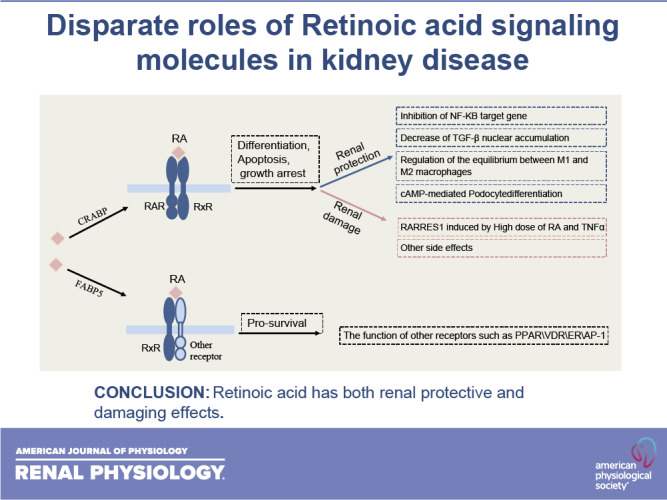
Keywords: glomerular disease, podocytes, retinoic acid, retinoic acid receptor responder protein 1, retinoic acid receptors
Abstract
Retinoid acid (RA) is synthesized mainly in the liver and has multiple functions in development, cell differentiation and proliferation, and regulation of inflammation. RA has been used to treat multiple diseases, such as cancer and skin disorders. The kidney is a major organ for RA metabolism, which is altered in the diseased condition. RA is known to have renal-protective effects in multiple animal models of kidney disease. RA has been shown to ameliorate podocyte injury through induction of expression of differentiation markers and regeneration of podocytes from its progenitor cells in animal models of kidney disease. The effects of RA in podocytes are mediated mainly by activation of the cAMP/PKA pathway via RA receptor-α (RARα) and activation of its downstream transcription factor, Kruppel-like factor 15. Screening of RA signaling molecules in human kidney disease has revealed RAR responder protein 1 (RARRES1) as a risk gene for glomerular disease progression. RARRES1, a podocyte-specific growth arrest gene, is regulated by high doses of both RA and TNF-α. Mechanistically, RARRES1 is cleaved by matrix metalloproteinases to generate soluble RARRES1, which then induces podocyte apoptosis through interaction with intracellular RIO kinase 1. Therefore, a high dose of RA may induce podocyte toxicity through upregulation of RARRES1. Based on the current findings, to avoid potential side effects, we propose three strategies to develop future therapies of RA for glomerular disease: 1) develop RARα- and Kruppel-like factor 15-specific agonists, 2) use the combination of a low dose of RAR-α agonist with phosphodiesterase 4 inhibitors, and 3) use a combination of RARα agonist with RARRES1 inhibitors.
NEW & NOTEWORTHY Retinoic acid (RA) exerts pleotropic cellular effects, including induction of cell differentiation while inhibiting proliferation and inflammation. These effects are mediated by both RA responsive element-dependent or -independent pathways. In kidneys, RA confers renoprotection by signaling through podocyte RA receptor (RAR)α and activation of cAMP/PKA/Kruppel-like factor 15 pathway to promote podocyte differentiation. Nevertheless, in kidney disease settings, RA can also promote podocyte apoptosis and loss through downstream expression of RAR responder protein 1, a recently described risk factor for glomerular disease progression. These disparate roles of RA underscore the complexity of its effects in kidney homeostasis and disease, and a need to target specific RA-mediated pathways for effective therapeutic treatments against kidney disease progression.
INTRODUCTION
Retinoid acid (RA), the metabolite of vitamin A, plays an essential role in organ development such as in the kidney (1), exerts potent effects on stem cell differentiation (2), and inhibits inflammation and cell proliferation (3). RA has been shown to have preventive and therapeutic effects in various experimental models of kidney diseases, such as lupus nephritis, acute and chronic Thy1.1 nephritis, acute and chronic antiglomerular basement membrane nephritis, and human immunodeficiency virus (HIV)-associated nephropathy (3–5). However, the role of RA in human kidney disease remains unclear, and clinical trials are lacking. The clinical translation of RA as a therapy for kidney disease has been hindered by its significant toxicity, including liver toxicity, hyperlipidemia, central nervous system abnormalities, reduced spermiogenesis, myalgia, arthralgias, and mucocutaneous side effects (6). Therefore, it is likely that RA has diverse or opposite effects by activating different downstream signaling pathways or tissue-specific pathways. A better understanding of its downstream signaling pathways and the target genes will help us to develop more specific therapies for kidney disease. In this review, we will address this important question by providing an overview of RA effects but focusing on the disparate roles of RA signaling molecules in kidney disease.
RA METABOLISM
Vitamin A, obtained from the diet, is initially absorbed by intestinal epithelium cells in the form of its precursors, mainly β-carotene. The precursors are then esterified into retinyl esters by lecithin retinol acyltransferase (LRAT) and incorporated into chylomicrons for absorption into the general circulation. The circulating chylomicrons are captured by hepatocytes, and carotene is hydrolyzed to retinol. Retinol binds to retinol-binding protein (RBP) in the bloodstream and is then transported by the retinoic acid 6 (STRA6) receptor from extracellular to the cytosol (1). The conversion of retinol into its active form, RA, requires two oxidation steps: 1) reversible conversion from retinol to retinaldehyde mediated by alcohol dehydrogenases and short-chain dehydrogenases and 2) irreversible conversion from retinal to RA catalyzed by retinaldehyde dehydrogenases (RALDHs), which are the key enzymes of RA synthesis (5). The clearance of RA is mediated predominantly by cytochrome P-450 family 26 enzymes (CYP26). The complex is filtered by the kidneys. The filtered retinol-RBP4 complex is reabsorbed in the proximal tubule cells by megalin (7). RBP4 is mainly synthesized in the liver and catabolized in the kidneys after glomerular filtration. RBP4 is complexed with transthyretin, a homotetrameric protein with a molecular weight of ∼55 kDa, to prevent renal loss (8). Cells in the intestinal epithelium can synthesize RA from retinol after metabolizing vitamin A into β-carotene, which can be directly released into the intestinal mucosa (Fig. 1) (9).
Figure 1.

Retinoic acid (RA) metabolism and signaling. A: vitamin A obtained from diet is metabolized into β-carotene, which is absorbed by intestinal epithelial cells and esterified into retinyl esters by the enzyme lecithin retinol acyltransferase (LRAT). B: retinyl esters are carried together with chylomicrons in the bloodstream; they are then captured by hepatocytes and hydrolyzed into retinol. C: retinol binds to retinol-binding protein (RBP) in the bloodstream and transported into the cytosol via the stimulated by retinoic acid 6 (STRA6) receptor. D: in the cytosol, RA is generated from retinol by two sequential reactions. Retinol is first oxidized into retinal by alcohol dehydrogenase (ADH) and then retinal is oxidized by the enzyme retinal dehydrogenase (RALDH) to generate RA. E: the function of RA depends on the type of RBP that RA binds. If the fatty acid-binding protein 5 (FABP5)/cellular retinol-binding protein II (CRABPII) ratio is low, RA binds mostly to CRABPII, which delivers it to the retinoic acid receptor (RAR)/retinoid X receptor (RXR) complex to regulate the transcription of target genes (proapoptotic genes) by binding to retinoic acid-responsive element (RAREs). When the FABP5-to-CRABPII ratio is high, all-trans-retinoic acid (ATRA) binds mostly to FABP5, which targets RA to other receptors, such as peroxisome proliferator-activated receptor-β/δ, activating mostly survival genes. F: the retinol-RBP4 complex is filtered by the kidneys and is reabsorbed in proximal tubule cells by the megalin-mediated pathway. CYP, cytochrome P-450. PPAR, peroxisome proliferator-activated receptor.
