


Keywords: hyperkalemia, phosphatase, potassium, thiazide, transport
Abstract
Phosphorylation of the thiazide-sensitive NaCl cotransporter (NCC) in the distal convoluted tubule (DCT) is altered rapidly in response to changes in extracellular K+ concentration ([K+]). High extracellular [K+] is believed to activate specific phosphatases to dephosphorylate NCC, thereby reducing its activity. This process is defective in the human disease familial hyperkalemic hypertension, in which extracellular [K+] fails to dephosphorylate NCC, suggesting an interplay between NCC-activating and NCC-inactivating switches. Here, we explored the role of STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) and intracellular Cl− concentration in the rapid effects of extracellular K+ on NCC phosphorylation. SPAK was found to be rapidly dephosphorylated in vitro in human embryonic kidney cells and ex vivo in kidney slices by high [K+]. Acute high-K+ challenge resulted in DCT1-specific SPAK dephosphorylation in vivo and dissolution of SPAK puncta. In line with the postulate of interplay between activating and inactivating switches, we found that the “on” switch, represented by with no lysine kinase 4 (WNK4)-SPAK, must be turned off for rapid NCC dephosphorylation by high [K+]. Longer-term WNK-SPAK-mediated stimulation, however, altered the sensitivity of the system, as it attenuated rapid NCC dephosphorylation due to acute K+ loading. Although blockade of protein phosphatase (PP)1 increased NCC phosphorylation at baseline, neither PP1 nor PP3, singly or in combination, was essential for NCC dephosphorylation. Overall, our data suggest that NCC phosphorylation is regulated by a dynamic equilibrium between activating kinases and inactivating phosphatases, with kinase inactivation playing a key role in the rapid NCC dephosphorylation by high extracellular K+.
NEW & NOTEWORTHY Although a great deal is known about mechanisms by which thiazide-sensitive NaCl cotransporter is phosphorylated and activated, much less is known about dephosphorylation. Here, we show that rapid dephosphorylation by high K+ depends on the Cl− sensitivity of with no lysine kinase 4 and the rapid dephosphorylation of STE20/SPS1-related proline-alanine-rich protein kinase, primarily along the early distal convoluted tubule.
INTRODUCTION
A K+ switch in the distal nephron of mammalian kidneys is now recognized to play a key role in both systemic K+ balance and dietary effects on blood pressure. A key element of this switch pathway involves thiazide-sensitive NaCl cotransporter (NCC; encoded by SLC12A3), a member of the cation Cl− cotransporter family whose activity is inversely correlated with extracellular K+ concentration ([K+]) (1–3). Plasma [K+] is tightly regulated between 3.5 mM and 5 mM in healthy humans and animals. When this switch is broken, as in the case of the human disease familial hyperkalemic hypertension, sustained hyperkalemia results along with the development of hypertension, highlighting the role of the switch in normal K+ homeostasis (4).
NCC is expressed at the apical surface of the renal distal convoluted tubule (DCT) (5, 6). It is activated by a cascade of upstream kinases. With no lysine (WNK) kinases activate STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) by phosphorylating Thr243 within the catalytic T loop and Ser383 within the regulatory S motif (7, 8). SPAK serves as the predominant terminal kinase that activates NCC by phosphorylating it on several key threonine residues that lie within its NH2-terminal cytoplasmic domain (9, 10). It is currently believed that K+ deprivation activates NCC by hyperpolarizing DCT cells, leading to Cl− exit (1, 11, 12). WNK kinases are known to be inhibited by Cl− (13, 14); thus, a reduction of intracellular Cl− should activate WNK and subsequently SPAK and NCC (1, 15).
In contrast to conditions of hypokalemia, raising plasma [K+] leads to a rapid reduction in phosphorylated (p)NCC abundance without a change in total NCC levels. This uncoupling has been suggested to reflect rapid NCC dephosphorylation (16–18), which is believed to be physiologically adaptive. Rapid high-K+-mediated dephosphorylation of NCC is expected to increase Na+ delivery to the aldosterone-sensitive distal nephron. Along this segment, electrogenic K+ secretion is driven by the transepithelial voltage derived from Na+ absorption via the epithelial Na+ channel (ENaC). Increased Na+ delivery into this segment is postulated to facilitate Na+ reabsorption and thereby K+ excretion (19).
Protein phosphatase (PP)1 (16, 20), calcineurin (also called PP3) (17, 21, 22), and PP4 (23) have each been suggested as contributing to dephosphorylation of NCC, but their roles in this process in vivo remain unclear. PP4 mRNA is expressed at extremely low levels in the DCT (20, 24), whereas PP1 and calcineurin are abundant (20). Data pertaining to the roles of these phosphatases in rapid NCC dephosphorylation by high K+ are not entirely consistent. PP1/PP2 inhibition by calyculin A blocked rapid high-[K+]-mediated dephosphorylation in kidney slices ex vivo in one study (18), whereas a subsequent study by the same group did not find an effect (16). Similarly, calcineurin inhibition has also led to conflicting findings (16, 17, 21).
Furthermore, the roles of SPAK dephosphorylation and even of intracellular Cl− in the rapid effects of high extracellular [K+] are unclear. Here, we explored the role of SPAK and binding of Cl− to WNK4 in the rapid effects of extracellular K+ on NCC phosphorylation status. We postulated that exposure to high concentrations of K+ would affect not only pNCC but also pSPAK. Furthermore, we hypothesized that the Cl− sensitivity of WNK kinases would play a role. The current study, therefore, aimed to determine whether SPAK dephosphorylation participates in high-K+-mediated NCC dephosphorylation.
METHODS
Antibodies
The antibodies used in this study are shown in Table 1.
Table 1.
Antibodies
| Antibody | Species | Source and Validation | Dilution for Western Blot | Dilution for Immunofluorescence |
|---|---|---|---|---|
| tNCC | Rabbit | Ellison Laboratory (46) | 1:5,000 | |
| pNCC (T53) | Rabbit | Ellison Laboratory (25) | 1:2,000 | |
| SPAK | Rabbit | McCormick Laboratory (42) | 1:5,000 | 1:5,000 |
| OxSR1 | Rabbit | (6) | 1:5,000 | |
| pSPAK (S383) | Rabbit | Millipore, 07-2273 | 1:1,000 | 1:100 |
| β-Actin | Rabbit | Abcam, ab8227 | 1:5,000 | |
| Myc | Rabbit | Sigma-Aldrich, C3956 | 1:1,000 | |
| Calcineurin α | Mouse | Sigma-Aldrich, C1956 | 1:1,000 | |
| PP1α | Rabbit | Epitomics, 1950-1 | 1:1,000 | |
| Parvalbumin | Guinea pig | Swant, GP72 | 1:2,000 | |
| Calbindin | Mouse | Swant, calbindin D-28, 300 | 1:2,000 |
NCC, NaCl cotransporter; OxSR1, oxidative stress responsive kinase 1; PP1α, α subunit of protein phosphatase 1; pNCC (T53), phosphorylated NaCl cotransporter (recognizes threonine 53); pOxSR1, phosphorylated oxidative stress responsive kinase 1; pSPAK (S383), phosphorylated SPAK (recognizes serine 383); SPAK, STE20/SPS-1-related proline/alanine-rich kinase; tNCC, total NCC.
Cell Culture
Human embryonic kidney (HEK)-293 cells were maintained in DMEM with high glucose, sodium pyruvate, and l-glutamine (GE Healthcare, Dallas, TX) supplemented with HEPES, 10% FBS, and penicillin-streptomycin (antibiotic-free media were used for transfection purposes). Cells were transfected with 0.5 μg of the indicated plasmids using lipofectamine 2000 (Invitrogen) on poly-l-lysine-coated plates. Transfection media were changed after 16–18 h. Next day, experiments examining the effects of [K+] were performed on the cells. Cells were incubated in 0 mM K+ basic Krebs media for 1 h followed by an incubation in 0 mM, 3 mM, or 10 mM K+ basic Krebs media for 1 h. The basic Krebs media composition was 1.8 mM CaCl2, 0.00025 mM Fe(NO3)3.9H2O, 0.8 mM MgSO4, 44.0 mM NaHCO3, 110.3 mM NaCl, 0.9 mM NaH2PO4, MEM Vitamin Solution (100×, Sigma), MEM Amino Acids Solution (50×, Gibco) supplemented with 0.4 mM glycine and 4.0 mM l-glutamine, 4.5 g/L d-glucose, and 1.0 mM sodium pyruvate, pH 7.4. [K+] = 0 mM, 3 mM, or 10 mM was achieved by adding the appropriate amount of KCl, and the balance anion and cation concentrations were maintained as invariant by adding NaCl. Proteins from cells were detergent extracted [0.5% Triton X-100, 10 mM Tris·HCl, 5 mM EGTA (pH 8.0), 2.5 mM MgCl2, 1 mM EDTA, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1% (wt/vol) Nonidet P-40, 270 mM sucrose, 0.1% (vol/vol) 2-mercaptoethanol, Roche complete tablet protease inhibitors, and 2× PhosSTOP phosphatase inhibitor]. Lysates were centrifuged at 13,000 g to remove nuclei and cell debris. Extracted proteins were subjected to SDS-PAGE and probed with the indicated antibodies.
