

Abstract
Acute respiratory distress syndrome (ARDS) is a common cause of mortality in patients with acute kidney injury (AKI). Inflammatory crosstalk from the kidney to the lung has been shown to contribute to lung injury after AKI, but anti-inflammatory therapies have not been proven beneficial in human studies. Recently, AKI was shown to alter mitochondria and related metabolic pathways in the heart, but the impact of AKI on lung metabolism has not been investigated to our knowledge. In this study, we evaluated the metabolomic profile of the lung following renal ischemia and reperfusion to identify novel pathways that may be modifiable. We randomized C57BL/6 mice to 20 minutes of bilateral renal arterial clamping or sham operation under ketamine/xylazine anesthesia. At 4 hours after reperfusion, we found a significant increase in markers of lung injury, as well as significant metabolomic changes across lung, kidney, plasma and bronchoalveolar lavage fluid (BALF) compared to shams. Comparative analyses revealed that the fatty acid oxidation pathway was the most significantly altered metabolic pathway, a finding which is consistent with mitochondrial dysfunction systemically and in the lung. These metabolomic changes correlated with the extracellular accumulation of the mitochondrial damage associated molecular patterns (mtDAMPs), mitochondrial DNA (mtDNA) and transcription factor A, mitochondria (TFAM). Finally, we found that intraperitoneal injection of renal mtDAMPs caused metabolomic changes consistent with mitochondrial dysfunction in the lung in vivo. Mitochondrial function and mtDAMPs warrant further investigation as potential therapeutic targets in preventing lung injury because of AKI.
Keywords: AKI, ARDS, metabolomics, mitochondria, mtDAMPs
INTRODUCTION
Acute kidney injury (AKI) occurs in 30% of critically ill patients, and the mortality rate of AKI in the intensive care unit (ICU) is ∼30–50% (1). Traditional management of AKI has focused on treating the direct complications of loss of renal function, such as volume overload, acidosis, and electrolyte derangements. Despite advances in traditional management, the mortality of AKI remains unacceptably high. Of note, the leading causes of death in AKI are non-renal complications (2), and recent literature has provided evidence that AKI is a systemic disease that increases mortality via detrimental effects on distant organs, particularly the lungs (3–6).
There are mechanisms of respiratory failure in AKI beyond volume overload, such as the development of lung injury (3–5), more commonly referred to as acute respiratory distress syndrome (ARDS). For example, animal models of ischemia-reperfusion (IR)-AKI have demonstrated that neutrophil infiltration and pulmonary edema develop in the lungs just 2 hours after renal injury (3, 5). Inflammatory crosstalk from the kidney to the lung via the systemic release of inflammatory mediators is a major driver of this pathology. Anti-inflammatory agents targeting specific mediators, such as IL-6 (5) and TNF-α (6), have mitigated lung injury in animal models, but no therapies have been proven beneficial in human studies. Investigations focused on novel mechanisms of respiratory failure because of AKI are urgently needed.
Mitochondrial dysfunction is a key mechanism of renal injury after IR (7), but few studies have investigated the impact of renal mitochondrial dysfunction on remote organs (8, 9). Importantly, critical elements of the functional response to lung injury, such as hypoxic vasoconstriction (10) and surfactant production (11), are controlled by mitochondria or related metabolic pathways. Recently, the cardiac metabolome was found to be impacted by renal ischemia (8), but the effect of AKI on lung metabolism has not been investigated to our knowledge. Mitochondrial dysfunction in the setting of hemodynamic stress promotes inflammation via the release of mitochondrial damage associated molecular patterns (mtDAMPs; 12), and the extracellular mtDAMPs, mitochondrial DNA (mtDNA) and transcription factor A, mitochondria (TFAM), have been shown to cause lung injury in vivo (13, 14). We hypothesized that IR-AKI would lead to metabolic derangements consistent with mitochondrial dysfunction in the lung, and correlate with the accumulation of extracellular mtDAMPs. To investigate this possibility, we evaluated the metabolome of the lung, kidney, plasma and bronchoalveolar lavage fluid (BALF) in mice exposed to renal IR. Additionally, we evaluated for mtDNA and TFAM in the plasma and BALF. Finally, we evaluated the impact of renal mtDAMPs on lung metabolism in vivo. Our goal was to identify metabolic pathways, metabolites, and mediators that may serve as biomarkers or targets for novel therapies focused on preventing lung injury because of AKI.
MATERIALS AND METHODS
Animals and Ischemia-Reperfusion (IR) Surgical Procedure
Experiments were performed ethically in accordance with international guidelines and were approved by the local animal care and use committees. Eight to twelve-week old C57BL/6 male mice were randomized to 20 min of bilateral renal arterial clamping or sham operation. Intraperitoneal injection of ketamine (100 mg/kg)/xylazine (8 mg/kg) was used for anesthesia, and core body temperatures were maintained between 36–37.5°C via heating pad. Kidneys were exposed via bilateral flank incisions, and renal ischemia was induced by clamping renal arteries with non-traumatic microvessel clamps. After 20 min of ischemia, the clamps were released, and reflow was verified by visual inspection. Sham operation was identical in all aspects, including dose and duration of anesthesia, except that the renal arteries were only exposed for 20 min rather than being clamped. All mice received 200 μL saline dripped over the open flanks during surgery and 30 μL saline/gram body weight via subcutaneous injection after surgery.
Preparation of Mitochondrial DAMPs and in Vivo Metabolomics
Mitochondria were isolated from mouse kidney using the Mitochondria Isolation Kit (MITOISO1, Millipore Sigma) according to manufacturer’s instructions, and mtDAMP solutions were prepared from mouse kidney as has been described previously from liver (14). Mitochondria suspensions were sonicated on ice at 100% amplitude for 30 s at 30 s intervals ten times. The disrupted mitochondrial suspensions were then centrifuged at 12,000 × g for 10 min at 4°C followed by 100,000 × g at 4°C for 30 min and the supernatant was collected as the final mtDAMP solution. For in vivo experiments, mice were sedated with inhaled isoflurane anesthesia before intraperitoneal injection of 50 µg/mL mtDAMP solution in 1 mL or 1 mL vehicle (normal saline).
Plasma and BALF Preparation
Blood samples were collected via inferior vena cava puncture at the time of harvest and were transferred to 1.5 mL tubes coated with EDTA. Whole blood samples were centrifuged at 300 × g for 20 min at room temperature to collect plasma. BALF was collected by instilling 800 µL of PBS into the trachea with steady pressure, followed by gentle aspiration. Plasma and BALF were then centrifuged at 3751 × g for 5 min to remove large debris before snap freezing and storage at −80°C.
