

Abstract
Airway submucosal gland serous cells are important sites of fluid secretion in conducting airways. Serous cells also express the cystic fibrosis (CF) transmembrane conductance regulator (CFTR). Protease-activated receptor 2 (PAR-2) is a G protein-coupled receptor that activates secretion from intact airway glands. We tested if and how human nasal serous cells secrete fluid in response to PAR-2 stimulation using Ca2+ imaging and simultaneous differential interference contrast imaging to track isosmotic cell shrinking and swelling reflecting activation of solute efflux and influx pathways, respectively. During stimulation of PAR-2, serous cells exhibited dose-dependent increases in intracellular Ca2+. At stimulation levels >EC50 for Ca2+, serous cells simultaneously shrank ∼20% over ∼90 s due to KCl efflux reflecting Ca2+-activated Cl− channel (CaCC, likely TMEM16A)-dependent secretion. At lower levels of PAR-2 stimulation (<EC50 for Ca2+), shrinkage was not evident due to failure to activate CaCC. Low levels of cAMP-elevating VIP receptor (VIPR) stimulation, also insufficient to activate secretion alone, synergized with low-level PAR-2 stimulation to elicit fluid secretion dependent on both cAMP and Ca2+ to activate CFTR and K+ channels, respectively. Polarized cultures of primary serous cells also exhibited synergistic fluid secretion. Pre-exposure to Pseudomonas aeruginosa conditioned media inhibited PAR-2 activation by proteases but not peptide agonists in primary nasal serous cells, Calu-3 bronchial cells, and primary nasal ciliated cells. Disruption of synergistic CFTR-dependent PAR-2/VIPR secretion may contribute to reduced airway surface liquid in CF. Further disruption of the CFTR-independent component of PAR-2-activated secretion by P. aeruginosa may also be important to CF pathophysiology.
Keywords: airway surface liquid, antimicrobial peptide, chronic rhinosinusitis, cilia, cystic fibrosis
INTRODUCTION
The secretion of airway surface liquid (ASL) is critical for the maintenance of mucociliary clearance (MCC) and for the ability to rid the airways of inspired pathogens and irritants (1–3). In conducting airways of large mammals like pigs and humans, submucosal exocrine glands (Fig. 1A) secrete a large percentage of fluid and mucus (1–4) as well as a battery of antimicrobial peptides (5, 6) that comprise the ASL. Knowledge of the regulation of submucosal gland secretion is essential for understanding lung fluid homeostasis, including how it is altered in diseases such as cystic fibrosis (CF) (4), chronic obstructive pulmonary disease (COPD) (7, 8), and asthma (7, 9–13).
Figure 1.

Submucosal exocrine gland serous acinar cells and protease-activated receptor 2 (PAR-2). A: diagram of anatomical structure of an airway submucosal gland, showing cystic fibrosis transmembrane conductance regulator (CFTR)-expressing serous cells at the distal acini, which have been demonstrated to be the origin of the majority of secreted fluid (14). B: differential interference contrast (DIC) and immunofluorescence images of middle turbinate submucosal gland stained with anti-PAR-2 antibody SAM11 (65). Scale bar is 50 µm. C: image of isolated serous acini. Scale bar is 25 µm. D: image of isolated acinar cells loaded with Ca2+ indicator fura-2. Scale bar is 10 µm.
Serous cells at the distal ends of the glands secrete the bulk of glandular fluid (14) and produce proteins important for airway defense, including lysozyme, lactoferrin, defensins, and Muc7 (4–6, 15, 16). Serous cells express the cystic fibrosis transmembrane conductance regulator (CFTR) secretory Cl− channel (17–22), which is activated by cAMP-dependent protein kinase A (PKA) phosphorylation. Intact glands from patients with CF fail to secrete fluid in response to agonists that elevate cAMP, such as vasoactive intestinal peptide (VIP) (23–27). Stimulation of isolated serous cells with VIP activates both conductive Cl− and HCO3− efflux, both likely through CFTR (12, 19). Serous cells thus likely contribute to both the volume and pH of glandular secreted fluid (14), and both parameters are likely altered in CF.
The volume of gland secretory cells may exceed the volume of surface goblet cells by as much as fifty times in cartilaginous airways (28–30). There has been recent interest in a rare surface epithelial cell type, termed the ionocyte, due to high CFTR expression in mouse airways (31, 32). However, mouse airways contain few submucosal glands (33). Furthermore, mouse submucosal glands exhibit reduced rates of fluid secretion compared with glands from larger animals (34, 35). In contrast to mice, fluid secretion is largely intact when the surface epithelium is stripped away in glandular pig bronchi (36–41), which are more histologically similar to human bronchi (1, 4, 33). Submucosal glands are clearly important for bulk airway fluid and mucus secretion in larger animals, regardless of the contributions of ionocytes, which have yet to be clarified in human airways. Occluded mucus-filled gland ducts, gland hypertrophy and hyperplasia, and gland infection are observed early in the lungs of patients with CF (42, 43) and CF pig models (44, 45), suggesting an important role for these glands in CF. Thus, a better knowledge of the mechanisms of serous cell secretion is important in understanding CF pathophysiology.
One major G protein-coupled receptor (GPCR) family driving robust secretion in salivary and pancreatic exocrine acinar cells are the protease-activated receptors (PARs) (46–49), which also play important roles in airway epithelial inflammation during asthma, allergy, and chronic rhinosinusitis (CRS) (49–53). PARs are activated by proteolytic cleavage of the extracellular N-terminus, which exposes a peptide sequence that acts as an intramolecular tethered ligand (54). Thrombin activates PAR-1, PAR-3, and PAR-4, whereas trypsin-like proteases activate PAR-2 and PAR-4 (52). PAR-2 may also be “disarmed” by some elastases and cathepsin G (55) via extracellular cleavage downstream of the activating site to remove the tethered ligand, preventing trypsin activation but not activation by peptides agonists (56). We demonstrated that protease-activated receptor 2 (PAR-2) is expressed basolaterally in primary sinonasal and bronchial ciliated cells on the surface epithelium, where it activates calcium-driven ciliary beat frequency (CBF) increases as well as CFTR-independent chloride secretion via elevation of intracellular Ca2+ (57, 58). Alterations of PAR-2 polarization with epithelial de-differentiation may contribute to enhanced sensitivity to inhaled proteases in allergic airway disease (57).
Although PAR-2 has been demonstrated to activate fluid secretion from intact airway submucosal exocrine glands of mouse trachea (59) and human inferior turbinate explants (60), the molecular mechanisms underlying PAR-2-induced secretion have not been thoroughly studied, including the effects of PAR-2 stimulation specifically on serous cells. Although PAR-2 can activate CFTR-independent gland secretion (59, 60), we hypothesized that CFTR may play an important role during combined PAR-2 and cAMP-elevating agonist stimulation, as observed with combined cholinergic (Ca2+-elevating) and VIPergic (cAMP-elevation) stimulation (19, 24). We also hypothesized that some host and pathogen proteases may have differential effects on PAR-2 activation of secretion, based on some prior studies described in the discussion.
To test these hypotheses and examine if and how PAR-2 contributes to serous cell fluid secretion, we quantitatively examined fluid secretion in living submucosal serous cells acutely isolated from human middle turbinate glands. We used minimally invasive optical imaging of intact serous cells that allowed study of both conductive and electroneutral channel and transporter pathways. We also cultured primary gland and surface epithelial cells at air-liquid interface (ALI) to quantitatively examine fluid and antimicrobial peptide secretion as well as ciliary beat frequency, respectively. The results below reveal mechanisms of human airway serous cell signaling, mechanisms of serous cell dysfunction related to CF, and mechanisms by which Pseudomonas aeruginosa infection may contribute to reduced airway surface liquid in CF through inactivation of PAR-2.
METHODS
Reagents
Fura-2-acetoxymethyl ester (AM), fluo-4-AM, BAPTA-AM, AlexaFluor-labeled secondary donkey anti-mouse, and donkey anti-rabbit antibodies were from Thermo Fisher Scientific (Waltham, MA). PAR-2 antagonists GB88 [IC50 ∼1–2 µM in HT29 cells (61–63)] was from MedChemExpress (Monmouth Junction, NJ). PAR-2 antagonist GB83 [IC50 ∼1–2 µM in HT29 cells (61–63)] was from Axon Medchem (Reston, VA). Unless indicated, cell culture reagents were from Gibco (Gaithersburg, MD). BTP2 (YM 58483; Cat. No. 3939), VIP6–28 (Cat. No. 1905), PAR-4 agonist AY-NH2 (Cat. No. 1487), and PAR-4 antagonist tcY-NH2 (Cat. No. 1488) were from Tocris (Bristol, UK). Phosphoramidon (Cat. No. 15113), VIP (Cat. No. 24996), cariporide (Cat. No. 16935), T16Ainh-A01 (Cat. No.18518), Eact (Cat. No. 29791), CaCCinh-A01 (Cat. No. 21922), AH 6809 (Cat. No. 14050), CFTRinh-172 (Cat. No. 15772), carbachol (CCh; Cat. No. 14486), charybdotoxin (CTX; Cat. No. 24115), and xestospongin C (XeC; Cat. No. 64950) were from Cayman Chemical (Ann Arbor, MI). Recombinant P. aeruginosa elastase produced in Escherichia coli (Cat. No. MBS1046200) was from MyBioSource (San Diego, CA).
Antibody for Na+/K+ ATPase (EP1845Y; Abcam Cat. No. ab76020) was validated for both immunofluorescence and Western blot using siRNA knockdown in a prior study (64). PAR-2 SAM-11 (mouse monoclonal, Abcam Cat. No. ab184673) was validated for immunofluorescence in a previous study (65) and here using siRNA knockdown (Dharmacon ON-TARGETplus SMARTpool Human F2RL1, Cat. No. L-005095-00-0005 and ON-TARGETplus nontargeting pool No. 1 Cat. No. D-001810-01-05). PAR-2 EPR180953 (rabbit monoclonal; Abcam Cat. No. ab180953) was previously validated for Western blot using siRNA (66) and shRNA (67) knockdown as well as siRNA knockdown in the present study. Glut1 (rabbit polyclonal; Cat. No. ab15309) as well as 2-furoyl-LIGRLO-NH2 (2FLI; Cat. No. ab120800) were from Abcam (Cambridge, MA).
Inorganic salts for buffers, S3226 (Cat. No. SML1996), soybean trypsin inhibitor (Cat. No. T6522), EGTA (Cat. No. E3889), TNFα (Cat. No. T6674), niflumic acid (Cat. No. N0630), bumetanide (Cat. No. B3023), dimethylamiloride (Cat. No. A4562), forskolin (Cat. No. F6886), H89 (Cat. No. B1427), 1-EBIO (Cat. No. SML0034), clotrimazole (Cat. No. C6019), trypsin (Cat. No. T4799), tryptase from human lung tissue (Cat. No. 650366-M), neutrophil elastase prepared from human blood (Cat. No. 324681), human thrombin (Cat. No. T9326), and all other reagents were from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise below.
Stocks solutions of all hydrophobic compounds were made at ≥1,000× in DMSO and frozen at −20°C. Working solutions were made up at the time of experiment with ≤0.1% DMSO; when compounds were dissolved in DMSO, control experiments contained an equal amount of DMSO to control for any effects of solvent alone. Heat inactivation of proteases was carried out for 20 min at 100°C, a time we found sufficient to inactivate trypsin (58) and others have found sufficient to inactivate P. aeruginosa elastase (68); after heat inactivation, protease solution was quenched on ice before being warmed to room temperature for use. An aliquot of the protease solution was saved before heat inactivation for use as a positive control for each experiment.
Solutions
The Krebs HCO3− buffer for isolated acinar cell experiments was based on the studies by Lee et al. (21, 22) and contained (in mM): 125 NaCl, 5 KCl, 1.2 MgCl2, 1.2 CaCl2, 1.2 NaH2PO4, 11 glucose, and 25 NaHCO3, gassed with 95% O2 + 5% CO2 for a pH of 7.4. Krebs HCO3−-free buffer contained (in mM): 125 NaCl, 5 KCl, 1.2 MgCl2, 1.2 CaCl2, 1.2 NaH2PO4, 11 glucose, 20 sucrose, 20 HEPES, gassed with 100% O2 and pH adjusted to 7.4 with NaOH. Hank’s balanced salt solution (HBSS) contained (in mM): 138 NaCl, 5.3 KCl, 0.4 KH2PO4, 0.24 NaHPO4, 0.41 MgSO4, 0.49 MgCl2, 1.8 CaCl2, 5.6 glucose, 20 HEPES with pH adjusted to 7.4 with NaOH. The Hank’s balanced salt solution (HBSS) used for tissue acquisition and Calu-3 Ca2+ experiments contained (in mM): 138 NaCl, 5.3 KCl, 0.4 KH2PO4, 0.34 NaHPO4, 0.41 MgSO4, 0.49 MgCl2, 1.8 CaCl2, 5.6 glucose, 20 HEPES pH 7.4.
We previously demonstrated that the activation of secretion by serous cells results in efflux of predominately K+ and Cl−, but also HCO3− (12, 19–22, 35). For experiments blocking the driving force for HCO3− and Cl− efflux [performed as described by McMahon et al. (12) and Lee et al. (21)], we assumed [HCO3−]i = 16 mM based on measured mean resting intracellular pH (pHi) of ∼7.2 (12, 21) using Henderson–Hasselbach with the pKa of CO2-HCO3− ≈ 6.1 (69, 70) and with highly permeant [CO2]o = [CO2]i = 1.2 mM in 5% CO2 gassed solution by Henry’s Law. With [HCO3−]o = 25 mM, the Nernst equilibrium potential for HCO3− (EHCO3−) ≈ 60 mV × log(16/25) = −12 mV. Mean resting [Cl−]i was previously measured at 65 mM (12). With 135 mM [Cl−]o in the Krebs buffer used, ECl− ≈ −19 mV by the Nernst equation as in the previous sentence. [K+]i was assumed to be 140 mM and with [K+]o = 5 mM, EK+ ≈ −87 mV. Using the Nernst equation, we calculated that setting [Cl−]o = 103 mM and [K+]o = 89 mM would set ECl− ≈ EK+ ≈ EHCO3− to reduce driving forces for efflux of cellular KCl and KHCO3. The solution used contained (in mM): 41 NaCl, 57 KCl, 32 KGluconate, 1.2 MgCl2, 1 CaCl2, 4 CaGluconate, 1.2 NaH2PO4, 11 glucose, 25 NaHCO3, pH 7.4 by gassing with 95% O2/5% CO2. As high K+ reduces the electrochemical driving force for Ca2+ influx due to plasma membrane depolarization, the high K+ solution contained 5 mM Ca2+o versus 1.2 mM Ca2+o in normal K+ solution. Solutions with 0-Na+ had isosmotic replacement of NaCl with N-methyl-d-glucamine (NMDG)-Cl, NaH2PO3 with KH2PO4, and NaHCO3− with NMDG-HCO3− as described by McMahon et al. (12) and Lee et al. (21).
Serous Cell Isolation
Primary serous acinar cells were isolated from human nasal middle turbinate as previously described (12, 19). Tissue was acquired from patients ≥18 yr of age undergoing surgery for sinonasal disease (e.g., CRS) or other medically necessary procedures (e.g., transnasal approaches to the skull base), carried out in accordance with The University of Pennsylvania guidelines regarding use of residual clinical material with Institutional review board approval (No. 800614) and with written informed consent obtained in accordance with the US Department of Health and Human Services code of federal regulation Title 45 CFR 46.116 and the Declaration of Helsinki. Exclusion criteria included history of systemic inheritable disease (e.g., granulomatosis with polyangiitis or systemic immunodeficiencies) with the exception of cystic fibrosis (CF). All patients with CF used in this study were genotyped as homozygous for the ΔF508 CFTR mutation. Comparisons between non-CF and CF cell secretion are valid, because acinar cells from both genotypes have identical resting [Cl−]i, resting pHi, intracellular pHi buffering capacity, and cAMP responses to VIP stimulation (12) as well as Ca2+ responses to PAR-2 stimulation (this study). The only parameter observed to differ is the Cl− efflux pathway during stimulation with cAMP agonists (12, 19).
Among non-CF patients, there is minimal patient-to-patient variability in the VIP-activated ion transport phenotype, as measured by cell shrinkage, once cells are removed from the tissue environment, as previously described (12, 19–22, 35). This allows disease-relevant in vitro manipulations (treatment with agonists ± inhibitors) with comparison of unmanipulated cells from the same patient as “control.” All experiments shown utilized cells from multiple patients as indicated in the figure legends. For logistical feasibility, some data points in each figure are independent experiments that used cells that originated from the same patient, as done in prior studies (12, 19–22, 35). An equal number of independent experiments, typically 2–3, was performed using cells from each patient to ensure that one patient could not skew results in any experiment. Serous cells isolated from 49 different patient turbinate samples were used in this study. Clinical characteristics of the patients from whom tissue was obtained are shown in Supplemental Table S1 (all Supplemental material is available at https://doi.org/10.6084/m9.figshare.13515098).
Isolated turbinate tissue was placed in HBSS containing 1% BSA, 2 mM l-glutamine, and 1× minimal essential media (MEM)-vitamins, amino acids, and nonessential amino acids. The epithelium was removed, and submucosal tissue was pulled off the bone. A volume of ∼500 µL submucosal tissue was mechanically minced with scissors in a 1.5-mL Eppendorf tube, then incubated for 90 min at room temperature in 10 mL HBSS with MEM supplements as aforementioned and mixed with 1 mg/mL Collagenase P (Roche), 10 µg/mL DNAse I (Roche), 0.01% soybean trypsin inhibitor with gentle shaking. After large tissue pieces were allowed to settle, gland acini were isolated from the overlaying digestion solution by centrifugation (30 s, 500 g), followed by washing with HBSS and further dispersal with 0.5 mg/mL collagenase P plus 0.01% soybean trypsin inhibitor for 45–60 min. Cells were pelleted (1 min, 800 g) and washed with HBSS before seeding onto glass coverslips for imaging or transwells for culturing as described (12).
Isolation of serous acinar cells from mouse nasal turbinate was carried out as described (21, 22, 35). PAR-2 knockout (par-2−/−) B6.cg-F2rl1tm1MSlb/J and isogenic wild type (WT) mice were purchased from Jackson Laboratory (Bar Harbor, ME) for other experimental purposes; residual nasal turbinate tissue was removed from mice euthanized by CO2 asphyxiation by B. Chen and N. Cohen (Philadelphia VA Medical Center and University of Pennsylvania) with Institutional Animal Care and Use Committee (IACUC) approval. Minced mouse nasal turbinate was digested for 60 min in 1 mg/mL collagenase P in solution as mentioned earlier, followed by centrifugation (1 min, 800 g) and washing with HBSS.
For air-liquid interface cultures, human acinar cells were resuspended in 50% minimal essential media plus Earle’s salts (MEME)/50% Ham’s F12K plus 20% FBS, 1× cell culture pen/strep supplement, gentamycin (100 μg/mL), and amphotericin B (2.5 μg/mL), modified from Finkbeiner et al. (71) and Fischer et al. (72) as described (12). Cells were seeded (∼3 × 105 cells/cm2) on transparent Falcon filters (No. 353095, 0.3 cm2, 0.4 μm pore size for antimicrobial peptide measurements or No. 353180, 0.9 cm2, 0.4 µm pore size for ASL height measurements) coated with human placental collagen. After confluence, the media were changed to 50% MEM + 50% Lonza bronchial epithelial basal media (BEBM) containing insulin (5 μg/mL), transferrin (5 μg/mL), hydrocortisone (0.5 μg/mL), triiodothyronine (20 ng/mL), and retinoic acid (50 nM) included with Lonza bronchial epithelial cell culture Singlequot supplements (without the EGF, epinephrine, bovine pituitary extract, or gentamycin/amphotericin supplements included), with added 2 mg/mL BSA, 2% NuSerum (Corning), and 1% cell culture penicillin/streptomycin supplement [modified from Finkbeiner et al. (71) and Fischer et al. (72)] as described (12). After 5 days of confluence, transepithelial electrical resistance (TEER) reached ∼300–500 Ω·cm2 and cells were fed with similar media except with NuSerum reduced to 0.5% on the basolateral side. The apical side was washed with PBS and exposed to air for 2–4 wk before use.
For siRNA experiments of primary cells, submerged cells were plated onto transwells at high density (100,000 cells per 0.3-cm2 transwell) in siRNA delivery media (Dharmacon Cat. No. B-005000-500) with Accell pooled siRNAs (Dharmacon non-targeting pool, Cat. No. D-001910-10-05; TMEM16A/ANO1 SMARTPool, Cat. No. E-027200-00-0005; BEST1 SMARTPool, Cat. No. E-019825-00-0005; or PAR-2 [F2RL1] SMARTPool, Cat. No. L-005095-00-0005) plus 2% NuSerum (Corning) for 72 h. Apical medium was removed and basolateral mediium was changed to differentiation media described in the previous paragraph for another 24 h, immediately followed by ASL height measurements (see Measurements of ASL Height).
Isolation and Culture of Surface Epithelial Cells
Air-liquid interface cultures of primary ciliated and goblet cells were cultured from surface epithelium of middle turbinate as described (57, 58, 73, 74). Cells were enzymatically dissociated and grown to confluence in 50% DMEM/Ham’s F-12 plus 50% bronchial epithelial basal media (BEBM, Lonza) for 7 days (57, 58, 73, 74). Dissociated cells were then seeded onto Corning Transwell filters coated with collagen, fibronectin, and bovine serum albumin. After 5–7 days, culture medium was removed from the upper compartment and cells were fed basolaterally with differentiation medium containing 50% DMEM and 50% BEBM plus Lonza B-ALI Singlequot supplements as provided but lacking the gentamycin and amphotericin (GA component). The medium was supplemented with 100 U/mL penicillin, 100 µg/mL streptomycin, and retinoic acid B-ALI inducer (Lonza, added fresh for each feeding) as described (57, 58, 73, 74). ALIs were cultured for 3 wk before use to ensure full polarization and differentiation of cilia. Mouse nasal septal cultures were grown similarly but differentiated in high-glucose (25 mM) DMEM with 2% NuSerum on the basolateral side, as previously described (58, 75).
Live Cell Imaging of Primary Serous Acinar Cells
Live cell simultaneous imaging of Ca2+ and serous cell volume were carried out as described (12, 20). After washing via gentle centrifugation and resuspension in HCO3− containing buffer, acinar cells were plated on CellTak (BD Biosciences/Corning)-coated glass coverslips and allowed to adhere for 10–20 min in 5% CO2. The isolation protocol yielded acini, single cells, and strings of cells that were identified based on visible morphology (size, polarized secretory granules, acinar structures) under differential interference contrast (DIC) optics. The rationale of using isosmotic changes in cell volume to track agonist-induced changes in secretory state in acinar cells is extensively described (22, 76, 77) and has been used for airway serous cells in prior studies (12, 19–22, 35). The relevance of these isosmotic volume changes to fluid secretion is also validated within this study.
Imaging was performed on an Olympus (Tokyo, Japan) IX-83 microscope with ×30 1.05 numerical aperture (NA) UPlanSApo silicone oil immersion objective. Excitation light for visible fluorophores was generated with an X-Cite 120 Boost LED (Excelitas Technologies, Waltham, MA). Excitation light for fura-2 was created with a xenon arc lamp (Lambda LS, Sutter Instruments, Novato, CA) with dual 340/380 nm excitation filters (Chroma Technology Corp., Bellows Falls, VT) switched with a filter wheel (Sutter Lambda 10-2) controlled by Metafluor (Molecular Devices, Sunnyvale, CA). Emission was captured with an ORCA Flash 4.0 sCMOS camera (Hamamatsu, Tokyo Japan). Microscope filters were from Chroma Technologies and were previously described (58, 73, 74). Cells were continuously perfused with 37°C solution heated with an inline heater (Warner Instruments, Hamden, CT); perfusion solution was gassed with 95% O2-5% CO2 or 100% O2 as appropriate in 37°C heated reservoirs. Calibration of fura-2 340/380 ratios only estimates global intracellular Ca2+ concentration ([Ca2+]i) and not localized [Ca2+] in microdomains (e.g., at the membrane within the vicinity of transporters and channels). Because this study does not depend on estimating global [Ca2+]i, and because fura-2 calibration can introduce error in some circumstances (78), we chose to report fura-2 data as background-subtracted 340/380 ratios rather than [Ca2+]i. Ratiometric 340/380 images were created by dividing the background-subtracted raw 340 nm excitation and 380 nm excitation fluorescence images using the image calculator in FIJI to create a 32-bit floating point image that was then scaled across the range of the ratio values by adjusting the min/max levels (no nonlinear adjustments applied) and intensity pseudocolored using the FIRE look-up-table.
Cell volume was estimated by taking the cross-sectional area of the cell as imaged by DIC to the 3/2 power (79, 80). DIC imaging with a high NA objective results in a shallow depth of field that allows optical sectioning through the center of the acinar cell with high-contrast cell edge visibility (79). We have found that this method yields volume measurements of cells and small acini faster than but indistinguishable from confocal 3D reconstructions (22). DIC images were acquired sequentially by computer controlled shuttering off of the fluorescence light, rotating of the DIC polarizer into position, and shuttering on transmitted light. Imaging data were collected and analyzed in Metafluor and/or FIJI (81).
As previously shown, serous cell shrinkage linearly tracks Cl− efflux (3, 19–22, 35), though some cellular HCO3− is also lost through the same conductive Cl− efflux pathways [CFTR (12) or TMEM16A (21)]. However, the relative magnitude HCO3− solute content lost during secretion is much smaller than Cl− content. A serous cell has resting [Cl−]i = ∼65 mM and loses >50% of cellular Cl− content (>40 meq/L) during ∼20% volume decrease (12, 19, 20, 22). The actual HCO3− content lost from the cell during secretion is much smaller; a ∼0.2 unit pHi change occurs with agonist-induced HCO3− efflux, causing [HCO3−]i to drop from ∼16 mM to 12 mM (calculated via Henderson–Hasselbach with 5% CO2). Considering the 20% volume loss but ignoring non-osmotically active volume for simplicity, this is a loss of cellular HCO3− content of (1 × 16 meq/L) – (0.8 × 12 meq/L) = ∼6.4 meq/L HCO3−. Thus, volume changes are primarily indicative of Cl− content changes. Although it is likely that conductive HCO3− secretion is affected in parallel with Cl− secretion as the efflux pathways are the same (CFTR or TMEM16A), any extrapolation of results here to HCO3− secretion is an assumption that requires confirmatory pHi imaging to track HCO3− secretion as previously done (12, 21).
Culture of the Calu-3 Serous-Like Cell Line and Imaging of Ca2+ with Fluo-4
Calu-3 cells were obtained directly from American Type Culture Collection (Manassas, VA) and cultured in Minimal Essential Media with Earle’s salts (Thermo Fisher Scientific), l-glutamine (Gibco), 10% FetalPlex fetal bovine serum complex (Gemini Bioproducts, West Sacramento, CA), and 1× cell culture Penicillin/Streptomycin mix (Gibco) as described (12, 82). Cells were confirmed to be mycoplasma negative (Cell Center Services Core, Department of Genetics, University of Pennsylvania) ∼5 passages before their use here. Cells were seeded onto transparent 0.4-µm pore Thincert 12-well cell culture inserts (Greiner BioOne) and grown with media on the apical and basolateral sides. After 5 days, confirmation of confluence was determined by measurement of transepithelial electrical resistance (TEER) > 300 Ω·cm2 measured via chop-stick style electrodes and an epithelial volt-ohmmeter (EVOM2 World Precision Instruments, Sarasota, FL). The apical side was then washed with PBS and exposed to air whereas the basolateral medium was changed to Lonza bronchial epithelial cell differentiation medium as for primary serous ALIs. Basolateral medium was changed three times per week.
Calu-3 cells were cultured for 3 wk for full differentiation/polarization before use. Cells were washed into HBSS and loaded with 10 µM Fluo-4 for 90 min followed by washing with HBSS. For pre-exposure to proteases or P. aeruginosa conditioned media (CM), cells were incubated in a separate dish for 5 min at room temperature. Cells were then transferred into a glass bottom dish (CellVis) containing only HBSS on the basolateral and apical sides for imaging. Cells were imaged using a standard green fluorescent protein (GFP)/FITC filter set (Semrock, Rochester, NY) on a Nikon microscope (×20 0.75 Plan Apo objective) equipped with a QImaging Retiga R1 camera and XCite 110 LED illumination system. Data were acquired with Micromanager (83). All experiments were performed at room temperature. A central field of view of each ALI was randomly chosen, and the entire field of view was used for measurement of Fluo-4 changes.
Western Blotting and Immunofluorescence
For PAR-2 Western blotting, ∼60 µg of isolated protein was loaded into each lane of a NuPage 4%–12% Bis-Tris gel. Separated proteins were transferred to nitrocellulose and blocked with 5% milk in 50 mM Tris, 150 mM NaCl, and 0.05% Tween-20. Anti-PAR-2 antibody (rabbit monoclonal; Abcam, Cambridge, MA) was used at 1:1,000 in 5% BSA in Tris-Tween for 1.5 h, followed by secondary goat anti-rabbit IgG-HRP at 1:10,000 for 1.5 h. Validation of this antibody for PAR-2 Western blot was carried out using Beas-2B immortalized bronchial cells cultured in Ham’s F12K media plus cell culture Pen/Strep mix and 10% FetalPlex. Cells were transfected with control or PAR-2 siRNA (as noted earlier) diluted in siRNA delivery media using Dharmafect transfection reagent. Cells were incubated for 72 h before lysis for Western blotting as described earlier. Beas-2B cells used were obtained directly from ATCC and confirmed to be mycoplasma negative, ∼6 passages, and <6 mo from their use here.