GENERAL EFFECTS OF RA ON IMMUNOMODULATION, DIFFERENTIATION OF STEM CELLS, AND ANTICANCER THERAPY
RA plays a critical role in the regulation of the immune system through maintaining mucosal and epithelial homeostasis and contributing to its anti-inflammatory function. RA can act on both innate and adaptive immune systems locally and systemically. RA modulates inflammatory responses at different mucosal sites and different tissues. For example, RA could promote Foxp3 regulatory T cell differentiation and immunoglobulin production in the intestinal mucosa, induce the homing of innate immune cells, and produce proinflammatory cytokines (1). RA can also induce T helper (Th)2 responses and inhibit Th1 responses to regulate airway inflammation (10, 11). Tazarotene is the most commonly used retinoid for topical treatment of psoriasis, an inflammatory skin disease associated with a large amount of leukocyte infiltration and characterized by marked hyperproliferation and altered differentiation of keratinocytes (12).
RA has been used frequently to induce stem cell differentiation. When RA is transported into the nucleus, it binds to the RA receptor (RAR)/retinoid X receptor (RXR) heterodimer complex to activate transcription of RA primary target genes that contain RA DNA response elements (RAREs). These direct target genes could activate their target genes to generate secondary responses, thereby inducing a transcription activation cascade. It has been shown that nuclear factor-1 (NF1), recruited by the RA-RXR/RAR complex, could stabilize the status of the open nucleosome structure of RA-responsive genes, leading to cell lineage-specific epigenetic modifications and transcription activation (2). Another study has demonstrated that RA can initiate embryonic stem cells to two-cell-like (a rare population of cells resembling two-cell stage embryos in pluripotent embryonic stem cell cultures that display molecular features of totipotency and broader developmental plasticity) reprogramming through a coordinated expression of double homeobox (Dux) and Dux B-like 1 (Duxbl1) in a RARE-dependent manner (13). Besides, RAR/RXRs can interact with other nuclear proteins to change the transcription status of genes, for example, RA administration to stem cells caused the rapid disassociation of polycomb group proteins from RA primary target genes (Hoxa1, Cyp26a1, RARβ2, etc.) to promote their transcription. Polycomb group proteins, which are widely distributed during embryogenesis and play a critical role in differentiation, can form large complexes of gene-silencing proteins (14, 15). RA could also induce stem cell differentiation via a RARE-independent pathway, such as through interactions with the Notch pathway (16, 17).
Treatment of patients with acute promyelocytic leukemia (APL) with all-trans-retinoic acid (ATRA) has been used for more than 20 years. Treatment with single-agent ATRA can achieve up to 80–90% complete remission in newly diagnosed and relapsed patients with APL (18). Significant side effects have been reported in these patients, which are likely from leukemia therapy (tumor cell lysis) rather than effects of ATRA. Well-known “RA syndrome,” which is a generalized severe capillary leakage syndrome with weight gain, fever, pleural effusions, fluid retention, and acute kidney injury (AKI) occurs in ∼25% of patients with ATRA treatment (19). AKI occurred in 71.4% of patients with RA syndrome (20). Renal injuries associated with ATRA treatment include tubular necrosis (21), granulomatous interstitial nephritis (22), renal infiltration by lymphocytes, cortical necrosis (20), and drug-induced nephrotic syndrome (23). The potential mechanism of ATRA-mediated side effect includes: 1) release of a variety of cytokines by differentiating blast cells such as IL-1β, IL-6, IL-8, and TNF-α; 2) a change in adhesive properties of blasts cells such as lymphocyte function-associated antigen 1, intercellular adhesion molecule-1, intercellular adhesion molecule-2, very late antigen 4, and vascular cell adhesion molecule 1; and 3) a hypercoagulable state due to upregulation of thrombomodulin and rapid correction of low fibrinogen levels in APL (24).
The use of retinoids in solid tumors remains controversial. Although many studies have shown that RA can inhibit cell proliferation and induce cell differentiation in different kinds of cancer cells such as lung cancer, gastric cancer, liver cancer, breast cancer, and colon cancer, the efficacy of RA in these cancers is variable and has not been fully documented in clinical trials (25). Multiple mechanisms have been proposed for explaining the resistance of RA in these cancers such as defective expression of RBPs, which mediate the delivery of RA to its nuclear receptors, decreased expression of enzymes critical for the synthesis of RA, and aberrant methylation of the RAR promoter (25). Therefore, the effects and mechanisms of RA in cancer require further validation.
RARS AND THEIR DOWNSTREAM SIGNALING PATHWAYS
RA can be catalyzed by isomerases into different isoforms, which express different binding affinities to the receptors. ATRA and 13-cis-RA (13cRA) bind and activate RARs, whereas 9-cis-RA (9cRA) binds and activates both RARs and RXRs. These isoforms can be interconverted to each other, and ATRA is the most abundant isoform (5). These retinoid nuclear receptors (RAR and RXR) have three isoforms (α, β, and γ) and form RAR/RXR heterodimers to be activated by RA. Cellular RBP (CRABP) is required for the nuclear translocation of RA, which then binds to RARs/RXRs, leading to the subsequent conformational change of this nuclear receptor complex and binding to RAREs to induce the transcription of the downstream target genes. This is the classical or genomic pathway of RA or the so-called RARE-dependent pathway. This RA pathway plays an important role in cell differentiation, cell cycle arrest, and induction of apoptosis (Fig. 1) (26).
Alternatively, RA can be transported to the nucleus by fatty acid-binding protein 5 (FABP5) to bind to RAR/RXR heterodimers, which then interact with other types of nuclear receptors or transcription factors such as peroxisome proliferator-activated receptor (PPAR), activator protein-1 receptor, estrogen receptor-α, liver X receptors, and vitamin D receptor to regulate their target gene expression (25). This is referred to as the nonclassical or nongenomic or RARE-independent pathway (Fig. 1). These pathways often regulate processes that oppose the classical pathway. For example, when ATRA binds to PPAR-β/δ receptors, it results in upregulation of prosurvival genes, which is opposite to the classical effect of RA (27). It has been shown that the CRABPII-to-FABP5 ratio is critical to determine whether RA activates RARE-dependent or RARE-independent pathways. If the CRABPII concentration is high, RA activates the RARE-dependent or classical pathway. If the FABP5 concentration is higher than CRABPII, RA activates the RARE-independent pathway (28). For example, in mammary tissue, when there is a high intratumor FABP5-to-CRABP-II ratio, ATRA binds mostly FABP5, which interacts with PPAR-β/δ, a receptor involved in mitogenic and antiapoptotic activities, to activate prosurvival genes (Fig. 1). When there is a low intratumor FABP5-to-CRABPII ratio, ATRA binds mostly to CRABPII, which binds to RARs/RXRs to activate the proapoptotic signal (29). It has been shown that Kruppel-like factor (KLF)2 might play a role in the shifting of ATRA signaling from the prooncogenic pathway to the anticarcinogenic pathway by regulating the FABP5-to-CRABPII ratio (30).
The role of RA in the regulation of NF-κB pathway has been shown in both positive and negative ways, although RA is known to have mostly anti-inflammatory effects (3). For example, bexarotene, a RARs/RXRs agonist, could inhibit the NF-κB pathway through the release of p300 coactivator from NF-κB target gene promoters to cause histone deacetylation (31). There is also evidence showing that RA and TNF-α could act synergistically to facilitate the binding of NF-κB to the promoter of target genes, enhancing polymerase II phosphorylation and activating gene transcription (32). Further studies are needed to determine which target genes of NF-κB are positively or negatively regulated by RA. Similarly, retinoid regulates the transforming growth factor (TGF)-β pathway both positively and negatively. For example, retinoids suppress TGF-β signaling by ubiquitin-mediated SMAD degradation and decreasing its nuclear accumulation (33). However, retinoids could also increase TGF-β activity by activating transcription of TGF-β1 and TGF-β2 genes (34). These findings suggest that retinoids may have distinct effects in specific tissues and cell types and with specific experimental conditions.