Metabolite Extraction
Plasma, BALF, lung, and kidney were harvested 4 hours after reperfusion. Metabolite extraction was performed as described previously with slight modifications (15). Briefly, 50 μL of plasma, BALF, or tissue homogenate (50 μL) were extracted with 200 μL of prechilled buffer (−20°C) containing methanol and acetonitrile (50:50, v/v). Both chemicals were purchased from Honeywell Burdick & Jackson (Catalog No. LC230 and LC015). The mixture was incubated on ice for 10 min and then centrifuged at 16,000 × g for 10 min at 4°C. The supernatant was then transferred to new tubes and stored at −80°C.
LC-MS/MS Analysis of Lung, Kidney, Plasma, and BALF
LC − MS/MS analysis was conducted using a UHPLC system (LC-20A, Shimadzu, Japan) coupled to a Qtrap 6500 hybrid triple quadrupole mass spectrometer (SCIEX) operated in both negative (ESI−) and positive (ESI+) (16). Briefly, 10 μL of extract was injected and the mixture was separated on a Mixed-Mode HILIC-1 column (4.6 × 250 mm, 5 μm, 120 Å) (Thermo Fisher, CA). Mobile phase A was 95% H2O with 20 mM ammonium formate and 5% acetonitrile (pH 4). Mobile phase B was 100% acetonitrile. The gradient was as follows: 0 min-95% B, 3 min-95% B, 3.1 min-85% B, 6.0 min-85% B, 6.1 min-75% B, 10 min-75% B, 15 min-0% B, 25 min-0%, 26 min-95% B, and 31 min-end. The flow rate was 300 μL/min. MS detection was conducted in scheduled multiple reaction monitoring (MRM) mode with fast polarity switching (20 ms). The ESI source conditions were as follows: electrospray voltage of −4500 V on negative mode and 5500 V on positive mode, source temperature of 500°C, curtain gas of 50, CAD gas of 11, gas 1 and gas 2 of 50 and 50 psi, respectively. The values of declustering potential (DP), entrance potential (EP), collision energy (CE) and collision cell exit potential (CXP) were optimized using the purified standards and set on a compound-dependent basis. A broad-spectrum, targeted metabolomics method was utilized to evaluate a total of 483 metabolites. The metabolites evaluated cover a broad range of chemical classes that are known to be essential in metabolism, and all were confirmed by authentic standards.
LC-MS/MS Measurement of Plasma Creatinine
Plasma creatinine 24 hours after IR or sham operation was used as a marker of kidney injury, and determined by LC-MS/MS as described previously (17). A 10 μL sample of plasma was deproteinated and diluted with heavy isotope-labeled internal standard (ISTD) in a single step by adding ISTD in 80% acetonitrile. Two microliters of diluted sample were then subjected to isocratic, HILIC HPLC with 10 mM ammonium acetate in 65% acetonitrile at 0.15 mL/min. Creatinine and d3-creatinine (ISTD) were detected by electrospray ionization tandem mass spectrometry MRM transitions 114 > 44 and 117 > 47, respectively. Quantitation was achieved by comparing results with a synthetic standard calibration curve (0, 0.2, 1, 5, 100 µg/ml).
BALF Cell Count and Lung Myeloperoxidase (MPO) Activity Assay
BALF cell count and lung MPO were used to determine the degree of lung inflammation after IR-AKI. BALF was obtained as described above, and the cell pellet resuspended in 200 µL of ice-cold PBS. Cell pellet suspension was cytospun onto a slide at 800 rpm for 3 min, and total cell count obtained using a Countess® II automated cell counter. MPO activity in lung tissue was determined via the mouse Myeloperoxidase Colorimetric Activity Assay Kit (ab105136, abcam) according to the manufacturer’s instructions. Fresh lung tissue (8 mg/sample) was used for each assay and samples were incubated for 120 min after the addition of reaction mix. Optical density of samples and standards was determined via a Spectra MAX 190 (Molecular Devices) Spectrophotometer at wavelength 412 nm.
Plasma and BALF mtDAMP Quantification
Plasma and BALF levels of mtDNA and TFAM were determined to identify the accumulation of mtDAMPs after IR-AKI. For mtDNA, plasma and BALF samples were centrifuged at 16,000 × g for 15 min before DNA isolation using the QIAamp DSP DNA Blood Mini Kit (No. 61104; Qiagen) as previously described (18). Primers for mouse NADH dehydrogenase 1 (ND1) (forward-5′-GGATCCGAGCATCTTATCCA-3′ and reverse- 5′-GGTGGTACTCCCGCTGTAAA-3′) were synthesized by Integrated DNA Technologies. Real-time quantitative PCR (qPCR) was performed using an Applied Biosystem’s StepONEPLUS system with the following program; 95°C for 5 min, followed by 40 cycles of 95°C for 15 s, 52°C for 30 s, and 72°C for 30 s. A standard curve was created using a plasmid ND1 customized by Blue Heron Biotechnologies. Plasma and BALF levels of TFAM were determined by the mouse mtTFA/TFAM ELISA Kit (#LS-F9115-1, lifespan Biosciences). All samples were incubated in coated 96-well ELISA plates for 24 h to optimize yield before wash cycles and the addition of conjugate and substrate solutions. After stop solution was added, optical density was read on a Spectra MAX 190 (Molecular Devices) Spectrophotometer at wavelength 450 nm and 540 nm for optical correction and standard curve was calculated.
Lung Wet/Dry Ratio and BALF Albumin
Wet/dry ratio of lung was used as an indicator of pulmonary edema, and BALF was used as a marker of alveolar-capillary membrane permeability. Fresh lung samples were blotted to remove excess blood before obtaining wet lung weight. Lung was then dried in oven at 70°C for 24 h before determining dry lung weight. BALF levels of albumin, were quantified via the Albumin ELISA Quantitation Set (E90-134, Bethyl Laboratories) according to the manufacturer’s guidelines with methods as described for TFAM quantification above.
Lung Mechanics Assessment
Lung elastance and pressure-curve curves were evaluated to identify differences in lung mechanics after IR-AKI. At the time of harvest, mice were sedated with ketamine (100 mg/kg)/xylazine (8 mg/kg) and tracheostomy was performed. Mice were then placed on the flexiVent® ventilator (SciReq) and ventilated at a tidal volume of 10 mL/kg, positive end expiratory pressure (PEEP) 2.5 cm H2O, and rate of 150 breaths/minute without supplemental oxygen for 15 min. A lung mechanics scan was performed which included an assessment of lung elastance and pressure-volume curves.
