

Abstract
Acetaminophen (n-acetyl-p-aminophenol, APAP) use in the neonatal intensive care unit is rapidly increasing. Although APAP-related hepatotoxicity is rarely reported in the neonatal literature, other end-organ toxicity can occur with toxic exposures. APAP-induced lung injury has been reported with toxic exposures in adults, but whether this occurs in the developing lung is unknown. Therefore, we tested whether toxic APAP exposures would injure the developing lung. Neonatal C57BL/6 mice (PN7, early alveolar stage of lung development) were exposed to a dose of APAP known to cause hepatotoxicity in adult mice (280 mg/kg, IP). This exposure induced significant lung injury in the absence of identifiable hepatic toxicity. This injury was associated with increased pulmonary expression of Cyp2e1, the xenobiotic enzyme responsible for the toxic conversion of APAP. Exposure was associated with increased pulmonary expression of antioxidant response genes and decreased pulmonary glutathione peroxidase activity level. Furthermore, we observed an increase in pulmonary expression of proinflammatory cytokines and chemokines. Lastly, we were able to demonstrate that this toxic APAP exposure was associated with a shift in pulmonary metabolism away from glycolysis with increased oxidative phosphorylation, a finding consistent with increased mitochondrial workload, potentially leading to mitochondrial toxicity. This previously unrecognized injury and metabolic implications highlight the need to look beyond the liver and evaluate both the acute and long-term pulmonary implications of APAP exposure in the perinatal period.
Keywords: acetaminophen, CYP2E1, liver injury, lung injury, neonate, paracetamol
INTRODUCTION
Acetaminophen (n-acetyl-p-aminophenol, APAP) use in the neonatal intensive care unit (NICU) is rapidly increasing. Recent studies in the neonatal population support the use of APAP to reduce opioid exposure (1) and to close the patent ductus arteriosus (PDA) (2). It is likely that the in-hospital safety profile specifically related to hepatic function contributes to the perception that this medication is safe and without unintended consequence in the NICU (2). It has been noted that the most commonly used dosing regimen to treat PDA (15 mg/kg q6 × 12 doses) “is twice as high as the previously recommended maximum dose in preterm infants and was adopted without dose ranging studies to establish safety and efficacy.” (2) The lack of these data have led to recent calls by experts to establish “data on human perinatal physiology and pharmacology (e.g., placental transfer and metabolism) and animal experimental studies (dose, models) relevant to the human setting” (1).
Following exposure, the majority of ingested APAP (80%–90%) is metabolized to nontoxic metabolites (3). However, a small percentage of APAP is metabolized by CYP2E1 to the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI). With toxic APAP exposure, CYP2E1 generated NAPQI increases, ultimately depleting detoxifying glutathione stores. Accumulating NAPQI covalently binds to intracellular proteins forming adducts, causing cellular dysfunction and death. Of note, mitochondrial proteins are particularly susceptible to APAP-adduct formation and the resulting mitochondrial dysfunction contributes to cell injury and death with toxic exposures (4, 5).
Clinical data demonstrate that APAP-induced hepatic toxicity rarely occurs in the neonatal period (3). Consistent with this observation, hepatocyte expression of CYP2E1 is significantly lower in neonatal compared with adult animals (6). Furthermore, although neonatal mice (<8 days old) do not demonstrate histologic or serologic evidence of liver injury following exposure to doses of APAP that cause injury in adult rodents, they experience mortality at similar or higher rates (7, 8). Importantly, whether other organs express CYP2E1, accumulate toxic metabolites, and contribute to mortality has not been explored.
The prior research supporting the conclusion that the developed lung is susceptible to APAP-induced injury is rigorous. CYP2E1 is expressed in the human and rodent lung (9). Both clinical (10–13) and preclinical reports (14–16) demonstrate that toxic APAP exposure causes lung injury in adults. Importantly, APAP-induced pulmonary injury occurs where CYP2E1 is expressed, including the bronchiolar epithelium (14, 16–18). We recently demonstrated that in the adult lung, toxic-APAP exposure induced both apoptotic death of bronchial epithelium as well as an emphysematous injury in the distal lung (19). However, whether neonatal animals are susceptible to APAP-induced lung injury is unknown. This critical gap in our understanding contributes to the increasing, unchecked, and potentially harmful APAP exposures occurring during fetal and early neonatal life. Therefore, to determine whether toxic APAP injure the developing lung, we exposed C57BL/6 mice in the early alveolar stage of lung development (PN7) to APAP (280 mg/kg, IP).
METHODS
Murine Model of Toxic Acetaminophen Exposure
Male C57BL/6 mice were exposed to APAP [280 mg/kg, intraperitoneal (IP); dissolved in phosphate-buffered saline] during the early alveolar stage of lung development (PN7). Mice were sacrificed 5 or 24 h after exposure, and tissue samples were collected as previously described (20). All procedures were approved by the IACUC at the University of Colorado (Aurora, CO) and care and handling of the animals was in accord with the National Institutes of Health guidelines for ethical animal treatment.
Histologic Evaluation of APAP-Induced Hepatic Injury
Histopathological scoring of fixed liver tissue was performed by a trained histologist as previously described (21).
Histologic Evaluation of APAP-Induced Pulmonary Injury
Measurements of mean linear intercept (MLI), a measurement of the mean distance in the air spaces, and airspace area (ASA) were performed on perfused, inflation-fixed lungs as previously described (20). Ten randomly selected nonoverlapping section per mouse were assessed for each individual replicate.
Histopathological scoring of the lungs was performed by a trained histologist blinded to the treatments or grouping of animals as previously reported (19). Twenty fields of view were scored and averaged for each individual replicate. Briefly, four semiquantitative criteria were used for this scoring. 1) The integrity of the respiratory and terminal bronchiole epithelium (0–3, normal to severe, as well as the presence or not of apoptotic epithelium in the airway lumen). 2) The quantity of macrophages found in the peripheral airway (0–3, none to lots with clumps). 3) The presence of peripheral lung emphysema with alveolar wall clubbing (0–2, none to lots). Scores were tallied, grouped, and finally unblinded by the individual with knowledge of the groupings. Histological images were captured on an Olympus BX51 microscope equipped with a 17 mp Olympus DP73 high-definition, color, digital camera using the Olympus CellSens software (Olympus, Waltham, MA). All composite images were cropped and assembled using Adobe Photoshop.
