

Abstract
Patient: Male, 9-year-old
Final Diagnosis: Weill-Marchesani syndrome
Symptoms: Joint stiffnes • myopia • short stature
Medication: —
Clinical Procedure: —
Specialty: Endocrinology and Metabolic • Genetics
Objective:
Rare disease
Background:
Short stature is the second most common reason for referral to a pediatric endocrinology clinic. Numerous genetic causes have been identified. Weill-Marchesani syndrome (WMS) is one of the rare genetic disorders that cause short stature. It is caused by homozygous mutations in the FBN1 gene, ADAMTS10 gene, ADAMTS17 gene, or LTBP2 gene. Despite genetic heterogeneity, WMS is clinically homogeneous. It is characterized by short stature, brachydactyly, joint stiffness, ocular abnormalities, mainly microspherophakia and glaucoma, and occasionally cardiac defects.
Case Report:
A 9-year-old boy had bilateral narrow-angle glaucoma with lens subluxation, elevated intraocular pressure, and severe myopia since early childhood. He had phenotypic dysmorphic features and radiological findings consistent with WMS. He underwent lensectomy and scleral-fixated intraocular lens implantation as well as drug treatment to control the intraocular pressure. He was a slow grower, and his growth parameters showed disproportionate short stature with brachydactyly and joint stiffness. Growth hormone provocation tests were subnormal with a peak value of 7.89 ng/mL.
Conclusions:
The constellation of clinical presentation, radiological findings, and the molecular examination confirmed a homozygous familial variant of the ADAMTS10 gene identified by carrier gene testing. This known familial variant creates a premature termination codon classified as a likely pathogenic cause of WMS. In this syndrome, glaucoma treatment is considered the greatest challenge. The disease-causing mechanism in WMS is not known but thought to be due to abnormal actin distribution and organization in fibroblasts as a result of impaired connections between extracellular matrix components and the cytoskeleton.
Keywords: ADAM Proteins, Child, Homozygote, Weill-Marchesani Syndrome
Background
Short stature is the most common reason for referral to a pediatric endocrinologist after diabetes management [1]. A child is considered to have short stature if his height is less than 2 standard deviations from the mean (2.3rd percentile) when compared with children of the same age, sex, and race. When the upper segment to lower segment ratio (US/LS ratio) is in concordance with chronological age, this child is assumed to have proportionate stature [2]. The mean US/LS ratio is 1.7 at birth in both sexes. This decreases to 1.0 at age 10, and further decreases to 0.92 at age 15 in boys and 0.95 at age 13 in girls [3, 4]. The US/LS ratio is determined by first measuring the lower segment from the upper border of the pubic symphysis to the floor while the patient is standing straight. Second, this length is subtracted from the standing height to determine the upper segment length. Finally, the upper segment length is divided by the lower segment length to calculate the ratio, which can then be compared with reference data [4]. However, severe short stature is defined as a height more than 3 standard deviations below the mean and requires urgent assessment if present [2]. Unexplained short stature, before it is termed idiopathic, warrants careful evaluation, especially to rule out any evidence of systemic, endocrine, nutritional, or chromosomal abnormalities [1].
With the recent and tremendous advances in genetic growth disorders, numerous genetic causes of short stature have been identified. They are highly heterogeneous and can cause a broad phenotypic spectrum from mild and isolated short stature to severe and syndromic short stature [1, 5, 6].
The impact of consanguinity and the prevalence of adverse genetic outcomes have been largely ignored for many years, but are now receiving due attention, with special consideration given to the degree of homozygosity in identifying genetic outcomes. Consanguinity is expected to have a greater impact on the etiology of complex diseases, particularly when rare autosomal recessive alleles are implicated [7]. In a recent study, Al-Gazali et al reported that genetic and congenital disorders are more prevalent in Arab countries than in non-Arab developed countries due to the high consanguinity rate (25–60%) [8]. However, the reported prevalence of consanguinity in Saudi Arabia is particularly high, at 57% [9, 10].
Weill-Marchesani syndrome (WMS) is a rare genetic disorder first described by Weill and then further delineated by Marchesani more than 80 years ago [11–13]. The mode of inheritance can be either autosomal dominant (AD), caused by homozygous mutations within the FBN1 gene, or autosomal recessive (AR), caused by homozygous mutations in the ADAMTS10, ADAMTS17, or LTBP2 genes. Despite clinical homogeneity, AD and AR WMS are genetically heterogeneous conditions [14–18].
