

Abstract
BACKGROUND AND PURPOSE: Jugular foraminal stenosis (JFS) or atresia (JFA) with collateral emissary veins (EV) has been documented in syndromic craniosynostosis. Disruption of EV during surgery can produce massive hemorrhage. Our purpose was to describe the prevalence of prominent basal emissary foramina (EF), which transmit enlarged EV, in syndromic craniosynostosis. Our findings were correlated with phenotypic and molecular diagnoses.
METHODS: We reviewed the medical records and imaging examinations of 33 patients with syndromic craniosynostosis and known fibroblast growth factor receptor (FGFR) mutations. All patients underwent CT and 14 MR imaging. The cranial base was assessed for size of occipitomastoid EF and jugular foramina (JF). Vascular imaging studies were available from 12 patients. A control group (n = 76) was used to establish normal size criteria for JF and EF.
RESULTS: Phenotypic classification included Crouzon syndrome (n = 10), crouzonoid features with acanthosis nigricans (n = 3), Apert syndrome (n = 10), Pfeiffer syndrome (n = 4), and clinically unclassifiable bilateral coronal synostosis (n = 6). EF ≥ 3 mm in diameter and JFS or JFA were identified in 23 patients with various molecular diagnoses. Vascular imaging in patients with JFS or JFA and enlarged EF revealed atresia or stenosis of the jugular veins and enlarged basal EV. JFA was seen in all patients with the FGFR3 mutation with crouzonoid features and acanthosis nigricans. Four patients had prominent EF without JFS. Six patients had normal JF and lacked enlarged EF.
CONCLUSION: Enlarged basal EF are common in syndromic craniosynostosis and are usually associated with JFS or JFA. Bilateral basilar venous atresia is most common in patients with the FGFR3 ala391glu mutation and crouzonoid features with acanthosis nigricans, but may be found in patients with FGFR2 mutations. Skull base vascular imaging should be obtained in patients with syndromic craniosynostosis with enlarged EF.
Premature fusion of the coronal sutures is the predominant feature of several distinct monogenic disorders (1). These autosomal dominant craniosynostoses are conventionally known by eponyms and phenotypic features such as the severity of craniofacial abnormality and associated limb anomalies (2). The syndromic craniosynostoses include Apert syndrome, Crouzon syndrome, Pfeiffer syndrome, and Saethre-Chotzen syndrome.
Crouzon syndrome is characterized by proptosis, maxillary hypoplasia, and the absence of major limb abnormalities. Once thought to be a variant of Crouzon syndrome (3), another disorder called crouzonoid and acanthosis nigricans is now considered to be a discrete entity (4, 5). Periapical cemental dyplasia of the jaws (6) and spinal stenosis have also been reported in crouzonoid patients with acanthosis nigricans (7).
Patients who have Pfeiffer syndrome share many of the craniofacial characteristics of Crouzon syndrome; however, they are more likely to have pansynostosis and limb deformities. Their toes and thumbs are often broad, and sometimes medially deviated, with cutaneous syndactyly (2, 5). Apert syndrome is characterized by syndactyly/symphalangism and severe cranial dysmorphism (2). However, there are many patients with bilateral coronal synostosis who do not fit easily into any of these categories or have features that overlap the eponymous subgroups (5).
Recently the mutational basis for all of the well-known syndromic craniosynostoses has been elucidated and has provided insights into the mechanisms by which these malformations arise (8, 9). These mutations involve genes coding for fibroblast growth factor receptors (FGFR1, 2, or 3), known to have specialized roles in skeletal development (5, 8, 10, 11).
In addition to deformity of the cranium, intracranial hypertension, hydrocephalus, hindbrain herniation (Chiari I malformation), and severe cranial base deformity have been described in syndromic craniosynostosis (12–18). Impaired venous drainage has been proposed as an important factor in the pathogenesis of intracranial hypertension and hydrocephalus. We hypothesize that enlarged emissary foramina (EF) and veins (EV) develop to provide collateral venous drainage because of jugular vein stenosis (JVS) or atresia (JVA). Patients with syndromic craniosynostosis usually undergo frontoorbital advancement in infancy, midfacial advancement in childhood, and may require posterior fossa cranial remodeling or cervicooccipital decompression for Chiari I malformations. Disruption of the enlarged EV may result in massive, sometimes fatal, intraoperative hemorrhage (12). In this study, we established normal ranges for sizes of EF and jugular foramina (JF) in a control group and documented the prevalence of enlarged EF in patients with various craniosynostoses. The findings of enlarged EF were correlated with enlarged EV and size of JF. We also attempted to correlate our findings with phenotypic and molecular diagnoses.
Methods
Patients
A list of patients with coronal synostosis and known phenotypic and molecular diagnoses was obtained from the records of the Craniofacial Center at Children's Hospital. Only those patients who had undergone thin-section CT of the cranial base were included in this study (n = 33). All patients had been classified clinically based on craniofacial features and the appearance of the hands and feet. Patients with Saethre-Chotzen syndrome or with synostosis involving only one cranial suture were not included in this study. This group of 33 included 22 male and 11 female patients, ranging in age from 1 month to 28 years at the time of the initial thin-section CT examination.
