

Keywords: collecting duct, distal convoluted tubule, Kir4.1/Kir5.1, renal K+ excretion
Abstract
Neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) regulates the expression of Kir4.1, thiazide-sensitive NaCl cotransporter (NCC), and epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron (ASDN), and Nedd4-2 deletion causes salt-sensitive hypertension. We now examined whether Nedd4-2 deletion compromises the effect of high-salt (HS) diet on Kir4.1, NCC, ENaC, and renal K+ excretion. Immunoblot analysis showed that HS diet decreased the expression of Kir4.1, Ca2+-activated large-conductance K+ channel subunit-α (BKα), ENaCβ, ENaCγ, total NCC, and phospho-NCC (at Thr53) in floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl) mice, whereas these effects were absent in kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice. Renal clearance experiments also demonstrated that Nedd4-2 deletion abolished the inhibitory effect of HS diet on hydrochlorothiazide-induced natriuresis. Patch-clamp experiments showed that neither HS diet nor low-salt diet had an effect on Kir4.1/Kir5.1 currents of the distal convoluted tubule in Nedd4-2-deficient mice, whereas we confirmed that HS diet inhibited and low-salt diet increased Kir4.1/Kir5.1 activity in Nedd4lflox/flox mice. Nedd4-2 deletion increased ENaC currents in the ASDN, and this increase was more robust in the cortical collecting duct than in the distal convoluted tubule. Also, HS-induced inhibition of ENaC currents in the ASDN was absent in Nedd4-2-deficient mice. Renal clearance experiments showed that HS intake for 2 wk increased the basal level of renal K+ excretion and caused hypokalemia in Ks-Nedd4-2-KO mice but not in Nedd4lflox/flox mice. In contrast, plasma Na+ concentrations were similar in Nedd4lflox/flox and Ks-Nedd4-2 KO mice on HS diet. We conclude that Nedd4-2 plays an important role in mediating the inhibitory effect of HS diet on Kir4.1, ENaC, and NCC and is essential for maintaining normal renal K+ excretion and plasma K+ ranges during long-term HS diet.
NEW & NOTEWORTHY The present study suggests that Nedd4-2 is involved in mediating the inhibitory effect of high salt (HS) diet on Kir4.1/kir5.1 in the distal convoluted tubule, NaCl cotransporter function, and epithelial Na+ channel activity and that Nedd4-2 plays an essential role in maintaining K+ homeostasis in response to a long-term HS diet. This suggests the possibility that HS intake could lead to hypokalemia in subjects lacking proper Nedd4-2 E3 ubiquitin ligase activity in aldosterone-sensitive distal nephron.
INTRODUCTION
Neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) is an E3 ubiquitin ligase expressed in the early distal convoluted tubule (DCT1) and aldosterone-sensitive distal nephron (ASDN) (1, 2) and plays a role in regulating the abundance of the epithelial Na+ channel (ENaC) in the ASDN and NaCl cotransporter (NCC) and Kir4.1 in the DCT (1, 3–9). Deletion of Nedd4-2 causes salt-sensitive hypertension due to high NCC and ENaC activity (5). Our previous in vitro study demonstrated that Nedd4-2 was associated with Kir5.1 at the COOH-terminus and regulated Kir4.1 ubiquitination, thereby inhibiting Kir4.1 activity (10). The role of Nedd4-2 in regulating Kir4.1 in the DCT was also strongly suggested by the finding that deletion of Nedd4-2 increased Kir4.1 expression and hyperpolarized the DCT membrane (8). In addition to Nedd4-2, dietary Na+ intake also regulates the basolateral Kir4.1/Kir5.1 channel in the DCT and NCC and ENaC in the ASDN such that high-salt (HS) diet inhibits, whereas low-salt (LS) diet stimulates, Kir4.1/Kir5.1, NCC, and ENaC (11–16). Since both factors, HS diet and Nedd4-2-activation, are involved in inhibiting Kir4.1/Kir5.1, NCC, and ENaC, the first aim of the present study was to test whether deletion of Nedd4-2 compromises the effect of HS intake on basolateral Kir4.1/Kir5.1 activity in the DCT and NCC and ENaC in the ASDN. Also, HS-induced inhibition of NCC and ENaC expression/activity in the ASDN is essential not only for inhibiting renal Na+ absorption but also for keeping adequate K+ excretion during increased dietary Na+ intake (17–19). For instance, HS-induced inhibition of NCC should increase Na+ and fluid volume delivery to the late ASDN. Thus, although HS diet inhibits ENaC activity in the ASDN, increased Na+ and fluid volume delivery to the late portion of the ASDN should offset the effect of low ENaC activity, thereby keeping a stable renal K+ excretion rate and maintaining K+ homeostasis (20, 21). Thus, ENaC and NCC work in concert for maintaining an adequate renal K+ excretion rate and K+ homeostasis despite variation of dietary Na+ intake (22). Indeed, increased dietary Na+ intake has no significant effect on plasma K+ concentration under physiological conditions (23). Since Nedd4-2 plays a role in inhibiting ENaC in the ASDN and NCC and Kir4.1 in the DCT, the three important components involved in regulating renal K+ excretion (18), it is conceivable that Nedd4-2 may play a role in maintaining K+ homeostasis. Thus, the second aim of the present study was to examine whether deletion of Nedd4-2 affects renal K+ excretion and K+ homeostasis during increasing dietary Na+ intake.
METHODS
Animals
We used floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl) mice as control mice [referred to as “wild type” (WT) mice] and kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice in our study (5). Briefly, mice expressing Pax8-rtTA and tet-on LC-1, which tightly drive Pax8 and Cre expression under tetracycline-dependent induction, were crossed with Nedd4lfl/fl mice to generate inducible Ks-Nedd4-2 KO mice. We fed 10- to 12-wk-old Pax8-cre-Nedd4lfl/fl mice with doxycycline (2 mg in 2% sucrose solution) for 2 wk, and mice were then allowed to recover for additional 2 wk. We randomly selected three mice from each group of peers to conduct Western blot analysis to confirm the deletion of Nedd4-2. We also fed Nedd4lfl/fl mice with doxycycline at the beginning of the study. Since doxycycline treatment did not have an effect on Kir4.1/Kir5.1 and NCC expression, we just treated control mice with sucrose (2%) late. Animals were housed in the New York Medical College animal facility with lights on at 7:00 AM and off at 7:00 PM. Mice had unlimited access to water and rodent chow. To study the effect of dietary Na+ intake on renal K+ excretion, mice were fed the control diet (0.8% K+ and 0.4% Na+), HS diet (4% Na+ and 0.8% K+), or LS diet (0.01%–0.02% Na+ and 0.8% K+) for 7–14 days. The HS (4%) diet (Cat. No. TD.92034) and LS diet (Cat. No. TD.90228) were purchased from Envigo-Teklad Diets (Madison, WI). All procedures were reviewed and approved by the Institutional Animal Care and Use Committee.
Genotyping
Tail DNA was PCR amplified. For Nedd4l, the forward and reverse primers were 5′- TGAGCTCATTGCTTCACTTCC-3′ and 5′- TTCATGCTCGAAGCCTTAGC-3′, respectively (230 bp for floxed Nedd4l and 150 bp for WT). For Pax8-rtTA, the forward and reverse primers were 5′- CCATGTCTAGACTGGACAAGA-3′ and 5′- CAGAAAGTCTTGCCATGACT-3′, respectively (a 220-bp product). For LC1-CRE, the primers were forward 5′- TTTCCCGCAGAACCTGAAGATG-3′ and reverse 5′- TCACCGGCATCAACGTTTTCTT-3′, respectively (a 190-bp product).