In addition, RA could affect the remodeling of chromatin to activate the transcription of genes. Also, RA could induce cAMP to trigger cell growth arrest, cell death, or differentiation (35).
RENAL-PROTECTIVE EFFECTS OF RA IN MULTIPLE KIDNEY DISEASES
Retinoids have been shown to exert both protective and preventive effects in various animal models of kidney disease through improving podocyte injury, inducing differentiation of kidney progenitor cells, and attenuating inflammation and apoptosis of kidney cells (5).
Proliferative Glomerular Diseases
HIV-associated nephropathy is characterized by glomerular epithelial cell (podocytes) proliferation and dedifferentiation. Our previous study suggested that ATRA administration reduced cell proliferation rate by causing G1 arrest and restored the expression of differentiation podocyte markers in HIV-1-infected podocytes (36). In vivo, we showed that ATRA attenuated albuminuria and improved renal function in HIV-1 transgenic mice (Tg26), an animal model of collapsing focal segmental glomerulosclerosis (FSGS) (37). In these mice, ATRA inhibits glomerular cell proliferation and induces podocyte differentiation (36). Anti-Thy1.1 nephritis is the mouse model of mesangial proliferative glomerulonephritis (GN) characterized by an increased number of mesangial cells in the glomeruli in the kidneys. RA has also been demonstrated to inhibit glomerular proliferation in anti-Thy1.1 nephritis, in which RA treatment attenuated albuminuria, glomerular cell proliferation, macrophage infiltration, capillary occlusion scores, and fibrin deposition (38). The authors attributed the beneficial effects of retinoids on glomerular damage to the marked reduction in renal TGF-β1 and TGF receptor II expression (39). Later, the same group showed that ATRA reduces the number of glomerular cells in mitoses by decreasing angiotensin II synthesis and angiotensin receptor expression in the anti-Thy1.1 nephritis model (40).
Nonproliferation Glomerular Diseases
Animal models with puromycine aminonucleoside (PAN) nephrosis mimic either human minimal change disease or FSGS based on the dosage. Suzuki and colleagues performed a PAN nephrosis rat model in which RALDH2, one of the key enzymes to produce ATRA, was markedly upregulated in podocytes, along with the reduction of glomerular expression levels of nephrin, indicating a compensative role of endogenous RA in repairing podocyte injury. Consistently, PAN nephrosis rats fed with a vitamin A-deficient diet had a delayed recovery from PAN nephrosis. In contrast, administration of ATRA ameliorated proteinuria, attenuated the effacement of the foot processes, and restored nephrin expression (41). We have previously shown that induction of retinol dehydrogenase 9 overexpression in cultured podocytes induces the expression of podocyte differentiation markers. Induction of retinol dehydrogenase 9 overexpression in podocytes in mice with adriamycin-induced nephropathy decreases proteinuria, attenuates kidney injury, and restores podocyte differentiation markers (42). These data suggest a critical role of endogenous RA synthesis in podocyte repairing in glomerular disease.
Inflammatory Kidney Disease
Nephrotoxic serum (NTS) nephritis or antiglomerular basement membrane nephritis represents an experimental immune complex GN resembling crescentic GN as seen in systemic lupus erythematosus, small vessel vasculitis, or Goodpasture syndrome, characterized by a large amount of inflammatory cell infiltration. Our previous study suggested that RA can improve renal function and ameliorate kidney injury under conditions of NTS-GN by protecting podocytes from injury (43). In addition, RA could also reduce the cytokine expression of infiltrating immune cells through its immunomodulatory effects (43). Consistently, Schlöndorff and colleagues have shown that ATRA administration could significantly reduce the proliferative response of the lymph nodes and spleen, endocapillary cell proliferation, and T cell and macrophage infiltration in mice with lupus nephritis (44). However, the serum anti-DNA titers and glomerular immune complex as well as complement deposition were not altered. Mechanically, RA treatment increased renal mRNA levels of TGF-β, which serves as an anti-inflammatory molecule in the lupus nephritis model (44).
Diabetic Kidney Disease
Studies have suggested that RA metabolism is altered in the diabetic kidney. Starkey and colleagues (45) took advantage of a global, quantitative, differential proteomic approach to indicate that RA metabolism is significantly dysregulated in diabetic kidneys and suggested that a shift in ATRA metabolism is a novel feature in type 2 diabetic renal disease. They found increased renal cortical tissue levels of retinol and RALDH1 in db/db versus control mice but significantly decreased levels of ATRA in association with a significant decrease in PPAR-β/δ mRNA (45). Also, plasma and kidney concentrations of RBP are markedly lower in streptozotocin-treated rats compared with controls, which may be due, at least in part, to the impaired metabolic transport from the liver (46). RA has been shown to have renoprotective effects in experimental models of diabetic kidney disease through an anti-inflammatory mechanism (47). Besides, RA may have beneficial effects on insulin sensitivity, the lipid profile, circulating adipocytokines (48), and pancreatic cell proliferation (49).
Other Chronic Kidney Disease Models
ATRA has been also demonstrated to have a therapeutic effect in other animal models with various kidney diseases including the mouse model with unilateral ureteral obstruction (50, 51) and the rat model with 5/6 nephrectomy (52). In the unilateral ureteral obstruction model, ATRA administration had both prophylactic effects and therapeutic effects as evidenced by a significant improvement in histological and immunological findings by limiting inflammation and fibrosis (50).
Acute Kidney Injury
Chiba and colleagues (53) have proven that RA signaling, an embryonic signaling pathway playing a role in kidney development, is repressed in the adult but is reactivated after AKI to reduce injury and enhance repair. They showed evidence that in both mouse and zebrafish models of AKI, RA signaling is reactivated to limit the extent of the injury and promote normal repair through regulation of the dynamic equilibrium between M1 and M2 macrophages (53). RA has also been shown to ameliorate both contrast-induced AKI and cisplatin-induced AKI through activating autophagy and inhibiting apoptosis (54, 55) or decreasing the oxidative status (56). In the ischemia-reperfusion AKI mouse model, RA exerted renoprotective effects through inhibiting the expression of nuclear hormone receptor 77, which mediated renal epithelial apoptosis through a conformational change of Bcl-2 and increasing protein levels of proapoptotic Bcl-xS (57).