Animals
Experiments were approved by the Oregon Health and Science University’s Animal Care and Use Committee (Protocol No. IP00286). Male C57/BL6 mice were fed for 1 day or 5 days with a K+-deficient diet (∼0.002% K+, Cat. No. TD.88239, Envigo) or a normal K+ diet (1.2% K+, Cat. No. 5LOD, lab diet); water was provided ad libitum. To assess the role of SPAK and oxidative stress-responsive kinase 1 (OxSR1) in dephosphorylation, we used SPAK-knockout (KO) mice and global SPAK/kidney-specific OxSR1 double-KO mice, which we have previously characterized extensively (9, 25).
For gavage, 12- to 15-wk-old male C57/BL6 mice weighing 25–30 g were gastric gavaged with 450 μL of either vehicle (2% sucrose) or high-K+ (3% or 6% KCl + 2% sucrose) solutions as previously reported (17). After 15 min, animals were injected with an anesthesia cocktail (ketamine-xylazine-acepromazine, 50/5/0.5 mg/kg). Blood was collected by cardiac puncture and used for electrolyte measurement by Chem8+ cartridge in an i-STAT analyzer (Abbot Point of Care, Princeton, NJ). One kidney was processed for Western blot analysis, and the other kidney was either perfused for immunofluorescence or confocal microscopy. Kidneys collected for Western blot analysis were snap-frozen in liquid nitrogen, followed by transfer to a −80°C freezer. Kidneys were homogenized in 1 mL of cold homogenization buffer containing 300 mM sucrose, 50 mM Tris·HCl (pH 7.4), 1 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 1 mM dithiothreitol, 1 mM PMSF, 1 mg/mL aprotinin, and 4 mg/mL leupeptin for Western blot analysis.
Norepinephrine Injection and Gavage
Twelve- to fifteen-week-old male control, SPAK KO, and SPAK/OxSR1 double-KO animals (9) weighing 25–30 g were intraperitoneally injected with 1.25 mg/kg norepinephrine (dissolved in saline) or vehicle. After 30 min, animals were gastric gavaged with 450 μL of either vehicle (2% sucrose) or high-K+ (3% KCl + 2% sucrose) solutions as described above in Animals. Blood and kidneys were collected and processed as described above in Animals.
Kidney Slices
Closely following the protocol developed by Shoda et al. (17), 300-μm kidney slices were serially cut using a Leica Vibratome 1000s in ice-cold 0 mM K+ buffer containing 108.5 mM NaCl, 25 mM NaHCO3, 1.0 mM NaH2PO4·1H2O, 2.5 mM CaCl2, 1.8 mM MgCl2, and 25.0 mM glucose. Evenly cut sections were then selected and incubated in 0 mM K+ buffer at 35°C for 1 h to stimulate NCC phosphorylation. Sections were then evenly divided into four groups and incubated at 35°C for 30 min in 0 mM, 3 mM, 6 mM, or 10 mM K+-containing buffers. Carbogen gas (95% CO2 and 5% O2) was bubbled through all buffers throughout the procedure. For phosphatase inhibitor experiments, slices were preincubated with 20 nM calyculin A (inhibits PP1 and PP4) (26), 10 μM tacrolimus (inhibits calcineurin) (16, 17, 21), or 200 nM fostreicin (inhibits PP4); all three were dissolved in DMSO in 0 mM K+ before the slices were incubated in 0 mM, 3 mM, 6 mM, and 10 mM K+-containing buffers containing the drug at the indicated concentrations. All incubation buffers were supplemented with 0.017% Tween. Kidney sections were then snap-frozen in liquid nitrogen for Western blot analysis.
Immunofluorescence and Confocal Microscopy
Mice were anesthetized with an anesthesia cocktail (ketamine-xylazine-acepromazine, 50/5/0.5 mg/kg), and kidneys were perfusion fixed by retrograde abdominal aortic perfusion of 3% paraformaldehyde in PBS (pH 7.4) as previously described (27). After cryoprotection with 30% sucrose and freezing, 5-μm sections were cut, washed in 1× PBS three times, incubated in 1× PBS with 0.5% Triton X-100 for 30 min, washed in PBS, and blocked in 5% milk or BSA in PBS for 1 h. Primary antibody in 5% milk or BSA in 1× PBS was added for overnight incubation at 4°C, followed by a wash in PBS (antibody information shown in Table 1). Sections were incubated for 1 h with 1:1,000 FITC-conjugated secondary antibody in blocking buffer and then washed three times with PBS. The last wash contained DAPI. Coverslips were mounted using ProLong Diamond Antifade Mountant. Confocal images were taken using identical laser power, pinhole, and contrast settings, within the linear range of the photodetector. For pSPAK intensity and SPAK puncta quantitation, a Leica Airyscan microscope was used. Signal quantitation for confocal images was performed using Fiji-ImageJ software (https://imagej.net/Fiji/Downloads) following previously described methods (28, 29). Analysis of the sections was performed by an investigator blinded to the condition.
Immunoblot Analysis
Protein (40 μg) was electrophoresed on 4%–15% Criterion-TGX Stain-free gels (Bio-Rad) and transferred to PVDF membranes using trans-blot-turbo (Bio-Rad). Membranes were blocked in 5% milk with PBS and Tween 20 for 1 h. Primary antibodies were diluted in blocking buffer and used to incubate the membrane. Antibody information, including dilutions, is shown in Table 1. Membranes were washed with blocking buffer followed by incubation with the appropriate secondary antibody diluted in blocking buffer (1:2,500). Blots were imaged by enhanced chemiluminescence using an apparatus that ensures linearity of signal detection (PXI digital imaging system, Syngene, Frederick, MA). Densitometry was performed using ImageJ (http://rsbweb.nih.gov/ij/).
Statistics
Quantitative data are shown with means (SD). Data were analyzed using GraphPad Prism and either the Mann–Whitney test for two groups or one-way ANOVA followed by Tukey’s multiple comparison test as indicated, unless otherwise noted.
RESULTS
Extracellular K+ Rapidly Dephosphorylates NCC and SPAK in Kidney Slices Ex Vivo
Penton et al. (30) established a model system to assess the effect of extracellular [K+] on NCC phosphorylation in kidney slices ex vivo, devoid of systemic confounders. Using ex vivo kidney slices, we confirmed that increasing extracellular [K+] from 0 mM to 3 mM, 6 mM, or 10 mM rapidly reduced pNCC abundance (Fig. 1A). Our data broadly recapitulate previous reports in both in vivo and in vitro systems that have demonstrated an inverse relationship between pNCC abundance and extracellular [K+] (Fig. 1B) (1, 2, 17, 18). Similar to kidney slice data previously reported by Penton et al. (16), the greatest decrease in pNCC abundance occurred between 0 Mm and 3 mM K+, whereas pNCC abundance was not significantly different when [K+] was raised from 3 mM to 6 mM or 10 mM (15, 16, 18, 31). Note that the apparent half-maximal values for pNCC dephosphorylation in kidney slices were much lower than those observed in whole animals or perfused kidneys. This observation mirrors that made by Penton et al. (16).
Figure 1.

Extracellular K+ rapidly dephosphorylates NaCl cotransporter (NCC) and STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) in kidney slices ex vivo. A: protein extracts from 300-μM kidney slice homogenates that were incubated in 0 mM K+ for 1 h followed by incubations in 0 mM, 3 mM, 6 mM, or 10 mM K+ for 30 min were immunoblotted for phosphorylated (p)NCC at Thr53 (pNCC-T53), NCC, and β-actin. B: plot depicting the densitometric ratios of pNCC-T53 to NCC analyzed by ANOVA (F = 30.77, P < 0.0001); individual differences, by Tukey’s test, are shown. C: extracts blotted for pSPAK at Ser383 (pS383-SPAK), full-length SPAK (FL-SPAK), and β-actin. D: plot depicting the densitometric ratios of pSPAK to SPAK, analyzed by ANOVA (F = 25.30, P < 0.0001); individual differences, by Tukey’s test, are shown. Statistical analysis was performed on data from kidney slices from five different mouse kidneys. Each solid black circle represents protein extract from an individual mouse. NS, not significant; pOxSR1-S335, phosphorylated oxidative stress responsive kinase 1 at Ser335.