Preparation of the DCT
Mice were euthanized by cervical dislocation, and the abdomen was opened to expose the left kidney. We perfused the left kidney with 2-mL L-15 medium (Life Technology) containing collagenase type 2 (250 U/mL) and then removed the collagenase-perfused kidney. The renal cortex was separated and further cut into small pieces for additional incubation in collagenase-containing L-15 media for 30–50 min at 37°C. The tissue was then washed three times with fresh L-15 medium and transferred to an ice-cold chamber for dissection. Isolated DCT tubules were placed on a small coverglass coated with poly-l-lysine, and the coverglass was placed on a chamber mounted on an inverted microscope.
Single-Channel Recording
We used freshly isolated DCTs of both male and female mice for conducting single K+ channel experiments. Since we did not detect significant difference regarding channel activity, data were pooled. Single-channel patch-clamp experiments for studying the 40-pS K+ channel (Kir4.1/Kir5.1) were performed in the basolateral membrane of both the DCT1 and late DCT (DCT2). Single K+ channel currents were recorded with an Axon200B amplifier (Axon), low-pass filtered at 1 kHz, and digitized by an Axon interface (Digidata 1332) with a sampling rate of 4 kHz. The pipette solution for the single-channel recording contained (in mM) 135 KCl, 2 MgCl2, 1 EGTA, and 10 HEPES (titrated with KOH to pH 7.4), and the bath solution contained (in mM) 135 NaCl, 5 KCl, 2 MgCl2, 1.8 CaCl2, 5 glucose, and 5 HEPES (titrated with NaOH to pH 7.4). For the calculation of channel numbers, we selected a channel recording of at least 10-min long. We determined the channel open probability (Po) from the channel number (N) and NPo (the product of N and Po), which was calculated from data samples of 60-s duration at steady state. NPo was determined using the following equation:
where ti is the fractional open time spent at each of the observed current levels. Channel conductance was determined by measuring the current amplitudes over several voltages.
Whole Cell Recording
We measured whole-cell K+ currents and K+ current reversal potential only in the DCT1 of male and female mice with an Axon 200A amplifier. To measure K+ current reversal potential, the tip of the pipette was filled with a pipette solution containing (in mM) 140 KCl, 2 MgCl2, 1 EGTA, and 10 HEPES (pH 7.4). The pipette was then backfilled with the pipette solution containing amphotericin B (20 μg/0.1 mL). The bath solution was the same as the one we used to perform single-channel recordings. For the measurement of whole-cell Ba2+-sensitive K+ current, the bath solution contained (in mM) 140 KCl, 2 MgCl2, 1.8 CaCl2, and 10 HEPES (pH 7.4). After forming a high-resistance seal (24), the membrane capacitance was monitored until the whole-cell patch configuration was formed. We measured whole-cell currents twice (before and after 0.1 mM Ba2+), and Ba2+-sensitive K+ currents were obtained by subtracting Ba2+-insensitive currents from total currents. The currents were low-pass filtered at 1 kHz and digitized by an Axon interface with a 4-kHz sampling rate (Digidata 1440A). Data were analyzed using pClamp software system 9.0 (Axon).
To study ENaC, we performed the patch-clamp experiments in split-open tubules in the last 100 µm of the DCT before the start of the connecting tubule (CNT) and in the cortical collecting duct (CCD) of male mice. Since the diameter of the DCT2 is normally larger than the CNT (25), this anatomic characterization has been used to determine the end of the DCT or the start of the CNT. However, since it was not always obvious to identify the beginning of the CNT, some experiments were actually performed in the early CNT. Thus, we refer that the study was performed in the DCT2/CNT. ENaC currents were determined by adding amiloride (10 µΜ) in the bath solution. The pipette solution contained (in mM) 125 K-gluconate, 15 KCl, 2 MgATP, 1 EGTA, and 10 HEPES (pH 7.4), and the bath solution contained (in mM) 130 Na-gluconate, 10 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, and 5 HEPES (pH 7.4). We filled the tip of the pipette first with the aforementioned solution without amphotericin B, and the pipette was then backfilled with the solution containing amphotericin B (20 μg/0.1 mL).
Measurement of Renal Na+ and K+ Excretion
Animals were anesthetized with a peritoneal injection of Inactin at 100 mg/kg, and mice were placed on a heated small blanket to maintain body temperature at 37°C. The trachea was cannulated to clear mucus, and a carotid artery was catheterized with PE-10 tubing for blood collection. A jugular vein was also cannulated for intravenous infusion, and the bladder was exposed for urine collections (catheterized via a suprapubic incision with a 10-cm piece of PE-10 tubing). After the completion of surgery, isotonic saline was given intravenously for 4 h (0.25–0.3 mL/1 h and total 1.0–1.2 mL of 0.9% saline) to replace surgical fluid losses and to maintain hemodynamics. Urine collections started 1 h after the infusion of 0.3 mL saline, and six total collections (every 30 min) were performed [two collections before and four collections after hydrochlorothiazide (HCTZ) at 30 mg/kg body wt]. To measure the basal level of urinary K+ excretion, urine samples were collected every 30 min for six total collections, and samples were pooled. Plasma and urine Na+ and K+ concentrations were measured using a dual-channel flame photometer with an internal lithium standard (Cole-Parmer Instrument, Vernon Hills, IL).
Immunoblot Analysis
Whole kidney protein extract (male mice only) was obtained from frozen kidneys homogenized in buffer containing 250 mM sucrose, 50 mM Tris·HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, and 1 mM DTT supplemented with phosphatase and protease inhibitor cocktails (Sigma). Protein (40–60 µg) was separated on 4%–12% (wt/vol) Tris-glycine gels (Thermo Fisher Scientific) and transferred to nitrocellulose membranes. Membranes were incubated for 1 h with LI-COR blocking buffer (PBS) and then incubated overnight at 4°C with NCC and phosphorylated (p)NCC. An Odyssey infrared imaging system (LI-COR) was used to capture the images at a wavelength of 680 or 800 nm.
Materials
HCTZ, Inactin, and amiloride were purchased from Sigma-Aldrich (St. Louis, MO), and the information regarding all antibodies used for Western blot analysis is shown in Table 1.
Table 1.
Antibodies used for Western blots
| Antibody | Species | Dilution for Western Blot | Source |
|---|---|---|---|
| pT53-NCC | Rabbit | 1:3,000 | PhosphoSolutions (8) |
| NCC | Rabbit | 1:2,000 | Millipore (8) |
| ENaCα | Rabbit | 1:1,000 | StressMarq (25) |
| ENaCβ | Rabbit | 1:1,000 | StressMarq (25) |
| ENaCγ | Rabbit | 1:1,000 | StressMarq (25) |
| Kir4.1 (APC-035-AG) | Rabbit | 1:200 | AlomoneLabs (26) |
| Kir5.1 | Rabbit | 1:200 | Alomone Labs (16) |
| BKα | Goat | 1:200 | Santa Cruz Biotechnology (27) |
| Nedd4-2 | Rabbit | 1.1,000 | Cell Signaling (8) |
| Actin | Rabbit | 1.1,000 | Cell Signaling (28) |
| GAPDH | Rabbit | 1:1,000 | Cell Signaling (28) |
BKα, large-conductance K+ channel α-subunit; ENaC, epithelial Na+ channel; NCC, NaCl cotransporter; Nedd4-2, neural precursor cell expressed developmentally downregulated protein 4-2; pT53-NCC, phospho-Thr53 NaCl cotransporter.
Statistical Analysis
We used software (Sigmaplot) for the statistical analysis. We used a t test for analyzing the values between two groups and used one-way or two-way ANOVA for analyzing the results of more than two groups. A Holm–Sidak test was used as post hoc analysis. P values of <0.05 were considered statistically significant. Data are presented as means ± SE.