BENEFICIAL EFFECTS OF RA IN PODOCYTES
Loss of podocytes, which are a critical component of the glomerular filtration barrier, is a key pathogenic process in the progression of glomerular disease. It has been reported that more than 20% of podocyte depletion is enough to initiate glomerulosclerosis (58). Recent studies have suggested that podocytes have a very low proliferative capacity. Thus, the potential role of progenitors in podocyte repair and regeneration has generated great interest. Studies have suggested that renal progenitor cells (RPCs), which line Bowman’s capsule of the adult human kidney, may play a critical role in the repair of podocyte injury (59, 60). The transitional cells expressing markers for both progenitor cells and podocytes are considered to be podocyte progenitors, responsible for the regeneration of new podocytes. Shankland et al. took advantage of PECrtTA/LC1/R26 reporter mice to demonstrate that parietal epithelial cells (PECs) can serve as adult podocyte progenitors following disease-induced podocyte depletion, as evidenced by labeled PECs migrating onto the glomerular tuft in FSGS (61). However, it is generally accepted that podocyte regeneration is very limited and not enough to repair podocyte injury in glomerular disease. We and others have previously demonstrated that RA cannot only restore podocyte differentiation markers in the diseased condition but also enhance the regeneration of PECs into podocytes (36, 62). Shankland et al. found that ATRA administration increases the number of glomerular epithelial transition cells [defined as cells double stained for PAX2 and Wilms’ tumor-1 (WT-1)] in mice with experimental membranous nephropathy (passive Heymann nephritis model) and FSGS (antiglomerular antibody model), indicating that PECs serve as a progenitor of podocytes (63). Later, Peired and colleagues (64) demonstrated that human serum albumin could sequester endogenous RA and reduce the effects of RA transdifferentiation of RPCs into podocytes. They found that retinol is lost through the damaged glomerular filtration barrier and can be transformed to RA by retinaldehyde dehydrogenases (aldehyde dehydrogenase 1 family member A1) within Bowman’s space. This locally generated RA could induce the differentiation of RPCs into podocytes. Blockade of RA synthesis by administration of the aldehyde dehydrogenase 1 family member A1 inhibitor disulfiram into adriamycin-induced nephropathy resulted in a marked increase of proteinuria, displayed fewer WT-1+ claudin1+ cells (a marker of podocytes and PECs), reduced podocyte numbers, and worsened glomerulosclerosis in mice. In contrast, treatment with RA promoted the differentiation of RPCs into podocytes, thus increasing podocyte numbers and decreasing proteinuria (Fig. 2). However, as the progressive increase of proteinuria due to podocyte damage, RA could be sequestered with albumin, which impairs the RPC response to RA to differentiate into podocytes (64). These interesting findings provide us a potential explanation on why reducing proteinuria by angiotensin-converting enzyme inhibitor or angiotensin receptor blockers could slow down the progression of chronic kidney disease (65).
Figure 2.

Cellular and molecular mechanisms of retinoic acid (RA) in podocytes. The protective effects of RA on podocytes depend on the types of injury. RA can inhibit podocyte proliferation and differentiation in mice with human immunodeficiency virus (HIV)-associated nephropathy (HIVAN) through activation of the cAMP/PKA pathway and upregulation of Wilms’ tumor-1 (WT-1) and Kruppel-like factor 15 (KLF15) expression. In mice with crescentic glomerulonephritis [nephrotoxic serum (NTS) nephritis] and focal segmental glomerulosclerosis (FSGS), RA stimulates podocyte regeneration from parietal epithelial cells (PECs) via activation of retinoic acid receptor (RAR)α, and this protective effect is impaired by albuminuria. However, a high dose of RA may induce retinoic acid receptor responder protein 1 (RARRES1) expression, which can cause podocyte apoptosis, a potential side effect of RA. ADR, adriamycin; DKD, diabetic kidney disease.
Consistent with these findings, we found that in NTS-GN mice, RA treatment increases the number of PECs expressing the podocyte marker with double staining of claudin-1 and nephrin, indicating that RA induced PEC transdifferentiation into podocytes. These findings were further confirmed by lineage-tracing experiments of PECs in NTS-GN mice treated or without RA. We confirmed that RA treatment induced the migration of PECs into glomerular tufts under the NTS condition, where they exhibited podocyte morphology and concomitantly expressed synaptopodin, indicating that RA promoted PEC transdifferentiation into podocytes in the NTS situation (Fig. 2) (43).
CELLULAR AND MOLECULAR MECHANISMS OF RA IN PODOCYTES
Our study suggested that ATRA administration reduces the cell proliferation rate and restores the expression of differentiation podocyte markers in HIV-1-infected podocytes in a cAMP-dependent manner (36). ATRA stimulates cAMP production in podocytes via activation of adenyl cyclase. Increased cAMP could inhibit cell proliferation through the regulation of cyclin-dependent kinase 4 phosphorylation (66) and activated PKA (67), which subsequently inhibited ERK1/2 phosphorylation, resulting in inhibition of cell proliferation (68, 69). In vivo, we found that treatment of mice with an inhibitor of phosphodiesterase 4, a degradation pathway of cAMP, enhances the protection against podocyte injury in Tg26 mice (70). These data suggest that RA signaling pathways in podocytes are likely via activation of the RARE-independent pathway, which is different from those activated in cancer cells (Fig. 2).
We also showed that RA binds to RARα to induce cAMP production in podocytes (36). Knockout of RARα induces more albuminuria and podocyte injury, whereas activation of RARα with its specific agonists ameliorates albuminuria and podocyte injury in Tg26 mice (37). These data suggest a critical role of RARα in glomerular disease. Since most side effects of RA are mediated by activation of RARγ, RARα-specific agonists could be potentially developed as novel drugs to treat glomerular disease without significant side effects. Therefore, we have developed a series of new RARα agonists to treat glomerular disease (71).
We also found that RA stimulates not only WT-1 but also KLF15, two key transcription factors to induce podocyte differentiation via activation of RARα (72). Both transcription factors bind to the promoters of several key podocyte differentiation genes to induce their expression. Knockout of KLF15 induces more podocyte and glomerular injury, whereas induction of KLF15 expression attenuates podocyte and glomerular injury (73). Therefore, KLF15 agonists could be potentially developed as a novel therapy for glomerular disease, as a downstream target gene of RA signaling (Fig. 2).
HARMFUL EFFECTS OF RA IN PODOCYTES
Retinoids have clearly been evidenced to have a therapeutic benefit in experimental models of various glomerular diseases. However, these findings have not been translated into clinical use for kidney disease, largely because of the lack of insight into the effects of RA in human kidneys in the setting of glomerular disease and because RA has significant toxicity in multiple tissues. To confirm whether our findings of RA signaling from the animal models translate to human disease, we took advantage of transcriptomic data sets from patients with primary glomerular disease collected by the Nephrotic Syndrome Study Network Consortium (NEPTUNE) and examined the expression of RA-related signaling molecules and target genes. We found that mRNA expression of RAR responder protein 1 [RARRES1; also known as tazarotene-induced gene (TIG1)] correlates significantly with clinical outcomes. However, to our surprise, the higher expression of RARRES1 was associated with worse renal survival, suggesting that RARRES1 is a risk gene for kidney disease (74).
RARRES1 was first identified to be highly upregulated in skin raft cultures by a RARβ/γ-selective retinoid, AGN190168 (75). ATRA can induce the expression of RARRES1 at a high dose (76). In addition, our study demonstrated that TNF-α induces the expression of RARRES1. As patients with chronic kidney disease have a marked increase of inflammation, we believe this is the major cause of the high expression of RARRES1 in the kidney of these patients (74).
Interestingly, we found that RARRES1 is expressed predominantly in podocytes, as shown by single-cell RNA-sequencing analysis of both human and mouse kidneys. Since RARRES1 is a growth arrest gene, we believe that RARRES1 plays a role in maintaining podocytes in quiescent status. Our studies suggested that RARRES1, a type 1 membrane protein, can be cleaved and released into supernatants as a highly glycosylated soluble form in cultured podocytes. We mapped the exact cleavage site of RARRES1, and the cleavage is likely mediated by matrix metalloproteinases (74). Soluble RARRES1 can be detected in the urine of mice with diabetic kidney disease and FSGS and is significantly increased in the urine of patients with diabetic kidney disease compared with healthy subjects (unpublished observations). Mechanistically, soluble RARRES1 can reenter into cells by endocytosis and suppresses intracellular RIO kinase 1 (RIOK1) function to induce apoptosis of podocytes through activation of the p53 pathway (Fig. 3). In vivo, we validated that RARRES1 wild-type mice developed albuminuria and podocyte injury, whereas RARRES1 cleavage mutant mice, which are unable to generate soluble RARRES1, did not have any renal phenotype (Fig. 2) (74).
Figure 3.