A possible mechanism of sustained NCC dephosphorylation by high [K+] involves not just activation of the “off” switch but also inactivation of the “on” switch by dephosphorylation of SPAK. Having established that our ex vivo model could broadly recapitulate previous studies, we used the same experimental samples for Western blot analysis but interrogated pSPAK (Fig. 1C). Like NCC, SPAK was rapidly dephosphorylated by increasing [K+]. NCC and SPAK, however, exhibited different sensitivity to extracellular [K+]. The relation between pSPAK and [K+] appeared linear (R2 = 0.96) for the same range of [K+] (32). Although a true EC50 value for SPAK dephosphorylation could not be estimated, as plateau values were not reached, the curve appeared shifted to the right compared with the pNCC curve. As antibodies directed toward pSPAK Ser373 also recognize phosphorylated OxSR1, we used SPAK KO and OxSR1 KO mouse kidney lysates to identify the specific bands that represent SPAK and OxSR1 (Supplemental Fig. S1; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.13180748.v1).
K+ Loading in Vivo Results in Rapid SPAK Dephosphorylation and Alters Intracellular SPAK Redistribution in the Mouse DCT1
Previous studies have shown that high K+ administered into the stomach by gavage, or intravenously, rapidly dephosphorylates NCC (17, 18, 31). Having observed SPAK dephosphorylation ex vivo in response to increasing [K+], we tested whether SPAK was rapidly dephosphorylated in vivo by high K+. C57BL/6J mice were acutely K+ loaded by gastric gavage. Fifteen minutes after gavage with 3% KCl, mice showed a significant increase in plasma [K+] (3.64 ± 0.57 mM vs. 6.23 ± 1.24 mM, vehicle vs. 3% K+ gavage, n = 8 animals in each group) and a corresponding decrease in NCC phosphorylation in the whole kidney homogenate (Fig. 2, A and B), as previously reported by others (16–18). pSPAK abundance, however, was essentially unchanged (Fig. 2, C and D). Although this lack of effect is at variance with the data from kidney slices, (Fig. 1) and previously reported by others (16), it was similar to the whole animal data reported by Uchida and colleagues (17). Although there are obvious technical and physiological differences in the dynamics of [K+] changes in vivo and in ex vivo kidney slices (18), one potential reason for a failure to detect an effect of plasma [K+] by kidney Western blot analysis is that SPAK is widely expressed along the nephron and SPAK dephosphorylation in vivo may not occur in every segment (25, 29). Therefore, we tested whether K+ gavage elicits preferential SPAK dephosphorylation along the DCT in vivo.
Figure 2.
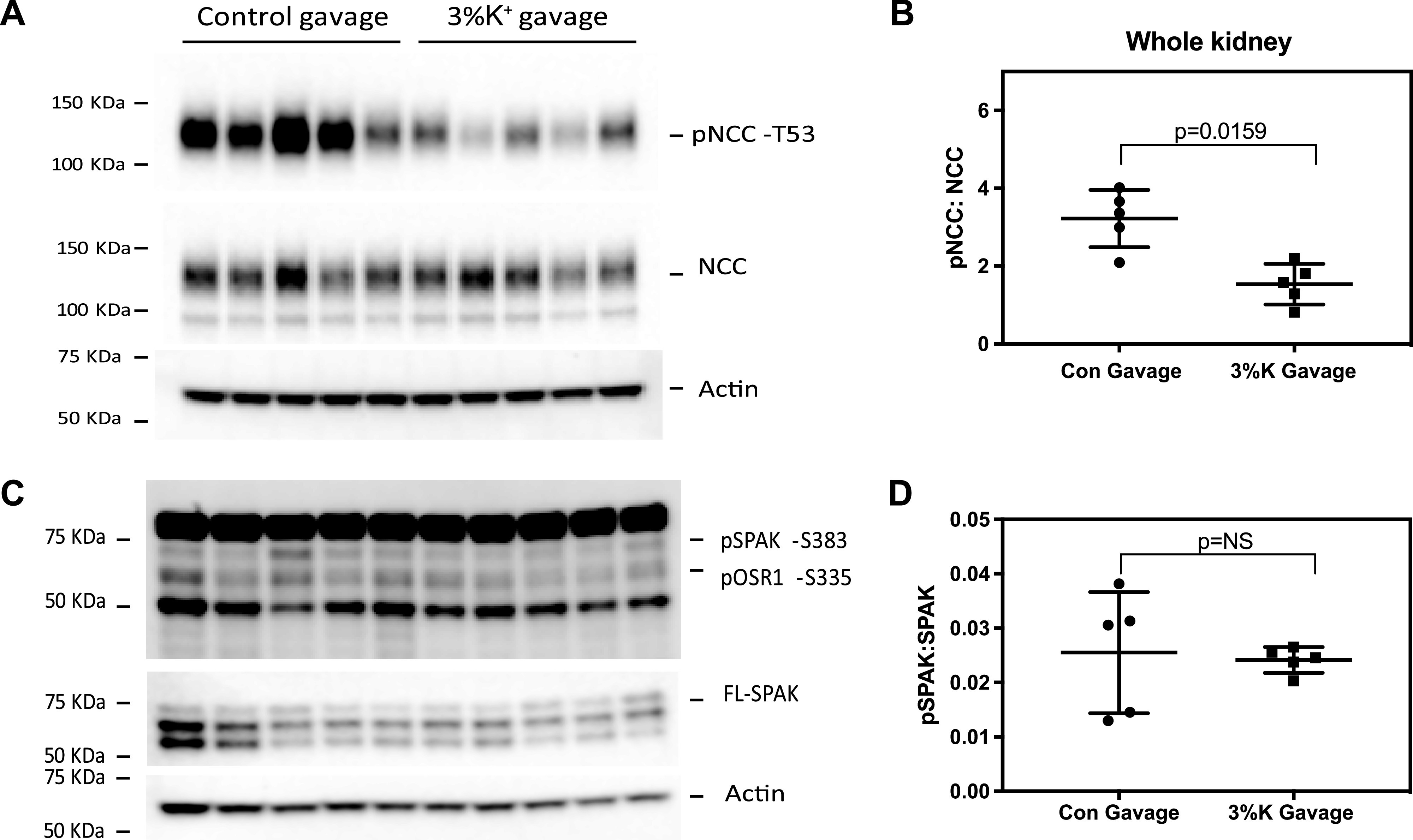
High-K+ gavage dephosphorylates NaCl cotransporter (NCC) and not STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) in kidney slices. C57/BL6 mice were gavaged with either control (2% sucrose) or 3% K+ (2% sucrose + 3% KCl) for 15 min. A: protein extracts from whole kidney homogenates were immunoblotted for phosphorylated (p)NCC at Thr53 (pNCC-T53), NCC, and β-actin. B: dot plot depicting the densitometric ratios of pNCC-T53 and NCC. C: blot of pSPAK at Ser383 (pSPAK-S383), full-length SPAK (FL-SPAK), and β-actin. D: dot plot depicting the densitometric ratios of pSPAK-S383 and FL-SPAK. Each solid black circle represents the immunoblot densitometric ratio from individual control gavaged mouse, whereas each solid black square represents the immunoblot densitometric ratio from individual 3% K+ gavaged mouse (means ± SD, n = 5 per group). Comparisons were performed with a Mann–Whitney test. P values are indicated. NS indicates P > 0.05 (not significant). pOxSR1-S335, phosphorylated oxidative stress responsive kinase 1 at Ser335.
Confocal images of perfused, paraformaldehyde-fixed kidney sections from C57BL/6J mice that were administered gastric gavage of either vehicle or 3% K+ were labeled with an antibody that recognizes pSPAK (and pOxSR1). DCT1 segments were identified by colabeling with an antibody against parvalbumin. pSPAK/pOxSR1 was found to be localized in apical segments of the DCT1 and in cytosolic punctate structures, as previously described (12). We observed an ∼36% decrease in the abundance of pSPAK/pOxSR1 in DCT1 segments of high-K+-gavaged mouse kidneys compared with vehicle-gavaged kidneys (Fig. 3, A and B). The decrease appeared to occur in both apical and cytosolic pools of pSPAK in DCT1 cells. In contrast, the signal from segments that do not express parvalbumin (likely DCT2, thick ascending limb, and connecting tubule) did not appear to diminish appreciably.