Immunofluorescence was carried out using PAR-2 mouse monoclonal antibody SAM11 (1:50 in DPBS + 1% BSA, 2% normal donkey serum, 0.2% saponin; Abcam) using previously described methods (20, 58). Dissociated or cultured cells were fixed with 4% formaldehyde followed by 3 min permeabilization with −20°C methanol and blocking with 1% BSA and 2% normal donkey serum in the presence of 0.25% saponin for 1 h. Imaging was performed on an Olympus IX83 microscope with spinning disk (Olympus DSU) ×60 (1.4 NA oil) objective, XCite 120 LED Boost excitation light source, Hamamatsu Orca Flash 4.0 sCMOS camera, and DAPI, FITC, TRITC, and Cy5 LED-optimized filter sets (Semrock). Paraffin-embedded nasal turbinate slices were stained for PAR-2 using SAM11 mouse monoclonal PAR-2 antibody and AlexaFluor488-conjugated secondary antibody. Imaging of tissue slices was done on an Olympus IX73 Fluoview laser scanning confocal microscope with ×60 (1.4 NA oil) objective and Argon ion laser. No offset, gain, or gamma alterations were applied. Validation of antibody staining for IF was carried out using PAR-2 and control siRNAs in primary cells as described above.
P. aeruginosa Culture, Preparation of Conditioned Medium, and Antibacterial Assays
Clinical strains used were previously described (12, 82). Briefly, patient sinonasal microbiology cultures were collected using BBL CultureSwab Plus transport system (Becton, Dickinson, and Co., Sparks, MD), grown overnight in Luria broth (LB) media, and speciated by the Philadelphia VA Medical Center microbiological laboratory. P. aeruginosa strain PAO1 (HER-1018; ATCC BAA-47) was from American Type Culture Collection and cultured in LB medium (Gibco/Thermo Fisher Scientific). PAO-JP2 (ΔlasI, ΔrhlI; Tcr, HgCl2r) (84, 85) was grown as previously described (86–88). Bacteria were grown with shaking at 37°C for two days to achieve a high-density plateau-phase culture (optical density [OD] > 2) to induce elastase production, then diluted with LB to OD 1. Bacteria were removed from the CM by centrifugation and CM was filtered sequentially through 0.4-µm and 0.2-µm syringe filters. Spotting of CM onto LB plates and incubation for 24 h at 37°C confirmed the absence of living bacteria in the CM. Dialyzed CM was obtained by incubating CM overnight in ∼500× volume of HBSS with Spectra/Por 1 regenerated cellulose 6–8 kDa molecular weight cut-off (MWCO) dialysis tubing at 4°C. This equilibrated salts likely removed small molecules like LPS as described (57). Note that molecular weights of P. aeruginosa elastase and thermolysin are both >30 kDa (89, 90) and thus should be largely retained by this dialysis. Single use aliquots of the dialyzed CM were frozen at −80°C.
Bacterial growth assays were carried out as previously described (12, 82, 91) similarly to a previously published protocol (87, 92). Cultures were washed copiously with PBS and transferred to antibiotic-free MEME for 48 h before use. Cultures were unstimulated or stimulated as indicated for 15 min on the basolateral side. Afterward, the surface of a 0.33-cm2 transwell filter was washed with 50 μL 25% saline. Although washing significantly dilutes the ASL fluid [normally ∼1 µL per cm2 of surface area (93)], washings retained antibacterial activity and were thus sufficient to be used for this assay. Note that protease concentrations used here (100 nM–1 µM) were insufficient to lift the confluent primary serous cells off of the transwell filter, which typically requires 0.25% trypsin (∼100 µM) for ≥15 min at 37°C. However, ASL was spun in at max speed for 5 min in a table-top picofuge to pellet any cells that might have lifted off the filter. ASL washings were pooled from multiple cultures stimulated under the same conditions and mixed with bacteria resuspended in 25% PBS, adjusted to 0.1 OD, then diluted 1:1,000 in 25% PBS. Bacteria/ASL mixture was incubated statically in a 96-well plate at 37°C for 2 h, followed by four serial 10-fold dilutions and spot plating 10 µL onto LB plates. After overnight incubation at 37°C, colony forming units (CFUs) were manually counted.
Enzyme-Linked Immunosubstrate Assays
Precoated enzyme-linked immunosubstrate assays (ELISAs) for lysozyme (Cat. No. OKCD01349) and lactoferrin (Cat. No. OKEH02822) were from Aviva Systems Biology (San Diego, CA) and carried out as per the manufacturer’s instructions with minor modifications. Briefly, wells precoated with capture antibody were blocked with 3% nonfat dry milk in PBS at 4°C for 1–2 h. After aspiration of fluid, wells were washed five times with 300 µL ELISA wash buffer (PBS + 0.5% Tween-20). Wells were incubated with manufacturer-supplied standards or ASL from cell cultures for 2 h at room temperature, followed by aspiration and washing as aforementioned. Biotinylated detection antibody was added in diluent buffer (supplied by the manufacturer) to each well, followed by incubation at 37°C for 1 h and subsequent aspiration and washing. Avidin-horseradish peroxidase conjugate was added, followed by incubation for 1 h at 37°C. After aspiration and six washes, a manufacturer-supplied substrate solution (3,3′,5,5′-Tetramethylbenzidine + acetic acid + H2O2) was added. Wells were incubated for ∼15 min at room temperature and absorbance was measured at 450 nm on a Tecan Spark 10M multimode Plate Reader. Comparisons were made if samples fell within the linear range of the standard curve.
Measurements of ASL Height
ASL height measurements were carried out largely as described previously (12, 94) at 37°C in a Tokai Hit stage top incubator in HCO3−-free conditions (100% O2 with basolateral HBSS buffered with 20 mM HEPES) using Texas red dextran (10,000 MW). Data presented are not corrected for refractive index mismatch, so ASL heights are approximate. When corrected for refractive index mismatch (1.52 ηoil/1.33 ηwater = ∼1.14), an observed change in ASL height of ∼30 µm over 15 min observed below with PAR-2 stimulation is ≈ 30/1.14 = 26 µm. Treating the ALI surface liquid as a cylinder, with volume = area (1 cm2 = 1 × 10−4 m2) × height, the ∼26 µm (2.6 × 10−5 m) change in ASL height of over 15 min equals a secretion volume of 2.6 × 10−5 m (height) × 1 × 10−4 m2 (area) ≈ 2.6 × 10−9 m3 ≈ 2.6 × 10−6 L. Thus, the cells secreted ∼2.6 µL/cm2 of fluid over 15 min, or ∼10 μL·cm−2·hr−1. These measurements of primary serous cells using the Texas red ASL technique are within an order of magnitude of measurements of Calu-3 serous-like cells using a “virtual gland” technique, which measured fluid secretion rates of 4 and 5.4 μL·cm−2·hr−1 when stimulated with forskolin and VIP, respectively (95). Thus, the ASL height measurements presented here are reasonable within the context of biological cellular fluid secretion capabilities.
PAR-2 Trio Arrestin-Binding Assay
PAR-2 Trio assay was modified from Ref. (96) as previously described (57). A549 cells were obtained directly from ATCC (confirmed mycoplasma negative ∼8 passages and <8 months before use) and grown in MEM plus 10% FetalPlex plus 1× cell culture pen/strep. Cells were seeded onto 8-well chamber slides and cotransfected with one plasmid encoding GFP β strands 1–9 (courtesy of Xiaokun Shu via Addgene No. 121684) and another containing PAR-2 or β2 adrenergic receptor (β2AR) fused to GFP β strand 11, arrestin fused to GFP β strand 10, and with mCherry as a transfection marker (Addgene plasmid No. 113609 or No. 113610, respectively). Cells were imaged 24 h after transfection at room temperature using standard GFP and TRITC filters on an Olympus IX83 microscope with ×40 (0.75 NA PlanFluor) objective. This assay allows direct visualization of PAR-2 activation and receptor interaction with β-arrestin, which allows formation of a complete GFP molecule and an increase in fluorescence. A549 cells were used over Calu-3 cells because of better sticking to glass without coating as well as much higher transfection efficiency. Cells were imaged over the course of 2 h using a programmable stage and the multiple stage positions function of Metamorph as described previously (57). Experiments were carried out at room temperature.
Measurement of Ciliary Beat Frequency
Whole-field ciliary beat frequency (CBF) was measured as previously described (58, 73, 74) using Sisson-Ammons Video Analysis software (97) on a Leica (Mannheim, Germany) DM I1 with ×20 0.8 NA objective with Basler (Ahrensburg Germany) A602f camera capturing images at 100 frames per second. Experiments were performed in HEPES-buffered HBSS at 30°C in atmospheric CO2. PAR-2 activating proteases were added to the basolateral side of the transwell in a 24-well glass bottom dish (CellVis, Mountain View, CA) as previously described (58).
Data Analysis and Statistics
One-way analysis of variance (ANOVA) was performed in Prism (GraphPad, La Jolla, CA) with appropriate post-tests as indicated; P < 0.05 was considered statistically significant. For comparisons of all samples within a data set, Tukey–Kramer post-test was used. For preselected pair-wise comparisons, Bonferroni post-test was performed. For comparisons to a single control group, Dunnett’s post-test was used. All other data analysis was performed in Microsoft Excel. For all figures, one asterisk or pound sign (* or #) indicates P < 0.05 and two asterisks or pound signs (** or ##) indicates P < 0.01 respectively; “n.s.” indicates no statistical significance. All data are presented as means ± SE. Traces shown are representative experiments. Bar graphs show individual data points from independent experiments on separate biological samples.
RESULTS
Human Nasal Turbinate Serous Cells Express PAR-2 That Drives Ca2+ Signaling
We used human middle turbinate submucosal gland serous cells, which are directly relevant to CF-related chronic rhinosinusitis (CRS), a significant complication in CF (98). Turbinate gland serous cells are also an approximation of serous cells from the lower airways. Histology suggests that nasal turbinate submucosal glands are similar to bronchial glands, though there may be differences in size and relative numbers of serous versus mucous cells (4, 15, 33). We previously showed that pig bronchial serous cell responses are nearly identical to human turbinate serous cells (19, 20), and thus nasal serous cell are likely a valid approximation of bronchial serous cells. We predominately focused on human cells for this study, as data from intact glands (4) and our own isolated cell studies (3, 19–22, 35) have established important differences between mouse serous cells and those from larger animals such as pigs and humans, including much smaller rates of CFTR-dependent cAMP-evoked secretion in mice.
We observed that human nasal middle turbinate glands exhibited strong immunofluorescence staining for PAR-2 (Fig. 1B), as previously reported for inferior turbinate glands (60). We isolated serous acini (Fig. 1C) from middle turbinate submucosa, proteolytically dissociated them into individual cells and small acinar clumps and loaded them with the ratiometric Ca2+ indicator fura-2 (Fig. 1D). We noted dose-dependent Ca2+ responses to the PAR-2 peptide agonist 2-furoyl-LIGRO-NH2 (2FLI; Fig. 2, A and B) as well as the PAR-2 activating protease trypsin (Fig. 2, C and D). After prolonged stimulation with trypsin, 2FLI had no further effect (Fig. 2C), supporting that trypsin and 2FLI act through the same pathway, likely PAR-2. Responses to trypsin were lost when the enzyme was heat inactivated for 20 min at 100°C or preincubated with a 2× molar excess of soybean trypsin inhibitor (Fig. 2D), demonstrating the Ca2+ response required enzyme activity. The response to trypsin was also inhibited by the phospholipase C (PLC) inhibitor U73122, but not by its inactive analog U73343 (Fig. 2, E and F). The Ca2+ response to trypsin was also blocked by PAR-2 antagonists GB83, GB88, or FSLLRY-NH2, but not PAR-4 antagonist tcY-NH2 (Fig. 2F), suggesting the Ca2+ response is dependent on PLC activation downstream of PAR-2 GPCR signaling.
Figure 2.
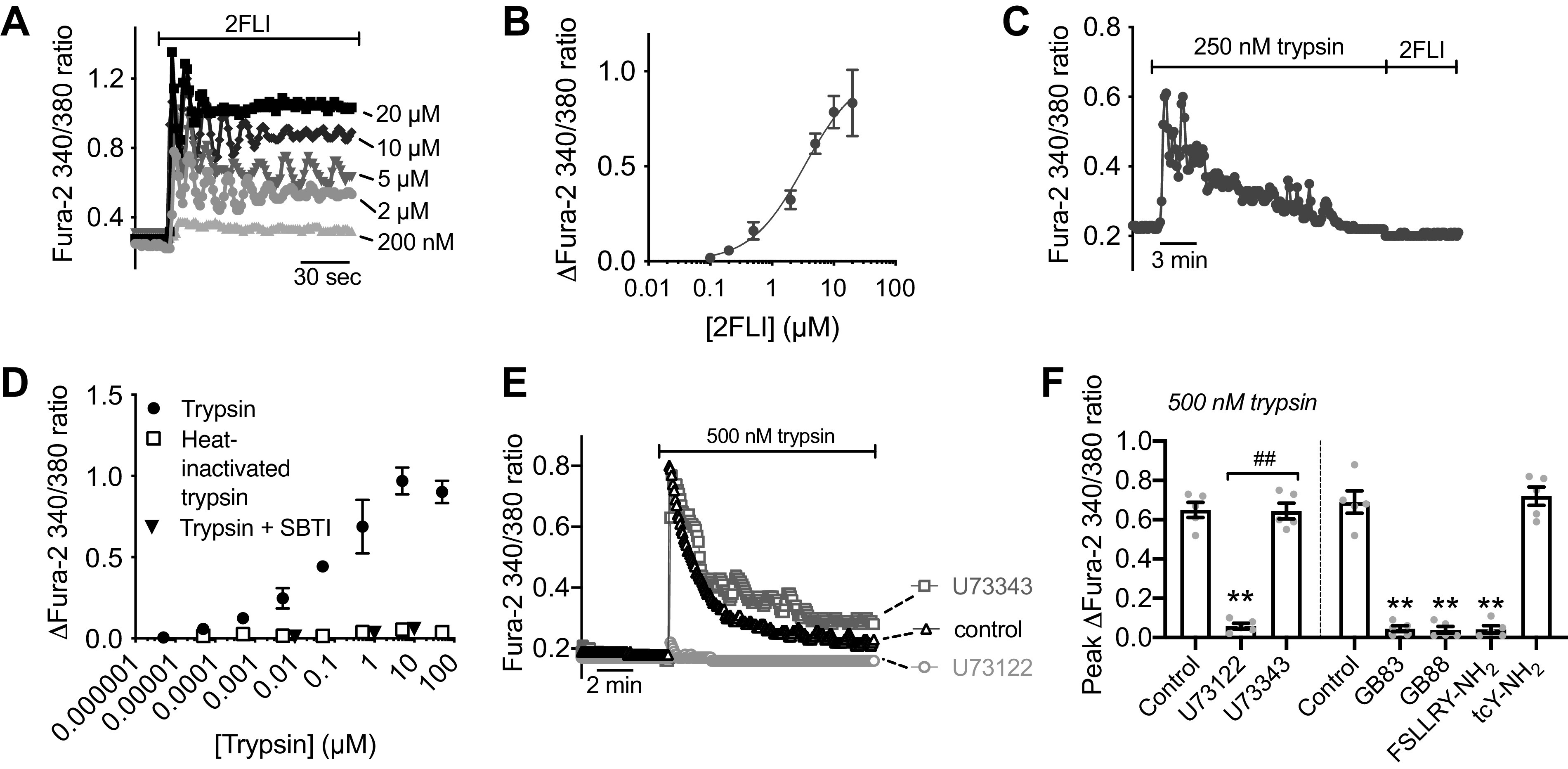
Activation of Ca2+ responses in submucosal gland serous acinar cells by protease-activated receptor 2 (PAR-2) peptide and protease activators. A: representative fura-2 Ca2+ traces from a single serous acinus stimulated with 200 nM to 20 µM PAR-2-activating peptide 2-Furoyl-LIGRLO-NH2 (2FLI) in increasing concentrations. Each stimulus was followed by 5 min perfusion with Krebs buffer for recovery. B: a nonlinear three parameter fit with constant slope (assuming a Hill coefficient of 1) of [2FLI] vs. peak fura-2 response (solid line) revealed apparent EC50 ≈ 3.4 µM for peak intracellular Ca2+. C: prolonged stimulation (>10 min) with trypsin to the point that the Ca2+ response decayed resulted in no further response with 2FLI. D: fura-2 340/380 ratio dose response during trypsin stimulation. Graph shows means ± SE of five (trypsin) or three [heat-inactivated trypsin or trypsin + soybean trypsin inhibitor ([SBTI)] individual experiments using cells from ≥3 patients. A nonlinear fit of the trypsin data revealed EC50 ≈ 0.04 µM. E: representative Ca2+ traces showing responses to trypsin after 30-min pretreatment with vehicle control (0.1% DMSO), U73122 (10 µM), or U73343 (10 µM). F: peak changes in fura-2 340/380 ratio (means ± SE) from experiments as in E were 0.65 ± 0.04 (control; 0.1% DMSO pretreatment only), 0.058 ± 0.01 (U73122 pretreatment; **P < 0.01 vs. control), and 0.64 ± 0.04 (U73343 pretreatment; ##P < 0.01 vs. U73122). In separate experiments with PAR-2 antagonists, peak fura-2 340/380 ratio changes (means ± SE) were 0.69 ± 0.06 (control; no antagonist), 0.045 ± 0.01 (PAR-2 antagonist GB83; 20 µM; **P < 0.01 vs. control), 0.038 ± 0.01 (PAR-2 antagonist GB88; 20 µM; **P < 0.01 vs. control), 0.042 ± 0.02 (PAR-2 antagonist FSLLRY-NH2; 50 µM; **P < 0.01 vs. control), 0.72 ± 0.05 (PAR-4 antagonist tcY-NH2; 50 µM). Significances by one-way ANOVA with Bonferroni post-test. Data points shown in graphs are 4–6 independent experiments per condition using independent cell samples obtained from ≥2 different patients.
Strong PAR-2 Stimulation Elicits CFTR-Independent Fluid Secretion, Likely Mediated by TMEM16A
To elucidate if these Ca2+ responses drive fluid secretion, we combined fura-2 Ca2+ imaging with DIC imaging to approximate cell volume from the cross-sectional area of single acinar cells and small acini. Exocrine acinar cells, including airway serous cells, can exhibit isosmotic changes in cell volume during secretagogue stimulation. These volume changes reflect salt and water flux due to activation of ion channels and transporters involved in fluid secretion (3, 76, 77, 80). Exocrine acinar cells can accumulate Cl− above electrochemical equilibrium [up to 65–70 mM (22, 99)]. Fluid secretion is typically driven by Cl− efflux out of the acinar cells into the gland lumen via apical membrane Cl− channels; this is balanced by counterion K+ efflux through K+ channels, typically localized basolaterally (3). This creates a lumen-negative transepithelial electrical potential then draws Na+ into the gland lumen via paracellular pathways. Osmotically obliged water follows the ions, resulting in secretion of a NaCl-rich isotonic primary fluid (3). Thus, activation of secretion can be accompanied by an initial net loss of KCl solute from the secretory cells. We previously showed that secretagogue stimulation of isolated serous cells activates shrinkage primarily reflecting conductive KCl and some KHCO3 efflux due to activation of K+ channels and secretory Cl− channels, either TMEM16A (20–22) or CFTR (12, 19) depending on the involvement of Ca2+ or cAMP, respectively.
Serous cells stimulated with a high level of PAR-2 agonist 2FLI (5 µM; a level higher than the observed EC50 for Ca2+) exhibited a ∼20% cell shrinkage that correlated with elevation of intracellular Ca2+ (Fig. 3, A–C). A PAR-4 agonist, AY-NH2, had no effect (Fig. 3, B and C). This shrinkage was not blocked by CFTRinh172 (Fig. 3D), which inhibits serous cell shrinkage in response to cAMP-elevating agonists like vasoactive intestinal peptide (VIP) (12, 19). Shrinkage in response to 5 µM 2FLI was instead blocked by CaCC inhibitors, including niflumic acid (100), T16Ainh-A01 (101), CaCCinh-A01 (102), and Ani9 (103) despite no reduction of the Ca2+ responses (Fig. 3, E–H). Although not specific for TMEM16A, these compounds are known TMEM16A blockers as well as blockers for some other CaCCs.
Figure 3.
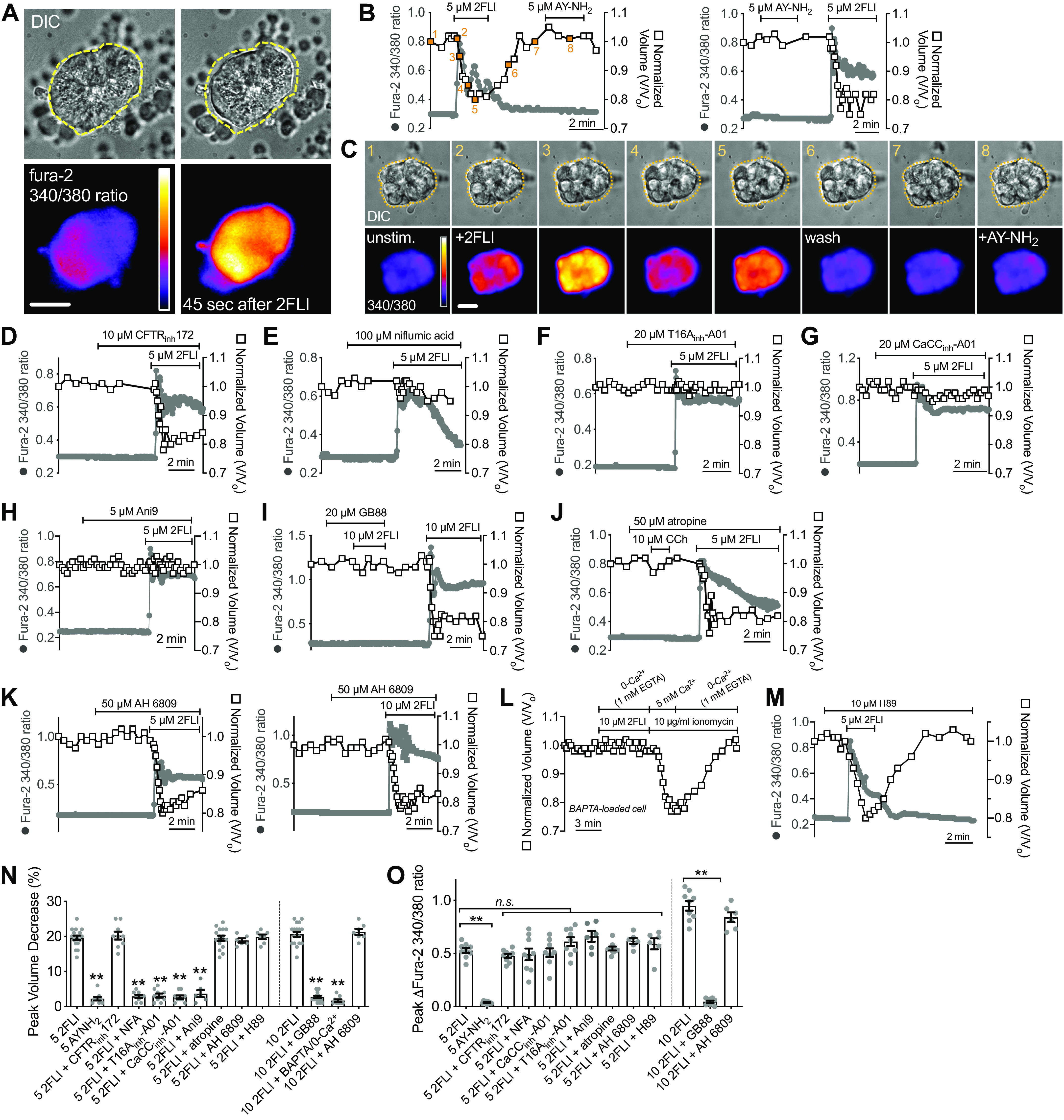
Protease-activated receptor 2 (PAR-2) receptor activation results in serous cell shrinkage that is dependent on Ca2+ and calcium-activated Cl− channel (CaCC) (likely TMEM16A) activity. A: representative image of serous cell volume and Ca2+ (fura-2 340/380 ratio) to 5 µM 2-Furoyl-LIGRLO-NH2 (2FLI). Representative traces (B) of Ca2+ (fura-2 340/380 ratio; gray solid circles) and volume (open squares) as well as differential interference contrast (DIC) and intensity pseudocolored ratiometric fura-2 images (C) showing responses during stimulation with 5 µM 2FLI or AY-NH2. Points from which the images are taken are denoted and numbered in orange (B, left trace). Scale bar in C is 10 µm. Representative Ca2+ and volume responses to 2FLI in the presence of CFTRinh172 (D), niflumic acid (E), T16Ainh-A01 (F), CaCCinh-A01 (G), and Ani9 (H). Representative Ca2+ and volume responses to 2FLI in the presence of GB88 (I), atropine (J), or AH 6809 (K). L: representative Ca2+ and volume responses in cells preloaded with 10 µM BAPTA-acetoxymethyl ester (AM) (30 min) stimulated in the absence of extracellular Ca2+ + 1 mM EGTA (0-Ca2+o). Note introduction of ionomycin and 5 mM extracellular Ca2+ at the end overcame Ca2+ buffering and caused shrinkage, whereas removal of Ca2+ in the continued presence of ionomycin caused cell swelling back to baseline. M: representative Ca2+ and volume trace during 2FLI stimulation in the presence of H89. N: bar graph showing peak volume decreases (means ± SE) from independent experiments as shown in B–M. Peak volume decreases (in %) were 19 ± 0.6 (5 µM 2FI), 2 ± 0.5 (5 µM AY-NH2; **P < 0.01 vs. 5 µM 2FLI), 20 ± 1 (5 µM 2FLI + CFTRinh172), 3 ± 0.4 (5 µM 2FLI + NFA; **P < 0.01 vs. 5 µM 2FLI), 3 ± 0.6 (5 µM 2 FLI + T16Ainh-A01; **P < 0.01 vs. 5 µM 2FLI), 3 ± 0.5 (5 µM 2FLI + CaCCinh-A01; **P < 0.01 vs. 5 µM 2FLI), 4 ± 1 (5 µM 2FLI + Ani9; **P < 0.01 vs. 5 µM 2FLI), 19 ± 0.6 (5 µM 2FLI + AH 6809), 20 ± 0.7 (5 µM 2FLI + H89), 19 ± 0.8 (5 µM 2FLI + atropine), 21 ± 0.7 (10 µM 2FLI), 3 ± 0.04 (10 µM 2FLI + GB-88; **P < 0.01 vs. 10 µM 2FL), 21 ± 0.9 (10 µM 2FLI + AH 6809), 2 ± 0.4 (10 µM 2FLI + BAPTA/EGTA; **P < 0.01 vs. 10 µM 2FL). O: bar graph showing peak fura-2 340/380 increases (means ± SE) from independent experiments as shown in B–L, which were 0.53 ± 0.02 (5 µM 2FI), 0.037 ± 0.004 (5 µM AY-NH2; **P < 0.01 vs. 5 µM 2FLI), 0.48 ± 0.02 (5 µM 2FLI + CFTRinh172), 0.49 ± 0.06 (5 µM 2FLI + NFA), 0.61 ± 0.04 (5 µM 2 FLI + T16Ainh-A01), 0.51 ± 0.04 (5 µM 2FLI + CaCCinh-A01), 0.66 ± 0.05 (5 µM 2FLI + Ani9), 0.62 ± 0.03 (5 µM 2FLI + AH 6809), 0.59 ± 0.05 (5 µM 2FLI + H89), 0.55 ± 0.02 (5 µM 2FLI + atropine), 0.95 ± 0.05 (10 µM 2FLI), 0.045 ± 0.01 (10 µM 2FLI + GB-88; P < 0.01 vs. 5 µM 2FLI), 0.84 ± 0.05 (10 µM 2FLI + AH 6809). Significances in L and M by one-way ANOVA with Bonferroni post-test comparing values to 5 µM or 10 µM 2FLI alone as respective controls. Bar graphs show data points and from 6 to 18 independent experiments using independent samples of cells from ≥3 patients. n.s., not significant.
Both 2FLI-activated shrinkage and Ca2+ were blocked by PAR-2 antagonist GB88 (Fig. 3I), but not by muscarinic antagonist atropine (Fig. 3J), which blocks shrinkage and Ca2+ responses to cholinergic agonist carbachol [CCh (20, 22)]. A prior study (104) suggested that prostaglandin release is involved in PAR-2-activated Cl− secretion in Calu-3 cells, a human bronchial adenocarcinoma line commonly used as a surrogate for serous cells. However, shrinkage in response to 2FLI was not inhibited by AH 6809, an EP and DP prostanoid receptor antagonist (Fig. 3K). Shrinkage was directly blocked by Ca2+ chelation (BAPTA-AM preloading plus extracellular Ca2+-free [0-Ca2+o]/1 mM EGTA conditions; Fig. 3L) but not by PKA inhibitor H89 (Fig. 3M). Peak shrinkage and Ca2+ responses are summarized in Fig. 3, N and O.
The aforementioned data suggest PAR-2 stimulation can activate Ca2+ to directly drive fluid secretion via CaCC (likely TMEM16A) and Ca2+-activated K+ channels. When serous cells were stimulated in the absence of extracellular Ca2+ (0-Ca2+o; no added Ca2+ plus 2 mM EGTA), both the Ca2+ and volume responses to 2FLI were more transient (Fig. 4, A and B), suggesting PAR-2 stimulation can activate release from intracellular Ca2+ stores that requires Ca2+ influx for sustained signaling. This was supported by a transient Ca2+ and shrinkage responses during stimulation in extracellular Ca2+-containing solution with store-operated calcium influx channel inhibitor BTP2 [YM58483 (105)] (Fig. 4C). Under 0-Ca2+o conditions, the transient Ca2+ response to 2FLI was inhibited by xestospongin C (XeC), an inhibitor of inositol trisphosphate receptors (IP3Rs) (106, 107) (Fig. 4D). These data are summarized in Fig. 4, E and F. Together, they support the key role of IP3-receptor Ca2+ release in the shrinkage response and suggest store-operated Ca2+ influx is necessary for sustaining secretion.
Figure 4.