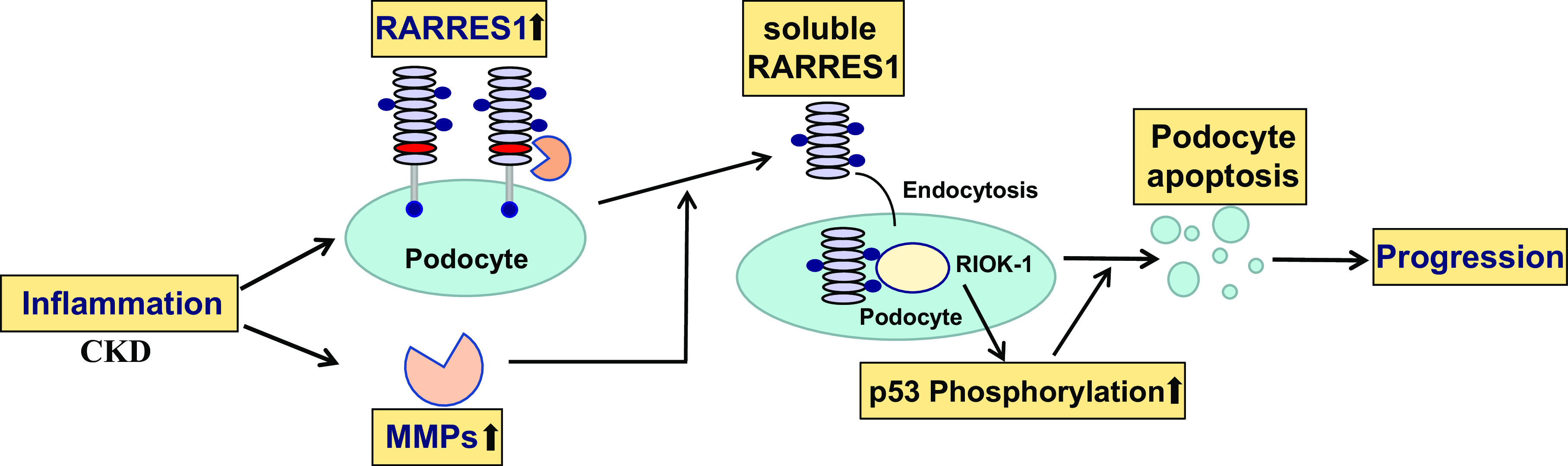
Summary of the mechanism by which retinoic acid receptor responder protein 1 (RARRES1) regulates podocyte apoptosis. In the diseased kidneys, inflammation, such as through TNF-α, induces expression of RARRES1, which is cleaved into its soluble form, probably mediated by inflammation-stimulated matrix metalloproteinases (MMPs). Soluble RARRES1 is then endocytosed and interacts with intracellular RIO kinase 1 (RIOK1), leading to the inactivation of RIOK1, thereby increasing p53 phosphorylation and podocyte apoptosis. Podocyte loss promotes the progression of kidney disease. CKD, chronic kidney disease.
These data suggest that, although RA has protective effects against podocyte injury and glomerular disease, a high dose of RA might be toxic by promoting apoptosis of podocytes. Therefore, the therapeutic window of RA might be limited in patients with glomerular disease. This may explain why the clinical trial did not show significant benefits (NCT00098020). Previous studies have shown that the systemic adverse effects of RA are mostly mediated by RA-induced apoptosis (4), and our study suggested that RARRES1 might also mediate the systemic adverse effects of RA.
THE PERSPECTIVE OF RA AS A THERAPY FOR GLOMERULAR DISEASE
The previous strong experimental evidence for the use of RA in kidney disease prompted the initiation of a National Institutes of Health-sponsored open-label randomized phase 2 clinical trial with the use of isotretinoin in the treatment of patients with collapsing glomerulopathy and FSGS (NCT00098020) in 2004. By December 13, 2017, only 12 patients were enrolled due to the risk of toxicity of RA. Among those patients, only six subjects completed the study. The baseline protein-to-creatinine ratio was 8.42 ± 4.69 g/g, whereas the protein-to-creatinine ratio at wk 24, the end point of the study, was 7.67 ± 5.36 g/g. No participant achieved complete remission or partial remission at 6 mo. This study suffered from small sample size due to the significant side effects of RA. The study was also designed to treat glomerular diseases that are resistant to conventional therapy. In addition, isotretinoin (13-cis-RA) is a pan agonist that can bind to all subtypes of RARs (5) and is often associated with irritation to the skin as one of the major side effects that are due to the RARγ subtype (77). Thus, Kinki University sponsored another open-label randomized phase 2 clinical trial with the use of tamibarotene for the treatment of steroid-refractory lupus nephritis (NCT01226147). Tamibarotene has a higher receptor selectivity toward the RARα and RARβ but not RARγ. However, the updated results of this clinical trial have not been available yet. Future clinical trials of RA for glomerular disease might pay attention to the following key points: more specific RARα agonists to avoid potential side effects mediated by RARγ, use of combination therapy with a low dose of RARα agonists and phosphodiesterase 4 inhibitors to avoid the side effects associated with a high dose of RA, focus on downstream targets of RA such as KLF15 agonists, and consider use of a combination of RA with RARRES1 inhibitors. However, these strategies need to be further validated in preclinical studies before moving to clinical trials in patients with kidney disease (Fig. 4).
Figure 4.

Therapeutic strategy of retinoic acid (RA) in kidney disease for future clinical trials. Future therapeutic strategies of RA for glomerular disease could include: 1) the development of retinoic acid receptor (RAR)α- and Kruppel-like factor 15 (KLF15)-specific agonists to avoid the side effects mediated by RARγ; 2) use of combination therapy with a low dose of RARα agonists and phosphodiesterase 4 (PDE4) inhibitors to avoid side effects associated with a high dose of RA; 3) possible use of a combination of RA with retinoic acid receptor responder protein 1 (RARRES1) inhibitors to avoid RARRES1-mediated podocyte toxicity.
In summary, the kidney is one of the major organs for RA metabolism, which is altered in the diseased kidney. RA has multiple cellular effects including induction of cell differentiation, antiproliferation, and anti-inflammation. The effects of RA are mediated by both RARE-dependent or RARE-independent pathway. The renal-protective effects of RA have been reported in many animal models. RA induces podocyte differentiation likely through a cAMP/PKA/KLF15-mediated pathway. RARα is the major receptor mediating the effects in podocytes. However, RARRES1, a downstream molecule of the RA signaling pathway, mediates podocyte toxicity and is a risk factor for glomerular disease progression. Due to the disparate roles of RA, it is challenging to use RA to treat kidney disease. Future studies are required to develop the molecules that could specifically activate RARα while avoiding the stimulation of RARRES1 expression for the treatment of kidney disease.
GRANTS
A.C. is supported by National Natural Science Foundation of China Grant 81800637. J.C.H. is supported by Veterans Affairs Merit Award IBX000345C and by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants 1R01DK078897, 1R01DK088541, and P01DK56492. K.L. is supported by NIDDK Grant R01DK117913.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.C. and Y.L. prepared figures; A.C., Y.L., K.L., and J.C.H. drafted manuscript; A.C., Y.L., Y.L., K.L., and J.C.H. edited and revised manuscript; A.C., Y.L., Y.L., K.L., and J.C.H. approved final version of manuscript.