Figure 3.
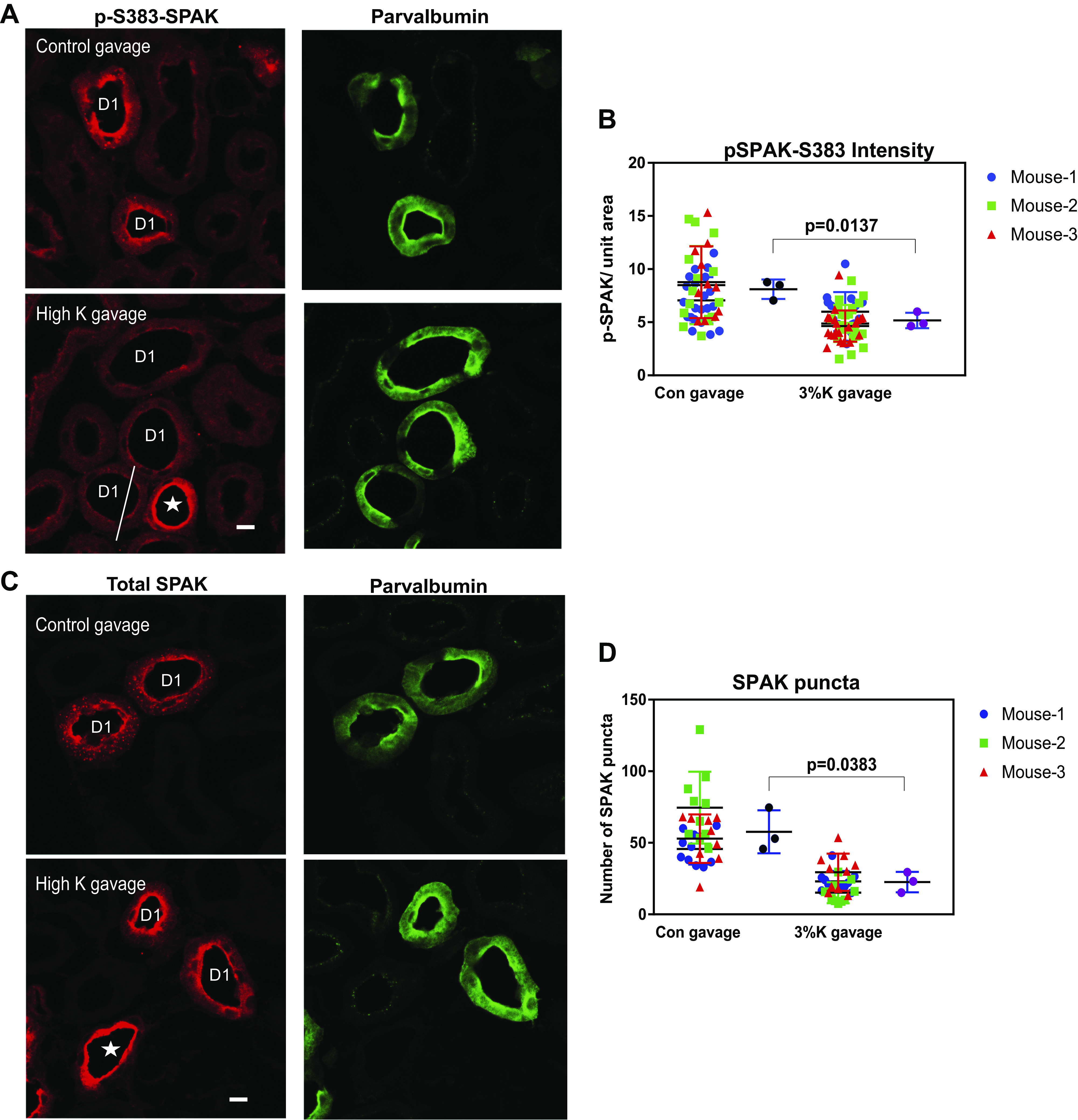
K+ loading in vivo results in rapid STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) dephosphorylation and alters intracellular SPAK localization in the mouse early distal convoluted tubule (DCT1). Shown is confocal microscopic analysis of perfusion-fixed C57/BL6 mouse kidney sections (5 µm) from mice gavaged with either control (Con; 2% sucrose) or high K+ (2% sucrose + 3% KCl) 15 min prior. A: representative ×63 image of localization of phosphorylated SPAK at Ser383 (pSPAK-S383) in control gavaged (top) and high-K+-gavaged (bottom) mouse kidneys, with parvalbumin from the same section at the right. D1, parvalbumin-positive DCT1. *Parvalbumin-negative, pSPAK-positive segment (likely the thick ascending limb). The transitional area in the mixed tubule is marked with a line. B: dot plot representing pSPAK signal intensity in DCT1 segments of control gavaged vs. high-K+-gavaged mice. The first and third groups from the left represent average pSPAK intensity in the DCT1 segment in an individual microscopic field, with each mouse represented by a colored shape, as shown. The second and fourth groups from the left represent the average of the total pSPAK signal from the DCT1 in each mouse gavaged with control solution (black circle) or high-K+ solution (purple circle). C: immunofluorescent micrograph of total SPAK in control gavaged (top) and high-K+-gavaged (bottom) mouse kidneys, with parvalbumin shown at the right. D: dot plot showing the number of SPAK puncta in DCT1 segments of control gavaged vs. high-K+-gavaged mice. The first and third groups from the left show the average number of SPAK puncta in DCT1 segments in an individual microscopic field, with each mouse represented by colored shapes. The second and fourth groups from the left represent the average number of SPAK puncta in the counted DCT1 of individual mice, gavaged with control solution (black circle), or high-K+ solution (purple circle). Magnification: ×63. Scale bars = 10 µm. Plots show means ± SD, with a t test for the mean values using Welch’s correction, as data were normally distributed.
In addition to SPAK phosphorylation, chronic K+ manipulation has been shown to affect total SPAK localization in previous studies (1, 28). We determined whether acute K+ loading rapidly altered total SPAK (phosphorylated and nonphosphorylated) localization in the DCT1. SPAK has been previously reported to localize in intracellular punctate structures in response to lowering of extracellular [K+] (1, 28). SPAK was observed to be localized in both the apical membrane and cytosolic punctate structures in the DCT1 of the control kidney cortex. K+ loading resulted in a rapid reduction of both size and number (approximately by 60%) of SPAK puncta in the DCT1 (Fig. 3, C and D). Under a low-power confocal objective (×10), total SPAK intensity was not different between the control gavaged and K+ gavaged kidney cortex (Supplemental Fig. S2; all Supplemental material is available at http://doi.org/10.6084/m9.figshare.13180667).
SPAK Activated by Cl−-Insensitive WNK4 Abrogates NCC Dephosphorylation by High K+ in HEK-293 Cells
Since low plasma K+ activates the WNK4-SPAK-NCC signaling cascade, at least in part, by reducing intracellular Cl− concentration ([Cl−]) (1, 11, 12, 33), we tested whether rendering WNK4 insensitive to inhibition by Cl− would prevent SPAK and NCC dephosphorylation by high K+. HEK-293 cells transfected with NCC, SPAK, and WNK4 showed maximal dephosphorylation of NCC at 3 mM extracellular K+ (Fig. 4, A and B). Similarly, transfected SPAK was maximally dephosphorylated by 3 mM K+ in cells transfected with SPAK and WNK4 (Fig. 4, C and D). Cl−-binding site-deficient (LLFF mutant) WNK4 increased NCC phosphorylation in HEK-293 cells transfected with NCC, SPAK, and WNK4-LLFF (Fig. 4E) and completely prevented dephosphorylation of SPAK and NCC at both 3 mM and 10 mM K+ (Fig. 4, A–D). This result confirms the importance of intracellular [Cl−] in the effects of high [K+] and suggests that SPAK must be dephosphorylated to permit NCC dephosphorylation. Of note, these experiments also confirmed endogenous expression of PP1 and calcineurin in HEK cells (Fig. 4E).
Figure 4.