Protease-activated receptor 2 (PAR-2) activated Ca2+ signaling and shrinkage is caused by both Ca2+ release from intracellular stores as well as Ca2+ influx. A: representative graph of Ca2+ (gray circles) and volume (open squares) showing sustained shrinkage and Ca2+ elevation in response to 10 µM 2-Furoyl-LIGRLO-NH2 (2FLI). B: representative graph of transient Ca2+ and volume responses in cells stimulated with 2FLI in the absence of extracellular Ca2+ (no added Ca2+ + 2 mM EGTA). C: representative graph of transient Ca2+ and volume in response to 2FLI in the presence of BTP2 (also known as YM58483). D: representative graph of Ca2+ and volume in cells stimulated with 2FLI xestospongin C (XeC) in the absence of extracellular Ca2+. E: bar graph showing peak fura-2 340/380 ratios from independent experiments as shown in A and B. Peak ratio values were 1.17 ± 0.07 (2FLI only), 1.08 ± 0.05 (2FLI in 0-Ca2+), 0.27 ± 0.04 (2FLI in 0-Ca2+ + XeC; P < 0.01 vs. 2FLI only), 1.10 ± 0.09 (2FLI + BTP2). Fura-2 ratio after 4 min was 0.61 ± 0.04 (2FLI only), 0.19 ± 0.02 (2FLI in 0-Ca2+; **P < 0.01 vs. 2FLI only), 0.25 ± 0.03 (2FLI + 0-Ca2+ + XeC; **P < 0.01 vs. 2FLI only), 0.20 ± 0.02 (2FLI + BTP2; **P < 0.01 vs. 2FLI only); n = 4 independent experiments each using cells from two individual patients. F: bar graph showing peak shrinkage from independent experiments as show in A and B. Peak shrinkages (in %) were 20 ± 0.1 (2FLI only), 18 ± 0.01 (2FLI in 0-Ca2+), 4 ± 1 (2FLI in 0-Ca2+ + XeC), 18 ± 1 (2FLI + BTP2). Shrinkage after 4 min was 18 ± 1 (2FLI only), 2 ± 1 (2FLI in 0-Ca2+), 2 ± 0.004 (2FLI in 0-Ca2+ + XeC), 3 ± 1 (2FLI + BTP2); n = 4 independent experiments each using cells from two individual patients. Significance in E and F determined by one-way ANOVA with Bonferroni post-test comparing values to 2FLI alone (control). All bar graphs show data points from independent experiments and means ± SE.
We saw similar cell shrinkage responses to trypsin (Fig. 5A), which activates PAR-2 and PAR-4, but not to thrombin (Fig. 5A), which activates PAR-4 only (49, 96). We also saw Ca2+ and shrinkage responses to neutrophil elastase purified from human blood (Fig. 5B) and tryptase purified from human lung (Fig. 5C). Tryptase-induced (Fig. 5D) and elastase-induced (Fig. 5E) Ca2+ and shrinkage responses were inhibited by PAR-2 antagonist GB88. Tryptase-induced Ca2+ and shrinkage were unaffected by PKA inhibitor H89 (Fig. 5F). In the presence of CaCCinh-A01, tryptase-induced Ca2+ responses were intact but shrinkage was absent (Fig. 5G). These data are summarized in Fig. 5H. We observed intact shrinkage responses to both 2FLI and trypsin in serous cells isolated from ΔF508 CFTR homozygous CF patient turbinates (Fig. 5, I and J), which have severely reduced CFTR Cl− permeability (12, 19), supporting a primary role for CaCC in these responses rather than CFTR.
Figure 5.
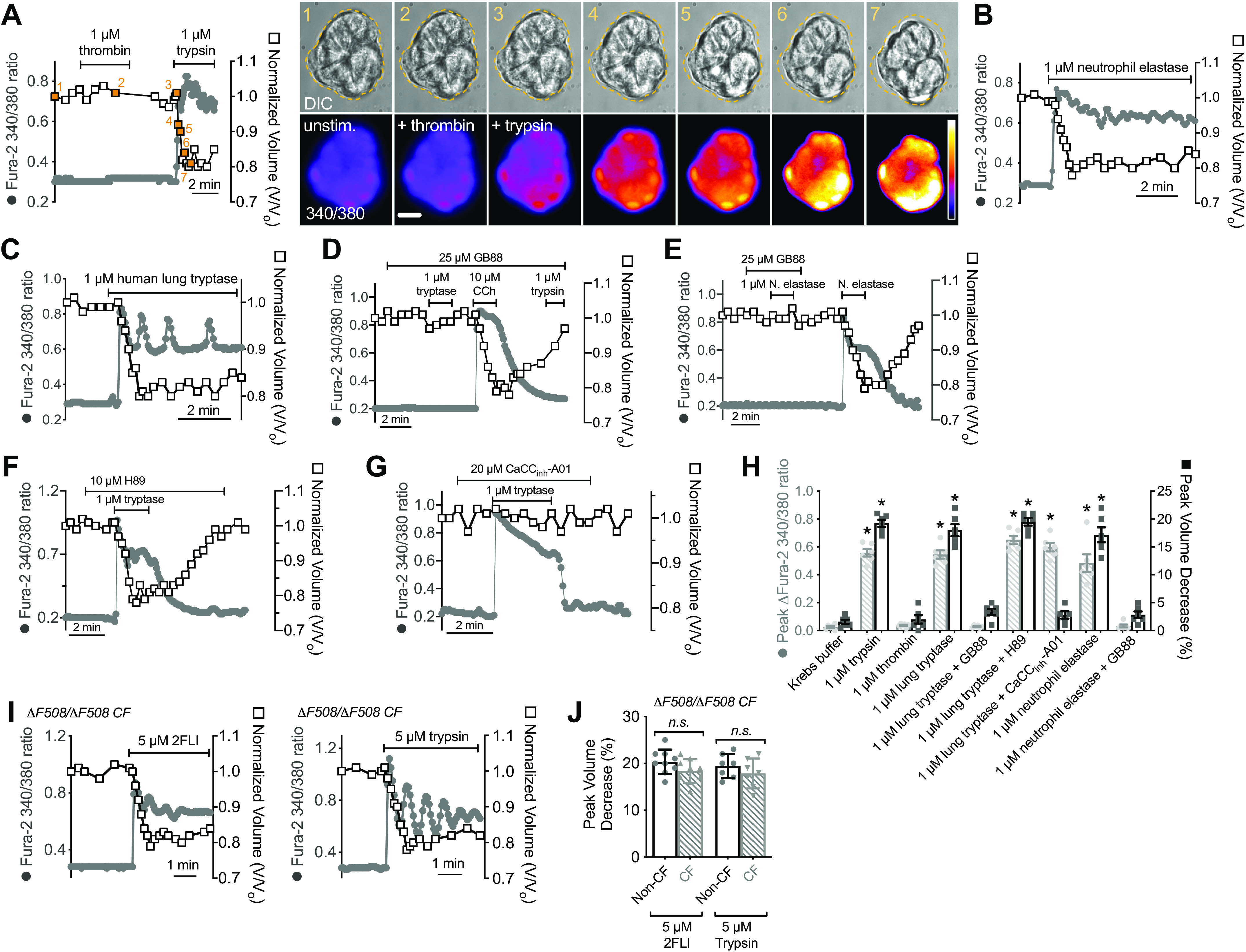
Activation of protease-activated receptor 2 (PAR-2)-dependent Ca2+ and shrinkage responses by trypsin and endogenous lung proteases. A: left is representative Ca2+ (gray circles) and volume (open squares) trace during stimulation with thrombin and trypsin. Right shows differential interference contrast (DIC) and intensity pseudocolored ratiometric fura-2 images from points indicated and numbered on the graph in orange. Dotted line on DIC image is cell outline at time 0 (image 1 of the sequence). Scale bar is 10 µm. Representative Ca2+ and volume traces during stimulation with neutrophil elastase (B), or human lung tryptase (C). Representative Ca2+ and volume traces during stimulation with tryptase (D), trypsin (D), neutrophil elastase (E), or carbachol (CCh; D) with GB88. Representative Ca2+ and volume traces during stimulation with tryptase with H89 (F) or CaCCinh-A01 (G). H: bar graphs showing peak change in fura-2 ratios (gray crossed bars) from independent experiments as shown in A–G; responses were 0.02 ± 0.01 (Krebs only), 0.56 ± 0.03 (trypsin), 0.04 ± 0.01 (thrombin), 0.54 ± 0.03 (human lung tryptase), 0.48 ± 0.06 (neutrophil elastase), 0.029 ± 0.01 (tryptase + GB88), 0.03 ± 0.01 (elastase + GB88), 0.65 ± 0.03 (tryptase + H89), 0.59 ± 0.03 (tryptase + CaCCinh-A01). Peak shrinkage (in %; black open bars) was 2 ± 1 (Krebs only), 19 ± 1 (trypsin), 2 ± 1 (thrombin), 18 ± 1 (tryptase), 17 ± 1 (neutrophil elastase), 3 ± 1 (tryptase ± GB88), 3 ± 0.1 (elastase + GB88), 20 ± 1 (tryptase + H89), 3 ± 1 (tryptase + CaCCinh-A01). Significance by one-way ANOVA with Dunnett’s post-test comparing all values to Krebs only control; n = 6 independent experiments using cells from ≥2 patients per condition; *P < 0.01. I: representative traces of Ca2+ and volume in cells from ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) homozygous cystic fibrosis (CF) patients stimulated with 2-Furoyl-LIGRLO-NH2 (2FLI) or trypsin alone. J: bar graph showing peak shrinkage (in %) in CF and non-CF patients; values were 20 ± 1 (non-CF, 2FLI), 18 ± 1 (CF, 2FLI), 19 ± 1 (non-CF, trypsin), 18 ± 1 (CF, trypsin); n = 9 and 7 independent experiments for 2FLI and trypsin, respectively using cells from ≥2 patients. No significant difference (n.s.) by one-way ANOVA with Bonferroni post-test with paired comparisons. All bar graphs show data points from independent experiments and means ± SE.
To further confirm the dependence of these responses on PAR-2, we tested Ca2+ and shrinkage responses in nasal turbinate serous cells from Wt and PAR-2 knockout (par-2−/−) mice. Wt mouse serous cells exhibited increased Ca2+ and shrinkage in response to trypsin (Fig. 6A), lung tryptase (Fig. 6B), or 2FLI (Fig. 6C), but not to PAR-4 activators thrombin (Fig. 6A) or AY-NH2 (Fig. 6C). The par-2−/− cells did not respond to trypsin (Fig. 6D), neutrophil elastase (Fig. 6D), tryptase (Fig. 6E), or 2FLI (Fig. 6F), nor did they respond to AY-NH2 (Fig. 6F), suggesting that PAR-4 was not functionally upregulated in the par2−/− serous cells over Wt cells, at least in terms of Ca2+ signaling.
Figure 6.
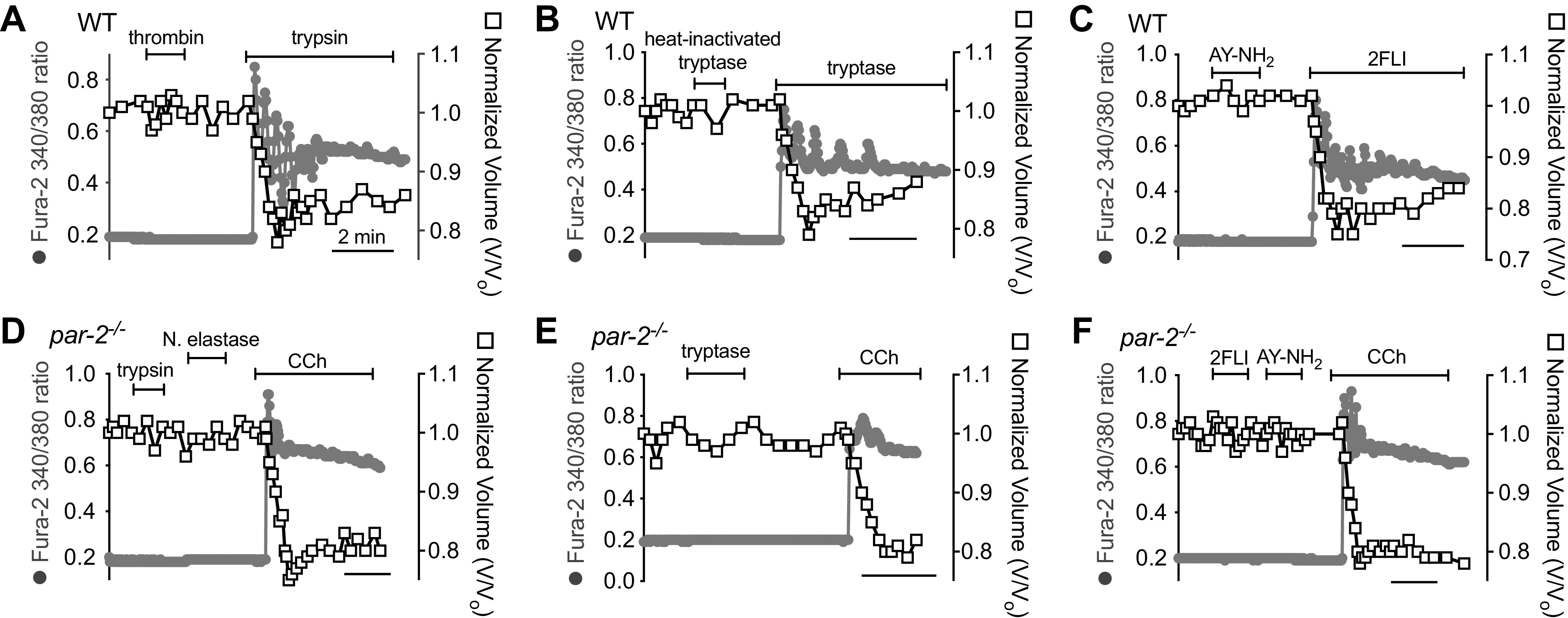
Ca2+ and shrinkage responses to trypsin, tryptase, and 2-Furoyl-LIGRLO-NH2 (2FLI) were eliminated in nasal turbinate serous acinar cells from protease-activated receptor 2 (PAR-2) knockout (par-2−/−) mice. Representative traces showing Ca2+ (gray circles) and volume (open squares) responses in nasal turbinate cells from Wt mice C57Bl/6 mice stimulated with 1 µM trypsin (A), 1 µM tryptase (B), 10 µM 2FLI (C), 1 µM thrombin (A), heat-inactivated 1 µM tryptase (B), or 10 µM AY-NH2 (C). Representative traces of cells from par-2−/− mice during stimulation with trypsin (D), tryptase (E), 2FLI (F), or carbachol (CCh, D–F). Results are representative of ≥3 experiments using cells from three separate mice of each genotype.
To further test that these cell shrinkage responses reflected Ca2+-activated secretion, as observed during stimulation with cholinergic agonists (12, 19–22, 35), we eliminated the driving force for conductive Cl− and HCO3− efflux using ion substitution to set EK+ = ECl− = EHCO3− We used our own prior measurements of [Cl−]i and [HCO3−]i in turbinate serous cells and assumed [K+]i was 140 mM, as described in methods. This concentration was empirically determined to have no acute de novo effect on [Ca2+]i and to maintain similar PAR-2 activated fura-2 responses (Fig. 7A). Under these high extracellular [K+] ([K+]o; 85 mM) conditions, human nasal serous cell shrinkage in response to 2FLI was blocked whereas Ca2+ responses were intact (Fig. 7A).
Figure 7.

Protease-activated receptor 2 (PAR-2)-induced cell shrinkage is due to conductive KCl efflux, whereas cells swelling is due to solute uptake via Na+K+2Cl− cotransporter (NKCC)1 and paired Na+/H+/anion exchanger (NHE/AE) activity. A: representative trace (left) of Ca2+ (gray circles) and volume (open squares) during stimulation with 10 µM 2-Furoyl-LIGRLO-NH2 (2FLI) in high K+o solution. Bar graph (right) shows peak fura-2 340/380 ratio (gray crossed bars) and shrinkage (%; black open bars) from six independent experiments in normal and high K+o. Peak fura-2 340/380 change was 0.54 ± 0.02 (normal K+o) vs. 0.53 ± 0.03 (high K+o). Peak shrinkage was 19 ± 1 (normal K+o) vs. 4 ± 0.7 (high K+o). Significance by one-way ANOVA with Bonferroni posttest comparing normal vs. high K+o for the two parameters. B: representative trace showing swelling with 100 µM niflumic acid after 2FLI-induced shrinkage despite elevated Ca2+ (left); effect was blocked in the absence of Na+o (right). Results are representative of four independent experiments using cells from ≥2 patients. C: representative traces showing transient Ca2+ and swelling during stimulation with 2FLI in 0-Ca2+o ± 100 µM bumetanide, 30 µM dimethylamiloride (DMA), or bumetanide + DMA. D: time to 50% volume recovery from experiments as in C was 150 ± 10 (control), 628 ± 25 (bumetanide; **P < 0.01 vs. control), 536 ± 41 (DMA; *P < 0.05 vs. control), 1119 ± 112 (bumetanide + DMA; **P < 0.01 vs. control; #P < 0.05 vs. bumetanide or DMA alone), 524 ± 63 (cariporide; **P < 0.05 vs. control), 188 ± 25 (S3226); n = 5 independent experiments each using cells from five individual patients. Significance determined by one-way ANOVA with Bonferroni posttest. E: model of secretion evoked during “strong” PAR-2 stimulation (>EC50 for Ca2+). PAR-2 activates Ca2+ signals that activate Ca2+-sensitive K+ channels and Cl− channels (likely TMEM16A) that mediate KCl efflux, reflected by isosmotic cell shrinkage. Secretion is likely sustained by secondarily active (Na+ gradient-dependent) Cl− influx via NKCC1 and paired anion (Cl−/HCO3−) exchange (AE) and dimethylamiloride-sensitive NHE activity as described for carbachol (CCh)-induced secretion (3). Upon removal of agonist and lowering of Ca2+, these pathways allow the acinar cells to accumulate Cl− above electrochemical equilibrium (3, 12), resulting in isosmotic swelling. CaCC, calcium-activated Cl− channel; n.s., not significant.
Further supporting the notion that shrunken cells are actively secreting, we found that when niflumic acid was applied to block Cl− efflux after PAR-2 stimulation, shrunken cells swelled back to resting volume despite Ca2+ remaining elevated (Fig. 7B). We hypothesized that this swelling reflected the activity of basolaterally localized Na+-coupled solute influx pathways that sustain secretion, such as the Na+K+2Cl− cotransporter (NKCC) and paired Na+/H+ (NHE) and Cl−/HCO3− (anion) exchange (AE) activity, as observed in airway serous and parotid acinar cells during cholinergic stimulation (3, 12, 20, 22, 99). Stimulated cell swelling during niflumic acid application was blocked when extracellular Na+ was isosmotically replaced with NMDG+ (0-Na+o conditions; Fig. 7B), demonstrating a requirement for Na+ influx.
To determine the molecular identities of the Na+-coupled solute influx pathways, we used a protocol for measuring cell swelling previously used for mouse turbinate serous cells (21). Cells were stimulated with 2FLI in the absence of extracellular Ca2+ (extracellular solution with no added Ca2+ + 1 mM EGTA to chelate trace Ca2+). Stimulation in 0-Ca2+o solution resulted in agonist/Ca2+-activated shrinkage that relaxed as Ca2+ rapidly returned to baseline in the absence of Ca2+ influx. Swelling under these conditions was inhibited ∼50% by NKCC inhibitor bumetanide and ∼50% by NHE inhibitor dimethylamiloride (DMA), and more fully reduced by both inhibitors combined (Fig. 7, C and D). Thus, solute influx likely reflects both NKCC (likely NKCC1) and paired NHE/AE activity, as previously described during cholinergic stimulation of serous cells (3) and parotid acinar cells (108).
The NHE1 isoform is expressed on the basolateral membranes of many cells, including parotid and submucosal gland acinar cells (21, 109), whereas NHE3 is expressed in the intestine on the apical membrane and endosomes (110). The NHE1-specific inhibitor cariporide blocked swelling similarly to DMA (Fig. 7D), whereas the NHE3 inhibitor S3226 did not significantly reduce swelling (Fig. 7D), as observed with cholinergic stimulation of serous cells (19, 20). The data up to this point suggest that stimulation of PAR-2 can activate Ca2+ signals that drive Cl− efflux through CaCC/TMEM16A, which is apically localized in intact gland acini (12, 20, 35). This Cl− secretion is likely supported by counterion K+ efflux through basolateral Ca2+-activated K+ channels. Thus, acinar cell shrinking during PAR-2 stimulation likely reflects activation of Cl− and fluid secretion (Fig. 7E). Secretion is likely sustained by NKCC1 and paired NHE/AE activity, localized basolaterally in intact glands (21). This can be visualized by cell swelling when solute efflux pathways are shut down via lowering of Ca2+ or pharmacological blockade (Fig. 7E).
Lower-Level PAR-2 Stimulation Does Not Activate Secretion Alone, but Can Synergize with cAMP-Elevating Receptors to Activate CFTR-Dependent Secretion
As noted, the ability of PAR-2 to activate Ca2+-driven CFTR-independent secretion is similar to muscarinic stimulation and distinct from secretion observed with activation of vasoactive intestinal peptide (VIP) receptors (VIPRs) or β2 adrenergic receptors (β2ARs) that drive secretion through cAMP/PKA activation of CFTR. However, these cAMP-elevating receptors also stimulate low-level cAMP-dependent Ca2+ signals that, while insufficient to activate CaCC/TMEM16A, are necessary to activate K+ channels for counterion flux (Fig. 8A) (12, 19, 35). Confirming our previous studies (19), we observed that low-level Ca2+ responses in human nasal serous cells to VIP stimulation were inhibited by PKA inhibitor H89 (Fig. 8B), suggesting they are downstream of PKA. As also expected based on prior studies (12, 19, 35), we found that cell shrinkage during direct adenylyl cyclase activation with forskolin (Fig. 8, C and D) or with VIP (Fig. 8E) was absent in ΔF508/ΔF508 CFTR cells despite intact cAMP-activated Ca2+ responses (Fig. 8F).
Figure 8.
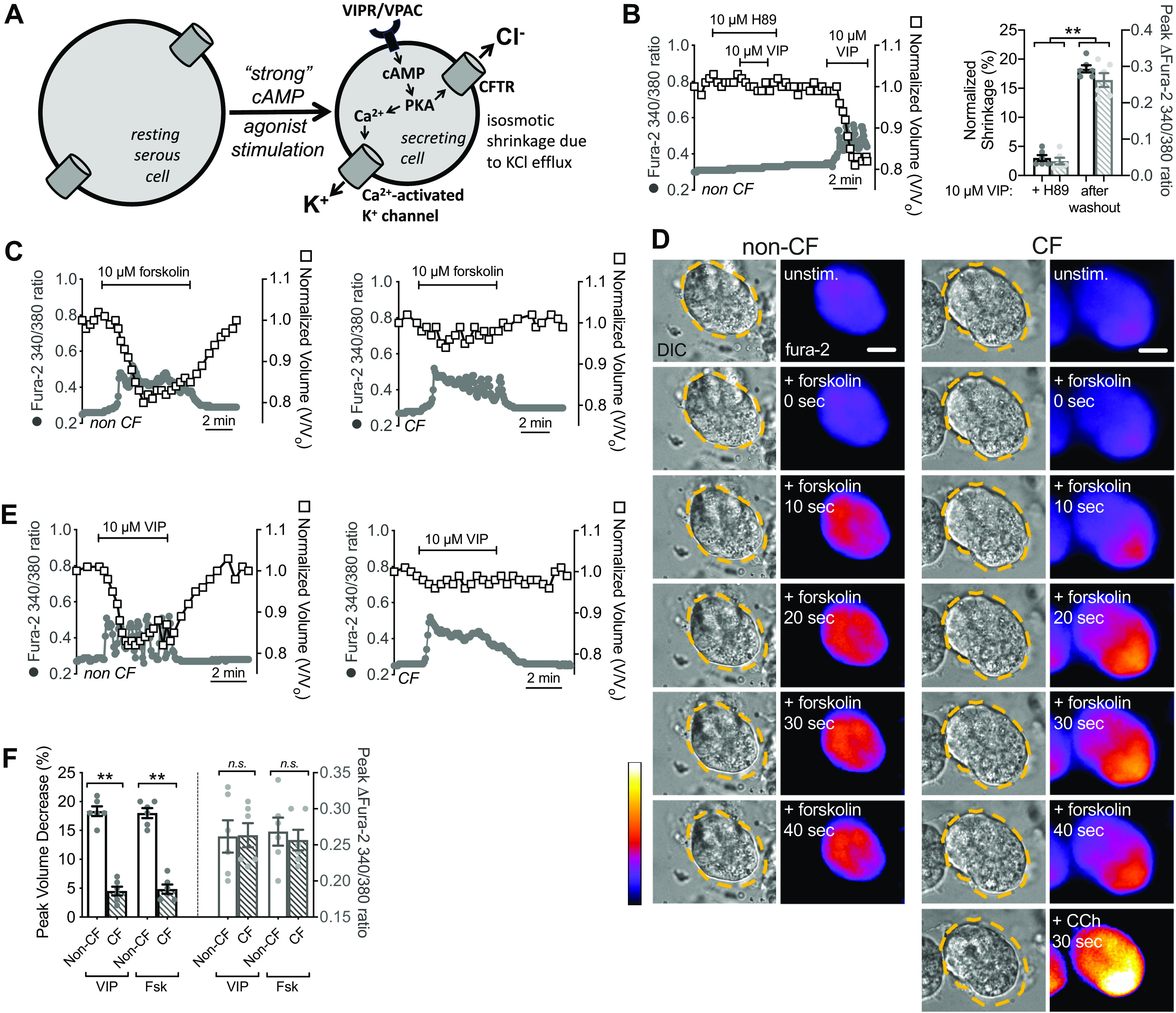
Secretion evoked by cAMP-elevating agonists is dependent on cystic fibrosis transmembrane conductance regulator (CFTR). A: diagram showing previously proposed model of cAMP-activated secretion (3, 12, 19, 35). Elevation of cAMP downstream of Gs-coupled G protein-coupled receptor (GPCRs) [vasoactive intestinal peptide receptor (VIPR) or β2Ars] causes protein kinase A (PKA)-dependent activation of CFTR as well as PKA-dependent low-level Ca2+ signaling required for K+ channel activation but insufficient to activate TMEM16A. Activation of both of these pathways results in KCl efflux and isosmotic cell shrinkage in isolated cells and acini. B: left shows representative Ca2+ (fura-2 340/380, gray circles) and volume (open squares) during vasoactive intestinal peptide (VIP) stimulation ± H89. Bar graph on right compares shrinkage and Ca2+ before and after H89. Peak shrinkages (in %) were 3 ± 1 (VIP + H89) and 18 ± 1 (VIP after H89 washout; **P <0.01). Peak fura-2 ratio changes were 0.04 ± 0.01 (VIP + H89) and 0.25 ± 0.02 (VIP after H89 washout; **P < 0.01); n = 6 independent experiments using cells from three individual patients. Significance by one-way ANOVA with Bonferroni post-test comparing each parameter between the two conditions; **P < 0.01. C: representative Ca2+ and volume traces in cells isolated from non-cystic fibrosis (CF, left) and ΔF508/ΔF508 CF cells (right) stimulated with forskolin. D: representative images showing cell volume (differential interference contrast, DIC) and low-level Ca2+ responses to 10 µM forskolin in cells from a non-CF (left) or ΔF508 CFTR homozygous CF patient (right). Note intact Ca2+ response but lack of shrinkage in CF cells. CF cells were subsequently stimulated with 10 µM carbachol (CCh) to confirm intact calcium-activated Cl− channel (CaCC)-dependent secretion. Scale bar is 10 µm. E: representative Ca2+ and volume traces in cells isolated from non-CF (left) and ΔF508/ΔF508 CF cells (right) stimulated with VIP. F: peak shrinkage (in %) was 18 ± 1 (non-CF, VIP), 5 ± 1 (CF, VIP; **P < 0.01 vs. non-CF), 18 ± 1 (non-CF, forskolin), 5 ± 1 (CF, forskolin; **P < 0.01 vs. non-CF). Peak change in fura-2 340/380 ratio was 0.26 ± 0.02 (non-CF, VIP), 0.26 ± 0.01 (CF, VIP), 0.27 ± 0.02 (non-CF, forskolin), 0.26 ± 0.01 (CF forskolin); n = 6 independent experiments using cells from three individual patients (two experiments per patient). Significance by one-way ANOVA with Bonferroni post-test with paired comparisons (non-CF vs. CF for each stimulation). Bar graphs show data points from independent experiments and means ± SE. n.s., not significant.
Ca2+ and cAMP agonists (namely CCh and VIP) can have synergistic effects on secretion in intact submucosal glands (24) and isolated serous cells (19), which we hypothesize occurs because of the common need for Ca2+-activated K+ channels to support robust Cl− secretion (Figs. 7E and 8A). As we described previously (19), a low level of VIP (100 nM) can activate CFTR but not Ca2+ signals, and thus cannot evoke robust secretion as visualized by shrinkage (Fig. 9A). Higher levels of VIP (10 µM) can engage Ca2+ and cause secretion, evidenced by shrinkage (Fig. 9A). Conversely, low levels of Ca2+ agonists like cholinergic agonist CCh (100 nM) can activate low-level Ca2+ responses that activate K+ channels but do not stimulate CaCC and thus do not support secretion (19). We found that a <EC50 level of 2FLI (500 nM; here termed “weak” stimulation of PAR-2) elicited lower Ca2+ signals than 5–10 µM 2FLI (>EC50 for Ca2+; here termed “strong” stimulation of PAR-2). This weak stimulation of PAR-2 was not effective at activating cell shrinkage (Fig. 9B). This was likely due to a failure to engage CaCC/TMEM16A, as weak stimulation of PAR-2 (500 nM 2FLI) in combination with TMEM16A activator Eact (101) allowed shrinkage to occur (Fig. 9C), suggesting Cl− conductance is rate limiting during 500 nM 2FLI stimulation. Note that while a prior study showed Eact is a direct elevator of [Ca2+]i via TRP channels in some airway cells (111), we did not observe elevation of Ca2+ by Eact in serous cells in either this current study or a prior one (12).