In Situ Hybridization for Cyp2e1 Expression
RNAScope detection was used to perform in situ hybridization according the manufacturer’s protocol (Advanced Cell Diagnostics, Hayward, CA). Briefly, formalin-fixed paraffin embedded mouse lungs were cut into 5-µm-thick tissue sections. Slides were deparaffinized in xylene, followed by rehydration in a series of ethanol washes. Following citrate buffer (Advanced Cell Diagnostics) antigen retrieval, slides were rinsed in deionized water, and immediately treated with protease (Advanced Cell Diagnostics) at 40°C for 30 min in a HybEZ hybridization oven (Advanced Cell Diagnostics). Probe directed against Cyp2e1 mRNA was applied at 40°C in the following order: target probes, preamplifier, amplifier; and label probe for 10 min. After each hybridization step, slides were washed two times in a washing buffer (Advanced Cell Diagnostics) at room temperature. Chromogenic detection was performed followed by counterstaining with hematoxylin QS (Vector Labs, Burlingame, CA). Staining was visualized using an Aperio CS2 whole slide scanner (Leica Biosystems, Buffalo Grove, IL). Cyp2e1 expression was quantified using Aperio Positive Cells Counting V9 protocol (Leica).
Isolation of mRNA, cDNA Synthesis, and Analysis of Relative mRNA Levels by RT-qPCR
Hepatic and pulmonary mRNA was isolated, cDNA synthesized, and relative mRNA levels were evaluated by quantitative real-time PCR using exon spanning primers normalized to 18S RNA levels, and quantification was performed using the cycle threshold (ΔΔCt) method as described previously (20) (Table 1).
Table 1.
List of genes and primers used for qPCR analysis
| Target | Assay ID |
|---|---|
| Cyp2e1 | Mm00491127_m1 |
| Il6 | Mm00446190_m1 |
| Gclc | Mm00802655_m1 |
| Cxcl10 | Mm00445235_m1 |
| Cxcl1 | Mm04207460_m1 |
| 18s | Mm03928990_g1 |
| Ddit3 | Mm01135937_g1 |
| Ccl2 | Mm00441242_m1 |
| Hmox1 | Mm00516005_m1 |
| Nqo1 | Mm01253561_m1 |
| Gpx3 | Mm00492427_m1 |
| Trxrd1 | Mm00443675_m1 |
Glutathione Peroxidase Activity Level
The activity level of glutathione peroxidase (GPx) was determined indirectly by a coupled reaction with glutathione reductase, as previously described (22).
Fluorescence Lifetime Imaging Microscopy
Fluorescence lifetime imaging microscopy (FLIM) was performed to detect local metabolic changes in 20 different areas in fresh right lower lungs using a Zeiss 780 laser-scanning confocal/multiphoton-excitation fluorescence microscope with a 34-Channel GaAsP QUASAR Detection Unit and nondescanned detectors for two-photon fluorescence (Zeiss, Thornwood, NY) equipped with an ISS A320 FastFLIM box and a titanium:sapphire Chameleon Ultra II (Coherent, Santa Clara, CA) as previously described (23, 24).
For the acquisition of FLIM images, fluorescence for reduced form of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) was detected simultaneously by two photon-counting photomultiplier tube (PMT) detectors (H7422p-40; Hamamatsu). Images of the lung were obtained with Vista Vision software by ISS in 256 × 256 format with a pixel dwell time of 6.3 µs/pixel and averaging over 30 frames. The number of pixels covered with lifetimes for free and bound NADH and FAD were calculated in SimFCS (LFD), and the values were normalized to the total number of pixels detected as previously described (13).
