

Abstract
Metacommunity theory provides a framework for how community patterns arise from processes across scales, which is relevant for understanding patterns in host-associated microbial assemblages. Microbial metacommunities may have important roles in host health through interactions with pathogens; however, it is unclear how pathogens affect host microbial metacommunities. Here, we studied relationships between a fungal pathogen and a host-associated microbial metacommunity. We hypothesized that a fungal pathogen of bats, Pseudogymnoascus destructans, correlates with a shift in metacommunity structure and changes in relationships between community composition, and factors shaping these assemblages, such as ecoregion. We sampled bat cutaneous microbial assemblages in the presence/absence of P. destructans and analyzed microbial metacommunity composition and relationships with structuring variables. Absence of P. destructans correlated with a metacommunity characterized by a common core microbial group that was lacking in disease positive bats. Additionally, P. destructans presence correlated with a change in the relationship between community structure and ecoregion. Our results suggest that the fungal pathogen intensifies local processes influencing a microbial metacommunity and highlights the importance of cutaneous microbial assemblages in host–pathogen interactions.
Subject terms: Community ecology, Ecology, Microbial ecology, Microbiome
Introduction
Elucidating how patterns of community structure relate to underlying structuring variables and processes of community assembly is a primary goal of community ecologists. Patterns observed at one scale of observation can be directly influenced by processes occurring at another scale1. For example, rescue effects describe the process by which species can persist in unfavorable local environments through dispersal from regional source populations2. Communities interacting between scales, including local and regional, form a spatial patchwork of taxa referred to as a metacommunity3.
Metacommunities are defined as groups of habitat patches, linked by species dispersal and interactions between taxa among these patches3,4. Both local and regional processes contribute to shaping metacommunity structure and the distribution of species across habitat patches3. By using pattern-based assessments, one can analyze species distributions along environmental and spatial gradients to diagnose metacommunity structure5,6.
Leibold and Mikkelson6 developed a framework to identify metacommunity patterns, called the Elements of Metacommunity Structure (EMS), which uses three metrics to describe metacommunity structures: coherence, turnover, and boundary clumping (Fig. 1). Coherence is measured as the number of embedded species absences from a site and describes the overall response of a community to an environmental or spatial gradient (Fig. 1a). Turnover is measured as the number of species replacements across samples. Boundary clumping describes clustering in species’ range boundaries and is a metric that defines how cohesive species ranges are across sites6. After determining the EMS, an idealized distributional pattern including hyperdispersed species loss, clumped species loss, evenly spaced, or Clementsian (Fig. 1b-e) can be used to describe metacommunity structure5,6. Clementsian structure (Fig. 1e) describes communities of species that have similar responses to environmental differences, resulting in discrete community boundaries5–7. Specifically, Clementsian metacommunities have positive coherence, turnover, and clumping, meaning there are less absences (positive coherence) but more frequent species replacements (positive turnover) than expected by chance alone. Additionally, Clementsian metacommunities have clumped species boundaries defined by positive boundary clumping. For example, Clementsian succession occurs when communities of species replace each other over time, with little overlap in community composition. Evenly spaced metacommunities (Fig. 1d) are similar to Clementsian, in that they have positive coherence and turnover, but they have hyperdispersed species boundaries as opposed to clumped species loss. Evenly spaced metacommunities still exhibit turnover, with species replacing each other across sites, however there are no distinct communities characteristic of Clementsian metacommunities. Nested patterns consist of less diverse assemblages making up subsets of more diverse communities5,6,8. Nested community structures result from positive coherence, but negative turnover, where species do not replace each other but rather, are lost from sites. For example, nested sites are made up of subsets of species from a much larger species pool. Nested metacommunities that exhibit positive clumping have clumped species loss (Fig. 1c) where species are lost from sites in groups. Those with negative clumping have hyperdispersed loss (Fig. 1b) where individual species are lost from sites. While EMS analyses provide descriptions of metacommunity structure, they do not reveal the variables responsible for such processes.
Figure 1.

The elements of metacommunity structure and their resulting patterns. Plus signs ( +) indicate a significantly positive relationship, whereas minus signs ( −) indicate a negative relationship. (a) metacommunity with checkerboard pattern. (b) nested metacommunity with hyperdispersed species loss. (c) nested metacommunity with clumped species loss. (d) evenly spaced metacommunity. (e) Clementsian metacommunity structure. (Modified from reference5 using the software Inkscape 1.0 www.inkscape.org).
Complimentary analyses are needed to elucidate the variables driving metacommunity structure. The influence of geographic distance on community structure is assessed using distance-decay models, which estimate the rate of species turnover along a gradient9. Positive relationships between community dissimilarity and geographic distance may indicate that species’ distributions are highly affected by dispersal limitation10, whereas, the lack of a relationship suggests that environmental filtering and species sorting may be more important for determining community structure11. Species sorting and environmental filtering emphasize the role of the local abiotic environment in determining what species can persist within an assemblage, resulting in assemblages correlating with local habitat factors, such as precipitation3. In addition, permutational multivariate methods are frequently implemented to understand the effects of environmental factors on community patterns. For instance, permutational models can distinguish differences in community composition and turnover across environmental variables, as well as the interactive effects of multiple environmental variables12. Interactive effects are especially important to consider when new structuring variables, such as invasive species or anthropogenic perturbations, are introduced to a metacommunity, as they might change the role of established structuring factors.
While macroorganismal metacommunities have been studied in some detail10,13,14, minimal work has focused on characterization of host-associated microbial metacommunities15–17. Understanding variation in structure of host-associated microbial metacommunities may be especially important given the role of host assemblages in pathogen defense18,19. For instance, the pathogenic fungus Pseudogymnoascus destructans, the causative agent of white-nose syndrome, was introduced into the United States in 2006, and is responsible for massive bat population declines20,21. Recently, declines have been shown to be highly variable across space, bat species, and time22. Tri-colored bats (Perimyotis subflavus) have shown recent population stabilizations possibly due in part to the presence of antifungal bacteria composing the cutaneous microbial assemblage22,23. Previous work has shown P. subflavus that are exposed to, but not invaded by P. destructans, have microbial assemblages enriched in antifungal bacterial taxa24. Determining the structure and drivers of bat cutaneous microbial metacommunities in relation to this fungal pathogen may improve our understanding of microbial metacommunity response to fungal invasion, as well as our understanding of assemblages that are resistant to fungal invasion.
Understanding how the presence of a fungal pathogen correlates with changes in both metacommunity structure and its relationship with structuring factors is likely important to understand the role of the host associated microbial assemblage in pathogen defense. The objectives of this study were to understand how the presence of a fungal pathogen correlates with the composition of a host-associated cutaneous microbial metacommunity and its structuring factors. We investigated the relationship between P. destructans and the cutaneous microbial metacommunity of hibernating P. subflavus across 48 sites in Tennessee, USA. We hypothesized that the presence of P. destructans would be correlated with a shift in 1) metacommunity structure and 2) relationships between structuring variables and community composition. We tested the first hypothesis using EMS, further informed by indicator operational taxonomic unit (OTU) and fungal pathogen load analyses. We tested the second hypothesis with distance-decay and permutational models to understand how spatial and environmental variables structure the bat cutaneous microbial assemblage in the presence/absence of a fungal pathogen.