Figure 9.
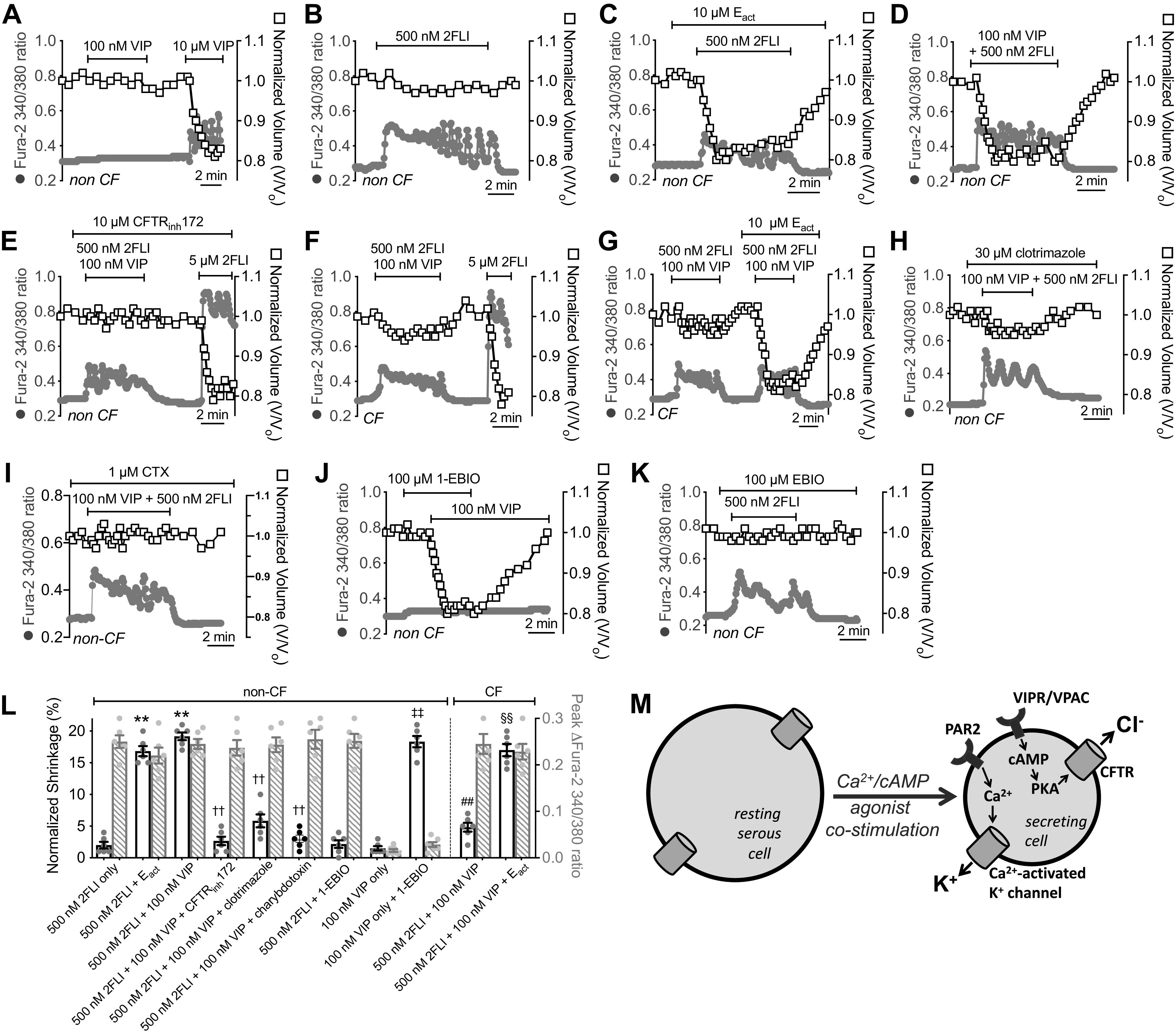
Synergistic cystic fibrosis transmembrane conductance regulator (CFTR)-dependent fluid secretion is observed with low-level protease-activated receptor 2 (PAR-2) and vasoactive intestinal peptide receptor (VIPR) activation. Representative traces of Ca2+ (gray circles) and volume (open squares) during stimulation with 100 nM or 10 µM vasoactive intestinal peptide (VIP) (A) or 500 nM 2-Furoyl-LIGRLO-NH2 (2FLI) (B). C: representative traces of Ca2+ and volume during stimulation with 500 nM 2FLI + Eact. Representative traces of Ca2+ and volume during stimulation with 500 nM 2FLI + 100 nM VIP in non-cystic fibrosis (CF) cells (D), Wt cells + CFTRinh172 (E), ΔF508/ΔF508 CF cells (F), or CF cells + Eact (G). Representative traces of Ca2+ and volume during stimulation 500 nM 2FLI + 100 nM VIP in non-CF cells + clotrimazole (H) or charybdotoxin (CTX; I). Representative traces of Ca2+ and volume during stimulation with 100 nM VIP + 1-EBIO (J) or 500 nM 2FLI + 1-EBIO (K). L: bar graph showing peak shrinkage (black open bars) in non-CF cells, which was (in %) 2 ± 0.5 (500 nM 2FLI only), 17 ± 0.8 (500 nM 2FLI + Eact; **P < 0.01 vs. 500 nM 2FLI only), 2 ± 0.6 (500 nM 2FLI + 1-EBIO), 19 ± 0.6 (500 nM 2FLI + 100 nM VIP; **P < 0.01 vs. 500 nM 2FLI only), 3 ± 0.7 (500 nM 2FLI + 100 nM VIP + CFTRinh172; ††P < 0.01 vs. 500 nM 2FLI + 100 nM VIP only), 6 ± 1 (500 nM 2FLI + 100 nM VIP + clotrimazole; ††P < 0.01 vs. 500 nM 2FLI + 100 nM VIP only), 3 ± 1; 500 nM 2FLI + 100 nM VIP + charybdotoxin; ††P < 0.01 vs. 500 nM 2FLI + 100 nM VIP only), 1.5 ± 0.3 (100 nM VIP only), 18 ± 0.9 (100 nM VIP + 1-EBIO; ‡‡P < 0.01 vs. 100 nM VIP only). Peak shrinkage in CF cells was (in %) 5 ± 0.7 (500 nM 2FLI + 100 nM VIP; ##P < 0.01 vs. 500 nM 2FLI + 100 nM VIP in non-CF cells) and 17 ± 1 (500 nM 2FLI + 100 nM VIP + Eact; §§P < 0.01 vs. 500 nM 2FLI + 100 nM VIP only in CF cells). Peak change in fura-2 340/380 ratio (gray crossed bars) was 0.25 ± 0.01 (500 nM 2FLI) 0.22 ± 0.02 (500 nM 2FLI + Eact) 0.25 ± 0.02 (500 nM 2FLI + 1-EBIO), 0.25 ± 0.01 (500 nM 2FLI + 100 nM VIP), 0.24 ± 0.02 (500 nM 2FLI + 100 nM VIP + CFTRinh172), 0.24 ± 0.01 (500 nM 2FLI + 100 nM VIP + clotrimazole), 0.26 ± 0.02 (500 nM 2FLI + 100 nM VIP + charybdotoxin), 0.02 ± 0.002 (100 nM VIP only), and 0.03 ± 0.005 (100 nM VIP + 1-EBIO). Peak change in CF cells was 0.25 ± 0.02 (500 nM 2FLI + 100 nM VIP) and 0.23 ± 0.02 (500 nM 2FLI + 100 nM VIP + Eact). All bars show data points from six independent experiments using cells from three patients and means ± SE. M: diagram of proposed synergistic secretion activated by low-level PAR-2 and VIPR stimulation. Weak stimulation of PAR-2 (<EC50 for Ca2+) can elicit Ca2+ responses sufficient to activate K+ channels but not calcium-activated Cl− channel (CaCC). Weak VIPR stimulation can elicit cAMP elevations that activate CFTR but not K+ channels. Combined, this can elicit CFTR-dependent secretion.
We hypothesized that CFTR could also support Cl− efflux during weak PAR-2 activation if cAMP-elevating receptors are simultaneously stimulated, as observed with weak cholinergic stimulation combined with weak VIPergic stimulation (19, 35). Costimulation with concentrations of VIP (100 nM) and 2FLI (500 nM) that are by themselves subsecretory (Fig. 9, A and B) synergistically activated secretion as visualized by cell shrinkage (Fig. 9D). This low-level VIPR + PAR-2-activated secretion is blocked by CFTRinh172 (Fig. 9E) and is absent in cells from patients with ΔF508/ΔF508 CF (Fig. 9F). A higher elevation of Ca2+ with stronger PAR-2 stimulation (5 µM 2FLI) can overcome this to activate CFTR-independent secretion, likely by engaging CaCC/TMEM16A (Fig. 9, E and F). Activating TMEM16A with small molecule Eact also allowed synergistic secretion to occur with 500 nM 2FLI + 100 nM VIP in ΔF508/ΔF508 CF cells (Fig. 9G).
In non-CF cells, the PAR-2 + VIPR synergistic secretion was blocked by clotrimazole and charybdotoxin (CTX) (Fig. 9, H and I), inhibitors of Ca2+-activated K+ channels in airway epithelial Calu-3 cells (112) and inhibitors of synergistic CCh + VIP secretion from intact submucosal glands (24). Treatment of serous cells with K+ channel activator 1-EBIO allowed cell shrinkage to occur with 100 nM VIP stimulation alone (Fig. 9J), supporting that K+ conductance is rate limiting with low-level VIPR stimulation. However, 1-EBIO did not affect cells stimulated with 500 nM 2FLI (Fig. 9K), supporting that Cl− conductance instead is rate limiting with low level PAR-2 stimulation. Peak shrinkage and Ca2+ responses are summarized in Fig. 9L. Together, our data support a model where low-level combined stimulation of PAR-2 and cAMP-elevating receptors like VIPRs synergizes to allow Cl− secretion through cAMP-activated CFTR, while Ca2+ downstream of PAR-2 activation allows counterion K+ flux (Fig. 9M).
It has been suggested that submucosal gland acini are tonically exposed to low levels of multiple agonists in the serosal fluid. Thus, synergistic responses that occur with lower-level costimulation of different types of receptors are likely more physiologically important than responses observed with large concentrations of agonists (24, 113). Proteases secreted by mast cells or other immune cells may be able to activate secretion at lower levels within the serosal milieu of other signaling molecules like VIP or prostaglandins that elevate cAMP. To test if this synergy can occur with low levels of proteases, we stimulated serous cells with trypsin and lung tryptase in combination with VIP. Trypsin and tryptase activated secretion at 1 µM but not 10 nM in both non-CF and CF (ΔF508/ΔF508) cells (Fig. 10, A and B). However, 10 nM trypsin or tryptase did activate secretion in combination with 100 nM VIP in non-CF but not in CF cells (Fig. 10, C–E). Heat-inactivated trypsin or tryptase at 10 nM in combination with 100 nM VIP did not activate secretion (Fig. 10, F and G), demonstrating a requirement for the enzyme activity. As with 2FLI, low-level 10 nM tryptase stimulation also activated secretion in combination with Eact (Fig. 10H), suggesting Cl− conductance is rate-limiting during 10 nM trypsin stimulation. These data are summarized in Fig. 10I, suggesting that low-level protease and VIP stimulation activates CFTR-dependent secretion.
Figure 10.
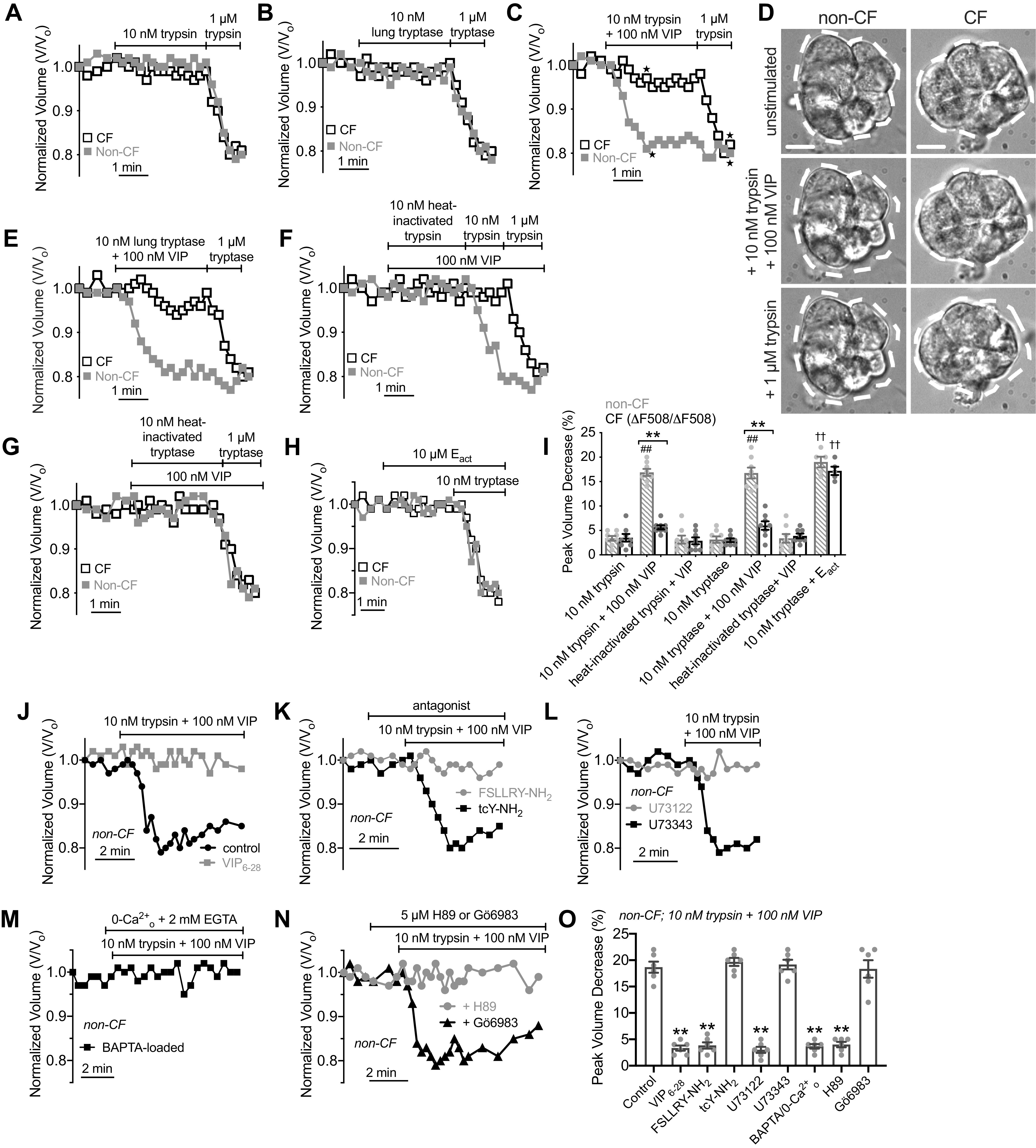
Synergistic, cystic fibrosis transmembrane conductance regulator (CFTR)-dependent fluid secretion is also observed with low-level protease and vasoactive intestinal peptide (VIP) stimulation requiring both cAMP/protein kinase A (PKA) and Ca2+. Representative cell volume traces of ΔF508/ΔF508 cystic fibrosis (CF, black open squares) and non-CF (gray solid squares) cells showing during stimulation with 10 nM or 1 µM trypsin (A) or tryptase (B). Representative cell volume traces (C) and differential interference contrast (DIC) images (D) during stimulation with 10 nM trypsin + VIP. Dotted lines represent outline of cell at time 0 (unstimulated conditions). Scale bar is 10 µm. First image in D shows time 0 on the trace; points from which subsequent images were taken are denoted with stars on graph in C. E: representative cell volume traces during stimulation with 10 nM tryptase + 100 nM VIP. To prevent degradation of VIP, trypsin and VIP were mixed from 2× solutions immediately before addition to the cells. Representative cell volume traces during stimulation with 100 nM VIP + 10 nM trypsin (E), tryptase (F), heat-inactivated trypsin (E), or heat-inactivated tryptase (F). H: representative volume traces with 10 nM trypsin + Eact. I: bar graph showing peak volume decreases (in %) in experiments as in A–G. Crossed gray bars are non-CF patients and black open bars are patients with CF. Data points are independent experiments with means ± SE, which were 3 ± 0.5 (trypsin, non-CF), 4 ± 1 (trypsin, CF), 17 ± 1 (trypsin + VIP, non-CF; ##P < 0.01 vs. trypsin alone), 4 ± 0.5 (trypsin + VIP, CF; **P < 0.01 vs. non-CF), 3.1 ± 1 (heat-inactivated trypsin + VIP, non-CF), 3 ± 1 (heat-inactivated trypsin + VIP, CF), 3 ± 1 (tryptase, non-CF), 3 ± 0.5 (tryptase, CF), 17 ± 1 (tryptase + VIP, non-CF; ##P < 0.01 vs. tryptase alone), 5 ± 1 (tryptase + VIP, CF; **P < 0.01 vs. non-CF), 3 ± 1 (heat-inactivated tryptase + VIP, non-CF), 4 ± 0.5 (heat-inactivated tryptase + VIP, CF), 19 ± 1 (tryptase + Eact, non-CF; ††P < 0.01 vs. tryptase only), 17 ± 1 (tryptase + Eact, CF; ††P < 0.01 vs. tryptase only). Significances by one-way ANOVA with Bonferroni post-test; n = 5–8 independent experiments per condition using separate samples of cells obtained from ≥2 different non-CF and ≥2 different ΔF508 CFTR homozygous CF patients. J and K: representative volume traces in non-CF cells during stimulation with 10 nM trypsin + 100 nM VIP ± VIP6–28 (10 µM), FSLLRY-NH2 (50 µM), or tcY-NH2 (50 µM). L: representative volume traces in non-CF cells during stimulation with 10 nM trypsin + 100 nM VIP ± 30 min pretreatment with 10 µM U73122 or U73343. M: representative volume trace in non-CF cells during stimulation with 10 nM trypsin + 100 nM VIP in BAPTA-loaded cells stimulated in 0-Ca2+o (2 mM EGTA) extracellular solution. N: representative volume traces showing inhibition of shrinkage responses in non-CF cells during stimulation with 10 nM trypsin + 100 nM VIP ± 10 µM H89 or Gö6983. O: bar graph showing peak volume decreases (in %; means ± SE) from experiments as in I–M, which were 19 ± 1 (control), 3 ± 1 (VIP6–28), 4 ± 1 (FSLLRY-NH2), 20 ± 1 (tcY-NH2), 3 ± 1 (U73122), 19 ± 1 (U73343), 4 ± 1 (BAPTA/0-Ca2+), 4 ± 1 (H89), 18 ± 2 (Gö6983). Significance determined by one-way ANOVA with Dunnett’s post-test with **P < 0.01 vs. control. Data points shown are independent experiments (n = 6 using independent samples of cells from ≥2 different non-CF patients).
In non-CF cells, the synergistic 10 nM trypsin + 100 nM VIP-activated secretion was blocked by VIP receptor antagonist VIP6–28 (Fig. 10J) or PAR-2 antagonist FSLLRY-NH2 (Fig. 10K) but not PAR-4 antagonist tcY-NH2 (Fig. 10K). The synergistic 10 nM trypsin + 100 nM VIP-activated secretion was also inhibited by PLC inhibitor U73122 but not inactive U73343 (Fig. 10L) and was blocked by Ca2+ chelation (Fig. 10M). Synergistic secretion was also blocked by PKA inhibitor H89 (Fig. 10N) but not PKC inhibitor Gö6983 (Fig. 10N). These data are summarized in Fig. 10O, supporting a requirement for both Ca2+ and cAMP downstream of PAR-2 and VIP receptors, respectively, in these synergistic secretory responses.
Cultures of Primary Serous Acinar Cells Secrete Fluid Synergistically in Response to Low Level PAR-2 and VIP Stimulation
An alternative explanation for the data aforementioned is that secretion does occur during low-level stimulation of PAR-2 or VIPR alone, but that solute influx and efflux pathways are balanced such that overt volume changes do not occur. However, 100 nM VIP or 100 nM CCh alone, which do not elicit cell shrinkage [here and in Ref. (19)] are also subsecretory concentrations in intact human airway glands (24). It was also demonstrated that acinar cell volume changes do occur in intact glands during activation of secretion, visualized by both cell shrinkage and gland lumen expansion (114). Thus, the low 100 nM VIP concentration is truly subsecretory, and the low 500 nM concentration of 2FLI used here that does not elicit cell shrinkage is also likely truly subsecretory.
However, to further test that these cell volume changes reflect secretion that would occur in the polarized environment of a gland acinus, we cultured primary serious acinar cells as previously described (12, 71, 72) at air-liquid interface (ALI) on transwell filters. These cells exhibited lateral membrane immunofluorescence with a PAR-2-directed antibody (Fig. 11A) and a band corresponding to the molecular weight of glycosylated PAR-2 protein was detected by Western blotting (Fig. 11B), fitting with functional data. PAR-2 siRNA knockdown experiments supported the specificity of IF and Western blot detection of PAR-2 by these antibodies (Supplemental Fig. S1). VIPR1 and VIPR2 expression was previously demonstrated in these cultured serous cells (12). We measured fluid secretion by tracking the height of airway surface liquid (ASL) on the apical side of these cultures as previously described (12) using Texas red-labeled dextran added in perfluorocarbon suspension (115). Basolateral stimulation (15 min) of these cultures with 5 µM but not 500 nM 2FLI increased ASL height in both non-CF and CF (ΔF508/(ΔF508) cultures (Fig. 11C). Likewise, stimulation with 1 µM but not 100 nM VIP caused increased ASL height, but only in non-CF cultures (Fig. 11C). Combined 500 nM 2FLI + 100 nM VIP synergistically increased ASL height in non-CF but not CF cultures (Fig. 11C). The increases in ASL height were significantly reduced by NKCC1 inhibitor bumetanide (Fig. 11C), supporting that the increased ASL height reflected fluid secretion. These data are summarized in Fig. 11D, and support conclusions aforementioned based on the data from dissociated primary cells.
Figure 11.

Polarized fluid secretion in air-liquid interface (ALI) cultures of primary nasal serous cells supports synergistic cystic fibrosis transmembrane conductance regulator (CFTR)-dependent secretion elicited by low-level protease-activated receptor 2 (PAR-2) and vasoactive intestinal peptide (VIP)ergic stimulation. A: confocal micrograph of methanol-fixed primary serous ALI showing immunofluorescence of Na+/K+ ATPase (magenta; Abcam ab76020 rabbit monoclonal antibody) and PAR-2 (cyan; SAM11 mouse monoclonal antibody). Both antibodies exhibited lateral membrane staining. Results representative of independent cultures from three patients. B: Western blot for PAR-2 (Abcam ab180953 rabbit monoclonal antibody) from ALIs from two separate patients. Results representative of four ALIs from four patients. C: representative orthogonal sections from confocal stacks [×60 1.4 numerical aperture (NA) oil objective, 0.3 µm step size] showing Texas red dextran-labeled airway surface liquid (ASL) in individual cultures from non-cystic fibrosis (CF, left) and ΔF508/ΔF508 CF cultures (right) after stimulation as indicated. Note z-axis scale bar is not calibrated for refractive index mismatch, as discussed in methods. D: bar graph of ASL heights from independent experiments as in C (n = 5 per condition using ALIs from five patients). ASL heights in non-CF cultures were (in µm) 10 ± 1 (unstimulated), 12 ± 2 [500 nM 2-Furoyl-LIGRLO-NH2 (2FLI)], 36 ± 5 (5 µM 2FLI; *P < 0.05 vs. unstimulated), 11 ± 1 (5 µM 2FLI + 50 µM GB83; †P < 0.05 vs. 5 µM 2FLI alone), 11 ± 1 (100 nM VIP), 34 ± 2 (1 µM VIP; *P < 0.05 vs. unstimulated), 41 ± 4 (500 nM 2FLI + 100 nM VIP; *P < 0.05 vs unstimulated; #P < 0.05 vs. either 500 nM 2FLI or 100 nM VIP alone), 15 ± 2 (500 nM 2FLI + 100 nM VIP + 100 µM bumetanide; ‡P < 0.05 vs. 500 nM 2FLI + 100 nM VIP without bumetanide). ASL heights in CF cultures were 9 ± 1 (unstimulated), 11 ± 1 (500 nM 2FLI), 31 ± 3 (5 µM 2FLI; *P < 0.05 vs. unstimulated), 13 ± 2 (5 µM 2FLI + 50 µM GB83; †P < 0.05 vs. 5 µM 2FLI alone), 12 ± 1 (100 nM VIP), 11 ± 2 (1 µM VIP), 10 ± 1 (500 nM 2FLI + 100 nM VIP). Significance by one-way ANOVA with Bonferroni post-test with paired comparisons. E: expression of TMEM16A (Ca2+-activated Cl− channel), BEST1 (Ca2+-activated Cl− channel), and UBC1 (housekeeping control) relative to GAPDH ± scrambled, TMEM16A, or BEST1 siRNAs (n = 4 ALIs, two each from two individual patients). Expression of TMEM16A was 0.10 ± 0.02 (scrambled siRNA), 0.025 ± 0.01 (TMEM16A siRNA; *P < 0.05 vs. scrambled), and 0.10 ± 0.02 (BEST1 siRNA). Expression of BEST1 was 0.03 ± 0.007 (scramble siRNA), 0.04 ± 0.006 (TMEM16A siRNA), 0.007 ± 0.003 (BEST1 siRNA; *P < 0.05 vs. scrambled). Expression of UBC1 was 0.60 ± 0.07 (scramble siRNA), 0.56 ± 0.05 (TMEM16A siRNA), and 0.61 ± 0.08 (BEST1 siRNA). Significance by one-way ANOVA with Bonferroni post-test. F: representative orthogonal sections of unstimulated (top) or 5 µM 2FLI stimulated ALIs treated with scrambled, TMEM16A, or BEST1 siRNAs. G: bar graph showing mean ASL heights from experiments as in F (n = 6 ALIs, three each from two individual patients). Mean ASL height was 10 ± 1 (unstimulated + scrambled siRNA), 32 ± 2 (5 µM 2FLI + scrambled siRNA; **P < 0.01 vs. unstimulated + scrambled siRNA), 11 ± 1 (unstimulated + TMEM16A siRNA), 16 ± 5 (5 µM 2FLI + TMEM16A siRNA; n.s. vs. unstimulated + TMEM16A siRNA; #P < 0.05 vs. 5 µM 2FLI + either scrambled or BEST1siRNA), 11 ± 1 (unstimulated + BEST1 siRNA), and 38 ± 2 (5 µM 2FLI + BEST1 siRNA; **P < 0.01 vs. unstimulated + BEST1 siRNA). Significance by one-way ANOVA with Bonferroni posttest. Data points in all bar graphs are independent experiments and error bars are SE.
We utilized this polarized transwell model to further test the role of TMEM16A in the CFTR-independent component of Ca2+-activated secretion. Cells were transfected with pooled siRNAs against either TMEM16A or bestrophin 1 (BEST1), another Ca2+-activated Cl− channel expressed in the airways (116, 117). Because confluent monolayers were resistant to siRNA transfection, submerged primary cells were lifted and seeded onto transwells at high density in the presence of siRNAs, to allow siRNAs to enter the cells before they became confluent, as described in methods. ASL measurements revealed a >50% decrease in stimulated ASL height with >50% knockdown of TMEM16A but not bestrophin 1 (Fig. 11, E–G), supporting that TMEM16A is a major component of the secretory CaCC in primary serous cells and agreeing with our own previous work suggesting strong expression of TMEM16A in porcine bronchial serous cells (20). Note that knockdown was measured at the RNA level by quantitative (q)PCR. We did not measure the level of protein knockdown in these experiments. We measured apical anion permeability using the Cl− sensitive dye 6-methoxy-N-(3-sulfopropyl)quinolinium [SPQ, as previously described (58)] in serous ALIs and found that it was reduced >60% with TMEM16A siRNA but not with BEST1 siRNA (Supplemental Fig. S2), supporting that the reduced fluid secretion with TMEM16A siRNA is indeed due to a reduced apical anion efflux.
Activation of PAR-2 Secretion by Proteases Is Inhibited by P. aeruginosa Secreted Products
Although it is generally accepted that PAR-2 is activated by house dust mite (46, 48, 49, 118), cockroach (119–121), and fungal proteases (12, 122–124), the effects of host and other pathogen proteases on PAR-2 activation is still unclear. Neutrophil elastase, P. aeruginosa elastase, and P. aeruginosa thermolysin have been reported to inhibit some PAR-2 cell lines (56, 125–127) and activate PAR-2 in others (128–134). “Disarming” of PAR-2 by P. aeruginosa elastase has been suggested to occur by cleaving downstream of the canonical trypsin activation site, removing the tethered ligand sequence (125, 126, 135). Differences in post-translational modification or purified commercially available enzymes may contribute to altered sensitivities of PAR-2 to proteases in different cell lines.
One way to clarify some discrepancies about the role of P. aeruginosa elastase on PAR-2 is to study PAR-2 function in acutely isolated primary cells obtained directly from human patients and to use proteases derived directly from cultured P. aeruginosa. We started with P. aeruginosa conditioned media (CM) that was dialyzed overnight against HBSS (6–8 kDa MWCO dialysis tubing) to remove small molecules and equilibrate salts as previously described (57). This dialyzed CM would be expected to retain most proteins, including proteases like elastase or thermolysin, which are >30 kDa (89, 90). We used laboratory strains PAO-1 and PAO-JP2 and clinical isolates of P. aeruginosa from patients with CRS. When primary human nasal serous cells were preincubated for 5 min in dialyzed PAO-1 CM (with copious washout following), activation of Ca2+ and cell shrinkage, reflecting secretion, was absent during subsequent stimulation with trypsin or tryptase but not 2FLI or CCh (Fig. 12A). The intact activation of secretion by 2FLI was nonetheless still inhibited by GB88, suggesting it was still mediated by functional PAR-2 receptors (Fig. 12B). If cells were pretreated with heat-inactivated dialyzed P. aeruginosa CM (20 min, 100°C), the Ca2+ and shrinkage responses to trypsin were intact (Fig. 12C). Likewise, if the PAO-1 CM was incubated with 5 µM protease inhibitor phosphoramidon (136, 137) before pretreatment of the serous cells, responses to trypsin remained intact (Fig. 12D). Preincubation with dialyzed CM from strain PAO-JP2 did not inhibit the responses to trypsin (Fig. 12E). PAO-JP2 lacks the lasR and rhlR quorum sensing transcriptional regulators that control elastase production (84, 85, 138), and elastase activity has been reported to be nearly abolished in this double mutant strain (85).