The medical records were reviewed for craniofacial and extremity anomalies, ventricular drainage procedures, posterior fossa decompression for chronic tonsillar herniation (CTH), and for excessive intraoperative bleeding.
A control population was assessed for the purpose of establishing normal values for the cross-sectional area of the JF at the skull base. The data were obtained from 77 CT scans of the temporal bone in 76 patients (46 male and 30 female patients) ranging in age from 12 days to 20 years. The clinical notes were assessed to ensure that none of the patients had a history of craniofacial abnormality. Clinical indications for temporal bone CT in this group included mastoiditis, hearing loss, otorrhea, cholesteatoma, and trauma.
CT Protocol
All study patients were imaged with CT of the head from the vertex to the hyoid bone by use of either a GE 9800 or Advantage CT scanner (General Electric Medical Systems; Milwaukee, WI). Patients were scanned with 1- to 3-mm conventional axial images (n = 22) or helical images (n = 11) with 3-mm increments at a pitch of 1:1. All patients in the control group were scanned with 1- to 3-mm conventional axial images of the temporal bones. Scans were obtained using 120 kV, 80–200 mA, a 1- to 2-s scan time, a 20- to 25-cm field of view (FOV), and a 512 × 512 matrix. Reformatted coronal and sagittal images and 3D images were obtained from all study patients. CT venography (CTV) was done (n = 3) using a helical acquisition with 3-mm increments at a pitch of 1:1, 30 seconds after commencing a bolus IV injection of 2cc/kg Ioversol 68% (w/v) (Optiray 320; Mallinckrodt, Medical Inc., St. Louis, MO).
MR Protocol
Fourteen patients underwent MR imaging of the brain on a 1.5-T system (General Electric Medical Systems, Milwaukee, WI) with a quadrature head coil. The imaging protocol consisted of sagittal conventional spin-echo T1-weighted images (450/14/1 [TR/TE/excitations]; 5-mm slice thickness; 1-mm interslice gap; 24-cm FOV; 256 × 192 matrix), axial fast spin- echo (FSE) T2-weighted images (4000/84/1; 5-mm slice thickness; 1-mm interslice gap; 22- to 24-cm FOV; 256 × 192 matrix), axial FSE proton density–weighted images (2000/17/1; 5-mm slice thickness; 1-mm interslice gap; 22- to 24-cm FOV; 256 × 192 matrix), or fluid-attenuated inversion recovery images (10004/175/1 ; TI, 2200 ms; 5-mm slice thickness; 1-mm interslice gap; 24-cm FOV; 256 × 192 matrix). Flow-sensitive pulse sequences included 2D time-of-flight (TOF) MR venography (MRV) (n = 2) (23/4.9/1 ; 60-degree flip angle; 49.5-mm slice thickness; 20 × 15-cm FOV; 256 × 192 matrix), 3D TOF MRV (n = 3) (53/6.9/1 ; 25-degree flip angle; 1-mm slice thickness; 22 × 16.5-cm FOV; 512 × 160 matrix), spoiled gradient-recalled imaging (n = 1) (24/6.9/2; 50-degree flip angle, 5-mm slice thickness; 1-mm interslice gap; 20 × 15-cm FOV; 256 × 160 matrix), or 2D phase- contrast MRV (38/9.3/1 ; 20-degree flip angle; 10-mm slice thickness; 0-mm interslice gap; 20-cm FOV; 256 × 128 matrix; 5-cm/s flow velocity). Contrast material was not used for MRV pulse sequences.
Conventional Angiography
Venous anatomy at the cranial base was evaluated in two patients with digital subtraction angiography.
Image Review and Analysis
Control Group
One pediatric neuroradiologist measured the cross-sectional area of the JF at the skull base in mm2 for children in the control group by use of a computer-generated free-hand trace method. Age, gender, and the sum of right and left JF were tabulated. The width of the right and left mastoid and central occipital EF were measured in mm.
The relationship between JF size and age in healthy children between 1 month and 16 years of age (n = 77) was then assessed using linear and nonlinear regression analysis. Several different linear and nonlinear statistical models were compared to determine the most accurate relationship for describing the data and to use this model to establish pediatric normal reference ranges. Goodness-of-fit, evaluated by R-squared, and mathematical simplicity were considered to be the important features in model selection. A log transformation (base 10) was applied to normalize and make the data amenable to Gaussian calculations. Two-way analysis of variance (ANOVA) using log units was performed to investigate the effects of age and gender on JF size and to test whether girls and boys shared a common rate of increase with age. Reference ranges were derived from the 95% individual prediction intervals from log-transformed variables by using least-squares regression analysis (19). Finally, predicted values and confidence intervals (CI) were converted back to the original units by calculating antilogs. Because one of the primary goals of developing the normal ranges was to evaluate the patients with abnormal findings in the study, as defined using the CT standard of reference, sensitivity and specificity of the ranges were determined using standard formulas with 95% CI approximated by Pratt's method (20). Herein, sensitivity corresponds to the proportion of patients in the study classified as having JF atresia (JFA) or stenosis (JFS), according to CT, who were correctly identified as having abnormal findings by the ranges. Specificity refers to the proportion of 77 normal findings correctly classified by the established reference ranges. Both sensitivity and specificity are expressed as percentages. For all statistical tests, two-tailed values of P <.05 were considered statistically significant. Analysis of the data was performed using the SPSS software package (version 10.0, SPSS Inc., Chicago, IL).
