

Abstract
Despite modest improvement in patient outcomes from recent advances in pharmacotherapy targeting fibrogenic signaling pathways, idiopathic pulmonary fibrosis (IPF) remains a major unsolved clinical problem. One reason for this is that available antifibrotic agents slow down but do not arrest fibrotic progression. To arrest fibrotic progression, its obligatory drivers need to be identified. We previously discovered that fibrogenic mesenchymal progenitor cells (MPCs) are key drivers of fibrotic progression in IPF, serving as cells of origin for disease-mediating myofibroblasts. IPF MPCs have high levels of nuclear S100A4, which interacts with the proteasome to promote p53 degradation and self-renewal. However, the mechanism underlying S100A4 accumulation in the nucleus of IPF MPCs remains unknown. Here we show that hyaluronan (HA) is present in the fibroblastic focus together with CD44-expressing MPCs and that ligation of CD44 by HA triggers S100A4 nuclear translocation to support IPF MPC self-renewal. The mechanism involves HA-mediated formation of a CD44/S100A4/transportin 1 complex, which promotes S100A4 nuclear import. In a humanized mouse model of pulmonary fibrosis, IPF MPC fibrogenicity was significantly attenuated by 1) knockdown of CD44 or 2) introduction of an S100A4 mutant construct that prevents S100A4 nuclear import. These data indicate that signaling through the HA/CD44/S100A4 axis is an integral component of IPF MPC fibrogenicity.
Keywords: CD44, hyaluronan, idiopathic pulmonary fibrosis, mesenchymal progenitor cells, S100A4
INTRODUCTION
Despite modest improvement in patient outcomes from recent advances in pharmacotherapy targeting fibrogenic signaling pathways, idiopathic pulmonary fibrosis (IPF) remains a major unsolved clinical problem (1, 2). One reason for this is that available antifibrotic agents slow down but do not arrest fibrotic progression (3). To arrest fibrotic progression, its obligatory drivers need to be identified. We have made several recent discoveries studying primary cells and extracellular matrix (ECM) from patients with IPF that illuminate a path forward toward understanding mechanisms driving the relentless progression of fibrosis following disease initiation (4–8). Fibrosis progression involves both cell-autonomous and ECM-driven mechanisms, and as in cancer, there is cooperation between autonomous cells and their microenvironment (9–18).
Cell-autonomous fibrogenicity was established when we discovered fibrogenic mesenchymal progenitor cells (MPCs) in the lungs of patients with IPF that are one source of IPF fibroblasts (6). Based on analysis of the MPC transcriptome from that initial report (GEO GSE97038), we determined that S100A4, also termed metastasin based on its ability to promote cancer cell metastasis (19–26), mediates IPF MPC fibrogenicity (7). We found that IPF MPCs have increased levels of nuclear S100A4, which promotes p53 degradation, IPF MPC self-renewal, and expansion of the fibrogenic MPC population. Importantly, xenografted IPF MPCs convert a bleomycin mouse model of self-limited, regressing lung fibrosis to a model of sustained fibrosis in an S100A4-dependent manner. Knockdown of S100A4 blocked IPF MPC fibrogenicity, proving that IPF MPCs are intrinsically fibrogenic and that S100A4 is necessary for IPF MPCs to manifest this fibrogenicity. However, the mechanism underlying the nuclear accumulation of S100A4 in IPF MPCs remains unknown and the precise mechanism(s) by which S100A4 mediates IPF MPC fibrogenicity remains unclear.
Our prior mass spectrometry studies to define the S100A4 nuclear interactome revealed that in addition to interacting with members of the proteasome, S100A4 exists in a complex with the HA receptor CD44 and transportin 1 within the nucleus of self-renewing IPF MPCs (7). CD44 nuclear import is mediated via transportin 1 (27). Prior studies have demonstrated that hyaluronan (HA) ligation of CD44 promotes CD44 internalization and its nuclear translocation (27, 28). We found that HA is enriched in the active cellular front of the fibroblastic focus together with CD44-expressing IPF MPCs (7). Although HA has been linked to IPF fibroblast motility/invasion (29), its role in regulating IPF MPCs has not been explored. Studies in cancer have revealed that HA is an integral component of cancer stromal tissue, and high HA levels are linked to cancer progression and poor patient survival (30, 31). The ability of HA to promote cancer progression relates to its capacity to promote cancer stem cell self-renewal. HA supports the stemness of cancer stem cells through its interactions with CD44 (32). This suggested to us that the HA-CD44 interaction may promote IPF MPC self-renewal. We have discovered that HA ligation of CD44 promotes S100A4 nuclear import and IPF MPC self-renewal. The mechanism involves HA/CD44-mediated formation of a CD44/S100A4/transportin 1 complex that accumulates in the nucleus. We further demonstrate that as IPF MPCs differentiate to IPF fibroblasts, nuclear S100A4 levels diminish, whereas cytoplasmic levels increase. This shift in S100A4 location during IPF MPC differentiation is associated with acquisition of a motile phenotype. We demonstrate that acquisition of the motile phenotype is S100A4 dependent and is associated with cytoplasmic S100A4 interaction with nonmuscle myosin IIA. To further elucidate the role of S100A4 in mediating IPF MPC fibrogenicity, we used an S100A4 mutant construct in which the nuclear localization sequence had been mutated, preventing S100A4 nuclear import. This mutation also impairs S100A4 association with nonmuscle myosin IIA and IPF fibroblast motility. Overexpression of the S100A4 mutant construct in IPF MPCs abrogated their ability to drive progression of fibrosis in a mouse xenograft model of fibrotic progression. Taken together, our data indicate that S100A4 mediates several cellular processes in IPF MPCs and their progeny that are crucial for fibrotic progression in a subcellular location-dependent manner.
MATERIALS AND METHODS
Study Approval
De-identified patient samples were obtained by our tissue procurement service (Bionet) under a waiver of written informed consent from the University of Minnesota Institutional Review Board (University of Minnesota IRB ID: 1504M68341). Animal protocols were approved and conducted in accordance with the University of Minnesota Institutional Animal Care and Use Committee regulations (Approval No. 1706–34890 A).
Primary Mesenchymal Cell Lines
Four primary lung mesenchymal cell lines were established from patients fulfilling diagnostic criteria for IPF including a pathological diagnosis of usual interstitial pneumonia (33). Cell lines were derived from lungs, characterized as mesenchymal cells, and cultivated as previously described (6, 7).
Isolation of Mesenchymal Progenitor Cells
IPF and control mesenchymal progenitor cells were isolated from primary IPF and control mesenchymal cell cultures at passage 0 (initial isolate before subcultivation) through passage 4. To isolate progenitors, primary IPF and control mesenchymal cells were labeled with mouse anti-human SSEA4 antibody conjugated to Alexa Fluor 647 (Clone MC-813-70; Cat. No. 560796; BD Biosciences, Franklin Lake, NJ) and mouse anti-human SSEA1 conjugated to PE (Clone MC480; Cat. No. 560142; BD Biosciences), as we previously described (34). Cells were sorted on a FACSAria Cell Sorter (BD Biosciences). Cells that were SSEA4+ and SSEA1− (relative to mouse IgG3 κ isotype control conjugated to Alexa Fluor 647 and mouse IgM κ isotype control conjugated to PE, respectively) (clone J606, Cat. No. 560803 and clone G155-228, Cat. No. 555584; BD Biosciences) were collected. To generate sufficient numbers of MPCs for the in vivo mouse studies, the SSEA4+ MPCs were expanded by culture in DMEM + 10% FCS for 7 days before use. The resulting MPC cultures were analyzed for SSEA4 expression by flow cytometry and for colony formation in vitro. Ninety-seven percent of day 7 MPCs were SSEA4+ and the cells formed colonies in methylcellulose, indicating retention of progenitor self-renewal properties.
Derivation of Progeny from Mesenchymal Progenitor Cells
IPF mesenchymal progenitor cells were isolated from IPF primary mesenchymal cell cultures by flow cytometry selecting for SSEA4+ cells. Progenitors were placed into tissue culture dishes and allowed to propagate and differentiate under tightly standardized conditions (DMEM + 10% FBS, 37°C, 10% CO2) for 21 days. The resultant cell population was designated IPF progeny (i.e., IPF fibroblasts).
Q-PCR
Analysis of S100A4 and HAS2 gene expression was conducted by quantitative polymerase chain reaction (Q-PCR) as previously described (7). Total RNA was isolated and reverse transcribed using a Taqman Reverse Transcriptase Reagent Kit (Roche) and primed with random hexamers. Primer sequences were selected using NCBI Primer-BLAST. Real-time PCR (Q-PCR) was performed using a LightCycler FastStart DNA MasterPLUS SYBR Green I Kit (Roche). Primer sequences were as follows: S100A4 forward: 5′-GTACGTGTTGATCCTGACTGCTGTCATGG-3′; S100A4 reverse: 5′-TCATTTCTTCCTGGGCTGCTTATCTGGG-3′. HAS2 forward: 5′- TGAACAAAACAGTTGCCCTTT-3′; HAS2 reverse: 5′-TTCCCATCTATGACCATGACAA-3′.
Samples were quantified at the log-linear portion of the curve using LightCycler analysis software and compared with an external calibration standard curve.
Self-Renewal Assay
Single cell suspensions of SSEA4+ IPF MPCs were incorporated into methylcellulose gels (StemCell Technologies, Inc., Vancouver, Canada) and maintained in Mesenchymal Stem Cell Serum-Free Medium Cell Therapy Systems (MSC SFM CTS; Thermo Scientific/Gibco, Rockford, IL) (37°C, 5% CO2, 1 wk). To assess the effect of HA on IPF MPC colony formation, the cells were incorporated into methylcellulose gels and then cultured in MSC SFM CTS containing various concentrations of 60-, 200-, and 1,000-kDa HA (Sodium Hyaluronate, Lifecore Biomedical, Chaska, MN). Enumeration of colonies was performed microscopically, and colony size was quantified by ImageJ.