Data Analysis and Statistics
For metabolomic data analysis, peak integration was performed in MultiQuant 3.0 (SCIEX) with manual inspection and adjustment based on the standards. After peak vetting, data were exported to excel files and data quality was checked again. The missing values were replaced by k-nearest neighbors (KNN) method. Data were log 2 transformed and auto-scaled before statistical analysis. Metabolomic analysis including partial least square analysis (PLS-DA), volcano plot analysis was performed in Metaboanalyst 4.0 (www.metaboanalyst.ca). PLS-DA model was cross-validated by leave-one-out cross-validation (LOOCV) method. VIP score > 1.5 in PLS-DA model was considered as statistically significant. For volcano plot analysis, the fold change threshold was set as 1.2 and the FDR-adjusted p-value was set as < 0.05. Pathway analysis was performed in Phyton 3.7.4 using our in-house database. The impact score of a pathway was calculated as the sum of the VIP scores for the significant metabolites normalized by the sum of VIP scores of all metabolites in that pathway. All non-metabolomic data were analyzed by one-way ANOVA using GraphPad Prism 8.0. Unless stated otherwise, results are presented as group means ± SEM with p < 0.05 used to determine significance.
RESULTS
Lung Injury and Functional Impairment after IR-AKI
Plasma creatinine was significantly increased after IR mice compared to shams (Fig. 1A). To evaluate lung inflammation after IR, we compared the total cell count in the BALF and lung MPO activity in IR mice compared to shams. Both BALF cell count and lung MPO activity were significantly increased after IR (Fig. 1B/C). Next, we evaluated the lung wet/dry weight and BALF albumin as indicators of pulmonary edema and alveolar-capillary leak, respectively. The wet/dry weight and BALF albumin levels were both significantly increased after IR compared with sham operation (Fig. 1D/E). Finally, we evaluated lung mechanics with the flexiVent® rodent ventilator in both groups. Lung elastance was significantly higher in IR mice (Fig. 1F), and there was a rightward shift in the pressure-volume curve with a significant reduction in volume measured at the end of inhalation (Fig. 1G).
Figure 1.

Lung injury after IR-AKI. A: Plasma creatinine, B: Cell count in BALF, C: MPO activity in lung tissue, D: lung wet/dry ratio, E: BALF albumin concentration, and F: Lung elastance are all significantly increased in IR mice compared to shams. G: Pressure-volume curves created from average values of curves from IR mice compared to shams shows a rightward shift of pressure volume curve and a significant reduction in the end inspiratory volume after IR. n = 4–6 in each group (*P < 0.05, **P< 0.01, ***P< 0.001).
Metabolomic Changes in the Lung after IR-AKI