Study Group
Two pediatric neuroradiologists independently reviewed the imaging findings in the study group. The widths of the right and left occipitomastoid and central occipital EF were measured on CT examinations. Measurements were obtained for the cross-sectional area of the JF at the skull base in 39 children. In four patients, the original CT scans could not be reloaded onto the workstation for JF area measurements. The JF were assessed as being normal, stenotic, or atretic. Sagittal reformatted CT images and/or sagittal T1-weighted MR images were used to document the presence or absence of tonsillar herniation. Ventricular size was assessed as being either normal or dilated, and termed ventriculomegaly if no ventricular drainage procedure had been done, or hydrocephalus if the patient had required ventricular shunting. The CTV, MRV, and conventional angiographic images were assessed independent of the CT bone window images for size and patency of the transverse and sigmoid sinuses and JV, and for the presence of prominent basal EV.
Results
Phenotypic Diagnosis
The patients were initially classified clinically as having Crouzon syndrome (n = 10), crouzonoid features with acanthosis nigricans (n = 3), Apert syndrome (n = 10), Pfeiffer syndrome (n = 4), or clinically nonspecific coronal synostosis and brachycephaly/turricephaly with or without midfacial retrusion and/or minor limb anomalies (n = 6).
Molecular Diagnosis
The results of genetic analysis are presented in Tables 1–6. All but one case had been labeled Crouzon syndrome and all cases labeled Pfeiffer syndrome (n = 13) had an FGFR2 point mutation. A wide phenotypic range was observed in patients with identical mutations (Tables). One patient who was diagnosed with Crouzon syndrome had FGFR3 Pro250Arg, the mutation most frequently encountered in the clinically unclassified patients (n = 3). All patients with the crouzonoid phenotype and acanthosis nigricans had the FGFR3 ala391glu mutation (n = 3). All Apert syndrome patients (n = 10) had one of the two known FGFR2 point mutations. The clinically unclassified patients had either an FGFR3 pro250arg (n = 3) or FGFR2 (n = 3) mutation.
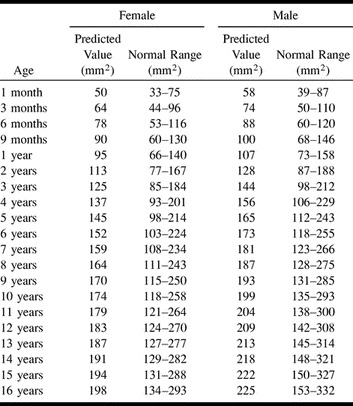
TABLE 1:
Predicted value and normal ranges for the sum of the cross-sectional area of the right and left jugular foramina according to age and gender
Clinical History
All patients underwent craniofacial procedures. One patient (Case 1, Fig 1) with crouzonoid and acanthosis nigricans developed excessive intraoperative hemorrhage during attempted craniotomy for repeat frontoorbital advancement. The procedure was abandoned. Twelve patients required ventricular shunting for hydrocephalus. Posterior fossa decompression for Chiari I malformation was required in five patients; in one, the dura was not opened to avoid the risk of bleeding from visibly enlarged basal veins at the level of the foramen magnum. In a second patient, prominent basal collateral veins were encountered and oversewn to avoid intraoperative bleeding. Another patient required an occipital craniotomy for relief of increased intracranial pressure. Very large veins were observed draining directly through the skull at the level of the torcular, and other large veins were seen along the course of the transverse sinus, and were associated with bleeding. To avoid laceration, a large vein at the torcular was clipped (Case 3, Fig 2). This patient subsequently developed a posttraumatic arteriovenous fistula that was treated surgically.
fig 1.

Case 1: Eight-year-old girl with crouzonoid features, acanthosis nigricans, and FGFR3 ala391glu mutation.
A, Helical CT shows bilateral JFA (arrows) and a large left mastoid emissary foramen (arrowheads).
B, Axial collapsed maximum intensity projection (MIP) from 3D TOF MRV (53/6.9/1 [TR/TE/excitations]) reveals atresia of the right transverse and sigmoid sinuses and the both internal JV (arrowheads). Large occipitomastoid EV arise from the left transverse and sigmoid sinuses (arrows).
fig 2.

Case 3: Three-year-old boy with crouzonoid features, acanthosis nigricans, and FGFR3 ala391glu mutation.
A, Axial CT shows a large central occipital emissary foramen (arrow). A small left mastoid emissary foramen is seen (arrowhead).
B, Axial CT 2 years after surgical clipping of the occipital emissary vein (arrowhead) shows marked enlargement of the left mastoid emissary foramen (arrow).
C, Digital subtraction cerebral angiogram, frontal projection, shows atresia of the right sigmoid sinus and lateral portion of the left sigmoid sinus (arrows) and atresia of both internal JV. There are large left mastoid EV arising from the proximal left sigmoid sinus (arrowheads).