Plasmids/Constructs
For gain of S100A4 function, we overexpressed S100A4 using a wild-type S100A4 construct (S100A4-GFP-tagged construct RG203035; Origene, Rockville, MD) in a lentiviral vector. Empty vector served as the control. For loss of S100A4 function, S100A4 was knocked down using S100A4 shRNA (pGIPZS100A4 shRNA; Addgene). Scrambled shRNA served as the control. To create the S100A4 nuclear localization sequence mutant construct, the S100A4 nuclear localization sequence 98PRKK101 was changed to 98PRAA101 by site-directed mutagenesis using wild-type S100A4 cDNA as a template, as previously described (35). The CD44 nuclear localization sequence mutant construct was constructed by site-directed mutagenesis using wild-type CD44S isoform cDNA as a template, as previously described (28). The putative nuclear localization sequence 292RRCGQKKK300 was changed to 292AAACGQAAA300. The mutant constructs were verified by DNA sequencing. Cells were transduced using a lentiviral vector containing mutant, wild-type, or empty vector constructs.
Western Blot and Immunoprecipitation
Western blots were performed as previously described (6–8). For immunoprecipitation, nuclear fractions were isolated by NE-PER nuclear and cytoplasmic extraction reagents. The samples were centrifuged at 12,000 g for 15 min at 4°C, and the lysates were precleared for 1 h at 4°C with protein A/G beads and immunoprecipitated for 2 h at 4°C with the appropriate primary antibody. The samples were processed for Western blot analysis. Primary antibodies used included S100A4 (Millipore, Cat. No. 07–2274), p53 (Santa Cruz, Cat. No. B1519), transportin 1 (Millipore, Cat. No. 05–1515), CD44 (Biolegend, Cat. No. 103006), and nonmuscle myosin IIA (NMMIIA) (Cell Signaling, Cat. No. 3403). Uncropped images of Western blots are provided in Supplemental Data (see https://doi.org/10.6084/m9.figshare.14132114).
Immunohistochemistry and Hyaluronan Staining on IPF Lung Tissue
Immunohistochemistry was performed on 4-µm paraffin-embedded serial-sectioned IPF lung tissue (n = 4 patients with IPF) and mounted on polylysine-coated slides. The sections were deparaffinized in xylene, rehydrated through a graded methanol series, quenched with 0.3% hydrogen peroxide in methanol, and immersed in a 98°C water bath for 30 min in citrate buffer (pH 6.0) for antigen retrieval. Sections were placed in 5% normal horse serum (Jackson Immunoresearch, West Grove, PA) to block nonspecific binding of secondary antibodies. A multiplex immunohistochemistry kit was used for antigen detection according to the manufacturer’s instructions (MULTIVIEW IHC Kit ADI-950-101-0001; Enzo Life Sciences, Farmingdale, NY). The tissue specimens were incubated overnight (18–20 h, 4°C) with the following primary antibodies: rabbit anti-human CD44 monoclonal antibody (1:800) (Cat. No. M3370, Spring Bioscience, Pleasanton, CA) and anti-human SSEA4 antibody (1:100) (Clone MC-813-70, Cat. No. 330402, Biolegend, San Diego, CA). Specimens were cover-slipped with a Prolong Antifade Kit (Invitrogen/Molecular Probes) and stored overnight at room temperature without light before image analysis. To assess proteoglycan distribution, including HA, in the IPF lung, Alcian Blue staining (Alcian Blue Stain Kit, American MasterTech Scientific, Lodi, CA) was performed according to the manufacturer’s instructions. HA staining using the hyaluronic acid-binding protein probe (Biotinylated HABP, Cat. No. 385911-50UG, Millipore, Billerica, MA) was used to assess HA staining patterns in the IPF lung. Deparaffinized sections were treated with 3% hydrogen peroxide for 10 min, 100% Background sniper for 1 h, 1 µg/mL biotinylated HABP for 16 h at 4°C, and streptavidin-HRP (Covance, SIG-32254) for 1 h. Tissue was developed with diaminobenzidine chromogen for 5 min, counterstained with hematoxylin, and cover-slipped. As a test of signal specificity, deparaffinized tissue was treated with hyaluronidase (16 U/mL, 30 min) (Calbiochem, Cat. No.. 389561) in a 37°C humidified oven before HABP treatment. To visualize IPF MPC pericellular HA, the cells were stained with biotinylated HABP. HABP was detected using streptavidin conjugated with Alexa Fluor 488 (Thermo Fisher Scientific).
Mouse Xenograft Model of Fibrotic Progression
To assess the role of S100A4 and CD44 in regulating the fibrogenicity of IPF MPCs in vivo, we utilized an IPF MPC-based mouse model of fibrotic progression as we previously described (7). NOD/SCID/IL2rγ/B2M (NSG) male and female mice (Jackson Laboratories), average 10 wk of age, were administered intratracheal bleomycin (1.25 U/kg) to establish self-limited lung fibrosis. To test the role of S100A4 in regulating IPF MPC fibrogenicity, 2 wk following bleomycin administration, IPF MPCs transduced with the S100A4 AA mutant construct, wild-type S100A4, or empty vector were suspended in PBS (106 cells/100 µL) and injected via the tail vein into the mice following our published protocol (7). Alternatively, to examine the role of CD44 in regulating IPF MPC fibrogenicity, IPF MPCs transduced with CD44 shRNA or scrambled shRNA were administered to the mice. Mice were euthanized 4 wk after adoptive transfer of human cells and the lungs were harvested. Collagen content was quantified in left lung tissue by the Sircol assay and served as the primary end point (7). The presence of lung fibrotic lesions by histological analysis served as the secondary end point. Histological (H&E, trichrome, and HABP staining) and immunohistochemical analyses were performed on paraffin-embedded and frozen right lung tissue. Cells positive for human procollagen (anti-human procollagen type I antibody, 1:500; Cat. No. 64409; Abcam; Cambridge, MA) were identified as human. IHC using the human procollagen antibody and S100A4 antibody (1:2,000; Cat. No. 07–2274; Millipore, Billerica, MA) was performed to assess the distribution of S100A4-expressing cells with human procollagen I-expressing cells.
RNA Sequencing Data
We used previously generated bulk RNA sequencing data (6) and single-cell RNA sequencing data (36). The complete datasets and protocols are deposited in the GEO (Accession No. GSE97038) and BioProject repository (Accession No. PRJNA641647), respectively.
Statistics
Comparisons of data among experiments were performed using the two-tailed Student’s t test. Experiments were independently replicated a minimum of three times. Data are expressed as means ± SE. P < 0.05 was considered significant.
RESULTS
CD44-Expressing MPCs Are Codistributed with Hyaluronan in the Active Fibrotic Front of Fibroblastic Foci
The IPF fibroblastic focus is a polarized structure containing a focus core and an active fibrotic front (7). The focus core consists of noncycling myofibroblasts actively synthesizing collagen. The active fibrotic front is a highly cellular region composed of cycling fibrogenic MPCs and their transit amplifying progeny, as identified by Ki67, SSEA4, and S100A4 expression (7). This suggests that the active fibrotic front harboring IPF MPCs can be conceptualized as a fibrogenic niche. The microenvironmental niche in which stem/progenitor cells reside has been shown to be a critical factor supporting stem/progenitor cell self-renewal. HA, a major determinant of tissue viscosity (34, 37), is an integral matrix component of the stem cell niche that regulates stemness and stem/progenitor cell self-renewal (38). HA is also an integral component of cancer stromal tissue, and high HA levels are linked to cancer progression and poor patient survival (39–45). Based on this information, we analyzed HA expression in the fibroblastic focus. We first performed Alcian Blue staining, which identifies glycosaminoglycans including HA. The fibroblastic focus stained intensely for glycosaminoglycans (Fig. 1A). We next performed IHC staining using the HA probe HABP. HABP staining demonstrated abundant HA in all regions of the fibroblastic focus (Fig. 1, B and C; brown stain), with the staining most intense in the focus core and less intense in the active fibrotic front. To verify the authenticity of the HA signal, the tissue was first treated with hyaluronidase and then stained using HABP. Hyaluronidase eliminated HABP staining, proving that abundant HA is present in the fibroblastic focus (Fig. 1D). This suggested to us that the presence of HA in the IPF fibroblastic focus may support the expansion of the fibrogenic mesenchymal progenitor cell pool, thus serving as an integral component of what we are designating the fibrogenic niche.
Figure 1.
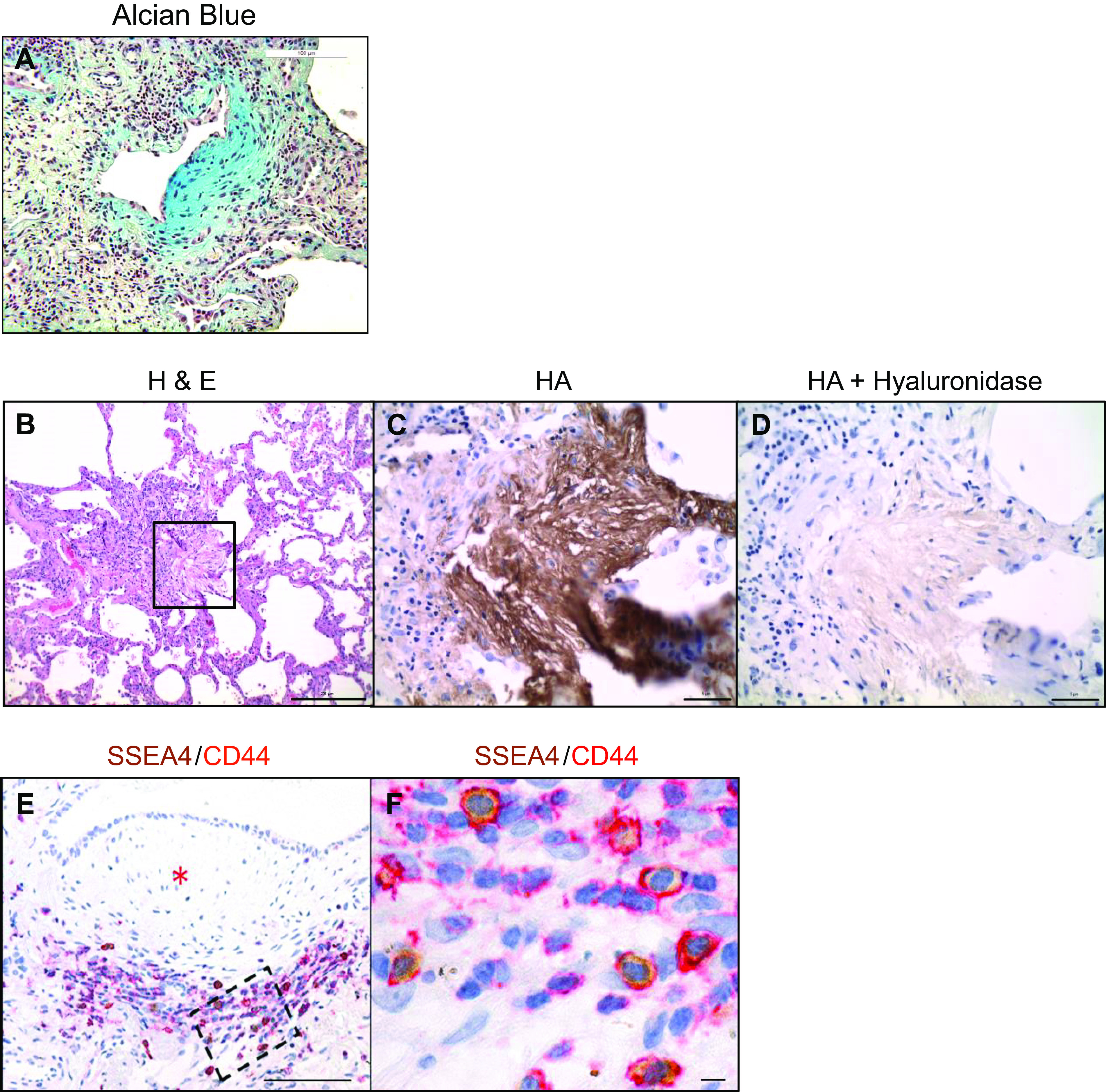
CD44-expressing IPF MPCs are codistributed with hyaluronan in the active fibrotic front of fibroblastic foci. A: Alcian Blue staining of a fibroblastic focus. Scale bar: 100 µm. B: H&E staining of a fibroblastic focus. Scale bar: 200 µm. C and D: high-power images of boxed region in B showing HABP staining (brown stain) (C) and hyaluronidase treatment followed by HABP staining (D). Scale bars: 5 µm. E: IHC double staining showing SSEA4-positive (brown) MPCs in the active fibrotic front expressing CD44. *Denotes focus (myofibroblast) core. Scale bar: 100 µm. F: high-power image of boxed region in active fibrotic front from E. Scale bar: 5 µm. HABP, hyaluronic acid-binding protein; H&E, hematoxylin-eosin; IHC, immunohistochemistry; IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells.