Cl−-insensitive with-no-lysine kinase 4 (WNK4) prevents NaCl cotransporter (NCC) and STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) dephosphorylation by high K+ in human embryonic kidney (HEK) cells. A: NCC and phosphorylated (p)NCC from cells transfected with SPAK, NCC, and wild-type (WT) WNK4 (left) or Cl−-insensitive WNK4-LLFF (right) and incubated with 0 mM, 3 mM, or 10 mM K+. B: quantification of the results shown in A. C: SPAK (myc tagged) and pSPAK at Ser383 (pSPAK-S383) from cells transfected with SPAK and WT WNK4 (left) or Cl−-insensitive WNK4-LLFF (right) and incubated with 0 mM, 3 mM, and 10 mM K+. D: quantification of the results shown in C. E: protein extracts from HEK cells transfected with NCC, SPAK, and either WT WNK4 or Cl−-insensitive WNK4-LLFF and blotted for pNCC, NCC, calcineurin α (CaNα), and protein phosphatase-1α (PP1α) subunits. Cells were incubated in DMEM. Individual values and means and SD are shown. Data were analyzed by ANOVA with a Tukey’s post hoc test. NS, not significant.
Effects of SPAK and OxSR1 Deletion on the Response to K+ Gavage
As these results suggested that dephosphorylation of SPAK might play an essential role in pNCC dephosphorylation, we examined the effects of K+ gavage in mice lacking SPAK and in mice lacking both SPAK and (in the kidney) OxSR1. The fundamental phenotypes of these mice have been previously described (9). As the abundance of pNCC is very low in mice lacking SPAK and extremely low in those lacking both SPAK and OxSR1, we used norepinephrine injection to increase pNCC abundance, as we have previously reported (34). Norepinephrine did not prevent dephosphorylation of NCC in response to K+ gavage (Fig. 5, A−C). In SPAK KO mice, K+ gavage appeared to induce a reduction in pNCC abundance, although this was not significant. In double-KO mice, there was no apparent change (Fig. 5, D−F). This highlights the key role that SPAK/OxSR1 plays in this process.
Figure 5.

K+ gavage causes decreases in phosphorylated (p)NaCl cotransporter (pNCC) levels in norepinephrine (NE)-injected STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) knockout (KO) mice. Mice were intraperitoneally injected with 1.25 mg/kg NE or vehicle. After 30 min, animals were gavaged with 450 μL of either vehicle (2% sucrose) or high K+ (3% KCl + 2% sucrose), and samples were collected after a further 15 min. A: blot of K+ gavage effects in control (Con) mice, mice lacking SPAK (SPAK KO), and mice lacking both SPAK and oxidative stress responsive kinase 1 (OxSR1; double KO [DKO]). B: quantification of the results shown in B and from plasma K+. C: blot confirming that NE increases pNCC abundance and does not abrogate the K+ effect. D: quantification of the results shown in C and results of plasma K+. Values are means ± SD. Data in B were analyzed by ANOVA with a Tukey’s multiple comparison test. Comparisons in D were made using a Mann–Whitney test. NS, not significant; WT, wild-type.
Long-Term SPAK Activation by Low-K+ Diet Prevents Rapid NCC Dephosphorylation by High K+ In Vivo
A recent study has suggested that the NCC dephosphorylation response differs based on short-term or long-term K+ treatment in transgenic mice expressing the same Cl−-insensitive, constitutionally activated WNK4; these mice dephosphorylated NCC when K+ loading was chronic, even though they failed to respond normally to acute K+ loading (11). To test whether chronic activation of NCC in vivo might alter the sensitivity to acute K+ loading, C57BL/6J mice were fed a low-K+ diet for 1 day or 5 days and then gavaged with either vehicle or high-K+ (3% KCl) solution. Mean plasma K+ after a 1-day low-K+ diet was 3.5 ± 0.34 mM. Fifteen minutes after high-K+ gavage, plasma [K+] was higher than baseline (5.16 ± 0.70 mM; Fig. 6A) and NCC phosphorylation was lower (Fig. 6, B and C). In contrast, mice fed a low-K+ diet for 5 days had much lower plasma K+ (2.49 ± 0.35 mM; Fig. 6A), and 3% K+ gavage, which increased plasma [K+] by ∼60% to 3.98 ± 0.32 mM, had no effect on pNCC abundance (Fig. 6, D and E). To determine whether the failure to dephosphorylate reflected that lower peak plasma [K+], we used a 6% K+ gavage. This increased plasma K+ levels to 5.22 ± 0.70 mM, similar to that of plasma [K+] in normal diet-fed mice after 3% K+ gavage. The 6% K+ gavage also did not lead to NCC dephosphorylation (Fig. 6, F and G) in 5-day low-K+ diet-fed mice. This suggests that NCC becomes insensitive to dephosphorylation in response to high-K+ gavage following long-term but not short-term dietary K+ restriction.
Figure 6.

Sustained activation of with-no-lysine kinase (WNK)-STE20/SPS1-related proline-alanine-rich protein kinase (SPAK) by 5-day low-K+ diet (LK) blocks NaCl cotransporter (NCC) dephosphorylation by high-K+ gavage. C57/BL6 mice were fed with a low-K+ diet for either 1 day or 5 days. A: plasma K+ values in animals gavaged with K+ after 1 day (open circle) or 5 days of low-K+ diet control (Con; open square). Means and SDs are indicated by bars. B: phosphorylated (p)NCC, NCC, and β-actin from kidneys of mice gavaged after being fed a low-K+ diet for 1 day. Quantification is shown on the right. C: pNCC-to-NCC ratios for mice treated for 5 days with a low-K+ diet and gavaged with 3% of K+. Quantification is shown on the right. D: pNCC and NCC from mice fed with a low-K+ diet for 5 days and gavaged with 6% K+. Quantification is shown on the right. Means and SDs are indicated by bars. Data were compared using ANOVA with a Tukey’s post hoc test (A) or Mann–Whitney test (B − D). NS, not significant.
Inhibition of PP1, PP2, PP4, or Calcineurin Does Not Block NCC Dephosphorylation by High K+
As suggested by others, the speed of NCC dephosphorylation suggests an active dephosphorylation process (16, 18). Therefore, we inquired whether inhibiting the known phosphatases would inhibit rapid NCC dephosphorylation by high K+. Using a similar kidney slice approach, we inhibited PP1/PP2 with calyculin A, PP4 with fostriecin, and calcineurin with tacrolimus.
As noted by others (16, 18), calyculin A markedly increased the abundance of pNCC when incubated at 3 mM K+ (Fig. 7, A and B), but it did not prevent dephosphorylation at 10 mM K+. The calcineurin inhibitor tacrolimus also increased the abundance of pNCC modestly when incubated at 3 mM K+, consistent with one report (17) but not another (16). As with calyculin A, however, it did not prevent dephosphorylation (Fig. 7, A and B). Kidney slices that were exposed simultaneously to calyculin A and tacrolimus did not show a significant additive increase in pNCC abundance compared with calyculin A- or tacrolimus-only treatment groups at 3 mM K+ (data not shown). Furthermore, combined tacrolimus and calyculin A treatment did not prevent the decrease in pNCC abundance when cells were incubated at 10 mM K+ (Fig. 7, C and D). As the maximal effect on pNCC was observed when [K+] was raised from 0 mM and 3 mM, the experiments were repeated using these concentrations (Fig. 7E). Again, the combination of calyculin A and tacrolimus did not prevent the reduction in pNCC to NCC when [K+] was increased. PP4 inhibition by fostreicin also did not increase NCC phosphorylation or block high-K+-mediated NCC dephosphorylation (Fig. 7, F and G). When viewed together with results from other groups, the current results suggest that, although both PP1 and PP3 play roles in determining the abundance of pNCC, other factors must also play a role.
Figure 7.