The glycolytic index was calculated for all experimental groups using the following equation:
Glycolytic Index = free NADH fraction/bound to enzyme NADH fraction as described previously (24, 25).
To assess mitochondrial activity (OXPHOS), FLIM-based optical redox ratios (Fluorescent Lifetime Redox Ratio = FLIRR) was calculated as follows:
FLIRR (OXPHOS) = bound to enzyme NADH fraction/bound to enzyme FAD fraction as previously described (25).
Statistical Analysis
For comparison between treatment groups, the null hypothesis that no difference existed between treatment means was tested by Student’s t test for two groups and two-way ANOVA for multiple groups with potentially interacting variables (time, APAP exposure), with statistical significance between and within groups determined by means of Bonferroni method of multiple comparisons (Prism, GraphPad Software, Inc). Statistical significance was defined as P < 0.05.
RESULTS
The PN7 Liver Is Resistant to Toxic APAP Exposures
Following APAP exposures known to injure the adult liver (280 mg/kg, IP) (19), the PN7 liver shows very little evidence of hepatic toxicity (Fig. 1, A–D). Blinded histopathologic evaluation revealed an elevated inflammatory infiltration score (Fig. 1E), which was driven by the presence of small foci of immune cells found in the liver parenchyma (Fig. 1D, yellow arrows). There was no difference in the sinusoidal dilation scores between control and APAP exposed (Fig. 1F), and no necrosis was noted in any animals and no score was tallied.
Figure 1.
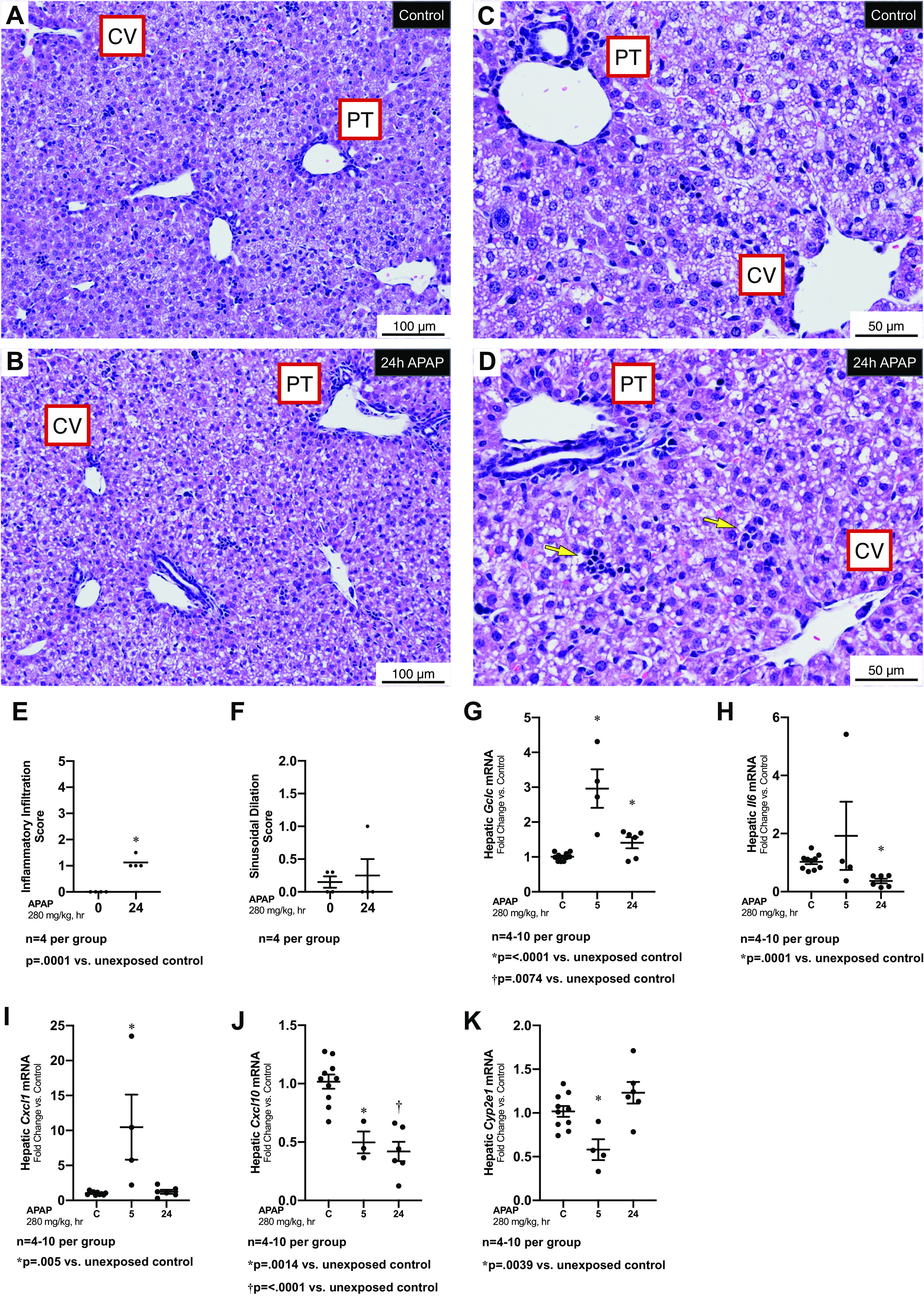
Hepatic response to APAP exposure (280 mg/kg, IP) at PN7. A–D: low (A and B) and high (C and D) magnification representative H&E-stained hepatic sections from unexposed (A, C) and exposed (C, D; APAP 280 mg/kg, IP; 24 h administered on PN7) C57BL/6 mice. Yellow arrows in D indicate small foci of immune cells. Examples of portal triad (PT) and central vein (CV) have been added. Internal scale bar 50 or 100 μm. E and F: blind histopathologic evaluation of H&E-stained hepatic sections scored for inflammatory infiltration (E) and sinusoidal dilatation (F). Data are expressed as mean ± SE. G–K: fold change in hepatic mRNA expression of Gclc (G), Il6 (H), Cxcl1 (I), Cxcl10 (J), and Cyp2e1 (K) following APAP exposure (280 mg/kg, IP; 24 h administered on PN7). Data expressed as mean ± SE. APAP, n-acetyl-p-aminophenol; H&E, hematoxylin-eosin; PN7, early alveolar stage of lung development.
In adult mice, APAP induces expression of Gclc, Il6, Cxcl1, and Cxcl10 (26, 27). In APAP-exposed PN7 mice, we found that hepatic expression of Gclc, Il6, Cxcl1, and Cxcl10 was not consistently significantly increased (Fig. 1, G–J). Finally, we found APAP exposures did not induce hepatic Cyp2e1 expression (Fig. 1K). These results demonstrate a transient and variable hepatic response to APAP exposure at PN7.
APAP Exposure Injures the Early Alveolar-Stage Lung