CD44 is a major HA receptor, and high levels of CD44 support cancer stem cell self-renewal (30). Therefore, we performed IHC double staining for CD44 and SSEA4 to examine CD44 and SSEA4 expression in the fibroblastic focus. CD44-expressing cells were found concentrated in the active fibrotic front, with few CD44-expressing cells in the focus (myofibroblast) core. Consistent with our prior IHC results (7), SSEA4-expressing MPCs resided at the perimeter of the focus core in the active fibrotic front. IHC double staining indicated that the SSEA4+ MPCs expressed CD44 (Fig. 1, E and F). These findings are consistent with our single-cell mRNA sequencing data, indicating that SSEA4+ IPF MPCs highly express CD44 (36). Taken together, our IHC studies demonstrate codistribution of HA with CD44-expressing MPCs in the active fibrotic front of the fibroblastic focus, suggesting that HA/CD44 interactions could be regulating IPF MPC function.
HA Ligation of CD44 Promotes IPF MPC Self-Renewal and S100A4 Nuclear Translocation
We have previously shown that the nucleus of IPF MPCs contains high levels of S100A4 that interact with the proteasome to promote p53 degradation and self-renewal (7). However, the mechanism by which S100A4 accumulates in the nucleus remains unclear. Since IPF MPCs exist in an ECM containing abundant HA, we first examined whether exogenous HA promotes IPF MPC cell self-renewal. HA size, which is a key determinant of its function, can vary greatly from large polymers to smaller fragments (46). Prior work indicates that in response to bleomycin-induced lung injury, there is an accumulation of smaller-molecular-weight (MW) HA fragments, whose persistence is associated with unremitting inflammation, impaired lung repair, and a profibrotic response (47, 48). Because there are an absence of data concerning the size(s) and concentration of HA present in IPF lung tissue during the course of the disease process, we first examined the effect of a range of HA sizes and concentrations on IPF MPC self-renewal. We tested various concentrations of low (60 kDa), intermediate (200 kDa), and high (1,000 kDa) MW forms of HA on self-renewal using a colony-forming assay. We found that the 60- and 200-kDa forms of exogenous HA increased IPF MPC self-renewal in a concentration-dependent manner (Fig. 2A). The 1,000-kDa form of HA did not significantly increase IPF MPC colony number at any concentration tested. We also quantified colony size of cells cultured with 800 µg/mL of 60-, 200-, or 1,000-kDa HA (Fig. 2B). Both low- and high-MW HA demonstrated a statistically significant increase in colony size. Because 60-kDa HA had the most robust effect on IPF MPC colony formation, we utilized 60-kDa HA at a concentration of 800 µg/mL for the remainder of experiments unless specified.
Figure 2.

HA ligation of CD44 promotes IPF MPC self-renewal and S100A4 nuclear translocation. A and B: IPF MPCs incorporated into methylcellulose gels were treated with various concentrations of 60-, 200-, and 1,000-kDa HA. Colony number (A) and size (B) were quantified at day 7 (n = 2 cell lines tested, IPF422, IPF429). C: IPF MPCs were treated with Hermes 3 CD44-activating antibody. S100A4 and p53 nuclear (nucl) and cytoplasmic (cyto) levels were quantified as a function of time by Western blot analysis. GAPDH and lamin served as loading controls (n = 2 cell lines tested, IPF422, IPF429; densitometry values summarizing Western blot data shown in right-hand graph). D: IPF MPCs were treated with HA (HA+) or no HA (HA−). Shown is Western blot analysis of nuclear fractions for S100A4 and p53. Lamin was used as a loading control (n = 2 cell lines tested, IPF422, IPF429; densitometry values summarizing Western blot data shown in graph below). E: IPF MPCs were pretreated with CD44-blocking antibody (BRIC 235; 5 µg/mL) or isotype control and then exposed to HA. Shown is Western blot analysis of nuclear fractions for S100A4, p53, and lamin (n = 2 cell lines, IPF422, IPF429; densitometry values summarizing Western blot data shown in graph below). Data are expressed as means ± SE. P values in B were determined by two-tailed Student’s t test. All experiments were performed using two cell lines with n ≥ 2 independent experiments for each experimental condition, except data generated by Western blot analyses were reproduced in three independent experiments. HA, hyaluronan; IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells.
Since IPF MPCs express CD44 and exist in a HA-rich environment, and HA promotes MPC self-renewal, we next examined whether ligation of the HA receptor CD44 would trigger S100A4 nuclear accumulation, thereby promoting self-renewal. Ligation of CD44 by the CD44-activating antibody Hermes 3 promoted S100A4 nuclear translocation within minutes of agonist addition followed by a decrease in p53 levels (Fig. 2C). Similarly, addition of HA led to a marked increase in nuclear S100A4 protein levels and a decrease in p53 levels (Fig. 2D). Because HA can bind to other cell surface receptors such as RHAMM, TLR2, and TLR4 (49), we tested the effect of CD44-blocking antibody on the ability of HA to promote S100A4 nuclear accumulation. Inhibition of HA/CD44 interaction by CD44-blocking antibody prevented HA-induced S100A4 nuclear translocation and preserved nuclear p53 levels (Fig. 2E). Since S100A4 promotes degradation of the negative cell cycle regulator p53, our data support the concept that HA ligation of CD44 triggers S100A4 nuclear accumulation, promoting p53 degradation and IPF MPC self-renewal.
Hyaluronan Synthesis and Release Promotes S100A4 Nuclear Accumulation and Supports IPF MPC Self-Renewal
In cancer, high levels of HA synthesis by HA-synthesizing enzymes and HA release from the cell surface support acquisition of cancer stem cell features and promote tumor aggressiveness (39, 44). Since HA is enriched in the fibroblastic focus and HA ligation of CD44 triggers S100A4 nuclear accumulation and promotes IPF MPC self-renewal, we examined HA synthesis and HA release in IPF MPCs. We found that IPF MPCs express hyaluronic acid synthase 2 (HAS2), a major HAS isoform expressed by other lung mesenchymal cells (29, 50). Consistent with this, visualization of cell surface/pericellular HA using the HABP probe revealed abundant cell surface/pericellular HA associated with IPF MPCs (Supplemental Fig. S1A; all Supplemental figures are available at https://doi.org/10.6084/m9.figshare.12988889). Treatment with hyaluronidase reduced the amount of cell surface/pericellular HA on IPF MPC (Supplemental Fig. S1B). We next examined HA synthesis as IPF MPCs differentiate to IPF fibroblasts. We have previously shown that IPF MPCs differentiate into IPF fibroblasts over 21 days in culture (6). HAS2 mRNA expression decreased 71% as IPF MPCs differentiated to IPF fibroblasts (Fig. 3A). In accord with this, IPF MPCs released relatively large amounts of HA, which decreased as the MPCs differentiated to IPF fibroblasts (Fig. 3B). Taken together, these data suggest that relatively high levels of HA synthesis and release support IPF MPC self-renewal.
Figure 3.