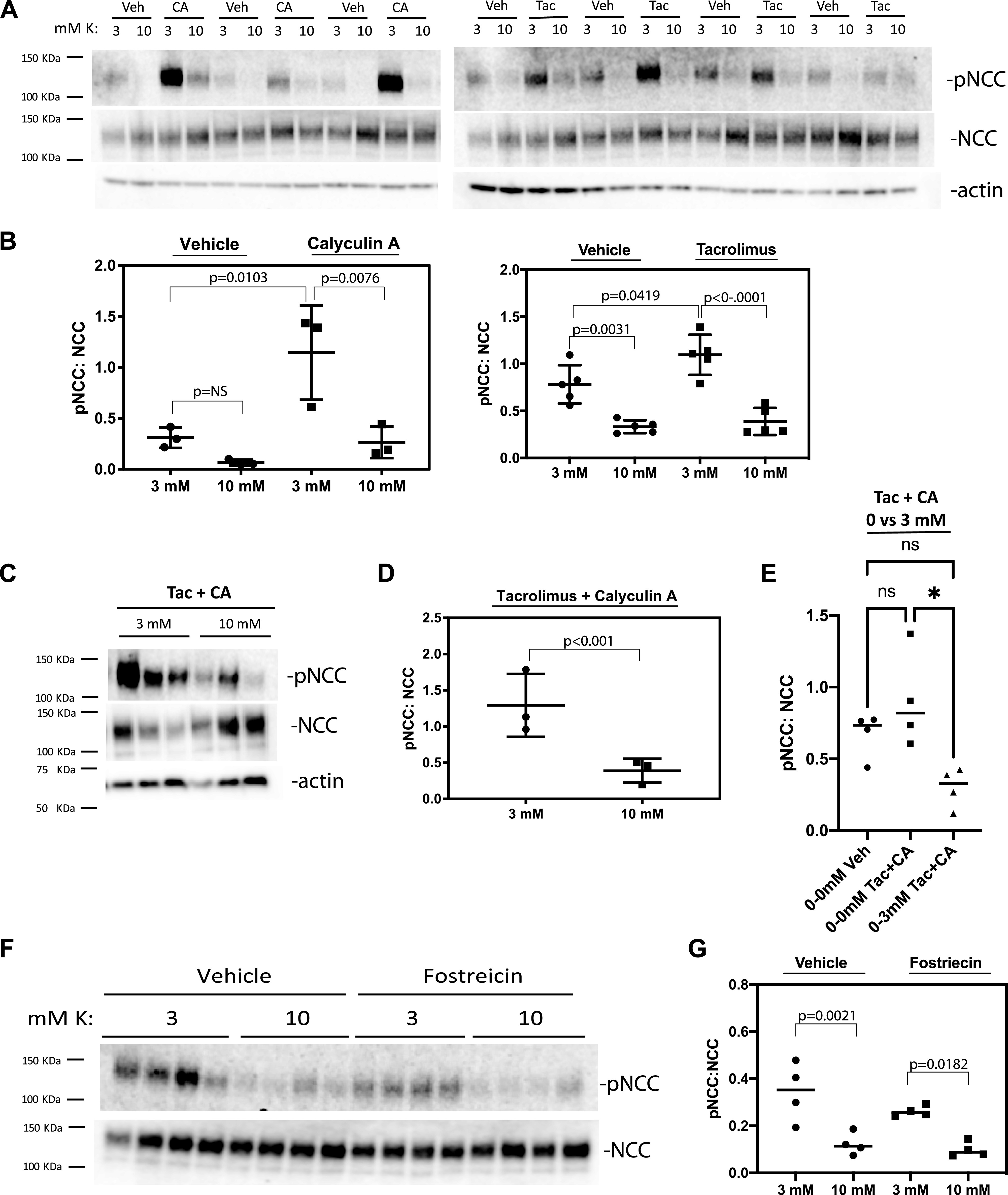
Inhibition of protein phosphatase (PP)1, PP2, PP4, or calcineurin does not block NaCl cotransporter (NCC) dephosphorylation by high K+. Kidney slices were preincubated in 0 mM K+ buffer containing vehicle (Veh) or serine/threonine phosphatase inhibitors followed by 30 min of incubation in 3 mM or 10 mM K+ buffers containing vehicle or inhibitor, respectively. A and C: protein extracts from vehicle (A)-treated or calyculin A (CA)-treated groups (left) or vehicle-treated or tacrolimus (Tac)-treated (right) groups. B: quantification of the results. The effects of K+ concentration and CA were significant by two-way ANOVA, as shown on the left; on the right, the effects of K+ concentraton and Tac were significant by two-way ANOVA, as shown. C: CA combined with Tac-treated slices. D: quantification of the Tac + CA experiments. E: effect of changing K+ concentration from 0 mM to 0 mM or 0 mM to 3 mM in the presence of vehicle or CA + Tac. F: blot of vehicle-treated or fostreicin-treated kidney slices. G: quantification of the fostreicin experiments. Means and SDs are shown. NS or ns, not significant; pNCC, phosphorylated NCC.
DISCUSSION
The current study examined the role of WNK4 and SPAK in modulating NCC dephosphorylation in situations of high extracellular K+. We found that SPAK was rapidly dephosphorylated by high K+ ex vivo in kidney slices and in vitro in HEK cells. Confocal experiments revealed that acute high-K+ challenge resulted in DCT1-specific SPAK dephosphorylation in vivo. Our data indicate that NCC phosphorylation levels are likely regulated by dynamic interactions between the “on” switches, mediated by kinases that phosphorylate and activate NCC, and “off” switches, mediated by phosphatases that dephosphorylate the transporter. We found that the “on” switch, represented by WNK4-SPAK, must be turned off for rapid NCC dephosphorylation by high K+, suggesting that a strongly activated “on” switch can override any dephosphorylation process. Our data show that WNK4-LLFF, which cannot be inactivated by Cl−, prevented rapid NCC dephosphorylation by K+; this finding is consistent with a report describing the effects of K+ gavage on mice expressing this same Cl−-insensitive WNK4 (3). Together, these data indicate that intracellular [Cl−] does play a central role in mediating, or permitting, K+-mediated dephosphorylation of NCC, in contrast to what has been suggested (16, 33). The current results also show that prolonged WNK-SPAK stimulation modifies the sensitivity of NCC to extracellular K+ in vivo. Longer-term dietary stimulation of the WNK4-SPAK pathway in mice attenuates the rapid NCC dephosphorylation due to acute K+ loading. Our examination of the known “off” switches regulating NCC phosphorylation showed that neither PP1 nor PP3, singly or in combination, was essential in NCC dephosphorylation due to high-K+ exposure. In Fig. 8, we propose a working model for these events.
Figure 8.

Simplified NaCl cotransporter (NCC) kinase and phosphatase cascade. A low intracellular Cl− environment, induced by low extracellular K+ concentration, activates with-no-lysine kinase 4 (WNK4) and vice versa. Activated WNK4 binds to and phosphorylates STE20/SPS1-related proline-alanine-rich protein kinase (SPAK). Activated SPAK binds to and phosphorylates NCC, thereby increasing its activity. Protein phosphatase (PP)1 can remove activating phosphates from both SPAK and NCC. PP1 Inhibitor 1 (I-1), which is activated by protein kinase A (PKA), suppresses PP1 activity (30). I-1, the inhibitor, is dephosphorylated by PP3 (calcineurin) (35), suggesting a mechanism for the effects of tacrolimus on NCC.
Cation Cl− transporters including NCC are regulated by similar phosphorylation cascades. NCC can be phosphorylated by SPAK kinase, which, in turn, is activated in the DCT primarily by WNK4, perhaps in concert with kidney-specific WNK1. Although OxSR1, which is homologous to SPAK, is also expressed along the DCT, its deletion alone does not reduce NCC abundance (9, 36), indicating that it plays only a secondary role under most circumstances. As shown in Fig. 8, exposure to low extracellular [K+] is a key physiological factor that activates this kinase cascade along the DCT, thereby increasing NCC activity. NCC activation, both by remodeling the distal nephron and by reducing Na+ delivery to the aldosterone-sensitive distal nephron, reduces electrogenic Na+ reabsorption and, therefore, K+ secretion. Thus, this kinase cascade plays a key role in the distal nephron K+ switch, a physiological control system that affects both [K+] and arterial pressure.
Although many details of the signaling cascade responsible for activating NCC have now been well established, there is less information about the “off” switch. Chronic exposure to diets that are high in K+ reverses all of the NCC-activating effects of low K+ intake, reducing the abundance of WNK4, SPAK, and pSPAK/OxSR1 (37). Loffing and colleagues, however, showed that even a rapid rise in plasma [K+] induced by gastrointestinal gavage also reduces the abundance of pNCC quickly (18), an effect suggested to facilitate rapid kaliuresis quickly (38). This effect has been replicated by others (17) and has also been observed in rats, where intravenous K+ infusion produces similar effects (31). The physiological relevance of rapid K+ loading by gavage is indicated by comparing our data (Figs. 5 and 6) with those of Rabelink et al. (39), who measured plasma [K+] following control (25 mmol) and high-K+ (100 mmol) meals; both raised plasma K+ acutely. The rise after a high-K+ meal (∼1 mM) was of a similar order of magnitude to that which occurred following high-K+ gavage. Penton et al. (16) confirmed that these effects are largely mediated by changes in extracellular [K+], by using ex vivo kidney slices and isolated DCT cells. They concluded, however, that the cellular mediators of rapid dephosphorylation were different from those involved in NCC activation. The effects of low extracellular [K+] on NCC phosphorylation are believed to be mediated largely by changes in intracellular [Cl−] (11, 12, 15, 33), but Penton et al. (16) suggested that the rapid dephosphorylation does not depend on alterations in intracellular [Cl−].