RESULTS
Deletion of Nedd4-2 Blunts the Effect of HS diet on Kir4.1/Kir5.1 in the DCT
We used single-channel recordings to examine the effect of HS or LS diets on the basolateral 40-pS K+ channel (Kir4.1/Kir5.1 heterotetramer) in Nedd4lfl/fl (WT; Fig. 1A) and Ks-Nedd4-2 KO mice (Fig. 1B). Although HS diet decreased (0.97 ± 0.17) and LS diet increased (2.07 ± 0.11) 40-pS K+ channel activity defined by NPo compared with normal salt (NS; 1.40 ± 0.09), as previously reported (Fig. 1, A and C) (16), deletion of Nedd4-2 not only increased the probability of finding the 40-pS K+ channel and increased NPo (Table 2 and Fig. 1C) but also abolished the effect of dietary Na+ intake on 40-pS K+ channel activity in the DCT (Fig. 1, B and C). The results are shown in Fig. 1C, demonstrating that 40-pS K+ channel activity (NPo) was 2.03 ± 0.15 (n = 11), 1.93 ± 0.10 (n = 12), and 2.0 ± 0.17 (n = 12) in Ks-Nedd4-2 KO mice on NS, HS diets (14 days), and LS diet (14 days), respectively.
Figure 1.

Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) abolishes the effect of dietary Na+ intake on the basolateral K+ channel in the distal convoluted tubule (DCT). A and B: representative single channel recordings showing basolateral K+ channel activity in the DCT of wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] mice (A) and kidney-specific Nedd2-4 knockout (Ks-Nedd4-2 KO) mice (B) on normal-salt (NS), high-salt (HS), or low-salt (LS) diets for 14 days, respectively. C: scatterplot summarizing the results of experiments in which Kir4.1/Kir5.1 channel activity in the DCT [defined by channel activity (NPo)] was measured in both male and female WT and Ks-Nedd4-2 KO mice on different Na+ diets for 14 days. The experiments were performed in cell-attached patches at a holding potential of 0 mV. The DCT was bathed in a solution containing 140 mM NaCl/5 mM KCl, and the pipette solution contained 145 mM K+. Significance was determined by two-way ANOVA.
Table 2.
Effect of Na+ diets on basolateral K+ channel activity in the distal convoluted tubule
| Na+ Diet Type | Numbers of Total Patches (Tubules) | Numbers of Patches with Kir4.1/Kir5.1 | Mean NPo/Patch | Mean Po |
|---|---|---|---|---|
| HS (WT) | 38 | 14 (37%)* | 0.97 ± 0.17 (n = 6)* | 0.34 ± 0.03* |
| NS (WT) | 35 | 18 (51%) | 1.40 ± 0.09 (n = 10) | 0.42 ± 0.02 |
| LS (WT) | 29 | 19 (66%)* | 2.07 ± 0.11 (n = 8) | 0.53 ± 0.04 |
| HS (Nedd4-2 KO) | 30 | 22 (73%)* | 1.93 ± 0.10 (n = 12) | 0.49 ± 0.02 |
| NS (Nedd4-2 KO) | 31 | 22 (71%)* | 2.03 ± 0.15 (n = 11) | 0.50 ± 0.02 |
| LS (Nedd4-2 KO) | 35 | 24 (69%)* | 2.00 ± 0.17 (n = 12) | 0.55 ± 0.04 |
Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 [Nedd4-2 knockout (KO)] abolished the effect of dietary Na+ intake on Kir4.1/Kir5.1. Single-channel recording was used to examine the effect of low salt (LS; 14 days) or high salt (HS; 14 days) on basolateral 40-pS K+ channels (Kir4.1/Kir5.1) in the early and late distal convoluted tubules of both male and female mice (data were pooled). Experiments were performed in cell-attached patches with 140 mM Na+/5 mM K+ in the bath and 145 mM K+ in the pipette. The patch-clamp holding potential was 0 mV. NPo is the product of channel open probability (Po) and channel number (N). NS, normal salt; WT, wild type.
Significant difference compared with the wild-type (WT) control (normal salt).
The notion that deletion of Nedd4-2 abolished HS-induced inhibition of Kir4.1/Kir5.1 in the DCT was also suggested in experiments in which we used perforated whole-cell recordings to examine Ba2+-sensitive K+ currents in the DCT1. Figure 2, A and B, shows a set of traces showing Ba2+-sensitive K+ currents measured with a step protocol and ramp protocol, respectively. Figure 2C shows a scatterplot summarizing the results measured at −60 mV in the DCT1 of mice on different Na+ diets. It is apparent that the effect of dietary Na+ intake on Kir4.1/Kir5.1 was absent in Ks-Nedd4-2 KO mice [HS (7 days): 2,060 ± 100 pA, n = 7; HS (14 days): 2,030 ± 80 pA, n = 16; NS: 1,850 ± 110 pA, n = 10; and LS (14 days): 1,990 ± 130 pA, n = 10] but not in WT mice [LS (14 days): 2,050 ± 135 pA, n = 6; HS (7 days): 640 ± 30 pA, n = 7; HS (14 days): 630 ± 41 pA, n = 7; and NS: 1,130 ± 83 pA, n = 5].
Figure 2.

Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) eliminates the effect of dietary Na+ intake on the basolateral K+ conductance in the distal convoluted tubule (DCT). A: set of traces showing Ba2+-sensitive K+ currents measured with a step protocol from −60 to 60 mV in the DCT of wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] mice and kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice on normal-salt (NS), high-salt (HS), and low-salt (LS) diets for 14 days, respectively. B: traces showing Ba2+-sensitive K+ currents measured with a ramp protocol (from −100 to 100 mV) in Ks-Nedd4-2 KO mice on NS, HS, and LS diets for 14 days, respectively. (A trace from a control mouse on NS is included as a reference.) The DCT was bathed in 140 mM KCl-containing solution, and the pipette solution also contained 140 mM KCl. C: scatterplot summarizing K+ currents measured at –60 mV in the DCT of male and female WT and KS-Nedd4-2 KO mice on HS for 7 days (HS-7d), HS for 14 days (HS-14d), NS, and LS for 14 days (LS-14d). Mean values and SEs are shown on the left of each column. Significance was determined by two-way ANOVA.
Since Kir4.1/5.1 activity determines the negativity of the DCT membrane potential (29), we next used the whole-cell recordings to measure the K+ current reversal potential (an index of DCT membrane potential) of the DCT1 in WT and Ks-Nedd4-2 KO mice on different Na+ diets for 14 days. Figure 3, A and B, shows typical traces of K+ current reversal potential of the DCT1 in WT or Ks-Nedd4-2 KO mice on HS, NS, and LS diets for 14 days, respectively. Figure 3C shows the results of the experiments. Although K+ current reversal potential in the DCT1 was more negative in Nedd4-2-deficient mice (−73 ± 0.6 mV, n = 11) than in WT mice on NS diet (−61 ± 0.5 mV, n = 5), neither HS diet (−71 ± 0.7 mV, n = 13) nor LS diet (−72 ± 1.1 mV, n = 8) significantly changed K+ current reversal potential of the DCT in Ks-Nedd4-2 KO mice compared with NS diet. In contrast, dietary Na+ intake altered the K+ current reversal potential of the DCT in WT mice (HS: −53 ± 1.5 mV, n = 6; LS: −72 ± 1.1 mV, n = 8). The notion that deletion of Nedd4-2 compromised the effect of HS diet on Kir4.1 was also suggested by Western blot analysis showing that HS diet decreased Kir4.1 expression by 30% ± 3% (n = 4) only in WT mice but not in Ks-Nedd4-2 KO mice (Fig. 4A). The effect of Nedd4-2 on HS-induced inhibition of Kir4.1 was specific since deletion of Nedd4-2 had no effect on Kir5.1 expression. Moreover, HS diet also decreased the expression of BKα in the kidney by 20% ± 3% (n = 4; Fig. 4A).