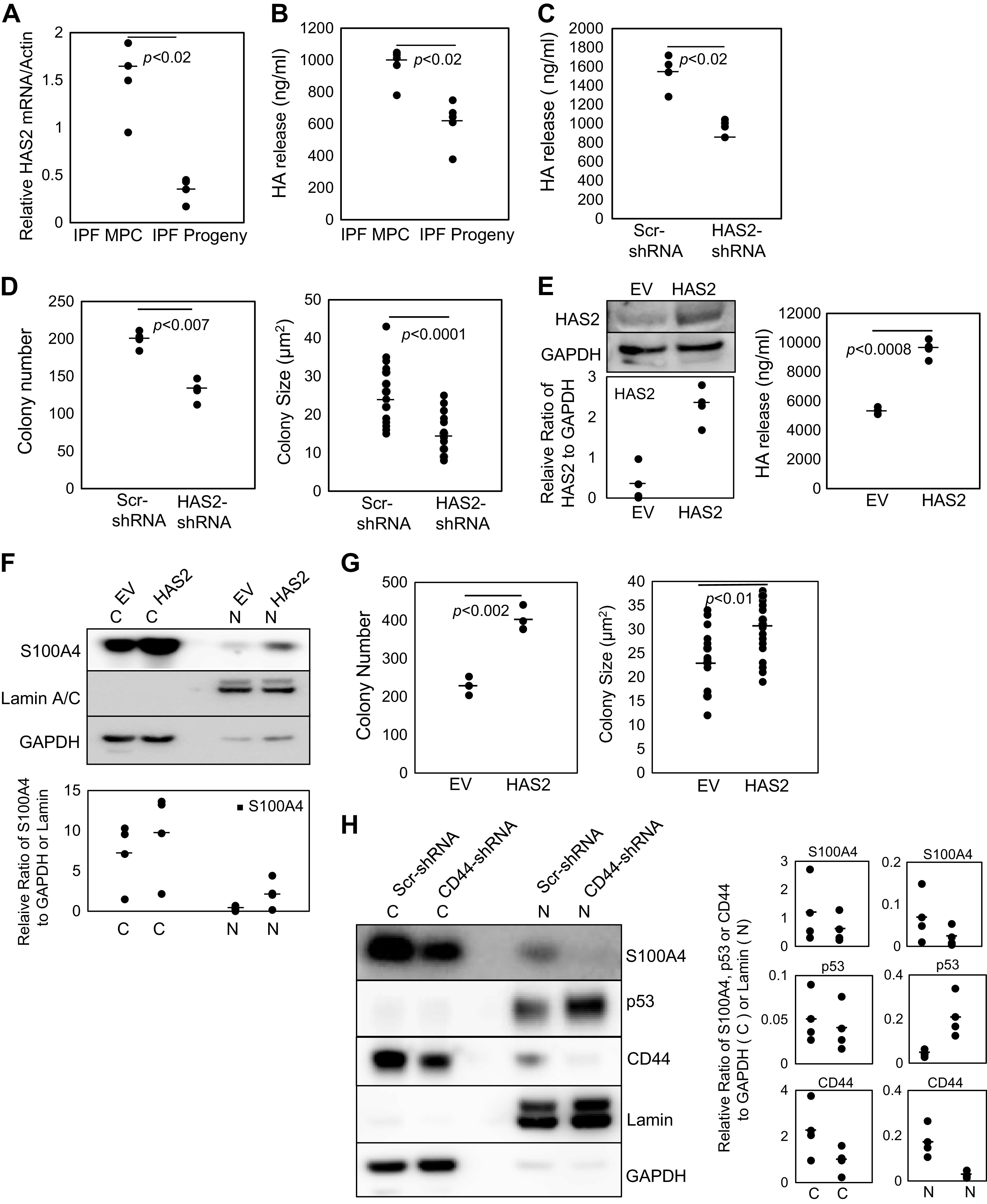
Hyaluronan synthesis and release promotes S100A4 nuclear accumulation and supports IPF MPC self-renewal. A: HAS2 expression was quantified in IPF MPCs and day 21 IPF MPC progeny by Q-PCR (n = 3 cell lines tested, IPF422, IPF424, IPF429). B: the amount of HAS2 released by IPF MPCs and IPF MPC progeny into the culture medium was quantified by ELISA (n = 4 cell lines tested, IPF422, IPF424, IPF429, IPF442). C and D: HAS2 was knocked down in IPF MPCs using HAS2 shRNA (HAS2-shRNA). Scrambled shRNA (Scr-shRNA) served as the control. HA release into cell culture medium was quantified by ELISA (n = 2 cell lines tested, IPF422, IPF429) (C). self-renewal was assessed in the colony-forming assay (n = 2 cell lines tested, IPF422, IPF429) (D). E–G: HAS2 was overexpressed in IPF MPCs (HAS2). Empty vector (EV) served as the control. HAS2 protein levels were quantified in IPF MPCs (E, left; densitometry values summarizing Western blot data shown in graph below). HAS2 released by IPF MPCs into the culture medium was quantified by ELISA (E, right). Nuclear (N) and cytoplasmic (C) S100A4 levels were quantified by Western blot analysis (F; densitometry values summarizing Western blot data shown in graph below). Lamin and GAPDH served as loading controls. Self-renewal was quantified using the colony-forming assay (G) (for E–G, n = 2 cell lines tested, IPF422, IPF424). H: HAS2 was overexpressed in IPF MPCs in which CD44 had been knocked down (CD44-shRNA). IPF MPCs overexpressing HAS2 and transduced with scrambled shRNA (Scr-shRNA) served as the control. S100A4, p53, and CD44 protein levels were quantified in nuclear (N) and cytoplasmic (C) fractions. Lamin and GAPDH served as loading controls (n = 2 cell lines tested, IPF422, IPF424; densitometry values summarizing Western blot data shown in right-hand graph). Data generated by Western blot analyses were reproduced in three independent experiments. Data are expressed as means ± SE. P values in A–E and G were determined by two-tailed Student’s t test. All experiments were performed using two to four cell lines with n ≥ 2 independent experiments for each experimental condition, except data generated by Western blot analyses were reproduced in three independent experiments. HA, hyaluronan; IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells; Q-PCR, quantitative polymerase chain reaction.
To examine the role of HA in regulating MPC function, we knocked down HAS2 in IPF MPCs. Knockdown of HAS2 in IPF MPCs reduced HA release from the cells and decreased self-renewal (Fig. 3, C and D). Conversely, overexpression of HAS2 in IPF MPCs increased HAS2 levels and increased the amount of HA released from the cells (Fig. 3E, left and right, respectively). This correlated with an increase in nuclear S100A4 levels and IPF MPC self-renewal (Fig. 3, F and G). Since ligation of CD44 by HA promotes S100A4 nuclear translocation, we knocked down CD44 in IPF MPCs overexpressing HAS2. CD44 knockdown inhibited S100A4 nuclear accumulation in IPF MPCs overexpressing HAS2 while increasing nuclear p53 protein levels (Fig. 3H). Together, these data support the concept that active HA synthesis and subsequent release into the pericellular microenvironment enhance the HA/CD44 interaction promoting CD44-dependent S100A4 nuclear accumulation, serving as an important mechanism of IPF MPC self-renewal.
HA/CD44 Interaction Promotes the Formation of a CD44/S100A4/transportin 1 Complex That Promotes S100A4 Nuclear Translocation and IPF MPC Self-Renewal
We next sought to determine the mechanism by which HA ligation of CD44 facilitates S100A4 nuclear accumulation. Prior work has shown that upon ligation, full-length CD44 is internalized and transported to the nucleus by transportin 1 (27), and internalized full-length CD44 can form a complex with other proteins, facilitating their nuclear translocation (28). Interestingly, our prior mass spectrometry studies identified both CD44 and transportin 1 as members of the S100A4 nuclear interactome (Table 1) (7). This suggested to us that in response to CD44 ligation by HA, cytoplasmic CD44 and S100A4 form a complex with transportin 1 and comigrate to the nucleus. To confirm our prior mass spectrometry results, we treated IPF MPCs with Hermes 3 CD44-activating antibody and examined for the formation of an S100A4/CD44/transportin 1 complex. Ligation of CD44 by CD44-activating antibody promoted the formation of a CD44/S100A4/transportin 1 complex found in both the cytoplasmic and nuclear fractions (Fig. 4A). These data support the hypothesis that S100A4 nuclear import stimulated by the HA/CD44 interaction is CD44 dependent.
Table 1.
S100A4 nuclear interactome: cell proliferation, adhesion, and migration-related proteins
| ACAN | CHD1 | FBXL13 | LASP1 | PRPF6 |
| ACTC1 | CKAP4 | FKBP1A | MAGI3 | PTBP1 |
| ACTG1 | CLIC4 | FLG | MAPK8IP3 | RAB1A |
| ACTN1 | CMYA5 | FLNA | MECOM | RAC1 |
| ACTR2 | COL1A2 | FUS | MIF | S100A4 |
| AHNAK | CORIN | GBF1 | MSN | SMARCA5 |
| ALB | COX5 | GCN1 | MYL9 | STOM |
| ALDOA | CRACR2A | GFAP | MYO1A | TNPO1 |
| ANXA1 | CTSB | GIPC1 | NEFM | TRP |
| ATP1A1 | DDX1 | GNAI2 | NOP56 | TSC22D1 |
| BSG | DES | GNAS | NUP54 | TTN |
| BZW2 | DHX9 | GOLGB1 | OVGP1 | TUBB |
| CARD10 | DOCK11 | GOT2 | PARP1 | VCP |
| CAV1 | DSP | GSTP1 | PCNA | VIM |
| CAVIN1 | EEF1B2 | H2AX | PDE4D | VPS13C |
| CCNB3 | EEF2 | HBB | PEX1 | |
| CCT5 | EIF4A2 | HDAC1 | PHB | |
| CD109 | EMD | HNRNPA2B1 | PKM | |
| CD44 | ENO1 | HSP90B1 | PLXNA4 | |
| CDSN | ERAL1 | ITGA3 | PPP2R1A |
Figure 4.