In contrast to control lungs (Fig. 2, A and B), lungs from APAP-exposed animals demonstrated bronchiolar injury including death and sloughing of some of the injured epithelium into the bronchial lumens (Fig. 2, B and C, yellow arrows). Furthermore, when compared with control lungs (Fig. 2E), the distal airspaces of lungs from APAP-exposed animals appear simplified and enlarged (Fig. 2F). We found that the scores for respiratory and terminal bronchial epithelial injury (Fig. 2G), peripheral lung macrophage load (Fig. 2H), and peripheral lung emphysema (Fig. 2I) were significantly elevated following APAP exposure. Lastly, mean linear intercept length (Fig. 2J) and airspace area (Fig. 2K) increased in APAP-exposed mice.
Figure 2.
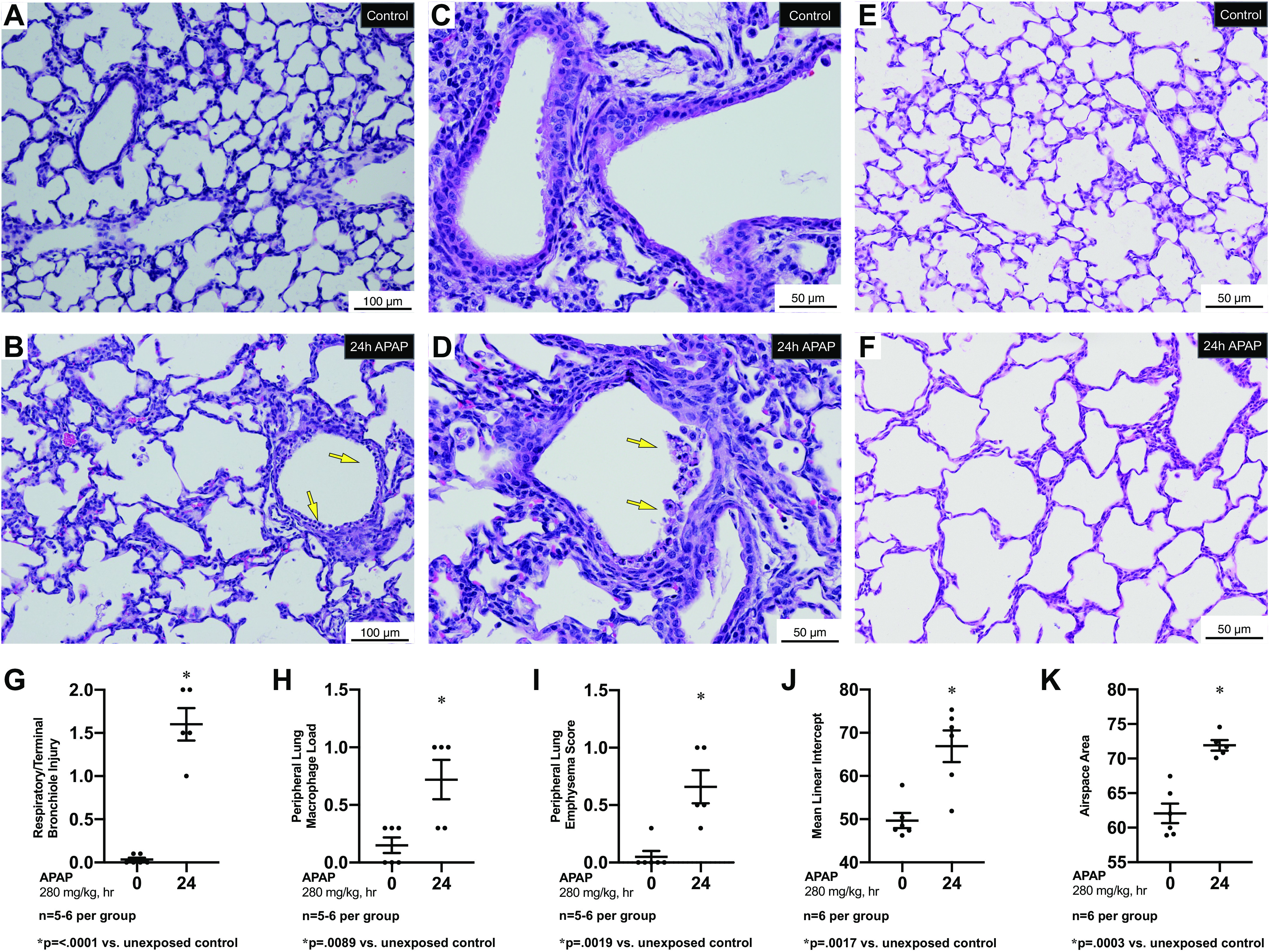
Pulmonary response to APAP exposure (280 mg/kg, IP) at PN7. A–D: low (A, B) and high (C, D) magnification representative H&E-stained proximal sections from unexposed (A, C) and exposed (C, D) (APAP 280 mg/kg, IP; 24 h administered on PN7) C57BL/6 mice. Yellow arrows indicate cell death and sloughing of a portion of the surface epithelium lining a bronchiole. Internal scale bar 50 or 100 μm E and F: high magnification representative H&E-stained distal sections from unexposed (E) and exposed (F) (APAP 280 mg/kg, IP; 24 h administered on PN7) C57BL/6 mice. Internal scale bar 50 or 100 μm. G–I: blind histopathologic evaluation of H&E-stained pulmonary sections from unexposed and exposed (APAP 280 mg/kg, IP; 24 h administered on PN7) C57BL/6 mice scored for respiratory/terminal bronchiole epithelial injury (G), peripheral lung macrophage load (H), and peripheral lung emphysema score (I). Data are expressed as mean ± SE; J and K: morphometric assessment of H&E-stained pulmonary sections from unexposed and exposed (APAP 280 mg/kg, IP; 24 h administered on PN7) C57BL/6 mice for mean linear intercept (J) and airspace area (K). Data are expressed as mean ± SE. APAP, n-acetyl-p-aminophenol; H&E, hematoxylin-eosin; PN7, early alveolar stage of lung development.
APAP Exposure Decreases Glutathione Peroxidase Activity and Induces an Oxidative Stress Responsive Gene Transcriptional Response
APAP decreases hepatic glutathione peroxidase (GPx) activity in adults (28) potentially due to APAP-adduct formation (29, 30). We found that pulmonary GPx activity decreased 24 h after APAP exposure (Fig. 3A). Furthermore, we assessed the pulmonary expression of the oxidant stress-related genes Gclc, Hmox1, Trxrd1, Nqo1, and Gpx3 at PN7. The expression of Gclc, Hmox1, Trxrd1, Nqo1, and Gpx3 all increased following APAP exposure (Fig. 3, B–F).
Figure 3.
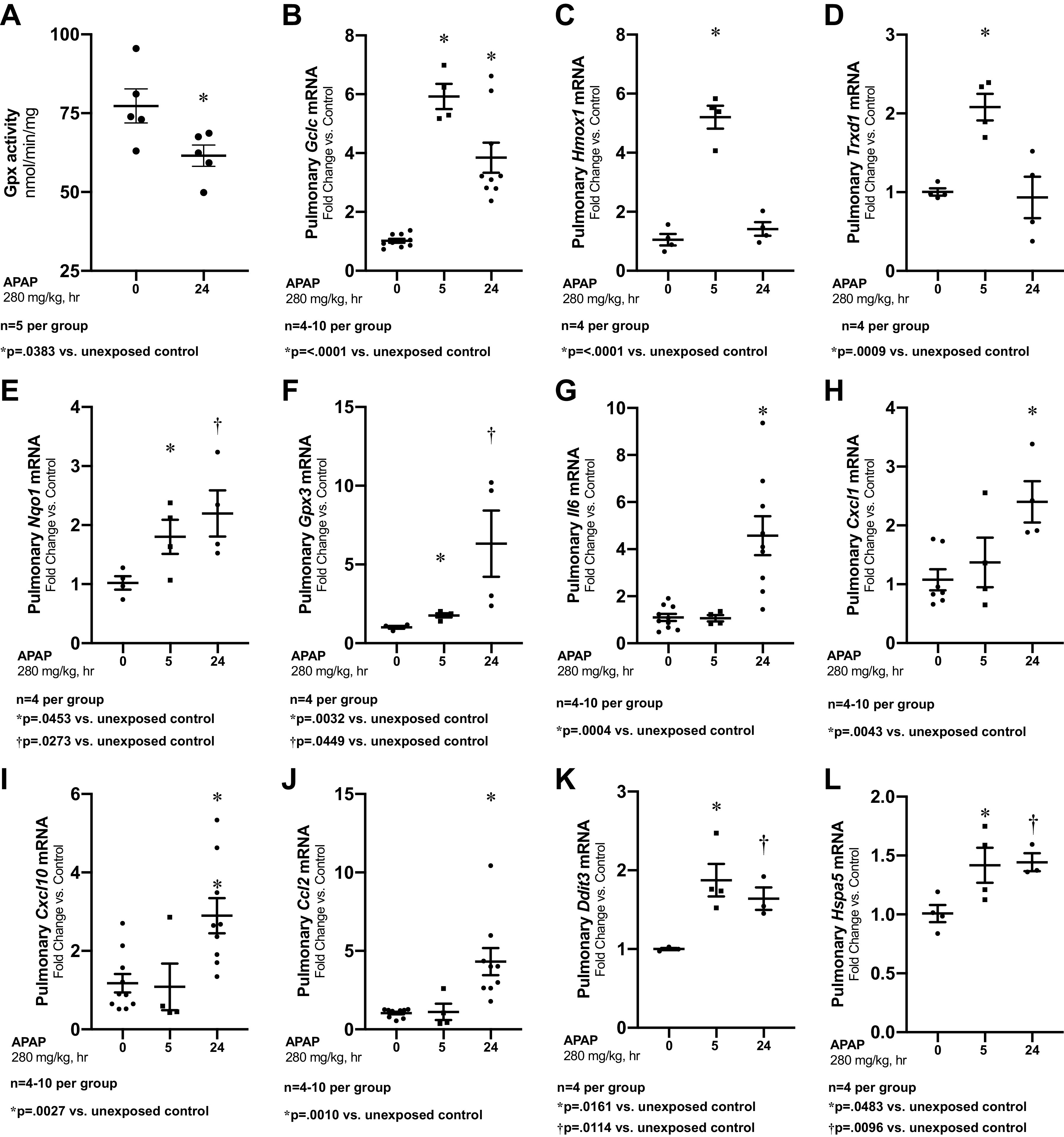
APAP induces oxidative stress and transcriptional pulmonary response. A: pulmonary glutathione peroxidase activity, by oxidation of NADPH per minute per milligram of protein. B–F: fold change in pulmonary mRNA expression of Gclc (B), Hmox1 (C), Trxrd1 (D), Nqo1 (E), and Gpx3 (F) following APAP exposure (280 mg/kg, IP; 24 h administered on PN7). Data expressed as mean ± SE. G–L: fold change in pulmonary mRNA expression of Il6 (G), Cxcl1 (H), Cxcl10 (I), Ccl2 (J), Ddit3 (K), and Hspa5 (L) and following APAP exposure (280 mg/kg, IP; 24 h administered on PN7). Data expressed as mean ± SE. APAP, n-acetyl-p-aminophenol; Gpx, glutathione peroxidase; PN7, early alveolar stage of lung development.