Figure 3.

Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) abolishes the effect of dietary Na+ intake in the distal convoluted tubule (DCT) membrane potential. A: perforated whole cell recordings showing K+ current (IK) reversal potential in the DCT of wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] mice on normal-salt (NS), high-salt (HS), and low-salt (LS) diets for 14 days, respectively. B: perforated whole cell recording showing the IK reversal potential in the DCT of kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice on NS, HS, and LS diets for 14 days, respectively. (A trace from a control mouse on NS is included as a reference.) C: scatterplot summarizing the results of experiments in which the IK reversal potential was measured in both male and female WT and Ks-Nedd4-2 KO mice on different Na+ diets for 14 days. Mean values and SEs are shown on the left of each column. For the measurement of IK reversal potential, the bath solution contained 140 mM NaCl and 5 mM KCl and the pipette solution had 140 mM KCl. Significance was determined by two-way ANOVA.
Figure 4.

High salt (HS) inhibits expression of Kir4.1, large-conductance K+ (BK) channels, and epithelial Na+ channels (ENaC). A, top: expression of Kir4.1, Kir5.1, and the BK channel α-subunit (BKα) in male wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] and kidney-specific neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) knockout (Ks-Nedd4-2 KO) mice on normal salt (NS) or HS for 14 days. A, bottom: bar graph showing normalized band density of Kir4.1, Kir5.1, and BKα. B, top: expression of ENaCα, ENaCβ, and ENaCγ in male WT and Ks-Nedd4-2 KO mice on NS or HS for 14 days. B, bottom: bar graph showing normalized band density of ENaCα, ENaCβ, and ENaCγ. Significance was determined by two-way ANOVA.
Deletion of Nedd4-2 Increased ENaC Activity in the DCT2/CNT and in the CCD
We next examined the effect of HS diet on ENaC expression in WT and Ks-Nedd4-2 KO mice (Fig. 4B). It is apparent that HS diet decreased the expression of ENaCβ (30 ± 3%, n = 4) and ENaCγ (28 ± 3%, n = 4) in WT mice compared with NS diet. Deletion of Nedd4-2 not only increased the expression of ENaCβ (125 ± 6% of the WT control, n = 4) and ENaCγ (130 ± 6% of the WT control, n = 4) but also abolished the effect of HS diet on ENaC in Ks-Nedd4-2 KO mice. Moreover, we confirmed that HS diet did not affect the expression of full-length ENaCα (5). We next used the patch-clamp method to examine the effect of HS diet on ENaC activity in the DCT2/CNT and in the CCD by measuring amiloride-sensitive whole-cell Na+ currents in WT and Ks-Nedd4-2 KO mice. Figure 5A shows a set of gap-free whole-cell recordings showing the effect of amiloride (10 µM) on Na+ currents of the DCT2/CNT in WT and Ks-Nedd4-2 KO mice on NS and HS diets for 14 days, respectively. Figure 5B shows a scatterplot showing the mean value and each data point of the experiments in which ENaC currents were measured at −60 mV. ENaC currents in the DCT2/CNT were 212 ± 10 pA/cell (n = 8) and 284 ± 31 pA/cell (n = 9) in WT and Ks-Nedd4-2 KO mice on NS diet, respectively. HS diet decreased ENaC currents in the DCT2/CNT only in WT mice but not in Ks-Nedd4-2 KO mice (WT: 88 ± 10 pA/cell, n = 7; Ks-Nedd4-2 KO: 266 ± 27 pA/cell, n = 7). We also examined ENaC currents in the CCD of WT and Ks-Nedd4-2 KO mice. Figure 6A shows two sets of gap-free recordings showing Na+ currents of the CCD in WT and Ks-Nedd4-2 KO mice on NS and HS diets for 14 days, respectively. Figure 6B shows a scatterplot showing the mean value and each data point of ENaC currents of the CCD measured at −60 mV. ENaC current in the CCD was 65 ± 5 pA/cell (n = 8) in WT but was 217 ± 17 pA/cell (n = 8) in Ks-Nedd4-2 KO mice. Also, HS intake did not inhibit ENaC in the CCD of Ks-Nedd4-2 KO mice (260 ± 18 pA/cell, n = 7), but it further decreased ENaC current to 31 ± 4 pA/cell in WT mice (n = 7). Thus, Nedd4-2 plays a key role in mediating the inhibitory effect of HS diet on ENaC activity in the DCT2/CNT and in the CCD.
Figure 5.

High salt (HS) inhibits epithelial Na+ channels (ENaC) in wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] mice but not kidney-specific neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) knockout (Ks-Nedd4-2 KO) mice. A: set of gap-free recordings showing amiloride-sensitive Na+ currents in the late distal convoluted tubule (DCT2)/connecting tubule (CNT) measured at −60 mV with the perforated whole cell patch in WT and Ks-Nedd4-2 KO mice on normal salt (NS) and HS (14 days; HS-14d), respectively. B: scatterplot summarizing the results of experiments in which amiloride (10 µM)-sensitive Na+ currents were measured at −60 mV with perforated whole cell recordings in the DCT2/CNT of male WT and Ks-Nedd4-2 KO mice on NS or HS diets for 14 days, respectively. Significance was determined by two-way ANOVA.
Figure 6.

Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) increases epithelial Na+ channel (ENaC) activity in the cortical collecting duct (CCD) and abolishes the effect of high salt (HS) on ENaC. A: set of gap-free recordings showing amiloride-sensitive Na+ currents in the CCD measured at −60 mV with the perforated whole cell patch in wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] and kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice on normal salt (NS) and HS (14 days; HS-14d), respectively. B: scatterplot summarizing the results of experiments in which amiloride (10 µM)-sensitive Na+ currents were measured at −60 mV with perforated whole cell recordings in the CCD of male WT and Ks-Nedd4-2 KO mice on NS or HS diets for 14 days, respectively. Mean values and SEs are shown on the left of each column. The pipette solution contained 125 mM K-gluconate, 15 mM KCl, 2 mM MgATP, 1 mM EGTA, and 10 mM HEPES (pH 7.4). The bath solution contained 130 mM Na-gluconate, 10 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 5 mM HEPES (pH 7.4). The addition of amiloride (10 µM) is indicated by an arrow. Significance was determined by two-way ANOVA.