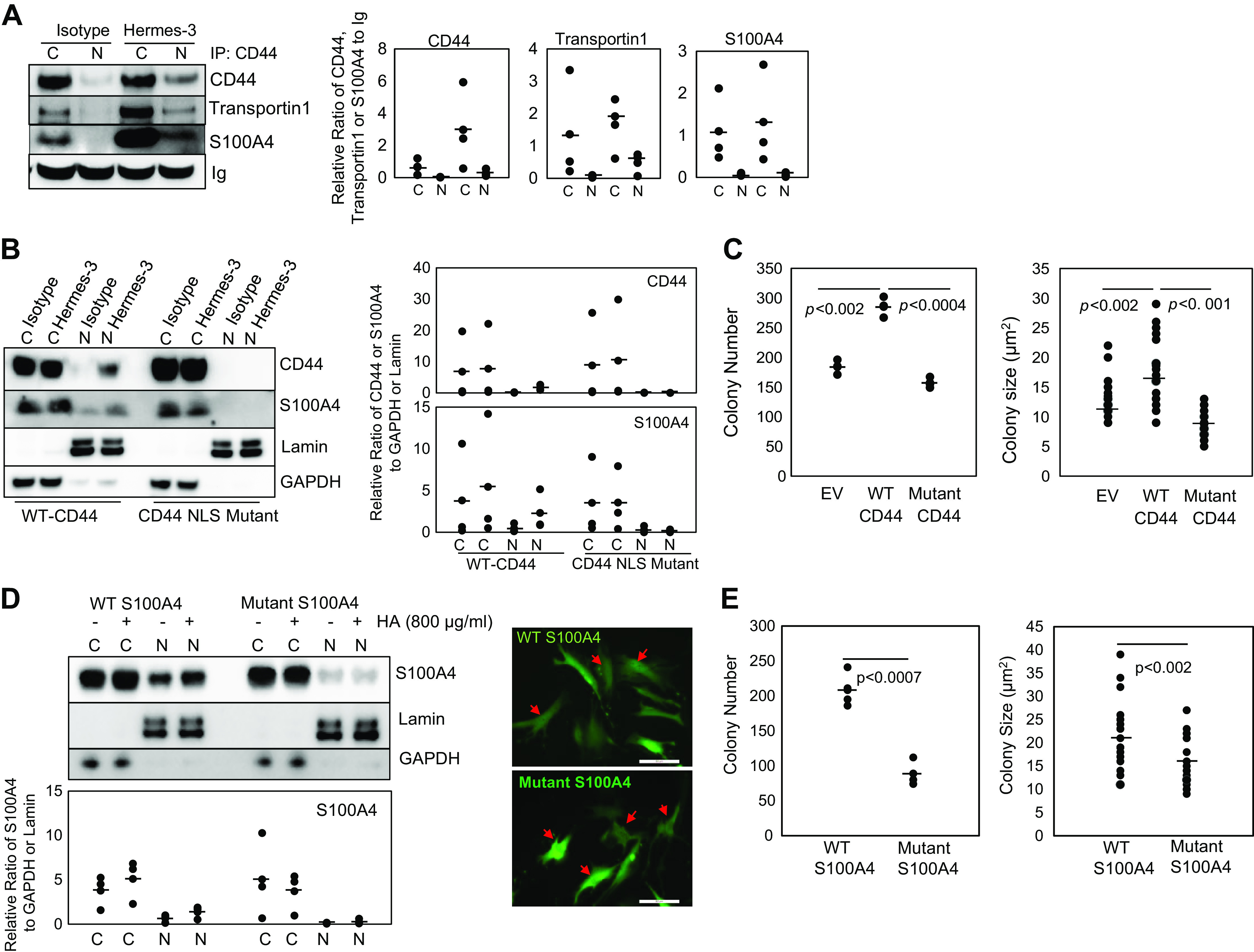
HA/CD44 interaction promotes the formation of a CD44/S100A4/transportin 1 complex that promotes S100A4 nuclear translocation and IPF MPC self-renewal. A: IPF MPCs were treated with Hermes 3 CD44-activating antibody or isotype control antibody. CD44 was immunoprecipitated from nuclear (N) and cytoplasmic (C) fractions and Western blot analysis for CD44, transportin 1, and S100A4 was performed (n = 2 cell lines tested, IPF422, IPF424; densitometry values summarizing Western blot data shown in right-hand graph). Ig served as the loading control. B: IPF MPCs were transduced with the CD44 NLS mutant construct (CD44 NLS mutant) or wild-type CD44 (WT-CD44). The cells were then treated with Hermes 3 CD44-activating antibody or isotype control antibody. CD44 and S100A4 protein levels in nuclear (N) and cytoplasmic (C) fractions were quantified by Western blot analysis. Lamin and GAPDH served as loading controls (n = 2 cell lines tested, IPF422, IPF429; densitometry values summarizing Western blot data shown in right-hand graph). C: IPF MPCs transduced with the CD44 mutant NLS construct, wild-type CD44, or empty vector (EV) were incorporated into methylcellulose gels to which HA had been added and colony number and size were quantified at day 7 (n = 2 cell lines tested, IPF422, IPF429). D: IPF MPCs transduced with the AA mutant S100A4 or wild-type S100A4 construct were treated with HA, and S100A4 in nuclear (N) and cytoplasmic (C) fractions were quantified by Western blot analysis (left). Lamin and GAPDH served as loading controls (n = 2 cell lines tested, IPF422, IPF424; densitometry values summarizing Western blot data shown in graph below). Confocal microscopic images of cells transduced with wild-type S100A4 or mutant S100A4 construct (right ). Scale bar: 50 µm. E: IPF MPCs expressing mutant or wild-type S100A4 were cultured in methylcellulose gels containing HA. Colony number and size were quantified at day 7 (n = 2 cell lines tested, IPF422, IPF424). Data generated by Western blot analyses were reproduced in three independent experiments. Data are expressed as means ± SE. P values in C and E were determined by two-tailed Student’s t test. All experiments were performed using two cell lines with n ≥ 2 independent experiments for each experimental condition, except data generated by Western blot analyses were reproduced in three independent experiments. HA, hyaluronan; IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells; NLS, nuclear localization sequence.
CD44 contains a nuclear localization sequence (NLS) 292RRRCGQKKK300. Prior studies have shown that a mutant CD44 NLS construct (292AAACGQAAA300) when engaged becomes internalized but fails to enter the nucleus (28). Therefore, to further analyze the role of CD44 in S100A4 nuclear import, we used the CD44 mutant NLS construct. Ectopically expressed wild-type CD44 (standard isoform) served as the control. We found that overexpression of the mutant CD44 NLS construct prevented the accumulation of both CD44 and S100A4 within the nucleus of IPF MPCs in response to treatment with Hermes 3 CD44-activating antibody (Fig. 4B). In contrast, cells transduced with wild-type CD44 displayed both S100A4 and wild-type CD44 (full-length CD44) nuclear accumulation in response to Hermes 3. We next examined the effect of the CD44 mutant NLS construct on IPF MPC self-renewal in response to treatment with HA. We found that IPF MPCs transduced with the CD44 NLS mutant construct displayed reduced colony formation compared with cells containing empty vector (Fig. 4C). In contrast, cells transduced with the CD44 wild-type construct displayed robust colony formation. These data suggest that the mutant CD44 construct, which is unable to enter the nucleus, sequesters S100A4 in the cytosol. Our data support the concept that HA interaction with CD44 triggers the formation of a CD44/S100A4/transportin 1 complex that facilitates S100A4 nuclear import and IPF MPC self-renewal.
Similar to CD44, S100A4 contains a monopartite nuclear localization sequence 98PRKK101 at its C-terminus (23). To verify the role of nuclear S100A4 in regulating IPF MPC self-renewal, we mutated the two C-terminal lysine residues to alanine and analyzed S100A4 nuclear translocation in response to HA in cells transduced with the mutated S100A4 construct. We found that in response to HA treatment, the mutant S100A4 construct largely failed to be transported to the nucleus (Fig. 4D, left) and remained in the cytoplasmic compartment (Fig. 4D, left and right bottom). In contrast, when IPF MPCs transduced with wild-type S100A4 were treated with HA, S100A4 underwent nuclear translocation (Fig. 4D, left) with high levels in the nucleus (Fig. 4D, right top). Importantly, IPF MPCs transduced with the mutant S100A4 construct displayed decreased colony formation in response to HA treatment compared with the control, indicating that inhibition of S100A4 nuclear transport decreased HA-mediated IPF MPC self-renewal (Fig. 4E). Taken together, our data indicate that HA ligation of CD44 promotes S100A4 nuclear accumulation and IPF MPC self-renewal. We demonstrate that the mechanism involves HA/CD44-mediated formation of a CD44/S100A4/transportin 1 complex the facilitates S100A4 nuclear import.
As IPF MPCs Differentiate to IPF Fibroblasts, S100A4 Shifts to a Cytoplasmic Location and Complexes with Nonmuscle Myosin IIA and the Cells Acquire an S100A4-Dependent Motile Phenotype
We have previously shown that IPF MPCs are fibrogenic and that S100A4 mediates their fibrogenicity. However, the precise mechanism(s) by which S100A4 mediates IPF MPC fibrogenicity is incompletely understood. In our prior report, we demonstrated that IPF MPCs proliferate and differentiate to IPF MPC progeny over 21 days in culture and acquire the IPF fibroblast phenotype (6). Our data indicate that as IPF MPCs differentiate to IPF MPC progeny, HAS2 expression and HA release decrease. Since high levels of HAS2 expression and HA release support S100A4 nuclear accumulation in IPF MPCs, we next sought to determine what happens to S100A4 nuclear and cytoplasmic levels as IPF MPC differentiate to IPF MPC progeny. We found that as IPF MPCs differentiate to IPF MPC progeny, nuclear S100A4 levels diminish, whereas cytoplasmic S100A4 levels increase (Fig. 5A). In IPF MPC progeny, cytoplasmic S100A4 is complexed with nonmuscle myosin IIA (Fig. 5B). Prior studies have shown that the interaction of S100A4 with nonmuscle myosin IIA stimulates cell motility by promoting the formation of forward cellular protrusions (24). We found that shifting of S100A4 into the cytoplasmic compartment as IPF MPCs differentiate correlates with acquisition of a motile phenotype (Fig. 5C). Knockdown of S100A4 in IPF MPC progeny abrogates their motility (Fig. 5D). Mutation of the two C-terminal lysine residues of S100A4 has been shown to decrease the affinity of S100A4 for nonmuscle myosin IIA, increasing the equilibrium dissociation constant (Kd) >20-fold from 91 to1,600 nM (35). Consistent with this, overexpression of the S100A4 mutant reduced S100A4 association with nonmuscle myosin IIA (Fig. 5E) and decreased cell motility (Fig. 5F) compared to cells transduced with wild-type S100A4. These data support the concept that acquisition of the motile phenotype during IPF MPC differentiation to IPF MPC progeny is S100A4 dependent and is contingent on S100A4 translocation to the cytoplasm, where it interacts with nonmuscle myosin IIA.
Figure 5.