APAP Exposure Induces Proinflammatory Transcriptional Changes in the Alveolar-Stage Lung
In contrast to the variable induction of proinflammatory genes observed in the liver, APAP induced significant increases in Il6, Cxcl1, Cxcl10, and Ccl2 expression (Fig. 3, G–J). We have previously reported that APAP exposure induced ER stress in the mature lung (19), and thus evaluated the expression of the ER stress-related markers Ddit3 (CHOP) and Hspa5 (BiP). We found that following APAP exposure induced pulmonary expression of Ddit3 (CHOP) and Hspa5 (BiP) (Fig. 3, K–L).
APAP Exposure Induces Cyp2e1 Expression in the Early Alveolar-Stage Lung
First, we sought to determine whether the APAP-metabolizing enzyme CYP2E1 was expressed in the early alveolar-stage lung. To investigate localization of Cyp2e1 mRNA expression, we performed RNAscope on pulmonary tissue obtained from mice on PN7. We demonstrate positively stained cells throughout the lung parenchyma in the early alveolar-stage murine lung (Fig. 4A). Two subtypes of the Cyp2e1-expressing cells were identified: the majority of the lung cells located within the developing mesenchyme had a positive signal (Fig. 4A, yellow arrows), and a small subset demonstrated a strong positive staining (Fig. 4A, red arrows).
Figure 4.
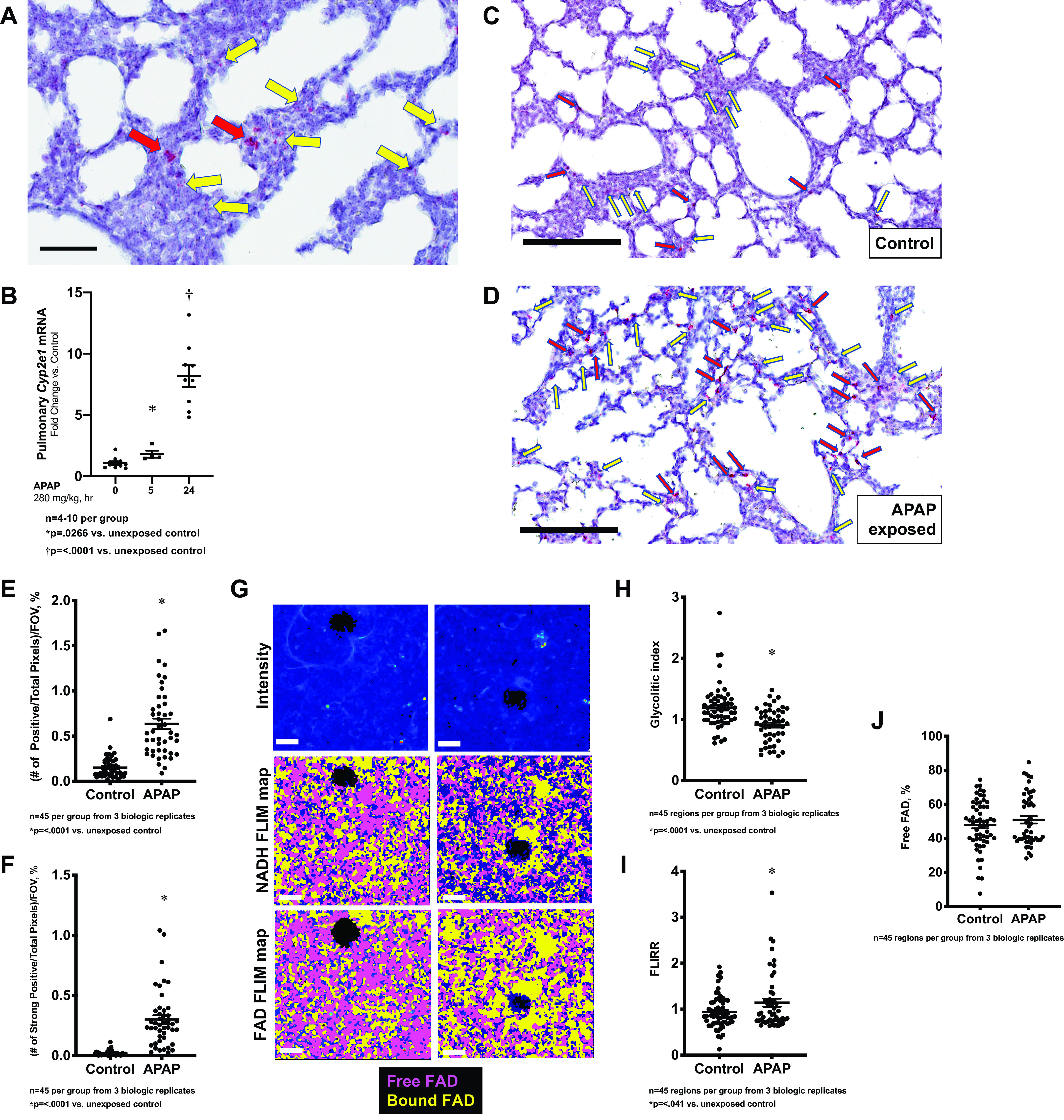
Pulmonary Cyp2e1 expression and metabolic changes following exposure to APAP exposure (280 mg/kg, IP) at PN7. A: in situ hybridization of PN7 murine lung specimens with Cyp2e1 (red). Yellow arrowheads depict positive staining cells, with red arrowheads depicting strongly positive staining cells. Bar is 60 µm. B: fold change in pulmonary Cyp2e1 mRNA expression following APAP exposure (280 mg/kg, IP; 5/24 h administered on PN7). Data expressed as mean ± SE. C and D: in situ hybridization of PN7 murine lung specimens with Cyp2e1 (red) in control (C) and APAP exposed (D; 280 mg/kg IP, 24 h). Yellow arrowheads depict positive staining cells, with red arrowheads depicting strongly positive staining cells. Bar is 200 µm. E: objective quantification of number of pixels with positive staining for Cyp2e1 normalized to total pixels per field of view, expressed as a percent. Data expressed as mean ± SE (N = 45 per time point). F: objective quantification of number of pixels with strong staining for Cyp2e1 normalized to total pixels per field of view, expressed as a percent. Data expressed as mean ± SE (N = 45 per time point). G: representative intensity images (top) depict distal lung parenchyma; their corresponding NADH (middle, free NADH—yellow, bound NADH—magenta) and FAD (bottom, free FAD—magenta, bound FAD—yellow) FLIM maps for control and APAP-treated tissues are shown. Quantified (H) glycolytic Index (free/bound NADH ratio), (I) FLIRR (bound NADH/bound FAD ratio), and (J) free FAD, expressed as a percent fraction. Data expressed as mean ± SE (N = 45 per time point). Bar is 20 mm. APAP, n-acetyl-p-aminophenol; FLIM, fluorescence lifetime imaging microscopy; PN7, early alveolar stage of lung development.