Imaging
Control Group
The best model for describing the relationship between JF size and age was a nonlinear power function revealing that JF size increased with age according to a curve rather than a straight line (R-squared = 0.80 for female patients, R-squared = 0.71 for male patients; P <.0001 in each case). There was a significant difference between female and male patients, which indicated a larger JF size for male patients (P = .015). Based on the interaction F-test in the two-way ANOVA, there was clearly no significant difference in the rate of increase in JF size with age (ie, slope) between female and male patients (P = .77). Because there was a gender effect, the pediatric reference ranges are provided separately for female and male patients (Table 1>). Using these ranges, 76 of the 77 normal scans were correctly classified as falling inside the normal range (specificity = 99%, 95% CI = 93–100%). Among the study group, JF area measurements were available in 19 of 23 children considered to have JFA or JFS. All of these were correctly classified as falling outside and below the normal range (sensitivity = 100%, 95% CI = 82–100%)
EF were usually not seen or measured approximately 1 mm in width (n=21) in patients younger than 2 years. In three children younger than 2 years, one mastoid EF measured up to 2 mm in width. EF were also more frequently not seen or measured approximately 1 mm in width (n = 32) in children 2 years or older. In 16 children, one mastoid EV measured up to 2 mm in width. In a small number of children (n = 5), all 9 years or older, one mastoid EF measured up to 3 mm in width. In the control group, the central occipital EF was not seen in 35 of 77 children and did not exceed 1 mm in width in any child.
Study Group
Either one (n = 5), two (n = 15) or three (n = 3) prominent EF ≥ 3 mm in diameter and bilateral JFS or JFA were identified in 23 patients. These patients were divided into three groups: group 1 had bilateral JFA (n = 5), group 2 had unilateral JFA and contralateral JFS (n = 5), and group 3 had bilateral JFS (n = 13). Group 4 had prominent EF without stenotic JF (n = 4). Group 5 had normal JF and lacked enlarged EF (n = 6) (Tables 2–6).
TABLE 2:
Group 1. Bilateral jugular foraminal atresia
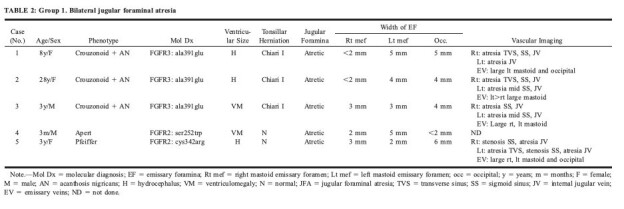
Group 1: Bilateral JFA with enlarged EF were found in all patients with crouzonoid features and acanthosis nigricans (n = 3), in Apert syndrome (n = 1), and in Pfeiffer syndrome (n = 1) (Table 2>). Conventional angiography (n = 2) or CTV (n = 2) was obtained in four of these patients and confirmed bilateral JVA (Fig 2C). Additional findings included atresia (n = 2) or stenosis (n = 1) of the right transverse sinus, atresia (n = 3), or stenosis (n = 1) of the right sigmoid sinus, atresia of the left transverse sinus (n = 1), and distal atresia (n = 2) or stenosis (n = 1) of the left sigmoid sinus (Fig 1B). Enlarged EV were seen in all of these patients, and one child had prominent duplicated occipital sinuses draining inferiorly from the torcular to the posterior condyloid canals (Case 5, Fig 3).
fig 3.

Case 5: Three-year-old girl with Pfeiffer syndrome and FGFR2 cys342arg mutation. Frontal MIP of a CT venogram shows occlusion of the transverse sinuses (arrowheads) and large duplicated occipital sinuses (arrows). fig 4. Case 20: Ten-year-old girl with Pfeiffer syndrome and FGFR2 ser354cys mutation. Helical CT shows a large right occipitomastoid emissary foramen (arrowhead). The right jugular foramen is stenotic, with prominent septations (arrow). The left JF appeared stenotic on a caudal image of the skull base (not shown)
Abnormal ventricular size characterized as hydrocephalus (n = 3) or ventriculomegaly (n = 2) was present in five patients. Chiari I malformation was found in all patients with crouzonoid features and acanthosis nigricans.
Group 2: Unilateral JFS and contralateral JFA was seen in Crouzon syndrome (n = 3), Apert syndrome (n = 1), and Pfeiffer syndrome (n = 1) (Table 3>). In one child (case 6), the left sigmoid plate appeared markedly thinned. MRV (n = 1) and CTV (n = 1) revealed atresia of one transverse sinus, stenosis or atresia of the ipsilateral sigmoid sinus, and atresia of the ipsilateral JV. This was right-sided in one patient and left- sided in the other. In both patients, there were large contralateral transverse and sigmoid sinuses with stenosis of the JV at the junction with the sigmoid sinuses and diversion of blood through very large mastoid EV.
TABLE 3:
Group 2. Unilateral jugular foraminal atresia and contralateral jugular foraminal stenosis
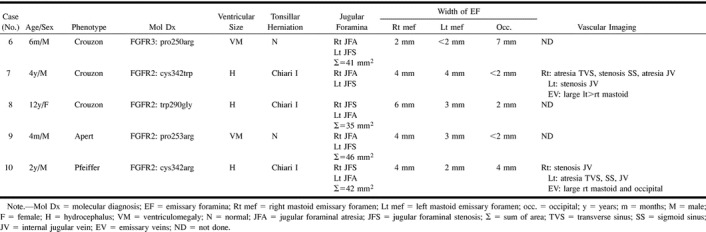
As in group 1, these patients had either hydrocephalus (n = 3) or ventriculomegaly (n = 2). Chiari I malformation was only seen in the children who had hydrocephalus.