Results
Of the 249 individuals of P. subflavus studied, quantitative PCR (qPCR) results indicated that there were 40 negative and 209 P. destructans positive bat individuals collected from 48 sites across three ecoregions (Interior, Ridge and Valley, and the South West Appalachians; Fig. 2a). All sites were determined to have at least one P. destructans positive bat. Post processing of high-throughput sequence data resulted in a mean read depth of 194,550 sequences per sample (14,899–2,968,637) reads and a total of 11,071 OTUs for P. destructans positive and 3370 OTUs for P. destructans negative bats.
Figure 2.

(a) Map of the study system, red dots indicate sample sites. Samples were collected across three Tennessee ecoregions (Interior Plateau in light green, South West Appalachians in red, and Ridge and Valley in olive green). Map produced using ArcGis 10.7.1. (https://desktop.arcgis.com/en/arcmap/) Copyright 1995–2018 Esri. All rights reserved. Published in the United States of America. Distance-decay relationships, comparing geographic distances between sites and (b) total beta diversity, (c) turnover, (d) nestedness, for P. destructans positive (dashed line and black dots) and P. destructans negative (solid line and grey dots) bats averaged by site. There is no significant relationship between geographic distance and community dissimilarity (GLM; SOR: z = 0.79, p > 0.05; SIM: z = 0.86, p > 0.05; SNE: z = 0.96, p > 0.05) or decay rates between disease states (p > 0.05) for any metric (ANOVA; SOR: p > 0.05, SIM: p > 0.05, SNE: p > 0.05). Graphs were produced using the ggplot2 package (version 3.3.2) in R25.
Metacommunity structure
Metacommunities for both P. destructans positive and negative bats showed positive coherence, with significantly (p ≤ 0.05) less embedded absences than expected based on null models (positive = 1,142,539 embedded absences, 1,376,131.98 ± 1943.23 expected; negative = 46,167 embedded absences, 52,651 ± 186.4 expected; Fig. 3, Table 1). The P. destructans positive metacommunity was characterized by significant positive turnover (p ≤ 0.05, 2.48e + 10 replacements; simulated mean 2.26e + 10 ± 2.25e + 08), while the P. destructans negative metacommunity had significant negative turnover (p ≤ 0.05, 42,579,240 replacements; 47,012,450 ± 1,005,206 expected replacements). Both P. destructans positive and negative metacommunities had significant clumping of species range boundaries (positive bats; Morisita’s index = 1.44, p ≤ 0.05; negative bats; Morisita’s index = 1.39, p ≤ 0.05). Together, these results indicate that the P. destructans positive metacommunity can be described as having a Clementsian structure (Fig. 1e), whereas, the P. destructans negative metacommunity had a nested structure with clumped species losses (Fig. 1c; ref.5).
Figure 3.

Site by species incidence matrix for OTUs on P. destructans positive/negative bats describing the actual metacommunity patterns. Black bars represent each OTUs (x-axis) range across bat samples (y-axis). EMS analysis suggests a Clementsian structure for P. destructans positive and a nested structure for P. destructans negative bat microbial metacommunities. Plots were produced using the Imagine function in the metacom package (version 1.5.3) in R26.
Table 1.
Results for the EMS analysis of bats across P. destructans status.
| P. destructans positive bats | ||
|---|---|---|
| Coherence | p value | |
| Absences | 1,142,539 | ≤ 0.0001 |
| Simulated mean | 1,376,131.9 (± 1943.2) | |
| Turnover | ||
| Turnover | 2.48 e + 10 | ≤ 0.0001 |
| Simulated mean | 2.26 e + 10 (± 2.25 e + 8) | |
| Boundary | ||
| Index | 1.44 | ≤ 0.0001 |
| P. destructans negative bats | ||
|---|---|---|
| Coherence | p value | |
| Absences | 46,167 | ≤ 0.0001 |
| Simulated mean | 52,651 (± 186.4) | |
| Turnover | ||
| Turnover | 42,579,240 | ≤ 0.0001 |
| Simulated mean | 47,012,450 (± 1,005,206) | |
| Boundary | ||
| Index | 1.39 | ≤ 0.0001 |
Results suggest Clementsian metacommunity structure for P. destructans positive bats, and a nested metacommunity structure for P. destructans negative bats5.
A total of 14 OTUs were identified as indicator taxa for P. destructans positive and 363 OTUs for negative bats (Supplemental Table). The group of indicator OTUs for the P. destructans negative bats represents the common taxa occurring across the individual microbial communities that contributed to the nestedness in metacommunity structure. OTUs indicative of P. destructans negative bats were significantly more abundant on P. destructans negative, relative to P. destructans positive bats (GLMM; z = − 62.84, p ≤ 0.05; Fig. 4a, Supplemental File 1E). Additionally, there was a significant negative relationship between log transformed fungal load and indicator taxa rarefied abundance (GLMM; z = − 10.78, p ≤ 0.05; Fig. 4b, Supplemental File 1E), with increased fungal load predictive of fewer indicator taxa. However, similar patterns were not found when analyzing the relationship between P. destructans load and the nested component (SNE) of averaged community dissimilarities (GLM; z = − 0.10, p > 0.05; Fig. 4c). Between site average community dissimilarity (SOR and SIM) was not related to the between site average difference in log transformed fungal load (GLM; SOR: z = 0.237, p > 0.05; SIM: z = 0.798, p > 0.05, Supplemental File 1E). When the compositional nature of the dataset was considered, patterns of indicator taxa abundance were identical to those observed with subsampled data (Supplemental File 1F).
Figure 4.
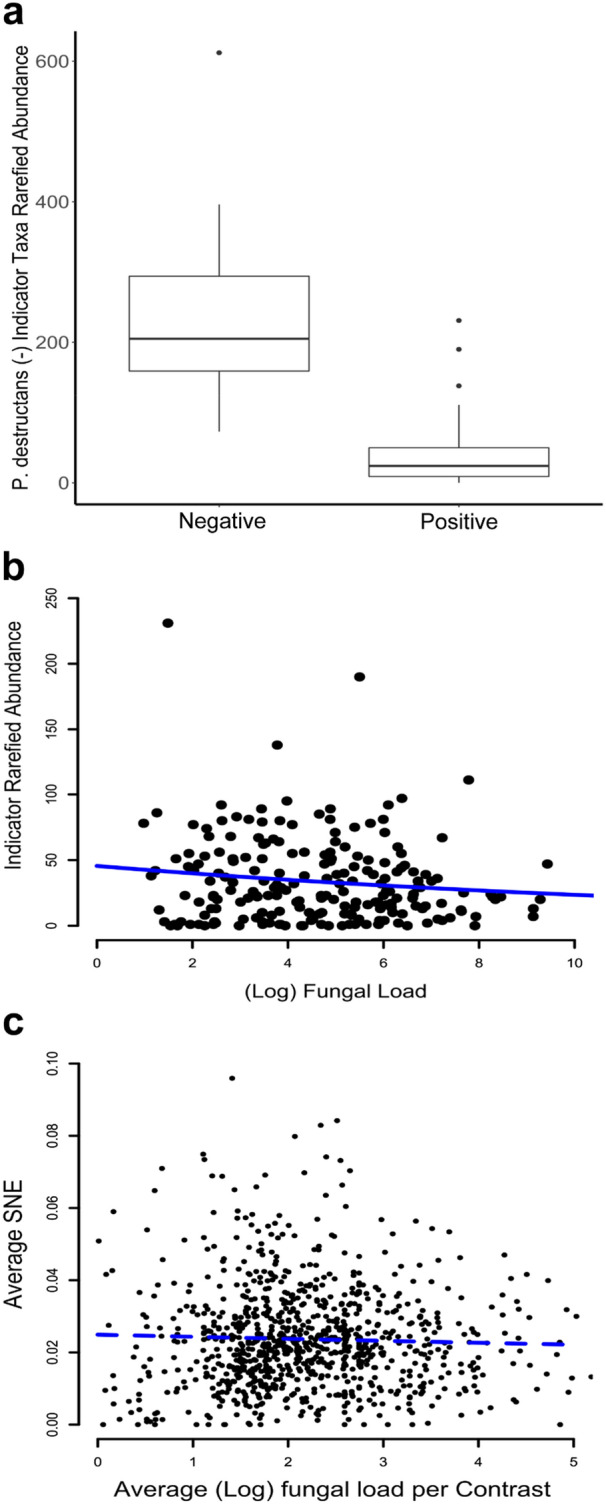
(a) Comparison of the rarefied abundance of P. destructans negative indicator taxa between P. destructans positive/negative samples. Indicator taxa are significantly more abundant within P. destructans negative samples (GLMM; z = − 62.84, p ≤ 0.05). (b) Comparison of the rarefied abundance of P. destructans negative indicator taxa by fungal load. There is a significantly negative relationship between indicator taxa rarefied abundance and amount of P. destructans present (GLMM; z = − 10.78, p ≤ 0.05). (c) Comparison of the (log) difference in average fungal load and average nestedness (SNE) of bats averaged by site. There is no significant relationship between similarity in fungal load and nestedness (GLM; z = − 0.10, p > 0.05). Graphs were produced using the ggplot2 package (version 3.3.2) in R25.