Here, we replicated the rapid effects of raising extracellular [K+] on pNCC abundance in vivo, in kidney slices, and in HEK-293 cells. We confirmed that SPAK is rapidly dephosphorylated by high extracellular [K+]. Confirming previous reports that indicated rapid SPAK dephosphorylation by high extracellular [K+], we detected a rapid reduction in pSPAK abundance in both kidney slices and HEK cells when exposed to high extracellular [K+] (16, 17). As previously reported by other groups, we could not detect such effects using immunoblots of whole kidney lysates (17). To explore the reasons for this difference between SPAK dephosphorylation behavior in vivo and ex vivo, we tested the hypothesis that SPAK dephosphorylation occurs in the highly [K+]-sensitive DCT1, but not in other segments. Indeed, we found that pSPAK abundance was reduced by high-K+ gavage only along the DCT1. Our observation suggests that, like NCC, dephosphorylation of SPAK was independent of systemic afferent cues. Importantly, we also detected DCT1-specific effects on SPAK distribution in vivo. Based on our data, we speculate that the activation status, i.e., the phosphorylation level of NCC, is a sum of the kinase and phosphatase activities. Altering exogenous conditions, such as increasing extracellular [K+], shifts the equilibrium toward dephosphorylation of the transporter, whereas decreasing extracellular [K+] shifts the equilibrium toward phosphorylation. The shifting of equilibrium toward dephosphorylation of NCC requires downregulation of both of the “on” switches, such as WNK4 and SPAK (Fig. 8). Our study indicates that phosphorylation or dephosphorylation of NCC is unlikely to be a standalone event.
Previous reports by Penton et al. (16) also found that high-K+ exposure reduced the abundance of pSPAK in ex vivo slices, but by incubating slices in a low-Cl− F, in which K+ effects remained, they concluded that changes in pSPAK abundance were not required for rapid dephosphorylation to occur. We used a different approach to seek evidence for a role of intracellular [Cl−] in rapid NCC dephosphorylation. Introduction of a Cl−-insensitive WNK4 mutant completely abrogated the effects of a high-[K+] medium on pNCC abundance in HEK cells. The conclusion that intracellular [Cl−] does play a key role is bolstered by the recent observation of Chen et al. (11) that mice expressing the same Cl−-insensitive WNK4 mutant are resistant to K+ gavage-induced NCC dephosphorylation. In addition, previous studies in mice expressing constitutively activated SPAK in the DCT1 showed elevated levels of NCC phosphorylation while having significantly elevated plasma [K+], suggesting that high plasma [K+] is not sufficient to dephosphorylate NCC in the background of activated SPAK (29); this is reminiscent of familial hyperkalemic hypertension, where the abundance of pNCC is high, despite hyperkalemia.
Interestingly, dephosphorylation of NCC in HEK cells and ex vivo occurs at a much lower extracellular [K+] than in animals or perfused kidneys (Figs. 5 and 6) (16). This difference may reflect differences in intracellular [Cl−] ex vivo. As noted, intracellular [Cl−] in HEK cells is ∼40 mM (1, 37), whereas that in DCT cells is ∼10−20 mM (12). Thus, the lower basal activity of WNK4 in HEK cells, owing to higher basal intracellular Cl−, might permit maximal dephosphorylation of NCC to occur at 3 mM K+ compared with 6−10 mM K+ in kidney slices.
The relation between plasma [K+] and pNCC abundance appears to be time dependent. When the pathway was stimulated by dietary K+ restriction for 5 days, high-K+ gavage was ineffective in reducing pNCC abundance, even when the blood [K+] increase was similar to that of 1-day dietary K+-restricted animals that were administered 3% KCl gavage. One possible explanation for this difference is that chronic dietary K+ restriction alters the basolateral membrane conductance of the DCT, thereby resetting the intracellular [Cl−] (40); such cell adaptation is likely to develop gradually and resolve slowly. In addition, other pathways may be reset, when dietary K+ intake is perturbed for longer periods. The current observation that longer-term changes in extracellular [K+] have effects beyond those observed acutely supports the observation of Chen et al. (11), who found that the WNK4 Cl−-insensitive mice are able to modulate NCC phosphorylation, but only when K+ loading is chronic.
Our results regarding rapid SPAK dephosphorylation by extracellular K+ resemble shared dephosphorylation mechanisms involving the kinase (SPAK) and a cation Cl− cotransporter substrate [Na+-K+-Cl− cotransporter (NKCC1)] observed by others. SPAK can be dephosphorylated directly by PP1 (6), an effect that is enhanced when the PP1 is scaffolded, either by NKCC1 or by another scaffolding protein (41, 42). NCC does not contain a consensus PP1-binding site along its NH2-terminus, unlike NKCC1. Yet, Loffing and colleagues recently reported that PP1 coimmunoprecipitates with the NH2-terminus of NCC (30). The effects of PP1 on SPAK occur very quickly, consistent with the timeframe observed here. The fact that SPAK was less sensitive to [K+] than was NCC in kidney slices suggests that SPAK dephosphorylation plays a permissive role, rather than a primary one. Yet, we did not observe dephosphorylation of NCC when cells were exposed to Cl−-insensitive WNK4, suggesting a complex relationship among rate constants for SPAK and NCC phosphorylation and dephosphorylation, as suggested in Fig. 8.
As observed by others, calyculin A, which can inhibit both PP1 and PP2, markedly increased the abundance of pNCC at baseline, consistent with a direct effect of PP1 on NCC, as suggested. Calyculin A, however, did not prevent dephosphorylation by 10 mM K+. There are several potential explanations for this observation. First, Gagnon et al. (41) reported a kinase-independent effect of PP1 on SPAK, one that would not be inhibited by calyculin A. Second, the concentration of calyculin A used might have inhibited PP1 incompletely, although we used the same calyculin A concentration (20 nM) recently used by Loffing and colleagues (16, 30). In contrast, this group previously used a higher concentration (50 nM) and found that pNCC abundance remained elevated when the extracellular [K+] bathing isolated kidney tubules was raised acutely (18). Finally, another PP may be involved. The calcineurin inhibitor tacrolimus also increased the abundance of pNCC at baseline, but, as previously reported by others, it also did not prevent dephosphorylation by 10 mM K+. Our findings are in line with the variable, relatively mild, and often delayed effects of calcineurin inhibitors on blood pressure of patients, suggesting an indirect role on NCC phosphorylation (21). One possibility, as suggested by a recent report (30), is that the effect may involve PP inhibitor 1, a canonical target of calcineurin (43), as suggested in Fig. 8. Calcineurin has also been shown to dephosphorylate Kelch-like protein 3, which increases its activity and leads to more degradation of WNK kinases (44). Furthermore, combined tacrolimus and calyculin A treatment did not prevent the decrease in pNCC abundance when cells were incubated at 10 mM K+. PP4 inhibition by fostriecin also appeared to be without effects on pNCC abundance, although it has been suggested that PP4 is key (23). When viewed together with results from other groups, the current results suggest that both PP1 and calcineurin play roles in modulating the abundance of pNCC, yet other mechanisms also appear to participate in the rapid dephosphorylation that occurs during exposure to high [K+].
Overall, our data suggest that NCC dephosphorylation is a complex process, likely reflecting simultaneous modifications of rate constants for each of the activation/inactivation reactions shown in Fig. 8. The current results suggest that SPAK dephosphorylation plays at least a permissive role in rapid NCC dephosphorylation. They suggest that changes in intracellular [Cl−] are required for dephosphorylation to occur. They do not, of course, exclude pathways that are independent of Cl−, as suggested by others. The current results also suggest that the DCT resets, during chronic changes in K+ balance, to alter the effects of a sudden change in plasma [K+]. This finding is consistent with the basolateral K+ channel resetting that we have previously demonstrated (27, 40, 45). When combined with K+-stimulated aldosterone secretion, these effects likely contribute to the kidney’s remarkable ability to eliminate a K+ load rapidly.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants R01DK051496 and R01DK054983, Veterans Affairs Grant 1I01BX002228, and a Fondation LeDucq Transatlantic Network of Excellence (to D.H.E.) as well as NIDDK Grant DK098141 (to J.A.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M., C.-L.Y., J.A.M., and D.H.E. conceived and designed research; A.M. and A.S. performed experiments; A.M., C.-L.Y., K.M., and D.H.E. analyzed data; A.M., C.-L.Y., and D.H.E. interpreted results of experiments; A.M. and D.H.E. prepared figures; A.M. drafted manuscript; A.M., C.-L.Y., J.A.M., K.M., A.S., and D.H.E. edited and revised manuscript; A.M., C.-L.Y., J.A.M., K.M., A.S., and D.H.E. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Dr. Charles Allen for generously allowing us to use laboratory space and equipment and Nathan Klett for technical assistance in conducting in vivo kidney slicing experiments. We also acknowledge Kayla Erspamer, Lauren Miller, and Brittany Gratreak for the excellent technical support.