Deletion of Nedd4-2 Abrogates HS-Induced Inhibition of NCC
Because deletion of Nedd4-2 abolished the effect of dietary Na+ intake on Kir4.1/Kir5.1 in the DCT, we speculated that HS-induced inhibition or LS-induced stimulation of NCC might also be compromised in Nedd4-2-deficeint mice. We confirmed that deletion of Nedd4-2 increased the expression of pNCC (160 ± 10% of the WT control, n = 4) and total NCC (145 ± 9% of the WT control, n = 4; Fig. 7A) (16). Although HS diet decreased and LS diet increased the expression of pNCC and total NCC in WT mice (Fig. 7B), as previously reported (16), dietary Na+ intake failed to alter the expression of pNCC and total NCC in Ks-Nedd4-2 KO mice (Fig. 7C). Figure 7D shows a bar graph showing the normalized band density of pNCC and total NCC in three male control mice and three male Ks-Nedd4-2 KO mice on LS and HS diets for 14 days compared with NS diet. It is apparent the dietary Na+ intake failed to alter the expression of pNCC and total NCC in Ks-Nedd4-2 KO mice. The notion that Nedd4-2 plays a role in mediating HS intake-induced inhibition of NCC function is also suggested by experiments in which HCTZ-induced renal Na+ excretion was examined in control and Ks-Nedd4-2 KO mice on different Na+ diets for 2 wk. The results are shown in Fig. 8A and demonstrate that HCTZ-induced natriuresis in Ks-Nedd4-2 KO mice on NS diet (2.33 ± 0.11 μeq/min/100 g body wt, n = 6) was significantly larger than in WT mice (1.23 ± 0.07 μeq/min/100 g body wt, n = 7). Moreover, neither LS diet increased Na+ excretion (2.45 ± 0.15 μeq/min/100 g body wt, n = 7) nor HS diet decreased Na+ excretion (2.46 ± 0.15 μeq/min/100 g body wt, n = 8) in Ks-Nedd4-2 KO mice. In contrast, HS diet decreased (0.82 ± 0.05 μeq/min/100 g body wt, n = 4) whereas LS diet increased (2.59 ± 0.10 μeq/min/100 g body wt, n = 4) Na+ excretion in Nedd4lfl/fl mice. The finding that HS diet for 14 days failed to inhibit the expression of pNCC and total NCC in Ks-Nedd4-2 KO mice was also recapitulated in Ks-Nedd4-2 KO mice supplemented with K+ in drinking water (1 g K-citrate/100 mL water). As shown in Fig. 8B, it is apparent that HS diet plus K+ supplement inhibited the expression of pNCC to 45 ± 5% and total NCC to 60 ± 6% in control mice compared with NS diet (n = 3). However, the effect of HS diet plus K+ supplement on pNCC and total NCC was absent in Ks-Nedd4-2 KO mice (n = 3), suggesting the role of Nedd4-2 in mediating the effect of HS diet on NCC.
Figure 7.

Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) abolishes the effect of dietary Na+ intake on NaCl cotransporter (NCC). A: Western blots showing the expression of phosphorylated (p)NCC and total (t)NCC in male floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl) and kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice on normal salt (NS). B and C: immunoblot (IB) showing the expression of pNCC (at Thr53) and tNCC in male Nedd4lfl/fl and Ks-Nedd4-2 KO mice on different Na+ diets for 14 days, respectively. D: two bar graphs summarizing normalized band density of pNCC and tNCC in control and Ks-Nedd4-2 KO mice on low-salt (LS) and high-salt (HS) diets compared with NS. *Significance determined by one-way ANOVA.
Figure 8.

Deletion of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) abolished the effect of high salt (HS) on NaCl cotransporter (NCC) activity/expression. A: hydrochlorothiazide (HCTZ)-induced net renal Na+ excretion (ENa) in floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl) and kidney-specific Nedd4-2 knockout (Ks-Nedd4-2 KO) mice on low-salt (LS), normal-salt (NS), and HS diets for 14 days. Significance was determined by two-way ANOVA. B: immunoblots showing expression of phosphorylated (p)NCC (at Thr53) and total (t)NCC in male Nedd4lfl/fl and Ks-Nedd4-2 KO mice on HS plus K+ supplement (1 g K-citrate in 100 mL drinking water for 14 days). C: two bar graphs summarizing the above results for control and Nedd4-2 KO mice. *Significance determined by two-way ANOVA.
Prolonged HS diet Caused Hypokalemia in Ks-Nedd4-2 KO Mice
Since NCC and ENaC play a role in affecting renal K+ excretion, we next used the renal clearance method to measure the basal level of renal K+ excretion in WT and Ks-Nedd4-2 KO mice on NS or HS diets for 7 or 14 days. Figure 9A shows a scatterplot demonstrating the basal level of renal K+ excretion in WT mice and Ks-Nedd4-2 KO mice on NS, HS (7 days) diets, and HS (14 days) diet, respectively. It is apparent that HS intake had no significant effect on the basal level of the renal K+ secretion rate in WT mice [NS: 0.21 ± 0.02 µeq/min/100 g body wt, n = 7; HS (7 days): 0.21 ± 0.03 µeq/min/100 g body wt, n = 4; and HS (14 days): 0.27 ± 0.02 µeq/min/100 g body wt, n = 6]. Deletion of Nedd4-2 had no significant effect on the basal level of renal K+ excretion compared with WT mice on NS diet (0.20 ± 0.02 µeq/min/100 g body wt, n = 8). However, HS diet for 14 days significantly increased the renal K+ excretion level in Ks-Nedd4-2 KO mice compared with NS diet [HS (7 days): 0.30 ± 0.04 µeq/min/100 g body wt, n = 7; HS (14 days): 0.43 ± 0.04 µeq/min/100 g body wt, n = 8]. As a consequence, Ks-Nedd4-2 KO mice on HS diet (14 days) were hypokalemic. Figure 9B shows a scatterplot showing the mean value and each data point of plasma K+ concentration. Plasma K+ concentrations in Ks-Nedd4-2 KO mice were 3.83 ± 0.04 mM (NS, n = 7), 3.66 ± 0.12 mM (14-day LS, n = 4), 3.53 ± 0.13 mM (7-day HS, n = 6), and 2.64 ± 0.10 mM (14-day HS, n = 13). HS-induced hypokalemia in Ks-Nedd4-2 KO mice was largely corrected by adding K+ supplement in the drinking water, and plasma K+ was 3.80 ± 0.08 mM (n = 4), suggesting that HS intake caused K+ wasting in Ks-Nedd4-2 KO mice. Dietary Na+ intake had no significant effect on plasma K+ levels in WT control mice [NS: 3.81 ± 0.07, n = 9; LS (14 days): 3.71 ± 0.08, n = 4; HS (7 days): 3.93 ± 0.06, n = 7; and HS (14 days): 3.64 ± 0.06, n = 4]. Thus, prolonged HS diet (14 days) significantly decreased plasma K+ concentration in Ks-Nedd4-2 KO mice. In contrast, HS intake had no significant effect on plasma Na+ concentration in both WT and Ks-Nedd4-2 KO mice (Fig. 9C). Thus, our data suggest that Nedd4-2 is required for maintaining plasma K+ concentration in normal ranges during long-term HS intake.
Figure 9.

Long-term high salt (HS) causes hypokalemia in kidney-specific neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) knockout (Ks-Nedd4-2 KO) mice. A: scatterplot showing the basal level of renal K+ excretion rate in male wild-type [WT; floxed neural precursor cell expressed developmentally downregulated gene 4-like (Nedd4lfl/fl)] mice and Ks-Nedd4-2 KO mice on normal salt (NS), HS (7 days; HS-7d), and HS (14 days; HS-14d), respectively. Mean values of each group are shown on the left side of column. B: scatterplot showing plasma K+ concentration in WT and Ks-Nedd4-2 KO mice on different Na+ diets. C: table showing the plasma Na+ concentration in WT and Ks-Nedd4-2 KO mice on different Na+ diets. *Significant difference compared with the other groups. We used two-way ANOVA to determine significance. LS-14d, low salt (14 days).