Relationship between community structure and structuring variables
All three measures of beta diversity lacked a distance-decay relationship (GLM; SOR: z = 0.79, p > 0.05; SIM: z = 0.86, p > 0.05; SNE: z = 0.96, p > 0.05; Fig. 2, Supplemental File 1E). There was no difference in the rate of decay between positive and negative bats for total beta diversity (ANOVA; SOR: p > 0.05, Fig. 2b), the turnover component of beta diversity (ANOVA; SIM: p > 0.05; Fig. 2c), or nestedness (ANOVA; SNE: p > 0.05 Fig. 2d). Multivariate dispersion was statistically different between P. destructans positive and negative bats for total beta diversity (betadisper; SOR: F1, 247 = 5.89, p ≤ 0.05, Supplemental File 1G). Interestingly, analyses of multivariate dispersion indicated that there was a significant interactive effect between P. destructans status and ecoregion for both total beta diversity and turnover (betadisper; SOR: F5,243 = 15.232, p ≤ 0.05; SIM: F5,243 = 8.646, p ≤ 0.05, Supplemental File 1G; Figs. 5a,b, 6). Post-hoc analysis of the interaction term for total beta diversity showed that dispersion was not different across ecoregions for P. destructans positive bats but varied for P. destructans negative bats (Fig. 5a). In general, dispersion was large in P. destructans positive bats, with negative bats within the Interior Plateau having significantly less dispersion (Fig. 5a), largely driven by a difference in the turnover component (Fig. 5b). PERMANOVA revealed that average community composition (multivariate centroids) differed between P. destructans status and ecoregion when analyzing both total dissimilarities and the turnover component, but not nestedness, and that these effects were independent of each other (Fig. 6, Table 2).
Figure 5.
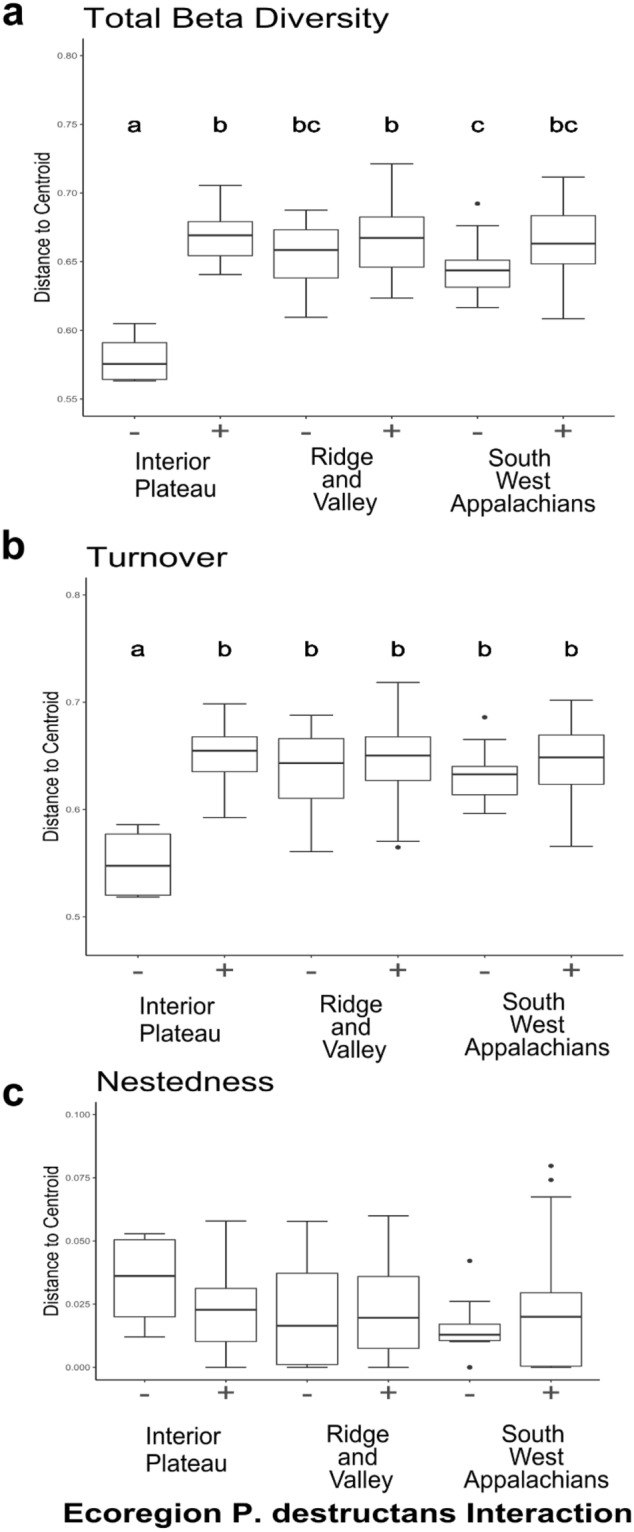
Betadisper analysis comparing beta diversity measured as multivariate dispersion across the interaction of ecoregion and P. destructans status, for (a) total beta diversity (SOR), (b) turnover (SIM), and (c) nestedness (SNE). Different lowercase letters indicate a significant difference (p ≤ 0.05) between groups, lowercase letters are missing from panel (c) due to lack of significant differences between groups. There is a significant interaction between P. destructans status and ecoregion for both total beta diversity as well as turnover (SOR: F5, 243 = 15.232, p ≤ 0.05; SIM: F5,243 = 8.646, p ≤ 0.05). Graphs were produced using the ggplot2 package (version 3.3.2) in R25.
Figure 6.

Non-metric multidimensional scaling ordination for. (a) P. destructans positive and (b) P. destructans negative bats across ecoregions (a) stress 0.17; (b) stress 0.14. There is a significant effect of P. destructans, year and ecoregion (p < 0.05), however, there is no significant interaction between P. destructans status and ecoregion. There is significant variation in dispersion across ecoregions for P. destructans negative bats (b). Graphs were produced using the ggplot2 package (version 3.3.2) in R25.