WMS is characterized by proportionate short stature; brachydactyly; joint stiffness; ocular abnormalities, including micro-spherophakia; shallow anterior chamber causing narrow-angle glaucoma; ectopia lentis; and, occasionally, cardiac defects. Rarely, retinitis pigmentosa has been presented [19], as have peripheral irio-corneal adhesions [20,21]. Of note, people with WMS may present with restricted mouth opening and may have difficulty in intubation, laryngeal stenosis, and postoperative laryngeal spasm that mandates careful preoperative assessment [22]. Short stature and brachydactyly are universal signs that manifest in almost all reported cases of WMS [15]. The relationship between short stature and WMS has received much less attention than studies of adverse genetic or ophthalmic abnormalities. There is no report in the literature on whether short stature in WMS is caused by a growth hormone deficiency or whether these patients respond to growth hormone therapy. In this article, we report a 9-year-old boy with WMS and discuss the various clinical manifestations, genetic testing, and treatment outcomes.
Case Report
A 9-year-old Saudi boy who had been born term, with appropriate birth weight (2.5 kg) following normal spontaneous delivery, had an unremarkable perinatal history except for neonatal jaundice due to G6PD deficiency that required several days of phototherapy.
Since early childhood, however, he was a slow feeder and a slow grower. At the age of 4, he presented with bilateral on and off ocular pain that lasted for 2 years. On examination, he was found to have bilateral narrow-angle glaucoma with the following findings: very shallow anterior chamber, lens subluxation, dense superficial punctate epithelial erosions throughout both corneas, more in the right eye, and elevated intraocular pressure (right eye, 26 mmHg; left eye, 27 mmHg). He was treated with antiglaucomatous treatment, namely brimonidine eye drops, under close monitoring. Two years later he developed poor vision and his intraocular pressure (IOP) was still high in both eyes. He underwent bilateral lensectomy with anterior vitrectomy and scleral-fixated intraocular lens (SF-IOL) implantation under general anesthesia. One year later, he presented with a ruptured globe of the right eye with visual acuity of 200/100, which required primary corneal laceration repair under general anesthesia. Anti-glaucoma treatment with brimonidine, timolol, and dorzolamide was continued. At 1-year follow-up, intraocular pressure had decreased to 13 mmHg in the right eye and 12 mmHg in the left eye. His best corrected visual acuity (BCVA) was 20/100 in the right eye and 20/60 in the left eye. The cornea and lens were clear in both eyes with shallow anterior chamber and no neovascularization of the iris (NVI). Fundus examination showed high myopia in both eyes, with a cup-to-disc (C/D) ratio of 0.4 (normal <0.5). A general physical examination of the patient demonstrated a prepubertal sexual maturity rating corresponding to Tanner stage I and disproportionate short stature with obvious dysmorphic features consistent with WMS. Growth parameters were as follows:
height: 103 cm (far below the third percentile; Z-score=−5.4); weight: 16 kg (far below the third percentile; Z-score=−4.6); body mass index: 15 kg/m2 (24.5th percentile); growth velocity rate: 2 cm/year (SD=−2.5); height age: 4 years; mid-parental height: 157.5 cm (0.35th percentile; Z-score=−2.7); bone age:
corresponding to 7.5 years of age (Greulich and Pyle Atlas of bone age); estimated mature height: 60% (Greulich and Pyle Atlas of Bone Age); predicted adult height: 137 cm (Bayley and Pinneau method); upper segment (US): 55 cm; lower segment (LS): 48 cm; US/LS: 1.14 (SD=+2); arm span (AS): 94.5 cm; AS minus height (94.5–103): −8.5 (more than 2 standard deviations below the mean) (Figure 1A–1C).
Figure 1.

(A) Height of our patient at ages 7 years and 9 years plotted against height for 2- to 20-year-old males (CDC Growth Chart). (B) Body mass index (BMI) of our patient at the age of 9 years plotted against BMI for 2- to 20-year-old males (CDC Growth Chart). (C) Arm span minus height: −8.5 cm; arm span divided by height: 0.92.