To determine if metabolic changes may contribute to lung injury after IR-AKI, we evaluated the metabolome in the lung after reperfusion. A clear separation was achieved between the IR-AKI and sham mice in lung tissue by PLS-DA, indicating the existence of dramatic metabolic differences between the two groups (Fig. 2A). Because PLS-DA is a supervised model, we then performed the leave-one-out cross-validation (LOOCV) to check the predictive ability of the PLS-DA model. We obtained a Q2 score of 0.65, which implied a high quality of the PLS-DA model (19). Out of 483 metabolites evaluated, 89 were significantly different between the groups with a fold change greater than 20%. Next, we performed univariate analysis to show the most significantly altered metabolites between IR and shams (Fig. 2B). Out of the top 25 significantly altered metabolites shown, 9 were medium and long-chain acylcarnitines from the fatty acid oxidation (FAO) pathway and 4 were prostaglandins or eicosanoids (Fig. 2B). No other pathway had more than 2 metabolites represented in the top 25. Pathway enrichment analysis also showed that FAO was the most altered pathway of the 23 pathways that were significantly altered (Fig. 2C). Further analysis by volcano plot showed that only D-glyceraldehyde from the glycolytic pathway was significantly decreased in the IR-AKI group compared to shams (Fig. 2D).
Figure 2.
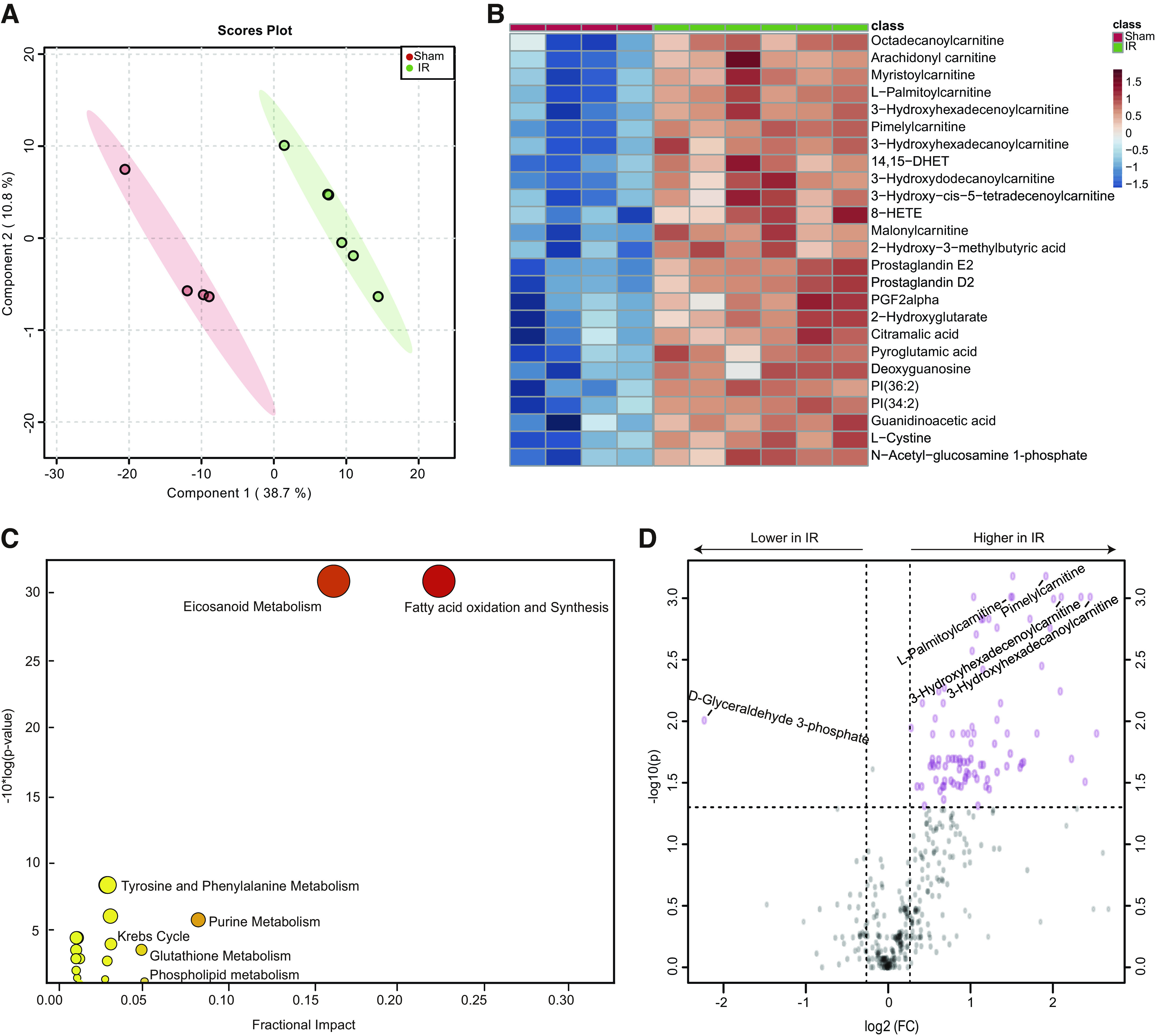
Metabolomic changes in lung tissue in response to IR-AKI. A: PLS-DA showed dramatically different metabolic profiles between IR and shams. B: Top 25 metabolites altered by univariate analysis between IR and shams. C: Pathway enrichment analysis of significantly altered metabolic pathways. D: Volcano plot analysis of the significantly increased or decreased metabolites. n = 4–6/group. Fold change > 1.2 and FDR-adjusted P-value < 0.05 considered significant.
Comparison of Altered Metabolic Pathways and Top 5 FAO Metabolites
Next, we sought to compare the key metabolic derangements in the lung to those found in the kidney, plasma, and BALF. Pathway enrichment analyses revealed that FAO metabolism was the most altered pathway in all tissues and fluids (Fig. 3, A–C). The ranking of the other major pathways altered, such as ceramide and phospholipid metabolism, were unique in each tissue and fluid. To determine if the FAO metabolites in plasma, lung, and BALF were simply a reflection of alterations in the kidney, we evaluated the metabolite peak area to determine the top 5 most altered FAO metabolites in the lung, kidney, plasma, and BALF. The top 5 FAO metabolites were predominately medium and long-chain acylcarnitines, and all were increased significantly (Fig. 4). The metabolites represented in the top 5 were unique in the lung, kidney, plasma, and BALF.
Figure 3.

Comparison of metabolic pathways altered in kidney, plasma, BALF, and lung. A–C: Pathway enrichment analysis of significantly altered metabolic pathways in the kidney, plasma, and BALF, respectively. n = 4–6/group.
Figure 4.
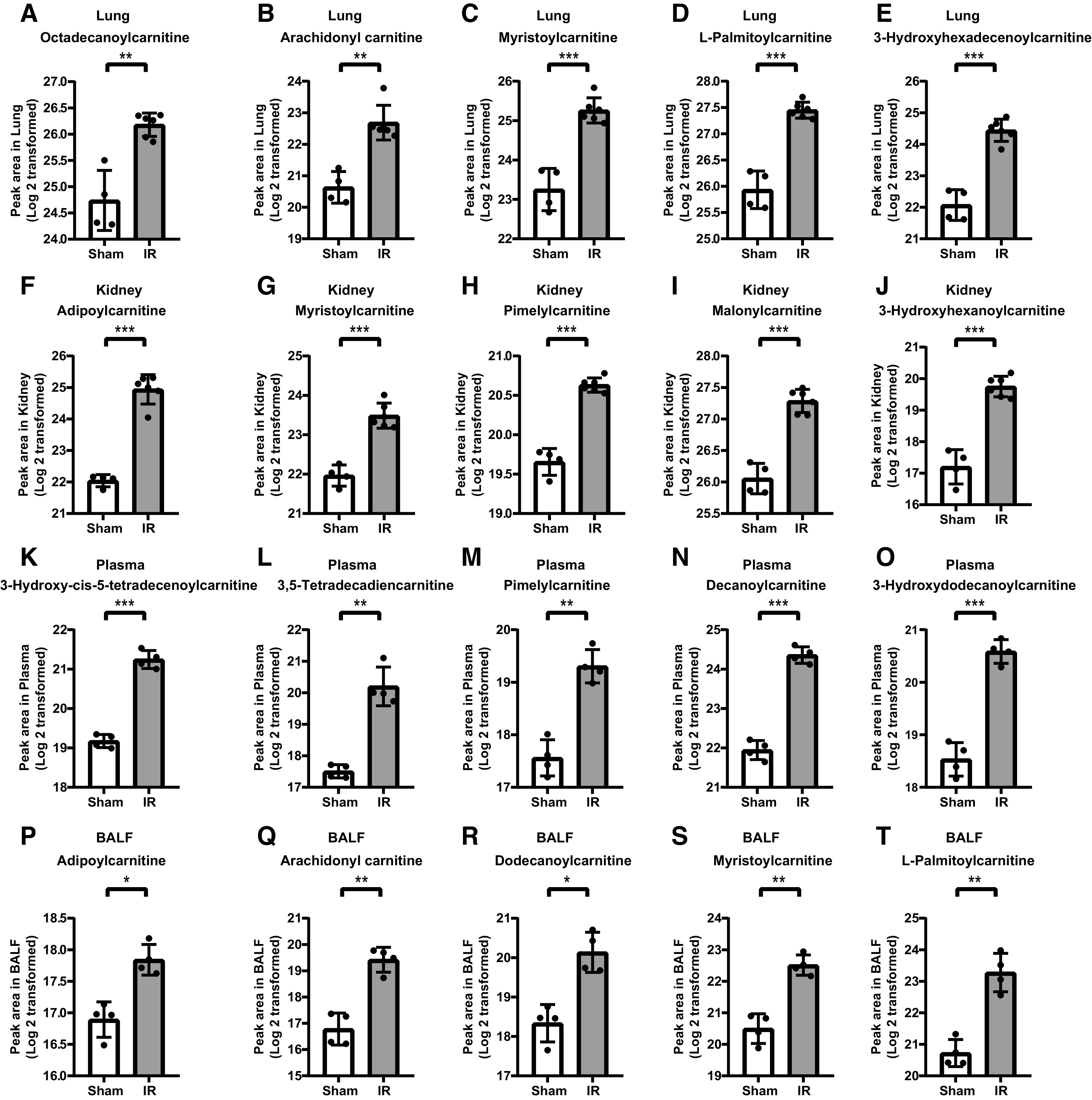
Comparison of top 5 FAO metabolites in the lung, kidney, plasma and BALF. A–E: Top 5 metabolites in lung, F–J: kidney, K–O: plasma, P–T: and BALF. n = 4–6 in each group (*P < 0.05, **P< 0.01, ***P< 0.001).