Table 2.
PERMANOVA results for total beta diversity (SOR), the turnover component of beta diversity (SIM), and the nested component of beta diversity (SNE).
| Df | Sums of squares | Mean squares | F test | R2 | p value | |
|---|---|---|---|---|---|---|
| Total beta diversity | ||||||
| P. destructans status | 1 | 0.746 | 0.746 | 1.7403 | 0.006 | 0.018* |
| Ecoregion | 2 | 1.427 | 0.713 | 1.664 | 0.012 | 0.011* |
| year | 1 | 0.795 | 0.794 | 1.8541 | 0.007 | 0.001*** |
| site | 45 | 24.357 | 0.541 | 1.2626 | 0.216 | 0.771 |
| P. destructans status:Ecoregion | 2 | 0.905 | 0.452 | 1.055 | 0.008 | 0.107 |
| Residuals | 197 | 84.454 | 0.428 | 0.749 | ||
| Total | 248 | 112.683 | 1 | |||
| Turnover | ||||||
| P. destructans status | 1 | 0.862 | 0.861 | 2.139 | 0.008 | 0.003** |
| Ecoregion | 2 | 1.591 | 0.795 | 1.975 | 0.014 | 0.033* |
| year | 1 | 0.605 | 0.605 | 1.502 | 0.005 | 0.012* |
| site | 45 | 23.712 | 0.526 | 1.308 | 0.221 | 0.72 |
| P. destructans status:Ecoregion | 2 | 0.876 | 0.437 | 1.087 | 0.008 | 0.22 |
| Residuals | 197 | 79.344 | 0.402 | 0.741 | ||
| Total | 248 | 106.99 | 1 | |||
| Nestedness | ||||||
| P. destructans status | 1 | − 0.007 | − 0.007 | − 7.92 | − 0.045 | 1 |
| Ecoregion | 2 | − 0.01 | − 0.005 | − 5.955 | − 0.067 | 0.347 |
| year | 1 | 0.009 | 0.009 | 11.111 | 0.063 | 0.105 |
| site | 45 | − 0.01 | − 0.0002 | − 0.269 | − 0.069 | 0.737 |
| P. destructans status:Ecoregion | 2 | − 0.0003 | − 0.0001 | − 0.22 | − 0.002 | 0.745 |
| Residuals | 197 | 0.174 | 0.0008 | 1.121 | ||
| Total | 248 | 0.156 | 1 | |||
There is a significant difference between P. destructans status, Ecoregion, and year for both SOR and SIM (p < 0.05).
Discussion
This study characterized the metacommunity structure of host microbial assemblages in the presence of a fungal pathogen. Overall, support was found for both of our hypotheses, we determined that the presence of P. destructans correlated with a change in cutaneous microbial metacommunity structure and loss of indicator OTUs from the core skin assemblage. Additionally, we found that the presence of P. destructans correlated with a change in relationship between community structure and an environmental structuring variable. These results suggest that the presence of P. destructans alters cutaneous microbial metacommunity structure by intensifying local processes, such as species sorting mechanisms or antagonistic species interactions.
The cutaneous microbial assemblages of P. destructans negative bats were characterized by a nested metacommunity structure with clumped species loss. The presence of numerous indicator taxa within negative bats further supported the inference of a nested metacommunity structure. Nested metacommunities have been observed in a variety of organisms, including Bryophytes14, macroinfauna27, and bats13, and likely represent variation in species-specific characteristics such as dispersal ability and tolerance to environmental conditions5. This is supported by previous work, which has shown the importance of host environment in shaping the cutaneous microbial assemblage12 and suggests that OTU-specific tolerances to host environmental conditions might drive the clumped OTU loss seen in P. destructans negative bats.
The core microbiome is defined as the taxonomic identity of the most common bacterial taxa within a system28. Pairing of the EMS and indicator analysis results for P. destructans positive bats suggested a loss of bacterial OTUs from the core microbiome. This loss might suggest alteration in community function and host defense against pathogens24,29,30. Nucleotide BLAST searches (based on ~ 250 bp region) revealed that indicator taxa identified in this study (Supplemental Table) were not genetically identical to cultured bacteria with in vitro anti-P. destructans activity identified in Grisnik et al.24. However, seven of 363 indicator taxa were identified to the same genera of anti-P. destructans bacteria identified previously24, including Nocardia, Rhodococcus, Streptomyces, Luteibacter, Lysobacter, and Sphingomonas. Each of these bacterial genera were detected on both positive and negative bats. Alternatively, bacteria with anti-fungal activity could have been gained to form the core microbiome of P. destructans negative bats, but additional work is required to mechanistically explain the correlational patterns found here. It is also important to acknowledge that approaches to understand assemblage function in vitro likely oversimplify complex inter- and intra-specific interactions at the community level, and further work to understand how bacterial function relates to fungal pathogenicity is warranted. The two most common phyla of indicator taxa were Proteobacteria (P. destructans positive n = 2 of 14, P. destructans negative n = 104 of 363) and Actinobacteria (P. destructans positive n = 12 of 14, P. destructans negative n = 95 of 363). Interestingly, the majority of indicator taxa for P. destructans positive bats are from the family Micrococcaceae (n = 11 of 14) which contains Micrococcus, a genus of well documented skin colonizers19. The more common indicator taxa of P. destructans negative bats include the phyla Planctomycetes (n = 35 of 363) and Acidobacteria (n = 28 of 363), both of which are commonly found in soils31,32. This might suggest increased interactions between cutaneous microbial assemblage and environmental microbes in the absence of P. destructans, however, manipulative studies would be required to address this question.
The cutaneous microbial assemblage of P. destructans positive bats exhibited turnover with boundaries clumped along an environmental gradient (Clementsian structure). Clementsian structure is known to be common in both free living14 and host-associated microbial assemblages15. Clementsian metacommunities can arise from antagonistic interactions preventing the coexistence of some taxa6,27. Interestingly, previous work showed an inverse relationship between P. destructans positive bats and bacteria that inhibited growth of P. destructans24, suggesting that antagonistic interactions might drive the shift to Clementsian metacommunity structure in microbial assemblages of positive bats.
The lack of a distance-decay relationship in microbial assemblages suggests either a lack of dispersal limitation or absence of species sorting mechanisms driving the assembly of bat cutaneous microbial assemblages. Since bat host environment (ecoregion) had a significant impact on average assemblage structure and there was no significant distance-decay relationship, we can conclude that dispersal limitation does not have a predominant role in the assembly of the cutaneous microbial assemblages of P. subflavus. Bacterial dispersal limitation is consistent with previous work that has shown a lack of population structure in Appalachian bat species33,34. This suggests that frequent roost switching and host dispersal may provide opportunities for microbial dispersal, and therefore, homogenization of bacterial assemblages across the region. Barriers to microbial dispersal between individual bats might be low, suggesting that the level of selection for microbial assemblage formation might be occurring at the colony level rather than the individual level35. Other studies have attributed environmental heterogeneity as the underlying driver of distance-decay relationships in microbial assemblages36–39. The lack of a distance-decay pattern driven by environmental heterogeneity could be due to the similarity of cave environments across our study system, as bats were sampled during the winter hibernation period, and not on a variety of summer/winter roost sites. Alternatively, variation within cave environments across the study system could result in a patchy distribution, rather than a geographically constrained gradient of environmental heterogeneity. The overall influence of host environment and species sorting mechanisms have been observed in the literature, as other studies have shown an influence of site on cutaneous microbial assemblage structure for a variety of host taxa12,40,41. Our results suggest the role of the host environment in shaping microbial communities through species sorting regardless of P. destructans status. The presence of P. destructans does not alter the rate (slope) of distance-decay in microbial assemblages across geographic space. In the context of community assembly, we found a lack of a P. destructans mediated change in dispersal limitation and/or species sorting in bacterial assemblage formation. Previous work has suggested an inverse pattern showing that as levels of disturbance increase, the rate of turnover within assemblages decreases, suggesting that disturbances can act as ecological filters42.