REFERENCES
- 1.Oliveira LD, Teixeira FME, Sato MN. Impact of retinoic acid on immune cells and inflammatory diseases. Mediators Inflamm 2018: 3067126, 2018. doi: 10.1155/2018/3067126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gudas LJ, Wagner JA. Retinoids regulate stem cell differentiation. J Cell Physiol 226: 322–330, 2011. doi: 10.1002/jcp.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mallipattu SK, He JC. The beneficial role of retinoids in glomerular disease. Front Med (Lausanne) 2: 16, 2015. doi: 10.3389/fmed.2015.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melnik BC. Overexpression of p53 explains isotretinoin's teratogenicity. Exp Dermatol 27: 91–93, 2018. doi: 10.1111/exd.13420. [DOI] [PubMed] [Google Scholar]
- 5.Xu Q, Lucio-Cazana J, Kitamura M, Ruan X, Fine LG, Norman JT. Retinoids in nephrology: promises and pitfalls. Kidney Int 66: 2119–2131, 2004. doi: 10.1111/j.1523-1755.2004.66002.x. [DOI] [PubMed] [Google Scholar]
- 6.Orfanos CE, Ehlert R, Gollnick H. The retinoids. A review of their clinical pharmacology and therapeutic use. Drugs 34: 459–503, 1987. doi: 10.2165/00003495-198734040-00003. [DOI] [PubMed] [Google Scholar]
- 7.Jing J, Isoherranen N, Robinson-Cohen C, Petrie I, Kestenbaum BR, Yeung CK. Chronic kidney disease alters vitamin A homeostasis via effects on hepatic RBP4 protein expression and metabolic enzymes. Clin Transl Sci 9: 207–215, 2016. doi: 10.1111/cts.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henze A, Frey SK, Raila J, Tepel M, Scholze A, Pfeiffer AFH, Weickert MO, Spranger J, Schweigert FJ. Evidence that kidney function but not type 2 diabetes determines retinol-binding protein 4 serum levels. Diabetes 57: 3323–3326, 2008. doi: 10.2337/db08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erkelens MN, Mebius RE. Retinoic acid and immune homeostasis: a balancing act. Trends Immunol 38: 168–180, 2017. doi: 10.1016/j.it.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Iwata M, Eshima Y, Kagechika H. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol 15: 1017–1025, 2003. doi: 10.1093/intimm/dxg101. [DOI] [PubMed] [Google Scholar]
- 11.Stephensen CB, Borowsky AD, Lloyd KCK. Disruption of Rxra gene in thymocytes and T lymphocytes modestly alters lymphocyte frequencies, proliferation, survival and T helper type 1/type 2 balance. Immunology 121: 484–498, 2007. doi: 10.1111/j.1365-2567.2007.02595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinstein GD, Koo JYM, Krueger GG, Lebwohl MG, Lowe NJ, Menter MA, Lew-Kaya DA, Sefton J, Gibson JR, Walker PS; Tazarotene Cream Clinical Study. Tazarotene cream in the treatment of psoriasis: two multicenter, double-blind, randomized, vehicle-controlled studies of the safety and efficacy of tazarotene creams 0.05% and 0.1% applied once daily for 12 weeks. J Am Acad Dermatol 48: 760–767, 2003.doi: 10.1067/mjd.2003.103. [DOI] [PubMed] [Google Scholar]
- 13.Tagliaferri D, Mazzone P, Noviello TMR, Addeo M, Angrisano T, Del Vecchio L, Visconte F, Ruggieri V, Russi S, Caivano A, Cantone I, De Felice M, Ceccarelli M, Cerulo L, Falco G. Retinoic acid induces embryonic stem cells (ESCs) transition to 2 cell-like state through a coordinated expression of Dux and Duxbl1. Front Cell Dev Biol 7: 385, 2020. doi: 10.3389/fcell.2019.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillespie RF, Gudas LJ. Retinoid regulated association of transcriptional co-regulators and the polycomb group protein SUZ12 with the retinoic acid response elements of Hoxa1, RARβ2, and Cyp26A1 in F9 embryonal carcinoma cells. J Mol Biol 372: 298–316, 2007. doi: 10.1016/j.jmb.2007.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laursen KB, Gudas LJ. Combinatorial knockout of RARα, RARβ, and RARγ completely abrogates transcriptional responses to retinoic acid in murine embryonic stem cells. J Biol Chem 293: 11891–11900, 2018. doi: 10.1074/jbc.RA118.001951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papi A, Guarnieri T, Storci G, Santini D, Ceccarelli C, Taffurelli M, De Carolis S, Avenia N, Sanguinetti A, Sidoni A, Orlandi M, Bonafe M. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ 19: 1208–1219, 2012. doi: 10.1038/cdd.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying M, Wang S, Sang Y, Sun P, Lal B, Goodwin CR, Guerrero-Cazares H, Quinones-Hinojosa A, Laterra J, Xia S. Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene 30: 3454–3467, 2011. doi: 10.1038/onc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayser S, Schlenk RF, Platzbecker U. Management of patients with acute promyelocytic leukemia. Leukemia 32: 1277–1294, 2018. doi: 10.1038/s41375-018-0139-4. [DOI] [PubMed] [Google Scholar]
- 19.Stahl M, Tallman MS. Differentiation syndrome in acute promyelocytic leukaemia. Br J Haematol 187: 157–162, 2019. doi: 10.1111/bjh.16151. [DOI] [PubMed] [Google Scholar]
- 20.Sastre A, Gago E, Banos M, Gomez E. [Acute renal failure in the transretinoic syndrome]. Nefrologia 27: 184–190, 2007. [PubMed] [Google Scholar]
- 21.Flombaum CD, Isaacs M, Reich L, Berman E, Warrell RP. Acute renal failure associated with the retinoic acid syndrome in acute promyelocytic leukemia. Am J Kidney Dis 27: 134–137, 1996. doi: 10.1016/S0272-6386(96)90041-4. [DOI] [PubMed] [Google Scholar]
- 22.Tomita N, Kanamori H, Fujita H, Maruta A, Naitoh A, Nakamura S, Ota Y, Nozue N, Kihara M, Ishigatsubo Y. Granulomatous tubulointerstitial nephritis induced by all-trans retinoic acid. Anticancer Drugs 12: 677–680, 2001. doi: 10.1097/00001813-200109000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Moradveisi B, Rashidi A, Alavi S, Eskandarifar A. Nephrotic syndrome in acute promyelocytic leukemia. Clin Kidney J 7: 424–425, 2014. doi: 10.1093/ckj/sfu062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Breen KA, Grimwade D, Hunt BJ. The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br J Haematol 156: 24–36, 2012. doi: 10.1111/j.1365-2141.2011.08922.x. [DOI] [PubMed] [Google Scholar]
- 25.Costantini L, Molinari R, Farinon B, Merendino N. Retinoic acids in the treatment of most lethal solid cancers. J Clin Med 9: 360, 2020. doi: 10.3390/jcm9020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connolly RM, Nguyen NK, Sukumar S. Molecular pathways: current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin Cancer Res 19: 1651–1659, 2013. doi: 10.1158/1078-0432.ccr-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf G. Retinoic acid as cause of cell proliferation or cell growth inhibition depending on activation of one of two different nuclear receptors. Nutr Rev 66: 55–59, 2008. doi: 10.1111/j.1753-4887.2007.00006.x. [DOI] [PubMed] [Google Scholar]