As IPF MPCs differentiate to IPF fibroblasts, S100A4 shifts to a cytoplasmic location and the cells acquire an S100A4-dependent motile phenotype. A: IPF MPCs were allowed to expand and differentiate in culture for 21 days. S100A4 expression in nuclear (N) and cytoplasmic (C) fractions of IPF MPCs and their day 21 progeny (IPF MPC progeny) was quantified by Western blot analysis. Lamin and GAPDH served as nuclear and cytoplasmic loading controls, respectively (n = 2 cell lines tested, IPF422, IPF424; densitometry values summarizing Western blot data shown in graph below). B: S100A4 was immunoprecipitated from cell lysates obtained from IPF MPC progeny. Western blot analysis for nonmuscle myosin IIA (NMMIIA) and S100A4 was performed. Control: IP using isotype control antibody (isotype) (n = 2 cell lines tested, IPF422, IPF424; densitometry values summarizing Western blot data shown in graph below). Ig served as the loading control. C: IPF MPC and IPF MPC progeny migration was quantified using the QCM cell migration assay (n = 3 cell lines tested, IPF422, IPF424, IPF442). D: IPF MPC progeny were transduced with S100A4 or scrambled (Scr) shRNA, and migration was quantified using the QCM cell migration assay (n = 2 cell lines tested, IPF422, IPF424). E and F. IPF MPC progeny were transduced with a lentiviral vector containing the S100A4 AA mutant construct or wild-type S100A4. S100A4 was immunoprecipitated from cell lysates and Western blot analysis for nonmuscle myosin IIA (NMMIIA) and S100A4 was performed (E) (n = 2 cell lines tested, IPF422, IPF424; densitometry values summarizing Western blot data shown in right-hand graph). Ig served as the loading control. Migration was quantified using the QCM Cell Migration Assay Kit (F) (n = 2 cell lines tested, IPF422, IPF424). Data generated by Western Blot analyses were reproduced in three independent experiments. Data are expressed as means ± SE. P values in C, D, and F were determined by two-tailed Student’s t test. All experiments were performed using two to three cell lines with n ≥ 2 independent experiments for each experimental condition, except data generated by Western Blot analyses were reproduced in three independent experiments. IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells.
S100A4 and CD44 Are Necessary for IPF MPC Fibrogenicity In Vivo
Our in vitro studies indicate that S100A4 promotes IPF MPC self-renewal and population expansion in a CD44-dependent manner and that a nuclear localization mutant inhibits this effect. To test whether this was also the case in vivo, we genetically modified IPF MPCs and tested them in a mouse xenograft model of nonresolving lung fibrosis. In this model, delivery of human IPF MPCs converts the bleomycin mouse model from a self-limited regressing model to a model of sustained fibrosis in an S100A4-dependent manner (7). Importantly, our in vitro data indicate that the S100A4 AA mutant construct has antagonistic effects on both IPF MPCs and their progeny. In IPF MPCs, the mutant construct prevents S100A4 nuclear accumulation and inhibits MPC proliferation. In IPF MPC progeny, the mutant construct displays decreased affinity for cytoplasmic nonmuscle myosin IIA and inhibits their motility. Mice receiving IPF MPCs transduced with the S100A4 AA mutant had a 42% and 27% decrease in lung collagen content compared with mice receiving IPF MPCs transduced with wild-type S100A4 or with empty vector, respectively (Fig. 6A). Assessment of fibrosis and collagen deposition by H& E and trichrome staining revealed decreased collagen deposition in mice receiving cells transduced with the S100A4 mutant construct compared with mice receiving IPF MPCs transduced with wild-type S100A4 or empty vector (Fig. 6, B–G). We performed IHC analysis of serial sections to further analyze the effect of the S100A4 mutant construct on IPF MPC fibrogenesis. We used a human-specific procollagen I antibody to identify human cells expressing procollagen in the mouse lung and an S100A4 antibody to assess whether the distribution of S100A4-expressing cells was congruent with human procollagen-expressing cells. Compared with mice receiving IPF MPCs overexpressing wild-type S100A4 or empty vector, the lungs of mice receiving IPF MPCs transduced with the S100A4 mutant construct displayed fewer numbers of human cells expressing procollagen and S100A4 (Fig. 6, H–M, and Supplemental Fig. S2, A–F). Importantly, in mice receiving IPF MPCs overexpressing wild-type S100A4, S100A4 positive cells codistributed with human procollagen-expressing cells in regions of extensive fibrosis (Fig. 6, I and L, and Supplemental Fig. S2, C and D). This is consistent with our in vitro data indicating that overexpression of wild-type S100A4 robustly supports IPF MPC self-renewal, whereas overexpression of the mutant construct inhibits self-renewal and suggests that overexpression of S100A4 promoted expansion of the IPF MPC population in vivo. Since our in vitro data indicate that IPF MPCs display relatively high HA synthesis and release, we used the HABP probe to visualize HA deposition. In mice receiving IPF MPCs transduced with the mutant construct, we found reduced lung HA staining compared with mice receiving IPF MPCs transduced with wild-type S100A4 or empty vector (Fig. 6, N–P and Supplemental Fig. S2, G–I). Of note, in mice receiving IPF MPCs transduced with wild-type S100A4, we found abundant HA staining in the regions of extensive fibrosis that contained numerous human IPF MPCs expressing procollagen. However, only sparse HA staining was apparent in uninvolved regions lacking human cells expressing procollagen. (Supplemental Fig. S3). This staining pattern suggests that the human IPF MPCs are synthesizing, releasing, and depositing HA as the fibrosis evolves.
Figure 6.

Lung fibrosis is attenuated in mice receiving IPF MPCs transduced with the NLS S100A4 mutant construct. NSG mice were treated with intratracheal bleomycin (1.25 U/kg). Fourteen days later, the mice received IPF MPCs (IPF429) transduced with the S100A4 AA mutant construct, wild-type S100A4, or empty vector via tail vein injection (106 cells/100 µL). There were 10 mice/group. Lungs were harvested at the end of week 6. A: collagen content was quantified in left lungs by the Sircol assay. Data are expressed as means ± SE. P values were determined by two-tailed Student’s t test. B–P: serial 4-µm sections of right lung tissue (B–G: scale bar 500 µm; H–P: scale bar 50 µm). Representative H&E and trichrome stains assessing fibrosis and collagen deposition, respectively (B–D and E–G). IHC using an antibody recognizing human procollagen to identify human cells and assess collagen synthesis (H–J) and an S100A4 antibody to assess the distribution of S100A4- and human procollagen-expressing cells (K–M). The HABP probe was used to assess HA distribution (N–P). HA, hyaluronan; HABP, hyaluronic acid-binding protein; IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells; NLS, nuclear localization sequence.
Since S100A4 promotes IPF MPC population expansion in a CD44-dependent manner, we also examined the effect of CD44 knockdown on IPF MPC fibrogenicity. Knockdown of CD44 inhibited IPF MPC fibrogenicity. Collagen content, fibrotic areas, and collagen deposition were all markedly diminished in mice receiving IPF MPCs in which CD44 had been knocked down compared with mice receiving cells transduced with scrambled shRNA (Fig. 7, A–E). IHC analysis demonstrated few human cells expressing procollagen in mouse lungs receiving IPF MPCs transduced with CD44-shRNA (Fig. 7F). In contrast, numerous human cells expressing procollagen in the areas of extensive fibrosis in mouse lungs receiving IPF MPCs transduced with scrambled shRNA (Fig. 7G). This supports the concept that CD44 is an obligatory component of the pathway regulating IPF MPC fibrogenicity. Taken together, the in vivo data established that both CD44 and S100A4 are necessary for IPF MPC fibrogenicity in vivo.
Figure 7.

CD44 is necessary for IPF MPC fibrogenicity in vivo. NSG mice were treated with intratracheal bleomycin (1.25 U/kg). Fourteen days later, the mice received IPF MPCs (IPF422) transduced with CD44 sh-RNA or scrambled shRNA. There were five mice/group. Lungs were harvested at the end of week 6. A: collagen content quantified was quantified in left lungs by the Sircol assay. Data are expressed as means ± SE. P value was determined by two-tailed Student’s t test. B–G: serial 4-µm sections of right lung tissue (B–E: scale bar = 500 µm; F and G: scale bar = 50 µm). Representative H&E (B and D) and trichrome stains (C and E) assessing fibrosis and collagen deposition, respectively. F and G: IHC using an antibody recognizing human procollagen to identify human cells and assess collagen synthesis. Shown are high-power views corresponding to the boxed regions in C and E. IPF, idiopathic pulmonary fibrosis; MPCs, mesenchymal progenitor cells.
DISCUSSION
The most complete theory of IPF disease initiation posits that epithelial injury emerges in the aging lung due to misfolded proteins and generation of endoplasmic reticulum stress (51–53). Data in support of this theory are compelling (54). Epithelial genes are altered in IPF (55), and an extensive body of work documents epithelial injury and attrition (56, 57). Failure of alveolar epithelial stem and progenitor cells to regenerate the epithelium sets the stage for fibrosis to develop (57). Activation of the transforming growth factor-β (TGF-β) pathway ensues, which in turn signals resident mesenchymal cells to differentiate into myofibroblasts and produce ECM (58). Although this theory accounts for disease initiation, it does not fully account for the stereotypical pattern of IPF fibrotic progression. Following principles that have emerged in the field of cancer biology, we made key discoveries that motivate our experimental approach to elucidate mechanisms driving fibrosis progression in IPF (4–8). In studies comparing primary mesenchymal cells and decellularized ECM from patients with IPF with those from patient-controls, we found that fibrosis progression involves both cell-intrinsic/autonomous and ECM-driven mechanisms. As in cancer, robustness of the disease process emerges from cooperation between autonomous cells and their microenvironment to create positive feedback and feed-forward loops (9–18).
Cell-autonomous fibrogenicity was established when we discovered fibrogenic mesenchymal progenitor cells (MPCs) in the lungs of patients with IPF that serve as one source of IPF fibroblasts (6). Based on analysis of the MPC transcriptome from that initial study, we reported that S100A4 mediates IPF MPC fibrogenicity (7). We demonstrated that IPF MPCs contain high nuclear levels of S100A4, which interacts with members of the proteasome to promote p53 degradation and MPC self-renewal. However, the mechanism underlying S100A4 nuclear accumulation in IPF MPCs remained unclear. Prior work indicates that HA ligation of CD44 promotes CD44 internalization and transport to the nucleus, a process facilitated by its interaction with transportin 1 (27, 28). Our prior mass spectrometry studies defining the S100A4 nuclear interactome revealed that in addition to interacting with members of the proteasome, S100A4 exists in a complex with CD44 and transportin 1, providing a possible clue to the mechanism underlying S100A4 nuclear import (7). These data suggested that the HA/CD44 axis may be an important regulator of S100A4 subcellular localization and IPF MPC self-renewal.