Results of permutational models indicated the role of the environment in shaping the bat cutaneous microbial assemblage. The PERMANOVA analysis indicates that the presence of P. destructans correlates with a difference in average community composition. Additionally, analysis of multivariate dispersion indicates that there is a significant interaction between P. destructans status and the environment, which suggests that the presence of P. destructans can alter the relationship between community structure and structuring variables, specifically the ecoregion where a bat is located. Of particular interest is the lack of significant differences in dispersion across ecoregions for P. destructans positive bats, despite significant differences for negative bats. In general, P. destructans positive bats have higher dispersion than negative bats. The presence of P. destructans within the Interior Plateau correlates with increased dispersion in the turnover component of beta diversity compared to negative bats within that ecoregion. When the analysis of multivariate dispersion is coupled with the lack of a distance-decay relationship, it suggests that local processes (such as antagonistic interactions or species sorting) may be stronger in the presence of P. destructans. Previous work has suggested that disturbance increases the importance of species sorting mechanisms through the filtering of species that cannot persist within the disturbed environment43.
We assessed metacommunity structure in cutaneous assemblages as they responded to the progression of fungal disease. There was no significant relationship between community similarity and fungal load, which serves as a proxy for disease progression. This suggests that the presence of P. destructans alone might be enough to alter the average microbial assemblage. Previous work has shown the opposite pattern with increasing fungal load being positively correlated with assemblage dissimilarity44. However, this study was done on salamanders infected with a chytrid fungus in a mesocosm setting, which could explain the conflicting results. Alternatively, due to the hierarchical structure of our data (bats nested within caves), the patterns we observed could be a result of site level averages rather than being representative of bacterial-fungal interactions on individual bats. While this is a valid concern, it has been shown that colony-level dynamics rather than individual identity better explain bat cutaneous microbial assemblage structure35, suggesting valid ecological patterns observed during this study. The presence of P. destructans may drive the formation of a unique assemblage through deterministic processes, but within a disease state category (P. destructans positive or negative) the variation might be best explained by stochastic or species-specific factors. While our study failed to find a relationship between assemblage-level similarity and fungal load, previous work has shown a correlation between pathogenic fungal load and specific bacterial taxa on bats24. Pseudogymnoascus destructans load may induce OTU-specific abundance responses but not influence overall assemblage similarity in terms of species presence. Future research at a fine-scale temporal resolution and quantifying microbial relative abundances is necessary to understand the effects of disease progression on microbial assemblage structure.
In order to understand interactions between host-associated microbial assemblages and pathogens it is important to take scale into account. The goal of this study was to elucidate if the presence of a fungal pathogen correlates with changes in metacommunity structure and the variables that structure these communities as they relate to assembly mechanisms. Results suggest that invasion of these communities by a fungal pathogen correlates with a shift in metacommunity structure likely driven by intrinsic factors that alter community assembly mechanisms. We hypothesize that the change in community structure is caused by increased strength of local processes within assemblages. Understanding how the host cutaneous microbial assemblage interacts with a fungal pathogen, specifically within a metacommunity context, is important for elucidating how this assemblage potentially protects hosts from pathogens. Future work should aim to better understand potential antagonistic interactions within microbial assemblages as they may help describe observed shifts in metacommunity structure and role in pathogen defense. Increased understanding of these antagonistic interactions has potential conservation implications, with recent interest in augmenting host cutaneous microbial assemblages with antifungal bacterial taxa, as a way to protect declining host populations24,30. Additionally, future experimental work in a controlled setting is required to confirm the community assembly processes structuring bat skin assemblages. Lastly, this study was conducted exclusively on bacterial communities, future work should incorporate other members of the host associated microbiome including viruses and fungi.
Methods
Sample collection
Swabs from 369 individuals of adult P. subflavus were collected during statewide surveys between December 2016 and March 2019 across 57 cave sites in Tennessee. After bioinformatics processing and quality control, 249 P. subflavus from 48 sites were statistically analyzed (see methods below and supplemental file 1D for metadata). Each bat had its cutaneous microbial assemblage sampled following the protocol outlined in Grisnik et al.24. Briefly we took five swab (sterile Puritan polyester tipped swabs, Puritan, Guilford Maine) strokes of each bat muzzle/ear and five from wings/fur while avoiding the mouth using one sterile swab per bat individual. Due to the conservation status of P. subflavus, when possible, bats were left hanging attached to their roost and swabbed without disturbing torpor. All samples were stored on ice in the field and permanently at − 20 °C until processing. This study was approved by the Tennessee Technological University Institutional Animal Care and Use Committee (TTU-16-17-003) and USFWS (2009-038). All methods were carried out following relevant guidelines and regulations. We isolated DNA from 369 bats using the Qiagen DNeasy PowerSoil HTP 96 kit following the manufacturer’s protocol. Each plate of 96 samples contained a single DNA extraction blank (n = 8 total blanks) to filter out kit-based contamination during bioinformatics processing and quantitative PCR reactions (see below). When setting up each DNA extraction plate and subsequent library preparation, the location of samples on each 96 well plate was randomized, in order to reduce biased effects of well-to-well contamination45. Extracted DNA was then used for molecular characterization of the microbial community, as well as, qPCR for the detection of P. destructans.
Characterization of microbial community
Each step of library preparation (DNA isolation, PCR setup, and post PCR processes) was separated into specific PCR cabinet hoods with designated pipettes to minimize environmental and/or cross-contamination. Pipettes were autoclaved, and UV crosslinked periodically throughout library preparation. Once isolated, DNA was concentrated, using an Eppendorf Vacufuge plus, to a final volume of ~ 25 µL. After concentration, PCR amplification and high-throughput sequencing was performed following a modified version of the Illumina 16S Metagenomic Sequencing Library Preparation protocol. Specifically, we targeted the V4 region of 16S rRNA marker using primers 806R/515F (ref.46). Each PCR reaction contained 12.5 µL MCLAB I-5 Hi-Fi taq mastermix, 1 µL of 806R (10 µM), 1 µL of 515F (10 µM), 5.5 µL PCR grade water, and 5 µL DNA template. PCR amplification was performed with an initial denaturation at 95 °C for 2 min, followed by 35 cycles of 98 °C for 10 s, 55 °C for 15 s, and 72 °C for 5 s, with a final extension cycle of 72 °C for 5 min. MAGBIO High-prep magnetic beads were used to remove primer/adapter dimers after amplicon PCR and indexing steps. Samples were quantified with a Promega Quantus Fluorometer then normalized, pooled at a 4 picomolar concentration, and loaded onto an Illumina MiSeq v2 flow cell. Sequencing was performed in eight separate runs each using a 500-cycle reagent kit (paired-end, 2 × 250 bp reads).