Extracellular mtDAMPs and Metabolomic Changes in the Lung in Vivo
We evaluated mtDNA and TFAM levels in plasma and BALF as evidence of extracellular mtDAMP accumulation in the setting of metabolomic derangements, as these mtDAMPS are known to cause lung injury in vivo (13, 14). We found that mtDNA and TFAM were increased significantly in plasma and BALF in the IR-AKI group compared to shams (Fig. 5). To determine if renal mtDAMPs may contribute to the metabolomic alterations we identified in the lung after IR-AKI, we evaluated the metabolomic profile of the lung in mice following intraperitoneal injection of mtDAMP solution isolated from the kidney. Again, a clear separation was achieved in lung tissue from mice exposed to mtDAMPs compared to vehicle by PLS-DA, indicating the existence of dramatic metabolic differences between the two groups (Fig. 6A). and we obtained a Q2 score of 0.46 which validated the PLS-DA model (19). Univariate analysis of the top 25 most significantly altered metabolites revealed 13 metabolites that were significantly increased and 12 metabolites that were significantly decreased in the mtDAMP group compared to vehicle (Fig. 6B). Most of the metabolites that were decreased in the mtDAMP group, such as urea, d-glyceraldehyde 3-phosphate and arginine, were metabolites broken down during anaerobic metabolism or those generated by mitochondrial metabolism. Pathway enrichment analysis showed that FAO was the second most altered pathway behind the phospholipid metabolism pathway (Fig. 6C), and all 5 of the FAO metabolites that were significantly altered were increased in the mtDAMP group compared to vehicle. Volcano analysis revealed that the 2 metabolites that showed the greatest fold-increase after mtDAMP exposure, 3-hydroxisovaleryl carnitine and 3-hydroxhexanoylcarnitine, are from the FAO pathway (Fig. 6D). The 2 metabolites that with the greatest fold-decrease in the mtDAMP group, phosphoenolpyruvic acid and D-glyceraldehyde 3-phosphate, are metabolized during glycolysis (Fig. 6D).
Figure 5.

Extracellular mitochondrial DNA and TFAM after IR-AKI. A, B: MtDNA in plasma and BALF and C, D: TFAM in plasma and BALF are increased in IR mice compared to shams. n = 5 in each group (*P < 0.05, **P< 0.01, ***P< 0.001).
Figure 6.
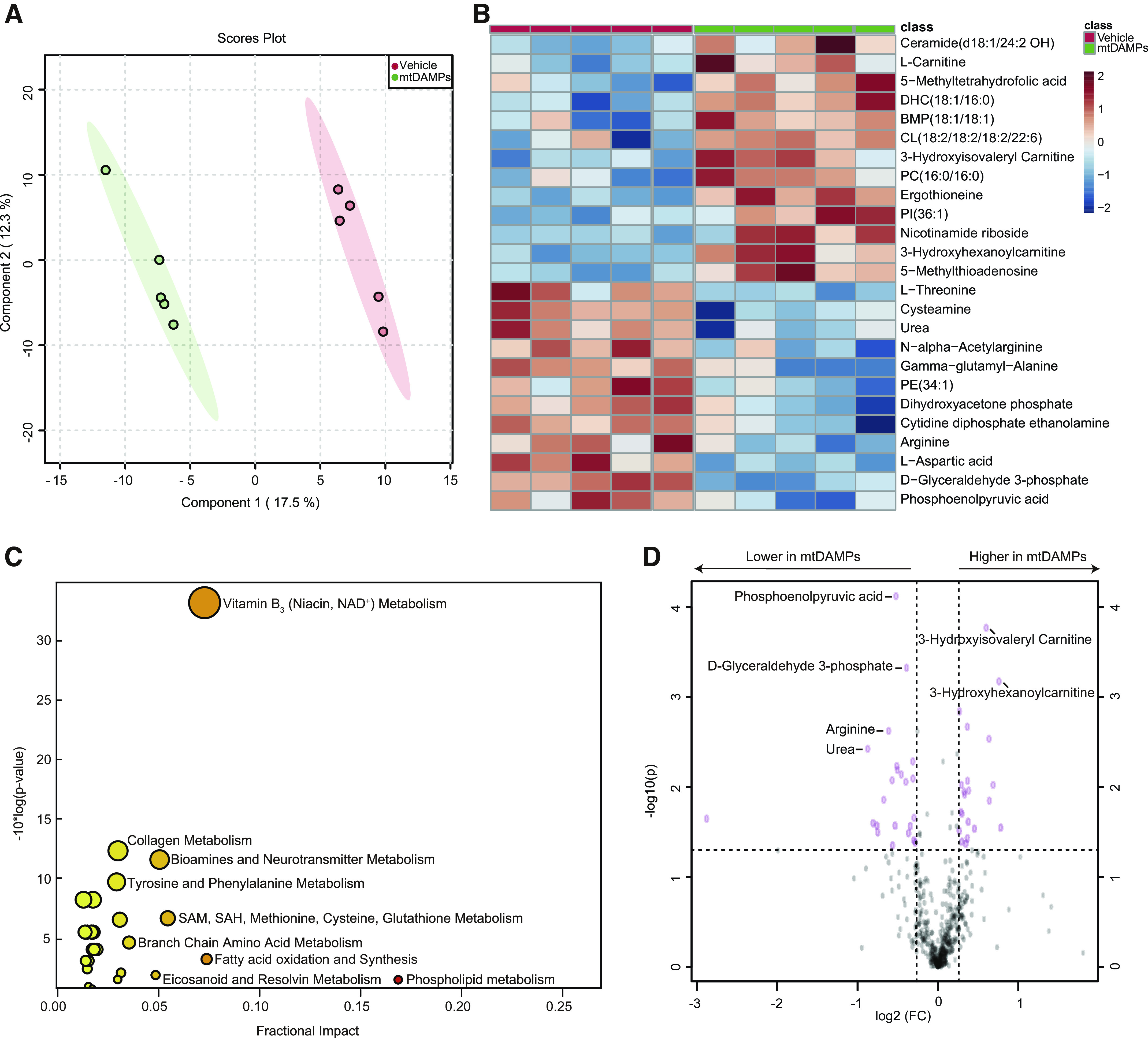
Metabolomic changes in lung tissue in response to renal mtDAMPs. A: PLS-DA showed dramatically different metabolic profiles between mice who received intraperitoneal injection of mtDAMPs compared to those who received vehicle. B, Top 25 metabolites altered by univariate analysis between mtDAMP and vehicle groups. C, Pathway enrichment analysis of significantly altered metabolic pathways. D, Volcano plot analysis of the significantly increased or decreased metabolites. n= 5/group. Fold change > 1.2 and FDR-adjusted P-value < 0.05 considered significant.