Figure 12.

Serous cell secretion in response to protease activation of protease-activated receptor 2 (PAR-2) is blocked by pretreatment with Pseudomonas aeruginosa conditioned media (CM). A: representative volume (open squares) and Ca2+ (fura-2 340/380; gray circles) traces showing responses to trypsin, tryptase, 2-Furoyl-LIGRLO-NH2 (2FLI), or carbachol (CCh) after exposure to P. aeruginosa PAO-1 CM. B: representative Ca2+ and volume traces during stimulation with 10 µM 2FLI after PAO-1 CM exposure + GB88. Representative Ca2+ and volume during stimulation with trypsin after pretreatment with heat-inactivated PAO-1 CM (C), PAO-1 CM treated with elastase inhibitor phosphoramidon (10 µM; D), or CM from PAO-JP2 (E). F: representative Ca2+ and volume traces during stimulation with 2FLI and trypsin after treatment with recombinant P. aeruginosa (P.a.) elastase (500 nM) ± phosphoramidon. G: representative Ca2+ and volume traces during acute exposure to recombinant P.a. elastase (1 μM) with subsequent stimulation with 1 μM trypsin or tryptase (left), 5 μM 2FLI (left), or 100 μM CCh. All experiments shown are representative of 3–5 independent experiments using cells from 3 to 5 patients.
We also tested recombinant P. aeruginosa elastase produced in E. coli. In serous cells pretreated with 100 nM P. aeruginosa elastase for 5 min, subsequent stimulation with trypsin did not elicit shrinkage or Ca2+ response whereas 2FLI did (Fig. 12F, left). If P. aeruginosa elastase was pretreated with 10 µM phosphoramidon before serous cell exposure, trypsin elicited both Ca2+ and cell shrinkage (Fig. 12F, right). Acute exposure to 1 µM P. aeruginosa elastase did not affect Ca2+ or cell shrinkage alone (Fig. 12G) but after 2 min exposure, there was no response to subsequent 1 µM trypsin or tryptase, whereas 2FLI (Fig. 12G, left) or CCh (Fig. 12G, right) activated both Ca2+ elevation and cell shrinkage. These data suggest that P. aeruginosa secreted products within the CM can inhibit serous cell secretion by inhibiting protease activation of PAR-2. However, the receptor is not completely destroyed, as it can still be activated by peptide agonist 2FLI. This supports the previously proposed “disarming” model discussed earlier (125, 126, 135).
We sought to confirm these observations using another human cell model. We used Calu-3 cells, an adenocarcinoma line often used as a serous cell surrogate due to high CFTR expression as well as expression of other serous cell markers like lactoferrin and lysozyme (6, 139–141). We observed that Calu-3 cells grown in submerged monolayers exhibited punctate lateral membrane staining for PAR-2 (Fig. 13A). We also observed an appropriately sized band for PAR-2 via Western blot in both submerged and ALI Calu-3 cultures (Fig. 13B). We previously showed expression of PAR-2 but not PAR-4 in Calu-3 cells by rtPCR (58). Calu-3 cells grown at ALI and loaded with Ca2+-indicator Fluo-4 exhibited basolateral, but not apical, responses to trypsin (Fig. 13, C and D). No response to apical or basolateral thrombin was observed (Fig. 13D). Similarly, we observed Ca2+ responses to basolateral 2FLI but not apical 2FLI or AY-NH2 (Fig. 13E).
Figure 13.
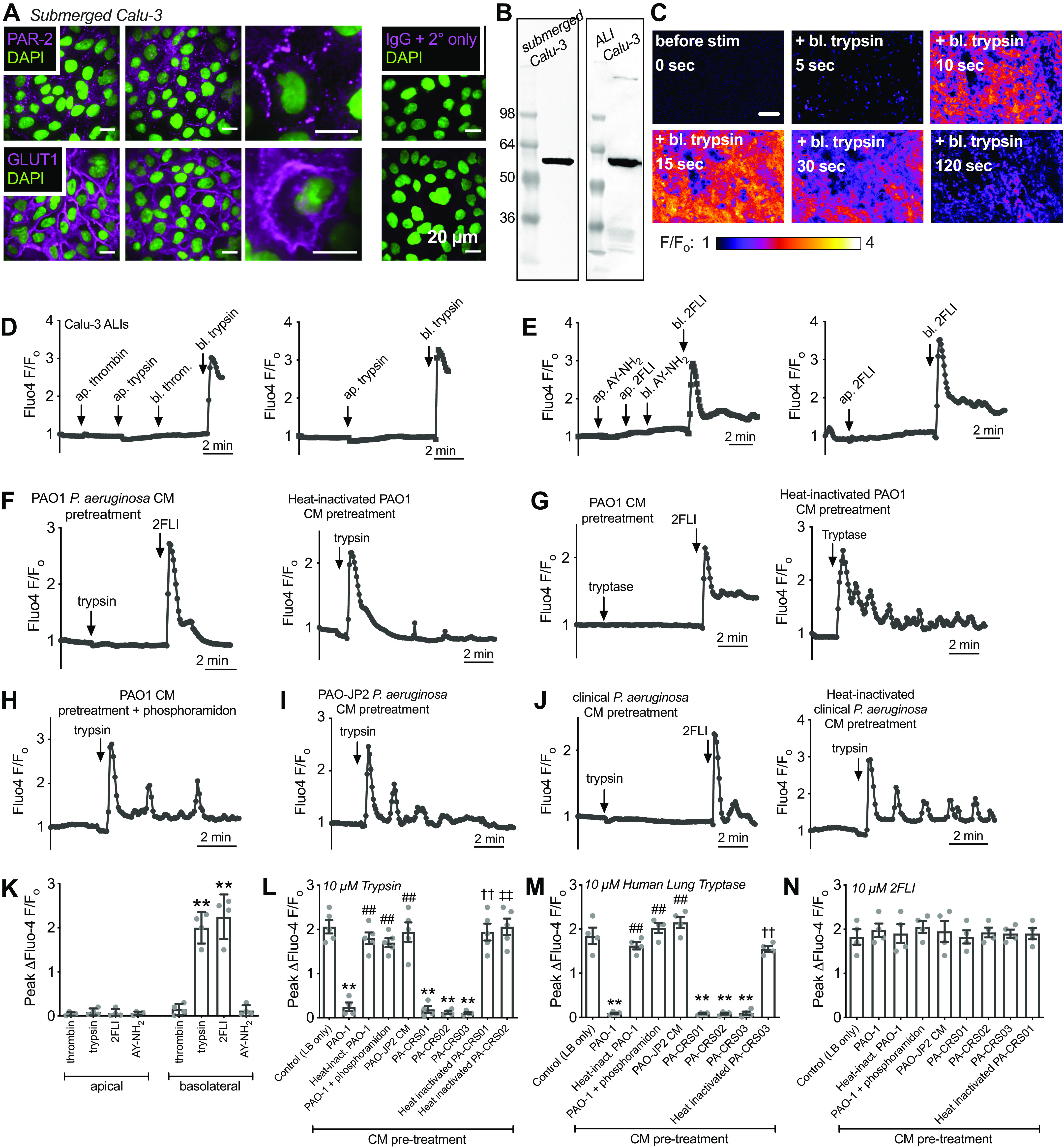
Ca2+ responses to protease protease-activated receptor 2 (PAR-2) activation is inhibited by Pseudomonas aeruginosa secreted products in Calu-3 air-liquid interfaces (ALIs). A: representative immunofluorescence image showing punctate lateral membrane staining of PAR-2 (SAM11) in methanol-fixed Calu-3s, with GLUT1 staining (Abcam SPM498/ab40084 mouse monoclonal) for comparison. B: Western (Abcam ab180953 rabbit monoclonal antibody) showing expression of PAR-2 in submerged (left) and ALI (right) Calu-3s. C: Fluo-4 intensity measurements of Calu-3 air-liquid interface cultures were made using the entire field of view as the region of interest, which thus averaged intensity over hundreds of cells grown on the transwell. Pseudo-colored F/Fo images are shown are shown for one representative experiment showing response to 10 µM basolateral trypsin application. Images were created using the average of the first five images of the series as baseline using “F div Fo” plugin from the ImageJ Cookbook plugin set (https://imagej.net/Cookbook). Background was estimated based on imaging a non-Fluo-4-loaded ALI at identical settings and subtracted before calculation of F/Fo. D: representative Ca2+ (Fluo-4 F/Fo) responses to basolateral trypsin (10 µM) but not apical thrombin (10 µM), apical trypsin, or basolateral thrombin. E: Fluo-4 traces showing Ca2+ response to basolateral 2-Furoyl-LIGRLO-NH2 (2FLI) (10 µM) but not apical 2FLI, apical AY-NH2 (10 µM), or basolateral AY-NH2. Representative Fluo-4 traces showing loss of responses to 10 µM trypsin (F) or 10 µM tryptase (G) after 5 min pretreatment with PAO-1 conditioned media (CM) but not heat-inactivated PAO-1 CM. 2FLI response was intact after PAO-1 CM pretreatment. Representative Fluo-4 traces showing intact trypsin response after pretreatment with PAO-1 CM + phosphoramidon (10 µM; H) or PAO-JP2 CM (I). J: Fluo-4 traces showing loss of 10 µM trypsin Ca2+ response with clinical P. aeruginosa (PAO-CRS01) CM pretreatment (left) intact trypsin response after heat-inactivated PAO-CRS01 pretreatment (right). K: bar graph of apical vs. basolateral responses from experiments as in E and F. Peak ΔFluo-4 F/Fo was 0.05 ± 0.02 (apical thrombin), 0.09 ± 0.04 (apical trypsin) 0.07 ± 0.04 (apical 2FLI), 0.06 ± 0.02 (apical AY-NH2), 0.15 ± 0.06 (basolateral thrombin), 2.0 ± 0.18 (basolateral trypsin; **P < 0.01 vs. apical trypsin), 2.3 ± 0.25 (basolateral 2FLI; **P < 0.01 vs. apical 2FLI), 0.12 ± 0.06 (basolateral AY-NH2); n = 4 independent experiments each. Significance by one-way ANOVA with Bonferroni post-test with paired comparisons. L: bar graph of peak Ca2+ responses with 10 µM trypsin from experiments as shown in G–K. Peak ΔFluo-4 F/Fo with 10 µM trypsin was 2.1 ± 0.15 (control LB only pretreatment), 0.25 ± 0.1 (PAO-1 CM; **P < 0.01 vs. LB only), 1.81 ± 0.13 (heat-inactivated PAO-1 CM; ##P < 0.01 vs. PAO-1 CM), 1.70 ± 0.1 (PAO-1 CM + phosphoramidon; ##P < 0.01 vs. PAO-1 CM), 1.94 ± 0.22 (PAO-JP2 CM; ##P < 0.01 vs. PAO-1 CM), 0.19 ± 0.08 (PA-CRS01 CM; **P < 0.01 vs. LB only), 0.12 ± 003 (PA-CRS02 CM; **P < 0.01 vs. LB only), 0.11 ± 0.02 (PA-CRS03 CM; **P < 0.01 vs. LB only), 1.94 ± 0.19 (heat-inactivated PA-CRS01 CM; ††P < 0.01 vs. PA-CRS01 CM), 2.06 ± 0.19 (heat-inactivated PA-CRS02 CM; ‡‡P < 0.01 vs. PA-CRS02 CM). Significance determined by one-way ANOVA with Bonferroni post-test; n = 4–5 independent experiments. M: bar graph of peak Ca2+ responses with 10 µM lung tryptase from experiments as shown in G–K. Peak ΔFluo-4 F/Fo with 10 µM tryptase was 1.85 ± 0.2 (control LB only pretreatment), 0.09 ± 0.02 (PAO-1 CM; **P < 0.01 vs. LB only control), 1.63 ± 0.09 (heat-inactivated PAO-1 CM; ##P < 0.01 vs. PAO-1 CM), 2.0 ± 0.11 (PAO-1 CM + phosphoramidon; ##P < 0.01 vs. PAO-1 CM), 2.2 ± 0.13 (PAO-JP2 CM; ##P < 0.01 vs. PAO-1 CM), 0.083 ± 0.01 (PA-CRS01 CM; **P < 0.01 vs. LB only control), 0.080 ± 0.02 (PA-CRS02 CM; **P < 0.01 vs. LB only control), 0.09 ± 0.04 (PA-CRS03 CM; **P < 0.01 vs. LB only control), 1.55 ± 0.06 (Heat-inactivated PA-CRS03 CM; ††P < 0.01 vs. PA-CRS03 CM). Significance determined by one-way ANOVA with Bonferroni post-test; n = 4–5 independent experiments. N: bar graph of peak Ca2+ responses with 10 µM 2FLI from experiments as shown in G–K. Peak ΔFluo-4 F/Fo with 10 µM 2FLI were 1.83 ± 0.18 (control LB only pretreatment), 1.98 ± 0.15 (PAO-1 CM), 1.90 ± 0.21 (heat-inactivated PAO-1), 2.05 ± 0.13 (PAO-1 CM + phosphoramidon), 1.95 ± 0.24 (PAO-JP2 CM), 1.83 ± 0.15 PA-CRS01, 1.93 ± 0.11 PA-CRS02, 1.91 ± 0.09 (PA-CRS03), 1.90 ± 0.13 (heat-inactivated PA-CRS01). No significant differences by one-way ANOVA; n = 4 experiments per condition. For all bar graphs, data points are independent experiments and error bars are SE. Note that pretreatment of cultures with dialyzed LB media alone had no effect on the magnitude of Ca2+ responses.
In Calu-3 ALIs, we observed that pretreatment with PAO-1 CM eliminated the response to basolateral trypsin or human lung tryptase, whereas the response to trypsin or tryptase was intact with heat-inactivated PAO-1 CM pretreatment (Fig. 13, F and G). The response to 2FLI was intact with PAO-1 CM pretreatment (Fig. 13F). The response to trypsin was also intact when PAO-1 CM was treated with phosphoramidon (Fig. 13H). PAO-JP2 CM also did not inhibit the response to trypsin (Fig. 13I). However, like PAO-1, pretreatment with CM from a clinical isolate of P. aeruginosa from a patient with CRS did inhibit the response to trypsin but not 2FLI, and this effect was eliminated by heat-inactivation of the CM (Fig. 13J). Peak Fluo-4 responses are summarized in Fig. 13, K–N and support that exposure to P. aeruginosa secreted products, likely a protease or multiple proteases, can disarm PAR-2 activation by host proteases but not completely destroy the ability of PAR-2 to signal, as it was still activated by 2FLI.
We wanted to examine PAR-2 activation even more directly to test this hypothesis that P. aeruginosa secreted products have direct effects on PAR-2. To do this, we used a fluorescent arrestin-binding-based assay based on a tripartite GFP (termed the “Trio assay” (96), shown in Fig. 14A), which we previously used to examine activation of PAR-2 by Aspergillus fumitagus-CM (57). In short, a GPCR, in this case PAR-2, is tagged with the 11th β strand of GFP and coexpressed with β-arrestin tagged with the 10th β strand of GFP and soluble GFP β strands 1–9. After receptor activation, phosphorylation of the receptor C-terminus by G-receptor kinases (GRKs) causes arrestin recruitment and formation of a complete GFP molecule. Activation of the GPCR, based on arrestin binding, is tracked by the increase in fluorescence. This assay is thus specific for the heterologously expressed GPCR and relatively independent of its downstream G-protein signaling partners.
Figure 14.

Visualization of protease protease-activated receptor 2 (PAR-2) activation by fluorogenic arrestin binding assay and inhibition by Pseudomonas aeruginosa secreted products. A: we used a fluorescence-based Trio assay to directly visualize PAR-2 activation by interaction with β-arrestin (96), which associates with activated and phosphorylated G protein-coupled receptors (GPCRs) (209). The assay is based on a tripartite green fluorescent protein (GFP), with the 11th β strand of GFP fused to the C-terminus of PAR-2, the 10th β strand of GFP fused to β-arrestin, and β strands 1–9 of GFP expressed as a soluble protein. Association of these components after GPCR activation results in the formation of a complete GFP molecule and increase in fluorescence. B: representative images of cells transfected with the PAR-2 Trio assay and simulated with 2-Furoyl-LIGRLO-NH2 (2FLI) (1 µM) at 0 and 90 min in the absence (left) or presence (right) of GB88 (10 µM). Note that mCherry is included on one of the PAR-2 trio plasmids as a transfection control. C: representative images showing PAR-2 activation at 90 min by agonist peptide SLIGR-NH2 (50 µM; left) but not scrambled LRGILS-NH2 (right). D: bar graph showing results from experiments as in B and C. Normalized increase in GFP fluorescence was 22.5 ± 3 (2FLI; B), 1.8 ± 0.3 (2FLI + GB88; **P < 0.01 vs. 2FLI alone; B), 18 ± 1. (SLIGRL-NH2; C), and 1.6 ± 0.4 (LRGILS-NH2; **P <0.01 vs. SLIGRL-NH2; C); n = 5 independent experiments per condition; each experiment imaged ∼30 cells. E: representative images of cells infected with β2 adrenergic receptor (β2AR) Trio assay and stimulated with β2AR agonist isoproterenol before and after PAO-1 conditioned media (CM) pretreatment as well as with 2FLI, trypsin, or SLIGRL-NH2. F: bar graph showing results from experiments as in E. Normalized increase in GFP fluorescence was 14 ± 2 (isoproterenol), 14 ± 1 (isoproterenol after PAO-1 CM pretreatment), 1.3 ± 0.2 (2FLI; **P < 0.01 vs isoproterenol), 1.7 ± 0.2 (trypsin), 1.8 ± 0.3 (SLIGRL-NH2). G: representative images of cells transfected with the PAR-2 Trio assay and simulated with trypsin after 5 min pretreatment with LB, PAO-1 CM, heat-inactivated PAO-1 CM, or PAO-JP2 CM. H: representative images of cells transfected with the PAR-2 Trio assay and simulated with trypsin after 5 min pretreatment with clinical P. aeruginosa (PA-CRS01 or PA-CRS02) CM or heat-inactivated CM. I: normalized increase in GFP fluorescence with 100 nM trypsin was 21 ± 1 (control; no pretreatment), 2 ± 1 (PAO-1 CM pretreatment; **P < 0.01 vs. control), 19 ± 2.1 (heat-inactivated PAO-1 CM pretreatment), 19 ± 1.2 (PAO-JP2 CM pretreatment), 2 ± 1 (PAO-CRS01 CM pretreatment; **P < 0.01 vs. control), 3 ± 1 (PAO-CRS02 CM pretreatment; **P < 0.01 vs. control), 17 ± 1 (heat-inactivated PAO-CRS01 CM pretreatment), 16 ± 1 (heat-inactivated PAO-CRS02 CM pretreatment). For all bar graphs, data points are independent experiments and error bars are SE.
The PAR-2 Trio assay components were expressed in A549 cells due to transfection efficiency and ability to grow the cells directly on glass. As originally described (96) and as we previously also observed (57), PAR-2 activation resulted in arrestin binding and an increase in fluorescence at 60–90 min after agonist stimulation at room temperature (Fig. 14B). The fluorescence that appeared was punctate and intracellular, suggesting that arrestin binding was associated with PAR-2 internalization, as previously described for PAR-2 (142) and other GPCRs (143). Activation of PAR-2 by 2FLI was inhibited by antagonist GB88 (Fig. 14B), confirming the role of PAR-2. PAR-2 agonist peptide SLIGRL-NH2, but not scrambled LRGILS-NH2, also increased GFP fluorescence, further confirming this reflected PAR-2 activation (Fig. 14C). These results are quantified in Fig. 14D. As a control, we expressed Trio components based on the β2AR (96) and saw an increase in GFP fluorescence with β2AR agonist isoproterenol but not 2FLI, trypsin, or SLIGRL-NH2 (Fig. 14E). There was no change in β2AR activation by isoproterenol after preincubation of cells with PAO-1 CM (Fig. 14E). These results are quantified in Fig. 14F, and support that we can measure PAR-2 activation by the PAR-2 Trio assay.
We found that a short (5 min) exposure to trypsin activated PAR-2 in cells pretreated with LB media, but not in cells pretreated with PAO-1-conditioned LB media (PAO-1 CM; Fig. 14G). Trypsin could still activate PAR-2 after exposure to heat-treated PAO-1 CM or CM from elastase-deficient strain PAO-JP2 (Fig. 14G). CM, but not heat-inactivated CM, from two clinical isolate strains of P. aeruginosa eliminated trypsin-dependent PAR-2 activation (Fig. 14H). These results are quantified in Fig. 14I. Together, the Trio assay data support the Ca2+ imaging and cell volume data aforementioned suggesting that P. aeruginosa secreted products can specifically reduce PAR-2 activation by proteases. We next considered how the ability to inactivate PAR-2 might improve the growth of P. aeruginosa in the airway.
Beyond fluid, serous cells secrete a variety of antimicrobial peptides like lysozyme, lactoferrin, defensins, and LL-37/cathelicidin (1, 2, 4). Secretion of proteins from exocrine acinar cells is typically a mixture of constitutive secretion and regulated secretion that is controlled by the same second messengers that control ion transport, namely Ca2+ and cAMP (144). PAR-2 is involved in amylase secretion by parotid acinar cells (47) and mucin secretion by bronchial cells (134, 145). We previously showed that ALI cultures of serous cells produce robust amounts of antimicrobial peptides (12). Primary serous ALIs had a higher baseline lysozyme secretion rate than Calu-3 cells (Fig. 15A), and also secreted lactoferrin in the absence of added stimulation (Fig. 15B). The lysozyme secretion level observed for Calu-3 cells was similar to previous observations from another group (6).
Figure 15.
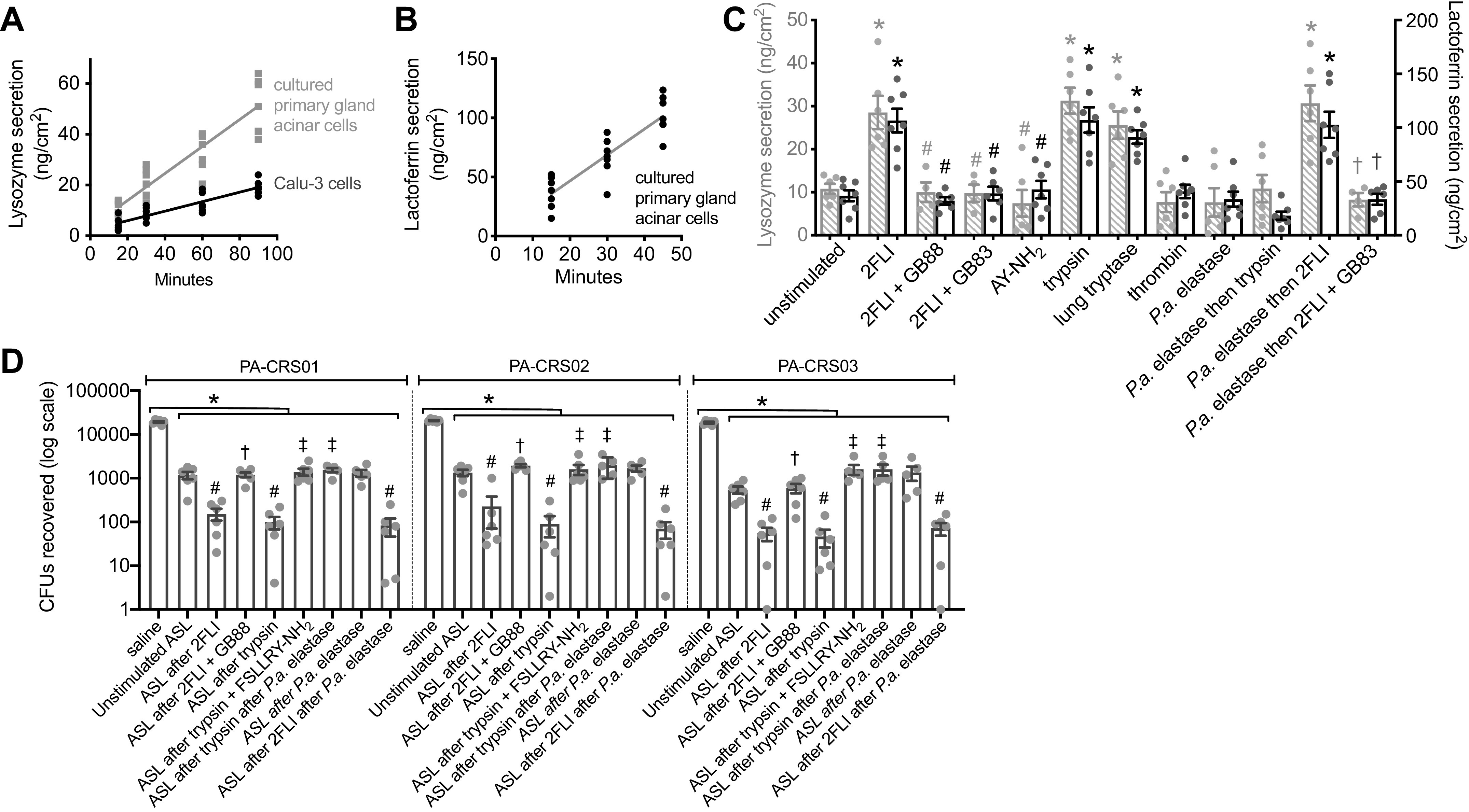
Inhibition of protease protease-activated receptor 2 (PAR-2)-activated antimicrobial peptide secretion by Pseudomonas aeruginosa (P.a.) secreted products. A: graph showing secretion of lysozyme [measured by ELISA as in Ref. (12)] in primary serous cell air-liquid interfaces (ALIs, gray squares) and Calu-3 ALIs (black circles); n = 3–5 independent experiments per time point per cell type. B: graph showing secretion of lactoferrin (measured by ELISA) in primary serous ALIs. C: bar graph from independent experiments showing apical lysozyme (gray crossed bars) and lactoferrin (black open bars) secretion after basolateral stimulation as indicated. Lysozyme secretion was 11 ± 1 (unstimulated), 29 ± 4 (10 µM 2-Furoyl-LIGRLO-NH2, 2FLI), 10 ± 2 (2FLI + 25 µM GB88), 9 ± 2 (2FLI + 25 µM GB83), 7 ± 3 (20 µM AY-NH2), 31 ± 3 (100 nM trypsin), 26 ± 3 (100 nM tryptase), 8 ± 2 (100 nM thrombin), 8 ± 3 (100 nM P.a. elastase), 11 ± 3 (P.a. elastase then trypsin), 31 ± 4 (P.a. elastase then 2FLI), 8 ± 2 (P.a. elastase then 2FLI + GB83). Lactoferrin secretion was 37 ± 5 (unstimulated), 107 ± 11 (2FLI), 32 ± 3 (2FLI + GB88), 39 ± 6 (2FLI + GB83), 43 ± 8 (AY-NH2), 107 ± 12 (trypsin), 91 ± 6 (tryptase), 41 ± 6 (thrombin), 34 ± 7 (P.a. elastase), 18 ± 4 (P.a. elastase then trypsin), 103 ± 12 (P.a. elastase then 2FLI), 33 ± 5 (P.a. elastase then 2FLI + GB83). Significances determined by one-way ANOVA with Bonferroni post-test with *P < 0.05 vs. unstimulated ASL, #P < 0.05 vs. 2FLI-stimluted ASL, †P < 0.05 vs. P.a. elastase then 2FLI; n = 6–7 independent experiments using cultures from ≥2 different patients. D: secretions from stimulated primary serous cell ALIs were collected and mixed with clinical P. aeruginosa as described in the text. Bar graph (note log scale) shows colony forming units (CFUs) remaining from independent experiments. PAO-CRS01 CFUs remaining were 19,333 ± 881 (saline only), 1,167 ± 223 (unstimulated ASL), 153 ± 46 [10 µM 2FLI-stimulated airway surface liquid (ASL)], 1,200 ± 148 (2FLI + 30 µM GB88), 99 ± 31 (100 nM trypsin), 1,392 ± 256 (trypsin + 100 µM FSLLRY-NH2), 1,680 ± 263 (100 nM P.a. elastase), 1,540 ± 175 (P.a. elastase then trypsin), 83 ± 37 (P.a. elastase then 2FLI). PAO-CRS02 CFUs remaining were 20,833 ± 601 (saline only), 1,325 ± 239 (unstimulated ASL), 227 ± 156 (2FLI-stimluated ASL), 1,933 ± 152 (2FLI + GB88), 90 ± 46 (trypsin), 1,598 ± 415 (trypsin + FSLLRY-NH2), 1,310 ± 240 (P.a. elastase), 1,990 ± 456 (P.a. elastase then trypsin), 70 ± 29 (P.a. elastase then 2FLI). PAO-CRS03 CFUs remaining were 18,667 ± 803 (saline only), 542 ± 100 (unstimulated ASL), 55 ± 19 (2FLI-stimluated ASL), 595 ± 142 (2FLI + GB88), 46 ± 20 (trypsin), 1,594 ± 412 (trypsin + FSLLRY-NH2), 1,350 ± 479 (P.a. elastase), 1,600 ± 451 (P.a. elastase then trypsin), 72 ± 23 (P.a. elastase then 2FLI). Significances determined by one-way ANOVA with Bonferroni post-test with *P < 0.05 vs. saline, #P < 0.05 vs. unstimulated ASL, †P < 0.05 vs. 2FLI stimulated ASL, and ‡P < 0.05 vs. trypsin-stimulated ASL; n = 5–6 independent experiments using cultures from ≥2 different patients. For all bar graphs, data points are independent experiments and error bars are SE.