REFERENCES
- 1.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vitzthum H, Seniuk A, Schulte LH, Muller ML, Hetz H, Ehmke H. Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J Physiol 592: 1139–1157, 2014. doi: 10.1113/jphysiol.2013.266924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK kinase signaling in ion homeostasis and human disease. Cell Metab 25: 285–299, 2017. doi: 10.1016/j.cmet.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 6.Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S. Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5: 331–344, 2007. doi: 10.1016/j.cmet.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 7.Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem 280: 42685–42693, 2005. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- 8.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferdaus MZ, Barber KW, Lopez-Cayuqueo KI, Terker AS, Argaiz ER, Gassaway BM, Chambrey R, Gamba G, Rinehart J, McCormick JA. SPAK and OSR1 play essential roles in potassium homeostasis through actions on the distal convoluted tubule. J Physiol 594: 4945–4966, 2016. doi: 10.1113/JP272311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 11.Chen JC, Lo YF, Lin YW, Lin SH, Huang CL, Cheng CJ. WNK4 kinase is a physiological intracellular chloride sensor. Proc Natl Acad Sci USA 116: 4502–4507, 2019. doi: 10.1073/pnas.1817220116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su XT, Klett NJ, Sharma A, Allen CN, Wang WH, Yang CL, Ellison DH. Distal convoluted tubule chloride concentration is modulated via potassium channels and transporters. Am J Physiol Renal Physiol 319: F534–F540, 2020. doi: 10.1152/ajprenal.00284.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bazua-Valenti S, Chavez-Canales M, Rojas-Vega L, Gonzalez-Rodriguez X, Vazquez N, Rodriguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, Garcia-Valdes J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+-Cl- cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2015. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, Loffing J. Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl- -dependent and independent mechanisms. J Physiol 594: 6319–6331, 2016. doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shoda W, Nomura N, Ando F, Mori Y, Mori T, Sohara E, Rai T, Uchida S. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int 91: 402–411, 2017. doi: 10.1016/j.kint.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 19.McCormick JA, Ellison DH. Distal convoluted tubule. Compr Physiol 5: 45–98, 2015. doi: 10.1002/cphy.c140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Picard N, Trompf K, Yang C-L, Miller RL, Carrel M, Loffing-Cueni D, Fenton RA, Ellison DH, Loffing J. Protein phosphatase 1 inhibitor-1 deficiency reduces phosphorylation of renal NaCl cotransporter and causes arterial hypotension. J Am Soc Nephrol 25: 511–522, 2014. doi: 10.1681/ASN.2012121202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoorn EJ, Walsh SB, McCormick JA, Furstenberg A, Yang CL, Roeschel T, Paliege A, Howie AJ, Conley J, Bachmann S, Unwin RJ, Ellison DH. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 17: 1304–1309, 2011. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazelle RA, McCully BH, Terker AS, Himmerkus N, Blankenstein K, Mutig K, Bleich M, Bachman S, Yang C-L, Ellison DH. Renal FKBP12 deletion attenuates tacrolimus-induced hypertension. J Am Soc Nephrol 27: 1456–1464, 2016. doi: 10.1681/ASN.2015040466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glover M, Mercier Zuber A, Figg N, O'Shaughnessy KM. The activity of the thiazide-sensitive Na(+)-Cl(-) cotransporter is regulated by protein phosphatase PP4. Can J Physiol Pharmacol 88: 986–995, 2010. doi: 10.1139/y10-080. [DOI] [PubMed] [Google Scholar]
- 24.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, Hartshorne DJ. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun 159: 871–877, 1989. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 27.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28: 1814–1825, 2017. doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Namboodiri HN, Nkashama LJ, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29: 499–509, 2018. doi: 10.1091/mbc.E17-08-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penton D, Moser S, Wengi A, Czogalla J, Rosenbaek LL, Rigendinger F, Faresse N, Martins JR, Fenton RA, Loffing-Cueni D, Loffing J. Protein phosphatase 1 inhibitor-1 mediates the cAMP-dependent stimulation of the renal NaCl cotransporter. J Am Soc Nephrol 30: 737–750, 2019. doi: 10.1681/ASN.2018050540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306: F1059–F1068, 2014. [Erratum in Am J Physiol Renal Physiol 10: F688, 2016]. doi: 10.1152/ajprenal.00015.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savir Y, Tu BP, Springer M. Competitive inhibition can linearize dose-response and generate a linear rectifier. Cell Syst 1: 238–245, 2015. doi: 10.1016/j.cels.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Q, Wu Y, Jonusaite S, Pleinis JM, Humphreys JM, He H, Schellinger JN, Akella R, Stenesen D, Kramer H, Goldsmith EJ, Rodan AR. Intracellular chloride and scaffold protein Mo25 cooperatively regulate transepithelial ion transport through WNK signaling in the malpighian tubule. J Am Soc Nephrol 29: 1449–1461, 2018. doi: 10.1681/ASN.2017101091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terker AS, Yang CL, McCormick JA, Meermeier NP, Rogers SL, Grossmann S, Trompf K, Delpire E, Loffing J, Ellison DH. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 64: 178–184, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hemmings HC , Jr., Nairn AC, Greengard P. DARPP-32, a dopamine- and adenosine 3':5'-monophosphate-regulated neuronal phosphoprotein. II. Comparison of the kinetics of phosphorylation of DARPP-32 and phosphatase inhibitor 1. J Biol Chem 259: 14491–14497, 1984. [PubMed] [Google Scholar]
- 36.Lin SH, Yu IS, Jiang ST, Lin SW, Chu P, Chen A, Sytwu HK, Sohara E, Uchida S, Sasaki S, Yang SS. Impaired phosphorylation of Na(+)-K(+)-2Cl(-) cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci USA 108: 17538–17543, 2011. doi: 10.1073/pnas.1107452108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamprecht G, Schaefer J, Dietz K, Gregor M. Chloride and bicarbonate have similar affinities to the intestinal anion exchanger DRA (down regulated in adenoma). Pflugers Arch 452: 307–315, 2006. doi: 10.1007/s00424-006-0049-6. [DOI] [PubMed] [Google Scholar]
- 38.McDonough AA, Youn JH. Need to quickly excrete K(+)? Turn off NCC. Kidney Int 83: 779–782, 2013. doi: 10.1038/ki.2012.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabelink TJ, Koomans HA, Hene RJ, Dorhout Mees EJ. Early and late adjustment to potassium loading in humans. Kidney Int 38: 942–947, 1990. doi: 10.1038/ki.1990.295. [DOI] [PubMed] [Google Scholar]
- 40.Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 93: 893–902, 2018. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gagnon KB, Delpire E. Multiple pathways for protein phosphatase 1 (PP1) regulation of Na-K-2Cl cotransporter (NKCC1) function: the N-terminal tail of the Na-K-2Cl cotransporter serves as a regulatory scaffold for Ste20-related proline/alanine-rich kinase (SPAK) AND PP1. J Biol Chem 285: 14115–14121, 2010. doi: 10.1074/jbc.M110.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gagnon KB, England R, Diehl L, Delpire E. Apoptosis-associated tyrosine kinase scaffolding of protein phosphatase 1 and SPAK reveals a novel pathway for Na-K-2C1 cotransporter regulation. Am J Physiol Cell Physiol 292: C1809–C1815, 2007. doi: 10.1152/ajpcell.00580.2006. [DOI] [PubMed] [Google Scholar]
- 43.Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol 47: 365–371, 2009. doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishizawa K, Wang Q, Li J, Yamazaki O, Tamura Y, Fujigaki Y, Uchida S, Lifton RP, Shibata S. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc Natl Acad Sci USA 116: 3155–3160, 2019. doi: 10.1073/pnas.1817281116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welling PA. Roles and regulation of renal K channels. Annu Rev Physiol 78: 415–435, 2016. doi: 10.1146/annurev-physiol-021115-105423. [DOI] [PubMed] [Google Scholar]
- 46.Bostanjoglo M, Reeves B, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998. [DOI] [PubMed] [Google Scholar]