DISCUSSION
The first main finding of the present study is to demonstrate that dietary Na+ intake fails to regulate basolateral K+ channel activity in the DCT of Ks-Nedd4-2 KO mice. The notion that Nedd4-2 plays a role in mediating the inhibitory effect of HS diet on basolateral K+ channel activity in the DCT is supported by three lines of evidence: 1) single-channel analysis demonstrated that HS intake did not decrease basolateral 40-pS K+ channel activity (NPo) in Ks-Nedd4-2 KO mice, 2) the HS intake-induced decrease in whole cell K+ currents in the DCT was blunted in Nedd4-2 deficient mice, and 3) HS intake did not decrease the negativity of the DCT membrane potential of Ks-Nedd4-2 KO mice. In addition, we also confirmed the previous finding that deletion of Nedd4-2 increased basolateral K+ conductance in the DCT and hyperpolarized the DCT membrane (8), suggesting the role of Nedd4-2 in regulating basolateral K+ channels in the DCT. Since Nedd4-2 is also present in the CCD (1), it is conceivable that deletion of Nedd4-2 might also activate Kir4.1/Kir5.1, thereby hyperpolarizing the CCD membrane and thereby increasing the driving force for K+ excretion. The basolateral K+ channel in the DCT and CCD is composed of Kir4.1 and Kir5.1 (30–33): while Kir4.1 is responsible for providing K+ conductance for the heterotetramer (26, 34, 35), Kir5.1 is a regulatory subunit for the heterotetramer (10, 30, 36–39). For instance, Kir5.1 has been shown to be associated with Nedd4-2 (10). Since deletion of Nedd4-2 abolished the effect of HS diet on Kir4.1/Kir5.1 activity in the DCT, this supports the notion that Nedd4-2 plays a role in determining basolateral Kir4.1/Kir5.1 in the DCT not only under control conditions (NS) but also in mediating the effect of HS intake on Kir4.1/Kir5.1 activity. In this regard, it has been previously reported that HS intake increases Nedd4-2 expression (1). The finding that the HS-induced decrease in Kir4.1 expression was abrogated in Ks-Nedd4-2 KO mice supports the role of Nedd4-2 in mediating HS-induced degradation of Kir4.1.
The view that Kir4.1/Kir5.1 activity in the DCT plays a key role in mediating the effect of Na+ intake on NCC is supported by the finding that deletion of Kir4.1 abolished the effect of Na+ intake on NCC (16). The possible mechanism by which basolateral K+ channel activity in the DCT mediates the effect of HS diet on NCC activity depends on with no lysine kinase (WNK), a key kinase regulating NCC expression/activity (40–46). Low Kir4.1/Kir5.1 activity in the DCT induced by HS should depolarize the DCT membrane and decrease Cl− exit through the basolateral Cl− channel (ClC-Kb), thereby increasing the intracellular Cl− concentration and inhibiting WNK (47). Consequently, HS should also inhibit STE20 proline-alanine-rich kinase (SPAK) or oxidative stress responsive kinase-1 (OSR1), two of WNK’s downstream kinases responsible for phosphorylating NCC (42, 48–50), thereby inhibiting pNCC expression. In this regard, it has been shown that WNK1 expression is increased in Ks-Nedd4-2 KO mice. Thus, deletion of Nedd4-2-induced stimulation of pNCC may be also due to activating WNK1 expression, thereby activating SPAK and NCC (6, 51). In the present study, we demonstrated that deletion of Nedd4-2 also compromises the effect of HS intake on NCC. We speculate that the lack of HS-induced inhibition of NCC expression/activity in Nedd4-2 KO mice might be related to the fact that deletion of Nedd4-2 also compromised the effect of HS diet on Kir4.1/Kir5.1 activity in the DCT.
Our present study also demonstrated that deletion of Nedd4-2 induces a larger increase in ENaC currents in the CCD than in the DCT2/CNT, especially in considering that principal cell size in the CCD is smaller than in the DCT2 (25). Although the role of Nedd4-2 in regulating ENaC activity is well established, most studies have been performed in the CCD but not in the DCT2 (13, 52, 53). The Nedd4-2 deletion-induced larger increase in ENaC currents of the CCD than that of the DCT2/CNT may be due to the fact that aldosterone or mineralocorticoid receptor plays a dominant role in regulating ENaC activity in the CCD but to a less degree in the DCT2/CNT (14, 25, 54). It is well established that aldosterone stimulates ENaC in the ASDN not only by transcriptional modulation but also by a post-translational mechanism (13, 53, 55–58). For instance, aldosterone-induced stimulation of serum and glucocorticoid-regulated kinase 1 (SGK1) increases existing ENaC activity by inhibiting Nedd4-2-dependent ENaC ubiquitination (4, 59). Thus, the aldosterone-SGK1-Nedd4-2 pathway plays a more dominant role in determining ENaC activity in the CCD than in the DCT2/CNT. Consequently, deletion of Nedd4-2 increased ENaC currents in the CCD more than in the DCT2/CNT since aldosterone plays a more important role in regulating ENaC in the CCD than in the DCT2 (14, 25). In this regard, high ENaC activity in the CCD may also indirectly stimulate Kir4.1/Kir5.1 in the DCT due to affecting K+ excretion and plasma K+.
The observation that HS-induced inhibition of ENaC currents was absent in Ks-Nedd4-2 KO mice suggests the role of Nedd4-2 in mediating the effect of HS diet on ENaC in both the DCT2 and CCD. HS-induced inhibition of NCC and ENaC not only plays a role in decreasing renal Na+ absorption but also in maintaining K+ homeostasis during increased Na+ intake because high Na+ delivery induced by NCC inhibition should offset the negative effect of low ENaC activity on K+ excretion in the ASDN. Despite the high NCC activity, we and others have demonstrated that Ks-Nedd4-2 KO mice are normokalemic rather than hyperkalemic under control (NS) conditions (5, 51). We think that the high ENaC activity in the ASDN of Ks-Nedd4-2 KO mice should compensate for the negative effect of high NCC activity on renal K+ excretion, thereby maintaining a normal plasma K+ level during NS or short-term HS diets. This notion is supported by previous findings and those of our present study (5): Ronzaud et al. observed that plasma K+ and urine K+ excretion in Ks-Nedd4-2 KO mice on NS or HS diets (3.2%) for 8 days were similar to control mice. We have also not found a significant difference regarding K+ excretion and plasma K+ concentration in Nedd4-2 KO mice on NS or HS diets (4%) for 7 days. However, the observation that the renal K+ excretion rate was significantly higher in Nedd4-2 KO mice on HS diet for 14 days than NS diet strongly suggests that Nedd4-2 activity is essential for maintaining a proper renal K+ excretion rate in response to long-term HS diet. We believe that the high ENaC activity in the CCD should be mainly responsible for renal K+ wasting in Ks-Nedd4-2 KO mice on long-term HS diet. In addition, it is conceivable that long-term HS intake should suppress the renin-angiotensin II system in the proximal tubule and in the thick ascending limb, thereby inhibiting K+ and Na+ absorption in both nephron segments (60). This should increase Na+ and fluid volume delivery to the ASDN despite the high NCC activity in Ks-Nedd4-2 KO mice, thereby stimulating ENaC-dependent and BK-dependent K+ excretion (21). This notion is supported by the observation that HS diet failed to inhibit BK in Nedd4-2 KO mice. Relevant to our finding is a report showing that K+ restriction decreased plasma K+ in Ks-Nedd4-2 KO mice more than in WT mice (51).