Quantitative PCR
To determine the presence or absence of P. destructans within a sample we followed the protocol outlined by Muller et al.47 to amplify the fungal intergenic spacer region (IGS). Each reaction was run in triplicate on an Agilent AriaMx Real-Time PCR system, and contained 5 µL 2 × Primetime MasterMix, 0.4 µL forward primer (20 µM), 0.4 µL reverse primer (20 µM), 0.1 µL probe (20 µM), 3.1 µL PCR grade water, and 1 µL sample DNA for a total of 10 µL per reaction. Thermocycling conditions included a 3-min activation step at 95 °C, then 50 cycles of 95 °C for 3 s and 60 °C for 30 s. Each plate included both a known concentration of synthetically made P. destructans DNA (gBlocks; Integrated DNA Technologies) to serve as a positive control and a no template negative control (run in triplicate) to account for within plate contamination. A positive sample was indicated by exponential amplification in triplicate with a Ct value of less than 4047,48. If samples did not test positive in triplicate, they were re-tested, and were considered positive if there was amplification in at least one of the three subsequent reactions49. In order to quantify P. destructans fungal load, qPCR reactions of a serial dilution of synthetic DNA was used to generate the standard curve equation y = − 0.2936x + 11.439, with x being the average Ct value for each sample run in triplicate, and y being the log DNA copy number.
Bioinformatic analysis
Amplicon sequencing reads were processed using mothur v1.42.1 (ref.50). A total of 48,442,995 raw data sequence reads were obtained from eight sequencing runs. Paired-end reads were assembled into contigs, and sequences containing homopolymers greater than 8 nucleotides or any ambiguous base calls were removed. We identified unique sequences and aligned them to the SILVA v123 bacterial reference database51. After alignment, sequences were trimmed to the V4 region and pre-clustered allowing for two-nucleotide differences between clusters. Chimera removal was then done using the vsearch function in mothur52. Sequences were classified into taxonomic lineages and reads identified as Archaea, Eukaryota, chloroplast, mitochondria, and unknown were removed. The cluster.split command in mothur was used to cluster sequences into operational taxonomic units at 97% similarity53. OTUs that appeared less than five times were considered rare and were removed from the dataset. Additionally, OTUs that were found within the DNA extraction blanks were also removed (n = 1669 OTUs). OTUs were selected as the focal taxonomic level rather than ASVs (amplicon sequence variants), as previous work has shown that there is negligible difference in ecological patterns observed when OTU or ASV data are analyzed54. In total 5,701,307 sequences (11.7%) passed all quality control steps. We compared final library sizes across all samples and found that they were significantly different (Kruskal–Wallis: χ2[2] = 83.98, p < 0.05). Therefore, the data were rarefied by subsampling each library at 1200 sequence reads55. Data were subsampled as previous work has shown that it is an effective way to account for variation in library size55. The final OTU × sample matrix included 268 samples of P. subflavus. Since we were interested in observing variation between P. destructans positive and negative bats over geographic distance we standardized the data so that geographic distances between sample sites were equal for P. destructans positive and negative P. subflavus. This resulted in a total of 249 P. subflavus (40 P. destructans negative and 209 positive) used for the statistical analysis described below. All mothur commands are included in the Supplemental File 1A for reproducibility purposes.
Statistical analyses
Previous work has shown that rare OTUs can skew the results of elements of metacommunity structure (EMS) analysis5. Prior to conducting analyses, all OTUs that summed to less than two were removed resulting in 12,603 OTUs in the complete OTU × sample matrix. All analyses were conducted in R 3.4.2 (ref.56) using α = 0.05 unless multiple comparisons were made, and thus Bonferroni adjusted.
We used the metacom package (version 1.5.3) in R26 to determine if the presence of P. destructans correlated with changes in metacommunity structure of cutaneous microbial assemblages as outlined in Leibold and Mikkelson6 and Presley et al.5 following the Elements of Metacommunity Structure (EMS) framework. We evaluated three EMS metrics (coherence, turnover, and boundary clumping) using a site-by-species presence/absence matrix to determine metacommunity structure. Coherence was assessed as the number of embedded species absences, or the number of gaps/interruptions in species distributions, within an ordinated community matrix (Fig. 1). The number of observed absences was compared to an expected number of absences determined through the formulation of a null distribution created from simulated matrices with 1000 iterations. Negative coherence describes a pattern of significantly more observed embedded absences than predicted by the null model, and a metacommunity perceived with a “checkerboard” appearance (Fig. 1a). A random metacommunity is identified when there is a non-significant difference between observed and expected embedded absences. Significantly less embedded absences indicate positive coherence, which is suggestive of species responding to a structuring gradient. The latter pattern requires further analysis of turnover and boundary clumping for more specific designation of metacommunity structure. Turnover, was assessed to describe the number of times a species is replaced by another between two sites. As with coherence, the number of turnover events observed is compared to the number of expected events using a null model prediction. If there are significantly more replacements than expected by chance, this represents positive turnover and signals a Clementsian (Fig. 1e) or evenly spaced (Fig. 1d) metacommunity structure. If there are significantly less replacements than expected, this represents negative turnover and signals a nested metacommunity structure. Boundary clumping was evaluated using Morisita’s index to describe how distinct blocks of species are clumped along a range boundary. A Morisita’s index significantly greater than one, indicates clumped species loss (positive turnover: Fig. 1e; negative turnover: Fig. 1c), whereas, an index value significantly fewer than one indicates evenly spaced, i.e. hyperdispersed, species loss (positive turnover: Fig. 1d; negative turnover: Fig. 1b). To assess significance for each EMS metric we used the default fixed-proportional null model (“r1”), 1000 permutations, and allowed for null matrices to have empty rows and columns26. EMS analysis was performed using the metacommunity function26 and analyses for P. destructans positive and negative samples were run separately. Metacommunity patterns were visualized using the Imagine function within the metacom package.
In order to further describe how P. destructans status correlated with changes in community structure we performed an indicator analysis using the multipatt function in R package indicspecies (version 1.7.9;57) on the presence-absence transformed OTU data table. We used a generalized linear mixed-effects model (GLMM) with the glmer function (package lme4;58) assuming a Poisson error structure, with site set as the random effect to account for nested data, to compare the rarefied abundance of significant indicator taxa of P. destructans negative bats across all samples. Additionally, we compared the amount of P. destructans present (number of copies using qPCR) to the rarefied abundance of indicator taxa using a GLMM assuming a Poisson error structure, with site set as the random effect. In order to explore the compositional nature of our dataset59, we repeated the above analyses following the pipeline outlined by Gloor et al.59 using a clr transformed dataset. Complete methods and code for this analysis are included in Supplemental File 1F.
To determine if fungal load influenced community dissimilarity, we converted fungal copy number to a distance matrix representing differences between samples using the dist function with Euclidean distances. This allowed for us to determine if bats having more similar fungal loads have more similar microbial assemblages. The resulting distance matrix was compared to total beta diversity (Sørensen dissimilarity: SOR), the turnover (Simpson dissimilarity: SIM), and nested (nestedness: SNE) components of total beta diversity (package betapart;60). To address the nested structure of the data, a dummy variable was created to describe the pairwise site level comparisons by grouping the samples by geographic distances into a categorical “site contrast” variable. Due to issues resulting in singular fit of mixed models, we then averaged both fungal load and beta diversity (SOR, SIM, and SNE) by the “site contrast” variable resulting in an average dissimilarity between two samples, thus removing the nested structure of the data (Supplemental File 1B). We then used a GLM (function glm) to compare average fungal load difference to average beta diversity metrics. The GLM was run assuming a binomial distribution with log transformed fungal load dissimilarity set as a fixed effect.