When we stimulated primary serous ALI cultures with basolateral 2FLI for 15 min, lysozyme secretion was acutely increased approximately two- to threefold (Fig. 15C). This did not occur in the presence of PAR-2 antagonists GB88 or GB83 (Fig. 15C). Trypsin and lung tryptase also increased lysozyme and lactoferrin secretion. PAR-4 activator AY-NH2 did not enhance lysozyme secretion, nor did thrombin or P. aeruginosa elastase (Fig. 15C). However, when cultures were preincubated for 5 min with P. aeruginosa elastase and then stimulated for 15 min with trypsin, there was no enhanced secretion (Fig. 15C). However, 2FLI was still able to increase secretion after P. aeruginosa elastase, and this was blocked by GB83 (Fig. 15C), suggesting PAR-2 is still able to be activated by peptide agonists but not by proteases after exposure to P. aeruginosa elastase.
To test if these changes in antimicrobial secretion correlated with a change in the ability of serous cell secretions to kill P. aeruginosa, we used a CFU counting assay previously described (12). Diluted ASL from unstimulated ALI cultures or ALIs stimulated with 10 µM 2FLI, 100 nM trypsin, or 100 nM recombinant P. aeruginosa elastase were mixed with ∼20,000 colony forming units (CFUs) of three different clinical P. aeruginosa isolates. Unstimulated ASL caused a >1 log reduction in CFUs, whereas ASL from ALIs stimulated with 10 µM 2FLI caused a >2 log reduction in CFUs (Fig. 15D). The enhancement of bacterial killing by 2FLI was blocked by GB88 (Fig. 15D). Trypsin stimulation also increased bacterial killing by >1 log, while this was blocked by PAR-2 antagonist FSLLRY-NH2 or 5 min preexposure to P. aeruginosa elastase (Fig. 15D). P. aeruginosa elastase alone has minimal effect and also had no effect on subsequent enhancement of bacterial killing by 2FLI (Fig. 15D).
Together, the data in Figs. 12–15 suggest that P. aeruginosa proteases can disarm PAR-2 against host protease activation. This may confer a survival advantage by reducing not only fluid secretion necessary for MCC but also antimicrobial secretion. We tested if P. aeruginosa CM could also prevent activation of PAR-2 in surface epithelial cells, where basolateral PAR-2 regulates ciliary beating (57, 58). Surface epithelial cells cultured at ALI as previously described (57, 58) exhibited apical cilia by immunofluorescence for β-tubulin IV (Fig. 16A). Basolateral trypsin (Fig. 16B) increased ciliary beat frequency (CBF). The effect of trypsin was blocked by preincubation with basolateral PAO-1 CM (Fig. 16C) but not by exposure to heat-treated PAO-1 CM (Fig. 16C), elastase-deficient PAO-JP2 CM (Fig. 16D), or PAO-1 CM mixed with protease inhibitor phosphoramidon (Fig. 16E). Trypsin had minimal effect on CBF when pre-exposed to recombinant P. aeruginosa elastase but still increased CBF when elastase was treated with phosphoramidon (Fig. 16F). Lung tryptase also increased CBF (Fig. 16G), and this effect was blocked by PAO-1 CM pretreatment but not by heat-inactivated PAO-1 CM pretreatment (Fig. 16H). PAR-2 agonist 2FLI but not PAR-4 agonist AY-NH2 increased CBF (Fig. 16I), and 2FLI-induced CBF increase was not affected by PAO-1 CM pretreatment (Fig. 16J), PAO-JP2 CM exposure (Fig. 16K), or recombinant P. aeruginosa elastase pretreatment (Fig. 16L). These results are summarized in Fig. 16M and suggest the P. aeruginosa inactivation of PAR-2 is not unique to serous cells but occurs in other airway cells as well, supporting broader implications of this effect.
Figure 16.
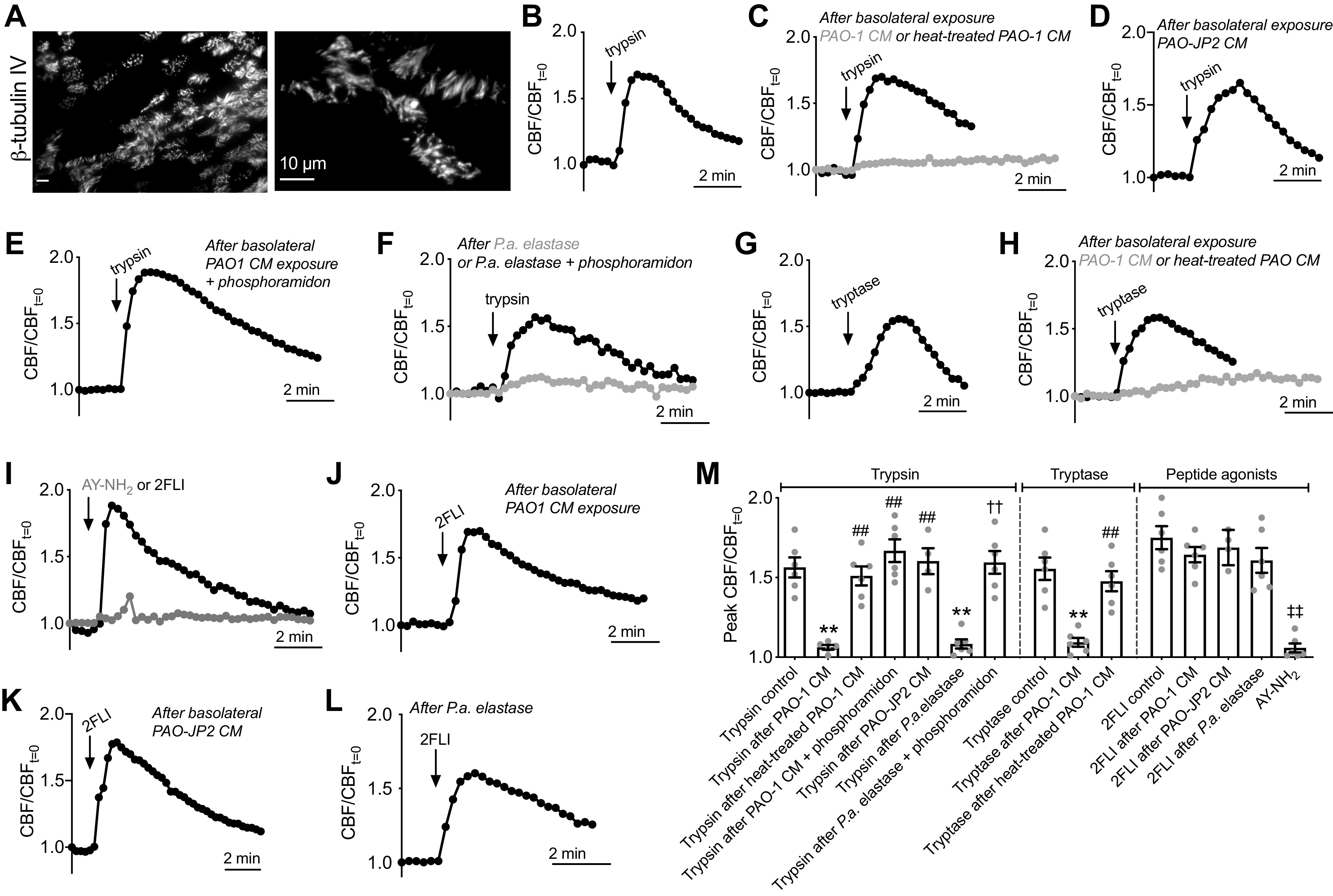
Exposure to Pseudomonas aeruginosa conditioned media (CM) also inhibits protease activation of protease-activated receptor 2 (PAR-2)-stimulated ciliary beat frequency (CBF) increase. A: immunofluorescence image of β-tubulin IV showing motile cilia on a surface epithelial air-liquid interface (ALI) culture [×60, 1.4 numerical aperture (NA) wide-field image; Abcam ab11315 mouse monoclonal antibody]. B: representative trace of CBF during stimulation with basolateral trypsin (5 µM) previously shown to be dependent on PAR-2 (57, 58). Representative CBF traces in response to basolateral trypsin after basolateral pre-exposure to PAO-1 CM (C; gray trace), heat-inactivated PAO-1 CM (C; black trace), PAO-JP2 CM (D), or PAO-1 CM treated with phosphoramidon (E). Mean raw starting CBF was not different after exposure to LB only (7.3 ± 0.2 Hz), PAO-1 CM (7.1 ± 0.3 Hz), or PAO-JP2 CM (7.5 ± 0.4 Hz), or PAO-1 CM + phosphoramidon (7.6 ± 0.2 Hz); P < 0.05 by one-way ANOVA with Bonferroni posttest. F: representative CBF traces after pre-exposure to P. aeruginosa (P.a.) elastase (500 nM; gray trace) ± phosphoramidon (black trace). G: representative CBF traces during stimulation with human lung tryptase (5 µM). H: representative tryptase responses PAO-1 CM (gray) or heat-treated PAO-1 CM (black). 2-Furoyl-LIGRLO-NH2 (2FLI) (10 µM) but not AY-NH2 (10 µM) also increased CBF (I), and 2FLI CBF increases were unaffected by PAO-1 CM (J), PAO-JP2 CM (K), or P.a. elastase (L). M: peak normalized CBF was 1.56 ± 0.06 (trypsin control), 1.06 ± 0.02 (trypsin after PAO-1 CM; **P < 0.01 vs. trypsin only control), 1.51 ± 0.06 (trypsin after heat-treated PAO-1 CM; ##P < 0.01 vs. trypsin after PAO-1 CM), 1.67 ± 0.07 (trypsin after PAO-1 CM + phosphoramidon; ##P < 0.01 vs. trypsin after PAO-1 CM), 1.60 ± 0.08 (trypsin after PAO-JP2 CM; ##P < 0.01 vs. trypsin after PAO-1 CM), 1.08 ± 0.03 (trypsin after P.a. elastase; **P < 0.01 vs. trypsin only control), 1.59 ± 0.07 (trypsin after P.a. elastase + phosphoramidon; ††P < 0.01 vs. trypsin after P.a. elastase), 1.56 (tryptase control), 1.09 ± 0.03 (tryptase after PAO-1 CM), 1.48 ± 0.06 (tryptase after heat-treated PAO-1 CM), 1.75 ± 0.07 (2FLI control), 1.64 ± 0.05 (2FLI after PAO-1 CM), 1.69 ± 0.06 (2FLI after PAO-JP2 CM), 1.61 ± 0.08 (2FLI after P.a. elastase), 1.06 (AY-NH2; ‡‡P < 0.01 vs. 2FLI only). Significance by one-way ANOVA with Bonferroni posttest; n = 5–6 independent experiments with cultures from ≥2 patients. Data points are independent experiments and error bars are SE.
DISCUSSION
In this study, we first found that PAR-2-activated Cl− and fluid secretion by airway submucosal gland serous cells is CFTR-independent during “strong” stimulation of PAR-2 (>EC50 for intracellular Ca2+ elevation). This secretion is inhibited by multiple compounds that block TMEM16A and is likely sustained by NKCC1 and paired AE/NHE activity, as previously reported for cholinergic-driven secretion (3, 19, 20, 22, 35). A diagram of PAR-2-activated, Ca2+-driven, CFTR-independent secretion is shown in Fig. 17A. However, weaker subsecretory protease stimulation of PAR-2 can synergize with low-level VIPergic stimulation to drive secretion through CFTR. Ca2+ from the PAR-2 signaling pathway is likely required to activate K+ channels whereas cAMP from the VIP receptor signaling pathway is required to activate CFTR. This was revealed by both pharmacology and tissue obtained from patients with CF with minimally functional CFTR. A diagram of synergistic low-level secretion is outlined in Fig. 17B. This is similar to low-level cholinergic stimulation (19, 24), but is nonetheless an important new observation for CF pathophysiology.
Figure 17.

Model of protease-activated receptor 2 (PAR-2)-induced fluid secretion in airway submucosal gland serous acinar cells. A: as described in the text, strong PAR-2 stimulation (>EC50 for Ca2+), results in activation of calcium-activated Cl− channel (CaCC) [likely TMEM16A apically expressed in serous acini (12, 20)] and K+ channels for counter ion flux. As described by the classic model of exocrine fluid secretion (3, 210), the net negative lumen potential generated by apical Cl− secretion likely draws Na+ into the gland lumen paracellularly, and osmotically obliged water moves either paracellularly or through aquaporins. Secretion is sustained by Na+K+2Cl- cotransporter (NKCC)1 and paired Na+/H+ exchange (NHE) and anion exchange (AE) activity, both of which require the Na+ gradient established by the Na+K+ ATPase. This secretion is intact in cystic fibrosis (CF) cells, agreeing with observations of intact glands that high doses of PAR-2 activating peptides stimulate cystic fibrosis transmembrane conductance regulator (CFTR)-independent secretion (59). However, this PAR-2 evoked section may be reduced by P. aeruginosa inactivation of PAR-2 during Pseudomonas aeruginosa infection in CF airways, possibly contributing to decreased gland fluid secretion in CF. B: during weaker PAR-2 stimulation (<EC50 for Ca2+), PAR-2 can activate Ca2+ responses that likely are sufficient for K+ channel activation but insufficient for activating CaCC. However, synergistic secretion can be activated by simultaneous low-level vasoactive intestinal peptide (VIP)ergic stimulation to activate CFTR downstream of cAMP. This is similar to VIPergic/cholinergic synergy described in intact glands (24) and isolated serous cells (19). This mechanism of secretion is lost in CF cells due to loss of CFTR function. Basolateral influx pathways are not shown in B for simplicity. VIPR, vasoactive intestinal peptide receptor.
We hypothesize that loss of this synergistic secretion in CF contributes to reduced airway hydration during inflammation when immune cell proteases like mast cell tryptase and neutrophil elastase are released. Lower-level synergistic agonist interactions are likely important for gland physiology in vivo (113), likely more so than the strong stimuli used in many physiology experiments to study the effects of single agonists. It is likely that other Ca2+-elevating GPCRs beyond muscarinic acetylcholine receptors and PAR-2 exhibit the same synergy with cAMP elevating receptors, and thus inhibition of secretion in patients with CF through loss of CFTR may be occurring during multiple types of stimulation. This requires further testing in intact gland models. Although prior studies using intact glands have concluded that PAR-2-activated gland secretion is CFTR-independent (59, 60), these studies used high concentrations of activating peptides (100 µM SLIGRL-NH2), which likely activated strong Ca2+ responses to engage CaCC and did not test lower levels of PAR-2 activation in combination with VIP.
The Ca2+ responses downstream of protease or peptide activation of PAR-2 likely reflect PLC activation, IP3 production, and engagement of the IP3R to activate release of ER Ca2+ stores, as shown in surface epithelial cells (58), with subsequent store-operated Ca2+ influx via resulting Stim1-Orai1 interaction. The exact nature of the Ca2+ influx pathway that keeps Ca2+ elevated during GPCR stimulation in serous cells remains to be fully determined in future studies. However, it appears likely to be store-operated Ca2+ entry, fitting with observations that TMEM16A is activated downstream of Orai1-dependent store-operated Ca2+ entry in other exocrine cells (146–148). The inhibition of Ca2+ influx by BTP-2 fits involvement of the Stim-Orai pathway in airway serous cells as well.
Future work is also needed to more fully characterize the molecular identities of the Cl− conductances existing in human airway serous cells. Notably, the pharmacological tools used here (T16Ainh-A01, CaCCinh-A01, niflumic acid, Ani9) and in a prior study [DIDS (12)] to block human serous cell CaCC are not completely specific for TMEM16A as they block other Cl− channels and have some off-target effects (149). However, TMEM16A is blocked by all four of these compounds (150, 151), and niflumic acid is a poorer inhibitor of bestrophin Ca2+-activated Cl− channels (151). We previously demonstrated strong expression of TMEM16A in pig bronchial serous cells (20). Moreover, strong data supports TMEM16A as the obligatory CaCC conductance mediating Cl− secretion in other exocrine acinar cells (152–156), though other Cl− channels do exist in acinar cells (157, 158). In aggregate, our data from both pharmacology and siRNA experiments suggest an important role for TMEM16A in airway serous cells, though a role for other Cl− channels cannot yet be ruled out.
Although the optical methods used here to study serous cell secretion cannot identify characteristics of specific ionic currents as directly as patch clamp experiments, our methods allow study of intact cells with intact signal transduction pathways. Cell volume measurements also allow interrogation of electroneutral pathways such as NKCC and NHE, one advantage over electrical measurements. However, electrophysiological data is needed to complement studies from intact cells and intact glands. Previous electrophysiological characterizations of airway gland serous cells are limited. Most work has focused on activation of whole cell K+ and Cl− currents downstream of Ca2+-elevating receptors (159–163). More electrophysiological studies to identify specific K+ and Cl− channels involved in serous cell secretion are needed, though strong data suggest that both CFTR and CaCC, likely TMEM16A, are the primary secretory Cl− channels during agonist stimulation.
Furthermore, future work is needed to understand how the high- versus low-global Ca2+ responses translate to activation of TMEM16A versus no activation of TMEM16A, respectively. Global measurements made via dyes like fura-2 do not reveal the full picture of localized Ca2+ responses. Is this a simple concentration effect where lower-level stimulation does not raise Ca2+ high enough in the vicinity of TMEM16A to activate it? Alternatively, are there subtle differences in the localizations of these Ca2+ responses (release, influx, or both) such that Ca2+ is only raised in the vicinity of TMEM16A during stronger PAR-2 stimulation? Polarized and localized Ca2+ signaling is an important regulator of function in other types of exocrine acinar cells, with mitochondrial buffering possibly playing an important role for segregating some Ca2+ responses to basolateral compartments (164). High-speed imaging studies combined with genetically encoded and plasma membrane-targeted or organelle-targeted Ca2+ indicators are needed to help resolve these mechanisms in future studies. However, although biologically interesting, the exact molecular mechanisms involved in the differential effects of the Ca2+ responses do not change the most important implications for tissue-level physiology. Lower levels of stimulation of Ca2+-coupled PAR-2 (this study) or muscarinic acetylcholine receptors (mAChRs) (19) can activate K+ channels whereas higher levels of stimulation can engage both K+ channels and CaCC. This creates opportunities for agonist synergy between cAMP-elevating CFTR-activating pathways and Ca2+-elevating K+ channel-activating pathways, as observed in serous cells [this study and (19)] and intact glands (24).
In intact glands, the maximal secretory rates of Ca2+ agonist-evoked fluid secretion are approximately three times greater than maximal cAMP-evoked fluid secretion rates (24, 113, 165, 166). Why then, if CFTR is part of a “minor” secretion pathway, is CFTR so important that its functional loss leads to gland duct plugging despite functional CaCC? An answer to this could be because Ca2+ agonists in vivo rarely reach the concentrations required to activate CaCC for sustained periods of time. The terminal axons innervating these glands penetrate into the basement membrane surrounding serous acinar cells and get very close (<1 µm) to their basolateral membranes (28). However, although release of a single large neurotransmitter granule has been calculated to be able to create local agonist concentrations of 1–10 µM, diffusion would likely dictate that this occurs for only a very brief (5–10 ms) period of time (167). We speculate that serous cells may spend the majority of their time in vivo being “tickled” by low concentrations of agonists and secreting more through CFTR than CaCC, as intracellular Ca2+ might rarely stay high enough to activate CaCC for prolonged periods of time.
However, our observations do support a therapeutic strategy of activating TMEM16A with a small molecule like Eact to allow TMEM16A to substitute for CFTR to restore defective Cl− secretion in CF during both strong cAMP stimulation as well as during low-level synergistic Ca2+/cAMP secretion. This idea has been proposed by several groups as a “CFTR mutation-agnostic” approach to hydrate airway mucus in patients with CF (168, 169). Our recent study (12) suggested that CaCC/TMEM16A activation by Eact can support similar levels of both Cl− and HCO3− secretion to CFTR in CF cells during strong VIP stimulation, and this likely occurs with synergistic cAMP/Ca2+ secretion as well. Such a strategy likely requires more specific TMEM16A-activating compounds, as Eact has off-target effects in some cells and/or may activate TMEM16A indirectly through altering Ca2+ (111). As we report in the results, we do not observe global Ca2+ elevations with Eact, and cell shrinkage is not activated by Eact alone, suggesting Ca2+-sensitive K+ channels are not activated alongside TMEM16A with Eact alone. However, this does not preclude highly localized Ca2+ elevations within the TMEM16A vicinity not visible with the relatively slow (∼5 s sampling frequency) wide-field fura-2 imaging used here. The exact mechanism of Eact function in serous cells remains to be determined.
We nonetheless hypothesize that TMEM16A might be a useful therapeutic target for serous cells as its activation would allow secretion only when endogenous agonists engage K+ channels, as activation of Cl− conductance alone does not promote secretion in the absence of counterion flux (12). Thus, this strategy may allow fluid secretion to remain under the control of physiological stimuli. Similar effects would be expected by CFTR corrector/potentiator combination therapies in many patients with CF (170). However, in some patients that cannot benefit from corrector/potentiator compounds (e.g., patients with CF with premature stop codon CFTR variants like G542X), TMEM16A activation may have therapeutic benefit by bypassing the requirement for CFTR.
We previously showed that reduction of VIP-induced cAMP responses by neuropeptide Y (NPY), which activates Gi-coupled NYP receptors, can reduce CFTR-dependent secretion (12). NPY is elevated in diseases like asthma (171–174), with NPY production upregulated in immune cells like macrophages (174, 175), submucosal neurons (176–179), or the epithelium itself (171). Reduction of this synergistic cAMP/Ca2+-evoked secretion in the presence of NPY by reduced cAMP levels may also contribute to reduced mucus hydration in the context of Th2 inflammation in asthma or chronic rhinosinusitis, where inflammatory proteases are likely prevalent (46, 51, 119, 123, 124, 180, 181).
Our data suggest that PAR-2 is rendered nonresponsive to proteases by P. aeruginosa secreted products, likely a heat-labile phosphoramidon-sensitive protease produced under control of the lasR/rhlR (acyl homoserine lactone) quorum sensing pathway. Others have suggested that P. aeruginosa elastase can “disarm” PAR-2 by cleaving off the tethered ligand sequence (125, 126, 135), though activation of PAR-2 by P. aeruginosa proteases has also been reported (182–184). The reasons for these discrepancies are unclear, but may involve alterations in receptor processing, expression, or dimerization, recombinant versus native P. aeruginosa elastase preparations used, strain origin of the elastase, or one of many factors. We tried to better address this by using primary cells and secreted products directly from airway P. aeruginosa isolates. Note that in almost all studies here, the trypsin or tryptase is never incubated directly with the P. aeruginosa CM or elastase. The cells are pretreated with CM and then CM is washed away. It is thus unlikely that the P. aeruginosa CM is inhibiting the actual trypsin or tryptase activity itself (e.g., by cleaving and inactivating the trypsin or tryptase). Likewise, a secreted heat-labile protease inhibitor could explain these results only if it sticks to the cells and is highly resistance to washout over many minutes but still binds to the subsequent applied proteases with substantial affinity. This seems unlikely. The more likely explanation is that P. aeruginosa CM affects the PAR-2 receptor rather than the activating protease, which fits with prior studies noted earlier in the discussion. Notably, PAR-2 protease activation is eliminated whereas peptide agonist (2FLI) activation is intact, supporting altered cleavage of the receptor and likely removal of the tethered ligand sequence.
The inhibition of PAR-2-activated secretion by P. aeruginosa likely has important implications for CF lung disease. P. aeruginosa infection is a major cause of lung function decline in patients with CF, significantly contributing to morbidity and mortality (185, 186). PAR-2 knockout mice have enhanced lung injury and inflammation during P. aeruginosa challenge (187), suggesting PAR-2 is important during pulmonary P. aeruginosa infection. PAR-2 inactivation affects both fluid secretion as well as antimicrobial peptide secretion, which is likely mediated by regulated vesicular secretion driven by the same Ca2+ pathway regulating TMEM16A-activated secretion, as observed in salivary acinar cells (188). Furthermore, this same inactivation extends to PAR-2 regulation of ciliary beat frequency, suggesting this has wide-ranging implications for airway physiology.
Our data here include CM from clinical P. aeruginosa strains isolated from patients with CRS as well as recombinant P. aeruginosa elastase produced in another Gram-negative bacteria (E. coli). The ability to pre-expose cells to P. aeruginosa and study PAR-2 activation suggests an apparent slow turn-over of activated PAR-2 at the plasma membrane, supported by the fact that arrestin binding and internalization of activated PAR-2 takes ∼60–90 min at room temperature. As PAR-2 has been demonstrated to exhibit arrestin-biased signaling under some conditions (131, 133, 189–191), the PAR-2 Trio assay (96) was important to confirm that we are really seeing inhibition of the activation of the receptor and not biasing of the receptor away from Ca2+ to arrestin by P. aeruginosa proteases.
Our data support other studies in various airway cells (128, 131–134, 192, 193) suggesting airway PAR-2 can be activated by immune cell elastase and tryptase. Prior studies have also suggested that neutrophil elastase activates protein secretion from cultured bovine (194) and human tracheal gland cells (195). However, as with P. aeruginosa elastase, conflicting studies suggest disarming of PAR-2 by neutrophil elastase (56) or activation of beta-arrestin-based signaling by neutrophil elastase (131). However, we saw PAR-2-dependent activation of Ca2+ responses with both neutrophil elastase and tryptase purified from human blood and lung, respectively. Overall, this supports a differential regulation of PAR-2 by host and some pathogen proteases. However, an important caveat is that we are using purified host proteases whose activity or specificity may have changed during the purification process. With P. aeruginosa, we use both purified protease as well as CM, which is an advantage of this aspect of the study.
Notably, we saw the Western blot band detected by the PAR-2 antibody used here ran at a slightly higher molecular weight in primary serous cells than Calu-3 cells (cf. Fig. 11B vs. Fig. 13B). We hypothesize this is due to PAR-2 glycosylation or palmitoylation, which have been reported to differ in various types of cells (196–199). Differences in N-linked glycosylation and/or sialylation of PAR-2 have been linked to activation by tryptase (198, 199). Similar alterations may affect responsiveness to elastases. This highlights the importance of working in primary cells, where PAR-2 processing through the secretory pathway is more likely to be similar to in vivo processing. Future biochemical characterization is needed to understand if differences in PAR-2 glycosylation or other post-translational modifications exist in different types of primary airway cells as well as if this translates to altered responses to host and/or pathogen proteases.
We found that PAR-2 expression is largely basolateral in both differentiated nasal surface epithelial cells (57, 58) and cultured serous cells (this study). Under healthy well-polarized conditions with intact epithelial tight junctions and barrier function, this is expected to create a spatial barrier between luminal P. aeruginosa secretions that may disarm PAR-2. However, compounds in cigarette smoke (57, 200) like cadmium (201) as well as the 3-oxo-dodecanoyl-homoserine lactone quorum-sensing molecule produced by P. aeruginosa (202–205) can disrupt tight junctions and epithelial integrity. Moreover, squamous metaplasia and poor epithelial polarization and/or barrier function is often observed in airway diseases like CF due to the strong inflammatory environment (42, 57, 206). The ability of P. aeruginosa to disarm PAR-2 to reduce the activation of fluid secretion and increased ciliary beating may depend on the breakdown of the epithelial barrier. P. aeruginosa proteases themselves may contribute to epithelial breakdown. To our knowledge, direct cleavage of airway tight junction proteins by P. aeruginosa proteases has not yet been directly demonstrated, but P. aeruginosa elastase LasB cleaves VE-cadherin to cause endothelial breakdown (207).
As PAR-2 drives ciliary beating as well as fluid and antimicrobial secretion, another major question is whether PAR-2 is itself a potential therapeutic target. The basolateral localization of PAR-2 could enhance its therapeutic potential via a systemically delivered activator that could access the receptor from the serosal fluid. A small molecule agonist that could activate both the uncleaved and disarmed version of the receptor would likely stimulate ciliary beating, lysozyme secretion, and CFTR-independent fluid secretion. However, PAR-2 is ubiquitously expressed in many tissues and drives diverse phenomenon like blood clotting and immune cell function (208). We hypothesize that undesired effects in other tissues may prohibit systemic targeting of PAR-2. Localized agonist delivery via nasal spray or nebulized inhaled delivery may be limited due to basolateral localization. Even if local delivery can be achieved, PAR-2 activation is also pro-inflammatory in airway and other cells (49, 51, 52), promoting a T-helper 2 phenotype similar to that observed in asthma and CRS (123). Proinflammatory effects could outweigh the beneficial effects of activating PAR-2 exogenously. Further in vitro and in vivo experimental work is required to better understand the biology of PAR-2 in the lung and if PAR-2 itself could be leveraged as a therapeutic target.
In summary, the data presented here use primary human airway serous cells to demonstrate that PAR-2 is involved in CFTR-independent secretion when cells are stimulated with high concentrations of PAR-2 agonist alone. However, when lower levels of PAR-2 stimulation are combined with low-level stimulation of receptors that elevate cAMP, CFTR-dependent secretion occurs. We observed PAR-2 activation in serous cells by human immune cell proteases but inhibition by P. aeruginosa secreted products, likely via P. aeruginosa elastase modification of the receptor itself. Disruption of the CFTR-dependent PAR-2 secretion pathway in CF and further disruption of the CFTR-independent PAR-2 secretion pathway with P. aeruginosa infection may be important to CF sinus and lung pathophysiology. Moreover, disruption of host protease-activated, PAR-2-dependent increases in ciliary beating by P. aeruginosa may also exacerbate mucociliary clearance defects in P. aeruginosa-infected patients.
SUPPLEMENTAL DATA
Supplemental Table S1 and Figs. S1 and S2: https://doi.org/10.6084/m9.figshare.13515098.
GRANTS
This work was funded by grants from the National Institutes of Health (R01DC016309) and Cystic Fibrosis Foundation Grant LEE19G0 (to R. J. Lee).