On a whole lung level, we found that APAP induced pulmonary Cyp2e1 mRNA expression (Fig. 4B). To obtain Cyp2e1 expressing cell locality and distribution, we performed RNAScope on pulmonary tissue obtained from mice following APAP exposure. Both subpopulations of Cyp2e1 expressing cells were increased after APAP exposure (Fig. 4, C and D; positive cells—yellow arrows, strong positive—red arrows). Objective assessment of this staining was performed by quantifying the number of positive (Fig. 4E) and strong positive pixels (Fig. 4F) and normalizing the total number of pixels in field of view. These results are in contrast to what is observed in the liver, where no induction of Cyp2e1 expression occurs after APAP exposure (Fig. 1K).
APAP Exposure Leads to the Local Metabolic Changes in the Early Alveolar-Stage Lung
FLIM was used to examine local metabolic changes in the early alveolar-stage murine lungs. The percentages for free and bound NADH and FAD were calculated for control and APAP-treated tissues. The glycolytic index (free to enzyme-bound NADH ratio) and FLIRR (enzyme-bound NADH to enzyme-bound FAD ratio) were used to estimate glycolysis and OXPHOS contributions to metabolic balance in control and injured lungs (Fig. 4, G–J). APAP-treated lungs demonstrated a significantly decreased glycolytic index (Fig. 4H) (1.29 ± 0.10% in control compared with 0.91 ± 0.04% in APAP-treated animals, P = 0.002) in comparison with control lungs, which was accompanied by a significant increase in OXPHOS (Fig. 4I; 0.95 ± 0.05% in control compared with 1.14 ± 0.09% in APAP-treated animals, P = 0.05). Free FAD fractions were similar in both control and APAP-treated lungs (Fig. 4J). Since free FAD is mostly produced in the electron transport chain (ETC) (31), similar to control, the free FAD fraction and increased FLIRR ratio in APAP indicate that most changes were induced in the tricarboxylic acid (TCA) cycle.
DISCUSSION
We found that C57BL/6 mice in the early alveolar stage of lung development (PN7) exposed to APAP (280 mg/kg, IP) had significant lung injury in the absence of hepatic toxicity. Through blinded histologic analysis and objective morphometric assessment of the lung, we demonstrate both airway and alveolar manifestations of toxic APAP exposure. This injury is associated with decreased GPx activity and increased pulmonary expression of oxidative stress response and proinflammatory genes. Additionally, and in contrast to the liver, APAP induces pulmonary Cyp2e1 expression. This increased expression is associated with a metabolic shift in pulmonary metabolism from glycolysis to increased oxidative phosphorylation, a finding consistent with increased mitochondrial workload.
Both clinical and preclinical studies prove that the developed lung is susceptible to APAP-induced injury. Clinically, case reports and case series have linked toxic APAP exposures to alveolar injury (10), pneumonitis (12), and ARDS (11). Preclinical data demonstrate that APAP-induced lung injury in the mature lung is consistent across multiple species including mice, rats, and pigs (14, 16–18, 32, 33). These studies convincingly demonstrate that toxic APAP exposure injure the mature lung, and our findings add to this body of literature by demonstrating that the developing lung is susceptible to the toxic effects of APAP. This injury is associated with transcriptional upregulation of Cyp2e1, ER stress response genes, and proinflammatory mediators. Here, we demonstrate that the developing lung responds to toxic APAP exposures, and that these exposures result in lung injury. However, the exact mechanisms linking these APAP exposure and lung injury and deserve further study. Possible mechanisms include death of a subset of cells important for lung development, ongoing oxidant and inflammatory stress, innate immune activation, or a combination of all or part of these factors.
Pericentral hepatocytes express the highest level of the toxin producing CYP2E1, leaving this cell population highly susceptible to NAPQI-induced injury. Prior work demonstrates APAP-induced pulmonary injury occurs in the mature lung where CYP2E1 is expressed, including the bronchiolar epithelium (14, 16–18) and the distal pulmonary epithelium (19). Very little is known about Cyp2e1 expression during development. Here, we report that Cyp2e1 is expressed in the early alveolar-stage lung, supporting the hypothesis that the developing lung has the capacity to metabolize APAP into toxic metabolites.
Interestingly, with toxic APAP exposures at PN7, pulmonary toxicity occurs in the absence of hepatic injury. These data add to the body of literature demonstrating that APAP-induced pulmonary injury is independent from hepatic toxicity. Following hepatectomy, APAP-induced pulmonary injury occurs in rats (34). Furthermore, liver-specific knockout of NADPH-cytochrome P450 reductase—the obligate redox partner for all P450 enzymes—abrogates APAP-induced hepatic injury, while pulmonary injury remains consistent (32). Finally, the mixed function oxidase inhibitor piperonyl butoxide attenuates APAP-induced hepatic injury while significant pulmonary injury still occurs (15).
Here, we have reported that APAP exposure induces a metabolic change in the developing lung. APAP-induced mitochondrial toxicity, and the resulting metabolic changes, are well documented in liver (4). In the early alveolar-stage lung, APAP exposures induce a shift from glycolytic metabolism to oxidative phosphorylation, a finding consistent with increased mitochondrial activity before the onset of toxicity. The disruption of mitochondrial respiration by APAP adducts plays a central role in APAP-induced hepatotoxicity (5). Further work is needed to determine whether these same mechanisms occur in the developing lung, and whether these changes have long-term implications. Additionally, although changes in pulmonary cellular metabolism have been associated with various pulmonary pathologies in the developed lung (35), the impact of these changes on the developing lung remains to be elucidated.