- 28.Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129: 723–733, 2007. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARβ/δ to RAR. Proc Natl Acad Sci USA 105: 7546–7551, 2008. doi: 10.1073/pnas.0709981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang W, Levi L, Banerjee P, Jain M, Noy N. Kruppel-like factor 2 suppresses mammary carcinoma growth by regulating retinoic acid signaling. Oncotarget 6: 35830–35842, 2015. doi: 10.18632/oncotarget.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cras A, Politis B, Balitrand N, Darsin-Bettinger D, Boelle PY, Cassinat B, Toubert M-E, Chomienne C. Bexarotene via CBP/p300 induces suppression of NF-κB-dependent cell growth and invasion in thyroid cancer. Clin Cancer Res 18: 442–453, 2012. doi: 10.1158/1078-0432.CCR-11-0510. [DOI] [PubMed] [Google Scholar]
- 32.Witcher M, Pettersson F, Dupere-Richer D, Padovani A, Summers-Deluca L, Baldwin AS, Miller WH Jr.. Retinoic acid modulates chromatin to potentiate tumor necrosis factor alpha signaling on the DIF2 promoter. Nucleic Acids Res 36: 435–443, 2008. doi: 10.1093/nar/gkm1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheng N, Xie Z, Wang C, Bai G, Zhang K, Zhu Q, Song J, Guillemot F, Chen Y-G, Lin A, Jing N. Retinoic acid regulates bone morphogenic protein signal duration by promoting the degradation of phosphorylated Smad1. Proc Natl Acad Sci USA 107: 18886–18891, 2010. doi: 10.1073/pnas.1009244107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Q, Kopp JB. Retinoid and TGF-β families: crosstalk in development, neoplasia, immunity, and tissue repair. Semin Nephrol 32: 287–294, 2012. doi: 10.1016/j.semnephrol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillemin M-C, Raffoux E, Vitoux D, Kogan S, Soilihi H, Lallemand-Breitenbach V, Zhu J, Janin A, Daniel M-T, Gourmel B, Degos L, Dombret H, Lanotte M, De The H. In vivo activation of cAMP signaling induces growth arrest and differentiation in acute promyelocytic leukemia. J Exp Med 196: 1373–1380, 2002. doi: 10.1084/jem.20021129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He JC, Lu T-C, Fleet M, Sunamoto M, Husain M, Fang W, Neves S, Chen Y, Shankland S, Iyengar R, Klotman PE. Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J Am Soc Nephrol 18: 93–102, 2007. doi: 10.1681/ASN.2006070727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ratnam KK, Feng X, Chuang PY, Verma V, Lu T-C, Wang J, Jin Y, Farias EF, Napoli JL, Chen N, Kaufman L, Takano T, D'Agati VD, Klotman PE, He JC. Role of the retinoic acid receptor-α in HIV-associated nephropathy. Kidney Int 79: 624–634, 2011. doi: 10.1038/ki.2010.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner J, Dechow C, Morath C, Lehrke I, Amann K, Waldherr R, Floege J, Ritz E. Retinoic acid reduces glomerular injury in a rat model of glomerular damage. J Am Soc Nephrol 11: 1479–1487, 2000. [DOI] [PubMed] [Google Scholar]
- 39.Morath C, Dechow C, Lehrke I, Haxsen V, Waldherr R, Floege J, Ritz E, Wagner J. Effects of retinoids on the TGF-β system and extracellular matrix in experimental glomerulonephritis. J Am Soc Nephrol 12: 2300–2309, 2001. [DOI] [PubMed] [Google Scholar]
- 40.Dechow C, Morath C, Peters J, Lehrke I, Waldherr R, Haxsen V, Ritz E, Wagner J. Effects of all-trans retinoic acid on renin-angiotensin system in rats with experimental nephritis. Am J Physiol Renal Physiol 281: F909–F919, 2001. doi: 10.1152/ajprenal.2001.281.5.F909. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki A, Ito T, Imai E, Yamato M, Iwatani H, Kawachi H, Hori M. Retinoids regulate the repairing process of the podocytes in puromycin aminonucleoside-induced nephrotic rats. J Am Soc Nephrol 14: 981–991, 2003. doi: 10.1097/01.asn.0000057857.66268.8f. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Dai Y, Chuang PY, He JC. Induction of retinol dehydrogenase 9 expression in podocytes attenuates kidney injury. J Am Soc Nephrol 25: 1933–1941, 2014. doi: 10.1681/ASN.2013111150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dai Y, Chen A, Liu R, Gu L, Sharma S, Cai W, Salem F, Salant DJ, Pippin JW, Shankland SJ, Moeller MJ, Ghyselinck NB, Ding X, Chuang PY, Lee K, He JC. Retinoic acid improves nephrotoxic serum-induced glomerulonephritis through activation of podocyte retinoic acid receptor α. Kidney Int 92: 1444–1457, 2017. doi: 10.1016/j.kint.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Lema GP, Lucio-Cazana FJ, Molina A, Luckow B, Schmid H, de Wit C, Moreno-Manzano V, Banas B, Mampaso F, Schlondorff D. Retinoic acid treatment protects MRL/lpr lupus mice from the development of glomerular disease. Kidney Int 66: 1018–1028, 2004. doi: 10.1111/j.1523-1755.2004.00850.x. [DOI] [PubMed] [Google Scholar]
- 45.Starkey JM, Zhao Y, Sadygov RG, Haidacher SJ, Lejeune WS, Dey N, Luxon BA, Kane MA, Napoli JL, Denner L, Tilton RG. Altered retinoic acid metabolism in diabetic mouse kidney identified by O isotopic labeling and 2D mass spectrometry. PLoS One 5: e11095, 2010. doi: 10.1371/journal.pone.0011095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jijon EM, Munoz RR, Reyes JL. Protective effects of retinoic acid on streptozotocin-induced type I diabetes. Pancreat Disord Ther 4: 2, 2014. doi: 10.4172/2165-7092.1000141. [DOI] [Google Scholar]
- 47.Han S-Y, So G-A, Jee Y-H, Han K-H, Kang Y-S, Kim H-K, Kang S-W, Han D-S, Han J-Y, Cha D-R. Effect of retinoic acid in experimental diabetic nephropathy. Immunol Cell Biol 82: 568–576, 2004. doi: 10.1111/j.1440-1711.2004.01287.x. [DOI] [PubMed] [Google Scholar]
- 48.Corbetta S, Angioni R, Cattaneo A, Beck-Peccoz P, Spada A. Effects of retinoid therapy on insulin sensitivity, lipid profile and circulating adipocytokines. Eur J Endocrinol 154: 83–86, 2006. doi: 10.1530/eje.1.02057. [DOI] [PubMed] [Google Scholar]
- 49.Ramirez-Moreno A, Quintanar Escorza MA, Garza RG, Hady K, Melendez Valenzuela AM, Marszalek JE, Sharara-Nunez I, Delgadillo-Guzman D. All-trans retinoic acid improves pancreatic cell proliferation on induced type 1 diabetic rats. Fundam Clin Pharmacol 34: 345–351, 2020. doi: 10.1111/fcp.12523. [DOI] [PubMed] [Google Scholar]
- 50.Kishimoto K, Kinoshita K, Hino S, Yano T, Nagare Y, Shimazu H, Nozaki Y, Sugiyama M, Ikoma S, Funauchi M. Therapeutic effect of retinoic acid on unilateral ureteral obstruction model. Nephron Exp Nephrol 118: e69–e78, 2011. doi: 10.1159/000322409. [DOI] [PubMed] [Google Scholar]
- 51.Schaier M, Jocks T, Grone H-J, Ritz E, Wagner J. Retinoid agonist isotretinoin ameliorates obstructive renal injury. J Urol 170: 1398–1402, 2003. doi: 10.1097/01.ju.0000084620.64255.b3. [DOI] [PubMed] [Google Scholar]
- 52.Morath C, Ratzlaff K, Dechow C, Schwenger V, Schaier M, Zeier B, Peters J, Tsukada M, Zouboulis CC, Waldherr R, Gross M-L, Ritz E, Zeier M, Wagner J. Chronic low-dose isotretinoin treatment limits renal damage in subtotally nephrectomized rats. J Mol Med (Berl) 87: 53–64, 2009. doi: 10.1007/s00109-008-0404-5. [DOI] [PubMed] [Google Scholar]