DISCLAIMERS
This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data interpretation, or decision to submit.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.B.M., R.M.C., and R.J.L. conceived and designed research; D.B.M., R.M.C., M.A.K., N.D.A., and R.J.L. performed experiments; D.B.M., R.M.C., M.A.K., N.D.A., J.N.P., and R.J.L. analyzed data; D.B.M., R.M.C., M.A.K., N.D.A., J.N.P., and R.J.L. interpreted results of experiments; D.B.M., R.M.C., and R.J.L. prepared figures; D.B.M. and R.J.L. drafted manuscript; D.B.M., R.M.C., M.A.K., N.D.A., J.N.P., and R.J.L. edited and revised manuscript; D.B.M., R.M.C., M.A.K., N.D.A., J.N.P., and R.J.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank M. Victoria (University of Pennsylvania) for excellent technical support during the study. We thank B. Chen and N. Cohen (University of Pennsylvania and Philadelphia VA Medical Center) for PAR-2 knockout mouse tissue and cell culture.
REFERENCES
- 1.Ballard ST, Inglis SK. Liquid secretion properties of airway submucosal glands. J Physiol 556: 1–10, 2004. doi: 10.1113/jphysiol.2003.052779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballard ST, Spadafora D. Fluid secretion by submucosal glands of the tracheobronchial airways. Respir Physiol Neurobiol 159: 271–277, 2007. doi: 10.1016/j.resp.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee RJ, Foskett JK. Ca2+ signaling and fluid secretion by secretory cells of the airway epithelium. Cell Calcium 55: 325–336, 2014. doi: 10.1016/j.ceca.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Widdicombe JH, Wine JJ. Airway gland structure and function. Physiol Rev 95: 1241–1319, 2015. doi: 10.1152/physrev.00039.2014. [DOI] [PubMed] [Google Scholar]
- 5.Joo NS, Evans IA, Cho HJ, Park IH, Engelhardt JF, Wine JJ. Proteomic analysis of pure human airway gland mucus reveals a large component of protective proteins. PLoS One 10: e0116756, 2015. doi: 10.1371/journal.pone.0116756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joo NS, Lee DJ, Winges KM, Rustagi A, Wine JJ. Regulation of antiprotease and antimicrobial protein secretion by airway submucosal gland serous cells. J Biol Chem 279: 38854–38860, 2004. doi: 10.1074/jbc.M407077200. [DOI] [PubMed] [Google Scholar]
- 7.Jeffery PK. Comparative morphology of the airways in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 150: S6–S13, 1994. doi: 10.1164/ajrccm/150.5_Pt_2.S6. [DOI] [PubMed] [Google Scholar]
- 8.Rogers DF. Mucus hypersecretion in chronic obstructive pulmonary disease. Novartis Found Symp 234: 65–77, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Chen FH, Samson KT, Miura K, Ueno K, Odajima Y, Shougo T, Yoshitsugu Y, Shioda S. Airway remodeling: a comparison between fatal and nonfatal asthma. J Asthma 41: 631–638, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Cluroe A, Holloway L, Thomson K, Purdie G, Beasley R. Bronchial gland duct ectasia in fatal bronchial asthma: association with interstitial emphysema. J Clin Pathol 42: 1026–1031, 1989. doi: 10.1136/jcp.42.10.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins HA, Cool C, Szefler SJ, Covar R, Brugman S, Gelfand EW, Spahn JD. Histopathology of severe childhood asthma: a case series. Chest 124: 32–41, 2003. doi: 10.1378/chest.124.1.32. [DOI] [PubMed] [Google Scholar]
- 12.McMahon DB, Carey RM, Kohanski MA, Tong CCL, Papagiannopoulos P, Adappa ND, Palmer JN, Lee RJ. Neuropeptide regulation of secretion and inflammation in human airway gland serous cells. Eur Respir J 55: 1901386, 2020. doi: 10.1183/13993003.01386-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodruff PG, Fahy JV. Airway remodeling in asthma. Semin Respir Crit Care Med 23: 361–367, 2002. doi: 10.1055/s-2002-34331. [DOI] [PubMed] [Google Scholar]
- 14.Wu JV, Krouse M, Wine JJ. Acinar origin of CFTR-dependent airway submucosal gland fluid secretion. Am J Physiol Lung Cell Mol Physiol 292: L304–L311, 2007. doi: 10.1152/ajplung.00286.2006. [DOI] [PubMed] [Google Scholar]
- 15.Groneberg DA, Peiser C, Dinh QT, Matthias J, Eynott PR, Heppt W, Carlstedt I, Witt C, Fischer A, Chung KF. Distribution of respiratory mucin proteins in human nasal mucosa. Laryngoscope 113: 520–524, 2003. doi: 10.1097/00005537-200303000-00023. [DOI] [PubMed] [Google Scholar]
- 16.Sharma P, Dudus L, Nielsen PA, Clausen H, Yankaskas JR, Hollingsworth MA, Engelhardt JF. MUC5B and MUC7 are differentially expressed in mucous and serous cells of submucosal glands in human bronchial airways. Am J Respir Cell Mol Biol 19: 30–37, 1998. doi: 10.1165/ajrcmb.19.1.3054. [DOI] [PubMed] [Google Scholar]
- 17.Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 2: 240–248, 1992. doi: 10.1038/ng1192-240. [DOI] [PubMed] [Google Scholar]
- 18.Jacquot J, Puchelle E, Hinnrasky J, Fuchey C, Bettinger C, Spilmont C, Bonnet N, Dieterle A, Dreyer D, Pavirani A. Localization of the cystic fibrosis transmembrane conductance regulator in airway secretory glands. Eur Respir J 6: 169–176, 1993. [PubMed] [Google Scholar]
- 19.Lee RJ, Foskett JK. cAMP-activated Ca2+ signaling is required for CFTR-mediated serous cell fluid secretion in porcine and human airways. J Clin Invest 120: 3137–3148, 2010. doi: 10.1172/JCI42992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee RJ, Foskett JK. Mechanisms of Ca2+-stimulated fluid secretion by porcine bronchial submucosal gland serous acinar cells. Am J Physiol Lung Cell Mol Physiol 298: 22, 2010. doi: 10.1152/ajplung.00342.2009. [DOI] [PubMed] [Google Scholar]
- 21.Lee RJ, Harlow JM, Limberis MP, Wilson JM, Foskett JK. HCO3(−) secretion by murine nasal submucosal gland serous acinar cells during Ca2+-stimulated fluid secretion. J Gen Physiol 132: 161–183, 2008. doi: 10.1085/jgp.200810017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee RJ, Limberis MP, Hennessy MF, Wilson JM, Foskett JK. Optical imaging of Ca2+-evoked fluid secretion by murine nasal submucosal gland serous acinar cells. J Physiol 582: 1099–1124, 2007. doi: 10.1113/jphysiol.2007.131995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho HJ, Joo NS, Wine JJ. Defective fluid secretion from submucosal glands of nasal turbinates from CFTR−/− and CFTR (deltaF508/deltaF508) pigs. PLoS One 6: e24424, 2011. doi: 10.1371/journal.pone.0024424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi JY, Joo NS, Krouse ME, Wu JV, Robbins RC, Ianowski JP, Hanrahan JW, Wine JJ. Synergistic airway gland mucus secretion in response to vasoactive intestinal peptide and carbachol is lost in cystic fibrosis. J Clin Invest 117: 3118–3127, 2007. doi: 10.1172/JCI31992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS. Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc Natl Acad Sci USA 98: 8119–8123, 2001. doi: 10.1073/pnas.131087598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joo NS, Irokawa T, Robbins RC, Wine JJ. Hyposecretion, not hyperabsorption, is the basic defect of cystic fibrosis airway glands. J Biol Chem 281: 7392–7398, 2006. doi: 10.1074/jbc.M512766200. [DOI] [PubMed] [Google Scholar]
- 27.Joo NS, Irokawa T, Wu JV, Robbins RC, Whyte RI, Wine JJ. Absent secretion to vasoactive intestinal peptide in cystic fibrosis airway glands. J Biol Chem 277: 50710–50715, 2002. doi: 10.1074/jbc.M208826200. [DOI] [PubMed] [Google Scholar]
- 28.Meyrick B, Reid L. Ultrastructure of cells in the human bronchial submucosal glands. J Anat 107: 281–299, 1970. [PMC free article] [PubMed] [Google Scholar]
- 29.Meyrick B, Sturgess JM, Reid L. A reconstruction of the duct system and secretory tubules of the human bronchial submucosal gland. Thorax 24: 729–736, 1969. doi: 10.1136/thx.24.6.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reid L. Measurement of the bronchial mucous gland layer: a diagnostic yardstick in chronic bronchitis. Thorax 15: 132–141, 1960. doi: 10.1136/thx.15.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov AM, Waghray A, Slyper M, Waldman J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560: 319–324, 2018. doi: 10.1038/s41586-018-0393-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plasschaert LW, Zilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560: 377–381, 2018. doi: 10.1038/s41586-018-0394-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi HK, Finkbeiner WE, Widdicombe JH. A comparative study of mammalian tracheal mucous glands. J Anat 197: 361–372, 2000. doi: 10.1046/j.1469-7580.2000.19730361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ianowski JP, Choi JY, Wine JJ, Hanrahan JW. Mucus secretion by single tracheal submucosal glands from normal and CFTR knock-out mice. J Physiol 580: 301–314, 2007. doi: 10.1113/jphysiol.2006.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee RJ, Foskett JK. Why mouse airway submucosal gland serous cells do not secrete fluid in response to cAMP stimulation. J Biol Chem 287: 38316–38326, 2012. doi: 10.1074/jbc.M112.412817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballard ST, Fountain JD, Inglis SK, Corboz MR, Taylor AE. Chloride secretion across distal airway epithelium: relationship to submucosal gland distribution. Am J Physiol Lung Cell Mol Physiol 268: L526–L531, 1995. doi: 10.1152/ajplung.1995.268.3.L526. [DOI] [PubMed] [Google Scholar]
- 37.Inglis SK, Corboz MR, Ballard ST. Effect of anion secretion inhibitors on mucin content of airway submucosal gland ducts. Am J Physiol Lung Cell Mol Physiol 274: L762–L766, 1998. doi: 10.1152/ajplung.1998.274.5.L762. [DOI] [PubMed] [Google Scholar]
- 38.Inglis SK, Corboz MR, Taylor AE, Ballard ST. Effect of anion transport inhibition on mucus secretion by airway submucosal glands. Am J Physiol Lung Cell Mol Physiol 272: L372–L377, 1997. doi: 10.1152/ajplung.1997.272.2.L372. [DOI] [PubMed] [Google Scholar]
- 39.Inglis SK, Corboz MR, Taylor AE, Ballard ST. In situ visualization of bronchial submucosal glands and their secretory response to acetylcholine. Am J Physiol Lung Cell Mol Physiol 272: L203–L210, 1997. doi: 10.1152/ajplung.1997.272.2.L203. [DOI] [PubMed] [Google Scholar]
- 40.Trout L, Gatzy JT, Ballard ST. Acetylcholine-induced liquid secretion by bronchial epithelium: role of Cl− and HCO3− transport. Am J Physiol Lung Cell Mol Physiol 275: L1095–L1099, 1998. doi: 10.1152/ajplung.1998.275.6.L1095. [DOI] [PubMed] [Google Scholar]
- 41.Trout L, King M, Feng W, Inglis SK, Ballard ST. Inhibition of airway liquid secretion and its effect on the physical properties of airway mucus. Am J Physiol Lung Cell Mol Physiol 274: L258–L263, 1998. doi: 10.1152/ajplung.1998.274.2.L258. [DOI] [PubMed] [Google Scholar]
- 42.Jeffery PK, Brain AP. Surface morphology of human airway mucosa: normal, carcinoma or cystic fibrosis. Scanning Microsc 2: 553–560, 1988. [PubMed] [Google Scholar]
- 43.Ornoy A, Arnon J, Katznelson D, Granat M, Caspi B, Chemke J. Pathological confirmation of cystic fibrosis in the fetus following prenatal diagnosis. Am J Med Genet 28: 935–947, 1987. doi: 10.1002/ajmg.1320280420. [DOI] [PubMed] [Google Scholar]
- 44.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, Stoltz DA, Welsh MJ. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345: 818–822, 2014. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie Y, Ostedgaard L, Abou Alaiwa MH, Lu L, Fischer AJ, Stoltz DA. Mucociliary transport in healthy and cystic fibrosis pig airways. Ann Am Thorac Soc 15: S171–S176, 2018. doi: 10.1513/AnnalsATS.201805-308AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cocks TM, Moffatt JD. Protease-activated receptor-2 (PAR2) in the airways. Pulm Pharmacol Ther 14: 183–191, 2001. doi: 10.1006/pupt.2001.0285. [DOI] [PubMed] [Google Scholar]
- 47.Kawabata A, Kuroda R, Nishida M, Nagata N, Sakaguchi Y, Kawao N, Nishikawa H, Arizono N, Kawai K. Protease-activated receptor-2 (PAR-2) in the pancreas and parotid gland: Immunolocalization and involvement of nitric oxide in the evoked amylase secretion. Life Sci 71: 2435–2446, 2002. [DOI] [PubMed] [Google Scholar]
- 48.Lan RS, Stewart GA, Henry PJ. Role of protease-activated receptors in airway function: a target for therapeutic intervention? Pharmacol Ther 95: 239–257, 2002. doi: 10.1016/s0163-7258(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 49.Peters T, Henry PJ. Protease-activated receptors and prostaglandins in inflammatory lung disease. Br J Pharmacol 158: 1017–1033, 2009. doi: 10.1111/j.1476-5381.2009.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HM, Kim HY, Kang HJ, Woo JS, Chae SW, Lee SH, Hwang SJ. Up-regulation of protease-activated receptor 2 in allergic rhinitis. Ann Otol Rhinol Laryngol 116: 554–558, 2007. doi: 10.1177/000348940711600712. [DOI] [PubMed] [Google Scholar]
- 51.Reed CE, Kita H. The role of protease activation of inflammation in allergic respiratory diseases. J Allergy Clin Immunol 114: 997–1008, 2004. doi: 10.1016/j.jaci.2004.07.060. [DOI] [PubMed] [Google Scholar]
- 52.Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol 34: 133–149, 2012. doi: 10.1007/s00281-011-0289-1. [DOI] [PubMed] [Google Scholar]
- 53.Sun L, Ye RD. Role of G protein-coupled receptors in inflammation. Acta Pharmacol Sin 33: 342–350, 2012. doi: 10.1038/aps.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bunnett NW. Protease-activated receptors: how proteases signal to cells to cause inflammation and pain. Semin Thromb Hemost 32: 039–048, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev 84: 579–621, 2004. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 56.Dulon S, Cande C, Bunnett NW, Hollenberg MD, Chignard M, Pidard D. Proteinase-activated receptor-2 and human lung epithelial cells: disarming by neutrophil serine proteinases. Am J Respir Cell Mol Biol 28: 339–346, 2003. doi: 10.1165/rcmb.4908. [DOI] [PubMed] [Google Scholar]
- 57.Carey RM, Freund JR, Hariri BM, Adappa ND, Palmer JN, Lee RJ. Polarization of protease-activated receptor 2 (PAR-2) signaling is altered during airway epithelial remodeling and deciliation. J Biol Chem 295: 6721–6740, 2020. doi: 10.1074/jbc.RA120.012710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McMahon DB, Workman AD, Kohanski MA, Carey RM, Freund JR, Hariri BM, Chen B, Doghramji LJ, Adappa ND, Palmer JN, Kennedy DW, Lee RJ. Protease-activated receptor 2 activates airway apical membrane chloride permeability and increases ciliary beating. FASEB J 32: 155–167, 2018. doi: 10.1096/fj.201700114RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee HJ, Yang YM, Kim K, Shin DM, Yoon JH, Cho HJ, Choi JY. Protease-activated receptor 2 mediates mucus secretion in the airway submucosal gland. PLoS One 7: e43188, 2012. doi: 10.1371/journal.pone.0043188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cho HJ, Lee HJ, Kim SC, Kim K, Kim YS, Kim CH, Lee JG, Yoon JH, Choi JY. Protease-activated receptor 2-dependent fluid secretion from airway submucosal glands by house dust mite extract. J Allergy Clin Immunol 129: 529–535, 2012. doi: 10.1016/j.jaci.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 61.Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, Fairlie DP. Novel agonists and antagonists for human protease activated receptor 2. J Med Chem 53: 7428–7440, 2010. doi: 10.1021/jm100984y. [DOI] [PubMed] [Google Scholar]
- 62.Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, Fairlie DP. Modulating human proteinase activated receptor 2 with a novel antagonist (GB88) and agonist (GB110). Br J Pharmacol 165: 1413–1423, 2012. doi: 10.1111/j.1476-5381.2011.01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yau MK, Liu L, Fairlie DP. Toward drugs for protease-activated receptor 2 (PAR2). J Med Chem 56: 7477–7497, 2013. doi: 10.1021/jm400638v. [DOI] [PubMed] [Google Scholar]
- 64.Lingemann M, McCarty T, Liu X, Buchholz UJ, Surman S, Martin SE, Collins PL, Munir S. The alpha-1 subunit of the Na+,K+-ATPase (ATP1A1) is required for macropinocytic entry of respiratory syncytial virus (RSV) in human respiratory epithelial cells. PLoS Pathog 15: e1007963, 2019. doi: 10.1371/journal.ppat.1007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adams MN, Pagel CN, Mackie EJ, Hooper JD. Evaluation of antibodies directed against human protease-activated receptor-2. Naunyn Schmiedebergs Arch Pharmacol 385: 861–873, 2012. doi: 10.1007/s00210-012-0783-6. [DOI] [PubMed] [Google Scholar]
- 66.Klinngam W, Fu R, Janga SR, Edman MC, Hamm-Alvarez SF. Cathepsin S alters the expression of pro-inflammatory cytokines and MMP-9, partially through protease-activated receptor-2, in human corneal epithelial cells. Int J Mol Sci 19: 3530, 2018. doi: 10.3390/ijms19113530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He L, Ma Y, Li W, Han W, Zhao X, Wang H. Protease-activated receptor 2 signaling modulates susceptibility of colonic epithelium to injury through stabilization of YAP in vivo. Cell Death Dis 9: 949, 2018. doi: 10.1038/s41419-018-0995-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kon Y, Tsukada H, Hasegawa T, Igarashi K, Wada K, Suzuki E, Arakawa M, Gejyo F. The role of Pseudomonas aeruginosa elastase as a potent inflammatory factor in a rat air pouch inflammation model. FEMS Immunol Med Microbiol 25: 313–321, 1999. doi: 10.1111/j.1574-695X.1999.tb01356.x. [DOI] [PubMed] [Google Scholar]
- 69.Roos A, Boron WF. Intracellular pH. Physiol Rev 61: 296–434, 1981. doi: 10.1152/physrev.1981.61.2.296. [DOI] [PubMed] [Google Scholar]
- 70.Weintraub WH, Machen TE. pH regulation in hepatoma cells: roles for Na-H exchange, Cl-HCO3 exchange, and Na-HCO3 cotransport. Am J Physiol Gastrointest Liver Physiol 257: G317–G327, 1989. doi: 10.1152/ajpgi.1989.257.3.G317. [DOI] [PubMed] [Google Scholar]
- 71.Finkbeiner WE, Zlock LT, Mehdi I, Widdicombe JH. Cultures of human tracheal gland cells of mucous or serous phenotype. In Vitro Cell Dev Biol Anim 46: 450–456, 2010. doi: 10.1007/s11626-009-9262-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer H, Illek B, Sachs L, Finkbeiner WE, Widdicombe JH. CFTR and calcium-activated chloride channels in primary cultures of human airway gland cells of serous or mucous phenotype. Am J Physiol Lung Cell Mol Physiol 299: L585–L594, 2010. doi: 10.1152/ajplung.00421.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freund JR, Mansfield CJ, Doghramji LJ, Adappa ND, Palmer JN, Kennedy DW, Reed DR, Jiang P, Lee RJ. Activation of airway epithelial bitter taste receptors by Pseudomonas aeruginosa quinolones modulates calcium, cyclic-AMP, and nitric oxide signaling. J Biol Chem 293: 9824–9840, 2018. doi: 10.1074/jbc.RA117.001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hariri BM, McMahon DB, Chen B, Freund JR, Mansfield CJ, Doghramji LJ, Adappa ND, Palmer JN, Kennedy DW, Reed DR, Jiang P, Lee RJ. Flavones modulate respiratory epithelial innate immunity: anti-inflammatory effects and activation of the T2R14 receptor. J Biol Chem 292: 8484–8497, 2017. doi: 10.1074/jbc.M116.771949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee RJ, Chen B, Redding KM, Margolskee RF, Cohen NA. Mouse nasal epithelial innate immune responses to Pseudomonas aeruginosa quorum-sensing molecules require taste signaling components. Innate Immun 20: 606–617, 2014. doi: 10.1177/1753425913503386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Foskett JK. [Ca2+]i modulation of Cl− content controls cell volume in single salivary acinar cells during fluid secretion. Am J Physiol Cell Physiol 259: C998–C1004, 1990. doi: 10.1152/ajpcell.1990.259.6.C998. [DOI] [PubMed] [Google Scholar]
- 77.Foskett JK, Melvin JE. Activation of salivary secretion: coupling of cell volume and [Ca2+]i in single cells. Science 244: 1582–1585, 1989. [Erratum in Science 245: 343, 1989]. doi: 10.1126/science.2500708. [DOI] [PubMed] [Google Scholar]
- 78.Fowler CJ, Tiger G. Calibration of Fura-2 signals introduces errors into measurement of thrombin-stimulated calcium mobilisation in human platelets. Clin Chim Acta 265: 247–261, 1997. doi: 10.1016/s0009-8981(97)00139-3. [DOI] [PubMed] [Google Scholar]
- 79.Foskett JK. Simultaneous Nomarski and fluorescence imaging during video microscopy of cells. Am J Physiol Cell Physiol 255: C566–C571, 1988. doi: 10.1152/ajpcell.1988.255.4.C566. [DOI] [PubMed] [Google Scholar]
- 80.Foskett JK, Wong MM, Sue AQG, Robertson MA. Isosmotic modulation of cell volume and intracellular ion activities during stimulation of single exocrine cells. J Exp Zool 268: 104–110, 1994. doi: 10.1002/jez.1402680206. [DOI] [PubMed] [Google Scholar]
- 81.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682, 2012. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hariri BM, McMahon DB, Chen B, Adappa ND, Palmer JN, Kennedy DW, Lee RJ. Plant flavones enhance antimicrobial activity of respiratory epithelial cell secretions against Pseudomonas aeruginosa. PLoS One 12: e0185203, 2017. doi: 10.1371/journal.pone.0185203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using microManager. Curr Protoc Mol Biol Chapter14: Unit14.20, 2010.doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pearson JP, Gray KM, Passador L, Tucker KD, Eberhard A, Iglewski BH, Greenberg EP. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc Natl Acad Sci USA 91: 197–201, 1994. doi: 10.1073/pnas.91.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pearson JP, Pesci EC, Iglewski BH. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J Bacteriol 179: 5756–5767, 1997. doi: 10.1128/jb.179.18.5756-5767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee RJ, Hariri BM, McMahon DB, Chen B, Doghramji L, Adappa ND, Palmer JN, Kennedy DW, Jiang P, Margolskee RF, Cohen NA. Bacterial d-amino acids suppress sinonasal innate immunity through sweet taste receptors in solitary chemosensory cells. Sci Signal 10: eaam7703, 2017. doi: 10.1126/scisignal.aam7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee RJ, Kofonow JM, Rosen PL, Siebert AP, Chen B, Doghramji L, Xiong G, Adappa ND, Palmer JN, Kennedy DW, Kreindler JL, Margolskee RF, Cohen NA. Bitter and sweet taste receptors regulate human upper respiratory innate immunity. J Clin Invest 124: 1393–1405, 2014. doi: 10.1172/JCI72094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee RJ, Xiong G, Kofonow JM, Chen B, Lysenko A, Jiang P, Abraham V, Doghramji L, Adappa ND, Palmer JN, Kennedy DW, Beauchamp GK, Doulias P-T, Ischiropoulos H, Kreindler JL, Reed DR, Cohen NA. T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J Clin Invest 122: 4145–4159, 2012. doi: 10.1172/JCI64240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beaufort N, Corvazier E, Hervieu A, Choqueux C, Dussiot M, Louedec L, Cady A, de Bentzmann S, Michel JB, Pidard D. The thermolysin-like metalloproteinase and virulence factor LasB from pathogenic Pseudomonas aeruginosa induces anoikis of human vascular cells. Cell Microbiol 13: 1149–1167, 2011. doi: 10.1111/j.1462-5822.2011.01606.x. [DOI] [PubMed] [Google Scholar]
- 90.Bever RA, Iglewski BH. Molecular characterization and nucleotide sequence of the Pseudomonas aeruginosa elastase structural gene. J Bacteriol 170: 4309–4314, 1988. doi: 10.1128/jb.170.9.4309-4314.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hariri BM, Payne SJ, Chen B, Mansfield C, Doghramji LJ, Adappa ND, Palmer JN, Kennedy DW, Niv MY, Lee RJ. In vitro effects of anthocyanidins on sinonasal epithelial nitric oxide production and bacterial physiology. Am J Rhinol Allergy 30: 261–268, 2016. doi: 10.2500/ajra.2016.30.4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y, Reenstra WW, Chidekel A. Antibacterial activity of apical surface fluid from the human airway cell line Calu-3: pharmacologic alteration by corticosteroids and beta(2)-agonists. Am J Respir Cell Mol Biol 25: 196–202, 2001. doi: 10.1165/ajrcmb.25.2.4211. [DOI] [PubMed] [Google Scholar]
- 93.Widdicombe JH. Regulation of the depth and composition of airway surface liquid. J Anat 201: 313–318, 2002. doi: 10.1046/j.1469-7580.2002.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee RJ, Chen B, Doghramji L, Adappa ND, Palmer JN, Kennedy DW, Cohen NA. Vasoactive intestinal peptide regulates sinonasal mucociliary clearance and synergizes with histamine in stimulating sinonasal fluid secretion. FASEB J 27: 5094–5103, 2013. doi: 10.1096/fj.13-234476. [DOI] [PubMed] [Google Scholar]
- 95.Irokawa T, Krouse ME, Joo NS, Wu JV, Wine JJ. A “virtual gland” method for quantifying epithelial fluid secretion. Am J Physiol Lung Cell Mol Physiol 287: L784–L793, 2004. doi: 10.1152/ajplung.00124.2004. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Q, Zheng YW, Coughlin SR, Shu X. A rapid fluorogenic GPCR-beta-arrestin interaction assay. Protein Sci 27: 874–879, 2018. doi: 10.1002/pro.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sisson JH, Stoner JA, Ammons BA, Wyatt TA. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc 211: 103–111, 2003. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- 98.Virgin FW, Rowe SM, Wade MB, Gaggar A, Leon KJ, Young KR, Woodworth BA. Extensive surgical and comprehensive postoperative medical management for cystic fibrosis chronic rhinosinusitis. Am J Rhinol Allergy 26: 70–75, 2012. doi: 10.2500/ajra.2012.26.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robertson MA, Foskett JK. Na+ transport pathways in secretory acinar cells: membrane cross talk mediated by [Cl−]i. Am J Physiol Cell Physiol 267: C146–C156, 1994. doi: 10.1152/ajpcell.1994.267.1.C146. [DOI] [PubMed] [Google Scholar]
- 100.Ferrera L, Caputo A, Galietta LJ. TMEM16A protein: a new identity for Ca(2+)-dependent Cl(−) channels. Physiology (Bethesda) 25: 357–363, 2010. doi: 10.1152/physiol.00030.2010. [DOI] [PubMed] [Google Scholar]
- 101.Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J 25: 4048–4062, 2011. doi: 10.1096/fj.11-191627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol 73: 758–768, 2008. doi: 10.1124/mol.107.043208. [DOI] [PubMed] [Google Scholar]
- 103.Seo Y, Lee HK, Park J, Jeon DK, Jo S, Jo M, Namkung W. Ani9, a novel potent small-molecule ANO1 inhibitor with negligible effect on ANO2. PLoS One 11: e0155771, 2016. doi: 10.1371/journal.pone.0155771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palmer ML, Lee SY, Maniak PJ, Carlson D, Fahrenkrug SC, O'Grady SM. Protease-activated receptor regulation of Cl− secretion in Calu-3 cells requires prostaglandin release and CFTR activation. Am J Physiol Cell Physiol 290: C1189–C1198, 2006. doi: 10.1152/ajpcell.00464.2005. [DOI] [PubMed] [Google Scholar]
- 105.Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H, Yamada T. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J Immunol 170: 4441–4449, 2003. doi: 10.4049/jimmunol.170.9.4441. [DOI] [PubMed] [Google Scholar]
- 106.De Smet P, Parys JB, Callewaert G, Weidema AF, Hill E, De Smedt H, Erneux C, Sorrentino V, Missiaen L. Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca(2+) pumps. Cell Calcium 26: 9–13, 1999. doi: 10.1054/ceca.1999.0047. [DOI] [PubMed] [Google Scholar]
- 107.Miyamoto S, Izumi M, Hori M, Kobayashi M, Ozaki H, Karaki H. Xestospongin C, a selective and membrane-permeable inhibitor of IP(3) receptor, attenuates the positive inotropic effect of alpha-adrenergic stimulation in guinea-pig papillary muscle. Br J Pharmacol 130: 650–654, 2000. doi: 10.1038/sj.bjp.0703358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Robertson MA, Foskett JK. Membrane crosstalk in secretory epithelial cells mediated by intracellular chloride concentration. Jpn J Physiol 44, Suppl 2: S309–S315, 1994. [PubMed] [Google Scholar]