Group 3: Bilateral JFS was noted in Crouzon syndrome (n = 4), Apert syndrome (n = 5), Pfeiffer syndrome (n = 1) (case 19, Fig 4), and in clinically unclassified patients (n = 3) (Table 4>). In two patients (cases 13, 18), the right sigmoid plate was markedly thinned or deficient and both right and left sigmoid plates were attenuated in one patient (case 19). MRV (n = 4) or CTV (n = 1) revealed bilateral JVS (n = 5) at the junction of the sigmoid sinus with the JV. Additional features included atresia of the left transverse sinus, stenosis of the left sigmoid sinus (n = 3), and stenosis of both right and left transverse and sigmoid sinuses (n = 1) (case 12, Fig 5). Enlarged occipitomastoid EV were seen extending from the sigmoid sinus or torcular to the occipital soft tissues (n = 5) (Figs 5 and 6).
TABLE 4:
Group 3. Bilateral jugular foraminal stenosis
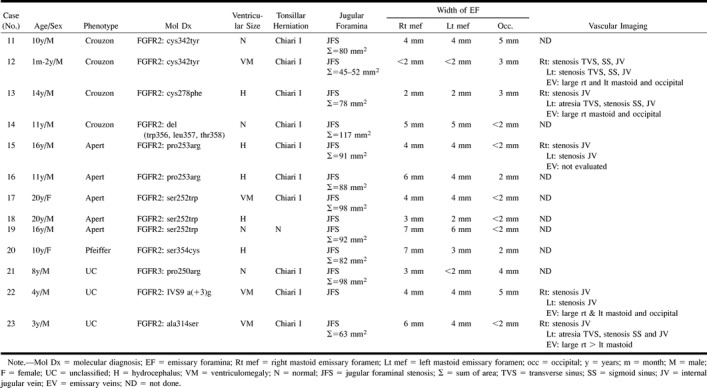
fig 5.

Case 12: A 21-month-old girl with Crouzon syndrome and FGFR2 cys342tyr mutation. Axial collapsed MIP from 3D TOF MRV (53/6.9/1) 20 months after the initial CT shows JFS. There is stenosis of both transverse and sigmoid sinuses and the internal JV (arrowheads). There are large, right, occipitomastoid EV (arrows). fig 6. Case 23: Three-year-old boy with clinically unclassified, bilateral, coronal synostosis and FGFR2 ala314ser mutation. Axial T1-weighted MR imaging (450/14/1) reveals flow voids within large bilateral mastoid EV (arrows). The JV are stenotic (arrowheads)
Hydrocephalus (n = 5) or ventriculomegaly (n = 4) were found in two thirds of group 3, with normal-sized ventricles (n = 4) in one third. Chiari I malformation was seen in association with hydrocephalus (n = 3), ventriculomegaly (n = 4), and normal-sized ventricles (n = 3). In two children who did not undergo MR imaging, the craniocervical junction was not adequately assessed on the sagittal reformatted CT images.
Group 4: Four patients had prominent EF without stenotic JF. Three children had bilaterally (cases 24, 26) or unilaterally (case 25) thinned or deficient sigmoid plates (Fig 7). MRV in one of these patients revealed bilateral stenosis at the junction of the JV with the sigmoid sinus (case 24, Fig 7). The torcular appeared irregular, and there was atresia of the left transverse sinus, with stenosis of the left sigmoid sinus. Large, central, occipital and right mastoid EV were also noted in this patient.
fig 7.

Case 24: Two-year-old girl with Crouzon syndrome and FGFR ser354cys mutation.
A, Axial CT shows that the JF are not stenotic (asterisks).
B, Magnified, oblique, frontal MIP from 3D TOF MRV (53/6.9/1) reveals a focal stenosis at the junction of the right sigmoid sinus with the internal JV (arrow). There is a large right mastoid EV (arrowheads).
Hydrocephalus (n = 1), or ventriculomegaly (n = 3) were seen in group 4. Chiari I malformation was documented in one child with hydrocephalus.
Group 5: Six patients had normal JF without enlarged EF. A dehiscent left jugular bulb was seen in one patient. No vascular imaging studies were obtained from these patients. Ventriculomegaly (n = 5) was more frequent than was the finding of normal-sized ventricles (n = 1). Chiari I malformation was seen in three patients, including one with normal-sized ventricles.
Discussion
Obstruction of dural and jugular venous outflow is associated with development of collateral venous circulation through mastoid and central occipital emissary vessels (21). Jugular venous obstruction and enlarged EV have been reported in association with syndromic craniosynostoses, but the prevalence of enlarged EF in association with syndromic craniosynostoses is unknown (12, 15, 22–24). In our series, 27 of 33 patients with syndromic craniosynostosis were found to have enlarged basal EF, usually but not invariably, in association with stenosis or atresia of the JF. Imaging of cranial venous anatomy in 12 patients correlated with the CT examinations of the skull base and confirmed the presence of enlarged EV and variable stenosis or atresia of the posterior fossa dural venous sinuses and JV.