The child, who wears eyeglasses, showed characteristic facial dysmorphisms in the form of a narrow palpebral fissure, long eyelashes, facial hypertrichosis, a wide depressed nasal bridge, and thin upper lips. Skeletal manifestations included disproportionate severe short stature (short limbs), brachydactyly (broad short fingers and toes), and clinodactyly, with an overlap of the second and third toes bilaterally. He had broad hands, joint stiffness, and a wide carrying angle of the elbow (Figure 2A–2C). The skeletal survey revealed a small middle phalanx of the little finger, causing a shortening of the little finger (Figure 3). Cardiac auscultation revealed no murmur and normal cardiac sounds.
Figure 2.

(A) Frontal and lateral photograph of the patient with WMS. (B) Photograph of the hand of the WMS patient showing short hands and stubby fingers (brachydactyly), as well as curvature of the fourth and little fingers (clinodactyly). (C) Photograph of the foot of the patient with WMS showing short toes and overlapping second and third toes. WMS – Weill-Marchesani syndrome.
Figure 3.
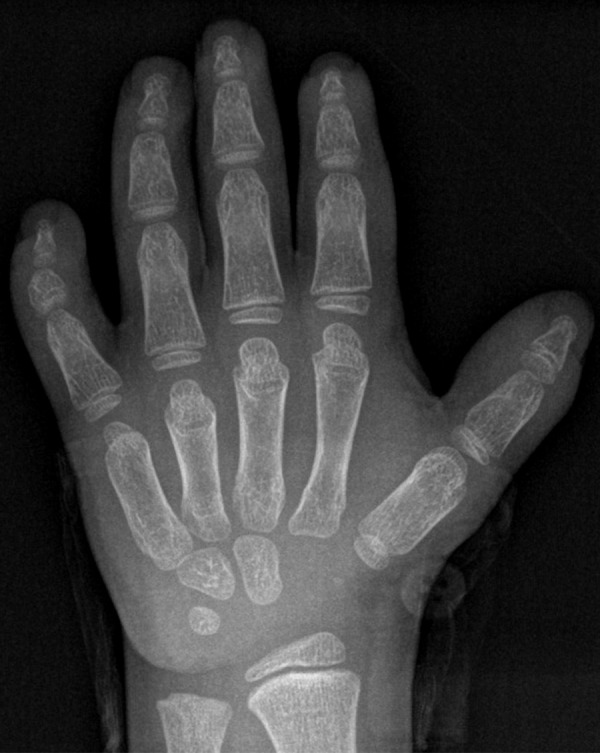
Frontal left-hand radiograph showing small middle phalanx of the little finger causing shortening and curvature of the little finger.
There was no developmental delay, he had normal cognitive functioning, and his school performance was excellent.
The parents of the patient were both healthy, but both were carriers of the same pathogenic variant of the disease. The father was short (156 cm; 0.19%, Z-score: −2.89), with small feet and hands, but had normal intelligence. The mother’s height was 146 cm (0.39%, Z-score: −2.66). The patient had 2 healthy sisters with normal growth and intelligence. He had 2 affected cousins with similar presentations to his own. Both were diagnosed with the same pathogenic variant in the ADAMTS10 gene (Figure 4).
Figure 4.

Pedigree of the family with Weill-Marchesani syndrome including 3 generations.
A likely pathogenic variant in the ADAMTS10 gene, c2050C>T p(Arg684*) had been previously detected in a homozygous state in a relative of the patient. Through targeted genetic testing in the patient, the same familial variant in the ADAMTS10 gene, c2050C>T p(Arg684*), was detected, also in a homozygous state. This mutation creates a premature stop codon that is classified as likely pathogenic (class 2) according to the recommendation of Centogene, LLC (Cambridge, MA, USA) and the American College of Medical Genetics. It is consistent with the genetic diagnosis of Weill-Marchesani syndrome type 1, which is inherited in an autosomal recessive manner.
Laboratory test results were normal, including thyroid function and celiac screening, echocardiogram was unremarkable, and growth hormone provocation tests showed a subnormal response with a peak of 7.8 ng/mL and a normal serum cortisol level.
Discussion
Weill-Marchesani syndrome is a rare connective tissue disorder caused by defects in multiple genes. Four types of WMS have been described. A homozygous mutation in the ADAMTS10 gene causes type I; a mutation in FBN1 causes type II; a mutation in LTBP2 causes type III; and type IV is caused by a mutation in ADAMTS17. Types I, III, and IV are inherited in an autosomal recessive manner, while type II is dominant [14,16,18,23]. Regardless of the mode of inheritance and genetic heterogeneity, the majority of affected children have similar clinical manifestations [23]. The clinical features in our case are similar to those presented in the literature except for the short stature, which was disproportionate.