DISCUSSION
Treatment of AKI remains largely supportive, and novel management strategies focused on the remote organ complications from AKI, such as ARDS, have great potential to improve outcomes. In this study, we utilized next-generation metabolomics to identify metabolic pathways that are altered in lung injury following IR-AKI to identify novel targets for future investigations. We found significant metabolomic alterations in the lung correlated with previously identified markers of lung injury after IR-AKI, including increased lung MPO activity, increased wet/dry ratio and BALF albumin, and impaired ventilatory mechanics. The FAO pathway was the most significantly altered metabolic pathway in the lung, kidney, plasma, and BALF after renal IR. The alterations in the FAO pathway were characterized by an increase in medium and long chain acylcarnitines which were uniquely altered in each tissue and fluid we evaluated. Extracellular acylcarnitines have been established as biomarkers of mitochondrial dysfunction (20, 21), thus our findings suggest that IR-AKI leads to systemic mitochondrial dysfunction in multiple organs and cell types, including the lung. These results correlated with an increase in the mtDAMPs, mtDNA and TFAM, in the plasma and BALF. Finally, we showed that renal mtDAMPs cause metabolomic alterations with an increase in FAO metabolites and a decrease in metabolites generated through mitochondrial metabolism in the lung in vivo. Together, our results suggest that metabolomic changes consistent with mitochondrial dysfunction and renal mtDAMPs contribute to lung injury because of IR-AKI.
Our findings are consistent with other studies that found systemic metabolic derangements and mitochondrial dysfunction in the heart following renal ischemia (8, 9). In addition to an increase in FAO metabolites, we anticipated to see a decrease in metabolites from the glycolytic pathway, amino acids, and urea cycle metabolites as cells shift away from mitochondrial metabolism as was demonstrated in the heart by Fox et al. (8). However, we found only D-glyceraldehyde from the glycolysis pathway to be significantly decreased whereas others were largely unchanged. It is unclear why we did not see more of these metabolites significantly impacted in the lung, but we speculate that we would see more of a decrease in these metabolites at different time points given metabolism is a dynamic process. Intriguingly, our experiments investigating the impact of renal mtDAMPs on the lung in vivo did show a significant decrease in many metabolites, such as amino acids and urea, that would be expected to be decreased in the setting of mitochondrial dysfunction. These results correlated with the anticipated increase in FAO metabolites that we also showed following IR-AKI. It is worth noting that there are differences in the degree of metabolomic changes, as well as the exact metabolites and pathways that were most significantly impacted by IR-AKI compared to renal mtDAMP administration. These differences suggest that circulating mtDAMPs are not solely responsible for the metabolomic changes induced by IR-AKI, and additional mechanisms are likely to be involved. Although not the focus of our study, we found that other metabolic pathways implicated in lung injury and repair, such as eicosanoids, phospholipids, and ceramides, were also significantly impacted by renal IR and by renal mtDAMPs in vivo.
Circulating mtDAMPs are established mediators of lung injury (14, 22), but the mechanisms that lead to extracellular mtDAMP accumulation are largely unknown. Renal mitochondrial dysfunction, as occurs during AKI, leads to mitochondrial fragmentation (9), and mitochondrial fragmentation may lead to the release of mtDAMPs from the mitochondrial membrane (18). We evaluated for the mtDAMPs, mtDNA and TFAM, in the plasma and BALF as these specific mtDAMPs were shown to cause lung injury in vivo in previous studies (13, 14). Circulating TFAM promotes neutrophil infiltration in the lung, and mtDNA activates pulmonary endothelial cells (23) and neutrophils in vitro (24). MtDNA has also been shown to be released into the BALF where it promotes apoptosis in AECs during the early stages of lung injury (25, 26). We found that both mtDNA and TFAM were significantly increased in plasma and BALF after IR-AKI. These results are consistent with clinical data from Johansson et al. (27) who showed that circulating mtDNA levels were associated with increased plasma acylcarnitines. MtDAMPs are known to accumulate in plasma following trauma (14) and sepsis (28) and the circulating concentration of mtDNA has been shown to predict mortality in critically ill patients with ARDS (29). However, mtDNA and TFAM accumulating in the lung and circulation after IR-AKI, as observed in the present study, has not been previously reported to our knowledge. Importantly, we also provide novel in vivo data that suggest that circulating mtDAMPs may directly contribute to alterations in lung metabolism that are consistent with mitochondrial dysfunction.
The major strength of our study lies in the association we establish between renal mitochondrial dysfunction, extracellular mtDAMPs, and pulmonary mitochondrial dysfunction in lung injury because of IR-AKI. This provides a rationale for future bench and translational studies targeting mitochondrial dysfunction and mtDAMPs in preventing lung injury because of AKI. For example, inhaled DNase was recently shown to decrease extracellular mtDNA accumulation in BALF and mitigate lung injury in animal models of ventilator associated pneumonia (30) and lung ischemia (31). Our results suggest that inhaled DNase would be predicted to have a similar impact in mitigating lung injury and pulmonary mitochondrial dysfunction after AKI(32). Importantly, inhaled DNase is a safe and commonly used therapy that would be readily available for translational investigations. Mitochondria are also the main source of ATP in the lung, and many normal lung functions are dependent on mitochondrial metabolism (33). For example, type II airway epithelial cells utilize FAO metabolism to generate surfactant, and the accumulation of long-chain acylcarnitines has been shown to inhibit pulmonary surfactant production in mice (11). Thus, mitochondrial dysfunction and impaired FAO metabolism may contribute to the impaired lung mechanics we found after IR-AKI. Recently, mitochondrial transplantation was shown to improve lung mechanics and mitigate lung inflammation after pulmonary ischemia (34), and our findings suggest that this therapeutic approach may be beneficial in preventing indirect lung injury because of renal ischemia.