We recently demonstrated that the fibroblastic focus in IPF is highly ordered with a distinct polarity, comprised by a myofibroblast-rich core synthesizing collagen and an active fibrotic front containing cycling MPCs (7). This suggests that the fibroblastic focus can be conceptualized as a fibrogenic niche that harbors and nurtures IPF MPCs and their progeny. Work identifying the stem/progenitor cell niche has determined that HA is an important component of the niche microenvironment supporting their self-renewal (34). Moreover, work in cancer has found that the cancer stromal microenvironment is enriched with HA and that HA supports cancer stem cell function (39–45). Similar to the cancer stromal microenvironment, our IHC studies demonstrate that the fibroblastic focus is enriched with HA, which codistributes with CD44-expressing IPF MPCs. Importantly, we demonstrate that HA supports IPF MPC self-renewal.
Our data support the concept that HA ligation triggers the formation of a CD44/S100A4/transportin 1 complex that mediates S100A4 nuclear import. First, we show that ligation of CD44 by HA or the CD44-activating antibody Hermes 3 promotes S100A4 nuclear translocation with minutes of agonist treatment. In contrast, inhibition of HA/CD44 interaction with CD44-blocking antibody prevented HA-induced S100A4 nuclear translocation and increased nuclear p53 levels. Since S100A4 promotes degradation of the negative cell cycle regulator p53, our data suggest that HA ligation of CD44 triggers S100A4 nuclear accumulation promoting p53 degradation and IPF MPC self-renewal. In addition, we demonstrate that the CD44-activating antibody promotes the formation of a CD44/S100A4/transportin 1 complex. To prove that CD44/S100A4 complex formation is important for S100A4 nuclear import, we used a CD44 mutant construct in which the nuclear localization sequence was mutated. Prior work has demonstrated that when ligated, the CD44 mutant construct is internalized but fails to enter the nucleus (28). Consistent with this, we demonstrate that in response to treatment with the Hermes 3 CD44-activating antibody, neither CD44 nor S100A4 accumulates in the nucleus of IPF MPCs transduced with the CD44 NLS mutant construct. In contrast, both CD44 and S100A4 accumulate in the nucleus when IPF MPCs containing the CD44 wild-type construct are treated with the CD44-activating antibody. These data provide strong evidence that the mechanism underlying high S100A4 nuclear levels in IPF MPCs involves HA ligation of CD44, which promotes the formation of a CD44/S100A4/transportin 1 complex facilitating S100A4 nuclear import.
Studies in the field of cancer have shown that high HA synthesis and release from the cell surface imparts tumorgenicity (39, 44). Tumor cells expressing high levels of both hyaluronan synthase and hyaluronidase display increased aggressiveness, indicating that HA turnover regulates tumor cell function. Prior studies have shown a role for HAS2 in IPF fibroblast migration and invasion (29). Here we demonstrate that IPF MPCs express HAS2, a major HAS isoform expressed by lung mesenchymal cells (29, 50); that HAS2 expression level decreases as IPF MPCs differentiate to IPF fibroblasts; and that HAS2 regulates IPF MPC self-renewal. Visualization of pericellular HA using the HABP probe revealed an abundant pericellular coat associated with IPF MPCs. Pericellular HA-CD44 interactions have been shown to regulate cancer cell tumorgenicity (59). We show that overexpression of HAS2 in IPF MPCs augments HA release from the cell, promotes S100A4 nuclear accumulation in a CD44-dependent manner, and increases IPF MPC self-renewal. These data suggest that HA synthesis and subsequent release into the pericellular microenvironment may be an important mechanism by which IPF MPCs maintain self-renewal.
Our data indicate that nuclear S100A4 mediates IPF MPC fibrogenicity via promoting self-renewal. Using an S100A4 nuclear localization sequence mutant construct, we demonstrate that S100A4 nuclear import is crucial for self-renewal. However, our data indicate that this is not the sole mechanism by which S100A4 mediates IPF MPC fibrogenicity. We have previously shown that IPF MPCs serve as a cell-of-origin for IPF fibroblasts (6). We have found that as the MPCs differentiate to IPF fibroblasts, nuclear S100A4 levels decrease, whereas cytoplasmic S100A4 levels increase, and S100A4 associates with nonmuscle myosin IIA. This association has been linked to cell motility. Consistent with this, we have found that IPF MPC progeny are more motile than IPF MPCs and that knockdown of S100A4 inhibits this motility. Thus, our data indicate that S100A4 is capable of mediating several cellular processes crucial for fibrotic progression in a subcellular location-dependent manner.
We have developed a mouse xenograft model of sustained interstitial lung fibrosis. In this model, mice are first treated with a low dose of bleomycin to induce lung injury. Two weeks later at the height of the self-limited bleomycin-induced injury/fibrotic response, human IPF MPCs are administered via tail vein. Four weeks after injection of IPF MPCs, the lungs are harvested. We have previously demonstrated that in mice receiving IPF MPCs, lung fibrosis is markedly worse with a fourfold increase in collagen deposition and there is extensive infiltration of human cells expressing procollagen in fibrotic regions (7). Furthermore, our in vitro studies indicate that as IPF MPCs differentiate to IPF MPC progeny over 21 days of culture, they acquire the IPF fibroblast phenotype (6). Although it is unclear what precisely happens to the IPF MPC differentiation state during the 4-wk period that the human IPF MPCs are within the bleomycin-injured mouse lung in vivo, some degree of MPC differentiation into collagen-producing IPF fibroblasts may occur. To further unravel the mechanism by which S100A4 regulates IPF MPC fibrogenicity, we used the S100A4 AA mutant construct. This construct has dual effects. In IPF MPCs, mutation of the NLS blocks S100A4 nuclear import, inhibiting self-renewal. In IPF MPC progeny, it inhibits S100A4 interaction with nonmuscle myosin IIA and disrupts motility. Using the xenograft model, here we show that the mutant S100A4 construct abrogates IPF MPC fibrogenicity, suggesting that inhibition of IPF MPC proliferation and IPF MPC progeny motility neutralizes IPF MPC fibrogenicity.
Identifying pathways critical for the maintenance and expansion of the fibrogenic IPF MPC pool within the active fibrotic front will facilitate the development of effective therapeutics that can arrest fibrotic progression.
SUPPLEMENTAL DATA
Supplemental uncropped images for figures: https://doi.org/10.6084/m9.figshare.14132114.
Supplemental Figs. S1, S2, and S3: https://doi.org/10.6084/m9.figshare.12988889.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants R01 HL125227 to C. A. Henke and R01 HL125236 to P. B. Bitterman and by funds provided by the O’Brien and Witowski families.
DISCLAIMERS
No role was played by the funding body in the design of the study, collection, analysis, and interpretation of data or in writing the manuscript.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.X. and C.A.H. conceived and designed research; H.X., J.H., K.S., L.Y., A.G., A.B., and E.R. performed experiments; H.X., C.A.H., L.Y., and P.B.B. analyzed data; H.X., C.A.H., and P.B.B. interpreted results of experiments; H.X. prepared figures; C.A.H. drafted manuscript; C.A.H., and P.B.B. edited and revised manuscript; H.X., C.A.H., and P.B.B. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the assistance of the Flow Cytometry Core Facility of the Masonic Cancer Center, a comprehensive cancer center designated by the National Cancer Institute, supported in part by P30 CA77598 and the University of Minnesota Imaging Center. We also acknowledge the Center for Mass Spectrometry and Proteomics.
REFERENCES
- 1.Noble PW. Idiopathic pulmonary fibrosis: natural history and prognosis. Clin Chest Med 27: S11–S16, 2006. doi: 10.1016/j.ccm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 174: 810–816, 2006. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 3.Raghu G, Selman M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am J Respir Crit Care Med 191: 252–254, 2015. doi: 10.1164/rccm.201411-2044ED. [DOI] [PubMed] [Google Scholar]
- 4.Herrera J, Beisang DJ, Peterson M, Forster C, Gilbertsen A, Benyumov A, Smith K, Korenczuk CE, Barocas VH, Guenther K, Hite R, Zhang L, Henke CA, Bitterman PB. Dicer1 deficiency in the IPF fibroblastic focus promotes fibrosis by suppressing microRNA biogenesis. Am J Respir Crit Care Med 198: 486–496, 2018. doi: 10.1164/rccm.201709-1823OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker MW, Rossi D, Peterson M, Smith K, Sikstrom K, White ES, Connett JE, Henke CA, Larsson O, Bitterman PB. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 124: 1622–1635, 2014. doi: 10.1172/JCI71386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xia H, Bodempudi V, Benyumov A, Hergert P, Tank D, Herrera J, Braziunas J, Larsson O, Parker M, Rossi D, Smith K, Peterson M, Limper A, Jessurun J, Connett J, Ingbar D, Phan S, Bitterman PB, Henke CA. Identification of a cell-of-origin for fibroblasts comprising the fibrotic reticulum in idiopathic pulmonary fibrosis. Am J Pathol 184: 1369–1383, 2014. doi: 10.1016/j.ajpath.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia H, Gilbertsen A, Herrera J, Racila E, Smith K, Peterson M, Griffin T, Benyumov A, Yang L, Bitterman PB, Henke CA. Calcium-binding protein S100A4 confers mesenchymal progenitor cell fibrogenicity in idiopathic pulmonary fibrosis. J Clin Invest 127: 2586–2597, 2017. doi: 10.1172/JCI90832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L, Herrera J, Gilbertsen AJ, Xia H, Smith K, Benyumov A, Bitterman PB, Henke CA. IL-8 mediates idiopathic pulmonary fibrosis mesenchymal progenitor cell fibrogenicity. Am J Physiol Lung Cell Mol Physiol 314: L127–L136, 2018. doi: 10.1152/ajplung.00200.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol 8: 42–54, 2011. doi: 10.1038/nrrheum.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, Moore BB, Martinez FJ, Niklason LE, White ES. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am J Respir Crit Care Med 186: 866–876, 2012. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diegelmann RF, Cohen IK, McCoy BJ. Growth kinetics and collagen synthesis of normal skin, normal scar and keloid fibroblasts in vitro. J Cell Physiol 98: 341–346, 1979. doi: 10.1002/jcp.1040980210. [DOI] [PubMed] [Google Scholar]