There are many factors that may alter either the venous or osseous anatomy in patients with syndromic craniosynostosis. During normal fetal development, morphologic changes have been observed in the formation of the dural venous sinuses of the posterior fossa. Toward the end of the 1st trimester, a narrowed segment termed the jugular sinus exists between the sigmoid sinus and the JV (25). The inner diameter of the jugular sinus usually remains at 1 to 2 mm until the 3rd trimester, resulting in a period of intraluminal hypertension with enlargement of the transverse sinus (from the 3rd to 7th fetal month), and the development of multiple EV to provide collateral extracranial venous drainage (25). Lumenal irregularity, segmental absence of the sinus (particularly the medial portion), or septations can accompany the changes in size of the transverse sinus, as noted in vascular imaging studies in some of our patients. The jugular bulb develops postnatally and is usually only seen after the age of 2 years. This should be taken into consideration when assessing JF size in children under 2 years. Our normal ranges for JF size take into account the effects of both age and gender. These ranges can be useful for sequential measurements of JF area in an individual patient over time. One boy with Crouzon syndrome (case 12) underwent CT at 1 month of age. Combined right and left JF area fell within normal limits for age and gender at that time. However, a repeat CT examination at 2 years of age revealed failure of interval growth of the JF, with area measurements well below the predicted normal range for age. As some of the children included in group 5 (normal JF, normal EF) were under 1 year, they may still be at risk for the development of JFS and enlarged EF, which could be detected on follow-up CT scans.
Extracranial drainage of the posterior fossa dural sinuses can occur from routes other than the JV, including drainage via the condylar veins or the marginal (occipital) sinus into the vertebral, paravertebral, and/or deep cervical veins; the mastoid EV; and the occipital EV into the occipital (or posterior auricular) veins (25). The usual size of the mastoid EV has been reported as 1 mm, with 10% over 2 mm in size, and only exceptionally as much as 4 mm in width (26). In our control group, EF were usually ≤ 2mm in width, and only five of 76 children, all older than 9 years, had one EF of approximately 3 mm in width.
We found that enlarged EV in patients with syndromic craniosynostosis are associated with obstruction of dural venous sinus outflow. Stenosis or atresia of the sigmoid sinus and JV can occur for a variety of factors. These include synostosis of cranial base sutures, resulting in JFS or JFA (27), and alteration in obliquity with canting of the petrous bone, turbulence and progressive focal stenosis of the JV, and bony encasement of the dural venous sinuses (12). The role of altered FGFR activity has yet to be determined. Evidence suggests that the synchondroses of the cranial base are completely fused at birth in Crouzon syndrome and fuse early in Apert syndrome (27). Despite the presence of collateral venous drainage, relative impairment of dural venous drainage can cause intracranial hypertension and impaired CSF resorption.
Ventriculomegaly (either stable or progressive) and/or dilatation of the subarachnoid spaces are relatively common findings in patients with syndromic craniosynostosis. Ventricular dilatation alone does not indicate raised intracranial pressure (18). Approximately 12% of these patients require surgical treatment for progressive ventricular dilatation (21). The presence of hydrocephalus in 11 of 23 patients with JFA or JFS (Groups 1–3) supports the importance of venous hypertension as a pathophysiologic mechanism in the development of hydrocephalus in patients with craniosynostosis (15). Hydrocephalus occurred in one patient (case 24) with a normal JF, enlarged EF, and focal JVS, but did not occur in any patient who had normal JF and lacked enlarged EF.
Under normal conditions, CSF pressure is higher than is the venous pressure within the superior sagittal sinus. This pressure gradient is necessary for CSF resorption (15). Elevated superior sagittal sinus venous pressure has been implicated in the pathogenesis of hydrocephalus in infants with a fixed obstruction of venous outflow, often seen in achondroplasia and syndromic craniosynostosis (15). Venous hypertension caused by JFS results in higher CSF pressure to maintain CSF outflow (15). In addition to venous hypertension and increased CSF hydrostatic pressure, lambdoid synostosis with crowding of the posterior fossa may increase venous outflow resistance by compressing the sigmoid sinus. Hindbrain herniation can also contribute to increased CSF outflow resistance (16, 23). Raised intracranial pressure is more likely with bilateral coronal synostosis and with increasing number of sutures involved (28). Despite correction of the cranial deformity, a small percentage of patients who undergo early craniotomy and decompression will develop symptoms of raised intracranial pressure (14).
The association of CTH with syndromic craniosynostosis has been described (13, 23, 29). CTH has been found in 72.7% of patents with Crouzon syndrome and 1.9% with Apert syndrome. We found CTH in 100% of patients with crouzonoid features and acanthosis nigricans, 90% with Crouzon syndrome, 66% with clinically unclassified coronal synostosis, and 30% with Apert or Pfeiffer syndrome. The higher incidence of CTH in Crouzon syndrome has been linked to the premature fusion of the lambdoid sutures in the first 2 years of life (23), or possibly to increased venous turgor in children with JFS and treated hydrocephalus (13).