Whether lower doses or repetitive exposures will injure the developing lung is unknown but require further study. There are reasons to hypothesize that the early alveolar-stage lung will be susceptible to the toxic effects of APAP even at lower doses. Following toxic APAP exposure, pulmonary glutathione stores are rapidly depleted from the adult lung (17, 19, 36). Glutathione stores are limited in the developing lung (37, 38), and further limited due to the acute transition to breathing air and oxidative challenges due to supplemental oxygen. We speculate the developing lung may be susceptible to architectural and functional impairments after repeated APAP exposure, and our future work will evaluate this after clinically relevant doses.
Our study has multiple limitations. Similar to other studies, this report is limited to a single, toxic dose of APAP. We add to this literature by reporting the response of the early alveolar-stage lung to this exposure. The response of the early alveolar-stage lung responds to APAP has not previously been reported, and our results confirm pulmonary toxicity. How the canalicular and saccular stage lung respond to APAP exposures deserves further study. Furthermore, whether lower doses consistent with clinical exposure cause lung injury is unknown. More work is needed to confirm the threshold to toxicity and the ultimate developmental implications of these early life exposures. Although we show the developing lung expresses Cyp2e1 and that expression increases with APAP exposure, we did not evaluate pulmonary homogenates for the presence of APAP adducts. Previous studies demonstrating APAP-induced lung injury were performed in male mice, and we have replicated that study design here. This is an important limitation that must be noted, especially as APAP-induced hepatic and renal injury is sex specified (39). More work is needed to determine whether PN7 female mice are similarly susceptible.
In conclusion, the early alveolar-stage lung expresses the APAP-metabolizing enzyme Cyp2e1. The early alveolar-stage lung manifests both bronchiolar and alveolar injury following exposure to a single, high dose APAP exposure. This injury is associated with decreased GPx activity, proinflammatory transcriptional changes, and a metabolic shift to oxidative phosphorylation. With APAP exposures increasing in the NICU, these findings have clinical implications. Our work adds urgency to the need to study the end-organ-specific implications on early life APAP exposures.
DATA AVAILABILITY
The data used to support the findings of this study are included within the article.
GRANTS
The FLIM imaging experiments were performed in the Advanced Light Microscopy Core part of NeuroTechnology Center at University of Colorado Anschutz Medical Campus supported in part by Rocky Mountain Neurological Disorders Core Grant P30 NS048154. This work was supported by NIH grant R01HL132941 and by Diabetes Research Center Pilot and Feasibility Award P30 DK116073 to C. J. Wright.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.D., L.G.S., and C.J.W. conceived and designed research; E.D., L.G.S., L.Z., R.D.D., D.B., T.S., B.B., and C.J.W. performed experiments; E.D., L.G.S., D.O., and C.J.W. analyzed data; E.D., L.G.S., D.O., and C.J.W. interpreted results of experiments; E.D., L.G.S., and C.J.W. prepared figures; C.J.W. drafted manuscript; E.D., L.G.S., D.O., T.S., and C.J.W. edited and revised manuscript; E.D., L.G.S., D.O., L.Z., R.D.D., D.B., T.S., B.B., and C.J.W. approved final version of manuscript.
REFERENCES
- 1.Allegaert K, van den Anker JN. Perinatal and neonatal use of paracetamol for pain relief. Semin Fetal Neonatal Med 22: 308–313, 2017. doi: 10.1016/j.siny.2017.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Jasani B, Weisz DE, McNamara PJ. Evidence-based use of acetaminophen for hemodynamically significant ductus arteriosus in preterm infants. Semin Perinatol 42: 243–252, 2018. doi: 10.1053/j.semperi.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Pacifici GM, Allegaert K. Clinical pharmacology of paracetamol in neonates: a review. Curr Ther Res Clin Exp 77: 24–30, 2015. doi: 10.1016/j.curtheres.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 44: 88–106, 2012. doi: 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramachandran A, Jaeschke H. Acetaminophen hepatotoxicity: a mitochondrial perspective. Adv Pharmacol 85: 195–219, 2019. doi: 10.1016/bs.apha.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hines RN. Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol 21: 169–175, 2007. doi: 10.1002/jbt.20179. [DOI] [PubMed] [Google Scholar]
- 7.Alhava E, Hassinen K, Nieminen E. Toxicity of paracetamol in relation to age in mice. Acta Pharmacol Toxicol (Copenh) 42: 317–319, 1978. doi: 10.1111/j.1600-0773.1978.tb02208.x. [DOI] [PubMed] [Google Scholar]
- 8.Hart JG, Timbrell JA. The effect of age on paracetamol hepatotoxicity in mice. Biochem Pharmacol 28: 3015–3017, 1979. doi: 10.1016/0006-2952(79)90602-6. [DOI] [PubMed] [Google Scholar]
- 9.Hukkanen J, Pelkonen O, Hakkola J, Raunio H. Expression and regulation of xenobiotic-metabolizing cytochrome P450 (CYP) enzymes in human lung. Crit Rev Toxicol 32: 391–411, 2002. doi: 10.1080/20024091064273. [DOI] [PubMed] [Google Scholar]
- 10.Price LM, Poklis A, Johnson DE. Fatal acetaminophen poisoning with evidence of subendocardial necrosis of the heart. J Forensic Sci 36: 930–935, 1991. [PubMed] [Google Scholar]
- 11.Baudouin SV, Howdle P, O'Grady JG, Webster NR. Acute lung injury in fulminant hepatic failure following paracetamol poisoning. Thorax 50: 399–402, 1995. doi: 10.1136/thx.50.4.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akashi S, Tominaga M, Naitou K, Fujisawa N, Nakahara Y, Hiura K, Hayashi S. Two cases of acetaminophen-induced pneumonitis. Nihon Kyobu Shikkan Gakkai Zasshi 35: 974–979, 1997. [PubMed] [Google Scholar]
- 13.McKeever TM, Lewis SA, Smit HA, Burney P, Britton JR, Cassano PA. The association of acetaminophen, aspirin, and ibuprofen with respiratory disease and lung function. Am J Respir Crit Care Med 171: 966–971, 2005. doi: 10.1164/rccm.200409-1269OC. [DOI] [PubMed] [Google Scholar]
- 14.Placke ME, Wyand DS, Cohen SD. Extrahepatic lesions induced by acetaminophen in the mouse. Toxicol Pathol 15: 381–387, 1987. doi: 10.1177/019262338701500401. [DOI] [PubMed] [Google Scholar]