To assess how the presence of P. destructans correlated with differences in the rate of turnover and patterns of distance-decay, we compared total beta diversity (SOR), the turnover (SIM), and nestedness (SNE) components of Sørensen diversity across geographic distances. Pairwise geographic distances between samples were computed as the Euclidian distance between sample points using the dist function in the package vegan61. Similar to the analysis comparing assemblage dissimilarity to fungal load difference, beta diversity (SOR, SIM, and SNE) was averaged by site contrast, in order to remove the nested structure of the dataset, and to accommodate issues of singular fit in the mixed models. The relationship between average community dissimilarity and average geographic distance (distance-decay relationship) was determined using a generalized linear model (GLM). GLM was performed assuming a binomial distribution using the glm function with geographic distance, P. destructans status, and the interaction between these variables being set as fixed effects and a Bonferroni adjusted p-value of 0.016. The analysis was conducted using type II sum of squares with the Anova function in the package car62 to account for unequal sample sizes across groups.
To elucidate how environmental variables influenced beta diversity across P. destructans status we compared variation in beta diversity, measured as multivariate dispersion (function betadisper, package vegan), across P. destructans status, ecoregion (specifically ecoregion level 3, as delineated by the Environmental Protection Agency), and P. destructans × ecoregion interaction. Ecoregion was selected as the environmental variable as it represents a composite variable encompassing multiple fine scale environmental factors. A Tukey’s post hoc test was then used to determine pairwise differences between the groups of the interactive effect. We used permutational multivariate analysis of variance (PERMANOVA) stratified by site with 999 permutations using the adonis function (package vegan) on SOR, SIM, and SNE dissimilarity metrics to assess the influence of ecoregion on average assemblage similarity. Explanatory variables included ecoregion, P. destructans status, the P. destructans × ecoregion interaction, as well as year and site as covariates accounting for data structure. The PERMANOVA assumption of homogeneity of variance was violated, however previous work63 has shown that PERMANOVA is robust to violations of this assumption when the variable with the greater sample size has a larger variance, as seen with our data. All R code is included in the Supplemental File 1C for reproducibility purposes.
Supplementary Information
Acknowledgements
We would like to thank TWRA, TNC, and TVA for assistance with field work. This work was approved by IACUC TTU-16-17-003 and USFWS 2009-038. Funding and support for this research project was provided by Tennessee Wildlife Resources Agency as State Wildlife Grant 32801-00803 to DMW. We thank the four anonymous reviewers for their helpful comments to improve this manuscript.
Author contributions
M.G., J.G., and D.M.W. conceived the study and performed statistical analyses. M.G. completed all molecular work. M.G. and D.M.W ran bioinformatics. All authors wrote the manuscript.
Data availability
All sequence data were submitted to GenBank SRA under the accession number PRJNA691025. All mothur code and all R code has been made publicly accessible in the supplemental file.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-91118-1.
References
- 1.Levin SA. The problem of pattern and scale in ecology. Ecology. 1992;73:1943–1967. doi: 10.2307/1941447. [DOI] [Google Scholar]
- 2.Brown JH, Kodric-Brown A. Turnover rates in insular biogeography: Effect of immigration on extinction. Ecology. 1977;58:445–449. doi: 10.2307/1935620. [DOI] [Google Scholar]
- 3.Leibold MA, et al. The metacommunity concept: A framework for multi-scale community ecology. Ecol. Lett. 2004;7:601–613. doi: 10.1111/j.1461-0248.2004.00608.x. [DOI] [Google Scholar]
- 4.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Presley SJ, Higgins CL, Willig MR. A comprehensive framework for the evaluation of metacommunity structure. Oikos. 2010;119:908–917. doi: 10.1111/j.1600-0706.2010.18544.x. [DOI] [Google Scholar]
- 6.Leibold MA, Mikkelson GM. Coherence, species turnover, and boundary clumping: Elements of metacommunity structure. Oikos. 2002;97:237–250. doi: 10.1034/j.1600-0706.2002.970210.x. [DOI] [Google Scholar]
- 7.Clements FE. Plant Succession: An Analysis of the Development of Vegetation. Washington, DC: Carnegie Institution of Washington; 1916. [Google Scholar]
- 8.Patterson BD, Atmar W. Nested subsets and the structure of insular mammalian faunas and archipelagos. Biol. J. Linn. Soc. 1986;28:65–82. doi: 10.1111/j.1095-8312.1986.tb01749.x. [DOI] [Google Scholar]
- 9.Nekola JC, White PS. The distance decay of similarity in biogeography and ecology. J. Biogeogr. 1999;26:867–878. doi: 10.1046/j.1365-2699.1999.00305.x. [DOI] [Google Scholar]
- 10.Tornero I, et al. Dispersal mode and spatial extent influence distance-decay patterns in pond metacommunities. PLOS ONE. 2018;13:e0203119. doi: 10.1371/journal.pone.0203119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heino J. The importance of metacommunity ecology for environmental assessment research in the freshwater realm. Biol. Rev. 2013;88:166–178. doi: 10.1111/j.1469-185X.2012.00244.x. [DOI] [PubMed] [Google Scholar]
- 12.Walker DM, et al. Variability in snake skin microbial assemblages across spatial scales and disease states. ISME J. 2019;13:2209–2222. doi: 10.1038/s41396-019-0416-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Presley SJ, Cisneros LM, Patterson BD, Willig MR. Vertebrate metacommunity structure along an extensive elevational gradient in the tropics: A comparison of bats, rodents and birds. Glob. Ecol. Biogeogr. 2012;21:968–976. doi: 10.1111/j.1466-8238.2011.00738.x. [DOI] [Google Scholar]
- 14.Heino J, et al. Elements of metacommunity structure and community-environment relationships in stream organisms. Freshw. Biol. 2015;60:973–988. doi: 10.1111/fwb.12556. [DOI] [Google Scholar]
- 15.Hernández-Gómez O, Hoverman JT, Williams RN. Cutaneous microbial community variation across populations of eastern hellbenders (Cryptobranchus alleganiensis alleganiensis) Front. Microbiol. 2017;8:1379. doi: 10.3389/fmicb.2017.01379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilber MQ, Jani AJ, Mihaljevic JR, Briggs CJ. Fungal infection alters the selection, dispersal and drift processes structuring the amphibian skin microbiome. Ecol. Lett. 2020;23:88–98. doi: 10.1111/ele.13414. [DOI] [PubMed] [Google Scholar]
- 17.Brown JJ, et al. Metacommunity theory for transmission of heritable symbionts within insect communities. Ecol. Evol. 2020;10:1703–1721. doi: 10.1002/ece3.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belden LK, Harris RN. Infectious diseases in wildlife: The community ecology context. Front. Ecol. Environ. 2007;5:533–539. doi: 10.1890/060122. [DOI] [Google Scholar]
- 19.Grice EA, Segre JA. The skin microbiome. Nat. Rev. Microbiol. 2011;9:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blehert DS, et al. Bat white-nose syndrome: An emerging fungal pathogen? Science. 2009;323:227. doi: 10.1126/science.1163874. [DOI] [PubMed] [Google Scholar]
- 21.Frick WF, Puechmaille SJ, Willis CKR. White-nose syndrome in bats. In: Voigt CC, Kingston T, editors. Bats in the Anthropocene: Conservation of Bats in a Changing World. New York: Springer; 2016. pp. 245–262. [Google Scholar]
- 22.Langwig KE, et al. Resistance in persisting bat populations after white-nose syndrome invasion. Philos. Trans. R. Soc. B. 2017;372:20160044. doi: 10.1098/rstb.2016.0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langwig KE, et al. Sociality, density-dependence and microclimates determine the persistence of populations suffering from a novel fungal disease, white-nose syndrome. Ecol. Lett. 2012;15:1050–1057. doi: 10.1111/j.1461-0248.2012.01829.x. [DOI] [PubMed] [Google Scholar]
- 24.Grisnik M, et al. The cutaneous microbiota of bats has in vitro antifungal activity against the white nose pathogen. FEMS Microbiol. Ecol. 2020;96:fiz193. doi: 10.1093/femsex/fitz193. [DOI] [PubMed] [Google Scholar]
- 25.Wickham H. ggplot2: Elegant Graphics for Data Analysis. R package version 3.2.2. https://CRAN.R-project.org/package=ggplot2 (2020).