Genetic linkage analysis has implicated the mutation of three members of the FGFR gene family as the underlying cause of several skeletal dysplasias and autosomal dominant craniosynostotic syndromes (3, 5, 8, 10). The mutation is thought to result in inappropriate receptor signaling that affects the growth and/or patterning of bone (2). FGFR 3 mutations have been reported in achondroplasia, thanatophoric dysplasia, hypochondroplasia, and in patients with craniosynostosis and highly variable phenotype.
There is considerable genetic heterogeneity in the coronal craniosynostotic syndromes, and the phenotype does not always predict the genotype. There is also phenotypic heterogeneity even in patients with identical mutations, probably the result of modulation by genes modifying the effects of the FGFR mutations and by environmental factors (5). The use of molecular diagnosis as a predictor of patient outcome has been evaluated, and there are at least three mutations that allow phenotypic prognostication of a patient with craniosynostosis. Point mutations at position 252 and 253 of FGFR 2 are always associated with the distinctive phenotype of Apert syndrome (30). The FGFR3 ala391glu mutation defines the molecular basis for patients with the crouzonoid phenotype and acanthosis nigricans, Chiari I malformation, JFA, and ventriculomegaly. This mutation is close to the predominant mutation for achondroplasia in which JFS and spinal stenosis are commonly seen (5).
In this study, the only mutation that appeared to correlate with severe basilar venous stenosis or atresia was FGFR3 ala391glu associated with crouzonoid features and acanthosis nigricans. The FGFR3 pro250arg mutation was most commonly seen in clinically unclassified patients and occurred in two of six patients who did not have enlarged EF. However, two of four patients with this mutation did have enlarged EF and JFS or JFA.
Conclusion
Enlarged basal EF are common in syndromic craniosynostosis and result from stenosis or atresia of the JF. The presence of normal JF does not preclude the possibility of focal JVS. Bilateral basilar venous atresia is most common in patients with the FGFR3 ala391glu mutation and crouzonoid features with acanthosis nigricans, but may be found in patients with FGFR2 mutations. Preoperative assessment of patients with syndromic craniosynostosis and enlarged EF should include assessment of the basilar venous drainage.
TABLE 5:
Group 4. Normal jugular foramina; prominent basal emissary foramina
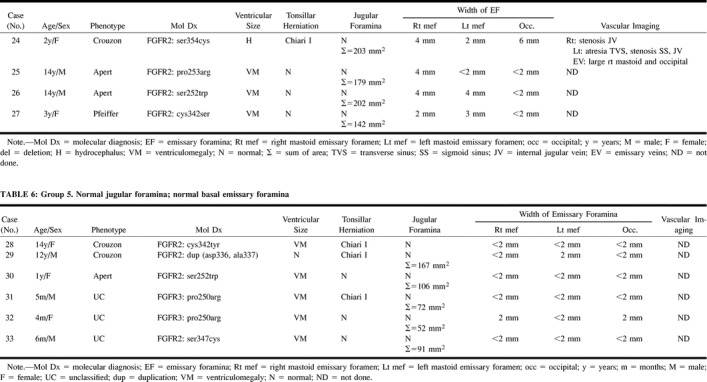
TABLE 6:
Group 5. Normal jugular foramina; normal basal emissary foramina

Footnotes
D.S. and U.M. were supported by a grant from the Deutsche Forschungsgemeinschaft (STE770/1–2).
Presented at the Annual Meeting of the American Society of Neuroradiology, Atlanta, GA, April 2000, and the American Society of Head and Neck Radiology, Washington, DC; May 2000.
Address reprint requests to Caroline D. Robson, MBChB, Department of Radiology, Children's Hospital, 300 Longwood Avenue, Boston, MA 02115
References
- 1.Wilkes D, Rutland P, Pulleyn LJ, et al. A recurrent mutation, ala391glu, in the transmembrane region of FGFR3 causes Crouzon syndrome and acanthosis nigricans. J Med Genet 1996;33:744-748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Moerlooze L, Dickson C. Skeletal disorders associated with fibroblast growth factor receptor mutations. Curr Opin Genet Dev 1997;7:378-385 [DOI] [PubMed] [Google Scholar]
- 3.Meyers GA, Orlow SJ, Munro IR, Przylepa KA, Jabs EW. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet 1995;11:462-464 [DOI] [PubMed] [Google Scholar]
- 4.Cohen MM, Jr. Let's call it “Crouzonodermoskeletal syndrome” so we won't be prisoners of our own conventional terminology [letter]. Am J Med Genet 1999;84:74. [PubMed] [Google Scholar]
- 5.Mulliken JB, Steinberger D, Kunze S, Muller U. Molecular diagnosis of bilateral coronal synostosis. Plast Reconstr Surg 1999;104:1603-1615 [DOI] [PubMed] [Google Scholar]
- 6.Suslak L, Glista B, Gertzman GB, Lieberman L, Schwartz RA, Desposito F. Crouzon syndrome with periapical cemental dysplasia and acanthosis nigricans: the pleiotropic effect of a single gene? Birth Defects Orig Artic Ser 1985;21:127-134 [PubMed] [Google Scholar]