- 15.Bartolone JB, Beierschmitt WP, Birge RB, Hart SG, Wyand S, Cohen SD, Khairallah EA. Selective acetaminophen metabolite binding to hepatic and extrahepatic proteins: an in vivo and in vitro analysis. Toxicol Appl Pharmacol 99: 240–249, 1989. doi: 10.1016/0041-008X(89)90006-9. [DOI] [PubMed] [Google Scholar]
- 16.Hart SG, Cartun RW, Wyand DS, Khairallah EA, Cohen SD. Immunohistochemical localization of acetaminophen in target tissues of the CD-1 mouse: correspondence of covalent binding with toxicity. Fundam Appl Toxicol 24: 260–274, 1995. doi: 10.1006/faat.1995.1029. [DOI] [PubMed] [Google Scholar]
- 17.Jeffery EH, Haschek WM. Protection by dimethylsulfoxide against acetaminophen-induced hepatic, but not respiratory toxicity in the mouse. Toxicol Appl Pharmacol 93: 452–461, 1988. doi: 10.1016/0041-008x(88)90048-8. [DOI] [PubMed] [Google Scholar]
- 18.Neff SB, Neff TA, Kunkel SL, Hogaboam CM. Alterations in cytokine/chemokine expression during organ-to-organ communication established via acetaminophen-induced toxicity. Exp Mol Pathol 75: 187–193, 2003. doi: 10.1016/s0014-4800(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 19.Sandoval J, Orlicky DJ, Allawzi A, Butler B, Ju C, Phan CT, Toston R, De Dios R, Nguyen L, McKenna S, Nozik-Grayck E, Wright CJ. Toxic acetaminophen exposure induces distal lung ER stress, proinflammatory signaling, and emphysematous changes in the adult murine lung. Oxid Med Cell Longev 2019: 7595126, 2019. doi: 10.1155/2019/7595126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen L, Castro O, De Dios R, Sandoval J, McKenna S, Wright CJ. Sex-differences in LPS-induced neonatal lung injury. Sci Rep 9: 8514, 2019. doi: 10.1038/s41598-019-44955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin-Murphy BV, Kominsky DJ, Orlicky DJ, Donohue TM Jr, Ju C. Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury. Hepatology 57: 1575–1584, 2013. doi: 10.1002/hep.26134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherlock LG, Sjostrom K, Sian L, Delaney C, Tipple TE, Krebs NF, Nozik-Grayck E, Wright CJ. Hepatic-specific decrease in the expression of selenoenzymes and factors essential for selenium processing after endotoxemia. Front Immunol 11: 595282, 2020. doi: 10.3389/fimmu.2020.595282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marwan AI, Shabeka U, Reisz JA, Zheng C, Serkova NJ, Dobrinskikh E. Unique heterogeneous topological pattern of the metabolic landscape in rabbit fetal lungs following tracheal occlusion. Fetal Diagn Ther 45: 145–154, 2019. doi: 10.1159/000487752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobrinskikh E, Al-Juboori SI, Shabeka U, Reisz JA, Zheng C, Marwan AI. Heterogeneous pulmonary response after tracheal occlusion: clues to fetal lung growth. J Surg Res 239: 242–252, 2019. doi: 10.1016/j.jss.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 25.Wallrabe H, Svindrych Z, Alam SR, Siller KH, Wang T, Kashatus D, Hu S, Periasamy A. Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci Rep 8: 79, 2018. doi: 10.1038/s41598-017-18634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J 16: 1227–1236, 2002. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- 27.Goldring CEP, Kitteringham NR, Elsby R, Randle LE, Clement YN, Williams DP, McMahon M, Hayes JD, Itoh K, Yamamota M, Park BK. Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology 39: 1267–1276, 2004. doi: 10.1002/hep.20183. [DOI] [PubMed] [Google Scholar]
- 28.Arnaiz SL, Llesuy S, Cutrin JC, Boveris A. Oxidative stress by acute acetaminophen administration in mouse liver. Free Radic Biol Med 19: 303–310, 1995. doi: 10.1016/0891-5849(95)00023-q. [DOI] [PubMed] [Google Scholar]
- 29.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem 273: 17940–17953, 1998. doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 30.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos 31: 1499–1506, 2003. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 31.Penjweini R, Roarke B, Alspaugh G, Gevorgyan A, Andreoni A, Pasut A, Sackett DL, Knutson JR. Single cell-based fluorescence lifetime imaging of intracellular oxygenation and metabolism. Redox Biol 34: 101549, 2020. doi: 10.1016/j.redox.2020.101549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu J, Cui H, Behr M, Zhang L, Zhang Q-Y, Yang W, Hinson JA, Ding X. In vivo mechanisms of tissue-selective drug toxicity: effects of liver-specific knockout of the NADPH-cytochrome P450 reductase gene on acetaminophen toxicity in kidney, lung, and nasal mucosa. Mol Pharmacol 67: 623–630, 2005. doi: 10.1124/mol.104.007898. [DOI] [PubMed] [Google Scholar]
- 33.Newsome PN, Henderson NC, Nelson LJ, Dabos C, Filippi C, Bellamy C, Howie F, Clutton RE, King T, Lee A, Hayes PC, Plevris JN. Development of an invasively monitored porcine model of acetaminophen-induced acute liver failure. BMC Gastroenterol 10: 34, 2010. doi: 10.1186/1471-230X-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breen K, Wandscheer JC, Peignoux M, Pessayre D. In situ formation of the acetaminophen metabolite covalently bound in kidney and lung. Supportive evidence provided by total hepatectomy. Biochem Pharmacol 31: 115–116, 1982. doi: 10.1016/0006-2952(82)90247-7. [DOI] [PubMed] [Google Scholar]
- 35.Liu G, Summer R. Cellular metabolism in lung health and disease. Annu Rev Physiol 81: 403–428, 2019. doi: 10.1146/annurev-physiol-020518-114640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen TS, Richie JP Jr, Lang CA. Life span profiles of glutathione and acetaminophen detoxification. Drug Metab Dispos 18: 882–887, 1990. [PubMed] [Google Scholar]
- 37.Berkelhamer SK, Farrow KN. Developmental regulation of antioxidant enzymes and their impact on neonatal lung disease. Antioxid Redox Signal 21: 1837–1848, 2014. doi: 10.1089/ars.2013.5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ofman G, Tipple TE. Antioxidants & bronchopulmonary dysplasia: beating the system or beating a dead horse? Free Radic Biol Med 142: 138–145, 2019. doi: 10.1016/j.freeradbiomed.2019.01.038. [DOI] [PubMed] [Google Scholar]
- 39.Hoivik DJ, Manautou JE, Tveit A, Hart SG, Khairallah EA, Cohen SD. Gender-related differences in susceptibility to acetaminophen-induced protein arylation and nephrotoxicity in the CD-1 mouse. Toxicol Appl Pharmacol 130: 257–271, 1995. doi: 10.1006/taap.1995.1031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are included within the article.