- 26.Dallas T. metacom: An R package for the analysis of metacommunity structure. Ecography. 2014;37:402–405. doi: 10.1111/j.1600-0587.2013.00695.x. [DOI] [Google Scholar]
- 27.Alves AT, Petsch DK, Barros F. Drivers of benthic metacommunity structure along tropical estuaries. Sci. Rep. 2020;10:1–12. doi: 10.1038/s41598-019-56847-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Risely A. Applying the core microbiome to understand host–microbe systems. J Anim. Ecol. 2020;89:1549–1558. doi: 10.1111/1365-2656.13229. [DOI] [PubMed] [Google Scholar]
- 29.Harris RN, et al. Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 2009;3:818–824. doi: 10.1038/ismej.2009.27. [DOI] [PubMed] [Google Scholar]
- 30.Lemieux-Labonté V, Simard A, Willis CK, Lapointe FJ. Enrichment of beneficial bacteria in the skin microbiota of bats persisting with white-nose syndrome. Microbiome. 2017;5:115. doi: 10.1186/s40168-017-0334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckley DH, Huangyutitham V, Nelson TA, Rumberger A, Thies JE. Diversity of Planctomycetes in soil in relation to soil history and environmental heterogeneity. Appl Environ Microbiol. 2006;72:4522–4531. doi: 10.1128/AEM.00149-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmermann J, Gonzalez JM, Saiz-Jimenez C, Ludwig W. Detection and phylogenetic relationships of highly diverse uncultured acidobacterial communities in altamira cave using 23s rRNA sequence analysis. Geomicrobiol. J. 2005;22:379–388. doi: 10.1080/01490450500248986. [DOI] [Google Scholar]
- 33.Wilder AP, Kunz TH, Sorenson MD. Population genetic structure of a common host predicts the spread of white-nose syndrome, an emerging infectious disease in bats. Mol. Ecol. 2015;24:5495–5506. doi: 10.1111/mec.13396. [DOI] [PubMed] [Google Scholar]
- 34.Martin, A. M. Historical Demography and Dispersal Patterns in the Eastern Pipistrelle Bat (Perimyotis subflavus). MS Thesis Grand Valley State University (2014).
- 35.Kolodny O, et al. Coordinated change at the colony level in fruit bat fur microbiomes through time. Nat Ecol. Evol. 2019;3:116–124. doi: 10.1038/s41559-018-0731-z. [DOI] [PubMed] [Google Scholar]
- 36.Fierer N, Jackson RB. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 2006;103:626–631. doi: 10.1073/pnas.0507535103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu L, Yang J, Yu Z, Wilkinson DM. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015;9:2068–2077. doi: 10.1038/ismej.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reche I, Pulido-Villena E, Morales-Baquero R, Casamayor EO. Does ecosystem size determine aquatic bacterial richness? Ecology. 2005;86:1715–1722. doi: 10.1890/04-1587. [DOI] [Google Scholar]
- 39.Hillebrand H, Watermann F, Karez R, Berninger UG. Differences in species richness patterns between unicellular and multicellular organisms. Oecologia. 2001;126:114–124. doi: 10.1007/s004420000492. [DOI] [PubMed] [Google Scholar]
- 40.Avena CV, et al. Deconstructing the bat skin microbiome: Influences of the host and the environment. Front. Microbiol. 2016;7:1–14. doi: 10.3389/fmicb.2016.01753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lemieux-Labonté V, Tromas N, Shapiro BJ, Lapointe FJ. Environment and host species shape the skin microbiome of captive neotropical bats. PeerJ. 2016;4:e2430. doi: 10.7717/peerj.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldenberg Vilar A, et al. Eutrophication decreases distance decay of similarity in diatom communities. Freshw. Biol. 2014;59:1522–1531. doi: 10.1111/fwb.12363. [DOI] [Google Scholar]
- 43.Chase JM. Drought mediates the importance of stochastic community assembly. Proc. Natl. Acad. Sci. U.S.A. 2007;104:17430–17434. doi: 10.1073/pnas.0704350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muletz-Wolz CR, Fleischer RC, Lips KR. Fungal disease and temperature alter skin microbiome structure in an experimental salamander system. Mol. Ecol. 2019;2:2917–3293. doi: 10.1111/mec.15122. [DOI] [PubMed] [Google Scholar]
- 45.Minich JJ, et al. Quantifying and understanding well-to-well contamination in microbiome research. MSystems. 2019;4:e00186–e219. doi: 10.1128/mSystems.00186-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caporaso JG, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller LK, et al. Bat white-nose syndrome: A real-time TaqMan polymerase chain reaction test targeting the intergenic spacer region of Geomyces destructans. Mycologia. 2013;105:253–259. doi: 10.3852/12-242. [DOI] [PubMed] [Google Scholar]
- 48.Janicki AF, et al. Efficacy of visual surveys for white-nose syndrome at bat hibernacula. PLoS ONE. 2015;10:e01333902015. doi: 10.1371/journal.pone.0133390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellison SL, English CA, Burns MJ, Keer JT. Routes to improving the reliability of low level DNA analysis using real-time PCR. BMC Biotechnol. 2006;6:33. doi: 10.1186/1472-6750-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schloss PD, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quast C, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012;41:590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schloss PD, Westcott SL. Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Appl. Environ. Microbiol. 2011;77:3219–3226. doi: 10.1128/AEM.02810-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Glassman, S.I., & Martiny, J.B. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. MSphere, 3, (2018). [DOI] [PMC free article] [PubMed]
- 55.Weiss S, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ (2020).
- 57.De Caceres, M., Jansen, F. & De Caceres, M.M. ‘indicspecies’. R package version 1.7.9. https://CRAN.R-project.org/package=indicspecies (2020).
- 58.Bates, D., Sarkar, D., Bates, M.D. & Matrix, L. The lme4 package. R package version 1–1.26. https://CRAN.R-project.org/package=lme4 (2020).
- 59.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: And this is not optional. Front Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baselga A, Orme CDL. betapart: An R package for the study of beta diversity. Methods Ecol. Evol. 2012;3:808–812. doi: 10.1111/j.2041-210X.2012.00224.x. [DOI] [Google Scholar]
- 61.Oksanen, J. et al. vegan: Community ecology package. R package version 2.5–2. https://CRAN.R-project.org/package=vegan (2019).
- 62.Fox, J. et al. ‘car’. R package version 2.1-4. https://CRAN.R-project.org/package=car (2016).
- 63.Anderson MJ, Walsh DC. PERMANOVA, ANOSIM, and the mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013;83:557–574. doi: 10.1890/12-2010.1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data were submitted to GenBank SRA under the accession number PRJNA691025. All mothur code and all R code has been made publicly accessible in the supplemental file.