- 7.Superti-Furga A, Locher ML, Steinlin M. Crouzon syndrome with acanthosis nigricans, spinal stenosis and desmo-osteoblastomas: pleiotropic effect of the FGFR-3 ALA-391-GLU mutation (abstract). J Craniomaxillofac Surg 1996;24::112 [Google Scholar]
- 8.Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 1994;8:98-103 [DOI] [PubMed] [Google Scholar]
- 9.Reardon W, Winter RM. The molecular pathology of syndromic craniosynostosis. Mol Med Today 1995;1:432-437 [DOI] [PubMed] [Google Scholar]
- 10.Park WJ, Theda C, Maestri NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet 1995;57:321-328 [PMC free article] [PubMed] [Google Scholar]
- 11.Muenke M, Schell U, Hehr A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet 1994;8:269-274 [DOI] [PubMed] [Google Scholar]
- 12.Thompson DN, Hayward RD, Harkness WJ, Bingham RM, Jones BM. Lessons from a case of kleeblattschadel. Case report [see comments]. J Neurosurg 1995;82:1071-1074 [DOI] [PubMed] [Google Scholar]
- 13.Francis PM, Beals S, Rekate HL, Pittman HW, Manwaring K, Reiff J. Chronic tonsillar herniation and Crouzon's syndrome. Pediatr Neurosurg 1992;18:202-206 [DOI] [PubMed] [Google Scholar]
- 14.Siddiqi SN, Posnick JC, Buncic R, et al. The detection and management of intracranial hypertension after initial suture release and decompression for craniofacial dysostosis syndromes. Neurosurgery 1995;36:703-708; discussion 708–709 [DOI] [PubMed] [Google Scholar]
- 15.Sainte-Rose C, LaCombe J, Pierre-Kahn A, Renier D, Hirsch JF. Intracranial venous sinus hypertension: cause or consequence of hydrocephalus in infants? J Neurosurg 1984;60:727-736 [DOI] [PubMed] [Google Scholar]
- 16.Cinalli G, Chumas P, Arnaud E, Sainte-Rose C, Renier D. Occipital remodeling and suboccipital decompression in severe craniosynostosis associated with tonsillar herniation. Neurosurgery 1998;42:66-71; discussion 71–73 [DOI] [PubMed] [Google Scholar]
- 17.Golabi M, Edwards MS, Ousterhout DK. Craniosynostosis and hydrocephalus. Neurosurgery 1987;21:63-67 [DOI] [PubMed] [Google Scholar]
- 18.Hanieh A, Sheen R, David DJ. Hydrocephalus in Crouzon's syndrome. Childs Nerv Syst 1989;5:188-189 [DOI] [PubMed] [Google Scholar]
- 19.Dixon WJ, Massey FJ. Introduction to Statistical Analysis.. 4 ed. New York: McGraw-Hill 1983;:92-95
- 20.Blyth CR. Approximate binomial confidence limits. J Am Stat Assoc 1986;81:843-855 [Google Scholar]
- 21.Cinalli G, Sainte-Rose C, Kollar EM, et al. Hydrocephalus and craniosynostosis. J Neurosurg 1998;88:209-214 [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Perez D, Vander Woude DL, Barnes PD, Scott RM, Mulliken JB. JFl stenosis in Crouzon syndrome. Pediatr Neurosurg 1996;25:252-255 [DOI] [PubMed] [Google Scholar]
- 23.Cinalli G, Renier D, Sebag G, Sainte-Rose C, Arnaud E, Pierre-Kahn A. Chronic tonsillar herniation in Crouzon's and Apert's syndromes: the role of premature synostosis of the lambdoid suture. J Neurosurg 1995;83:575-582 [DOI] [PubMed] [Google Scholar]
- 24.Tokumaru AM, Barkovich AJ, Ciricillo SF, Edwards MS. Skull base and calvarial deformities: association with intracranial changes in craniofacial syndromes [see comments]. AJNR Am J Neuroradiol 1996;17:619-630 [PMC free article] [PubMed] [Google Scholar]
- 25.Okudera T, Huang YP, Ohta T, et al. Development of posterior fossa dural sinuses, emissary veins, and jugular bulb: morphological and radiologic study. AJNR Am J Neuroradiol 1994;15:1871-1883 [PMC free article] [PubMed] [Google Scholar]
- 26.Boyd GI. The emissary foramina in the cranium of man and the anthropoids. J Anat 1930;65:108-121 [PMC free article] [PubMed] [Google Scholar]
- 27.Kreiborg S, Marsh JL, Cohen MM, Jr, et al. Comparative three-dimensional analysis of CT-scans of the calvaria and cranial base in Apert and Crouzon syndromes. J Craniomaxillofac Surg 1993;21:181-188 [DOI] [PubMed] [Google Scholar]
- 28.Renier D, Marchac D. Craniofacial surgery for craniosynostosis: functional and morphological results. Ann Acad Med Singapore 1988;17:415-426 [PubMed] [Google Scholar]
- 29.Saldino RM, Steinbach HL, Epstein CJ. Familial acrocephalosyndactyly (Pfeiffer syndrome). Am J Roentgenol Radium Ther Nucl Med 1972;116:609-622 [DOI] [PubMed] [Google Scholar]
- 30.Wilkie AO, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome [see comments]. Nat Genet 1995;9:165-172 [DOI] [PubMed] [Google Scholar]