Faivre et al described the clinical manifestations occurring in WMS based on 128 observations of WMS from 63 families. The characteristic features were as follows: Ophthalmological features included microspherophakia (84%), ectopia lentis (73%), myopia (94%), glaucoma (80%), and cataract (23%). Skeletal manifestations included short stature (98%), brachydactyly (98%), and joint restriction (62%). Other manifestations included cardiac abnormalities (24%) and mental retardation (13%) [15].
In the present case, the child had bilateral glaucoma since the age of 2 years, which was thought to be congenital and was treated supportively. However, later supportive treatment failed to control the high IOP, and then he was treated with lensectomy plus anterior vitrectomy and SF-IOL implantation in both eyes. Glaucoma management poses a great challenge in patients with WMS, as they have a high incidence of recurrent glaucoma, which may lead to angle adhesion and trabecular meshwork damage, resulting in chronically elevated IOP [21,24]. Sirisha et al, in a recent study, reported that half of all patients with glaucoma require surgical intervention and that trabeculectomy has moderate success. Moreover, one should be cautious before trabeculectomy, especially in the presence of lens subluxation, as a shallow anterior chamber or malignant glaucoma may develop [25]. Laser iridotomy has been performed as a treatment option, but it does not necessarily halt the disease progression and surgical removal of the lens may ultimately be necessary [26]. Although the child presented after one year with a ruptured globe of the right eye requiring immediate surgery and corneal laceration repair, his condition was successfully controlled with lensectomy and SFIOL implantation in both eyes plus drug treatment.
The child was noted as short with broad hands and short fingers and toes. He had a disproportionate short stature caused by short legs, as indicated by the ratio between the upper and lower segments, which was 1.14. The trunk to long bones ratio was also disproportionate, as objectified by the arm span to height ratio, which was more than 2 standard deviations below the mean [11].
Initial assessment of a short child should include accurate measurement of height, weight, and estimation of growth velocity plotted on the appropriate chart. Calculation of the midparenteral height and bone age is required to estimate target adult height. Ratios of upper body segment to lower body segment and arm span to height are helpful in determining whether the short child has proportionate or disproportionate body proportions.
Disproportions may be due to discrepancies between the limbs and trunk or between specific limb segments. Disproportionate shortening of the proximal limbs (humerus and femur) causes rhizomelia, whereas disproportionate shortening of the middle parts of the limbs (radius/ulna and tibia/fibula) results in mezomelia. Acromelia results from disproportionately shortened distal limbs (hands and feet). Thus, evaluation for disproportionality provides important information to help narrow the differential diagnosis, including the possibility of skeletal dysplasia [4].
A provocative growth hormone test is indicated when a child has severe short stature, defined as a height more than 3 standards deviations below the mean, a height more than 1.5 standard deviations below the mid-parental height, a height that is more than 2 standard deviations below the mean plus a height velocity over one year that is more than 1 standard deviation below the mean for children under 2 years of age or a decrease in relative height of more than 0.5 standard deviations over 1 year for children over 2 years of age [2,4]. In the absence of short stature, a height velocity greater than 2 standard deviations below the mean over one year, or greater than −1.5 standard deviations over 2 years often mandates pituitary assessment of growth hormone production [4].
In the present case, the child had severe short stature with a height more than 3 standard deviations below the mean with a Z-score of −5.4, and his growth velocity was 2 cm/year (−2.5 standard deviations). This is considered a slow-velocity rate and alone is indicative of the need for growth evaluation [27]. His estimated mature height was 60% of the average adult height, his predicted adult height was 137 cm, and there was no significant bone delay as the bone age was 7.5 years; within 2.5 standard deviations from chronological age. A patient’s bone age is considered delayed when it is >2.5 standard deviations from the mean.
It is difficult to assess pituitary growth hormone (GH) production, because GH secretion is pulsatile, and its surge occurs at the point of slow-wave EEG rhythms during phases 3 and 4 of sleep. Thus, measurement of random serum GH is therefore useless in diagnosing GH deficiency. Instead, physiological (such as exercise or fasting) and pharmacological (such as levodopa, clonidine, glucagon, propranolol, arginine, or insulin) stimuli are used to assess GH production [2,4,28]. Provocative GH testing has several pitfalls. However, to improve specificity, these provocative tests are performed in the fasting state and the child must fail 2 different tests to be diagnosed with growth hormone deficiency (GHD) [28].