- 109.Robertson MA, Woodside M, Foskett JK, Orlowski J, Grinstein S. Muscarinic agonists induce phosphorylation-independent activation of the NHE-1 isoform of the Na+/H+ antiporter in salivary acinar cells. J Biol Chem 272: 287–294, 1997. [PubMed] [Google Scholar]
- 110.Zachos NC, Tse M, Donowitz M. Molecular physiology of intestinal Na+/H+ exchange. Annu Rev Physiol 67: 411–443, 2005. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 111.Genovese M, Borrelli A, Venturini A, Guidone D, Caci E, Viscido G, Gambardella G, di Bernardo D, Scudieri P, Galietta LJV. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J Physiol 597: 5859–5878, 2019. doi: 10.1113/JP278784. [DOI] [PubMed] [Google Scholar]
- 112.Devor DC, Singh AK, Lambert LC, DeLuca A, Frizzell RA, Bridges RJ. Bicarbonate and chloride secretion in Calu-3 human airway epithelial cells. J Gen Physiol 113: 743–760, 1999. doi: 10.1085/jgp.113.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wine JJ. Parasympathetic control of airway submucosal glands: central reflexes and the airway intrinsic nervous system. Auton Neurosci 133: 35–54, 2007. doi: 10.1016/j.autneu.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Choi JY, Khansaheb M, Joo NS, Krouse ME, Robbins RC, Weill D, Wine JJ. Substance P stimulates human airway submucosal gland secretion mainly via a CFTR-dependent process. J Clin Invest 119: 1189–1200, 2009. [Erratum in J Clin Invest 120: 931–932, 2010]. doi: 10.1172/JCI37284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Worthington EN, Tarran R. Methods for ASL measurements and mucus transport rates in cell cultures. Methods Mol Biol 742: 77–92, 2011. doi: 10.1007/978-1-61779-120-8_5. [DOI] [PubMed] [Google Scholar]
- 116.Barro Soria R, Spitzner M, Schreiber R, Kunzelmann K. Bestrophin-1 enables Ca2+-activated Cl− conductance in epithelia. J Biol Chem 284: 29405–29412, 2009. doi: 10.1074/jbc.M605716200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barro-Soria R, Schreiber R, Kunzelmann K. Bestrophin 1 and 2 are components of the Ca(2+) activated Cl(−) conductance in mouse airways. Biochim Biophys Acta 1783: 1993–2000, 2008. doi: 10.1016/j.bbamcr.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 118.Cottrell GS, Amadesi S, Schmidlin F, Bunnett N. Protease-activated receptor 2: activation, signalling and function. Biochem Soc Trans 31: 1191–1197, 2003. doi: 10.1042/bst0311191. [DOI] [PubMed] [Google Scholar]
- 119.Arizmendi NG, Abel M, Mihara K, Davidson C, Polley D, Nadeem A, El Mays T, Gilmore BF, Walker B, Gordon JR, Hollenberg MD, Vliagoftis H. Mucosal allergic sensitization to cockroach allergens is dependent on proteinase activity and proteinase-activated receptor-2 activation. J Immunol 186: 3164–3172, 2011. doi: 10.4049/jimmunol.0903812. [DOI] [PubMed] [Google Scholar]
- 120.Page K, Strunk VS, Hershenson MB. Cockroach proteases increase IL-8 expression in human bronchial epithelial cells via activation of protease-activated receptor (PAR)-2 and extracellular-signal-regulated kinase. J Allergy Clin Immunol 112: 1112–1118, 2003. doi: 10.1016/j.jaci.2003.08.050. [DOI] [PubMed] [Google Scholar]
- 121.Polley DJ, Mihara K, Ramachandran R, Vliagoftis H, Renaux B, Saifeddine M, Daines MO, Boitano S, Hollenberg MD. Cockroach allergen serine proteinases: Isolation, sequencing and signalling via proteinase-activated receptor-2. Clin Exp Allergy 47: 946–960, 2017. doi: 10.1111/cea.12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boitano S, Flynn AN, Sherwood CL, Schulz SM, Hoffman J, Gruzinova I, Daines MO. Alternaria alternata serine proteases induce lung inflammation and airway epithelial cell activation via PAR2. Am J Physiol Lung Cell Mol Physiol 300: L605–L614, 2011. doi: 10.1152/ajplung.00359.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Homma T, Kato A, Bhushan B, Norton JE, Suh LA, Carter RG, Gupta DS, Schleimer RP. Role of Aspergillus fumigatus in triggering protease-activated receptor-2 in airway epithelial cells and skewing the cells toward a T-helper 2 bias. Am J Respir Cell Mol Biol 54: 60–70, 2016. doi: 10.1165/rcmb.2015-0062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matsuwaki Y, Wada K, White T, Moriyama H, Kita H. Alternaria fungus induces the production of GM-CSF, interleukin-6 and interleukin-8 and calcium signaling in human airway epithelium through protease-activated receptor 2. Int Arch Allergy Immunol 158, Suppl 1: 19–29, 2012. doi: 10.1159/000337756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dulon S, Leduc D, Cottrell GS, D'Alayer J, Hansen KK, Bunnett NW, Hollenberg MD, Pidard D, Chignard M. Pseudomonas aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell Mol Biol 32: 411–419, 2005. doi: 10.1165/rcmb.2004-0274OC. [DOI] [PubMed] [Google Scholar]
- 126.Nomura K, Obata K, Keira T, Miyata R, Hirakawa S, Takano K, Kohno T, Sawada N, Himi T, Kojima T. Pseudomonas aeruginosa elastase causes transient disruption of tight junctions and downregulation of PAR-2 in human nasal epithelial cells. Respir Res 15: 21, 2014. doi: 10.1186/1465-9921-15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ubl JJ, Grishina ZV, Sukhomlin TK, Welte T, Sedehizade F, Reiser G. Human bronchial epithelial cells express PAR-2 with different sensitivity to thermolysin. Am J Physiol Lung Cell Mol Physiol 282: L1339–L1348, 2002. doi: 10.1152/ajplung.00392.2001. [DOI] [PubMed] [Google Scholar]
- 128.Bakele M, Lotz-Havla AS, Jakowetz A, Carevic M, Marcos V, Muntau AC, Gersting SW, Hartl D. An interactive network of elastase, secretases, and PAR-2 protein regulates CXCR1 receptor surface expression on neutrophils. J Biol Chem 289: 20516–20525, 2014. doi: 10.1074/jbc.M114.575803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dwyer TM, Farley JM. Human neutrophil elastase releases two pools of mucinlike glycoconjugate from tracheal submucosal gland cells. Am J Physiol Lung Cell Mol Physiol 278: L675–L682, 2000. doi: 10.1152/ajplung.2000.278.4.L675. [DOI] [PubMed] [Google Scholar]
- 130.Liu C, Li Q, Zhou X, Kolosov VP, Perelman JM. Human airway trypsin-like protease induces mucin5AC hypersecretion via a protease-activated receptor 2-mediated pathway in human airway epithelial cells. Arch Biochem Biophys 535: 234–240, 2013. doi: 10.1016/j.abb.2013.02.013. [DOI] [PubMed] [Google Scholar]
- 131.Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, DeFea KA, Bouvier M, Hollenberg MD. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem 286: 24638–24648, 2011. doi: 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yamaguchi R, Yamamoto T, Sakamoto A, Narahara S, Sugiuchi H, Yamaguchi Y. Neutrophil elastase enhances IL-12p40 production by lipopolysaccharide-stimulated macrophages via transactivation of the PAR-2/EGFR/TLR4 signaling pathway. Blood Cells Mol Dis 59: 1–7, 2016. doi: 10.1016/j.bcmd.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 133.Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, Bunnett NW. Neutrophil elastase activates protease-activated receptor-2 (PAR2) and transient receptor potential vanilloid 4 (TRPV4) to cause inflammation and pain. J Biol Chem 290: 13875–13887, 2015. doi: 10.1074/jbc.M115.642736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhou J, Perelman JM, Kolosov VP, Zhou X. Neutrophil elastase induces MUC5AC secretion via protease-activated receptor 2. Mol Cell Biochem 377: 75–85, 2013. doi: 10.1007/s11010-013-1572-3. [DOI] [PubMed] [Google Scholar]
- 135.Chignard M, Pidard D. Neutrophil and pathogen proteinases versus proteinase-activated receptor-2 lung epithelial cells: more terminators than activators. Am J Respir Cell Mol Biol 34: 394–398, 2006. doi: 10.1165/rcmb.2005-0250TR. [DOI] [PubMed] [Google Scholar]
- 136.Kessler E, Spierer A. Inhibition by phosphoramidon of Pseudomonas aeruginosa elastase injected intracorneally in rabbit eyes. Curr Eye Res 3: 1075–1078, 1984. doi: 10.3109/02713688409011755. [DOI] [PubMed] [Google Scholar]
- 137.Morihara K, Tsuzuki H. Phosphoramidon as an inhibitor of elastase from Pseudomonas aeruginosa. Jpn J Exp Med 48: 81–84, 1978. [PubMed] [Google Scholar]
- 138.Pearson JP, Passador L, Iglewski BH, Greenberg EP. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci USA 92: 1490–1494, 1995. doi: 10.1073/pnas.92.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Babu PB, Chidekel A, Shaffer TH. Protein composition of apical surface fluid from the human airway cell line Calu-3: effect of ion transport mediators. Clin Chim Acta 347: 81–88, 2004. doi: 10.1016/j.cccn.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 140.Dubin RF, Robinson SK, Widdicombe JH. Secretion of lactoferrin and lysozyme by cultures of human airway epithelium. Am J Physiol Lung Cell Mol Physiol 286: L750–L755, 2004. doi: 10.1152/ajplung.00326.2003. [DOI] [PubMed] [Google Scholar]
- 141.Ramesh Babu PB, Chidekel A, Utidjian L, Shaffer TH. Regulation of apical surface fluid and protein secretion in human airway epithelial cell line Calu-3. Biochem Biophys Res Commun 319: 1132–1137, 2004. doi: 10.1016/j.bbrc.2004.05.101. [DOI] [PubMed] [Google Scholar]
- 142.DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol 148: 1267–1281, 2000. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115: 455–465, 2002. [DOI] [PubMed] [Google Scholar]
- 144.Messenger SW, Falkowski MA, Groblewski GE. Ca(2)(+)-regulated secretory granule exocytosis in pancreatic and parotid acinar cells. Cell Calcium 55: 369–375, 2014. doi: 10.1016/j.ceca.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lin KW, Park J, Crews AL, Li Y, Adler KB. Protease-activated receptor-2 (PAR-2) is a weak enhancer of mucin secretion by human bronchial epithelial cells in vitro. Int J Biochem Cell Biol 40: 1379–1388, 2008. doi: 10.1016/j.biocel.2007.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Concepcion AR, Feske S. Regulation of epithelial ion transport in exocrine glands by store-operated Ca(2+) entry. Cell Calcium 63: 53–59, 2017. doi: 10.1016/j.ceca.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Concepcion AR, Vaeth M, Wagner LE II, Eckstein M, Hecht L, Yang J, Crottes D, Seidl M, Shin HP, Weidinger C, Cameron S, Turvey SE, Issekutz T, Meyts I, Lacruz RS, Cuk M, Yule DI, Feske S. Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function. J Clin Invest 126: 4303–4318, 2016. doi: 10.1172/JCI89056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sharma A, Ramena G, Yin Y, Premkumar L, Elble RC. CLCA2 is a positive regulator of store-operated calcium entry and TMEM16A. PLoS One 13: e0196512, 2018. doi: 10.1371/journal.pone.0196512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Boedtkjer DM, Kim S, Jensen AB, Matchkov VM, Andersson KE. New selective inhibitors of calcium-activated chloride channels—T16A(inh) -A01, CaCC(inh) -A01 and MONNA—what do they inhibit? Br J Pharmacol 172: 4158–4172, 2015. doi: 10.1111/bph.13201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bradley E, Fedigan S, Webb T, Hollywood MA, Thornbury KD, McHale NG, Sergeant GP. Pharmacological characterization of TMEM16A currents. Channels (Austin) 8: 308–320, 2014. doi: 10.4161/chan.28065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu Y, Zhang H, Huang D, Qi J, Xu J, Gao H, Du X, Gamper N, Zhang H. Characterization of the effects of Cl(−) channel modulators on TMEM16A and bestrophin-1 Ca(2)(+) activated Cl(−) channels. Pflugers Arch 467: 1417–1430, 2015. doi: 10.1007/s00424-014-1572-5. [DOI] [PubMed] [Google Scholar]
- 152.Catalan MA, Kondo Y, Pena-Munzenmayer G, Jaramillo Y, Liu F, Choi S, Crandall E, Borok Z, Flodby P, Shull GE, Melvin JE. A fluid secretion pathway unmasked by acinar-specific TMEM16A gene ablation in the adult mouse salivary gland. Proc Natl Acad Sci USA 112: 2263–2268, 2015. doi: 10.1073/pnas.1415739112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kondo Y, Nakamoto T, Jaramillo Y, Choi S, Catalan MA, Melvin JE. Functional differences in the acinar cells of the murine major salivary glands. J Dent Res 94: 715–721, 2015. doi: 10.1177/0022034515570943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kunzelmann K, Kongsuphol P, Aldehni F, Tian Y, Ousingsawat J, Warth R, Schreiber R. Bestrophin and TMEM16-Ca(2+) activated Cl(−) channels with different functions. Cell Calcium 46: 233–241, 2009. doi: 10.1016/j.ceca.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 155.Ousingsawat J, Martins JR, Schreiber R, Rock JR, Harfe BD, Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J Biol Chem 284: 28698–28703, 2009. doi: 10.1074/jbc.M109.012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Romanenko VG, Catalan MA, Brown DA, Putzier I, Hartzell HC, Marmorstein AD, Gonzalez-Begne M, Rock JR, Harfe BD, Melvin JE. TMEM16A encodes the Ca2+-activated Cl− channel in mouse submandibular salivary gland acinar cells. J Biol Chem 285: 12990–13001, 2010. doi: 10.1074/jbc.M109.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, Baggs R, Shull GE, Melvin JE. Loss of hyperpolarization-activated Cl(−) current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem 277: 23604–23611, 2002. doi: 10.1074/jbc.M202900200. [DOI] [PubMed] [Google Scholar]
- 158.Srivastava A, Romanenko VG, Gonzalez-Begne M, Catalan MA, Melvin JE. A variant of the Ca2+-activated Cl channel Best3 is expressed in mouse exocrine glands. J Membr Biol 222: 43–54, 2008. doi: 10.1007/s00232-008-9098-4. [DOI] [PubMed] [Google Scholar]
- 159.Liu H, Farley JM Sr.. Prostaglandin E2 enhances acetylcholine-induced, Ca2+-dependent ionic currents in swine tracheal mucous gland cells. J Pharmacol Exp Ther 322: 501–513, 2007. doi: 10.1124/jpet.107.120154. [DOI] [PubMed] [Google Scholar]
- 160.Murakami K, Tamada T, Nara M, Muramatsu S, Kikuchi T, Kanehira M, Maruyama Y, Ebina M, Nukiwa T. Toll-like receptor 4 potentiates Ca2+-dependent secretion of electrolytes from swine tracheal glands. Am J Respir Cell Mol Biol 45: 1101–1110, 2011. doi: 10.1165/rcmb.2011-0020OC. [DOI] [PubMed] [Google Scholar]
- 161.Muramatsu S, Tamada T, Nara M, Murakami K, Kikuchi T, Kanehira M, Maruyama Y, Ebina M, Nukiwa T, Ichinose M. Flagellin/TLR5 signaling potentiates airway serous secretion from swine tracheal submucosal glands. Am J Physiol Lung Cell Mol Physiol 305: L819–L830, 2013. doi: 10.1152/ajplung.00053.2013. [DOI] [PubMed] [Google Scholar]
- 162.Phillips JE, Hey JA, Corboz MR. Tachykinin NK3 and NK1 receptor activation elicits secretion from porcine airway submucosal glands. Br J Pharmacol 138: 254–260, 2003. doi: 10.1038/sj.bjp.0705029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sasamori K, Sasaki T, Takasawa S, Tamada T, Nara M, Irokawa T, Shimura S, Shirato K, Hattori T. Cyclic ADP-ribose, a putative Ca2+-mobilizing second messenger, operates in submucosal gland acinar cells. Am J Physiol Lung Cell Mol Physiol 287: L69–L78, 2004. doi: 10.1152/ajplung.00454.2003. [DOI] [PubMed] [Google Scholar]
- 164.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70: 273–299, 2008. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 165.Joo NS, Saenz Y, Krouse ME, Wine JJ. Mucus secretion from single submucosal glands of pig. Stimulation by carbachol and vasoactive intestinal peptide. J Biol Chem 277: 28167–28175, 2002. doi: 10.1074/jbc.M202712200. [DOI] [PubMed] [Google Scholar]
- 166.Wine JJ, Joo NS, Choi JY, Cho HJ, Krouse ME, Wu JV, Khansaheb M, Irokawa T, Ianowski J, Hanrahan JW, Cuthbert AW, Tran KV. Measurement of fluid secretion from intact airway submucosal glands. Methods Mol Biol 742: 93–112, 2011. doi: 10.1007/978-1-61779-120-8_6. [DOI] [PubMed] [Google Scholar]
- 167.Pack RJ, Richardson PS, Smith IC, Webb SR. The functional significance of the sympathetic innervation of mucous glands in the bronchi of man. J Physiol 403: 211–219, 1988. doi: 10.1113/jphysiol.1988.sp017246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Amaral MD, Beekman JM. Activating alternative chloride channels to treat CF: friends or foes?: report on the meeting of the Basic Science Working Group in Dubrovnik, Croatia. J Cyst Fibros 19: 11–15, 2020. [DOI] [PubMed] [Google Scholar]
- 169.Li H, Salomon JJ, Sheppard DN, Mall MA, Galietta LJ. Bypassing CFTR dysfunction in cystic fibrosis with alternative pathways for anion transport. Curr Opin Pharmacol 34: 91–97, 2017. doi: 10.1016/j.coph.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 170.Gentzsch M, Mall MA. Ion channel modulators in cystic fibrosis. Chest 154: 383–393, 2018. doi: 10.1016/j.chest.2018.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li S, Koziol-White C, Jude J, Jiang M, Zhao H, Cao G, Yoo E, Jester W, Morley MP, Zhou S, Wang Y, Lu MM, Panettieri RA Jr, Morrisey EE. Epithelium-generated neuropeptide Y induces smooth muscle contraction to promote airway hyperresponsiveness. J Clin Invest 126: 1978–1982, 2016. doi: 10.1172/JCI81389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu Y, Andiappan AK, Lee B, Ho R, Lim TK, Kuan WS, Goh DY, Mahadevan M, Sim TB, Wang Y, Van Bever HP, Rotzschke O, Larbi A, Ng TP. Neuropeptide Y associated with asthma in young adults. Neuropeptides 59: 117–121, 2016. doi: 10.1016/j.npep.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 173.Lu Y, Van Bever HP, Lim TK, Kuan WS, Goh DY, Mahadevan M, Sim TB, Ho R, Larbi A, Ng TP. Obesity, asthma prevalence and IL-4: roles of inflammatory cytokines, adiponectin and neuropeptide Y. Pediatr Allergy Immunol 26: 530–536, 2015. doi: 10.1111/pai.12428. [DOI] [PubMed] [Google Scholar]
- 174.Makinde TO, Steininger R, Agrawal DK. NPY and NPY receptors in airway structural and inflammatory cells in allergic asthma. Exp Mol Pathol 94: 45–50, 2013. doi: 10.1016/j.yexmp.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schwarz H, Villiger PM, von Kempis J, Lotz M. Neuropeptide Y is an inducible gene in the human immune system. J Neuroimmunol 51: 53–61, 1994. doi: 10.1016/0165-5728(94)90128-7. [DOI] [PubMed] [Google Scholar]
- 176.Baraniuk JN, Castellino S, Lundgren JD, Goff J, Mullol J, Merida M, Shelhamer JH, Kaliner MA. Neuropeptide Y (NPY) in human nasal mucosa. Am J Respir Cell Mol Biol 3: 165–173, 1990. doi: 10.1165/ajrcmb/3.2.165. [DOI] [PubMed] [Google Scholar]
- 177.Chanez P, Springall D, Vignola AM, Moradoghi-Hattvani A, Polak JM, Godard P, Bousquet J. Bronchial mucosal immunoreactivity of sensory neuropeptides in severe airway diseases. Am J Respir Crit Care Med 158: 985–990, 1998. doi: 10.1164/ajrccm.158.3.9608104. [DOI] [PubMed] [Google Scholar]
- 178.Fang SY, Shen CL, Ohyama M. Presence of neuropeptides in human nasal polyps. Acta Otolaryngol 114: 324–328, 1994. doi: 10.3109/00016489409126064. [DOI] [PubMed] [Google Scholar]
- 179.Luts A, Uddman R, Alm P, Basterra J, Sundler F. Peptide-containing nerve fibers in human airways: distribution and coexistence pattern. Int Arch Allergy Immunol 101: 52–60, 1993. doi: 10.1159/000236498. [DOI] [PubMed] [Google Scholar]
- 180.Walker JK, DeFea KA. Role for beta-arrestin in mediating paradoxical beta2AR and PAR2 signaling in asthma. Curr Opin Pharmacol 16: 142–147, 2014. doi: 10.1016/j.coph.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yoshida T, Matsuwaki Y, Asaka D, Hama T, Otori N, Moriyama H. The expression of protease-activated receptors in chronic rhinosinusitis. Int Arch Allergy Immunol 161, Suppl 2: 138–146, 2013. doi: 10.1159/000350386. [DOI] [PubMed] [Google Scholar]
- 182.Caminero A, McCarville JL, Galipeau HJ, Deraison C, Bernier SP, Constante M, Rolland C, Meisel M, Murray JA, Yu XB, Alaedini A, Coombes BK, Bercik P, Southward CM, Ruf W, Jabri B, Chirdo FG, Casqueiro J, Surette MG, Vergnolle N, Verdu EF. Duodenal bacterial proteolytic activity determines sensitivity to dietary antigen through protease-activated receptor-2. Nat Commun 10: 1198, 2019. doi: 10.1038/s41467-019-09037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kida Y, Higashimoto Y, Inoue H, Shimizu T, Kuwano K. A novel secreted protease from Pseudomonas aeruginosa activates NF-kappaB through protease-activated receptors. Cell Microbiol 10: 1491–1504, 2008. doi: 10.1111/j.1462-5822.2008.01142.x. [DOI] [PubMed] [Google Scholar]
- 184.Kida Y, Taira J, Yamamoto T, Higashimoto Y, Kuwano K. EprS, an autotransporter protein of Pseudomonas aeruginosa, possessing serine protease activity induces inflammatory responses through protease-activated receptors. Cell Microbiol 15: 1168–1181, 2013. doi: 10.1111/cmi.12106. [DOI] [PubMed] [Google Scholar]
- 185.Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, Duan K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm Med 16: 174, 2016. doi: 10.1186/s12890-016-0339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Langan KM, Kotsimbos T, Peleg AY. Managing Pseudomonas aeruginosa respiratory infections in cystic fibrosis. Curr Opin Infect Dis 28: 547–556, 2015. [Erratum in Curr Opin Infect Dis 29: 98, 2016]. doi: 10.1097/QCO.0000000000000217. [DOI] [PubMed] [Google Scholar]
- 187.Moraes TJ, Martin R, Plumb JD, Vachon E, Cameron CM, Danesh A, Kelvin DJ, Ruf W, Downey GP. Role of PAR2 in murine pulmonary pseudomonal infection. Am J Physiol Lung Cell Mol Physiol 294: L368–L377, 2008. doi: 10.1152/ajplung.00036.2007. [DOI] [PubMed] [Google Scholar]
- 188.Suzuki A, Iwata J. Molecular regulatory mechanism of exocytosis in the salivary glands. Int J Mol Sci 19: 3208, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Jiang Y, Yau MK, Kok WM, Lim J, Wu KC, Liu L, Hill TA, Suen JY, Fairlie DP. Biased signaling by agonists of protease activated receptor 2. ACS Chem Biol 12: 1217–1226, 2017. doi: 10.1021/acschembio.6b01088. [DOI] [PubMed] [Google Scholar]
- 190.Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K, Hollenberg MD. Agonist-biased signaling via proteinase activated receptor-2: differential activation of calcium and mitogen-activated protein kinase pathways. Mol Pharmacol 76: 791–801, 2009. doi: 10.1124/mol.109.055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, Kocan M, Sostegni S, Haerteis S, Baraznenok V, Henderson I, Lindstrom E, Guerrero-Alba R, Valdez-Morales EE, Liedtke W, McIntyre P, Vanner SJ, Korbmacher C, Bunnett NW. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J Biol Chem 289: 27215–27234, 2014. [Erratum in J Biol Chem 289: 35858, 2014]. doi: 10.1074/jbc.M114.599712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Aoki M, Yamaguchi R, Yamamoto T, Ishimaru Y, Ono T, Sakamoto A, Narahara S, Sugiuchi H, Hirose E, Yamaguchi Y. Granulocyte-macrophage colony-stimulating factor primes interleukin-13 production by macrophages via protease-activated receptor-2. Blood Cells Mol Dis 54: 353–359, 2015. doi: 10.1016/j.bcmd.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 193.Lieu T, Savage E, Zhao P, Edgington-Mitchell L, Barlow N, Bron R, Poole DP, McLean P, Lohman RJ, Fairlie DP, Bunnett NW. Antagonism of the proinflammatory and pronociceptive actions of canonical and biased agonists of protease-activated receptor-2. Br J Pharmacol 173: 2752–2765, 2016. doi: 10.1111/bph.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Nadel JA. Role of enzymes from inflammatory cells on airway submucosal gland secretion. Respiration 58, Suppl 1: 3–5, 1991. doi: 10.1159/000195961. [DOI] [PubMed] [Google Scholar]
- 195.Maizieres M, Kaplan H, Millot JM, Bonnet N, Manfait M, Puchelle E, Jacquot J. Neutrophil elastase promotes rapid exocytosis in human airway gland cells by producing cytosolic Ca2+ oscillations. Am J Respir Cell Mol Biol 18: 32–42, 1998. doi: 10.1165/ajrcmb.18.1.2841. [DOI] [PubMed] [Google Scholar]
- 196.Botham A, Guo X, Xiao YP, Morice AH, Compton SJ, Sadofsky LR. Palmitoylation of human proteinase-activated receptor-2 differentially regulates receptor-triggered ERK1/2 activation, calcium signalling and endocytosis. Biochem J 438: 359–367, 2011. doi: 10.1042/BJ20101958. [DOI] [PubMed] [Google Scholar]
- 197.Compton SJ. Glycosylation and proteinase-activated receptor function. Drug Dev Res 59: 350–354, 2003. doi: 10.1002/ddr.10303. [DOI] [Google Scholar]
- 198.Compton SJ, Renaux B, Wijesuriya SJ, Hollenberg MD. Glycosylation and the activation of proteinase-activated receptor 2 (PAR(2)) by human mast cell tryptase. Br J Pharmacol 134: 705–718, 2001. doi: 10.1038/sj.bjp.0704303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Compton SJ, Sandhu S, Wijesuriya SJ, Hollenberg MD. Glycosylation of human proteinase-activated receptor-2 (hPAR2): role in cell surface expression and signalling. Biochem J 368: 495–505, 2002. doi: 10.1042/BJ20020706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tamashiro E, Xiong G, Anselmo-Lima WT, Kreindler JL, Palmer JN, Cohen NA. Cigarette smoke exposure impairs respiratory epithelial ciliogenesis. Am J Rhinol Allergy 23: 117–122, 2009. doi: 10.2500/ajra.2009.23.3280. [DOI] [PubMed] [Google Scholar]
- 201.Cao X, Lin H, Muskhelishvili L, Latendresse J, Richter P, Heflich RH. Tight junction disruption by cadmium in an in vitro human airway tissue model. Respir Res 16: 30, 2015. doi: 10.1186/s12931-015-0191-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Eum SY, Jaraki D, Bertrand L, Andras IE, Toborek M. Disruption of epithelial barrier by quorum-sensing N-3-(oxododecanoyl)-homoserine lactone is mediated by matrix metalloproteinases. Am J Physiol Gastrointest Liver Physiol 306: G992–G1001, 2014. doi: 10.1152/ajpgi.00016.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Schwarzer C, Fu Z, Patanwala M, Hum L, Lopez-Guzman M, Illek B, Kong W, Lynch SV, Machen TE. Pseudomonas aeruginosa biofilm-associated homoserine lactone C12 rapidly activates apoptosis in airway epithelia. Cell Microbiol 14: 698–709, 2012. doi: 10.1111/j.1462-5822.2012.01753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Schwarzer C, Ravishankar B, Patanwala M, Shuai S, Fu Z, Illek B, Fischer H, Machen TE. Thapsigargin blocks Pseudomonas aeruginosa homoserine lactone-induced apoptosis in airway epithelia. Am J Physiol Cell Physiol 306: C844–C855, 2014. doi: 10.1152/ajpcell.00002.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Vikstrom E, Tafazoli F, Magnusson KE. Pseudomonas aeruginosa quorum sensing molecule N-(3 oxododecanoyl)-l-homoserine lactone disrupts epithelial barrier integrity of Caco-2 cells. FEBS Lett 580: 6921–6928, 2006. doi: 10.1016/j.febslet.2006.11.057. [DOI] [PubMed] [Google Scholar]
- 206.Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev 54: 1359–1371, 2002. doi: 10.1016/s0169-409x(02)00144-8. [DOI] [PubMed] [Google Scholar]
- 207.Golovkine G, Faudry E, Bouillot S, Voulhoux R, Attree I, Huber P. VE-cadherin cleavage by LasB protease from Pseudomonas aeruginosa facilitates type III secretion system toxicity in endothelial cells. PLoS Pathog 10: e1003939, 2014. doi: 10.1371/journal.ppat.1003939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wojtukiewicz MZ, Hempel D, Sierko E, Tucker SC, Honn KV. Protease-activated receptors (PARs)—biology and role in cancer invasion and metastasis. Cancer Metastasis Rev 34: 775–796, 2015. doi: 10.1007/s10555-015-9599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Eichel K, von Zastrow M. Subcellular organization of GPCR signaling. Trends Pharmacol Sci 39: 200–208, 2018. doi: 10.1016/j.tips.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Nauntofte B. Regulation of electrolyte and fluid secretion in salivary acinar cells. Am J Physiol Gastrointest Liver Physiol 263: G823–G837, 1992. doi: 10.1152/ajpgi.1992.263.6.G823. [DOI] [PubMed] [Google Scholar]