There are also some limitations to our study that are worth noting. For example, the IR-AKI model we utilized does not directly reflect the types of AKI encountered in human subjects with critical illness. Even in ischemic AKI there is unlikely to be a complete cessation of blood flow, and the mechanisms involved in AKI in the critically ill are complex and multifactorial. We utilized the IR model to evaluate the effect of isolated kidney injury on the lung, but it is important to consider these translational limitations in subsequent studies. Metabolism is a dynamic process; therefore, we cannot comment on the metabolomic profile in later stages of lung injury. We chose to focus on the 4-h time point where lung injury has been found to peak in prior studies of IR-AKI (3, 5). Our study also does not provide insights into the tissue or cellular source of mtDAMPs. We investigated the impact of renal mtDAMPS on the lung under the assumption that mtDAMPS are released into the circulation directly from renal cells. However, the systemic effects of renal IR could cause mtDAMP release from circulating immune cells or other remotely injured organs. Finally, we do not demonstrate a direct link between metabolomic alterations induced by circulating mtDAMPs and lung injury. Still, the detrimental impact of extracellular mtDAMPs regarding lung injury and inflammation has been established in prior studies (14, 22), and we provide a strong foundation for subsequent investigations.
In conclusion, we present evidence for mitochondrial dysfunction, as characterized by altered FAO metabolism and increased medium and long chain acylcarnitines, in the lung and systemic circulation after IR-AKI. We also show an increase in extracellular mtDAMPs after IR-AKI, and that renal mtDAMPs contribute to metabolomic changes consistent with mitochondrial dysfunction in the lung. These findings provide a rationale for bench and translational studies using therapies that are already available, such as inhaled DNase or mitochondrial transplantation, to prevent lung injury and improve mortality because of AKI.
GRANTS
This work was supported by R01 DK107852 (P.S.), VA Merit BX002175 (P.S.), VA CDA 2 IK2BX004338 01 (M.H.), American Society of Nephrology Ben J. Lipps Fellowship Award (M.H.), American Thoracic Society Unrestricted Critical Care Grant (M.H.), NIH R01 HL147326 (L.C.A.) and UCSD Grant RS169R (L.C.A.), Shandong Provincial Natural Science Foundation no. ZR2017MH075 (J.W.), and resources and support from the UAB/UCSD O'Brien Core Center for Acute Kidney Injury Research (NIH P30 DK 079337).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.H., J.W., K.L., Y.L., L.E.C.A., A.M., and P.S. conceived and designed research; M.H., J.W., K.L., Y.L., P.G., T.M., and A. Moshensky performed experiments; M.H., J.W., K.L., Y.L., P.G., T.M., A. Moshensky, M.A., L.E.C.A., A. Malhotra, and P.S. analyzed data; M.H., J.W., K.L., Y.L., P.G., A. Moshensky, M.A., L.E.C.A., A. Malhotra, and P.S. interpreted results of experiments; M.H., J.W., and P.G. prepared figures; M.H. drafted manuscript; M.H., J.W., K.L., Y.L., P.G., T.M., A. Moshensky, M.A., L.E.C.A., A. Malhotra, and P.S. edited and revised manuscript; M.H., J.W., K.L., Y.L., P.G., T.M., A. Moshensky, M.A., and L.E.C.A., A. Malhotra, and P.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Some of the results have been presented as abstracts at American Society of Nephrology and American Thoracic Society Annual Meetings.
REFERENCES
- 1.Mandelbaum T, Scott DJ, Lee J, Mark RG, Malhotra A, Waikar SS, Howell MD, Talmor D. Outcome of critically ill patients with acute kidney injury using the Acute Kidney Injury Network criteria. Crit Care Med 39: 2659–2664, 2011. doi: 10.1097/CCM.0b013e3182281f1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chertow GM, Christiansen CL, Cleary PD, Munro C, Lazarus JM. Prognostic stratification in critically ill patients with acute renal failure requiring dialysis. Arch Intern Med 155: 1505–1511, 1995. [PubMed] [Google Scholar]
- 3.Deng J, Hu X, Yuen PS, Star RA. Alpha-melanocyte-stimulating hormone inhibits lung injury after renal ischemia/reperfusion. Am J Respir Crit Care Med 169: 749–756, 2004. doi: 10.1164/rccm.200303-372OC. [DOI] [PubMed] [Google Scholar]
- 4.Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, Rabb H. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol 293: F30–F40, 2007. doi: 10.1152/ajprenal.00023.2007. [DOI] [PubMed] [Google Scholar]
- 5.Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int 74: 901–909, 2008. doi: 10.1038/ki.2008.314. [DOI] [PubMed] [Google Scholar]
- 6.White LE, Santora RJ, Cui Y, Moore FA, Hassoun HT. TNFR1-dependent pulmonary apoptosis during ischemic acute kidney injury. Am J Physiol Lung Cell Mol Physiol 303: L449–L459, 2012. doi: 10.1152/ajplung.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jesinkey SR, Funk JA, Stallons LJ, Wills LP, Megyesi JK, Beeson CC, Schnellmann RG. Formoterol restores mitochondrial and renal function after ischemia-reperfusion injury. J Am Soc Nephrol 25: 1157–1162, 2014. doi: 10.1681/ASN.2013090952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox BM, Gil HW, Kirkbride-Romeo L, Bagchi RA, Wennersten SA, Haefner KR, Skrypnyk NI, Brown CN, Soranno DE, Gist KM, Griffin BR, Jovanovich A, Reisz JA, Wither MJ, D'Alessandro A, Edelstein CL, Clendenen N, McKinsey TA, Altmann C, Faubel S. Metabolomics assessment reveals oxidative stress and altered energy production in the heart after ischemic acute kidney injury in mice. Kidney Int 95: 590–610, 2019. doi: 10.1016/j.kint.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sumida M, Doi K, Ogasawara E, Yamashita T, Hamasaki Y, Kariya T, Takimoto E, Yahagi N, Nangaku M, Noiri E. Regulation of mitochondrial dynamics by dynamin-related protein-1 in acute cardiorenal syndrome. J Am Soc Nephrol 26: 2378–2387, 2015. doi: 10.1681/ASN.2014080750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, Dokic D, Ball MK, Schumacker PT. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med 187: 424–432, 2013. doi: 10.1164/rccm.201207-1294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otsubo C, Bharathi S, Uppala R, Ilkayeva OR, Wang D, McHugh K, Zou Y, Wang J, Alcorn JF, Zuo YY, Hirschey MD, Goetzman ES. Long-chain acylcarnitines reduce lung function by inhibiting pulmonary surfactant. J Biol Chem 290: 23897–23904, 2015. doi: 10.1074/jbc.M115.655837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255, 2012. [Erratum in Nature 490: 292, 2012]. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaung WW, Wu R, Ji Y, Dong W, Wang P. Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med 30: 199–203, 2012. doi: 10.3892/ijmm.2012.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107, 2010. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li K, Naviaux JC, Bright AT, Wang L, Naviaux RK. A robust, single-injection method for targeted, broad-spectrum plasma metabolomics. Metabolomics 13: 122, 2017. doi: 10.1007/s11306-017-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li K, Wang X, Pidatala VR, Chang CP, Cao X. Novel quantitative metabolomic approach for the study of stress responses of plant root metabolism. J Proteome Res 13: 5879–5887, 2014. doi: 10.1021/pr5007813. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi N, Boysen G, Li F, Li Y, Swenberg JA. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int 71: 266–271, 2007. doi: 10.1038/sj.ki.5002033. [DOI] [PubMed] [Google Scholar]
- 18.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szymańska E, Saccenti E, Smilde AK, Westerhuis JA. Double-check: validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics 8: 3–16, 2012. doi: 10.1007/s11306-011-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knottnerus SJG, Bleeker JC, wüst RCI, Ferdinandusse S, L IJ, Wijburg FA, Wanders RJA, Visser G, Houtkooper RH. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord 19: 93–106, 2018. doi: 10.1007/s11154-018-9448-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGill MR, Li F, Sharpe MR, Williams CD, Curry SC, Ma X, Jaeschke H. Circulating acylcarnitines as biomarkers of mitochondrial dysfunction after acetaminophen overdose in mice and humans. Arch Toxicol 88: 391–401, 2014. doi: 10.1007/s00204-013-1118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Deng S, Zhao S, Ai Y, Zhang L, Pan P, Su X, Tan H, Wu D. Intra-peritoneal administration of mitochondrial DNA provokes acute lung injury and systemic inflammation via Toll-like receptor 9. Int J Mol Sci 17: 1425, 2016. doi: 10.3390/ijms17091425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Ma Z, Tang ZL, Stevens T, Pitt B, Li S. CpG DNA-mediated immune response in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 287: L552–L558, 2004. doi: 10.1152/ajplung.00436.2003. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34: 55–59, 2010. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- 25.Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci 16: 21486–21519, 2015. doi: 10.3390/ijms160921486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szczesny B, Marcatti M, Ahmad A, Montalbano M, Brunyánszki A, Bibli SI, Papapetropoulos A, Szabo C. Mitochondrial DNA damage and subsequent activation of Z-DNA binding protein 1 links oxidative stress to inflammation in epithelial cells. Sci Rep 8: 914, 2018. doi: 10.1038/s41598-018-19216-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johansson PI, Nakahira K, Rogers AJ, McGeachie MJ, Baron RM, Fredenburgh LE, Harrington J, Choi AMK, Christopher KB. Plasma mitochondrial DNA and metabolomic alterations in severe critical illness. Crit Care 22: 360, 2018. doi: 10.1186/s13054-018-2275-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H. Role of Mitochondrial DNA in Septic AKI via Toll-Like Receptor 9. J Am Soc Nephrol 27: 2009–2020, 2016. doi: 10.1681/ASN.2015040376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faust HE, Reilly JP, Anderson BJ, Ittner CAG, Forker CM, Zhang P, Weaver BA, Holena DN, Lanken PN, Christie JD, Meyer NJ, Mangalmurti NS, Shashaty MGS. Plasma mitochondrial DNA levels are associated with ARDS in trauma and sepsis patients. Chest 157: 67–76, 2020. doi: 10.1016/j.chest.2019.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmons JD, Freno DR, Muscat CA, Obiako B, Lee YL, Pastukh VM, Brevard SB, Gillespie MN. Mitochondrial DNA damage associated molecular patterns in ventilator-associated pneumonia: Prevention and reversal by intratracheal DNase I. J Trauma Acute Care Surg 82: 120–125, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mallavia B, Liu F, Lefrançais E, Cleary SJ, Kwaan N, Tian JJ, Magnen M, Sayah DM, Soong A, Chen J, Saggar R, Shino MY, Ross DJ, Derhovanessian A, Lynch JP , 3rd, Ardehali A, Weigt SS, Belperio JA, Hays SR, Golden JA, Leard LE, Shah RJ, Kleinhenz ME, Venado A, Kukreja J, Singer JP, Looney MR. Mitochondrial DNA stimulates TLR9-dependent neutrophil extracellular trap formation in primary graft dysfunction. Am J Respir Cell Mol Biol 62: 364–372, 2020. doi: 10.1165/rcmb.2019-0140OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hepokoski ML, Bellinghausen AL, Bojanowski CM, Malhotra A. Can we DAMPen the cross-talk between the lung and kidney in the ICU? Am J Respir Crit Care Med 198: 1220–1222, 2018. doi: 10.1164/rccm.201712-2573RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massaro GD, Gail DB, Massaro D. Lung oxygen consumption and mitochondria of alveolar epithelial and endothelial cells. J Appl Physiol 38: 588–592, 1975. doi: 10.1152/jappl.1975.38.4.588. [DOI] [PubMed] [Google Scholar]
- 34.Moskowitzova K, Orfany A, Liu K, Ramirez-Barbieri G, Thedsanamoorthy JK, Yao R, Guariento A, Doulamis IP, Blitzer D, Shin B, Snay ER, Inkster JAH, Iken K, Packard AB, Cowan DB, Visner GA, Del Nido PJ, McCully JD. Mitochondrial transplantation enhances murine lung viability and recovery after ischemia-reperfusion injury. Am J Physiol Lung Cell Mol Physiol 318: L78–L88, 2020. doi: 10.1152/ajplung.00221.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]