There is no precise cutoff, but a peak GH concentration of 20 mU/L (10 ng/mL) excludes GHD, and GHD is confirmed when the peak is <7.5 mU/L (3 ng/mL). Partial GHD can be defined as a peak GH concentration of >7.5 mU/L but <15 mU/L (3–10 ng/mL) [29].
Our patient had a subnormal response to provocative growth hormone tests (peak GH 7.89 ng/mL) using 2 pharmacological stimuli (glucagon and clonidine). Although priming with gonadal steroids is recommended in children older than 8 years and classified as Tanner stage ≤2, we did not perform it in this case because the patient was prepubertal and his skeletal age was 7 years.
Globally, malnutrition is the most common cause of poor growth, with insufficient dietary intake often linked to food insecurity and poverty [4]. As presented in this case, the patient’s weight was far below the third percentile, although no apparent cause was identified, as his food intake was adequate.
However, within this clinical area, evidence for the efficacy of growth hormone treatment in WMS is lacking. Because the parents were very concerned about his stature, we offered him a trial of growth hormone treatment, and we will assess his response. The present article provides a comparison between the presented case and genetic variants in the ADAMTS10 gene, which have been reported in the previous literature to act as mutations.
The present case was similar in clinical features to previously reported cases, except for the disproportionate stature [15]. Also, here, we report the familial genetic variant; we describe a homozygous mutation in ADAMTS10 in the consanguineous Saudi family. This highlights the variability of the disease genotype and emphasizes the importance of offering genetic counseling, including preimplantation genetic diagnosis, to the families.
Conclusions
Weill-Marchesani Syndrome is caused by homozygous pathogenic variants in the ADAMTS10 gene, which should be considered in the differential diagnosis of children with short stature, whether it is proportional or disproportional short stature associated with ophthalmic disorders, especially in areas with a high prevalence of consanguinity such as Saudi Arabia. Management should involve a multidisciplinary team that includes several specialties. Glaucoma is the main presenting symptom and the most difficult to treat. Drug treatment and surgery are usually required to control intraocular pressure.
Footnotes
Conflict of Interest
None.
References:
- 1.Rosenbloom AL. Idiopathic short stature: Conundrums of definition and treatment. Int J Pediatr Endocrinol. 2009;2009:470378. doi: 10.1155/2009/470378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anil Bhansali AA, Parthan G, Gogate Y. Disorders of growth and development: Diagnosis and treatment. In: Anil Bhansali AA, Parthan G, Gogate Y, editors. Clinical Rounds in Endocrinology – Volume II – Pediatric Endocrinology. India: Springer, India; 2016. pp. 45–70. [Google Scholar]
- 3.Turan S, Bereket A, Omar A, et al. Upper segment/lower segment ratio and armspan-height difference in healthy Turkish children. Acta Paediatr. 2005;94(4):407–13. doi: 10.1111/j.1651-2227.2005.tb01909.x. [DOI] [PubMed] [Google Scholar]
- 4.Jorge AAL, Grimberg A, Dattani MT, Baron J. Section III: Childhood and adolescent endocrinology; Chapter 11: Disdorders of Chidhood Growth. In: Sperling M, editor. Sperling Pediatric Endocrinology. 5th ed. Philadelphia: Elsevier; 2020. pp. 299–356. [Google Scholar]
- 5.Dauber A, Rosenfeld RG, Hirschhorn JN. Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014;99(9):3080–92. doi: 10.1210/jc.2014-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jee YH, Baron J, Nilsson O. New developments in the genetic diagnosis of short stature. Curr Opin Pediatr. 2018;30(4):541–47. doi: 10.1097/MOP.0000000000000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bittles AH, Black ML. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci USA. 2010;107(Suppl. 1):1779–86. doi: 10.1073/pnas.0906079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Gazali L, Hamamy H, Al-Arrayad S. Genetic disorders in the Arab world. BMJ (Clinical research ed) 2006;333(7573):831–34. doi: 10.1136/bmj.38982.704931.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Mouzan MI, Al Salloum AA, Al Herbish AS, et al. Consanguinity and major genetic disorders in Saudi children: A community-based cross-sectional study. Ann Saudi Med. 2008;28(3):169–73. doi: 10.5144/0256-4947.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Hazmi MA, Al-Swailem AR, Warsy AS, et al. Consanguinity among the Saudi Arabian population. J Med Genet. 1995;32(8):623–26. doi: 10.1136/jmg.32.8.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beighton P, Beighton G, MARCHESANI . Oswald. In: Beighton G, editor. The Person Behind the Syndrome. London: Springer London; 1997. pp. 114–15. [Google Scholar]
- 12.Marchesani O. Brachydaktylie und angeborene kugellines als systemerkrankung. Klin Monatsbl Augenheilkd. 2020;103:392–406. [in German] [Google Scholar]
- 13.Weill G. Ectopie du cristallin et malformations générales. Ann Ocul. 1932:169. [in French] [Google Scholar]
- 14.Dagoneau N, Benoist-Lasselin C, Huber C, et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75(5):801–6. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faivre L, Dollfus H, Lyonnet S, et al. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet A. 2003;123a(2):204–7. doi: 10.1002/ajmg.a.20289. [DOI] [PubMed] [Google Scholar]
- 16.Faivre L, Gorlin RJ, Wirtz MK, et al. In frame fibrillin-1 gene deletion in auto-somal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40(1):34–36. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Autosomal Recessive Weill-Marchesani Syndrome [database on the Internet]. 2006.