- 53.Chiba T, Skrypnyk NI, Skvarca LB, Penchev R, Zhang KX, Rochon ER, Fall JL, Paueksakon P, Yang H, Alford CE, Roman BL, Zhang M-Z, Harris R, Hukriede NA, de Caestecker MP. Retinoic acid signaling coordinates macrophage-dependent injury and repair after AKI. J Am Soc Nephrol 27: 495–508, 2016. doi: 10.1681/ASN.2014111108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu J, Zheng C, Wan X, Shi M, McMillan K, Maique J, Cao C. Retinoic acid alleviates cisplatin-induced acute kidney injury through activation of autophagy. Front Pharmacol 11: 987, 2020. doi: 10.3389/fphar.2020.00987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J, Wan X, Zhang H, Li W, Ma M, Pan B, Liang X, Cao C. Retinoic acid attenuates contrast-induced acute kidney injury in a miniature pig model. Biochem Biophys Res Commun 512: 163–169, 2019. doi: 10.1016/j.bbrc.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 56.Yucel C, Yucel EE, Arslan FD, Ekmekci S, Kisa E, Ulker V, Ucar M, Ilbey YO, Celik O, Basok BI, Kozacioglu Z. All-trans retinoic acid prevents cisplatin-induced nephrotoxicity in rats. Naunyn Schmiedebergs Arch Pharmacol 392: 159–164, 2019. doi: 10.1007/s00210-018-01603-0. [DOI] [PubMed] [Google Scholar]
- 57.Balasubramanian S, Jansen M, Valerius MT, Humphreys BD, Strom TB. Orphan nuclear receptor Nur77 promotes acute kidney injury and renal epithelial apoptosis. J Am Soc Nephrol 23: 674–686, 2012. doi: 10.1681/ASN.2011070646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trimarchi H. Podocyturia: potential applications and current limitations. World J Nephrol 6: 221–228, 2017. doi: 10.5527/wjn.v6.i5.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berger K, Schulte K, Boor P, Kuppe C, van Kuppevelt TH, Floege J, Smeets B, Moeller MJ. The regenerative potential of parietal epithelial cells in adult mice. J Am Soc Nephrol 25: 693–705, 2014. doi: 10.1681/ASN.2013050481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wanner N, Hartleben B, Herbach N, Goedel M, Stickel N, Zeiser R, Walz G, Moeller MJ, Grahammer F, Huber TB. Unraveling the role of podocyte turnover in glomerular aging and injury. J Am Soc Nephrol 25: 707–716, 2014. doi: 10.1681/ASN.2013050452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eng DG, Sunseri MW, Kaverina NV, Roeder SS, Pippin JW, Shankland SJ. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int 88: 999–1012, 2015. doi: 10.1038/ki.2015.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaughan MR, Pippin JW, Griffin SV, Krofft R, Fleet M, Haseley L, Shankland SJ. ATRA induces podocyte differentiation and alters nephrin and podocin expression in vitro and in vivo. Kidney Int 68: 133–144, 2005. doi: 10.1111/j.1523-1755.2005.00387.x. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Pippin JW, Vaughan MR, Krofft RD, Taniguchi Y, Romagnani P, Nelson PJ, Liu Z-H, Shankland SJ. Retinoids augment the expression of podocyte proteins by glomerular parietal epithelial cells in experimental glomerular disease. Nephron Exp Nephrol 121: e23–e37, 2012. doi: 10.1159/000342808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peired A, Angelotti ML, Ronconi E, la Marca G, Mazzinghi B, Sisti A, Lombardi D, Giocaliere E, Bona MD, Villanelli F, Parente E, Ballerini L, Sagrinati C, Wanner N, Huber TB, Liapis H, Lazzeri E, Lasagni L, Romagnani P. Proteinuria impairs podocyte regeneration by sequestering retinoic acid. J Am Soc Nephrol 24: 1756–1768, 2013. doi: 10.1681/ASN.2012090950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carney EF. Glomerular disease: albuminuria inhibits podocyte regeneration. Nat Rev Nephrol 9: 554, 2013. doi: 10.1038/nrneph.2013.159. [DOI] [PubMed] [Google Scholar]
- 66.Rocha AS, Paternot S, Coulonval K, Dumont JE, Soares P, Roger PP. Cyclic AMP inhibits the proliferation of thyroid carcinoma cell lines through regulation of CDK4 phosphorylation. Mol Biol Cell 19: 4814–4825, 2008. doi: 10.1091/mbc.e08-06-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stork PJS, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12: 258–266, 2002. doi: 10.1016/S0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 68.Koga Y, Tsurumaki H, Aoki-Saito H, Sato M, Yatomi M, Takehara K, Hisada T. Roles of cyclic AMP response element binding activation in the ERK1/2 and p38 MAPK signalling pathway in central nervous system, cardiovascular system, osteoclast differentiation and mucin and cytokine production. Int J Mol Sci 20: 1346, 2019. doi: 10.3390/ijms20061346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmitt JM, Stork PJ. PKA phosphorylation of Src mediates cAMP’s inhibition of cell growth via Rap1. Mol Cell 9: 85–94, 2002. doi: 10.1016/S1097-2765(01)00432-4. [DOI] [PubMed] [Google Scholar]
- 70.Zhong Y, Wu Y, Liu R, Deng Y, Mallipattu SK, Klotman PE, Chuang PY, He JC. Roflumilast enhances the renal protective effects of retinoids in an HIV-1 transgenic mouse model of rapidly progressive renal failure. Kidney Int 81: 856–864, 2012. doi: 10.1038/ki.2011.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhong Y, Wu Y, Liu R, Li Z, Chen Y, Evans T, Chuang P, Das B, He JC. Novel retinoic acid receptor alpha agonists for treatment of kidney disease. PLoS One 6: e27945, 2011. doi: 10.1371/journal.pone.0027945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mallipattu SK, Liu R, Zheng F, Narla G, Ma'ayan A, Dikman S, Jain MK, Saleem M, D'Agati V, Klotman P, Chuang PY, He JC. Kruppel-like factor 15 (KLF15) is a key regulator of podocyte differentiation. J Biol Chem 287: 19122–19135, 2012. doi: 10.1074/jbc.M112.345983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo Y, Pace J, Li Z, Ma'ayan A, Wang Z, Revelo MP, Chen E, Gu X, Attalah A, Yang Y, Estrada C, Yang VW, He JC, Mallipattu SK. Podocyte-specific induction of Kruppel-Like factor 15 restores differentiation markers and attenuates kidney injury in proteinuric kidney disease. J Am Soc Nephrol 29: 2529–2545, 2018. doi: 10.1681/ASN.2018030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen A, Feng Y, Lai H, Ju W, Li Z, Li Y, Wang A, Hong Q, Zhong F, Wei C, Fu J, Guan T, Liu B, Kretzler M, Lee K, He JC. Soluble RARRES1 induces podocyte apoptosis to promote glomerular disease progression. J Clin Invest 130: 5523–5535, 2020. doi: 10.1172/JCI140155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagpal S, Patel S, Asano AT, Johnson AT, Duvic M, Chandraratna RA. Tazarotene-induced gene 1 (TIG1), a novel retinoic acid receptor-responsive gene in skin. J Invest Dermatol 106: 269–274, 1996. doi: 10.1111/1523-1747.ep12340668. [DOI] [PubMed] [Google Scholar]
- 76.Chandraratna RA. Rational design of receptor-selective retinoids. J Am Acad Dermatol 39: S124–S128, 1998. doi: 10.1016/S0190-9622(98)70308-1. [DOI] [PubMed] [Google Scholar]
- 77.Lazzeri E, Peired AJ, Lasagni L, Romagnani P. Retinoids and glomerular regeneration. Semin Nephrol 34: 429–436, 2014. doi: 10.1016/j.semnephrol.2014.06.009. [DOI] [PubMed] [Google Scholar]