- 12.Galambos JT, Hollingsworth MA Jr, Falek A, Warren WD, McCain JR. The rate of synthesis of glycosaminoglycans and collagen by fibroblasts cultured from adult human liver biopsies. J Clin Invest 60: 107–114, 1977. doi: 10.1172/JCI108746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang D, Correa-Gallegos D, Christ S, Stefanska A, Liu J, Ramesh P, Rajendran V, De Santis MM, Wagner DE, Rinkevich Y. Two succeeding fibroblastic lineages drive dermal development and the transistion from regeneration to scarring. Nat Cell Biol 20: 422–431, 2018. doi: 10.1038/s41556-018-0073-8. [DOI] [PubMed] [Google Scholar]
- 14.Pierce EM, Carpenter K, Jakubzick C, Kunkel SL, Flaherty KR, Martinez FJ, Hogaboam CM. Therapeutic targeting of CC ligand 21 or CC chemokine receptor 7 abrogates pulmonary fibrosis induced by the adoptive transfer of human pulmonary fibroblasts to immunodeficient mice. Am J Pathol 170: 1152–1164, 2007. doi: 10.2353/ajpath.2007.060649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, Weissman IL, Longaker MT. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348: aaa2151, 2015. doi: 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodemann HP, Muller GA. Abnormal growth and clonal proliferation of fibroblasts derived from kidneys with interstitial fibrosis. Proc Soc Exp Biol Med 195: 57–63, 1990. doi: 10.3181/00379727-195-43118. [DOI] [PubMed] [Google Scholar]
- 17.Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest 95: 1861–1868, 1995. doi: 10.1172/JCI117866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 205: 1659–1672, 2008. doi: 10.1084/jem.20080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Luther G, Zhang W, Nan G, Wagner ER, Liao Z, Wu N, Zhang H, Wang N, Wen S, He Y, Deng F, Zhang J, Wu D, Zhang B, Haydon RC, Zhou L, Luu HH, He TC. The E-F hand calcium-binding protein S100A4 regulates the proliferation, survival and differentiation potential of human osteosarcoma cells. Cell Physiol Biochem 32: 1083–1096, 2013. doi: 10.1159/000354508. [DOI] [PubMed] [Google Scholar]
- 20.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 proteins. Curr Mol Med 13: 24–57, 2013. doi: 10.2174/156652413804486214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a mediator of metastasis. J Biol Chem 281: 677–680, 2006. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 22.Harris MA, Yang H, Low BE, Mukherjee J, Guha A, Bronson RT, Shultz LD, Israel MA, Yun K. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res 68: 10051–10059, 2008. [Erratum in Cancer Res 69: 6005, 2009]. doi: 10.1158/0008-5472.CAN-08-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanojia D, Zhou W, Zhang J, Jie C, Lo PK, Wang Q, Chen H. Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics 12: 3407–3415, 2012. doi: 10.1002/pmic.201200103. [DOI] [PubMed] [Google Scholar]
- 24.Li ZH, Bresnick AR. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res 66: 5173–5180, 2006. doi: 10.1158/0008-5472.CAN-05-3087. [DOI] [PubMed] [Google Scholar]
- 25.Lo JF, Yu CC, Chiou SH, Huang CY, Jan CI, Lin SC, Liu CJ, Hu WY, Yu YH. The epithelial-mesenchymal transition mediator S100A4 maintains cancer-initiating cells in head and neck cancers. Cancer Res 71: 1912–1923, 2011. doi: 10.1158/0008-5472.CAN-10-2350. [DOI] [PubMed] [Google Scholar]
- 26.Yan X-L, Jia Y-L, Chen L, Zeng Q, Zhou J-N, Fu C-J, Chen H-X, Yuan H-F, Li Z-W, Shi L, Xu Y-C, Wang J-X, Zhang X-M, He L-J, Zhai C, Yue W, Pei X-T. Hepatocellular carcinoma-associated mesenchymal stem cells promote heptocarcinoma progression: role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatol 57: 2274–2286, 2013.doi: 10.1002/hep.26257. [DOI] [PubMed] [Google Scholar]
- 27.Janiszewska M, De Vito C, Le Bitoux MA, Fusco C, Stamenkovic I. Transportin regulates nuclear import of CD44. J Biol Chem 285: 30548–30557, 2010. doi: 10.1074/jbc.M109.075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol 185: 949–957, 2009. doi: 10.1083/jcb.200812060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, Wogensen L, Yamaguchi Y, Noble PW. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med 208: 1459–1471, 2011. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chanmee T, Ontong P, Kimata K, Itano N. Key roles of hyaluronan and its CD44 receptor in the stemness and survival of cancer stem cells. Front Oncol 5: 180, 2015. doi: 10.3389/fonc.2015.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee IC, Chuang CC, Wu YC. Niche mimicking for selection and enrichment of liver cancer stem cells by hyaluronic acid-based multilayer films. ACS Appl Mater Interfaces 7: 22188–22195, 2015. doi: 10.1021/acsami.5b04436. [DOI] [PubMed] [Google Scholar]
- 32.Yan Y, Zuo X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med 4: 1033–1043, 2015. doi: 10.5966/sctm.2015-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med 161: 646–664, 2000. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 34.Falcone SJ, Berg RA. Crosslinked hyaluronic acid dermal fillers: a comparison of rheological properties. J Biomed Mater Res A 87: 264–271, 2008. doi: 10.1002/jbm.a.31675. [DOI] [PubMed] [Google Scholar]
- 35.Ismail TM, Fernig DG, Rudland PS, Terry CJ, Wang G, Barraclough R. The basic C-terminal amino acids of calcium-binding protein S100A4 promote metastasis. Carcinogenesis 29: 2259–2266, 2008. doi: 10.1093/carcin/bgn217. [DOI] [PubMed] [Google Scholar]
- 36.Beisang DJ, Smith K, Yang L, Benyumov A, Gilbertsen A, Herrera J, Lock E, Racila E, Forster C, Sandri BJ, Henke CA, Bitterman PB. Single cell sequencing reveals that lung mesenchymal progenitor cells in IPF exhibit pathological features early in their differentiation trajectory. Sci Rep 10: 11162, 2020. doi: 10.1038/s41598-020-66630-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kreger S, Voytik-Harbin S. Hyaluronan concentration within a 3D collagen matrix modulates matrix viscoelasticity, but not fibroblast response. Matrix Biol 28: 336–346, 2009. doi: 10.1016/j.matbio.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solis MA, Chen YH, Wong TY, Bittencourt VZ, Lin YC, Huang LLH. Hyaluronan regulates cell behavior: a potential niche for stem cells. Biochem Res Int 2012: 346972, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bharadwaj AG, Kovar JL, Loughman E, Elowsky C, Oakley GG, Simpson MA. Spontaneous metastasis of prostate cancer is promoted by excess hyaluronan synthesis and processing. Am J Pathol 174: 1027–1036, 2009. doi: 10.2353/ajpath.2009.080501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bharadwaj AG, Rector K, Simpson MA. Inducible hyaluronan production reveals differential effects on prostate tumor cell growth and tumor angiogenesis. J Biol Chem 282: 20561–20572, 2007. doi: 10.1074/jbc.M702964200. [DOI] [PubMed] [Google Scholar]
- 41.Chanmee T, Ontong P, Mochizuki N, Kongtawelert P, Konno K, Itano H. Excessive hyaluronan production promotes acquisition of cancer stem cell signatures through the coordinated regulation of Twist and the transforming growth factor β (TGFβ)-Snail signaling axis. J Biol Chem 289: 26038–26056, 2014. doi: 10.1074/jbc.M114.564120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAtee CO, Barycki JJ, Simpson MA. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv Cancer Res 123: 1–34, 2014. doi: 10.1016/B978-0-12-800092-2.00001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwertfeger KL, Cowman MK, Telmer PG, Turley EA, McCarthy JB. Hyaluronan, inflammation, and breast cancer progression. Front Immunol 6: 236, 2015. doi: 10.3389/fimmu.2015.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson MA. Concurrent expression of hyaluronan biosynthetic and processing enzymes promotes growth and vascularization of prostate tumors in mice. Am J Pathol 169: 247–257, 2006. doi: 10.2353/ajpath.2006.060032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang L, Underhill CB, Chen L. Hyaluronan on the surface of tumor cells is correlated with metastatic behavior. Cancer Res 55: 428–433, 1995. [PubMed] [Google Scholar]
- 46.Cyphert JM, Trempus CS, Garantziotis S. Size matters: molecular weight specificity of hyaluronan effects in cell Biology. Int J Cell Biol 2015: 563818, 2015., doi: 10.1155/2015/563818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by toll-like receptors and hyaluronan. Nat Med 11: 1173–1179, 2005. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 48.Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW. Resolution of lung inflammation by CD44. Science 296: 155–158, 2002. doi: 10.1126/science.1069659. [DOI] [PubMed] [Google Scholar]
- 49.Tolg C, McCarthy JB, Yazdani A, Turley EA. Hyaluronan and RHAMM in wound repair and the cancerization of stromal tissues. Biomed Res Int 2014: 103923, 2014. doi: 10.1155/2014/103923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol 23: 435–461, 2007. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 51.Noble PW, Barkauskas CE, Jiang D. Pulmonary fibrosis: patterns and perpetrators. J Clin Invest 122: 2756–2762, 2012. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Noble PW, Homer RJ. Idiopathic pulmonary fibrosis: new insights into pathogenesis. Clin Chest Med 25: 749–758, 2004. doi: 10.1016/j.ccm.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Selman M, King TE, Pardo A; American College of Chest Physicians. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 134: 136–151, 2001. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 54.Wolters PJ, Blackwell TS, Eickelberg O, Loyd JE, Kaminski N, Jenkins G, Maher TM, Molina-Molina M, Noble PW, Raghu G, Richeldi L, Schwarz MI, Selman M, Wuyts WA, Schwartz DA. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancer Respir Med 6: 154–160, 2018. doi: 10.1016/S2213-2600(18)30007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwartz DA. Idiopathic pulmonary fibrosis is a complex genetic disorder. Trans Am Clin Climatol Assoc 127: 34–45, 2016. [PMC free article] [PubMed] [Google Scholar]
- 56.Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis. Aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med 190: 867–878, 2014. doi: 10.1164/rccm.201403-0509PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D, Noble PW. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat Med 22: 1285–1293, 2016. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH, Sheppard D. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19: 1617–1624, 2013. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu RL, Sedlmeier G, Kyjacova L, Schmaus A, Philipp J, Thiele W, Garvalov BK, Sleeman JP. Hyaluronic acid-CD44 interactions promote BMP4/7 – dependent Id1/3 expression in melanoma cells. Sci Rep 8: 14913, 2018. [Erratum in Sci Rep 9: 19220, 2019]. doi: 10.1038/s41598-018-33337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]