- 18.Karoulias SZ, Beyens A, Balic Z, et al. A novel ADAMTS17 variant that causes Weill-Marchesani syndrome 4 alters fibrillin-1 and collagen type I deposition in the extracellular matrix. Matrix Biol. 2020;88:1–18. doi: 10.1016/j.matbio.2019.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Jethani J, Mishra A, Shetty S, et al. Weill-Marchesani syndrome associated with retinitis pigmentosa. Indian J Ophthalmol. 2007;55(2):142–43. doi: 10.4103/0301-4738.30711. [DOI] [PubMed] [Google Scholar]
- 20.Morales J, Al-Sharif L, Khalil DS, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85(5):558–68. doi: 10.1016/j.ajhg.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujiwara H, Takigawa Y, Ueno S, et al. Histology of the lens in the Weill-Marchesani syndrome. Br J Ophthalmol. 1990;74(10):631–34. doi: 10.1136/bjo.74.10.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karabiyik L. Airway management of a patient with Weill-Marchesani syndrome. J Clin Anesth. 2003;15(3):214–16. doi: 10.1016/s0952-8180(02)00506-8. [DOI] [PubMed] [Google Scholar]
- 23.Marzin P C-DV, Tsilou E. Weill-Marchesani syndrome. USNational Library of Medicine [Internet] 2013. pp. 1993–2021. [cited 2007 2007 Nov 1 [Updated 2020 Dec 10]]. https://www.ncbi.nlm.nih.gov/books/NBK1114/
- 24.Guo H, Wu X, Cai K, et al. Weill-Marchesani syndrome with advanced glaucoma and corneal endothelial dysfunction: A case report and literature review. BMC Ophthalmol. 2015;15:3. doi: 10.1186/1471-2415-15-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senthil S, Rao HL, Hoang NT, et al. Presenting features and treatment outcomes. J Glaucoma. 2014;23(4):262–67. doi: 10.1097/IJG.0b013e3182707437. [DOI] [PubMed] [Google Scholar]
- 26.Taylor JN. Weill-Marchesani syndrome complicated by secondary glaucoma. Case management with surgical lens extraction. Aust NZJ Ophthalmol. 1996;24(3):275–78. doi: 10.1111/j.1442-9071.1996.tb01592.x. [DOI] [PubMed] [Google Scholar]
- 27.Kelly A, Winer KK, Kalkwarf H, et al. Age-based reference ranges for annual height velocity in US children. J Clin Endocrinol Metab. 2014;99(6):2104–12. doi: 10.1210/jc.2013-4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicol LE, Allen DB, Czernichow P, et al. Chapter 2: Normal growth and growth disorders. In: Kappy MS, Allen DB, Geffner ME, editors. Pediatric practice endocrine. 2 ed. New York: McGraw-Hill Education; 2014. pp. 433–39. [Google Scholar]
- 29.Brook CGD, Brown RS. Chapter13: Tests and normal values in pediatric endocrinology. In: Brook CGD, Brown RS, editors. Handbook of Clinical Pediatric Endocrinology. USA: Blackwell Publishing; 2008. pp. 224–24. [Google Scholar]