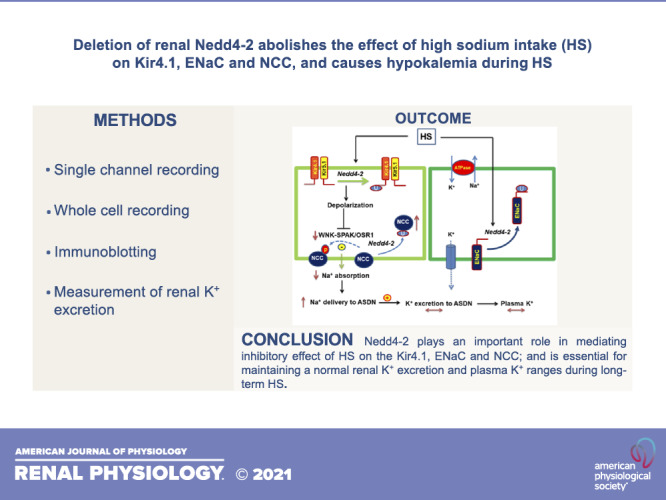
Figure 10 shows a cell scheme illustrating the role of Nedd4-2 in regulating renal Na+ and K+ transport in the ASDN under physiological conditions. HS intake should inhibit Kir4.1/Kir5.1 in the DCT by stimulating a Nedd4-2-dependent mechanism. HS-induced inhibition of Kir4.1/Kir5.1 and NCC in the DCT should decrease renal Na+ absorption in the DCT, and this should play a role in preventing salt-sensitive hypertension. The inhibition of Kir4.1/Kir5.1 and NCC should also increase Na+ delivery to the ASDN, thereby offsetting the negative effect of low ENaC activity on renal K+ excretion in the ASDN during increased dietary Na+ intake. Thus, by inhibiting NCC and ENaC simultaneously, Nedd4-2 plays a role not only in stimulating Na+ excretion but also in preventing excessive K+ excretion during salt loading. In summary, our present study has three novel aspects regarding Nedd4-2 function in the DCT1 and ASDN: 1) Nedd4-2 is involved in mediating the HS-induced inhibition of Kir4.1 expression/activity and NCC expression/activity; 2) Nedd4-2 is required for HS-induced inhibition of ENaCβ, ENaCγ, and BKα expression and ENaC activity; and 3) Nedd4-2 prevents excessive renal K+ excretion during long-term HS diet. We conclude that Nedd4-2 plays an important role in mediating the effect of HS diet on Kir4.1/Kir5.1 activity in the DCT and NCC and ENaC in the ASDN and also in maintaining K+ homeostasis during long-term HS intake.
Figure 10.

Cell scheme illustrating the role of neural precursor cell expressed developmentally downregulated protein 4-2 (Nedd4-2) in regulating the effect of high salt (HS) intake on Kir4.1/Kir5.1, NaCl cotransporter (NCC), and epithelial Na+ channel (ENaC) in the aldosterone-sensitive distal nephron (ASDN) and the role of Kir4.1/Kir5.1, NCC, and ENaC in regulating renal K+ excretion of the ASDN during HS. The solid line and dotted line represent enhanced or decreased pathways, respectively. Lines with double arrows indicate unchanged parameters. OSR1, oxidative stress responsive kinase-1; SPAK, STE20 proline-alanine-rich kinase; WNK, with no lysine kinase.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK115366 (to D.-H.L.) and DK54983 (to W.-H.W.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
W-H.W. and D-H.L. conceived and designed research; D-D.Z., X-P.D., Y.X., P.W., Z-X.G., and D-H.L. performed experiments; D-D.Z., X-P.D., Y.X., Z-X.G., W-H.W., and D-H.L. analyzed data; D-D.Z., X-P.D., W-H.W., and D-H.L. interpreted results of experiments; D-D.Z., X-P.D., Y.X., P.W., Z-X.G., W-H.W., and D-H.L. prepared figures; W-H.W. and D-H.L. drafted manuscript; W-H.W. and D-H.L. edited and revised manuscript; D-D.Z., X-P.D., Y.X., P.W., Z-X.G., W-H.W., and D-H.L. approved final version of manuscript.
REFERENCES
- 1.Loffing-Cueni D, Flores SY, Sauter D, Daidie D, Siegrist N, Meneton P, Staub O, Loffing J. Dietary sodium intake regulates the ubiquitin-protein ligase Nedd4-2 in the renal collecting system. J Am Soc Nephrol 17: 1264–1274, 2006. doi: 10.1681/asn.2005060659. [DOI] [PubMed] [Google Scholar]
- 2.Staub O, Yeger H, Plant PJ, Kim H, Ernst SA, Rotin D. Immunolocalization of the ubiquitin-protein ligase Nedd4 in tissues expressing the epithelial Na+ channel (ENaC). Am J Physiol Cell Physiol 272: C1871–C1880, 1997. doi: 10.1152/ajpcell.1997.272.6.c1871. [DOI] [PubMed] [Google Scholar]
- 3.Arroyo JP, Lagnaz D, Ronzaud C, Vazquez N, Ko BS, Moddes L, Ruffieux-Daidie D, Hausel P, Koesters R, Yang B, Stokes JB, Hoover RS, Gamba G, Staub O. Nedd4-2 modulates renal Na+-Cl− cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J Am Soc Nephrol 22: 1707–1719, 2011. doi: 10.1681/asn.2011020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20: 7052–7059, 2001. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, Staub O. Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657–665, 2013. doi: 10.1172/jci61110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, Chang YPC, Butterworth MB, NrM P-S, Hallows KR, Staub O, Subramanya AR. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action. J Clin Invest 125: 3433–3448, 2015. doi: 10.1172/jci75245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snyder PM. The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr Rev 23: 258–275, 2002. doi: 10.1210/edrv.23.2.0458. [DOI] [PubMed] [Google Scholar]
- 8.Wu P, Su XT, Gao ZX, Zhang DD, Duan XP, Xiao Y, Staub O, Wang WH, Lin DH. Renal tubule Nedd4-2 deficiency stimulates Kir4.1/Kir5.1 and thiazide-sensitive NaCl cotransporter in distal convoluted tubule. J Am Soc Nephrol 31: 1226–1242, 2020. doi: 10.1681/asn.2019090923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou R, Patel SV, Snyder PM. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J Biol Chem 282: 20207–20212, 2007. doi: 10.1074/jbc.m611329200. [DOI] [PubMed] [Google Scholar]
- 10.Wang MX, Su XT, Wu P, Gao ZX, Wang WH, Staub O, Lin DH. Kir5.1 regulates Nedd4-2-mediated ubiquitination of Kir4.1 in distal nephron. Am J Physiol Renal Physiol 315: F986–F996, 2018. doi: 10.1152/ajprenal.00059.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1//SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008. doi: 10.1038/ki.2008.451. [DOI] [PubMed] [Google Scholar]
- 12.Frindt G, Ergonul Z, Palmer LG. Na channel expression and activity in the medullary collecting duct of rat kidney. Am J Physiol Renal Physiol 292: F1190–F1196, 2007. doi: 10.1152/ajprenal.00399.2006. [DOI] [PubMed] [Google Scholar]
- 13.Loffing J, Pietri L, Aregger F, Bloch-Faure M, Ziegler U, Meneton P, Rossier BC, Kaissling B. Differential subcellular localization of ENaC subunits in mouse kidney in response to high- and low-Na diets. Am J Physiol Renal Physiol 279: F252–F258, 2000. doi: 10.1152/ajprenal.2000.279.2.f252. [DOI] [PubMed] [Google Scholar]
- 14.Nesterov V, Dahlmann A, Krueger BK, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299, 2012. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 15.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu P, Gao ZX, Su XT, Wang MX, Wang WH, Lin DH. Kir4.1/Kir5.1 activity is essential for dietary sodium intake-induced modulation of Na-Cl cotransporter. J Am Soc Nephrol 30: 216–227, 2019. doi: 10.1681/asn.2018080799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frindt G, Palmer LG. K+ secretion in the rat kidney: Na+ channel-dependent and -independent mechanisms. Am J Physiol Renal Physiol 297: F389–F396, 2009. doi: 10.1152/ajprenal.90528.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 10: 1050–1060, 2015. doi: 10.2215/cjn.08580813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandberg MB, Maunsbach AB, McDonough AA. Redistribution of distal tubule Na+-Cl− cotransporter (NCC) in response to a high-salt diet. Am J Physiol Renal Physiol 291: F503–F508, 2006. doi: 10.1152/ajprenal.00482.2005. [DOI] [PubMed] [Google Scholar]
- 20.Liu W, Morimoto T, Woda C, Kleyman TR, Satlin LM. Ca2+ dependence of flow-stimulated K secretion in the mammalian cortical collecting duct. Am J Physiol Renal Physiol 293: F227–F235, 2007. doi: 10.1152/ajprenal.00057.2007. [DOI] [PubMed] [Google Scholar]
- 21.Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol 280: F786–F793, 2001. doi: 10.1152/ajprenal.2001.280.5.f786. [DOI] [PubMed] [Google Scholar]
- 22.Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant 34: iii2–iii11, 2019. doi: 10.1093/ndt/gfz206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young DB, Jackson TE, Tipayamontri U, Scott RC. Effects of sodium intake on steady-state potassium excretion. Am J Physiol Renal Physiol 246: F772–F778, 1984. doi: 10.1152/ajprenal.1984.246.6.f772. [DOI] [PubMed] [Google Scholar]
- 24.Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, KAB-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett 388: 11–15, 1996. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- 25.Wu P, Gao Z, Zhang D, Duan X, Terker AS, Lin D, Ellison DH, Wang W. Effect of angiotensin II on ENaC in the distal convoluted tubule and in the cortical collecting duct of mineralocorticoid receptor deficient mice. J Am Heart Assoc 9: e014996, 2020. doi: 10.1161/jaha.119.014996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang C-L, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28: 1814–1825, 2017. doi: 10.1681/asn.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng D, Nan H, Wang W, Yan L, Du P, Zuo L, Zhang K, Zhao M, Cui G. Expression and alteration of BKCa channels in the sphincter of Oddi’s from rabbits with hypercholesterolemia. Channels (Austin) 11: 236–244, 2017. doi: 10.1080/19336950.2017.1279369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu P, Gao ZX, Zhang DD, Su XT, Wang WH, Lin DH. Deletion of Kir5.1 impairs renal ability to excrete potassium during increased dietary potassium intake. J Am Soc Nephrol 30: 1425–1438, 2019. doi: 10.1681/asn.2019010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su XT, Ellison DH, Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K+ excretion. Am J Physiol Renal Physiol 316: F582–F586, 2019. doi: 10.1152/ajprenal.00412.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol 294: F1398–F1407, 2008. doi: 10.1152/ajprenal.00288.2007. [DOI] [PubMed] [Google Scholar]
- 31.Zaika O, Palygin O, Tomilin V, Mamenko M, Staruschenko A, Pochynyuk O. Insulin and IGF-1 activate Kir4.1/5.1 channels in cortical collecting duct principal cells to control basolateral membrane voltage. Am J Physiol Renal Physiol 310: F311–F321, 2016. doi: 10.1152/ajprenal.00436.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. Src-family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10. J Biol Chem 288: 26135–26146, 2013. doi: 10.1074/jbc.m113.478453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, Geurts AM, Hodges MR, Staruschenko A. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight 2: e92331, 2017. doi: 10.1172/jci.insight.92331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang M-X, Cuevas CA, Su X-T, Wu P, Gao Z-X, Lin D-H, McCormick JA, Yang C-L, Wang W-H, Ellison DH. Potassium (K+) intake modulates NCC activity via the K+ channel, Kir4.1. Kidney Int 93: 893–902, 2018. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casamassima M, D'Adamo MC, Pessia M, Tucker SJ. Identification of a heteromeric interaction that influences the rectification, gating, and pH sensitivity of Kir4.1/Kir5.1 potassium channels. J Biol Chem 278: 43533–43540, 2003. doi: 10.1074/jbc.m306596200. [DOI] [PubMed] [Google Scholar]
- 37.Pessia M, Imbrici P, D'Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol 532: 359–367, 2001. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J Physiol 525: 587–592, 2000. doi: 10.1111/j.1469-7793.2000.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tucker SJ, Imbrici P, Salvatore L, D'Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem 275: 16404–16407, 2000. doi: 10.1074/jbc.c000127200. [DOI] [PubMed] [Google Scholar]
- 40.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet 20: 855–866, 2011. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang C-L, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277: 50812–50819, 2002. doi: 10.1074/jbc.m208108200. [DOI] [PubMed] [Google Scholar]
- 44.Rodan AR. WNK-SPAK/OSR1 signaling: lessons learned from an insect renal epithelium. Am J Physiol Renal Physiol 315: F903–F907, 2018. doi: 10.1152/ajprenal.00176.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanya AR, Yang CL, McCormick JA, Ellison DH. WNK kinases regulate sodium chloride and potassium transport by the aldosterone-sensitive distal nephron. Kidney Int 70: 630–634, 2006. doi: 10.1038/sj.ki.5001634. [DOI] [PubMed] [Google Scholar]
- 46.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010. doi: 10.1681/asn.2009121295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piala AT, Moon TM, Akella R, He HX, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphosphorylation. Sci Signal 7: ra41, 2014. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+: Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2555–2563, 2017. doi: 10.1681/asn.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thastrup J, Rafiqi F, Vitari A, Pozo-Guisado E, Deak M, Mehellou Y, Alessi D. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441: 325–337, 2012. doi: 10.1042/bj20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Qusairi L, Basquin D, Roy A, Rajaram RD, Maillard MP, Subramanya AR, Staub O. Renal tubular ubiquitin-protein ligase NEDD4-2 is required for renal adaptation during long-term potassium depletion. J Am Soc Nephrol 28: 2431–2442, 2017. doi: 10.1681/asn.2016070732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill WG, Butterworth MB, Wang H, Edinger RS, Lebowitz J, Peters KW, Frizzell RA, Johnson JP. The epithelial sodium channel (ENaC) traffics to apical membrane in lipid rafts in mouse cortical collecting duct cells. J Biol Chem 282: 37402–37411, 2007. doi: 10.1074/jbc.m704084200. [DOI] [PubMed] [Google Scholar]
- 53.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC α, β and γ subunit proteins in rat kidney. J Clin Invest 104: R19–R23, 1999. doi: 10.1172/jci7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kemendy AE, Kleyman TR, Eaton DC. Aldosterone alters the open probability of amiloride-blockable sodium channels in A6 epithelia. Am J Phyiol Cell Physiol 263: C825–C837, 1992. doi: 10.1152/ajpcell.1992.263.4.c825. [DOI] [PubMed] [Google Scholar]
- 55.Ecelbarger CA, Kim GH, Wade JB, Knepper MA. Regulation of the abundance of renal sodium transporters and channels by vasopressin. Exp Neurol 171: 227–234, 2001. doi: 10.1006/exnr.2001.7775. [DOI] [PubMed] [Google Scholar]
- 56.Kamynina E, Staub O. Concerted action of ENaC, Nedd4-2, and Sgk1 in transepithelial Na+ transport. Am J Physiol Renal Physiol 283: F377–F387, 2002. doi: 10.1152/ajprenal.00143.2002. [DOI] [PubMed] [Google Scholar]
- 57.Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: correlation between aENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+. J Am Soc Nephrol 14: 1107–1115, 2003. doi: 10.1097/01.asn.0000061777.67332.77. [DOI] [PubMed] [Google Scholar]
- 58.Stockand JD. New ideas about aldosterone signaling in epithelia. Am J Physiol Renal Physiol 282: F559–F576, 2002. doi: 10.1152/ajprenal.00320.2001. [DOI] [PubMed] [Google Scholar]
- 59.Pearce D, Kleyman TR. Salt, sodium channels, and SGK1. J Clin Invest 117: 592–595, 2007. doi: 10.1172/jci31538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev 98: 1627–1738, 2018. doi: 10.1152/physrev.00038.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]